You might also like
- Synthesis and Characterization of Cnt/Ce-Tio Nanocomposite For Phenol DegradationDocument8 pagesSynthesis and Characterization of Cnt/Ce-Tio Nanocomposite For Phenol DegradationŞebnem Gül İlarslanNo ratings yet
- Diamond & Related Materials: SciencedirectDocument10 pagesDiamond & Related Materials: SciencedirectŞebnem Gül İlarslanNo ratings yet
- Journal of Photochemistry and Photobiology A: ChemistryDocument9 pagesJournal of Photochemistry and Photobiology A: ChemistryIntenNo ratings yet
- Belhadjltaief 2016Document9 pagesBelhadjltaief 2016Blessing HarvestNo ratings yet
- Bor 8Document8 pagesBor 8petru apopeiNo ratings yet
- 1-s2.0-S0926860X05003054-mainDocument11 pages1-s2.0-S0926860X05003054-mainŞebnem İlarslanNo ratings yet
- 1 s2.0 S2213343720302657 MainDocument9 pages1 s2.0 S2213343720302657 MainThu Trang NguyenNo ratings yet
- Enhanced Visible-Light Photocatalytic Performance of Electrospun Carbon-Doped TiO2 Halloysite Nanotube Hybrid NanofibersDocument7 pagesEnhanced Visible-Light Photocatalytic Performance of Electrospun Carbon-Doped TiO2 Halloysite Nanotube Hybrid NanofibersBachi NanoNo ratings yet
- 27 KTXTDocument14 pages27 KTXTHoàng NhưNo ratings yet
- S027288422102722XDocument9 pagesS027288422102722Xahmad.faizal.2005136No ratings yet
- Zn-Cu Promoted TiO2 Photocatalyst For CO2 Reduction With H2O Under UV LightDocument9 pagesZn-Cu Promoted TiO2 Photocatalyst For CO2 Reduction With H2O Under UV Lightleilasalimleal_27406No ratings yet
- Accepted Manuscript: Journal of Photochemistry and Photobiology A: ChemistryDocument39 pagesAccepted Manuscript: Journal of Photochemistry and Photobiology A: ChemistryYon tinyayaNo ratings yet
- A TiO2 Coated Carbon Aerogel Derived From Bamboo Pulp Fibers For Enhanced Visible Light Photo-Catalytic Degradation of Methylene BlueDocument12 pagesA TiO2 Coated Carbon Aerogel Derived From Bamboo Pulp Fibers For Enhanced Visible Light Photo-Catalytic Degradation of Methylene BlueANH TRẦN HUỆNo ratings yet
- Gaurav PaperDocument10 pagesGaurav PaperGaurav KumarNo ratings yet
- Jurnal Modifikasi TiO2Document18 pagesJurnal Modifikasi TiO2Nadia fadlNo ratings yet
- Recent Developments of Tio - Based Photocatalysis in The Hydrogen Evolution and Photodegradation: A ReviewDocument16 pagesRecent Developments of Tio - Based Photocatalysis in The Hydrogen Evolution and Photodegradation: A ReviewwaniNo ratings yet
- Applied Catalysis B: EnvironmentalDocument7 pagesApplied Catalysis B: EnvironmentalDaniel MontalvoNo ratings yet
- Synergetic Photocatalytic-Activity Enhancement of Lanthanum Doped TiO2 On Halloysite Nanocomposites For Degradation of Organic DyeDocument8 pagesSynergetic Photocatalytic-Activity Enhancement of Lanthanum Doped TiO2 On Halloysite Nanocomposites For Degradation of Organic DyeBachi NanoNo ratings yet
- Sonochemical Synthesis of Tio2 Nanoparticles On Graphene For Use As PhotocatalystDocument9 pagesSonochemical Synthesis of Tio2 Nanoparticles On Graphene For Use As PhotocatalystBryanda ReyesNo ratings yet
- Application of Tio2-Mounted Activated Carbon To The Removal of Phenol From WaterDocument8 pagesApplication of Tio2-Mounted Activated Carbon To The Removal of Phenol From WaterA. M. SHAREQUENo ratings yet
- 3221 - Deposition of TiO2 and Ag TiO2 Thin Films by The Polymeric Precursor MethodDocument8 pages3221 - Deposition of TiO2 and Ag TiO2 Thin Films by The Polymeric Precursor MethodAttila BartiNo ratings yet
- Hydroxyl Radical RoleDocument10 pagesHydroxyl Radical RoleGRagaNo ratings yet
- Photocatalytic degradation of phenol using MWNT-titania compositesDocument8 pagesPhotocatalytic degradation of phenol using MWNT-titania compositesŞebnem Gül İlarslanNo ratings yet
- 1 Enhanced Photocatalytic Activity of TiO2-Zeolite Composite For Abatement of Pollutants (Zeolita Natural)Document8 pages1 Enhanced Photocatalytic Activity of TiO2-Zeolite Composite For Abatement of Pollutants (Zeolita Natural)VANESANo ratings yet
- Research Article Tio /Diazonium/Graphene Oxide Composites: Synthesis and Visible-Light-Driven Photocatalytic Degradation of Methylene BlueDocument16 pagesResearch Article Tio /Diazonium/Graphene Oxide Composites: Synthesis and Visible-Light-Driven Photocatalytic Degradation of Methylene BlueVõ Thắng NguyênNo ratings yet
- Photocatalytic CO2 Conversion to Methanol Using Cu2O/Graphene/TNA HeterostructureDocument10 pagesPhotocatalytic CO2 Conversion to Methanol Using Cu2O/Graphene/TNA HeterostructureDina PradinaNo ratings yet
- Ultrasonics Sonochemistry: Lei Zhu, Ze-Da Meng, Chong-Yeon Park, Trisha Ghosh, Won-Chun OhDocument7 pagesUltrasonics Sonochemistry: Lei Zhu, Ze-Da Meng, Chong-Yeon Park, Trisha Ghosh, Won-Chun OhSobhy Sayed IbrahimNo ratings yet
- Photocatalytic Degradation of Organic Dyes With Manganese-Doped Zno NanoparticlesDocument7 pagesPhotocatalytic Degradation of Organic Dyes With Manganese-Doped Zno NanoparticlesГулинурNo ratings yet
- Low Inrıdıum-Doped TiO2 Nanostructure For Promısıng Photocatalyst In Hexane TreatmentDocument15 pagesLow Inrıdıum-Doped TiO2 Nanostructure For Promısıng Photocatalyst In Hexane TreatmentIJAERS JOURNALNo ratings yet
- J Jallcom 2015 09 241Document8 pagesJ Jallcom 2015 09 241THƯ NGUYỄN MINHNo ratings yet
- 10 1016@j Seppur 2020 116520Document10 pages10 1016@j Seppur 2020 116520khue tranNo ratings yet
- A Comparative Study of Sodium Hydrogen Titanate Nanotubes 2017 Journal of CDocument10 pagesA Comparative Study of Sodium Hydrogen Titanate Nanotubes 2017 Journal of CDes RiliNo ratings yet
- Metal organic framework-derived C-doped ZnO-TiO2 nanocompositeDocument11 pagesMetal organic framework-derived C-doped ZnO-TiO2 nanocompositeم.م حيدر محمودNo ratings yet
- Reduction CO2Document7 pagesReduction CO2angala9804No ratings yet
- GO-TiO2 2016Document5 pagesGO-TiO2 2016Rafif QuthronadaNo ratings yet
- Bor 2Document8 pagesBor 2petru apopeiNo ratings yet
- Nitrogen and Fluorine Codoped TiO2 MDocument8 pagesNitrogen and Fluorine Codoped TiO2 Mpetru apopeiNo ratings yet
- Role of activated carbon features on photocatalytic degradationDocument5 pagesRole of activated carbon features on photocatalytic degradationA. M. SHAREQUENo ratings yet
- A Review On Tio - Based Composites For Superior Photocatalytic ActivityDocument10 pagesA Review On Tio - Based Composites For Superior Photocatalytic ActivityKawaiiBunnehSukiNo ratings yet
- 1 s2.0 S0022286022011541 MainDocument14 pages1 s2.0 S0022286022011541 MainGUO YUNo ratings yet
- Catalysts: Heterogeneous Photocatalysis: Recent Advances and ApplicationsDocument35 pagesCatalysts: Heterogeneous Photocatalysis: Recent Advances and Applicationseva indriyaniNo ratings yet
- Improved Photoelectric Performance Via Fabricated Heterojunction g-C3N4 TiO2 HNTs Loaded Photocatalysts For Photodegradation of CiprofloxacinDocument45 pagesImproved Photoelectric Performance Via Fabricated Heterojunction g-C3N4 TiO2 HNTs Loaded Photocatalysts For Photodegradation of CiprofloxacinBachi NanoNo ratings yet
- Sattar 2019Document8 pagesSattar 2019ImranNo ratings yet
- Novel Removal of Diazinon Pesticide by AdsorptionDocument7 pagesNovel Removal of Diazinon Pesticide by AdsorptionAlissom GomesNo ratings yet
- Nano-Mno Decoration of Tio Microparticles To Promote Gaseous Ethanol Visible PhotoremovalDocument12 pagesNano-Mno Decoration of Tio Microparticles To Promote Gaseous Ethanol Visible PhotoremovalRaul NicoaraNo ratings yet
- 1-s2.0-S000862231100621X-mainDocument9 pages1-s2.0-S000862231100621X-mainŞebnem İlarslanNo ratings yet
- Synthesis of Tio Mgo Mixed Metal Oxide Nanoparticles Via A Solgel Method and Studies On Their Optical PropertiesDocument15 pagesSynthesis of Tio Mgo Mixed Metal Oxide Nanoparticles Via A Solgel Method and Studies On Their Optical PropertiesNanda Hamzaini ZainNo ratings yet
- Hydrothermal Synthesis and Characterization of Cu2+/F-Co-Doped Titanium Dioxide (Tio2) Nanotubes As Photocatalyst For Methyl Orange DegradationDocument11 pagesHydrothermal Synthesis and Characterization of Cu2+/F-Co-Doped Titanium Dioxide (Tio2) Nanotubes As Photocatalyst For Methyl Orange DegradationWulan PurnamasariNo ratings yet
- Journal of Environmental Chemical Engineering: SciencedirectDocument9 pagesJournal of Environmental Chemical Engineering: SciencedirectmissaouiNo ratings yet
- 1 s2.0 S0167732220370574 MainDocument10 pages1 s2.0 S0167732220370574 MainHarshithNo ratings yet
- Deveraiji 2018Document12 pagesDeveraiji 2018Marcella BallaNo ratings yet
- 2-s2.0-84961120848 (1) Abbas ImportantDocument9 pages2-s2.0-84961120848 (1) Abbas Importantfarah al-sudaniNo ratings yet
- Applied Surface ScienceDocument12 pagesApplied Surface ScienceArdiansyah TaufikNo ratings yet
- Khedr 2017Document25 pagesKhedr 2017frankdebruin261No ratings yet
- Crafting Carbon Sphere-Titania Core-Shell Interfacial Structure To AchieveDocument10 pagesCrafting Carbon Sphere-Titania Core-Shell Interfacial Structure To Achievevarforex1No ratings yet
- Titanium Dioxide Photocatalysis: Fundamentals and Application On PhotoinactivationDocument39 pagesTitanium Dioxide Photocatalysis: Fundamentals and Application On PhotoinactivationNikhi Maria RajuNo ratings yet
- SSRN Id3844936Document27 pagesSSRN Id3844936Rahul SahaNo ratings yet
- Effect of TiO2 Nanoparticle Agglomeration on Photocatalytic PerformanceDocument6 pagesEffect of TiO2 Nanoparticle Agglomeration on Photocatalytic PerformanceYuniNo ratings yet
- 1 s2.0 S0043135410008663 MainDocument9 pages1 s2.0 S0043135410008663 MainAjit Kumar DhankaNo ratings yet
- Artiglia 2013Document10 pagesArtiglia 2013petru apopeiNo ratings yet
- ChatDocument1 pageChatpetru apopeiNo ratings yet
- Tio2 VizibilDocument14 pagesTio2 Vizibilpetru apopeiNo ratings yet
- Articol DDocument10 pagesArticol Dpetru apopeiNo ratings yet
- Activated Ccarbon and TiO2 ReviewDocument9 pagesActivated Ccarbon and TiO2 Reviewjuan davidNo ratings yet
- Degradation of Emerging Pollutants in Water Under Solar IrradiationDocument11 pagesDegradation of Emerging Pollutants in Water Under Solar Irradiationpetru apopeiNo ratings yet
- 1 s2.0 S030438940600255X MainDocument7 pages1 s2.0 S030438940600255X Mainpetru apopeiNo ratings yet
- 1 s2.0 S0304389410015517 MainDocument9 pages1 s2.0 S0304389410015517 Mainpetru apopeiNo ratings yet
- Meeting Saved ChatDocument2 pagesMeeting Saved Chatpetru apopeiNo ratings yet
- 1 s2.0 S0926860X08000963 MainDocument9 pages1 s2.0 S0926860X08000963 Mainpetru apopeiNo ratings yet
- 1 s2.0 S0957582011000735 MainDocument9 pages1 s2.0 S0957582011000735 Mainpetru apopeiNo ratings yet
- Composting For A New Generation - Latest Techniques For The Bin and BeyondDocument309 pagesComposting For A New Generation - Latest Techniques For The Bin and Beyondpetru apopei100% (1)
- BSB 457 - Manual On Composting - RODocument76 pagesBSB 457 - Manual On Composting - ROpetru apopeiNo ratings yet
- 1 s2.0 S0926337308002099 MainDocument8 pages1 s2.0 S0926337308002099 Mainpetru apopeiNo ratings yet
- Compost Everything - The Good Guide To Extreme Composting by David The GoodDocument109 pagesCompost Everything - The Good Guide To Extreme Composting by David The Goodpetru apopei0% (1)
- Journal Pre-Proof: Science of The Total EnvironmentDocument98 pagesJournal Pre-Proof: Science of The Total Environmentpetru apopeiNo ratings yet
- 1998 - Iron Powder, Graphite and Activated Carbon As Catalysts For The Oxidation of 4-Chlorophenol With H2O2 in Aqueous Solution - LuckingDocument8 pages1998 - Iron Powder, Graphite and Activated Carbon As Catalysts For The Oxidation of 4-Chlorophenol With H2O2 in Aqueous Solution - Luckingpetru apopeiNo ratings yet
- The Secret Life of CompostDocument149 pagesThe Secret Life of Compostpetru apopeiNo ratings yet
- Operating Instructions: Part 3: Analysis Specifications For The Available Test Kits AppendicesDocument136 pagesOperating Instructions: Part 3: Analysis Specifications For The Available Test Kits Appendicespetru apopeiNo ratings yet
- Journal of The Taiwan Institute of Chemical EngineersDocument10 pagesJournal of The Taiwan Institute of Chemical EngineersDwiNo ratings yet
- Energy Nexus: Saswat Mahapatra, Md. Hibzur Ali, Kundan SamalDocument17 pagesEnergy Nexus: Saswat Mahapatra, Md. Hibzur Ali, Kundan Samalpetru apopeiNo ratings yet
- EPA Handbook on Advanced Photochemical Oxidation ProcessesDocument97 pagesEPA Handbook on Advanced Photochemical Oxidation ProcessesCristianAtanasiuNo ratings yet
- Water Research: Marta O. Barbosa, Nuno F.F. Moreira, Ana R. Ribeiro, Manuel F.R. Pereira, Adri An M.T. SilvaDocument23 pagesWater Research: Marta O. Barbosa, Nuno F.F. Moreira, Ana R. Ribeiro, Manuel F.R. Pereira, Adri An M.T. Silvapetru apopeiNo ratings yet
- 1998 - Immobilisation of Iron Ions On Nafion and Its Applicabyility To The Photo-Fenton Method - MaletzkyDocument11 pages1998 - Immobilisation of Iron Ions On Nafion and Its Applicabyility To The Photo-Fenton Method - Maletzkypetru apopeiNo ratings yet
- Nanotechnology Safety Concerns RevisitedDocument18 pagesNanotechnology Safety Concerns Revisitedpetru apopeiNo ratings yet
- 1998 - Wet Oxidation of Acetic Acid by Hydrogen Oxidation Catalyzed by Transition Metal-Exchange NaY Zeolites - LarachiDocument4 pages1998 - Wet Oxidation of Acetic Acid by Hydrogen Oxidation Catalyzed by Transition Metal-Exchange NaY Zeolites - Larachipetru apopeiNo ratings yet
- Titanium Dioxide-Modified Activated CarbonDocument8 pagesTitanium Dioxide-Modified Activated Carbonpetru apopeiNo ratings yet
- 1997 - Wet Oxidation of Phenol by Hydrogen Peroxide - The Key Role of PH On The Catalytic Behaviour of Fe-ZSM-5 - FajerwergDocument8 pages1997 - Wet Oxidation of Phenol by Hydrogen Peroxide - The Key Role of PH On The Catalytic Behaviour of Fe-ZSM-5 - Fajerwergpetru apopeiNo ratings yet
- 1996 - Wet Oxidation of Phenol by Hydrogen Peroxide Using Heterogeneous Catalysis Fe-ZSM-5-a Promising Catalyst - FajerwergDocument7 pages1996 - Wet Oxidation of Phenol by Hydrogen Peroxide Using Heterogeneous Catalysis Fe-ZSM-5-a Promising Catalyst - Fajerwergpetru apopeiNo ratings yet
- Engineering Development of A PhotocatalyticDocument12 pagesEngineering Development of A Photocatalyticpetru apopeiNo ratings yet
- PCV CableDocument12 pagesPCV CableMahamud MusaNo ratings yet
- Indus Lecture 2 PartialDocument69 pagesIndus Lecture 2 PartialIris Jean Mosquera100% (1)
- Diagnosti ControladorDocument198 pagesDiagnosti ControladorMONTACARGAS AVS100% (1)
- Verify Geometrically That c×a+b = c×a + c×b Using ParallelogramsDocument4 pagesVerify Geometrically That c×a+b = c×a + c×b Using ParallelogramsblehboNo ratings yet
- Process Dynamic and Control Lecture NoteDocument120 pagesProcess Dynamic and Control Lecture NoteUb UsoroNo ratings yet
- Ultra-Pod FASTON Fully Insulated Receptacles and Tabs: Application SpecificationDocument13 pagesUltra-Pod FASTON Fully Insulated Receptacles and Tabs: Application SpecificationBrian RuttleNo ratings yet
- Midterm Quiz 3 StaticsDocument4 pagesMidterm Quiz 3 StaticsFerdinand Limba ĪīNo ratings yet
- Dual Band Electromagnetic Band Gap EBG StructureDocument4 pagesDual Band Electromagnetic Band Gap EBG StructureNhật Minh TạNo ratings yet
- GLOBE PFG-red planetary geared vane motors for industrial applicationsDocument26 pagesGLOBE PFG-red planetary geared vane motors for industrial applicationsTarcio TomNo ratings yet
- Chapter 3 256857Document45 pagesChapter 3 256857신재우No ratings yet
- Energy Systems and Resources ExplainedDocument22 pagesEnergy Systems and Resources ExplainedTayani Tedson KumwendaNo ratings yet
- AASHTO w-ASTM Equivs 2019-08-26Document11 pagesAASHTO w-ASTM Equivs 2019-08-26ayag allanNo ratings yet
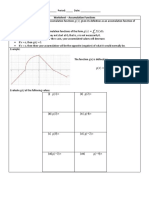
- Worksheet - Accumulation FunctionsDocument3 pagesWorksheet - Accumulation FunctionssteveNo ratings yet
- Air-Stable and Volatile Bis (Pyridylalkenolato) - Sanjay MathurDocument5 pagesAir-Stable and Volatile Bis (Pyridylalkenolato) - Sanjay MathurGanesh BabuNo ratings yet
- Automatic Bottle Washing MachineDocument14 pagesAutomatic Bottle Washing Machineheisnoble100% (1)
- Dynamic Mechanical Analysis - WikipediaDocument8 pagesDynamic Mechanical Analysis - WikipediamohsenNo ratings yet
- Impulse and Frequency Response: What They Are and How They RelateDocument3 pagesImpulse and Frequency Response: What They Are and How They RelatenazmulNo ratings yet
- INSTRUMENTATION AND CONTROL FUNDAMENTALSDocument53 pagesINSTRUMENTATION AND CONTROL FUNDAMENTALSDaniel D Danso100% (1)
- Belzona 1121: Product Specification SheetDocument2 pagesBelzona 1121: Product Specification SheetQuy RomNo ratings yet
- 1946 Shanley The Column Paradox J Aero SciDocument1 page1946 Shanley The Column Paradox J Aero Scitraders joeNo ratings yet
- FCC Crystal Phonon ModelingDocument4 pagesFCC Crystal Phonon ModelingLuis SerranoNo ratings yet
- ADOR Booklet F Web FDocument132 pagesADOR Booklet F Web FbadesharamkNo ratings yet
- Synchronous MachinesDocument13 pagesSynchronous MachinesKayalvizhi SelvamNo ratings yet
- Electromagnetism Course Notes (PHYS09060) 2017/18Document98 pagesElectromagnetism Course Notes (PHYS09060) 2017/18KeKa TVNo ratings yet
- Air-Conditioning: R.K. RajputDocument905 pagesAir-Conditioning: R.K. RajputAnkit RanjanNo ratings yet
- Pos FormatDocument6 pagesPos FormatChelmarie CuracheaNo ratings yet
- Thomson Clutches Brakes CatalogDocument152 pagesThomson Clutches Brakes CatalogElectromateNo ratings yet
- SohanDocument7 pagesSohanShuvayan GhoshNo ratings yet
- Ib Chem PrepDocument26 pagesIb Chem PrepSaketh VuppalapatiNo ratings yet
- Maryland Boiler Safety:: Compressed Air TanksDocument2 pagesMaryland Boiler Safety:: Compressed Air TanksMohammed YcfssNo ratings yet