You might also like
- Near Infrared Spectroscopy in Food AnalysisDocument14 pagesNear Infrared Spectroscopy in Food AnalysisHridyesh Pandey33% (3)
- Vacuum Engineering Calculations, Formulas, and Solved ExercisesFrom EverandVacuum Engineering Calculations, Formulas, and Solved ExercisesRating: 4.5 out of 5 stars4.5/5 (2)
- Method Report for GPT- GISAN AssayDocument1 pageMethod Report for GPT- GISAN AssayHussein N. FarhatNo ratings yet
- More NMR Problems 13a CetakDocument33 pagesMore NMR Problems 13a CetakRona Prima LarasatiNo ratings yet
- Spectroscopy: Ass Spectrometry Agnetic Spin ResonanceDocument30 pagesSpectroscopy: Ass Spectrometry Agnetic Spin ResonanceLisbeth Roos RoosNo ratings yet
- CH-103-4th LectureDocument14 pagesCH-103-4th LectureDhruv DhirawaniNo ratings yet
- UV-Visible Spectrophotometric Method and Validation of Organic CompoundsDocument4 pagesUV-Visible Spectrophotometric Method and Validation of Organic Compoundsianatul khafidlahNo ratings yet
- Refractive Index Enhancement With Vanishing Absorption in An Atomic VaporDocument11 pagesRefractive Index Enhancement With Vanishing Absorption in An Atomic VaporHebert BritoNo ratings yet
- Measurement of dynamic light scattering intensity in hydrogelsDocument16 pagesMeasurement of dynamic light scattering intensity in hydrogelsEuwan Tyrone PriasNo ratings yet
- Topic 20 NotesDocument21 pagesTopic 20 NotesAmmaarah PatelNo ratings yet
- "If It Were Done... Then Twere Well It Were Done Quickly": Chemical KineticsDocument14 pages"If It Were Done... Then Twere Well It Were Done Quickly": Chemical KineticsWoshi WoNo ratings yet
- UV - Visible Spectroscopy in Physical ChemistryDocument4 pagesUV - Visible Spectroscopy in Physical ChemistryMahmood Mohammed AliNo ratings yet
- Methods For Experimental Determination of Diffusion Current in PolarographyDocument5 pagesMethods For Experimental Determination of Diffusion Current in PolarographySmruthi SuvarnaNo ratings yet
- B.SC - Ii Paper-B (Optics and Lasers) : Submitted by Dr. Sarvpreet Kaur Assistant Professor PGGCG-11, ChandigarhDocument27 pagesB.SC - Ii Paper-B (Optics and Lasers) : Submitted by Dr. Sarvpreet Kaur Assistant Professor PGGCG-11, Chandigarhsuresh_c_pattar673No ratings yet
- Chapter - 2 Instrumental Methods of AnalysisDocument30 pagesChapter - 2 Instrumental Methods of Analysisdivya chouhanNo ratings yet
- Measurement of Refractive IndexDocument10 pagesMeasurement of Refractive IndexKarl Roderno100% (1)
- Module 6: Reaction Kinetics and Dynamics Lecture 27: Experimental Methods in Chemical KineticsDocument6 pagesModule 6: Reaction Kinetics and Dynamics Lecture 27: Experimental Methods in Chemical KineticsHamit RanaNo ratings yet
- Molecular Spectroscopy 2022Document120 pagesMolecular Spectroscopy 2022Lesedi mmabatho MashabelaNo ratings yet
- Nd: YAG Laser Document AnalysisDocument43 pagesNd: YAG Laser Document AnalysisMi Sinziana100% (1)
- Physical Lec 10-14Document13 pagesPhysical Lec 10-14rupayandaripaNo ratings yet
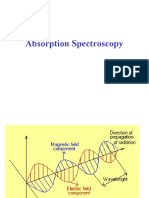
- Absorption SpectrosDocument55 pagesAbsorption SpectrosMahalakshmi SahasranamanNo ratings yet
- Principles of Gamma-ray Spectroscopy and Nuclear Forensics ApplicationsDocument4 pagesPrinciples of Gamma-ray Spectroscopy and Nuclear Forensics ApplicationsMohammed Khaja Moinuddin SiddiquiNo ratings yet
- DLS - Basic Theory & ApplicationDocument6 pagesDLS - Basic Theory & ApplicationFábio Pohhi KrahôNo ratings yet
- General RadioactivityDocument24 pagesGeneral RadioactivitytounessiNo ratings yet
- Testing Conductivity Detectors Used in Liquid and Ion ChromatographyDocument6 pagesTesting Conductivity Detectors Used in Liquid and Ion ChromatographyLuigi HernándezNo ratings yet
- QC 1 5Document13 pagesQC 1 5Isabel EsquijoNo ratings yet
- Absorption SpectrosDocument55 pagesAbsorption SpectrosfayvourajNo ratings yet
- MergeResult 2024 02 12 10 11 52Document69 pagesMergeResult 2024 02 12 10 11 52antarcia00No ratings yet
- Photocatalytic ReactorsDocument8 pagesPhotocatalytic ReactorsJoaquin LanderosNo ratings yet
- Notes SpectraDocument8 pagesNotes SpectraSUDIPTA SADHUKHANNo ratings yet
- E 1511 - 93 - Rte1mtetotmDocument5 pagesE 1511 - 93 - Rte1mtetotmOh No PotatoNo ratings yet
- Dynamic Light Scattering: Applications To Food Systems: D. G. Dalgleish and F. R. Hallett#Document13 pagesDynamic Light Scattering: Applications To Food Systems: D. G. Dalgleish and F. R. Hallett#Javier carretero mendozaNo ratings yet
- Light Scattering by Polymer Solutions: Light Waves - A Brief ReviewDocument23 pagesLight Scattering by Polymer Solutions: Light Waves - A Brief ReviewYashanshu GautamNo ratings yet
- Spectroscopy NotesDocument19 pagesSpectroscopy NotesNeerajNo ratings yet
- Laboratory Report - Module 2 (Refractometry)Document37 pagesLaboratory Report - Module 2 (Refractometry)Jeremy Kyle Edson AustriaNo ratings yet
- Revised Preliminary Introduction of SpectrosDocument29 pagesRevised Preliminary Introduction of SpectrosRevathiNo ratings yet
- URIECA Chemistry 5.35 Module 2: Synthesis of Coordination Compounds and KineticsDocument19 pagesURIECA Chemistry 5.35 Module 2: Synthesis of Coordination Compounds and KineticsFabian MelinaoNo ratings yet
- Dipole MomentDocument12 pagesDipole MomentSujalNo ratings yet
- Us1817 GratingSpectrometerDocument8 pagesUs1817 GratingSpectrometerAnonymous McruKTclNo ratings yet
- Determinising Shaping of Single Photon Temporal Wave Function G Rempe PRL 2019 Paper ID Quantum PM PhysRevLett.123.133602Document5 pagesDeterminising Shaping of Single Photon Temporal Wave Function G Rempe PRL 2019 Paper ID Quantum PM PhysRevLett.123.133602Raktim HalderNo ratings yet
- Module 1 Notes 1chem Mescenotes - inDocument63 pagesModule 1 Notes 1chem Mescenotes - inHafizNo ratings yet
- EXPERIMENT 8. Monolayer Characterization: Contact Angles, Reflection Infrared Spectroscopy, and EllipsometryDocument9 pagesEXPERIMENT 8. Monolayer Characterization: Contact Angles, Reflection Infrared Spectroscopy, and EllipsometryavniNo ratings yet
- Saturated Absorption Spectroscopy Experiment (SASDocument26 pagesSaturated Absorption Spectroscopy Experiment (SASPrakash ShanbogNo ratings yet
- Absorption / Transmission / Reflection SpectrosDocument6 pagesAbsorption / Transmission / Reflection SpectrosArmin Anwar ArminNo ratings yet
- Definitions and Concepts: 2.1 Stoichiometric CoefficientsDocument9 pagesDefinitions and Concepts: 2.1 Stoichiometric CoefficientsĐạt DokonNo ratings yet
- Amperometry: Working PrincipleDocument10 pagesAmperometry: Working PrincipleAbdulbar kelilNo ratings yet
- Circular Dichroism of Protein: PC3267 Updated in Jan. 2007Document7 pagesCircular Dichroism of Protein: PC3267 Updated in Jan. 2007Chellam Gayathri SubashNo ratings yet
- Absorbance and TransmittanceDocument38 pagesAbsorbance and TransmittanceHemchandra K Mahajan0% (1)
- Visible and Ultraviolet SpectrophotometryDocument41 pagesVisible and Ultraviolet SpectrophotometryalaahamadeinNo ratings yet
- G. Gallot Et Al - Non-Monotonic Decay of Transient Infrared Absorption in Dilute HDO/D2O SolutionsDocument5 pagesG. Gallot Et Al - Non-Monotonic Decay of Transient Infrared Absorption in Dilute HDO/D2O SolutionsMddl2aNo ratings yet
- SPECTROPHOTOMETER Practical Handout 2nd Year MBBSDocument7 pagesSPECTROPHOTOMETER Practical Handout 2nd Year MBBSIMDCBiochem100% (1)
- Lecture (4) : Reaction RatesDocument21 pagesLecture (4) : Reaction RatesAhmed AbdullaNo ratings yet
- Detectors - Evaporative Light ScatteringDocument6 pagesDetectors - Evaporative Light ScatteringgdgdfgNo ratings yet
- SpectrumDocument6 pagesSpectrumDarku CrowwNo ratings yet
- Infrared Spectroscopy Peak InterpretationDocument10 pagesInfrared Spectroscopy Peak Interpretationfouad elferdiNo ratings yet
- Infrared Spectroscopy of Chemical CompoundsDocument19 pagesInfrared Spectroscopy of Chemical CompoundsashenafiNo ratings yet
- Dispersion de Luz, PM PolimerosDocument19 pagesDispersion de Luz, PM PolimerosCarol ChagollaNo ratings yet
- NONLINEAR SPECTROSCOPYDocument43 pagesNONLINEAR SPECTROSCOPYThien Phu Nguyen NguyenNo ratings yet
- Alpha, Beta, and Gamma RadiationDocument5 pagesAlpha, Beta, and Gamma RadiationMalaya Kumar BhoiNo ratings yet
- Name of Student: Refractive IndexDocument4 pagesName of Student: Refractive IndexAddan JavidNo ratings yet
- Photocatalytic Reactors: M. Bouchy and O. ZahraaDocument8 pagesPhotocatalytic Reactors: M. Bouchy and O. Zahraavishal kumarNo ratings yet
- Macromolecular Microsymposium — 16: Main Lectures Presented at the Sixteenth Microsymposium on Macromolecules (Advances in Scattering Methods), Prague, 12 - 16 July 1976From EverandMacromolecular Microsymposium — 16: Main Lectures Presented at the Sixteenth Microsymposium on Macromolecules (Advances in Scattering Methods), Prague, 12 - 16 July 1976B. SedláčekNo ratings yet
- Chapter 2 EssayDocument4 pagesChapter 2 EssayarjunNo ratings yet
- Ujikom - Sop Spektronik 20 PDFDocument2 pagesUjikom - Sop Spektronik 20 PDFArmyAdisNo ratings yet
- App Note 323 ThermoDocument8 pagesApp Note 323 ThermoLoreto VillegasNo ratings yet
- Magnify Microscope ExperimentsDocument11 pagesMagnify Microscope ExperimentsozlemNo ratings yet
- Macroram: Remote Raman Measurements Using An External ProbeDocument2 pagesMacroram: Remote Raman Measurements Using An External ProbechiralicNo ratings yet
- PFGE Process Infographic-HDocument1 pagePFGE Process Infographic-Hqonita imma irfaniNo ratings yet
- Weizo DSRDocument1 pageWeizo DSRAshish SharmaNo ratings yet
- Types and Range of Applications For Liquid ChromatographyDocument24 pagesTypes and Range of Applications For Liquid ChromatographyHưng Trần VănNo ratings yet
- Dna ProfilingDocument19 pagesDna ProfilingAaron Janapin MirandaNo ratings yet
- T3 Rapid Quantitative Test COA - F2311630AADDocument1 pageT3 Rapid Quantitative Test COA - F2311630AADg64bt8rqdwNo ratings yet
- DNA Extraction Lab ReportDocument7 pagesDNA Extraction Lab ReportNazurah IbrahimNo ratings yet
- Informes de LaboratorioDocument17 pagesInformes de Laboratoriojessica vanessaNo ratings yet
- Practice Problem Set 7 Applications of UV Vis Absorption Spectroscopy9Document6 pagesPractice Problem Set 7 Applications of UV Vis Absorption Spectroscopy9Edna Lip AnerNo ratings yet
- What Is Flow CytometryDocument8 pagesWhat Is Flow CytometryRuksanurNo ratings yet
- VCE Chemistry Unit 3 2013 Chapter 7 - UV-Visible SpectrosDocument33 pagesVCE Chemistry Unit 3 2013 Chapter 7 - UV-Visible SpectrosLyn JiangNo ratings yet
- Priyanka Anjali (Medico) : Department of MicrobiologyDocument26 pagesPriyanka Anjali (Medico) : Department of Microbiologytummalapalli venkateswara raoNo ratings yet
- Mass Spectrometer HistoryDocument3 pagesMass Spectrometer HistorygvikrantNo ratings yet
- InstrumentalDocument46 pagesInstrumentalBetül MaşlakNo ratings yet
- Chromatography Experiment: By: Veda Purnitha S2Document10 pagesChromatography Experiment: By: Veda Purnitha S2Veda PurnithaNo ratings yet
- Fluorescence Spectroscopy and Imaging: Basic Principles and Sources of ContrastDocument78 pagesFluorescence Spectroscopy and Imaging: Basic Principles and Sources of Contrastraul reyesNo ratings yet
- Near-infrared spectroscopy data transferDocument5 pagesNear-infrared spectroscopy data transferhhNo ratings yet
- Analysis of Green Tea Extracts (Catechins) by HPLC-UVDocument2 pagesAnalysis of Green Tea Extracts (Catechins) by HPLC-UVmunmun islamNo ratings yet
- FullDocument542 pagesFullSoumajit DasNo ratings yet
- SEM Principles and ApplicationsDocument46 pagesSEM Principles and ApplicationsElkin ZapataNo ratings yet
- Pressure Source For Flash Chromatography: As A A To and As 4Document1 pagePressure Source For Flash Chromatography: As A A To and As 4NgọcKhánhÔngNo ratings yet
- Biochem Lab Report Experiment 1Document5 pagesBiochem Lab Report Experiment 1marymcauley6No ratings yet
- Instrumentation of Ftir and Its Herbal ApplicationsDocument8 pagesInstrumentation of Ftir and Its Herbal ApplicationsMurugesan JeevaNo ratings yet
- Fiche Technique - Dino-Lite - Am4515zt4Document3 pagesFiche Technique - Dino-Lite - Am4515zt4Chamakhi AmeurNo ratings yet