You might also like
- 11 Molecular DockingDocument6 pages11 Molecular DockingCarissa IslaNo ratings yet
- The Respiratory System - Freebie GuideDocument4 pagesThe Respiratory System - Freebie GuideIndustria Quimica0% (1)
- Structural Biology: What Does 3D Tell Us?Document20 pagesStructural Biology: What Does 3D Tell Us?Prakash KumarNo ratings yet
- Protein Structural Motifs: Doug Brutlag Professor Emeritus Biochemistry & Medicine (By Courtesy)Document100 pagesProtein Structural Motifs: Doug Brutlag Professor Emeritus Biochemistry & Medicine (By Courtesy)Joan Manuel Tovar CasallasNo ratings yet
- CENG3300 Lecture 4Document25 pagesCENG3300 Lecture 4huichloemailNo ratings yet
- Introduction To Structural DatabasesDocument10 pagesIntroduction To Structural Databasessumit mahajanNo ratings yet
- Protein Data Bank IntroductionDocument43 pagesProtein Data Bank IntroductionRigel_TNo ratings yet
- CIF-file and ValidationDocument25 pagesCIF-file and Validationbasco costasNo ratings yet
- Basic Introduction To SIESTA: Emilio ArtachoDocument48 pagesBasic Introduction To SIESTA: Emilio ArtachoRasool FarajiNo ratings yet
- Lecture Topic: Protein Databases: Topics CoveredDocument67 pagesLecture Topic: Protein Databases: Topics CoveredS ARUNANo ratings yet
- BazaamiloidaDocument5 pagesBazaamiloidaIvanaNo ratings yet
- Bioinformatics and Structural Genomics: John IonidesDocument69 pagesBioinformatics and Structural Genomics: John IonidesatpharateNo ratings yet
- Unit-5 BioinformaticsDocument13 pagesUnit-5 Bioinformaticsp vmuraliNo ratings yet
- CompimaDocument26 pagesCompimajeremytoh89No ratings yet
- Bioinformatics: Md. Fazlul Karim PatwaryDocument98 pagesBioinformatics: Md. Fazlul Karim Patwary1898Siddikur RahmanNo ratings yet
- How To Use The PDB: 1) What Is Protein Data Bank (PDB) ?Document4 pagesHow To Use The PDB: 1) What Is Protein Data Bank (PDB) ?Sumit HalderNo ratings yet
- GKN 860Document5 pagesGKN 860Recky PatalaNo ratings yet
- Lecture-5-Information-retrieval-from-databasesDocument22 pagesLecture-5-Information-retrieval-from-databasesVeer khadeNo ratings yet
- Proteins Bioinfo LatestDocument45 pagesProteins Bioinfo Latestpeace loverNo ratings yet
- 1 - em Structures - Ardan PatwardhanDocument50 pages1 - em Structures - Ardan PatwardhanRigel_TNo ratings yet
- Protein Structure and FunctionDocument52 pagesProtein Structure and FunctionbharatNo ratings yet
- Database: Formats and FunctionsDocument103 pagesDatabase: Formats and FunctionsSuraj VermaNo ratings yet
- Unit 6 - BioinformaticsDocument41 pagesUnit 6 - BioinformaticsLeonNo ratings yet
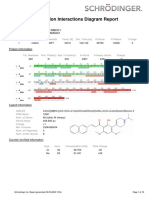
- Simulation Interactions Diagram Report: Jobname Entry TitleDocument10 pagesSimulation Interactions Diagram Report: Jobname Entry TitlepaiaravindNo ratings yet
- Bioinformatics Databases - An IntroductionDocument10 pagesBioinformatics Databases - An IntroductionVarshika SinghNo ratings yet
- CSDDocument14 pagesCSDNandni JhaNo ratings yet
- MD Tutorial Using GromacsDocument22 pagesMD Tutorial Using GromacsSeagal AsjaliNo ratings yet
- Biol BDs SingaporeDocument24 pagesBiol BDs Singaporestevekct10No ratings yet
- Technical DescriptionDocument15 pagesTechnical DescriptionElena VenegoniNo ratings yet
- Lec2 DatabasesDocument135 pagesLec2 DatabasesDeepali SinghNo ratings yet
- Bale Supplement-Supplementary MaterialsDocument46 pagesBale Supplement-Supplementary Materialsbhoomika_basuNo ratings yet
- DNA and Protein Databases for Molecular BiologyDocument31 pagesDNA and Protein Databases for Molecular Biologyaditya.2352700No ratings yet
- PAM Blosum: Assignment 1 Bioinformatics (DSE 1)Document9 pagesPAM Blosum: Assignment 1 Bioinformatics (DSE 1)Maithili Joshi100% (1)
- Protein 3dDocument86 pagesProtein 3dSitiHamidatulAliyahNo ratings yet
- Biological Databases GuideDocument31 pagesBiological Databases GuideSir RutherfordNo ratings yet
- Bioinfo - S1 2021 - L9 - Protein Structure - 1 SlideDocument87 pagesBioinfo - S1 2021 - L9 - Protein Structure - 1 SlideHuynh Ngoc Da ThaoNo ratings yet
- Resumen Unidad 1 y 2 BioinformaticaDocument14 pagesResumen Unidad 1 y 2 BioinformaticaPaulette GuerreroNo ratings yet
- Text Database Search SystemsDocument54 pagesText Database Search SystemsshikhaNo ratings yet
- Pertemuan 7 Kimkom Teori Desain Obat Berbasis LiganDocument83 pagesPertemuan 7 Kimkom Teori Desain Obat Berbasis LiganGalan Rizqi YanuarNo ratings yet
- 3.input Files For PDDocument5 pages3.input Files For PDDeepak Kumar S NadigerNo ratings yet
- We Are Intechopen, The World'S Leading Publisher of Open Access Books Built by Scientists, For ScientistsDocument20 pagesWe Are Intechopen, The World'S Leading Publisher of Open Access Books Built by Scientists, For ScientistsNaidan DensmaaNo ratings yet
- Biological DatabasesDocument39 pagesBiological DatabasesKasun BandaraNo ratings yet
- SCOP and CATH DatabaseDocument22 pagesSCOP and CATH DatabaseAishwarya Dharan100% (3)
- MOF diversity understanding metal-organic framework ecosystemDocument52 pagesMOF diversity understanding metal-organic framework ecosystemZijun DengNo ratings yet
- Biophysics and Structural Bioinformatics IDocument34 pagesBiophysics and Structural Bioinformatics IMaddy IlNo ratings yet
- Protein Analysis & Proteomics TechniquesDocument62 pagesProtein Analysis & Proteomics TechniquesNgan TranNo ratings yet
- Data Types: CMPS401 Class Notes (Chap06) Page 1 / 35 Dr. Kuo-Pao YangDocument35 pagesData Types: CMPS401 Class Notes (Chap06) Page 1 / 35 Dr. Kuo-Pao YangLuchin ZamoraNo ratings yet
- The Sumo Server: 3D Search For Protein Functional Sites: Bioinformatics November 2005Document3 pagesThe Sumo Server: 3D Search For Protein Functional Sites: Bioinformatics November 2005Ruboto BotoNo ratings yet
- Dali TutorialDocument19 pagesDali TutorialADITI KONARNo ratings yet
- Journal of Cheminformatics: Orchem - An Open Source Chemistry Search Engine For OracleDocument11 pagesJournal of Cheminformatics: Orchem - An Open Source Chemistry Search Engine For Oraclehoremheb1No ratings yet
- Biological Databases Pharmamatrix Workshop 2010: - Philip - Ishwar V. HosamaniDocument28 pagesBiological Databases Pharmamatrix Workshop 2010: - Philip - Ishwar V. Hosamanitri sutrianiNo ratings yet
- Building Molecular ModelsDocument4 pagesBuilding Molecular ModelsValine Cysteine MethionineNo ratings yet
- Proclust:: Improved Clustering of Protein Sequences With An Extended Graph-Based ApproachDocument58 pagesProclust:: Improved Clustering of Protein Sequences With An Extended Graph-Based ApproachJJ EBNo ratings yet
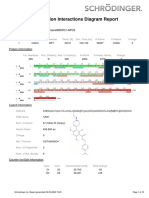
- Simulation Interactions Diagram Report: Jobname Entry TitleDocument10 pagesSimulation Interactions Diagram Report: Jobname Entry TitlepaiaravindNo ratings yet
- Serves ListDocument34 pagesServes Listgopi100% (1)
- Bioinformatics Seminar3rdOct18Document25 pagesBioinformatics Seminar3rdOct18subhasree majumderNo ratings yet
- Handbook of Inorganic Substances-De Gruyter (2018)Document1,817 pagesHandbook of Inorganic Substances-De Gruyter (2018)Samyak DholeNo ratings yet
- Basic Tutorial DockThor 1.0 6Document17 pagesBasic Tutorial DockThor 1.0 6Emmanuel MarinhoNo ratings yet
- PyMol PracticalDocument7 pagesPyMol PracticalTom FlemingNo ratings yet
- Protein Modeling: Protein Structure Prediction Other TopicsDocument76 pagesProtein Modeling: Protein Structure Prediction Other Topicsuma-chenNo ratings yet
- 3.1 Artifact OptimiserDocument120 pages3.1 Artifact Optimiseralex joNo ratings yet
- Tables and Bar ChartsDocument7 pagesTables and Bar ChartsVictoria FedoseevaNo ratings yet
- MidtermDocument3 pagesMidtermTrisha MondonedoNo ratings yet
- Reverse Phrase Action Camera LightsDocument40 pagesReverse Phrase Action Camera LightsDINDO AzucenaNo ratings yet
- Comparative Study of Organic Solvents For Extraction of Copper From Ammoniacal Carbonate Solution Hu2010Document6 pagesComparative Study of Organic Solvents For Extraction of Copper From Ammoniacal Carbonate Solution Hu2010mtanaydinNo ratings yet
- Tabcalcs.com general equations sheetDocument2 pagesTabcalcs.com general equations sheetRamadan RashadNo ratings yet
- Sponge BobDocument4 pagesSponge BobchabriesNo ratings yet
- Anchor Systems.: Hilti. Outperform. OutlastDocument49 pagesAnchor Systems.: Hilti. Outperform. Outlastthapa786mNo ratings yet
- Catalogus 2010 ENGELSDocument52 pagesCatalogus 2010 ENGELShacikadiNo ratings yet
- Hero LetterDocument3 pagesHero LetterArunachalam Muthiah0% (1)
- Sabp X 002Document18 pagesSabp X 002Hassan MokhtarNo ratings yet
- Fair Directory 02-2016Document44 pagesFair Directory 02-2016Ravichandran SNo ratings yet
- Introduction To The Philosophy of The Human PersonDocument14 pagesIntroduction To The Philosophy of The Human PersonDan Niel50% (2)
- Sliding, Overturning, Bearing Pressure and Bending Reinforcement Calculations for Retaining WallDocument4 pagesSliding, Overturning, Bearing Pressure and Bending Reinforcement Calculations for Retaining WallAbdul Aziz Julkarnain ZulkifliNo ratings yet
- Hearing Aid InformationDocument22 pagesHearing Aid InformationDeepakRodeyNo ratings yet
- Computational Models For Trunk Trajectory Planning and Load Distribution: A Test-Bed For Studying Various Clinical Adaptation and Motor Control Strategies of Low Back Pain PatientsDocument13 pagesComputational Models For Trunk Trajectory Planning and Load Distribution: A Test-Bed For Studying Various Clinical Adaptation and Motor Control Strategies of Low Back Pain PatientsOTorresGonzalezNo ratings yet
- FLIR Blackfly Users ManualDocument53 pagesFLIR Blackfly Users ManualPavan Kumar BittuNo ratings yet
- Meralco Bill 330370940102 04142023Document2 pagesMeralco Bill 330370940102 04142023Jha CruzNo ratings yet
- Lecture No.3 Part 1 (Fan)Document6 pagesLecture No.3 Part 1 (Fan)Mohsen HassanNo ratings yet
- Thermal Performance of Air-Cooled Condensing Units by CFD SimulationDocument2 pagesThermal Performance of Air-Cooled Condensing Units by CFD SimulationFauziah JeraiNo ratings yet
- Studyguide TracksDocument75 pagesStudyguide TracksAnonymous FabB2WJl485% (13)
- Owner S Manual Subaru LegacyDocument20 pagesOwner S Manual Subaru LegacyLexBgNo ratings yet
- A Thing of Beauty Analysis by Sharvaree S. ChavanDocument12 pagesA Thing of Beauty Analysis by Sharvaree S. Chavansharu11No ratings yet
- Tooth Development, Eruption & Applied Aspects: Saurabh Roy 09.03.2016Document95 pagesTooth Development, Eruption & Applied Aspects: Saurabh Roy 09.03.2016reema aslamNo ratings yet
- STP1236 Eb.1415051 1 PDFDocument208 pagesSTP1236 Eb.1415051 1 PDFpaolaNo ratings yet
- General Biology 1: Go Fast, or Slow Down?Document23 pagesGeneral Biology 1: Go Fast, or Slow Down?Mikhael OiraNo ratings yet
- Category D Fluid ServiceDocument2 pagesCategory D Fluid Serviceaslam.ambNo ratings yet
- BrosurDocument68 pagesBrosurKiki Xhui7No ratings yet
- CD 0400 CH 4 X 100 ML: For in Vitro Diagnostic Use Only. LinearityDocument1 pageCD 0400 CH 4 X 100 ML: For in Vitro Diagnostic Use Only. LinearityNguyễn ThơiNo ratings yet