You might also like
- Pharmacy Service Management For L-III COURSE SYLLABUSDocument2 pagesPharmacy Service Management For L-III COURSE SYLLABUSDawit BirhanuNo ratings yet
- Manage Pharm - Service Chapter 4 EDDocument27 pagesManage Pharm - Service Chapter 4 EDDawit BirhanuNo ratings yet
- Manage Pharm - Service Chapter-2Document43 pagesManage Pharm - Service Chapter-2Dawit BirhanuNo ratings yet
- Introduction To Pharmacy ManagementDocument27 pagesIntroduction To Pharmacy ManagementDawit BirhanuNo ratings yet
- Manage Pharm - Service Chapter-1Document34 pagesManage Pharm - Service Chapter-1Dawit BirhanuNo ratings yet
- University of Gondar College of Natural and Computational Sciences Department of BiologyDocument76 pagesUniversity of Gondar College of Natural and Computational Sciences Department of BiologyDawit Birhanu100% (1)
- Lay Armachiho EthnobotanyDocument88 pagesLay Armachiho EthnobotanyDawit BirhanuNo ratings yet
- 4.3 Alkyhalide PreparationDocument23 pages4.3 Alkyhalide PreparationDawit BirhanuNo ratings yet
- Ethnobotanical KontaDocument15 pagesEthnobotanical KontaDawit BirhanuNo ratings yet
- University of Gondar College of Natural and Computational Sciences Department of BiologyDocument76 pagesUniversity of Gondar College of Natural and Computational Sciences Department of BiologyDawit BirhanuNo ratings yet
- 4.10 Epoxidation RXNDocument6 pages4.10 Epoxidation RXNDawit BirhanuNo ratings yet
- 4.7 Hofmann RXNDocument4 pages4.7 Hofmann RXNDawit BirhanuNo ratings yet
- Ketones Can Be Converted Into Esters Via The Insertion of An Oxygen Atom Baeyer-Villiger OxidationDocument3 pagesKetones Can Be Converted Into Esters Via The Insertion of An Oxygen Atom Baeyer-Villiger OxidationDawit BirhanuNo ratings yet
- 4.5 OxymercurationDocument6 pages4.5 OxymercurationDawit BirhanuNo ratings yet
- 4.5 Oxidative cleveage-KMnO4 & O3Document21 pages4.5 Oxidative cleveage-KMnO4 & O3Dawit BirhanuNo ratings yet
- 4.1.1 Protic Vs Aprotic SolventDocument36 pages4.1.1 Protic Vs Aprotic SolventDawit BirhanuNo ratings yet
- 4.2 Hydration of AlkenesDocument33 pages4.2 Hydration of AlkenesDawit BirhanuNo ratings yet
- Proteins As Drug Targets: EnzymesDocument19 pagesProteins As Drug Targets: EnzymesDawit BirhanuNo ratings yet
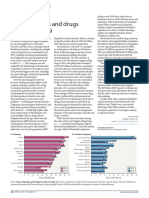
- Top Companies and Drugs by Sales in 2019: Market WatchDocument1 pageTop Companies and Drugs by Sales in 2019: Market WatchDawit BirhanuNo ratings yet
- Presented By: Anand Sagar Tiwari M.Pharm (Second Semester)Document56 pagesPresented By: Anand Sagar Tiwari M.Pharm (Second Semester)Dawit BirhanuNo ratings yet
- 1.2.protocol V5Document73 pages1.2.protocol V5Dawit BirhanuNo ratings yet
- 1.0.clinical Study ProtocolDocument97 pages1.0.clinical Study ProtocolDawit BirhanuNo ratings yet
- Structure Based Drug DesignDocument61 pagesStructure Based Drug DesignDawit BirhanuNo ratings yet
- 1.MedChem-L1 - 9Document327 pages1.MedChem-L1 - 9Dawit Birhanu100% (1)
- Retrospective Evaluation of Single Patient InvestiDocument9 pagesRetrospective Evaluation of Single Patient InvestiDawit BirhanuNo ratings yet
- 1.1..clinical Study ProtocolDocument54 pages1.1..clinical Study ProtocolDawit BirhanuNo ratings yet
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Unit 6: Enzymes: Image Modified From " ," by Openstax College, BiologyDocument4 pagesUnit 6: Enzymes: Image Modified From " ," by Openstax College, BiologyAbraham BanjoNo ratings yet
- End Term ALLDocument31 pagesEnd Term ALLJulie Anne CristalesNo ratings yet
- H2 Chem March Common Test:: Topics Tested ForDocument2 pagesH2 Chem March Common Test:: Topics Tested ForPrince MikeNo ratings yet
- OrganicDocument3 pagesOrganickaifiiNo ratings yet
- Lecture 9 - Collection and Analysis of Rate DataDocument13 pagesLecture 9 - Collection and Analysis of Rate DataSabrina AzharNo ratings yet
- Adiabatic Reactors Final Lab Group 1-ADocument22 pagesAdiabatic Reactors Final Lab Group 1-AHaris SheikhNo ratings yet
- CY 1001 - Structure, Bonding, and Reactivity: Organic ChemistryDocument22 pagesCY 1001 - Structure, Bonding, and Reactivity: Organic ChemistryIron BusterNo ratings yet
- Retrosynthetic Analysis - Wikipedia, The Free EncyclopediaDocument4 pagesRetrosynthetic Analysis - Wikipedia, The Free EncyclopediaALI RAZANo ratings yet
- Selective Formation of Light Olefins by Catalytic Cracking of Naphtha Components Over ZSM-5 Zeolite CatalystsDocument10 pagesSelective Formation of Light Olefins by Catalytic Cracking of Naphtha Components Over ZSM-5 Zeolite CatalystsLeonan Dos Santos RodriguesNo ratings yet
- ENZYMESDocument18 pagesENZYMESJennifer DesRochersNo ratings yet
- Consequetive or Sequential Reaction: Chemical KineticsDocument19 pagesConsequetive or Sequential Reaction: Chemical Kineticsrishabh mishraNo ratings yet
- Chapter Four Major Organic ReactionsDocument63 pagesChapter Four Major Organic ReactionsdagmawiNo ratings yet
- 3 - MS - Chemical KineticsDocument11 pages3 - MS - Chemical KineticsJems ChaudharyNo ratings yet
- Experiment 1Document5 pagesExperiment 1ain sufizaNo ratings yet
- Order of Degeneration +M First Method-Initial RateDocument9 pagesOrder of Degeneration +M First Method-Initial RateZeina Abi FarrajNo ratings yet
- Stem 12... Chapter Test On Chemical Kinetics.Document3 pagesStem 12... Chapter Test On Chemical Kinetics.Caryl Ann C. SernadillaNo ratings yet
- Chapter 8 Reactions of AlcoholsDocument12 pagesChapter 8 Reactions of AlcoholsRoberto SIlvaNo ratings yet
- Stirred Tank by MAUREENDocument27 pagesStirred Tank by MAUREENJimNo ratings yet
- Tetrahedron Letters: Payal Malik, Debashis ChakrabortyDocument3 pagesTetrahedron Letters: Payal Malik, Debashis ChakrabortyBagusChandraMahardhikaNo ratings yet
- Carbonyl ChemsketchDocument3 pagesCarbonyl ChemsketchJenica RiaNo ratings yet
- Redox ReactionDocument13 pagesRedox ReactionforbeskaNo ratings yet
- Modeling & Simulation of Methanol Synthesis From SyngasDocument32 pagesModeling & Simulation of Methanol Synthesis From Syngasshanku_bietNo ratings yet
- Chemical Kinetics TestDocument5 pagesChemical Kinetics Testrajneesh kumarNo ratings yet
- Chapter Tutorial KineticsDocument12 pagesChapter Tutorial KineticsDr.CharinNo ratings yet
- Enzyme WorksheetDocument3 pagesEnzyme Worksheetchinmayee chitturu100% (1)
- 115 Sample Exam 1Document6 pages115 Sample Exam 1bebo4gpaNo ratings yet
- Concept MapDocument1 pageConcept MapMonis Diana Abu BakarNo ratings yet
- Ib Chem IaDocument9 pagesIb Chem IaFrank Lala0% (1)
- Reactor Non IsothermicsDocument9 pagesReactor Non IsothermicsMelgi159No ratings yet
- Replacement of Gas Phase With Liquid HexamineDocument6 pagesReplacement of Gas Phase With Liquid HexaminePradhita Ramdani HNo ratings yet