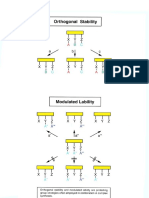
You might also like
- Transition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesFrom EverandTransition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesNo ratings yet
- Transition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesFrom EverandTransition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesNo ratings yet
- Protecting Groups M.S.Document28 pagesProtecting Groups M.S.Jonathan OswaldNo ratings yet
- Protecting Group Lbs Chem ProjectDocument21 pagesProtecting Group Lbs Chem ProjectVirendra Singh RajputNo ratings yet
- 04 - The - Logic - of - SynthesisDocument31 pages04 - The - Logic - of - SynthesisamelNo ratings yet
- 6L-Protecting GroupsDocument17 pages6L-Protecting GroupsCarlos Javier Orellana OrtizNo ratings yet
- Dionysios A. Papaioannou Professor of Organic Chemistry Department of Chemistry University of PatrasDocument90 pagesDionysios A. Papaioannou Professor of Organic Chemistry Department of Chemistry University of PatrasDiomedes DNo ratings yet
- Protection and Deprotection PDFDocument50 pagesProtection and Deprotection PDFKartik RanaNo ratings yet
- Protection of Functional Group - AbhayDocument15 pagesProtection of Functional Group - AbhayABHAY VISHWAKARMANo ratings yet
- Protecting Reagent TCIDocument12 pagesProtecting Reagent TCItech PSNo ratings yet
- Functional Group ProtectionDocument19 pagesFunctional Group ProtectionAhmad AhmadNo ratings yet
- Dionysios A. Papaioannou Professor of Organic Chemistry Department of Chemistry University of PatrasDocument95 pagesDionysios A. Papaioannou Professor of Organic Chemistry Department of Chemistry University of PatrasΑδαμαντία ΣτριμενοπούλουNo ratings yet
- UnitIII ProtectinggroupsDocument7 pagesUnitIII ProtectinggroupsMasoodNo ratings yet
- UnitIII ProtectinggroupsDocument8 pagesUnitIII ProtectinggroupsTharun KorraNo ratings yet
- Protecting Group HandoutDocument5 pagesProtecting Group HandoutRafaelle Sanvictores SilongNo ratings yet
- Protecting Groups in Organic SynthesisDocument27 pagesProtecting Groups in Organic SynthesisNur Asmi MNo ratings yet
- Angew Chem Int Ed - 2020 - Schulthoff - The Formosalides Structure Determination by Total SynthesisDocument9 pagesAngew Chem Int Ed - 2020 - Schulthoff - The Formosalides Structure Determination by Total Synthesiscaiohenriquelins1998No ratings yet
- Protecting Groups 2Document1 pageProtecting Groups 2Dushyant PatelNo ratings yet
- UnitIII ProtectinggroupsDocument8 pagesUnitIII ProtectinggroupsAnandarup GoswamiNo ratings yet
- Use of Carbonyl Group Addition EliminatiDocument19 pagesUse of Carbonyl Group Addition EliminatiMichaelNo ratings yet
- Chemical Process Design and Optimization: Chapter 2 - Synthesis of Process Flow DiagramDocument33 pagesChemical Process Design and Optimization: Chapter 2 - Synthesis of Process Flow DiagramLam DesmondNo ratings yet
- Heuristics For Process Synthesis: Ref: Seider, Seader and Lewin (2004), Chapter 5Document46 pagesHeuristics For Process Synthesis: Ref: Seider, Seader and Lewin (2004), Chapter 5María Carrillo De AlbaNo ratings yet
- Protection Groups in Organic PDFDocument67 pagesProtection Groups in Organic PDFToàn MinhNo ratings yet
- CHEN 4460 - Process Synthesis, Simulation and OptimizationDocument28 pagesCHEN 4460 - Process Synthesis, Simulation and Optimizationmppatilmayur1679No ratings yet
- Modern Organic Synthesis An IntroductionDocument37 pagesModern Organic Synthesis An IntroductionHanh DuongNo ratings yet
- Concepts in Organic SynthesisDocument1 pageConcepts in Organic SynthesisDushyant kumarNo ratings yet
- Banik 1992Document4 pagesBanik 1992Farschad AnsariNo ratings yet
- Protecting Groups For Organic SynthesisDocument31 pagesProtecting Groups For Organic SynthesisFranco CentNo ratings yet
- Lecture8 Macromol DesignDocument48 pagesLecture8 Macromol DesignfilippoNo ratings yet
- Heuristic SDocument31 pagesHeuristic Sjesi5445No ratings yet
- 2022 Int J Mol Sci 7152 (Lőrinczi)Document13 pages2022 Int J Mol Sci 7152 (Lőrinczi)Lázár LászlóNo ratings yet
- Chapter5Document92 pagesChapter5fNo ratings yet
- Introduction To Functional Group Protection-1Document7 pagesIntroduction To Functional Group Protection-1ALI HAMZANo ratings yet
- Green Chemistry CHP 1-17Document59 pagesGreen Chemistry CHP 1-17Basu GargNo ratings yet
- ChemistrySelect - 2016 - Uranga - Theoretical and Experimental Study of The Antioxidant Behaviors of 5 O CaffeoylquinicDocument8 pagesChemistrySelect - 2016 - Uranga - Theoretical and Experimental Study of The Antioxidant Behaviors of 5 O CaffeoylquinicErica SpinnenhirnNo ratings yet
- Combustion Unit 2 3 5Document62 pagesCombustion Unit 2 3 5shivkumarNo ratings yet
- 05 Protecting GroupsDocument30 pages05 Protecting GroupsManupal YadavNo ratings yet
- GreenDocument16 pagesGreenGopal SharmaNo ratings yet
- A. Title of ExperimentDocument4 pagesA. Title of ExperimentBang KaiNo ratings yet
- Peptide Synthesis: Amino Acids, Peptides Proteins, The Royal Society of Chemistry, 2003Document54 pagesPeptide Synthesis: Amino Acids, Peptides Proteins, The Royal Society of Chemistry, 2003philkaroyanNo ratings yet
- European Polymer Journal: Christian Hahn, Sebastian Wesselbaum, Helmut Keul, Martin MöllerDocument11 pagesEuropean Polymer Journal: Christian Hahn, Sebastian Wesselbaum, Helmut Keul, Martin MöllerShanti Astuti MustafaNo ratings yet
- Binkley1989. Photoremovable Hydroxyl Group ProtectionDocument5 pagesBinkley1989. Photoremovable Hydroxyl Group Protectioncristina.moya724No ratings yet
- SLCT 201801951Document4 pagesSLCT 201801951shruti guptaNo ratings yet
- Handbook of Heterogeneous CatalystDocument3,865 pagesHandbook of Heterogeneous Catalystminh tri phan100% (2)
- Catalytic Dehydration of Alcohols: University of Warsaw Faculty of Chemistry Chemical Technology DivisionDocument12 pagesCatalytic Dehydration of Alcohols: University of Warsaw Faculty of Chemistry Chemical Technology DivisionSpandan GhoshalNo ratings yet
- Abdulrahman Al-Sumait University.: Heterocyclic Compounds (8-Membered Ring)Document10 pagesAbdulrahman Al-Sumait University.: Heterocyclic Compounds (8-Membered Ring)Ali Issa OthmanNo ratings yet
- NIH Public Access: Author ManuscriptDocument10 pagesNIH Public Access: Author ManuscriptElisabeta StamateNo ratings yet
- Anie 201706312Document6 pagesAnie 201706312Tùng LêNo ratings yet
- An Elegant Example of Chemoselective Reaction: Resonance October 2008Document13 pagesAn Elegant Example of Chemoselective Reaction: Resonance October 2008krima riamrrrNo ratings yet
- Uv Visible SpectrosDocument7 pagesUv Visible SpectrosFernanda Stuani PereiraNo ratings yet
- Chalcone Synthesis, Structure DiversityDocument13 pagesChalcone Synthesis, Structure DiversityDini Elsi ANo ratings yet
- Separation of Reagents PDFDocument6 pagesSeparation of Reagents PDFRah%No ratings yet
- QB PDFDocument18 pagesQB PDFShivani0% (1)
- Heuristics For ProcessDocument46 pagesHeuristics For ProcessMuhammad SyafiieNo ratings yet
- Triton Ficha TecnicaDocument2 pagesTriton Ficha TecnicaBruno FilipeNo ratings yet
- PDFDocument20 pagesPDFThirunavuk KarasuNo ratings yet
- D Spore K ( - ') : Efinitions of Eyphrases RXN Type SDocument42 pagesD Spore K ( - ') : Efinitions of Eyphrases RXN Type SB C RaviNo ratings yet
- Friedel Crafts AcylationDocument3 pagesFriedel Crafts AcylationM Zeeshan aliNo ratings yet
- Organic Chemistry III Laboratory: NMR Verification of Diastereoselective Reduction of Substituted CyclohexanonesDocument4 pagesOrganic Chemistry III Laboratory: NMR Verification of Diastereoselective Reduction of Substituted Cyclohexanonesungu_sakuraNo ratings yet
- Sustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeFrom EverandSustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeNo ratings yet
- 1 Contents IndexDocument16 pages1 Contents IndexOlivier LeogageNo ratings yet
- OrganoMetSolv PDFDocument4 pagesOrganoMetSolv PDFHung Quoc DangNo ratings yet
- DMF ExcipientDocument65 pagesDMF ExcipientOlivier LeogageNo ratings yet
- Regulatory Sciences API Drug Development 04152015Document34 pagesRegulatory Sciences API Drug Development 04152015Olivier LeogageNo ratings yet
- Risk Assessment of Genotoxic Impurities in New Chemical Entities: Strategies To Demonstrate ControlDocument10 pagesRisk Assessment of Genotoxic Impurities in New Chemical Entities: Strategies To Demonstrate Controlharlan777No ratings yet
- Protection For The Alkyne - CHDocument6 pagesProtection For The Alkyne - CHOlivier LeogageNo ratings yet
- Reactivities, Reagents, and Reactivity ChartsDocument47 pagesReactivities, Reagents, and Reactivity ChartsOlivier LeogageNo ratings yet
- Protection For The Thiol GroupDocument40 pagesProtection For The Thiol GroupOlivier LeogageNo ratings yet
- A Rationale For Determining, Testing, and Controlling Specific Impurities in Pharmaceuticals That Possess Potential For GenotoxicityDocument14 pagesA Rationale For Determining, Testing, and Controlling Specific Impurities in Pharmaceuticals That Possess Potential For GenotoxicitytvvsagarNo ratings yet
- Primal Quest - Essentials - Digital v1.03Document30 pagesPrimal Quest - Essentials - Digital v1.03Olivier LeogageNo ratings yet
- Azeotropic dataIIDocument104 pagesAzeotropic dataIIah_liNo ratings yet
- Wm10 - Josh, An Origin Story: Special RulesDocument1 pageWm10 - Josh, An Origin Story: Special RulesOlivier LeogageNo ratings yet
- Evans Pka TableDocument6 pagesEvans Pka Tablethales94No ratings yet
- Ring of The Dragon One-Shot Adventure-2-1Document22 pagesRing of The Dragon One-Shot Adventure-2-1Olivier LeogageNo ratings yet
- Ring of The Dragon One-Shot Adventure-2-1Document22 pagesRing of The Dragon One-Shot Adventure-2-1Olivier LeogageNo ratings yet
- The Last of Us RPG v0.5.0Document182 pagesThe Last of Us RPG v0.5.0Olivier Leogage100% (1)
- Ork!Document64 pagesOrk!Stephen Yendle100% (1)
- Random Encounters Vol1 Issue3 Oct18 Low ResDocument70 pagesRandom Encounters Vol1 Issue3 Oct18 Low ResOlivier Leogage100% (1)
- Underwater City: Quest Quest - Zombicide ZombicideDocument1 pageUnderwater City: Quest Quest - Zombicide ZombicideOlivier LeogageNo ratings yet
- Chibi 2022 Le - Grand - Imagier Partie04Document109 pagesChibi 2022 Le - Grand - Imagier Partie04Olivier LeogageNo ratings yet
- All Hallows EvilDocument6 pagesAll Hallows EvilOlivier Leogage100% (1)
- Fantasy CraftDocument402 pagesFantasy CraftAnonymous tQ5oaQ2p9No ratings yet
- Random Encounters Vol1 Issue2 Sept18 Low ResDocument70 pagesRandom Encounters Vol1 Issue2 Sept18 Low ResOlivier Leogage100% (1)
- Random Encounters Vol 2 Issue 8 August 19 Low ResDocument70 pagesRandom Encounters Vol 2 Issue 8 August 19 Low ResOlivier LeogageNo ratings yet
- Random Encounters Vol1 Issue1 Aug18 Low ResDocument70 pagesRandom Encounters Vol1 Issue1 Aug18 Low ResOlivier Leogage100% (2)
- Random Encounters Vol1 Issue4 Nov18 Low ResDocument70 pagesRandom Encounters Vol1 Issue4 Nov18 Low ResOlivier Leogage50% (2)
- Random Encounters Vol1 Issue5 Dec18 Low ResDocument66 pagesRandom Encounters Vol1 Issue5 Dec18 Low ResOlivier Leogage100% (1)
- Random Encounters Vol 2 Issue 9 September 19 Low ResDocument68 pagesRandom Encounters Vol 2 Issue 9 September 19 Low ResOlivier LeogageNo ratings yet
- Chemistry 2pointsDocument4 pagesChemistry 2pointsjovanniNo ratings yet
- Iligan Medical Center College: Activity 1 (GC)Document4 pagesIligan Medical Center College: Activity 1 (GC)Krizza Gales DemecilloNo ratings yet
- B.tech Question Paper-2017Document3 pagesB.tech Question Paper-2017aryanr2233No ratings yet
- Sikaplan WP 1100-15HLDocument2 pagesSikaplan WP 1100-15HLalparslan GÜRENo ratings yet
- Applications of Basic Magnesium Sulfate Cement in Civil EngineeringDocument6 pagesApplications of Basic Magnesium Sulfate Cement in Civil EngineeringPrashant GaradNo ratings yet
- UV Vis NIR Reference SetsDocument44 pagesUV Vis NIR Reference Setsjljimenez1969No ratings yet
- TPR2Document10 pagesTPR2WILFREDO ROMAN PAUCARNo ratings yet
- 2015 U.S. NATIONAL Chemistry Olympiad: Local Section Exam Olympiad Examinations Task ForceDocument8 pages2015 U.S. NATIONAL Chemistry Olympiad: Local Section Exam Olympiad Examinations Task ForceNajmusawwa Aulia RahmahNo ratings yet
- Row Schott Technical Glasses View 2020-04-14Document80 pagesRow Schott Technical Glasses View 2020-04-14Oleg ChernovNo ratings yet
- MATERIAL SAFETY DATA SHEET Dust CleanerDocument2 pagesMATERIAL SAFETY DATA SHEET Dust CleanerHarry LaksonoNo ratings yet
- Chap 2 Material Balance Non-Reactive SystemDocument40 pagesChap 2 Material Balance Non-Reactive SystemAndreas LarssonNo ratings yet
- Basf Masterpolyheed 3532ae TdsDocument2 pagesBasf Masterpolyheed 3532ae TdsVisJadNo ratings yet
- DLL Q1 Lesson 5 Concentration of SolutionsDocument3 pagesDLL Q1 Lesson 5 Concentration of SolutionsMa. Elizabeth Cusi100% (1)
- Physioex Lab Report: Pre-Lab Quiz ResultsDocument3 pagesPhysioex Lab Report: Pre-Lab Quiz ResultsNicole de LeonNo ratings yet
- CoAs... Hidroxipropilmetilcelulosa (HPMC) (Methocel K100M Premium-2208) - CoA L-1EF0909 (Spectrum)Document1 pageCoAs... Hidroxipropilmetilcelulosa (HPMC) (Methocel K100M Premium-2208) - CoA L-1EF0909 (Spectrum)Maikel Perez NavarroNo ratings yet
- PDF Astm E1444 E1444m 16e1 Standard Practice For Magnetic Particle TestingDocument31 pagesPDF Astm E1444 E1444m 16e1 Standard Practice For Magnetic Particle TestingTHIRU.SNo ratings yet
- Controllable Synthesis of MN O Nanowires and Application in The Treatment of Phenol at Room TemperatureDocument9 pagesControllable Synthesis of MN O Nanowires and Application in The Treatment of Phenol at Room TemperaturesonficyusNo ratings yet
- IR Spectra AnalysisDocument37 pagesIR Spectra AnalysisdevoydouglasNo ratings yet
- ASTM F639-09 Standard Specification For Polyethylene Plastics For Medical ApplicationsDocument3 pagesASTM F639-09 Standard Specification For Polyethylene Plastics For Medical ApplicationsJoãoNo ratings yet
- Co3O4 NanotubesDocument3 pagesCo3O4 NanotubesKishoreBabu SNo ratings yet
- Paracetamol From AminophenolDocument47 pagesParacetamol From AminophenolProbablyсвдсNo ratings yet
- Replacing Part of Cement With Waste Material - ReviewDocument7 pagesReplacing Part of Cement With Waste Material - ReviewInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Organozinc CompoundDocument8 pagesOrganozinc Compoundamanuel tafeseNo ratings yet
- Technical SpecificationDocument6 pagesTechnical SpecificationAyse ClkNo ratings yet
- Concept Strengthening Sheet (CSS-06) Based On AIATS-06 (TYM) - ChemistryDocument6 pagesConcept Strengthening Sheet (CSS-06) Based On AIATS-06 (TYM) - ChemistryDev SoniNo ratings yet
- Moudling Operation GSIC Process: MouldingDocument13 pagesMoudling Operation GSIC Process: MouldingBalakumaran MurugesanNo ratings yet
- Lecture 3Document27 pagesLecture 3Dhiraj SharmaNo ratings yet
- Plan Design Lab SampleDocument3 pagesPlan Design Lab SampleDumissa MelvilleNo ratings yet
- Summative Exams - Term End Paper-G8Document12 pagesSummative Exams - Term End Paper-G8Samanvitha RaaviNo ratings yet
- The Periodic Ta-Wps OfficeDocument3 pagesThe Periodic Ta-Wps OfficeAlan Gandidze MotifNo ratings yet