You might also like
- Orthodontically Driven Corticotomy: Tissue Engineering to Enhance Orthodontic and Multidisciplinary TreatmentFrom EverandOrthodontically Driven Corticotomy: Tissue Engineering to Enhance Orthodontic and Multidisciplinary TreatmentFederico BrugnamiNo ratings yet
- Scanning Electron Microscopy of Bone: Methods in Molecular Medicine February 2003Document11 pagesScanning Electron Microscopy of Bone: Methods in Molecular Medicine February 2003kyuleen05No ratings yet
- Paper58 Mechanical Properties Hierarchical Structure Bone Med Eng Phys 20 (2) Rho Et Al 1998Document11 pagesPaper58 Mechanical Properties Hierarchical Structure Bone Med Eng Phys 20 (2) Rho Et Al 1998leksremeshNo ratings yet
- Biomimetic Porous Titanium Scaffolds For Orthopedic and Dental ApplicationsDocument36 pagesBiomimetic Porous Titanium Scaffolds For Orthopedic and Dental ApplicationsSocarisNo ratings yet
- 337 PDFDocument6 pages337 PDFBui Trung TuyenNo ratings yet
- Materials: Metallic Scaffolds For Bone RegenerationDocument43 pagesMaterials: Metallic Scaffolds For Bone RegenerationWookyoung LeeNo ratings yet
- Acta Biomaterialia: Rohit Khanna, Kalpana S. Katti, Dinesh R. KattiDocument11 pagesActa Biomaterialia: Rohit Khanna, Kalpana S. Katti, Dinesh R. KattiRohit KhannaNo ratings yet
- Research Article Comparison of Structural, Architectural and Mechanical Aspects of Cellular and Acellular Bone in Two Teleost FishDocument11 pagesResearch Article Comparison of Structural, Architectural and Mechanical Aspects of Cellular and Acellular Bone in Two Teleost FishcamilaNo ratings yet
- 1 - 2020 - Water-Lipid Interactions in Native Bone by High-Resolution Solid-State NMR Spectroscopy - CompressedDocument7 pages1 - 2020 - Water-Lipid Interactions in Native Bone by High-Resolution Solid-State NMR Spectroscopy - CompressedNidhi TiwariNo ratings yet
- Rho 1998Document11 pagesRho 1998Catherine NocuaNo ratings yet
- Collagen TreatmentDocument8 pagesCollagen TreatmentAlmiraNadiaNo ratings yet
- Aplikasi Penggunaan Micro CTDocument7 pagesAplikasi Penggunaan Micro CTZannuraini AZNo ratings yet
- 4b PDFDocument43 pages4b PDFsbhushanarya2546No ratings yet
- BoneTissueandBiomaterialDesignBasedontheAnisotropicMicrostreucture-TakayoshiNAKANOetalAdvances in Metallic BiomaterDocument32 pagesBoneTissueandBiomaterialDesignBasedontheAnisotropicMicrostreucture-TakayoshiNAKANOetalAdvances in Metallic BiomaterCatherine NocuaNo ratings yet
- Materials Science & Engineering C: SciencedirectDocument9 pagesMaterials Science & Engineering C: SciencedirectSantiago MorenoNo ratings yet
- Artificial Bone: Jump To Navigation Jump To SearchDocument8 pagesArtificial Bone: Jump To Navigation Jump To SearchIzabella LázárNo ratings yet
- LID201112052004Document10 pagesLID201112052004Gia BảoNo ratings yet
- Obs-13 240122 184549Document51 pagesObs-13 240122 184549Edison MesacheNo ratings yet
- 1 s2.0 S8756328207006205 MainDocument12 pages1 s2.0 S8756328207006205 Mainhakan sonsuzlukluNo ratings yet
- Osteoblasts, Osteoclasts, and Osteocytes: Unveiling Their Intimate-Associated Responses To Applied Orthodontic ForcesDocument12 pagesOsteoblasts, Osteoclasts, and Osteocytes: Unveiling Their Intimate-Associated Responses To Applied Orthodontic ForcesMariya NazimNo ratings yet
- Kim, Chang, Jung - 2010 - The Finite Element Analysis of A Fractured Tibia Applied by Composite Bone Plates Considering Contact ConditioDocument10 pagesKim, Chang, Jung - 2010 - The Finite Element Analysis of A Fractured Tibia Applied by Composite Bone Plates Considering Contact ConditioMorteza AtaeiNo ratings yet
- Chapter 1Document21 pagesChapter 1Fernando Melo GarciaNo ratings yet
- Resorptive Changes of Maxillary and Mandibular Bone Structures in Removable Denture WearersDocument5 pagesResorptive Changes of Maxillary and Mandibular Bone Structures in Removable Denture WearersAhmed SamirNo ratings yet
- Gillies Et Al ANZ J. Surg. 2007 - 77 69-72Document4 pagesGillies Et Al ANZ J. Surg. 2007 - 77 69-72rmg1198sNo ratings yet
- Tissue Changes1Document22 pagesTissue Changes1Ishtiaq HasanNo ratings yet
- ATWOODDocument15 pagesATWOODwaf51No ratings yet
- 00-Age Variations in The Properties of Human Tibial Trabecular Bone and CartilageDocument48 pages00-Age Variations in The Properties of Human Tibial Trabecular Bone and CartilageYONATAN FABIAN TOVAR TOVARNo ratings yet
- Analysis of Bone Tissue Mechanical Properties: Diana Mil I), Jadranka Keros and Andrija Bo (NjakDocument7 pagesAnalysis of Bone Tissue Mechanical Properties: Diana Mil I), Jadranka Keros and Andrija Bo (Njaksamuel rajNo ratings yet
- Articulo2 2Document34 pagesArticulo2 2DanielaSofiaRubianoBlancoNo ratings yet
- Bone Composite AnalysisDocument10 pagesBone Composite AnalysiskritiNo ratings yet
- Review On Calcium-And Magnesium-Based Silicates For Bone Tissue Engineering ApplicationsDocument17 pagesReview On Calcium-And Magnesium-Based Silicates For Bone Tissue Engineering ApplicationsWeihao CaiNo ratings yet
- Vật Liệu & Kỹ Thuật Y Sinh: Biomedical Materials and EngineeringDocument57 pagesVật Liệu & Kỹ Thuật Y Sinh: Biomedical Materials and EngineeringkhuongdaihuynhNo ratings yet
- Advancing Cancer DiagnosticsDocument17 pagesAdvancing Cancer DiagnosticsinzamamyoonusNo ratings yet
- Materials 14 04224 v3Document16 pagesMaterials 14 04224 v3B01Aadarsh SharmaNo ratings yet
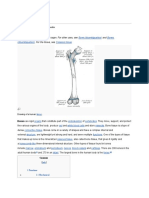
- This Article Is About The Skeletal Organ. For Other Uses, See and - For The Tissue, SeeDocument16 pagesThis Article Is About The Skeletal Organ. For Other Uses, See and - For The Tissue, SeeLouie George NeriNo ratings yet
- Osteocytic Osteolysis: Time For A Second Look?: ReviewDocument7 pagesOsteocytic Osteolysis: Time For A Second Look?: ReviewShahid HussainNo ratings yet
- Osteon: Structure Investigative Applications See Also References Bibliography External LinksDocument3 pagesOsteon: Structure Investigative Applications See Also References Bibliography External LinksAlan FosterNo ratings yet
- Porose Qualidade Ossea New England 2010Document12 pagesPorose Qualidade Ossea New England 2010Ben-Hur AlbergariaNo ratings yet
- Bone Substitute Materials in Implant DentistryDocument34 pagesBone Substitute Materials in Implant DentistryYun De LinNo ratings yet
- Bone Graft and Gbr1Document34 pagesBone Graft and Gbr1Dr. Nachammai NagarajanNo ratings yet
- 1 s2.0 S0968432813001510 MainDocument8 pages1 s2.0 S0968432813001510 MainEmon BaruaNo ratings yet
- 10 1016@j Mtla 2019 100573 PDFDocument36 pages10 1016@j Mtla 2019 100573 PDFimen mehriNo ratings yet
- PCB File 2Document25 pagesPCB File 2Sahil JethwaniNo ratings yet
- Bone Remodeling Process: Mechanics, Biology, and Numerical ModelingFrom EverandBone Remodeling Process: Mechanics, Biology, and Numerical ModelingNo ratings yet
- Calcium Phosphate-Based Osteoinductive Materials: Racquel Zapanta LegerosDocument12 pagesCalcium Phosphate-Based Osteoinductive Materials: Racquel Zapanta LegerosIrsyaNo ratings yet
- Infrared Spectroscopy Reveals Both Qualitative and Quantitative Differences in Equine Subchondral Bone During MaturationDocument9 pagesInfrared Spectroscopy Reveals Both Qualitative and Quantitative Differences in Equine Subchondral Bone During MaturationYevgeniya KobrinaNo ratings yet
- Pengaruh Milling Time Terhadap Sifat Fisik Dan Mekanik Hidroksiapatit Berbasis Batu KapurDocument7 pagesPengaruh Milling Time Terhadap Sifat Fisik Dan Mekanik Hidroksiapatit Berbasis Batu Kapuragus priyantoNo ratings yet
- BP MecanismDocument14 pagesBP MecanismAdryana StefyNo ratings yet
- Abel Chapter OneDocument6 pagesAbel Chapter OnebryanbotofficialmailNo ratings yet
- Morphologic and Nanomechanical Characterization of Bone Tissue Growth AroundDocument8 pagesMorphologic and Nanomechanical Characterization of Bone Tissue Growth AroundTina TinaNo ratings yet
- Tereos - Structure of PresentationDocument72 pagesTereos - Structure of PresentationSumitNo ratings yet
- Composite Structures: Hassan Mehboob, Seung-Hwan ChangDocument8 pagesComposite Structures: Hassan Mehboob, Seung-Hwan ChangRafael ZanettiNo ratings yet
- Differentiating Bone Osteonal Turnover Rates by Density Fractionation Validation Using The Bomb C Atmospheric PulseDocument9 pagesDifferentiating Bone Osteonal Turnover Rates by Density Fractionation Validation Using The Bomb C Atmospheric PulseJuan PerezNo ratings yet
- Surface Texturing of Implant MaterialsDocument17 pagesSurface Texturing of Implant MaterialsYashi VermaNo ratings yet
- Normal Bone Anatomy and PhysiologyDocument9 pagesNormal Bone Anatomy and PhysiologyindrapratisthaNo ratings yet
- 1 s2.0 S0927776521005609 MainDocument11 pages1 s2.0 S0927776521005609 Mainhoney10978No ratings yet
- Silicate Nanocomposites Enhance Stem Cell OsteogenesisDocument15 pagesSilicate Nanocomposites Enhance Stem Cell OsteogenesisRuchi ShahNo ratings yet
- Cellular Mechanisms of Bone RemodelingDocument9 pagesCellular Mechanisms of Bone RemodelingSilvia AlexandraNo ratings yet
- FabriationDocument8 pagesFabriationBendaud bataborNo ratings yet
- Bone Erosion Rheumatoid Arthritis Mechanisms Diagnosis&TreatmentDocument9 pagesBone Erosion Rheumatoid Arthritis Mechanisms Diagnosis&TreatmentMaría RodríguezNo ratings yet
- INSISIVDocument7 pagesINSISIVLansky TrimeilanaNo ratings yet
- Jurnal XRD AnalisisDocument12 pagesJurnal XRD AnalisisLansky TrimeilanaNo ratings yet
- Ha 4 FiDocument6 pagesHa 4 FiLansky TrimeilanaNo ratings yet
- Journal of Forensic and Legal Medicine: Research PaperDocument5 pagesJournal of Forensic and Legal Medicine: Research PaperAnonymous jqztWYNo ratings yet
- X-ray Diffraction Techniques for Polymer AnalysisDocument37 pagesX-ray Diffraction Techniques for Polymer AnalysisYuvaraj DhandapaniNo ratings yet
- 02 BMFG 1213 - Atomic Structure and Bonding PDFDocument48 pages02 BMFG 1213 - Atomic Structure and Bonding PDFHalizah RamthanNo ratings yet
- Design and Synthesis 2D Coordination Networks - Crcl2 (Pyz) 2 and CR (Oso2Ch3) 2 (Pyz) 2 (Pyz Pyrazine) Framework MagnetsDocument11 pagesDesign and Synthesis 2D Coordination Networks - Crcl2 (Pyz) 2 and CR (Oso2Ch3) 2 (Pyz) 2 (Pyz Pyrazine) Framework MagnetsMercyJatindroNo ratings yet
- AssignmentI InorgDocument3 pagesAssignmentI InorgCreative ThinkerNo ratings yet
- (WEEK 1) (Chemistry) Preparatory Learning ActivityDocument6 pages(WEEK 1) (Chemistry) Preparatory Learning Activity12 STEM Kyla Jean TantoyNo ratings yet
- Hybrid nanomembranes: A case where micro and nano complement each other through self-assemblyDocument36 pagesHybrid nanomembranes: A case where micro and nano complement each other through self-assemblysfantNo ratings yet
- ATOMIC STRUCTURE & PERIODIC TABLEDocument25 pagesATOMIC STRUCTURE & PERIODIC TABLEstan AB6IXNo ratings yet
- MSE 510 - Microstructural Characterization Techniques - Lecture 22Document29 pagesMSE 510 - Microstructural Characterization Techniques - Lecture 22Aneek M. NoorNo ratings yet
- Assignment 1 SCOD 2Document11 pagesAssignment 1 SCOD 2Benzene Free Fire100% (1)
- Optoelectronics SyllabusDocument4 pagesOptoelectronics SyllabusabdulbabulNo ratings yet
- Essentials of QUANTUM MECHANICSDocument4 pagesEssentials of QUANTUM MECHANICSliv2luvNo ratings yet
- Midterm MTE111 S2014 With SolutionDocument7 pagesMidterm MTE111 S2014 With SolutionVarij GosineNo ratings yet
- DCAM PART 66 CAT B1.1 MODULE 4 ELECTRONIC FUNDAMENTALS SEMICONDUCTORSDocument114 pagesDCAM PART 66 CAT B1.1 MODULE 4 ELECTRONIC FUNDAMENTALS SEMICONDUCTORSwmfadzli7265100% (2)
- Class 7 - 26thaugust1Document31 pagesClass 7 - 26thaugust1AADESH GUPTANo ratings yet
- NCERT Solutions For Class 11 Chemistry 15may Chapter 3 Classification of Elements and Periodicity in PropertiesDocument18 pagesNCERT Solutions For Class 11 Chemistry 15may Chapter 3 Classification of Elements and Periodicity in PropertiesChanchal KumariNo ratings yet
- Renormalization Made Easy, BaezDocument11 pagesRenormalization Made Easy, BaezdbranetensionNo ratings yet
- SummerSch Intro Lewis 2015Document115 pagesSummerSch Intro Lewis 2015Sareh SabetNo ratings yet
- Chapter 5 SolutionDocument11 pagesChapter 5 SolutiongglrNo ratings yet
- Engineering Physics - D. K. BhattacharyaDocument85 pagesEngineering Physics - D. K. Bhattacharyanisar akramNo ratings yet
- Ch4 Answers Asal Chem WBDocument5 pagesCh4 Answers Asal Chem WBamelia.szygendaNo ratings yet
- Introduction to MRI: Understanding Magnetic Properties & Relaxation TimesDocument16 pagesIntroduction to MRI: Understanding Magnetic Properties & Relaxation TimesjuhiNo ratings yet
- 5 Recovery RecrystallizationDocument116 pages5 Recovery Recrystallizationoğuz cantürkNo ratings yet
- Chemistry 1 11 Q2 M1Document14 pagesChemistry 1 11 Q2 M1Gaelle Mariposa100% (1)
- PH211 2019 1 NaCl Singlecrystals PhyweDocument7 pagesPH211 2019 1 NaCl Singlecrystals PhyweNANDEESH KUMAR K MNo ratings yet
- Integrated Electronics - Millman HalkiasDocument957 pagesIntegrated Electronics - Millman HalkiasLEKSHMI ANILKUMARNo ratings yet
- Chapter # 16 Electromagnetics Waves and ElectronicsDocument6 pagesChapter # 16 Electromagnetics Waves and ElectronicsSIR USMAN KHAN100% (1)
- Effect of Temperature On Photovoltaic Solar Energy ConversionDocument9 pagesEffect of Temperature On Photovoltaic Solar Energy ConversionAlina AlexandriucNo ratings yet
- Solid StateDocument42 pagesSolid StateVIMAL MEHTANo ratings yet
- Superconductivity - Theory and ApplicationsDocument358 pagesSuperconductivity - Theory and ApplicationsKosygin LeishangthemNo ratings yet
- Introduction To Silvaco ATHENA Tool and Basic Concepts in Process Modeling 1Document18 pagesIntroduction To Silvaco ATHENA Tool and Basic Concepts in Process Modeling 1engahmed25No ratings yet