You might also like
- Concepts in Pharmaceutical Science Midterm PSC G100 Fall, 2005, Dr. Loring's SectionDocument4 pagesConcepts in Pharmaceutical Science Midterm PSC G100 Fall, 2005, Dr. Loring's Sectionapi-19439852No ratings yet
- Thia Zoli Dined I OnesDocument6 pagesThia Zoli Dined I OnesSoumyadeep MondalNo ratings yet
- 10 1016@j BMCL 2019 126712Document10 pages10 1016@j BMCL 2019 126712Omar Nassir MoftahNo ratings yet
- International Journal of Pharmtech Research: Sweety Saini, Chandana Majee, Gunosindhu Chakraborthy, SalahuddinDocument14 pagesInternational Journal of Pharmtech Research: Sweety Saini, Chandana Majee, Gunosindhu Chakraborthy, SalahuddinHarshadbhai S.PrajapatiNo ratings yet
- Medicinal Chemistry & Drug Discovery: Dr. Peter Wipf Department of Chemistry University of PittsburghDocument69 pagesMedicinal Chemistry & Drug Discovery: Dr. Peter Wipf Department of Chemistry University of PittsburghKhushi KhanNo ratings yet
- Therapeutic Review Exploring Antimicrobial PotentiDocument19 pagesTherapeutic Review Exploring Antimicrobial PotentiAdithya BallalNo ratings yet
- Chemistry SynopsisDocument7 pagesChemistry SynopsisDr Prashant Shihora100% (1)
- Marine Natural Products Against TuberculosisDocument16 pagesMarine Natural Products Against TuberculosisAndreea MoalesNo ratings yet
- molecules-20-03821Document20 pagesmolecules-20-03821putryapurnomo.21No ratings yet
- Repaglinide: Chemical Name: (S) - (Document4 pagesRepaglinide: Chemical Name: (S) - (Rhenso Victor Albites CondoriNo ratings yet
- Research ArticleDocument7 pagesResearch Articleana ulfaNo ratings yet
- Novel Heterocyclyl Linked Benzaldehydes and Anilines for Biologically Active CompoundsDocument7 pagesNovel Heterocyclyl Linked Benzaldehydes and Anilines for Biologically Active CompoundsDr. Bharat SutharNo ratings yet
- Pharmacological and Synthetic Profile of Benzothiazepine: A ReviewDocument9 pagesPharmacological and Synthetic Profile of Benzothiazepine: A ReviewRao Zikar IslamNo ratings yet
- N-Demethylation of Alkaloids: A Key Transformation in Drug SynthesisDocument13 pagesN-Demethylation of Alkaloids: A Key Transformation in Drug SynthesisPhuongNo ratings yet
- Practical Approach For The Determination of ResponDocument8 pagesPractical Approach For The Determination of ResponPu ZhaoNo ratings yet
- Zeocin ManDocument24 pagesZeocin ManThaís Paiva Porto de SouzaNo ratings yet
- Pharmaceuticals: Nonsteroidal Anti-Inflammatory Drugs (Nsaids) : Progress in Small Molecule Drug DevelopmentDocument20 pagesPharmaceuticals: Nonsteroidal Anti-Inflammatory Drugs (Nsaids) : Progress in Small Molecule Drug Developmentlachi nadiaNo ratings yet
- Synthesis and Biological Evaluation of New 4-CarboxylDocument21 pagesSynthesis and Biological Evaluation of New 4-Carboxylglreddy09No ratings yet
- Alkaloid S1Document142 pagesAlkaloid S1Faisal MNo ratings yet
- MOLECULES IN 3DDocument6 pagesMOLECULES IN 3DtacosanchezbrayanNo ratings yet
- Prof Dai Hibbs David - Hibbs@sydney - Edu.au 9351 6005 RM N519Document27 pagesProf Dai Hibbs David - Hibbs@sydney - Edu.au 9351 6005 RM N519Sophia LiuNo ratings yet
- Design Synthesis of Novel N Prenylated Indole-3-CaDocument16 pagesDesign Synthesis of Novel N Prenylated Indole-3-Ca275 DILIP RAYGORNo ratings yet
- Synthesis, Characterization and Bioassay of Novel Substituted 1 - (3 - (1,3-Thiazol-2-Yl) Phenyl) - 5-OxopyrrolidinesDocument20 pagesSynthesis, Characterization and Bioassay of Novel Substituted 1 - (3 - (1,3-Thiazol-2-Yl) Phenyl) - 5-OxopyrrolidinesAna-Maria CiobotaruNo ratings yet
- Monomers Oligomers Polymers MacromoleculesDocument6 pagesMonomers Oligomers Polymers MacromoleculesQuar Tul AinNo ratings yet
- 1 s2.0 S0960894X09013183 mmc1Document7 pages1 s2.0 S0960894X09013183 mmc1Gourab SahaNo ratings yet
- New losartan-hydrocaffeic acid hybrids as dual antihypertensive-antioxidant drugsDocument12 pagesNew losartan-hydrocaffeic acid hybrids as dual antihypertensive-antioxidant drugsmiss missNo ratings yet
- ARTICULO - Dual Drugs - 2012 PDFDocument12 pagesARTICULO - Dual Drugs - 2012 PDFPablo Maldonado MuñozNo ratings yet
- Chem SketchDocument17 pagesChem SketchUsman GhaniNo ratings yet
- Mcmurry PDFDocument119 pagesMcmurry PDFRicardo SierraNo ratings yet
- A Systematic Investigation For The Preparation of A Novel Polycyclic Ring Systems by Reductive Cyclization With LAHDocument29 pagesA Systematic Investigation For The Preparation of A Novel Polycyclic Ring Systems by Reductive Cyclization With LAHVenugopal Rao VeeramaneniNo ratings yet
- Synthesis and Biological Evaluation of Some Thiazolyl and Thiadiazolyl Derivatives of 1 H-Pyrazole As Anti-Inflammatory Antimicrobial AgentsDocument8 pagesSynthesis and Biological Evaluation of Some Thiazolyl and Thiadiazolyl Derivatives of 1 H-Pyrazole As Anti-Inflammatory Antimicrobial AgentsWalid EbaiedNo ratings yet
- 6597363Document8 pages6597363Nandhy AgusNo ratings yet
- Synthesis and Antibacterial Activity of 35methyl1phenyl1h123triazol4yl6aryl7h124triazolo34b134thiadiazineDocument10 pagesSynthesis and Antibacterial Activity of 35methyl1phenyl1h123triazol4yl6aryl7h124triazolo34b134thiadiazineFinn NelsonNo ratings yet
- On The Electrophilic Reactivities of Acarbonyl Heterocylces and ArenesDocument7 pagesOn The Electrophilic Reactivities of Acarbonyl Heterocylces and ArenesFinn NelsonNo ratings yet
- 1a-Introduction To Pharmaceutical Chemistry and Drug ClassificationDocument23 pages1a-Introduction To Pharmaceutical Chemistry and Drug ClassificationsedasunkarNo ratings yet
- Karboksilne Kisline: Analiza in Nadzor Zdravil Fakulteta Za Farmacijo Oktober 2009Document57 pagesKarboksilne Kisline: Analiza in Nadzor Zdravil Fakulteta Za Farmacijo Oktober 2009api-3814389No ratings yet
- Chem 44.1 Special SynthesisDocument86 pagesChem 44.1 Special SynthesisCarlo Joseph Moskito100% (2)
- Cyclohexane 1,3 DioneDocument1 pageCyclohexane 1,3 DionetreemaddogNo ratings yet
- Preparation of MaleimideDocument11 pagesPreparation of MaleimideDotsha Raheem100% (1)
- Substrate-B reference compoundDocument2 pagesSubstrate-B reference compoundsauronsauronNo ratings yet
- Synthesis of MoleculesDocument25 pagesSynthesis of MoleculesAmit GohriNo ratings yet
- Harigopal S Sawarkar 2020 SYNTHESIS AND ANTIMICROBIAL ACTIVITIES OF NOVEL 1,3,4-THIADIAZOLE BEARING CARBOXAMIDES DERIVATIVESDocument10 pagesHarigopal S Sawarkar 2020 SYNTHESIS AND ANTIMICROBIAL ACTIVITIES OF NOVEL 1,3,4-THIADIAZOLE BEARING CARBOXAMIDES DERIVATIVESshubha shrivastavaNo ratings yet
- B EnzymesDocument33 pagesB Enzymesshashikant shingdilwarNo ratings yet
- Progress Report of Anamica T3Document2 pagesProgress Report of Anamica T3Jonathan HicksNo ratings yet
- Oxidative Stress Regulation On Endothelial CellsDocument15 pagesOxidative Stress Regulation On Endothelial CellsHelenaNo ratings yet
- ExplainerDocument17 pagesExplainerbruhstfuxdNo ratings yet
- Bioisosterismo - RevisãoDocument27 pagesBioisosterismo - RevisãoCLARA VITORIA CAVALCANTE CARVALHONo ratings yet
- ###Med-chem-Questions On Beta Lactam AntibioticsDocument3 pages###Med-chem-Questions On Beta Lactam AntibioticsDave DM100% (2)
- Acylated Flavone Glycosides From The Roots of Saussurea LappaDocument17 pagesAcylated Flavone Glycosides From The Roots of Saussurea LappaTuan NguyenNo ratings yet
- Aggoun Etal 2021 JMolStruct PreprintDocument38 pagesAggoun Etal 2021 JMolStruct PreprintAman AmanNo ratings yet
- Synthesis of N, N-diethyl-N-aminopropylpoly (Oxyethylene) AmineDocument39 pagesSynthesis of N, N-diethyl-N-aminopropylpoly (Oxyethylene) AmineJhon SandovalNo ratings yet
- Antisense Oligonucleotide Biotechnology, Applications and FutureDocument29 pagesAntisense Oligonucleotide Biotechnology, Applications and FuturesurojitarpitaNo ratings yet
- DM pp21-40Document20 pagesDM pp21-40MLUNGISI MkhwanaziNo ratings yet
- An Overview of Alkaloids: Their Classification, Extraction, Isolation, and Role in PlantsDocument49 pagesAn Overview of Alkaloids: Their Classification, Extraction, Isolation, and Role in PlantsmanishaNo ratings yet
- Activity 3 - The Cahn-Ingold-Prelog Rules - 0Document5 pagesActivity 3 - The Cahn-Ingold-Prelog Rules - 0Naved AnjoomNo ratings yet
- GlysocideDocument70 pagesGlysocideAlan K MhamadNo ratings yet
- Lectures in Heterocyclic ChemistryDocument130 pagesLectures in Heterocyclic ChemistryNorman FerdinalNo ratings yet
- E12 AtqDocument5 pagesE12 AtqCharlene InaoNo ratings yet
- Talaz 2010Document9 pagesTalaz 2010LuckyNo ratings yet
- Farmakokinetika Vitamin C PDFDocument20 pagesFarmakokinetika Vitamin C PDFKonsul Dosen PembibingNo ratings yet
- Piroxicamarticle PDFDocument7 pagesPiroxicamarticle PDFPriska FebiolaNo ratings yet
- Structural Elucidation of Small Organic Molecules by 1d 2d and Multi Dimensional Solution NMR Spectroscopy 2155 9872.S11 001Document8 pagesStructural Elucidation of Small Organic Molecules by 1d 2d and Multi Dimensional Solution NMR Spectroscopy 2155 9872.S11 001PriyosetyokoNo ratings yet
- Chapter 2 Priyosetyoko 24030120420007Document16 pagesChapter 2 Priyosetyoko 24030120420007PriyosetyokoNo ratings yet
- Jurnal AntioxidantDocument32 pagesJurnal AntioxidantPriyosetyokoNo ratings yet
- Air, Water and Land Pollution: Chromatographic Methods For Environmental AnalysisDocument90 pagesAir, Water and Land Pollution: Chromatographic Methods For Environmental AnalysisPriyosetyokoNo ratings yet
- HPLC For UndipDocument93 pagesHPLC For UndipPriyosetyokoNo ratings yet
- Basics of LCMSDocument24 pagesBasics of LCMSvkontoyianni1446No ratings yet
- Basics of LCMSDocument24 pagesBasics of LCMSvkontoyianni1446No ratings yet
- Msds Dietil EterijijDocument6 pagesMsds Dietil EterijijPriyosetyokoNo ratings yet
- Air, Water and Land Pollution: Chromatographic Methods For Environmental AnalysisDocument90 pagesAir, Water and Land Pollution: Chromatographic Methods For Environmental AnalysisPriyosetyokoNo ratings yet
- Bioorganic Synthesis - An Introduction (PDFDrive)Document453 pagesBioorganic Synthesis - An Introduction (PDFDrive)PriyosetyokoNo ratings yet
- 108 364 1 PBDocument6 pages108 364 1 PBPriyosetyokoNo ratings yet
- Sodium Acetate MsdsDocument5 pagesSodium Acetate MsdsMehmet Besim SacilikNo ratings yet
- Methanol Energy BalanceDocument6 pagesMethanol Energy BalancePriyosetyoko100% (2)
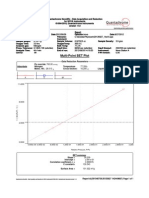
- Graph - Multi-Point BET Plot - 20120627 - 1kel 6Document1 pageGraph - Multi-Point BET Plot - 20120627 - 1kel 6PriyosetyokoNo ratings yet
- 008x TF-compressed PDFDocument66 pages008x TF-compressed PDFJosé Francisco Blanco VillalbaNo ratings yet
- Narrative Report For Environmental Protection Awareness ProgramDocument3 pagesNarrative Report For Environmental Protection Awareness ProgramFam ValerianoNo ratings yet
- BiochemistryDocument410 pagesBiochemistryCaptainReeham79% (14)
- Lab Technician Interview QuestionsDocument3 pagesLab Technician Interview QuestionsCandra AprizalNo ratings yet
- It ThesisDocument59 pagesIt Thesisroneldayo62100% (2)
- 2019 Cre Ii L24-26Document32 pages2019 Cre Ii L24-26Aman PrasadNo ratings yet
- BSC 6yh Sem Kuk SyllabusDocument8 pagesBSC 6yh Sem Kuk SyllabusVicky ChaharNo ratings yet
- Balauro Worksheet Protein SynthesisDocument4 pagesBalauro Worksheet Protein SynthesisHami BalauroNo ratings yet
- NEUROPHYSIOLOGYDocument224 pagesNEUROPHYSIOLOGYKheliwi100% (2)
- Determination of HHV of Diesel FuelDocument7 pagesDetermination of HHV of Diesel FuelAdrian Soriaga LontocNo ratings yet
- SDS Body Mist GenericDocument4 pagesSDS Body Mist Genericsabuyexpress.worldwideNo ratings yet
- MiniProject Stage 3 - Process Dynamic & ControlDocument8 pagesMiniProject Stage 3 - Process Dynamic & ControlFarihah Eyfa100% (2)
- Dosing PumpsDocument2 pagesDosing PumpsSherlockNo ratings yet
- MT12550FTDocument1 pageMT12550FTJuan carlosNo ratings yet
- Journal 9 PDFDocument45 pagesJournal 9 PDFRuzengulalebih ZEta's-ListikNo ratings yet
- Japanning 103, Traditional Japanning, The BlacDocument8 pagesJapanning 103, Traditional Japanning, The BlacFredy Alvarez LucasNo ratings yet
- Bomba Dosificadora Iwaki PDFDocument29 pagesBomba Dosificadora Iwaki PDFMaykolth BarrantesNo ratings yet
- DrillersManual Chapters 1 12Document192 pagesDrillersManual Chapters 1 12Hugo MoralesNo ratings yet
- IB Chemistry HL Summer AssignmentDocument4 pagesIB Chemistry HL Summer AssignmentVal B100% (1)
- Sensory and nutritional value of flatbread with banana peelsWheat flourBanana peels12.2010.1011.203.201.2111.201.500.800.601.20Document7 pagesSensory and nutritional value of flatbread with banana peelsWheat flourBanana peels12.2010.1011.203.201.2111.201.500.800.601.20Bagas Aryo SasongkoNo ratings yet
- Design Manual For Waste Stabilization Ponds in India BY MARA D.Document135 pagesDesign Manual For Waste Stabilization Ponds in India BY MARA D.cherogonyaNo ratings yet
- Ad 01202010214Document14 pagesAd 01202010214theijesNo ratings yet
- Item Codes for Pokémon Items & ModifiersDocument13 pagesItem Codes for Pokémon Items & ModifiersIsaccNo ratings yet
- WWW - Ncbi.nlm - Nih.gov Pubmed 15151274Document2 pagesWWW - Ncbi.nlm - Nih.gov Pubmed 15151274Ethan MorganNo ratings yet
- Product UserManual Pulsarlube V EnglishDocument2 pagesProduct UserManual Pulsarlube V EnglishTiago LimaNo ratings yet
- Storage ProteinDocument3 pagesStorage ProteinprincessicyjulietNo ratings yet
- Biology Cells Graphic OrganizerDocument1 pageBiology Cells Graphic OrganizerMaci StackhouseNo ratings yet
- Daftar Lazim Obat Dewasa Dan Anak, Bayi: AntibiotikDocument8 pagesDaftar Lazim Obat Dewasa Dan Anak, Bayi: AntibiotikkunkunNo ratings yet
- 2012 - Cosmetic Ingredient Review - Amended Safety Assessment of Alkyl Esters As Used in CosmeticsDocument83 pages2012 - Cosmetic Ingredient Review - Amended Safety Assessment of Alkyl Esters As Used in CosmeticsymiyazyNo ratings yet