You might also like
- ..Digestion of ProteinDocument21 pages..Digestion of Proteinaneeda shabirNo ratings yet
- Amino Acids Metabol Synth of UreaDocument32 pagesAmino Acids Metabol Synth of UreaAnastasiafynn100% (1)
- Common Medical/Dental Abbreviations and Acronyms: PharmacopoeiaDocument9 pagesCommon Medical/Dental Abbreviations and Acronyms: PharmacopoeiaFatima naserNo ratings yet
- Amino Acids and ProteinsDocument48 pagesAmino Acids and ProteinsSteph VeeNo ratings yet
- Protein and Amino Acid MetabolismDocument52 pagesProtein and Amino Acid MetabolismRisky OpponentNo ratings yet
- cc2 Lectures AllDocument256 pagescc2 Lectures AllJayson Dagohoy SudioNo ratings yet
- General Protein MetabolismDocument72 pagesGeneral Protein MetabolismHafizie SyahmanNo ratings yet
- Alanine Aminotransferase (Alt-Gpt)Document1 pageAlanine Aminotransferase (Alt-Gpt)Risqon Anjahiranda Adiputra50% (2)
- Biochem HomeworkDocument13 pagesBiochem Homeworkfcukingfranztastik50% (2)
- Amino Acid Metabolism NotesDocument152 pagesAmino Acid Metabolism NotesAkhilesh TiwariNo ratings yet
- EnzymesDocument60 pagesEnzymesRia AlcantaraNo ratings yet
- AST and ALTDocument7 pagesAST and ALTTrisha NavarceNo ratings yet
- Reading: Stryer Edition 6: Chapters 23 and 24. Figures in This Document Are FromDocument36 pagesReading: Stryer Edition 6: Chapters 23 and 24. Figures in This Document Are FromAbeer ShalabyNo ratings yet
- Notes in BiochemistryDocument13 pagesNotes in Biochemistrylian jaeNo ratings yet
- Amino Acid Catabolism1Document5 pagesAmino Acid Catabolism1rolwinNo ratings yet
- Topic: Protein Metabolism: Biochemistry & Biophysics: Paper-V, Unit-9Document14 pagesTopic: Protein Metabolism: Biochemistry & Biophysics: Paper-V, Unit-9aayushi tejwaniNo ratings yet
- Unit 1 Pharmacognosy and Phytochemistry 2 Wutp2iDocument45 pagesUnit 1 Pharmacognosy and Phytochemistry 2 Wutp2iNehal ArzuNo ratings yet
- Presented by "Prof. Dr. Masoom Khan" Lecture Notes. No. 678Document41 pagesPresented by "Prof. Dr. Masoom Khan" Lecture Notes. No. 678Qamar QamarNo ratings yet
- C10 Protein and Amino Acid MetabolismDocument8 pagesC10 Protein and Amino Acid MetabolismSoraya D. Al-ObinayNo ratings yet
- Amino Acid MetabolismDocument15 pagesAmino Acid Metabolismshanto.tn98No ratings yet
- Metabolism of Essential AADocument7 pagesMetabolism of Essential AA48 Syeda KainatNo ratings yet
- Biochem ReviewerDocument16 pagesBiochem ReviewerHennessy PerezNo ratings yet
- 6 Proteins Their Digestion & AbsorptionsDocument18 pages6 Proteins Their Digestion & AbsorptionshodaNo ratings yet
- Protein MetaboilisamDocument18 pagesProtein MetaboilisamSumit PandyaNo ratings yet
- Amino Acid MetabolismDocument10 pagesAmino Acid MetabolismatifzeaNo ratings yet
- WUCHS Protein Lecture Note For Medical StudentsDocument361 pagesWUCHS Protein Lecture Note For Medical StudentsZelalem bekeleNo ratings yet
- 03 - Metabolisme Protein - MonogastrikDocument25 pages03 - Metabolisme Protein - MonogastrikHerni Bustam100% (1)
- Unit 14:-Biomoulecules: ProjectDocument4 pagesUnit 14:-Biomoulecules: ProjectAnkit KumarNo ratings yet
- MBC 203 - Protein MetabolismDocument66 pagesMBC 203 - Protein MetabolismEniola DaramolaNo ratings yet
- Biological MoleculesDocument6 pagesBiological MoleculessanaullahNo ratings yet
- Protein Notes (Dipesh Routh)Document7 pagesProtein Notes (Dipesh Routh)Dipesh RouthNo ratings yet
- Amino Acid Catabolism-Part-1: Biochemistry For Medics - Lecture Notes Professor (DR.) Namrata ChhabraDocument43 pagesAmino Acid Catabolism-Part-1: Biochemistry For Medics - Lecture Notes Professor (DR.) Namrata Chhabrashree devNo ratings yet
- Amino AcidsDocument32 pagesAmino AcidsWakjira GemedaNo ratings yet
- Biochemistery:: Asparagine Is The 1Document12 pagesBiochemistery:: Asparagine Is The 1Naima AmjadNo ratings yet
- Amino Acids Metabolism Final For Pharm 2014Document57 pagesAmino Acids Metabolism Final For Pharm 2014Getu LuchesaNo ratings yet
- Cellular Metabolism Overview of MetabolismDocument4 pagesCellular Metabolism Overview of MetabolismNathan Louis PalacioNo ratings yet
- PARAS, Mark Heinrich B. - Homework 3 - Lecture Biochemistry CHEM 40 BSN 1EDocument3 pagesPARAS, Mark Heinrich B. - Homework 3 - Lecture Biochemistry CHEM 40 BSN 1EMark Heinrich B. ParasNo ratings yet
- Amino AcidsDocument7 pagesAmino Acidsrida khanNo ratings yet
- Protein MetabolismDocument23 pagesProtein MetabolismSafura IjazNo ratings yet
- Unit 3 DeaminationDocument27 pagesUnit 3 DeaminationAsjad HassanNo ratings yet
- Unit 3 EnzymesDocument18 pagesUnit 3 Enzymeskamsjain6802No ratings yet
- Protein DenaturationDocument12 pagesProtein DenaturationAhmed Elhakim100% (2)
- Unit 8 Digestion and AbsorptionDocument33 pagesUnit 8 Digestion and AbsorptionGlory MuedaNo ratings yet
- Foods and Nutrition Paper OneDocument119 pagesFoods and Nutrition Paper OneNaduku EridadiNo ratings yet
- Degradacion Aminoacidos y ProteinasDocument52 pagesDegradacion Aminoacidos y ProteinasEncina Abrigo Andrés PabloNo ratings yet
- Overview of MetabolismDocument13 pagesOverview of Metabolismgabby chaanNo ratings yet
- 12 Amino Acid Metabolism: 12.1 Metabolic Uses of Amino AcidsDocument22 pages12 Amino Acid Metabolism: 12.1 Metabolic Uses of Amino Acidsluis jose sanchezNo ratings yet
- Absortion AaDocument30 pagesAbsortion Aaandres herreraNo ratings yet
- Introduction Amino Acid Matabolism and CatabolismDocument45 pagesIntroduction Amino Acid Matabolism and CatabolismAboubakar Moalim Mahad moh'd100% (1)
- Amino AcidDocument24 pagesAmino AcidAdisa YellowNo ratings yet
- Removal of Nitrogen From Amino AcidsDocument34 pagesRemoval of Nitrogen From Amino AcidsSafura IjazNo ratings yet
- Note On Digestion of Protein 2021Document11 pagesNote On Digestion of Protein 2021Charity NwaogbeNo ratings yet
- Essential Amino AcidsDocument3 pagesEssential Amino AcidsNurshamimi JaafarNo ratings yet
- Protein MetabolismDocument6 pagesProtein MetabolismGandesa LangNo ratings yet
- 15BT103 Biochem-UNIT 3Document53 pages15BT103 Biochem-UNIT 3Adityanair RA1711009010128No ratings yet
- 12.protein - Rista Ajeng Mitasari - 18030194066 - PKU2018Document31 pages12.protein - Rista Ajeng Mitasari - 18030194066 - PKU2018Rista AjengNo ratings yet
- Biochem MidtermsDocument4 pagesBiochem MidtermschristinejeancenabreNo ratings yet
- Pathways in Amino Acids and Protein Metabolism 2Document76 pagesPathways in Amino Acids and Protein Metabolism 2godfreybwerere13No ratings yet
- Biomolecules of LifeDocument9 pagesBiomolecules of LifeAbhishek PantheeNo ratings yet
- Catabolism of Proteins and Amino Acid NitrogenDocument6 pagesCatabolism of Proteins and Amino Acid Nitrogenyel5buscatoNo ratings yet
- Amino Acids are linked by peptide bonds to form formed by linking the α-carboxyl group of one amino acid to the α-amino group of another amino acid with a peptide bond (also called an amide bond)Document2 pagesAmino Acids are linked by peptide bonds to form formed by linking the α-carboxyl group of one amino acid to the α-amino group of another amino acid with a peptide bond (also called an amide bond)Noviemae TuandaNo ratings yet
- كيمياء حياتية المرحلة الرابعة 2Document31 pagesكيمياء حياتية المرحلة الرابعة 2Abdala Al-MawlaNo ratings yet
- Protein, Nitrogen Katabolisme Dan Siklus UreaDocument35 pagesProtein, Nitrogen Katabolisme Dan Siklus UreaAnonymous QCMhA4wNgBNo ratings yet
- Intro To Proteins MetabolismDocument10 pagesIntro To Proteins MetabolismAbdul RaufNo ratings yet
- Macromolecules - Are Large Molecules Composed of Thousands of Covalently Connected AtomsDocument6 pagesMacromolecules - Are Large Molecules Composed of Thousands of Covalently Connected AtomsBon Joey BernestoNo ratings yet
- Proteins and Amino AcidsDocument9 pagesProteins and Amino AcidsNatanga GodfreyNo ratings yet
- Chemistry Form One EatwellDocument1 pageChemistry Form One Eatwellmistic JayNo ratings yet
- Monograph of GingerDocument2 pagesMonograph of Gingermistic JayNo ratings yet
- Emploi de Temps 2021-2022Document31 pagesEmploi de Temps 2021-2022mistic JayNo ratings yet
- BOJ Monetory StatmentDocument5 pagesBOJ Monetory Statmentmistic JayNo ratings yet
- Test For LipidDocument3 pagesTest For Lipidmistic JayNo ratings yet
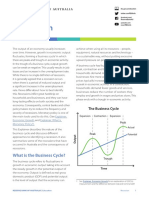
- Recession: What Is The Business Cycle?Document6 pagesRecession: What Is The Business Cycle?mistic JayNo ratings yet
- Clinical EnzymologyDocument23 pagesClinical EnzymologyKishore KaranNo ratings yet
- Hepatoprotective Activity of Ocimum Americanum L LDocument7 pagesHepatoprotective Activity of Ocimum Americanum L LNurul Rachman NasutionNo ratings yet
- Jamu The Indonesian Traditional Herbal MedicineDocument22 pagesJamu The Indonesian Traditional Herbal MedicineAlvian VianNo ratings yet
- Mind Map: Amino Acid MetabolismDocument1 pageMind Map: Amino Acid MetabolismAbbey Ayala100% (1)
- Technical Bulletin LDX - ALT-ASTDocument2 pagesTechnical Bulletin LDX - ALT-ASTTriadi Arif MaulanaNo ratings yet
- A Colorimetric Method For The Determination of Serum Glutamic Oxalacetic and Glutamic Pyruvic TransaminasesDocument8 pagesA Colorimetric Method For The Determination of Serum Glutamic Oxalacetic and Glutamic Pyruvic TransaminasesTuan NguyenNo ratings yet
- Humatrol N: Target Values / Sollwerte / Valores Asignados/ Valeurs CiblesDocument2 pagesHumatrol N: Target Values / Sollwerte / Valores Asignados/ Valeurs CiblesHussein N. FarhatNo ratings yet
- PI-e-ALAT IFCC-5Document2 pagesPI-e-ALAT IFCC-5Delphi CrackerNo ratings yet
- Amino Acid Metabolism All LecturesDocument18 pagesAmino Acid Metabolism All Lecturesmizare29gNo ratings yet
- Edited by K.: Enzyme Catalysis in Organic SynthesisDocument1,583 pagesEdited by K.: Enzyme Catalysis in Organic SynthesisSankar AdhikariNo ratings yet
- Serodos Plus: Target Values / Sollwerte / Valores Meta/ Valeurs SouhaitéesDocument3 pagesSerodos Plus: Target Values / Sollwerte / Valores Meta/ Valeurs SouhaitéesAniket dubeyNo ratings yet
- Diagnostic CentreDocument14 pagesDiagnostic CentreJonas AsiaNo ratings yet
- Amino Acid MetabolismDocument23 pagesAmino Acid MetabolismZ ZNo ratings yet
- Project 1 Day 1Document339 pagesProject 1 Day 1Mohamed NagyNo ratings yet
- Abnormal Labs-Full TableDocument6 pagesAbnormal Labs-Full TableGERIMAIA CRUZNo ratings yet
- CH 18 Cholesterol Metabolism: Biochem Block 3 NotesDocument38 pagesCH 18 Cholesterol Metabolism: Biochem Block 3 NotesJoseph KimNo ratings yet
- 577-Article Text-1423-1-10-20191201 PDFDocument6 pages577-Article Text-1423-1-10-20191201 PDFfahmiNo ratings yet
- C10 Protein and Amino Acid MetabolismDocument8 pagesC10 Protein and Amino Acid MetabolismSoraya D. Al-ObinayNo ratings yet
- Nitrogen MetabolismDocument36 pagesNitrogen Metabolism202210034No ratings yet
- Aminotransferase For Commercial: Chiral ChemistryDocument2 pagesAminotransferase For Commercial: Chiral ChemistryVenkata Suryanarayana GorleNo ratings yet
- MC 2 Notes (Midterm)Document4 pagesMC 2 Notes (Midterm)Francine Dominique CollantesNo ratings yet
- WO 2021/250702 Al: 16 December 2021 (16.12.2021)Document22 pagesWO 2021/250702 Al: 16 December 2021 (16.12.2021)rgNo ratings yet
- Eclipta AlbaDocument5 pagesEclipta Albakonversi stfi16No ratings yet