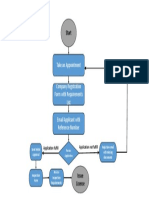
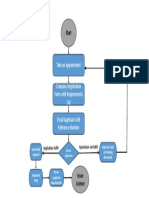
You might also like
- Procedure For Complaint Handling-AOCDocument3 pagesProcedure For Complaint Handling-AOCMohamed EzzatNo ratings yet
- Sop VigilanceDocument7 pagesSop VigilanceJane BrownNo ratings yet
- Sample SoP For Vigilance System PDFDocument4 pagesSample SoP For Vigilance System PDFhitham shehataNo ratings yet
- Procedure for Medical Device AlertsDocument4 pagesProcedure for Medical Device AlertsMohamed EzzatNo ratings yet
- SOP For Handling of Market ComplaintDocument32 pagesSOP For Handling of Market Complaintsubbu_281No ratings yet
- SOP For Maintenance of Medical DevicesDocument2 pagesSOP For Maintenance of Medical DevicesMuhammad NisarNo ratings yet
- Procedure For Recall - AOCDocument6 pagesProcedure For Recall - AOCMohamed EzzatNo ratings yet
- Complaint Handling 60.60 23JUN2020Document11 pagesComplaint Handling 60.60 23JUN2020Abdul RahmanNo ratings yet
- PMS PMCF CER RelationshipDocument1 pagePMS PMCF CER RelationshipMohammed HammedNo ratings yet
- QP19-Vigilance Report - CE MarkDocument18 pagesQP19-Vigilance Report - CE Markanusha shankarNo ratings yet
- Sop Post Market SurveillanceDocument3 pagesSop Post Market SurveillanceBiolytic LifesciencesNo ratings yet
- HSA Post-Market Device Vigilance RequirementsDocument8 pagesHSA Post-Market Device Vigilance RequirementsSubashiиy PяabakaяaиNo ratings yet
- Rules On Vigilance of Medical Devices PDFDocument8 pagesRules On Vigilance of Medical Devices PDFsks27974No ratings yet
- PSUR Action Plan and Follow Up (R1)Document6 pagesPSUR Action Plan and Follow Up (R1)Bo Ram KimNo ratings yet
- Clinical Evluation GiudenceDocument20 pagesClinical Evluation GiudenceabcNo ratings yet
- Psur Apr 2021Document14 pagesPsur Apr 2021Mauro CostaNo ratings yet
- Medical Devices - SOP 3 - Acceptance of Medical DevicesDocument6 pagesMedical Devices - SOP 3 - Acceptance of Medical DevicesCaalaa Dabalaa LamuuNo ratings yet
- SOP RECALL ALMED in EnglishDocument5 pagesSOP RECALL ALMED in EnglishMuztahid RivaiNo ratings yet
- Post-Market Surveillance Research ProjectDocument6 pagesPost-Market Surveillance Research ProjectAKSNo ratings yet
- Sop Capa PDFDocument3 pagesSop Capa PDFnakul tiwariNo ratings yet
- Guide Psur Apr 2021Document55 pagesGuide Psur Apr 2021ifrahNo ratings yet
- Sop PsurDocument8 pagesSop PsurGehan El Hefney100% (1)
- 1Document11 pages1Jonathan McloughlinNo ratings yet
- Template PMS Report TL01 V01Document9 pagesTemplate PMS Report TL01 V01Sandra SilvaNo ratings yet
- Risk Assessment 2Document5 pagesRisk Assessment 2Aditya Agrawal100% (2)
- Risk Management ISO 14971 - 2019 r10v1.0 (01-08-20)Document84 pagesRisk Management ISO 14971 - 2019 r10v1.0 (01-08-20)AelindahNo ratings yet
- Post Market Surveillance: Global Guidance For Adverse Event Reporting For Medical DevicesDocument37 pagesPost Market Surveillance: Global Guidance For Adverse Event Reporting For Medical DevicesSachin KumarNo ratings yet
- Pharmaceutical Product Complaints:: Causes Behind The Market ComplaintsDocument15 pagesPharmaceutical Product Complaints:: Causes Behind The Market ComplaintsKakon AhmedNo ratings yet
- Template For PSUR - MPVDocument6 pagesTemplate For PSUR - MPV-MPV Thảo - QAQC100% (2)
- SG2 N79 R11Document13 pagesSG2 N79 R11David OngNo ratings yet
- Medical Devices Benefit Risk ManagementDocument7 pagesMedical Devices Benefit Risk ManagementSteven KingNo ratings yet
- Risk Benefit FinalDocument7 pagesRisk Benefit FinalgoaltechNo ratings yet
- Post Marketing SurveillanceDocument15 pagesPost Marketing SurveillanceSundar Karuna100% (1)
- Adverse Event ReportingDocument2 pagesAdverse Event ReportingSanjay NavaleNo ratings yet
- Periodic Safety Update Report: ProductDocument5 pagesPeriodic Safety Update Report: ProductMauro CostaNo ratings yet
- Periodic Safety Report PsurDocument12 pagesPeriodic Safety Report PsurKrishna Mohan.p.rNo ratings yet
- Medical DeviceDocument8 pagesMedical DeviceHarshvardhan ChoudharyNo ratings yet
- Characteristics and Risk Analysis Report - Template v.1.0Document10 pagesCharacteristics and Risk Analysis Report - Template v.1.0Sarfraz EPIC ConsultingNo ratings yet
- Medical Device Technical Specification: TS-01: Good Distribution Practice For Medical Devices - RequirementsDocument33 pagesMedical Device Technical Specification: TS-01: Good Distribution Practice For Medical Devices - RequirementsSeanNo ratings yet
- Signal Assessment Report Template - enDocument8 pagesSignal Assessment Report Template - eneviyanaNo ratings yet
- Reporting Adverse Events for Medical Device TrialsDocument7 pagesReporting Adverse Events for Medical Device TrialssachinNo ratings yet
- Annex 5-Risk Management PlanDocument5 pagesAnnex 5-Risk Management PlanalexNo ratings yet
- Standard Operating Procedure: 1. PurposeDocument8 pagesStandard Operating Procedure: 1. PurposeElave SaberNo ratings yet
- SOP On Data Integrity and Security ProcedureDocument6 pagesSOP On Data Integrity and Security Procedureislampes9No ratings yet
- Post-Marketing Surveillance PDFDocument10 pagesPost-Marketing Surveillance PDFscjofyWFawlroa2r06YFVabfbajNo ratings yet
- Periodic Safety Update Report SummaryDocument11 pagesPeriodic Safety Update Report SummaryBo Ram KimNo ratings yet
- BT200 - Clinical Evaluation ReportDocument5 pagesBT200 - Clinical Evaluation ReportfjvillamunozNo ratings yet
- EU PMS PSUR Requirements MDR PDFDocument9 pagesEU PMS PSUR Requirements MDR PDFHiral PatelNo ratings yet
- PSUR Template for ATMPsDocument12 pagesPSUR Template for ATMPsKrishna Mohan.p.rNo ratings yet
- PGC.7.1-01-Risk Management ENG # 2021 05 12 # V8.1Document34 pagesPGC.7.1-01-Risk Management ENG # 2021 05 12 # V8.1MarcBenetPozo100% (1)
- 2a. Class A Compression DeviceDocument31 pages2a. Class A Compression DeviceCedric Bonneau100% (1)
- Work Instructions Key Activities When Screening Electronic Reaction Monitoring Reports Ermrs New - enDocument7 pagesWork Instructions Key Activities When Screening Electronic Reaction Monitoring Reports Ermrs New - enVladimir KostovskiNo ratings yet
- Works For Sanofi AventisDocument6 pagesWorks For Sanofi AventisMohit AroraNo ratings yet
- Regulatory RequirementsDocument24 pagesRegulatory RequirementsNikhilesh Naik100% (1)
- 227 20 Unique Device Indentifier UDI For Medical Devices Used in The Gas IndustryDocument16 pages227 20 Unique Device Indentifier UDI For Medical Devices Used in The Gas IndustryMauro CostaNo ratings yet
- Med-Info: Biological EvaluationDocument4 pagesMed-Info: Biological EvaluationRand OmNo ratings yet
- Spentys - Technical File (Face Shield Mask)Document20 pagesSpentys - Technical File (Face Shield Mask)hitham shehataNo ratings yet
- As Far As Possible - en ISO 14971Document19 pagesAs Far As Possible - en ISO 14971Kanwal Jit Singh100% (1)
- Procedure Conducting Pharmacovigilance Inspections Requested CVMP enDocument10 pagesProcedure Conducting Pharmacovigilance Inspections Requested CVMP enJasper Buss HubNo ratings yet
- Resolution No. (24) For 2021Document5 pagesResolution No. (24) For 2021Mohamed EzzatNo ratings yet
- HCF-Guidelines-Proposal Template-2021Document5 pagesHCF-Guidelines-Proposal Template-2021Mohamed EzzatNo ratings yet
- Medical Devices Violations GuideDocument5 pagesMedical Devices Violations GuideMohamed EzzatNo ratings yet
- 20+ Years Operations Manager Seeks New ChallengeDocument2 pages20+ Years Operations Manager Seeks New ChallengeMohamed EzzatNo ratings yet
- Medical Devices Authorized Representatives Check ListDocument1 pageMedical Devices Authorized Representatives Check ListMohamed EzzatNo ratings yet
- MD Companies RegistrationDocument50 pagesMD Companies RegistrationMohamed EzzatNo ratings yet
- Medical Device Registration and Licensing Proposal for NHRADocument2 pagesMedical Device Registration and Licensing Proposal for NHRAMohamed EzzatNo ratings yet
- Inspection Request FormDocument1 pageInspection Request FormMohamed EzzatNo ratings yet
- Your Company Proposal For Ar/Mdr: For Healthcare Facility and Medical Devices Companies in BahrainDocument1 pageYour Company Proposal For Ar/Mdr: For Healthcare Facility and Medical Devices Companies in BahrainMohamed EzzatNo ratings yet
- Process of Authorized Representative RegistrationDocument1 pageProcess of Authorized Representative RegistrationMohamed EzzatNo ratings yet
- Aged Ostafa Haaban: Isa Town, Kingdom of Bahrain +973 66344660Document1 pageAged Ostafa Haaban: Isa Town, Kingdom of Bahrain +973 66344660Mohamed EzzatNo ratings yet
- Adverse Event ProcedureDocument1 pageAdverse Event ProcedureMohamed EzzatNo ratings yet
- No Description No. of Staff No. of Hour: (All Applications Will Be Reviewed Twice For Safety and Quality Purposes)Document1 pageNo Description No. of Staff No. of Hour: (All Applications Will Be Reviewed Twice For Safety and Quality Purposes)Mohamed EzzatNo ratings yet
- Process of Authorized Representative RegistrationDocument1 pageProcess of Authorized Representative RegistrationMohamed EzzatNo ratings yet
- MDD - 11163-2017-CE-KOR-NA-PS Rev. 6.0Document4 pagesMDD - 11163-2017-CE-KOR-NA-PS Rev. 6.0Mohamed EzzatNo ratings yet
- ISO13485 - 10317-2017-AQ-KOR-NA-PS Rev. 2.0. - 20210413Document1 pageISO13485 - 10317-2017-AQ-KOR-NA-PS Rev. 2.0. - 20210413Mohamed EzzatNo ratings yet
- Proposal Ar and MDRDocument3 pagesProposal Ar and MDRMohamed EzzatNo ratings yet
- T4Document23 pagesT4Nicholas MutuaNo ratings yet
- WPI Log 2017.03.26 17.17.44Document4 pagesWPI Log 2017.03.26 17.17.44Mohamed EzzatNo ratings yet
- WPI Log 2017.03.26 17.33.58Document5 pagesWPI Log 2017.03.26 17.33.58Mohamed EzzatNo ratings yet
- Notice to Pay AR Q2 2021 Fees by 28 September 2021Document2 pagesNotice to Pay AR Q2 2021 Fees by 28 September 2021Mohamed EzzatNo ratings yet
- Review Report For RenewDocument5 pagesReview Report For RenewMohamed EzzatNo ratings yet
- S W T O: Swot AnalysisDocument7 pagesS W T O: Swot AnalysisMohamed EzzatNo ratings yet
- Comunicaciones Industriales by Luis MARTINEZ Vicente GUERRERO Ramon YUSTE and Alfaomega Grupo EditorDocument1 pageComunicaciones Industriales by Luis MARTINEZ Vicente GUERRERO Ramon YUSTE and Alfaomega Grupo EditorSalvador Yana RocaNo ratings yet
- Field Safety Notice for Ammar Optician Medical DevicesDocument3 pagesField Safety Notice for Ammar Optician Medical DevicesMohamed EzzatNo ratings yet
- Adverse Event ProcedureDocument1 pageAdverse Event ProcedureMohamed EzzatNo ratings yet
- Description Data Provided: This File Is For Educational Purposes Only. E&OEDocument1 pageDescription Data Provided: This File Is For Educational Purposes Only. E&OEMohamed EzzatNo ratings yet
- ISO 9001 Requirements ChecklistDocument9 pagesISO 9001 Requirements ChecklistMohamed EzzatNo ratings yet
- Supplementary Exercise 3 - Summary StatisticsDocument3 pagesSupplementary Exercise 3 - Summary StatisticsMohamed EzzatNo ratings yet
- Ammar Optician Item ListingDocument2 pagesAmmar Optician Item ListingMohamed EzzatNo ratings yet
- Advanced Operating Systems Fall 2023 AustinDocument11 pagesAdvanced Operating Systems Fall 2023 AustinnntuonNo ratings yet
- 4. Implementing IS-IS over IPv6 on IOS-XE and IOS-XRDocument14 pages4. Implementing IS-IS over IPv6 on IOS-XE and IOS-XRachmmygn9No ratings yet
- 2sc3055 PDFDocument3 pages2sc3055 PDFLuis CansinoNo ratings yet
- A Childrens Guide To Python ProgrammingDocument10 pagesA Childrens Guide To Python ProgrammingEdwin BolivarNo ratings yet
- CI SMD - HST 2-Input OR Gate - NC7ST32 - Marking Code - T32BDocument7 pagesCI SMD - HST 2-Input OR Gate - NC7ST32 - Marking Code - T32BsamuelborgesNo ratings yet
- Q:2 Draw and Explain A Model For Thinking About Ethical, Social and Political Issues?Document5 pagesQ:2 Draw and Explain A Model For Thinking About Ethical, Social and Political Issues?Shahbaz ArtsNo ratings yet
- Synopsis (Customer Billing System)Document3 pagesSynopsis (Customer Billing System)Akash Vashisht50% (2)
- Over Current Protection AnnexDocument20 pagesOver Current Protection Annexryumadmax100% (1)
- C21F60S C21F65 CL21F60S CL21F65: Instructions For UseDocument22 pagesC21F60S C21F65 CL21F60S CL21F65: Instructions For UseJerry MitchellNo ratings yet
- Digital Visual Effects in Cinema PDFDocument271 pagesDigital Visual Effects in Cinema PDFBalaram Anand100% (2)
- IZAR RE 434 PULSE - Installation Guide EN - FRDocument12 pagesIZAR RE 434 PULSE - Installation Guide EN - FRLosp PacmanNo ratings yet
- Daewoo DVN-14F6N, DVN-20F6N Chassis CN-140 TV VCR PDFDocument142 pagesDaewoo DVN-14F6N, DVN-20F6N Chassis CN-140 TV VCR PDFplatonvhmNo ratings yet
- JS2 Computer Studies ExaminationDocument2 pagesJS2 Computer Studies ExaminationEjiro Ndifereke80% (5)
- Counter-Strike 1.6 Servers - Page 49 PDFDocument1 pageCounter-Strike 1.6 Servers - Page 49 PDFMilot AvdyliNo ratings yet
- Yearly Status Report - 2018-2019: Data of The InstitutionDocument53 pagesYearly Status Report - 2018-2019: Data of The InstitutionVignesh DhuruvanNo ratings yet
- Software Construction Coding Methods Reviews StandardsDocument17 pagesSoftware Construction Coding Methods Reviews StandardsPavithra M PaviNo ratings yet
- Microcontroller Based Automatic Power Factor Correction in MinesDocument52 pagesMicrocontroller Based Automatic Power Factor Correction in MinesDdumbaNo ratings yet
- Computer Network SlidesDocument24 pagesComputer Network SlidespsnvaratharajanNo ratings yet
- Build Your Own Guitar Pedal: An Introduction To Electronics: Matt GrayDocument100 pagesBuild Your Own Guitar Pedal: An Introduction To Electronics: Matt GrayAhmed OmarNo ratings yet
- GST Outward ReportDocument31 pagesGST Outward ReportDiptimayeeGuptaNo ratings yet
- Holosun's P.IDDocument3 pagesHolosun's P.IDTwobirds Flying PublicationsNo ratings yet
- China Domestic Style Test ProcessDocument2 pagesChina Domestic Style Test ProcessPopper JohnNo ratings yet
- Serverless Architectures The Evolution of Cloud ComputingDocument9 pagesServerless Architectures The Evolution of Cloud ComputingciaoNo ratings yet
- 3FE77120AAAAFMZZA V1 ISAM R6.4 Customer Release NoteDocument55 pages3FE77120AAAAFMZZA V1 ISAM R6.4 Customer Release Notehadh2311.srovtcNo ratings yet
- Prediction of Heart Disease Using Machine Learning: Aditi Gavhane Gouthami KokkulaDocument4 pagesPrediction of Heart Disease Using Machine Learning: Aditi Gavhane Gouthami KokkulaVishakha UbaleNo ratings yet
- Listas Iptv LatinoDocument25 pagesListas Iptv Latinoandrew SR0% (1)
- Ilovepdf Merged AllDocument47 pagesIlovepdf Merged AllAkash VakkayilNo ratings yet
- Cisco Content Services SwitchDocument210 pagesCisco Content Services SwitchKishoNo ratings yet
- Test 1-Ces525 Question EndorsedDocument4 pagesTest 1-Ces525 Question EndorsedLAYLA SALIKIN CHE ROSMINNo ratings yet
- FALSE POSITIVES C14-Ajuste.exe abincn.dll FILESDocument1 pageFALSE POSITIVES C14-Ajuste.exe abincn.dll FILESsivakumarNo ratings yet