You might also like
- CME 301 - Mass Transfer Differential Equations of Mass Transfer Example ProblemsDocument9 pagesCME 301 - Mass Transfer Differential Equations of Mass Transfer Example ProblemsNajmul Puda PappadamNo ratings yet
- Group TheoryDocument204 pagesGroup Theorysohel rehanNo ratings yet
- Quantum Mechanical Operators and Wavefunctions: "Well Behaved" Functions, Have The Following PropertiesDocument21 pagesQuantum Mechanical Operators and Wavefunctions: "Well Behaved" Functions, Have The Following Propertiessmiling personNo ratings yet
- Faddeev-Senjanovic Quantization of Supersymmetrical Electrodynamical SystemDocument7 pagesFaddeev-Senjanovic Quantization of Supersymmetrical Electrodynamical SystemgermanNo ratings yet
- 3 - Angular MomentumDocument7 pages3 - Angular MomentumJoe AchkarNo ratings yet
- A Spin Chain Primer: Rafael I. NepomechieDocument16 pagesA Spin Chain Primer: Rafael I. NepomechieMichael StoneNo ratings yet
- HW4Document3 pagesHW4TosinNo ratings yet
- 6.2 - The Linear Stark Effect - Physics LibreTextsDocument3 pages6.2 - The Linear Stark Effect - Physics LibreTextsKartikKumarNo ratings yet
- .GOD SEND Mass All ChothasDocument89 pages.GOD SEND Mass All ChothasSadia HasanNo ratings yet
- Che112f 2000 ExamDocument3 pagesChe112f 2000 Exammh sepahdarNo ratings yet
- Harmonic Oscillator, A, A, Fock Space, Identicle Particles, Bose/FermiDocument15 pagesHarmonic Oscillator, A, A, Fock Space, Identicle Particles, Bose/FermiJosé JiménezNo ratings yet
- The Simple Harmonic OscillatorDocument44 pagesThe Simple Harmonic OscillatorDebora PaskarinaNo ratings yet
- 2D NMR-6Document54 pages2D NMR-6lazarusjamison1No ratings yet
- 1706 05175Document11 pages1706 05175kaosanhumamNo ratings yet
- Computer Graphics CheatsheetDocument5 pagesComputer Graphics Cheatsheet강우현No ratings yet
- 7-3 Example 7-2Document5 pages7-3 Example 7-2Oum ChhayNoyNo ratings yet
- SUN AlgebraDocument1 pageSUN AlgebraCarlosSanMillanNo ratings yet
- Pres 2006Document17 pagesPres 2006csreyeroNo ratings yet
- Straight Line Iit Fee Adv.Document15 pagesStraight Line Iit Fee Adv.PaperNo ratings yet
- Coherent States: Phy851 Fall 2009Document16 pagesCoherent States: Phy851 Fall 2009Ravi KumarNo ratings yet
- University of Waterloo Chemistry 254: Thermodynamics Term Test 1 Friday 29 May 2009 12:30-1:20 PMDocument5 pagesUniversity of Waterloo Chemistry 254: Thermodynamics Term Test 1 Friday 29 May 2009 12:30-1:20 PMArvin DalisayNo ratings yet
- Chemistry EquilibriumDocument97 pagesChemistry EquilibriumoluwadamilreibrahimNo ratings yet
- ps3 PDFDocument3 pagesps3 PDFantonioNo ratings yet
- 7-2 Example 7-1Document4 pages7-2 Example 7-1Oum ChhayNoyNo ratings yet
- Lec5 Forward Kinematics-ExamplesDocument33 pagesLec5 Forward Kinematics-ExamplesbalkyderNo ratings yet
- Thermodynamics 2Document5 pagesThermodynamics 2Charlotte HooperNo ratings yet
- Test 1 RevDocument6 pagesTest 1 RevSAYAN BAGCHINo ratings yet
- 07-Absorption For HAP and VOCcontrolDocument118 pages07-Absorption For HAP and VOCcontrolTakeshi Tanohuye TanohuyeNo ratings yet
- Lecture 13. Dissipation of Gravity Waves: G V P V V T V VDocument15 pagesLecture 13. Dissipation of Gravity Waves: G V P V V T V VsaryNo ratings yet
- Undersaturated Oil-Gas Simulation - Impes Type Solution: o Os GS So oDocument9 pagesUndersaturated Oil-Gas Simulation - Impes Type Solution: o Os GS So oAmir MNo ratings yet
- Universidad Militar Nueva Granada Mecánica de Sólidos Taller # 2 Presentado Por: Daniel Alfonso Bernal Gómez Código: 5800943Document5 pagesUniversidad Militar Nueva Granada Mecánica de Sólidos Taller # 2 Presentado Por: Daniel Alfonso Bernal Gómez Código: 5800943Daniel BernalNo ratings yet
- An Algorithmists Toolkit Lecture 1Document5 pagesAn Algorithmists Toolkit Lecture 1Alex YuNo ratings yet
- Northwestern University Thesis Template 1Document20 pagesNorthwestern University Thesis Template 1ranisingh4760No ratings yet
- The Experimental Study of The Hydrodynamic Effects On Seismic Response of BridgesDocument11 pagesThe Experimental Study of The Hydrodynamic Effects On Seismic Response of Bridgesalexandrhc20No ratings yet
- Lecture 4: Use of Symmetry & Skewed SupportsDocument10 pagesLecture 4: Use of Symmetry & Skewed SupportsRamachandra ReddyNo ratings yet
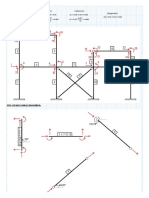
- DATOS: Unidades en Metros y Toneladas (Vigas) (Columnas) (Diagonales)Document13 pagesDATOS: Unidades en Metros y Toneladas (Vigas) (Columnas) (Diagonales)Yamil Edu Bravo TorresNo ratings yet
- Partial Molar PropertiesDocument5 pagesPartial Molar PropertiesRojo JohnNo ratings yet
- Commun Nonlinear Sci Numer Simulat: Maria V. Demina, Nikolay A. KudryashovDocument12 pagesCommun Nonlinear Sci Numer Simulat: Maria V. Demina, Nikolay A. Kudryashov饒英仿No ratings yet
- Simple Variation Method For The Hydrogen MoleculeDocument6 pagesSimple Variation Method For The Hydrogen Moleculetudor8sirbuNo ratings yet
- Ps1 SolutionDocument4 pagesPs1 Solutionshafin099No ratings yet
- 16.6 Half-Range Fourier Series For Functions Defined Over Range LDocument17 pages16.6 Half-Range Fourier Series For Functions Defined Over Range LwibisonoashariNo ratings yet
- Helium Atom, Approximate Methods: 22nd April 2008Document17 pagesHelium Atom, Approximate Methods: 22nd April 2008Julian David Henao EscobarNo ratings yet
- Assignment 2Document3 pagesAssignment 2Sanjay MauryaNo ratings yet
- Stat Mech HW6Document8 pagesStat Mech HW6andre123hadadNo ratings yet
- Materiel ModellingDocument14 pagesMateriel ModellingGanesh KumarNo ratings yet
- Super Chemistry Practice Final Exam CH222Document9 pagesSuper Chemistry Practice Final Exam CH222Nesrine LaradjiNo ratings yet
- 4.1. Stoichiometry SummaryDocument3 pages4.1. Stoichiometry SummaryWilliam TsuiNo ratings yet
- Producción de FormaldehídoDocument4 pagesProducción de FormaldehídoAstrid Altamirano LuisNo ratings yet
- Chemical Equilibrium Part III-1 PDFDocument6 pagesChemical Equilibrium Part III-1 PDFEstivenson Vasquez CNo ratings yet
- 49theoreticaltour1answer Eng PDFDocument9 pages49theoreticaltour1answer Eng PDFRay TanNo ratings yet
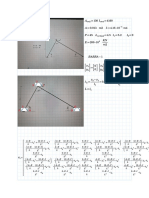
- Ejercicio #1 Corregido Practica PorticosDocument5 pagesEjercicio #1 Corregido Practica PorticosRODRIGO MARIO OROZA HERBASNo ratings yet
- Fulltext PDFDocument10 pagesFulltext PDFMuhammad FarooqNo ratings yet
- 1 Lewis Structures Part IDocument10 pages1 Lewis Structures Part IresodfortoNo ratings yet
- Writing and Balancing Chemical Equations: Equation. Consider As An Example The Reaction Between One Methane Molecule (CHDocument5 pagesWriting and Balancing Chemical Equations: Equation. Consider As An Example The Reaction Between One Methane Molecule (CHMegan CabahugNo ratings yet
- Week 8Document6 pagesWeek 8vivaline AchiengNo ratings yet
- Solución Parcial Mecánica de FluidosDocument5 pagesSolución Parcial Mecánica de FluidosIñigoNo ratings yet
- Rates of Chemical ReactionDocument48 pagesRates of Chemical ReactionAlexis Justin YapNo ratings yet
- Exercise 4 Outlet Temperature and Composition of A WGS ReactorDocument7 pagesExercise 4 Outlet Temperature and Composition of A WGS ReactorMiguelCardonaSalazarNo ratings yet
- Atomic Physics 12 PagesDocument12 pagesAtomic Physics 12 Pagessadam hussainNo ratings yet
- Ope¦rateurs maximaux monotones et semi-groupes de contractions dans les espaces de HilbertFrom EverandOpe¦rateurs maximaux monotones et semi-groupes de contractions dans les espaces de HilbertNo ratings yet
- Annexure-74. (B.sc. (Hons) Maths (REVISED)Document86 pagesAnnexure-74. (B.sc. (Hons) Maths (REVISED)hmingthansangiNo ratings yet
- Varimax RotationDocument47 pagesVarimax RotationpriyankaindoriaNo ratings yet
- Tutorial PDFDocument81 pagesTutorial PDFFilipe SaboiaNo ratings yet
- IEOR 160 Partial Lecture Notes 2017Document33 pagesIEOR 160 Partial Lecture Notes 2017Nate ArmstrongNo ratings yet
- Identification of Hydrological Neighborhoods Using Canonical Correlation AnalysisDocument19 pagesIdentification of Hydrological Neighborhoods Using Canonical Correlation AnalysisNibeditaNo ratings yet
- Numerical Methods 2 Marks PDFDocument61 pagesNumerical Methods 2 Marks PDFHarshithaNo ratings yet
- Briot Bouquet's Theorem High DimensionDocument18 pagesBriot Bouquet's Theorem High DimensionCarlos Antonio SalazarNo ratings yet
- Math Sport 2015 ProceedingsDocument223 pagesMath Sport 2015 ProceedingsDaniel GórkaNo ratings yet
- Slide Mode ControlDocument215 pagesSlide Mode ControlMaryamRazzaqNo ratings yet
- Math 1104Document4 pagesMath 1104Sultan Al-ZaabiNo ratings yet
- Spurious Modes in Two-Dimensional Isoparametric ElementsDocument13 pagesSpurious Modes in Two-Dimensional Isoparametric ElementskramaseshanNo ratings yet
- Role of Inter-Hemispheric ConnectionsDocument10 pagesRole of Inter-Hemispheric ConnectionsValentina SotoNo ratings yet
- Complex Networks: Topology and DynamicsDocument129 pagesComplex Networks: Topology and DynamicsPrem PanigrahiNo ratings yet
- PDFDocument238 pagesPDFolgajoylabajoNo ratings yet
- Full Download pdf of (eBook PDF) Biocalculus: Calculus, Probability, and Statistics for the Life Sciences all chapterDocument43 pagesFull Download pdf of (eBook PDF) Biocalculus: Calculus, Probability, and Statistics for the Life Sciences all chapteranichborlon100% (4)
- MentaryDocument3 pagesMentary曹明睿No ratings yet
- Factor Analysis PCA PDFDocument32 pagesFactor Analysis PCA PDFPrithvirajNo ratings yet
- Diagonalization MatrixDocument30 pagesDiagonalization Matrixkhatua.deb87No ratings yet
- Labour ProductivityDocument23 pagesLabour ProductivityAlaaGaballaNo ratings yet
- Implicit Analysis Intro 02-2011Document50 pagesImplicit Analysis Intro 02-2011Jhony GolombieskiNo ratings yet
- Matrix Structural Analysis 2nd EditionDocument483 pagesMatrix Structural Analysis 2nd EditionJaime Brown100% (17)
- Eigenfunctions of Discrete Time LTI SystemsDocument2 pagesEigenfunctions of Discrete Time LTI SystemsKrsto ProrokovicNo ratings yet
- Ma580 BookDocument101 pagesMa580 BookJosephNo ratings yet
- ECTS Guide 2011 Final2-1-1Document81 pagesECTS Guide 2011 Final2-1-1Lia TouskaNo ratings yet
- R19 (20-21 Admitted) FinalDocument45 pagesR19 (20-21 Admitted) FinalBaskar KNo ratings yet
- Lecture Notes For ECON 631Document556 pagesLecture Notes For ECON 631Zen OrtegaNo ratings yet
- Online Workshop On Mathematical Sciences For Csir-Net/Set/GateDocument95 pagesOnline Workshop On Mathematical Sciences For Csir-Net/Set/GateJayshri HuddarNo ratings yet
- Questions & Answers On State Variable Analysis and DesignDocument23 pagesQuestions & Answers On State Variable Analysis and Designkibrom atsbha100% (1)
- MathDocument22 pagesMathHeryVandoroNo ratings yet