You might also like
- VolvoTD70 Service Manual EngineDocument110 pagesVolvoTD70 Service Manual EngineHeikki Alakontiola87% (47)
- Good Clinical Practice GuideFrom EverandGood Clinical Practice GuideRating: 5 out of 5 stars5/5 (1)
- ZHINENG Qigong Breathing Exercises ZBEDocument12 pagesZHINENG Qigong Breathing Exercises ZBELong Le100% (4)
- S 264 Validation Spreadsheet ApplicationsDocument14 pagesS 264 Validation Spreadsheet ApplicationsNeoNo ratings yet
- Computer System Validation Risk Assessment ToolDocument3 pagesComputer System Validation Risk Assessment Toolcpkakope100% (1)
- CSV SopDocument1 pageCSV SopjeetNo ratings yet
- SOP For Computer System Validation in Pharmaceutical IndustryDocument8 pagesSOP For Computer System Validation in Pharmaceutical IndustryDeepakNo ratings yet
- Empower 3 21 CFR Part 11 Compliance Assessment RevA November 2019Document14 pagesEmpower 3 21 CFR Part 11 Compliance Assessment RevA November 2019Nur AcarNo ratings yet
- Computer System Validation (CSV) - Ajay KulkarniDocument66 pagesComputer System Validation (CSV) - Ajay KulkarniDeepak Jha33% (3)
- V Model - GampDocument2 pagesV Model - GampNitin KashyapNo ratings yet
- CSV Good Documentation and Test Practices For GXPDocument16 pagesCSV Good Documentation and Test Practices For GXPDivyendu DasNo ratings yet
- Data Integrity ChecklistDocument12 pagesData Integrity ChecklistprakashNo ratings yet
- Selogica and Gestica: Pioneering and Intuitive Control Technology "Made by ARBURG"Document32 pagesSelogica and Gestica: Pioneering and Intuitive Control Technology "Made by ARBURG"Ahmed SamirNo ratings yet
- Biocontamination Control for Pharmaceuticals and HealthcareFrom EverandBiocontamination Control for Pharmaceuticals and HealthcareRating: 5 out of 5 stars5/5 (1)
- Regulatory Aspects of Pharmaceutical Quality System: Brief IntroductionFrom EverandRegulatory Aspects of Pharmaceutical Quality System: Brief IntroductionNo ratings yet
- Data Integrity and Compliance: A Primer for Medical Product ManufacturersFrom EverandData Integrity and Compliance: A Primer for Medical Product ManufacturersNo ratings yet
- Nonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsFrom EverandNonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsWilliam J. BrockNo ratings yet
- Cost-Contained Regulatory Compliance: For the Pharmaceutical, Biologics, and Medical Device IndustriesFrom EverandCost-Contained Regulatory Compliance: For the Pharmaceutical, Biologics, and Medical Device IndustriesNo ratings yet
- Portfolio, Program, and Project Management in the Pharmaceutical and Biotechnology IndustriesFrom EverandPortfolio, Program, and Project Management in the Pharmaceutical and Biotechnology IndustriesPete HarpumNo ratings yet
- Practical Approaches to Method Validation and Essential Instrument QualificationFrom EverandPractical Approaches to Method Validation and Essential Instrument QualificationNo ratings yet
- Spreadsheetvalidation101 - Createandvalidatefda-Compliantms ExcelspreadsheetsDocument121 pagesSpreadsheetvalidation101 - Createandvalidatefda-Compliantms Excelspreadsheetszombiecorp100% (1)
- Computer System Validation in The Perspective of TDocument7 pagesComputer System Validation in The Perspective of Tttugce29No ratings yet
- S 285 Risk Qualification InfstructureDocument10 pagesS 285 Risk Qualification InfstructureMohamed SallamNo ratings yet
- CSV Risks Requirements Tests and TraceabilityDocument44 pagesCSV Risks Requirements Tests and TraceabilitymonsepackNo ratings yet
- Periodic ReviewDocument6 pagesPeriodic ReviewJorge GutierrezNo ratings yet
- PICS - Guidance On GP For SC in GXP EnvironmentsDocument54 pagesPICS - Guidance On GP For SC in GXP EnvironmentsTrilok Chander ManthaNo ratings yet
- GAMP 5 Risk Based Validation GuideDocument5 pagesGAMP 5 Risk Based Validation Guidevignan50No ratings yet
- Empower 3 EU Annex 11 Compliance Assessment Rev1 November 2019Document12 pagesEmpower 3 EU Annex 11 Compliance Assessment Rev1 November 2019Nur AcarNo ratings yet
- Computer System Validation (CSV) : Comparisons Between (GMP VS CGMP, GLP VS GCP, 21 CFR PART 11 VS EU 11)Document4 pagesComputer System Validation (CSV) : Comparisons Between (GMP VS CGMP, GLP VS GCP, 21 CFR PART 11 VS EU 11)T 1No ratings yet
- Comparison of FDA Part 11 and EU Annex 11Document5 pagesComparison of FDA Part 11 and EU Annex 11marco_fmNo ratings yet
- QRM For Computer SystemsDocument51 pagesQRM For Computer SystemsbaluchakpNo ratings yet
- FDA Compliance Digest Part 11 System Compliance Assessment ChecklistDocument3 pagesFDA Compliance Digest Part 11 System Compliance Assessment ChecklistSairam KishoreNo ratings yet
- 1 - Introduction To Computerized Systems Validation - For ReviewDocument41 pages1 - Introduction To Computerized Systems Validation - For Reviewpate malabananNo ratings yet
- Guidelines For The Development and Validation of Spreadsheets PDFDocument21 pagesGuidelines For The Development and Validation of Spreadsheets PDFraj kumar chaudharyNo ratings yet
- Computer System ValidationDocument2 pagesComputer System ValidationVishal VakilNo ratings yet
- FORM-000249184 DI Equipment Software AssessmentDocument6 pagesFORM-000249184 DI Equipment Software AssessmentSebastian LopezNo ratings yet
- FastVal User Requirement Template PDFDocument6 pagesFastVal User Requirement Template PDFViệt ThắngNo ratings yet
- Eres Annex 11 Eu GMP SiemensDocument30 pagesEres Annex 11 Eu GMP SiemenshuykhiemNo ratings yet
- Impact Analysis of Software RequirementDocument8 pagesImpact Analysis of Software RequirementsudheerNo ratings yet
- Excel Spreadsheet Validation SpecificationDocument8 pagesExcel Spreadsheet Validation Specificationjohn DevinsNo ratings yet
- Validation of MS Excel SpreadsheetsDocument5 pagesValidation of MS Excel SpreadsheetsNicolas Quero CarvajalNo ratings yet
- Empower 3 Enterprise or Workgroup Software For ServersDocument52 pagesEmpower 3 Enterprise or Workgroup Software For ServersMohamed Sallam100% (2)
- Writing An Effective 483 ResponseDocument27 pagesWriting An Effective 483 ResponseJanna Hendrix Babson100% (1)
- Data Integrity Global Quality StandardDocument10 pagesData Integrity Global Quality StandardSebastian LopezNo ratings yet
- 10.1-GDIR Data IntegrityDocument12 pages10.1-GDIR Data IntegritySebastian LopezNo ratings yet
- URS, Validation Phases ConceptDocument41 pagesURS, Validation Phases ConceptSyed Ghazanfar AliNo ratings yet
- Computersystemsvalidation BlogspotDocument7 pagesComputersystemsvalidation BlogspotNitin KashyapNo ratings yet
- Computer Sytems ValidationDocument92 pagesComputer Sytems ValidationmaikaNo ratings yet
- GAMP 5 Risk-Based ApproachDocument29 pagesGAMP 5 Risk-Based ApproachLia LiawatiNo ratings yet
- Guidelines For The Validation of Computerised SystemsDocument19 pagesGuidelines For The Validation of Computerised SystemsSisly LópezNo ratings yet
- Ema Process ValidationDocument15 pagesEma Process Validationdrs_mdu48100% (1)
- Good Manufacturing Practices For Pharmaceutical: A Plan Far Total Quality Control Irani Manufacturer To ConsomerDocument18 pagesGood Manufacturing Practices For Pharmaceutical: A Plan Far Total Quality Control Irani Manufacturer To ConsomerJosé Luis Rosales100% (1)
- 4 - Case Study On A Risk-Based Approach To Validation - For ReviewDocument49 pages4 - Case Study On A Risk-Based Approach To Validation - For Reviewpate malabanan100% (1)
- Computer System Validation ICHDocument67 pagesComputer System Validation ICHDoan Chi ThienNo ratings yet
- Regulatory framework for quality by designDocument26 pagesRegulatory framework for quality by designhdmnauNo ratings yet
- 3BSE077527 en FDA 21 CFR Part 11 Electronic Records and Signatures - Solutions For The Life Sciences IndustryDocument12 pages3BSE077527 en FDA 21 CFR Part 11 Electronic Records and Signatures - Solutions For The Life Sciences IndustryNeoNo ratings yet
- A Study of Computerized System ValidationDocument12 pagesA Study of Computerized System ValidationTenzin Tashi100% (1)
- Applying Lean Principles to Pharmaceutical Lab DesignDocument35 pagesApplying Lean Principles to Pharmaceutical Lab DesignReman A YehyaNo ratings yet
- Spreadsheet Design, Verification and Validation, Use and Storage of Single-User Workbook Files in US FDADocument25 pagesSpreadsheet Design, Verification and Validation, Use and Storage of Single-User Workbook Files in US FDAvg_vvg100% (1)
- How The Different GLP GMP GCPDocument14 pagesHow The Different GLP GMP GCPawang_timur100% (3)
- Laboratory Investigation RequirementsDocument16 pagesLaboratory Investigation RequirementsSebastian LopezNo ratings yet
- A Very Fruitful CSV DocumentDocument16 pagesA Very Fruitful CSV DocumentkushalNo ratings yet
- Top 25 Lean ToolsDocument2 pagesTop 25 Lean ToolsAnonymous dv2IR074100% (1)
- MEE WH Production EngineerDocument1 pageMEE WH Production EngineerAhmed SamirNo ratings yet
- MEE WH Production Engineer-1Document1 pageMEE WH Production Engineer-1Ahmed SamirNo ratings yet
- Overview VideosDocument1 pageOverview Videosnabila OktavianiNo ratings yet
- TPM and Oee: Mauri e O'BrieDocument51 pagesTPM and Oee: Mauri e O'Brieprashanth191182No ratings yet
- The Manufacturer's Guide To Succeeding With AI 2Document13 pagesThe Manufacturer's Guide To Succeeding With AI 2Ahmed SamirNo ratings yet
- Ahmed Samir Mohamed: Data Analysis ProfessionalDocument1 pageAhmed Samir Mohamed: Data Analysis ProfessionalAhmed SamirNo ratings yet
- DownloadDocument1 pageDownloadAhmed SamirNo ratings yet
- برنامج تحكمDocument16 pagesبرنامج تحكمMohamedHazemNo ratings yet
- FFP Guidelines. V383531256Document20 pagesFFP Guidelines. V383531256Денис АкуляковNo ratings yet
- C Comp R (Pany Refere (1992 y Exp Ence 2-201 Perie List 17) NceDocument35 pagesC Comp R (Pany Refere (1992 y Exp Ence 2-201 Perie List 17) NceAhmed SamirNo ratings yet
- Quantitative Results in One-Hour: Correlation (Pearson, Kendall, Spearman)Document6 pagesQuantitative Results in One-Hour: Correlation (Pearson, Kendall, Spearman)Ahmed SamirNo ratings yet
- Design TipsDocument30 pagesDesign Tipsrvejner5253No ratings yet
- Social Media Adv PDFDocument28 pagesSocial Media Adv PDFAndreea-Madalina CucuNo ratings yet
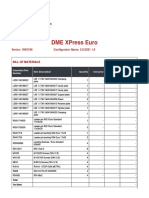
- Dme Xpress Euro: Series: 196X196 Configurator Name: 5/2/2021 1.0Document3 pagesDme Xpress Euro: Series: 196X196 Configurator Name: 5/2/2021 1.0Ahmed SamirNo ratings yet
- Ineos Polypropylene Processing GuideDocument18 pagesIneos Polypropylene Processing GuideLe Toan100% (1)
- PR Checklist Stage2 FinalDocument9 pagesPR Checklist Stage2 FinalAhmed SamirNo ratings yet
- CONFERENCE CALENDAR 1989-1990Document7 pagesCONFERENCE CALENDAR 1989-1990Ahmed SamirNo ratings yet
- Industrial Engineering DepartmentDocument23 pagesIndustrial Engineering DepartmentAhmed SamirNo ratings yet
- 2.008 03im 05S PDFDocument13 pages2.008 03im 05S PDFsilversky09No ratings yet
- 2.008 Design & Manufacturing IIDocument9 pages2.008 Design & Manufacturing IIAmit_DhimanjNo ratings yet
- PR Checklist Stage4 FinalDocument7 pagesPR Checklist Stage4 FinalAhmed SamirNo ratings yet
- PR Checklist Stage1 FinalDocument8 pagesPR Checklist Stage1 FinalAhmed SamirNo ratings yet
- PR Checklist Stage3 FinalDocument7 pagesPR Checklist Stage3 FinalAhmed SamirNo ratings yet
- D - M - E Mold Cooling: Reducing Cycle Time Through Cooling InnovationDocument26 pagesD - M - E Mold Cooling: Reducing Cycle Time Through Cooling InnovationAhmed SamirNo ratings yet
- PR Checklist Stage5 FinalDocument6 pagesPR Checklist Stage5 FinalAhmed SamirNo ratings yet
- Troubleshooting Guide: Injection MoldingDocument15 pagesTroubleshooting Guide: Injection MoldingGufran AhmedNo ratings yet
- PR Checklist Stage6 FinalDocument5 pagesPR Checklist Stage6 FinalAhmed SamirNo ratings yet
- Oil Jacket Design and Simulation AnalysisDocument7 pagesOil Jacket Design and Simulation AnalysisAhmed SamirNo ratings yet
- Chapter 1 F&B ManagementDocument59 pagesChapter 1 F&B ManagementMohd Faizal Bin AyobNo ratings yet
- If You Have Guts Then Dare To Be DIFFERENTDocument20 pagesIf You Have Guts Then Dare To Be DIFFERENTChirag Saiya (PHILOSOPHER) - SPIRITUAL Speaker and Writer100% (1)
- Dokumen - Tips Daewoo Service Manual Instrument Cluster Matiz-2023Document23 pagesDokumen - Tips Daewoo Service Manual Instrument Cluster Matiz-2023urexalg AlgériaNo ratings yet
- Wire Rope Slings Si 2 - 2 EmmDocument2 pagesWire Rope Slings Si 2 - 2 EmmheppyfaebanffNo ratings yet
- AISC Design Guide 27 - Structural Stainless SteelDocument159 pagesAISC Design Guide 27 - Structural Stainless SteelCarlos Eduardo Rodriguez100% (1)
- Spain-Final ProjectDocument29 pagesSpain-Final Projectapi-264431075No ratings yet
- RT-137 - A1 Onboard Repair Kits and Order NumbersDocument6 pagesRT-137 - A1 Onboard Repair Kits and Order Numbers郑开元No ratings yet
- Oil Keeper Job at PacrimDocument2 pagesOil Keeper Job at Pacrimwinda chairunissaNo ratings yet
- Ganesha PancharatnamDocument2 pagesGanesha PancharatnamrajiprakNo ratings yet
- Cerebrospinal Fluid Penetration of Amikacin in Children With Community-Acquired Bacterial MeningitisDocument3 pagesCerebrospinal Fluid Penetration of Amikacin in Children With Community-Acquired Bacterial MeningitisDanny JacobusNo ratings yet
- C CA AS SC CL Liin Niiq QU UE E // C CA AS SE ER RE EP PO OR RT TDocument7 pagesC CA AS SC CL Liin Niiq QU UE E // C CA AS SE ER RE EP PO OR RT TG Virucha Meivila IINo ratings yet
- 17EEX01-FUNDAMENTALS OF FIBRE OPTICS AND LASER INSTRUMENTATION SyllabusDocument2 pages17EEX01-FUNDAMENTALS OF FIBRE OPTICS AND LASER INSTRUMENTATION SyllabusJayakumar ThangavelNo ratings yet
- July 28Document8 pagesJuly 28thestudentageNo ratings yet
- Crystal-Phase Transitions and Photocatalysis in Supramolecular Scaffolds (2017)Document8 pagesCrystal-Phase Transitions and Photocatalysis in Supramolecular Scaffolds (2017)Louis Peronet VergaraNo ratings yet
- Applications of Proton Exchange Membrane Fuel CellDocument20 pagesApplications of Proton Exchange Membrane Fuel CellRiri SasyNo ratings yet
- Service Manual: Side by Side S20B RSB21-A/GDocument16 pagesService Manual: Side by Side S20B RSB21-A/GjicutuNo ratings yet
- Sri Venkateswara Caterers Tiffin MenuDocument4 pagesSri Venkateswara Caterers Tiffin MenuJPDGLNo ratings yet
- PFC Katalog PPDocument128 pagesPFC Katalog PPbmsinghNo ratings yet
- CHAP2 Bioavailability of Metals David Jhon LeventhalDocument9 pagesCHAP2 Bioavailability of Metals David Jhon LeventhalCrisThian PaucaNo ratings yet
- Anatomía Relacional de Los Músculos MiméticosDocument5 pagesAnatomía Relacional de Los Músculos MiméticosLizeth Conde OrozcoNo ratings yet
- Market Sorvey On PlywoodDocument19 pagesMarket Sorvey On PlywoodEduardo MafraNo ratings yet
- Ijpcr 22 309Document6 pagesIjpcr 22 309Sriram NagarajanNo ratings yet
- A Review of Bempedoic Acid - A New Drug For Old ProblemDocument6 pagesA Review of Bempedoic Acid - A New Drug For Old ProblemVõ Ngọc Bích VânNo ratings yet
- 12 Chemistry Aldehydes Ketones and Carboxylic Acids Test 04 PDFDocument1 page12 Chemistry Aldehydes Ketones and Carboxylic Acids Test 04 PDFShreyash KolekarNo ratings yet
- KOLHAN UNIVERSITY B.Sc Zoology Semester I Exam 2021 Provisional Marks CardDocument2 pagesKOLHAN UNIVERSITY B.Sc Zoology Semester I Exam 2021 Provisional Marks CardSmritiNo ratings yet
- Journal Review - Philosophical and Contextual Issue in Nursing Theory DevelopmentDocument4 pagesJournal Review - Philosophical and Contextual Issue in Nursing Theory DevelopmentJULIE ANN PARANo ratings yet
- National Ehealth: "Moving Towards Efficient Healthcare"Document30 pagesNational Ehealth: "Moving Towards Efficient Healthcare"nurul fatma diyanaNo ratings yet
- Lecture Notes 12&13 Phylum ApicomplexaDocument20 pagesLecture Notes 12&13 Phylum ApicomplexaAmirr4uddinNo ratings yet