You might also like
- The Coatings Expert - 2019Document76 pagesThe Coatings Expert - 2019Eyad Alsheiikh100% (1)
- Cyanide Leaching of GoldDocument11 pagesCyanide Leaching of GoldAzizul HakimNo ratings yet
- Cement Chemistry L1Document23 pagesCement Chemistry L1Bharat Singh100% (2)
- Hnology and Livelihood Education: Quarter 1 - Module 2: CookeryDocument15 pagesHnology and Livelihood Education: Quarter 1 - Module 2: Cookeryroderic v. perezNo ratings yet
- Phase DiagramDocument3 pagesPhase DiagramTing TCNo ratings yet
- BS EN ISO 5175-1 (2017) FBA'sDocument22 pagesBS EN ISO 5175-1 (2017) FBA'sPC100% (1)
- Acs JCTC 1c00791Document12 pagesAcs JCTC 1c00791Saeed AbdNo ratings yet
- Comparative Study of The Conductivity of Synthesized Bivalent Vanadates CaV2O6 and MnV2O6Document5 pagesComparative Study of The Conductivity of Synthesized Bivalent Vanadates CaV2O6 and MnV2O6Noremozachhom JahidNo ratings yet
- Ag BridgedDocument11 pagesAg BridgedpramodNo ratings yet
- Chatterjee 2023 Nanotechnology 34 495705Document8 pagesChatterjee 2023 Nanotechnology 34 495705Tania ChatterjeeNo ratings yet
- Impurity of Mg2Si - AlDocument7 pagesImpurity of Mg2Si - AlJasonLopezNo ratings yet
- Zhang 2020Document7 pagesZhang 2020AZ AnimalNo ratings yet
- ACS Catalysis - 2012 - Pd-Modified Tungsten Carbide For Methanol Electro-Oxidation From Surface Science Studies To Electrochemical EvaluDocument8 pagesACS Catalysis - 2012 - Pd-Modified Tungsten Carbide For Methanol Electro-Oxidation From Surface Science Studies To Electrochemical EvaluAli HafeezNo ratings yet
- Acsami 8b02425Document10 pagesAcsami 8b02425AKASHNo ratings yet
- Su 2018Document11 pagesSu 2018غسان بشيرNo ratings yet
- Optimization of Layered Cathode Materials For LithDocument26 pagesOptimization of Layered Cathode Materials For LithadilNo ratings yet
- First Principles Molecular Dynamics Simulation of Solution: Masaru HirataDocument4 pagesFirst Principles Molecular Dynamics Simulation of Solution: Masaru Hiratarushabh yeshwanteNo ratings yet
- Palladium Acetate Trimer - Understanding Its Ligand-Induced Dissociation Thermochemistry AMKDocument10 pagesPalladium Acetate Trimer - Understanding Its Ligand-Induced Dissociation Thermochemistry AMKRaian Yousuf TanmoyNo ratings yet
- Zhang 2014Document11 pagesZhang 2014ITK ChoirNo ratings yet
- Microbial Fuel Cell: A Prospective Sustainable Solution For Energy and Environmental CrisisDocument3 pagesMicrobial Fuel Cell: A Prospective Sustainable Solution For Energy and Environmental CrisisSaravanan PichiahNo ratings yet
- Solution Phase N-Doping of C60 and PCBM Using Tetrabutylammonium FluorideDocument5 pagesSolution Phase N-Doping of C60 and PCBM Using Tetrabutylammonium FluorideEXITOFORTUNANo ratings yet
- 立體選擇性探討 丙交酯與鋅和鎂的β-二氨基配合物的聚合:立體控制和機理。Document10 pages立體選擇性探討 丙交酯與鋅和鎂的β-二氨基配合物的聚合:立體控制和機理。hungNo ratings yet
- Application of NiCo-based Metal-Organicframeworks (MOFs) As An Advanced Electrodematerial For SupercapacitorDocument7 pagesApplication of NiCo-based Metal-Organicframeworks (MOFs) As An Advanced Electrodematerial For Supercapacitorsatyajit beheraNo ratings yet
- Convergent and Divergent Paired Electrodeposition of Metal-Organic Framework Thin FilmsDocument13 pagesConvergent and Divergent Paired Electrodeposition of Metal-Organic Framework Thin Filmsyookarina8No ratings yet
- Synthesis of Vesicle-Like Mgfe O /graphene 3D Network Anode Material With Enhanced Lithium Storage PerformanceDocument8 pagesSynthesis of Vesicle-Like Mgfe O /graphene 3D Network Anode Material With Enhanced Lithium Storage PerformanceLE Thi LyNo ratings yet
- (Done) Hollow Porous Co3O4 - NC@rGO Derived From Reuleaux Tetrahedral ZIF-67 As A Promising Anode Material For LDocument10 pages(Done) Hollow Porous Co3O4 - NC@rGO Derived From Reuleaux Tetrahedral ZIF-67 As A Promising Anode Material For Luser728384940No ratings yet
- C0JM04058D 4861..4868Document8 pagesC0JM04058D 4861..4868Aryan Singh LatherNo ratings yet
- Evaluation of MG Compounds As Coating Materials in MG BatteriesDocument10 pagesEvaluation of MG Compounds As Coating Materials in MG Batteriestolentino.jason1222No ratings yet
- Optical Photocatalytic Electrochemical Magnetic DiDocument19 pagesOptical Photocatalytic Electrochemical Magnetic DiTasawer Shahzad AhmadNo ratings yet
- Pardikar 2023 J. Phys. Energy 5 022002Document33 pagesPardikar 2023 J. Phys. Energy 5 022002ErnestNo ratings yet
- Yao 2018 Joule MG CLDocument14 pagesYao 2018 Joule MG CLAndrewSpeedyFeetNo ratings yet
- Chemical Science: Edge ArticleDocument6 pagesChemical Science: Edge Articlenityananada ChemNo ratings yet
- Polymer Testing: Jing Cao, Na Wen, Yuying ZhengDocument8 pagesPolymer Testing: Jing Cao, Na Wen, Yuying Zhengshade shaperNo ratings yet
- The Preparation of Calcium Pimelate Modified OMNT From Natural CA-montmorillonite and Its Application as Β-nucleating Agent for PolypropyleneDocument8 pagesThe Preparation of Calcium Pimelate Modified OMNT From Natural CA-montmorillonite and Its Application as Β-nucleating Agent for Polypropyleneshade shaperNo ratings yet
- MnO2 Phase SelectionDocument10 pagesMnO2 Phase SelectionJacob SchmidtNo ratings yet
- Paper 04Document12 pagesPaper 04Yov KosalNo ratings yet
- Supercacitores 4Document17 pagesSupercacitores 4marcosNo ratings yet
- J Jhazmat 2018 06 025Document85 pagesJ Jhazmat 2018 06 025mahsaNo ratings yet
- Angew Chem Int Ed - 2022 - Liu - Assembling Metal Organic Layer Composites For High Performance Electrocatalytic CO2Document6 pagesAngew Chem Int Ed - 2022 - Liu - Assembling Metal Organic Layer Composites For High Performance Electrocatalytic CO2pandiaraj1988No ratings yet
- Chemcomm: CommunicationDocument3 pagesChemcomm: CommunicationbsjsjsjNo ratings yet
- Mechanochemically Synthesized Pyrite and Its ElectDocument11 pagesMechanochemically Synthesized Pyrite and Its ElectBen HarropNo ratings yet
- Calculation Studies On Point Defects in Perovskite Solar CellsDocument10 pagesCalculation Studies On Point Defects in Perovskite Solar CellsUmu HanifahNo ratings yet
- 1 s2.0 S0254058423011380 MainDocument15 pages1 s2.0 S0254058423011380 Mainmy.local.address.infoNo ratings yet
- Electrochimica ActaDocument9 pagesElectrochimica ActareiroslesNo ratings yet
- Paper: Oxygen-Defect-Dependent Ferromagnetism and Strain Modulation in Free-Standing Two-Dimensional Tio MonolayersDocument9 pagesPaper: Oxygen-Defect-Dependent Ferromagnetism and Strain Modulation in Free-Standing Two-Dimensional Tio MonolayersFrijaKim E 21No ratings yet
- C 2 JM 14410 GDocument6 pagesC 2 JM 14410 Gcesafilho.idtNo ratings yet
- 10 1002@slct 201702197Document10 pages10 1002@slct 201702197SAID MAULANANo ratings yet
- Ge 2016Document7 pagesGe 2016tvph.novo.kinhbacNo ratings yet
- Braband Et Al 2021 Relativity As A Synthesis Design Principle A Comparative Study of 3 2 Cycloaddition ofDocument8 pagesBraband Et Al 2021 Relativity As A Synthesis Design Principle A Comparative Study of 3 2 Cycloaddition ofAssania NazidaNo ratings yet
- Jacs 1c13368Document10 pagesJacs 1c13368Евгения ИконниковаNo ratings yet
- Topochemical Synthesis of Phase-Pure Mo2AlB2 Through Staging MechanismDocument4 pagesTopochemical Synthesis of Phase-Pure Mo2AlB2 Through Staging MechanismunnisazureNo ratings yet
- Ab Initio Study of Point Defects in Magnesium Oxide: Loughborough University Institutional RepositoryDocument12 pagesAb Initio Study of Point Defects in Magnesium Oxide: Loughborough University Institutional RepositoryPhilmona SamuelNo ratings yet
- in Situ Growth of Cop Wrapped by Carbon Nanoarray Like Architecture Onto Nitrogen Doped Ti3C2 PT Based Catalyst For Efficient Methanol Oxidation W Zhan L Ma M Gan Full ChapterDocument34 pagesin Situ Growth of Cop Wrapped by Carbon Nanoarray Like Architecture Onto Nitrogen Doped Ti3C2 PT Based Catalyst For Efficient Methanol Oxidation W Zhan L Ma M Gan Full Chapterdaniel.taylor716100% (9)
- Effect of Buffer Layers On Performance of Organic Photovoltaic Devices Based On Copper Phthalocyanine-Perylene Dye HeterojunctionDocument6 pagesEffect of Buffer Layers On Performance of Organic Photovoltaic Devices Based On Copper Phthalocyanine-Perylene Dye HeterojunctionAnonymous cYpEVvoNo ratings yet
- Modification in Photovoltaic and Photocatalytic Properties of Bismuth Ferrites by Tailoring Band-Gap and Ferroelectric PropertiesDocument13 pagesModification in Photovoltaic and Photocatalytic Properties of Bismuth Ferrites by Tailoring Band-Gap and Ferroelectric Propertiesmanjeet redduNo ratings yet
- MoS2 MS Rai PDFDocument12 pagesMoS2 MS Rai PDFSafaa ArouhNo ratings yet
- Coordination Chemistry in Magnesium Battery Electrolytes How Ligands Affect Their PerformanceDocument7 pagesCoordination Chemistry in Magnesium Battery Electrolytes How Ligands Affect Their PerformanceWaqasskhanNo ratings yet
- Mondal Et Al 2020 Genetic Algorithm Driven Force Field Parameterization For Molten Alkali Metal Carbonate and HydroxideDocument11 pagesMondal Et Al 2020 Genetic Algorithm Driven Force Field Parameterization For Molten Alkali Metal Carbonate and HydroxideNavin kumarNo ratings yet
- 2016 CH4-stored HeadGordon ChemSciDocument16 pages2016 CH4-stored HeadGordon ChemSciAlejandra AwimbaweNo ratings yet
- Effect of Cs On The Stability and PhotovoltaicDocument7 pagesEffect of Cs On The Stability and Photovoltaica s m mosabbirNo ratings yet
- Batteries Supercaps - 2020 - Zhang - Carbon Composite Anodes With Tunable Microstructures For Potassium Ion BatteriesDocument8 pagesBatteries Supercaps - 2020 - Zhang - Carbon Composite Anodes With Tunable Microstructures For Potassium Ion Batteriesxcr66123No ratings yet
- Bioresource Technology: Shaoqiang Yang, Boyang Jia, Hong LiuDocument6 pagesBioresource Technology: Shaoqiang Yang, Boyang Jia, Hong LiuFPS CONo ratings yet
- Electrochimica Acta: Bin Qin, Qun Wang, Xiaohua Zhang, Xiaoling Xie, Li'e Jin, Qing CaoDocument9 pagesElectrochimica Acta: Bin Qin, Qun Wang, Xiaohua Zhang, Xiaoling Xie, Li'e Jin, Qing CaoJanethAlcalaNo ratings yet
- Presence of Gap States at Cu/Tio Anatase Surfaces: Consequences For The Photocatalytic ActivityDocument7 pagesPresence of Gap States at Cu/Tio Anatase Surfaces: Consequences For The Photocatalytic ActivityIsmael Antonio González RamirezNo ratings yet
- MarschallDocument20 pagesMarschallgasweb19No ratings yet
- Accepted Manuscript: Ceramics InternationalDocument30 pagesAccepted Manuscript: Ceramics InternationalMuhammad UsmanNo ratings yet
- Sno 2Document32 pagesSno 2Muhammad UsmanNo ratings yet
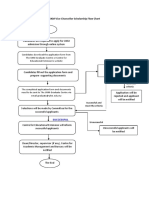
- UKM Vice-Chancellor Scholarship Flow ChartDocument1 pageUKM Vice-Chancellor Scholarship Flow ChartMuhammad UsmanNo ratings yet
- Cee Mno2 FTIRDocument4 pagesCee Mno2 FTIRMuhammad UsmanNo ratings yet
- Joint Entrance Test (Jet) - 2019: General Instructions For Admission ToDocument27 pagesJoint Entrance Test (Jet) - 2019: General Instructions For Admission ToKd Gurjar KhuriNo ratings yet
- Entropy and The 2nd & 3rd Laws of ThermodynamicsDocument8 pagesEntropy and The 2nd & 3rd Laws of ThermodynamicsJoanne SalresNo ratings yet
- Ferrous & Non-Ferrous AlloysDocument22 pagesFerrous & Non-Ferrous AlloysHarsh V Ashok0% (1)
- Neopor TMDocument2 pagesNeopor TMNurettinAlpNo ratings yet
- Determination of Heavy Metals in Fish Tissues WateDocument8 pagesDetermination of Heavy Metals in Fish Tissues WateADDY NARAYANNo ratings yet
- Flame Resistant FinishesDocument32 pagesFlame Resistant FinishesNishu JalotiaNo ratings yet
- Eric Ed259126Document167 pagesEric Ed259126NguyenDinhLyNo ratings yet
- 3.2 Understanding Pressure in LiquidDocument16 pages3.2 Understanding Pressure in LiquidzoulezNo ratings yet
- SPE 29694 Seafloor Monitoring For Synthetic-Based Mud Discharged in The Western Gulf of MexicoDocument19 pagesSPE 29694 Seafloor Monitoring For Synthetic-Based Mud Discharged in The Western Gulf of MexicoAminNo ratings yet
- Articulo 2Document8 pagesArticulo 2luisNo ratings yet
- Molecular Mass Transport: 3.1 Introduction of Mass TransferDocument34 pagesMolecular Mass Transport: 3.1 Introduction of Mass Transfergaurav sriNo ratings yet
- Factors Affecting The Rate of Loss of Heat of A LiquidDocument4 pagesFactors Affecting The Rate of Loss of Heat of A Liquidionschemistry44No ratings yet
- Inhibition of Mild Steel Corrosion in 0.25 M Sulphuric Acid Solution by Imatinib MesylateDocument9 pagesInhibition of Mild Steel Corrosion in 0.25 M Sulphuric Acid Solution by Imatinib Mesylatehadi ebrahimfathNo ratings yet
- Welding Procedure Specification - Kel 3Document4 pagesWelding Procedure Specification - Kel 3Helmi KusumaNo ratings yet
- MSDS - Ecoglass1Document5 pagesMSDS - Ecoglass1Syed Abdul RahmanNo ratings yet
- Aramv22 N1 P1 12Document12 pagesAramv22 N1 P1 12hamedpdmsNo ratings yet
- MSDS - Ultimeg 2000-372Document6 pagesMSDS - Ultimeg 2000-372Azka DeliaNo ratings yet
- KP - Method - Description-Dragica Vasileska PDFDocument51 pagesKP - Method - Description-Dragica Vasileska PDFombraga1896No ratings yet
- Physics II Dec 2002Document2 pagesPhysics II Dec 2002api-3782519No ratings yet
- C5 ExerciseDocument18 pagesC5 ExerciseHƯƠNG NGUYỄN LÊ NGỌCNo ratings yet
- Bioethanol Dehydration SelectedDocument9 pagesBioethanol Dehydration Selectedkaran rajputNo ratings yet
- REVIEW 570rev 1Document9 pagesREVIEW 570rev 1James Stephen ArriolaNo ratings yet
- Capabilities Chart: GeneralDocument3 pagesCapabilities Chart: GeneralFernando Acevedo FernandezNo ratings yet
- Grade 10 - Chemical ReactionDocument3 pagesGrade 10 - Chemical ReactionMaxineNo ratings yet