You might also like
- Basic Concepts in GeneticsDocument27 pagesBasic Concepts in Geneticsmohamed.puntforensicsNo ratings yet
- By: Muhammad Iqbal Khan SS Biology Thanks For Anna TzemachDocument28 pagesBy: Muhammad Iqbal Khan SS Biology Thanks For Anna TzemachRabiya KianiNo ratings yet
- Dasar Genetika+molekulerDocument45 pagesDasar Genetika+molekulerAGASTI MERCU SUANo ratings yet
- B. Concepts in Genetics335Document14 pagesB. Concepts in Genetics335owenspros5No ratings yet
- GenPop 2 - Pola Pewarisan SifatDocument29 pagesGenPop 2 - Pola Pewarisan SifatMonicha Yhuyhen SafitriNo ratings yet
- 1.2 Mendels Laws and More TerminologiesDocument18 pages1.2 Mendels Laws and More TerminologiesshrivatsapandeNo ratings yet
- CH4 Patho D&R AgamDocument28 pagesCH4 Patho D&R AgamS Balagopal SivaprakasamNo ratings yet
- 2a Inheritance CDocument9 pages2a Inheritance Capi-238529408No ratings yet
- 3 Genetics Lecture - Principles of HeredityDocument53 pages3 Genetics Lecture - Principles of HeredityAMIRA HELAYELNo ratings yet
- Genes and Inheritance: GCE Study Buddy BiologyDocument38 pagesGenes and Inheritance: GCE Study Buddy BiologyPeace FulNo ratings yet
- Lecture 11 - Genetics IIIDocument3 pagesLecture 11 - Genetics IIIbesillysillyNo ratings yet
- BIO SCI 97 Midterm Study GuideDocument15 pagesBIO SCI 97 Midterm Study GuidefsadfNo ratings yet
- Patterns of Single-Gene InheritanceDocument55 pagesPatterns of Single-Gene InheritanceinakiNo ratings yet
- Heredity and Hereditary DiseasesDocument31 pagesHeredity and Hereditary DiseasesMarcelo SicaNo ratings yet
- GeneticsDocument29 pagesGeneticsbabaaijaz01No ratings yet
- General Biology 2 Second SemesterDocument49 pagesGeneral Biology 2 Second SemesterRuthie TabanguilNo ratings yet
- Inborn Error of MetabolismDocument59 pagesInborn Error of MetabolismMadhu Sudhan PandeyaNo ratings yet
- Chapter 15 NotesDocument5 pagesChapter 15 NotesjonlimeNo ratings yet
- Genetics and EvolutionDocument6 pagesGenetics and EvolutionCherry Grace Articulo DabuconNo ratings yet
- Heredity Class 10 NotesDocument4 pagesHeredity Class 10 Notesashlyyyyyy33No ratings yet
- Genetics 23 24Document75 pagesGenetics 23 24Marlon CanilangNo ratings yet
- 1 GeneticsDocument36 pages1 GeneticszahuuNo ratings yet
- Notes On Inheritance CP 5Document34 pagesNotes On Inheritance CP 5Ryan AhmedNo ratings yet
- Genetic and ChromosomesDocument16 pagesGenetic and ChromosomesJay PornelaNo ratings yet
- Wa0010.Document5 pagesWa0010.Se YiNo ratings yet
- REV #5 - Introduction To GeneticsDocument3 pagesREV #5 - Introduction To GeneticsCatalytic AintNo ratings yet
- GeneticsDocument84 pagesGeneticsMarylan FerolinoNo ratings yet
- Mendel HuGeneticsDocument26 pagesMendel HuGeneticsEmi PhillipsNo ratings yet
- Lec 11Document20 pagesLec 11LabibNo ratings yet
- Genetic Disease - 230903 - 101420Document66 pagesGenetic Disease - 230903 - 101420ShadowStormNo ratings yet
- Genetic DiseasesDocument66 pagesGenetic DiseasesKeziah HérmosoNo ratings yet
- 91 Human Genetics 3 With AnnopdfDocument35 pages91 Human Genetics 3 With Annopdfnikhil naniNo ratings yet
- Genetics 1Document64 pagesGenetics 1Charles Joseph MaldiaNo ratings yet
- HeredityDocument19 pagesHeredityichan0001No ratings yet
- Genbio - Unit 2 - Genetics - l1 Mendelian and Post Mendelian GeneticsDocument40 pagesGenbio - Unit 2 - Genetics - l1 Mendelian and Post Mendelian GeneticsErika Jane BulanhaguiNo ratings yet
- Genetics in OrthoDocument58 pagesGenetics in OrthoAthira JithendranathNo ratings yet
- Game To GenesisDocument48 pagesGame To GenesisJovie Esquivias NicolasNo ratings yet
- Meiosis and Genetic DiversityDocument25 pagesMeiosis and Genetic DiversityBasla MalikNo ratings yet
- CYTOGENETICS NotesDocument6 pagesCYTOGENETICS NotesAndriaNo ratings yet
- Fundamentals of GeneticsDocument53 pagesFundamentals of GeneticsvladyNo ratings yet
- CL 12 Bio Chapter-5 Principle of Inheritance and VariationDocument53 pagesCL 12 Bio Chapter-5 Principle of Inheritance and Variationjackieaj093No ratings yet
- Chapter 10 Patterns of InheritanceDocument4 pagesChapter 10 Patterns of Inheritancesherlynmamac98No ratings yet
- 4 2018 11 27!08 57 45 PMDocument6 pages4 2018 11 27!08 57 45 PMA HNo ratings yet
- Unit 3 Genetics & Heredity: Biology 30 Mr. OosteromDocument93 pagesUnit 3 Genetics & Heredity: Biology 30 Mr. OosteromSa HilNo ratings yet
- Basic Concepts in GeneticsDocument28 pagesBasic Concepts in GeneticsFarooq HussainNo ratings yet
- Chromosomes: Magdalena F. Natividad, PHD Dean, School of Medical Lab Science Feu-NrmfDocument24 pagesChromosomes: Magdalena F. Natividad, PHD Dean, School of Medical Lab Science Feu-NrmfCharm AngelesNo ratings yet
- Mendelian Genetics and ChromosomesDocument48 pagesMendelian Genetics and Chromosomesreginedelamarquez14No ratings yet
- Genetics NotesDocument26 pagesGenetics Notesapi-655732711No ratings yet
- Heredity and EvolutionDocument65 pagesHeredity and EvolutionAbdul KarimNo ratings yet
- NOTES - CH 15 - Sex Determination - Linkage - SlideshowDocument59 pagesNOTES - CH 15 - Sex Determination - Linkage - Slideshowkaren milloNo ratings yet
- Biology Chapter11 Introductiontogenetics 091122213400 Phpapp01Document78 pagesBiology Chapter11 Introductiontogenetics 091122213400 Phpapp01Osama AlrawabNo ratings yet
- Genetics Course 3 M-1Document161 pagesGenetics Course 3 M-1Shiko LoveNo ratings yet
- Basic Concepts of GeneticsDocument27 pagesBasic Concepts of GeneticsNishant UpadhyayNo ratings yet
- GeneticsDocument27 pagesGeneticsRose HaliliNo ratings yet
- Variation F 5Document8 pagesVariation F 5Angie Kong Su MeiNo ratings yet
- Topic 16 Inherited Change 2021-22 A Level Biology 9700 Notes by Mr. ADEEL AHMADDocument23 pagesTopic 16 Inherited Change 2021-22 A Level Biology 9700 Notes by Mr. ADEEL AHMADADEEL AHMAD100% (1)
- An Introduction To Human Genetics JassimDocument16 pagesAn Introduction To Human Genetics Jassimdr_47839666No ratings yet
- Developmental Psy 1Document26 pagesDevelopmental Psy 1Sonia SoansNo ratings yet
- Genetic Information: - GeneDocument7 pagesGenetic Information: - GeneShahriar ShamimNo ratings yet
- Acute LeukemiasDocument79 pagesAcute LeukemiasSravani PeddagangannagariNo ratings yet
- RJ Flower The MediatorsDocument55 pagesRJ Flower The Mediatorstamuno7No ratings yet
- 6 InfectionsDocument71 pages6 InfectionsSravani PeddagangannagariNo ratings yet
- Thesis IntroductionDocument155 pagesThesis IntroductionSravani PeddagangannagariNo ratings yet
- Anatomy& EmbryologyDocument13 pagesAnatomy& EmbryologySravani PeddagangannagariNo ratings yet
- CONFERENCE PAPER FinalDocument29 pagesCONFERENCE PAPER FinalSravani PeddagangannagariNo ratings yet
- Soft ppt3Document99 pagesSoft ppt3Sravani PeddagangannagariNo ratings yet
- Revised Basic Course Workshop Government Medical College, KadapaDocument56 pagesRevised Basic Course Workshop Government Medical College, KadapaSravani PeddagangannagariNo ratings yet
- Educational Networking For GrowthDocument44 pagesEducational Networking For GrowthSravani PeddagangannagariNo ratings yet
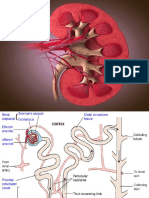
- Renal SystemDocument21 pagesRenal SystemSravani PeddagangannagariNo ratings yet
- Pyelonephritis: Dr.P.SravaniDocument70 pagesPyelonephritis: Dr.P.SravaniSravani Peddagangannagari100% (1)
- Bone Marrow ExaminationDocument88 pagesBone Marrow ExaminationSravani PeddagangannagariNo ratings yet
- BF254 BF255Document3 pagesBF254 BF255rrr2013No ratings yet
- SRM 7 EHP 4 Release Notes PDFDocument18 pagesSRM 7 EHP 4 Release Notes PDFMOHAMMED SHEHBAAZNo ratings yet
- José Guadalupe PosadaDocument19 pagesJosé Guadalupe PosadaJudy Baca100% (1)
- Stating Like and DislikesDocument2 pagesStating Like and DislikesDavid ArdiantoNo ratings yet
- AC7140 Rev CDocument73 pagesAC7140 Rev CRanga100% (1)
- Paso de Blas Lying in Clinic For NewDocument5 pagesPaso de Blas Lying in Clinic For NewNaheed Dean MustafaNo ratings yet
- L15 PDFDocument15 pagesL15 PDFlesNo ratings yet
- Loch ChildrenDocument4 pagesLoch ChildrenLauro De Jesus FernandesNo ratings yet
- Lecture 5: Triangulation Adjustment Triangulation: in This Lecture We Focus On The Second MethodDocument5 pagesLecture 5: Triangulation Adjustment Triangulation: in This Lecture We Focus On The Second MethodXogr BargarayNo ratings yet
- Combat Storm - Shipping ContainerDocument6 pagesCombat Storm - Shipping ContainermoiNo ratings yet
- NUFLO Low Power Pre-Amplifier: SpecificationsDocument2 pagesNUFLO Low Power Pre-Amplifier: SpecificationsJorge ParraNo ratings yet
- Arrhythmia - WikipediaDocument76 pagesArrhythmia - WikipediaJamesNo ratings yet
- Portfolio Eelco Maan - 06-2017Document25 pagesPortfolio Eelco Maan - 06-2017tungaas20011No ratings yet
- One God One People February 2013Document297 pagesOne God One People February 2013Stig DragholmNo ratings yet
- Module 1Document64 pagesModule 1Jackyson RajkumarNo ratings yet
- Expansions Meet Health Care Needs: Economists Question Trump Plan FiguresDocument10 pagesExpansions Meet Health Care Needs: Economists Question Trump Plan FiguresThe Daily Tar HeelNo ratings yet
- BZY Series Tension Meter ManualDocument29 pagesBZY Series Tension Meter ManualJORGE SANTANDER0% (1)
- Buy Wholesale China Popular Outdoor Football Boot For Teenagers Casual High Quality Soccer Shoes FG Ag Graffiti Style & FootballDocument1 pageBuy Wholesale China Popular Outdoor Football Boot For Teenagers Casual High Quality Soccer Shoes FG Ag Graffiti Style & Footballjcdc9chh8dNo ratings yet
- Easy NoFap EbookDocument37 pagesEasy NoFap Ebookசரஸ்வதி சுவாமிநாதன்No ratings yet
- HBS - Zara Fast Fashion Case Write UpDocument4 pagesHBS - Zara Fast Fashion Case Write Upaaronhwalton100% (1)
- ISO 11064-12000 Ergonomic Design of Control Centres - Part 1 Principles For The Design of Control Centres by ISO TC 159SC 4WG 8Document6 pagesISO 11064-12000 Ergonomic Design of Control Centres - Part 1 Principles For The Design of Control Centres by ISO TC 159SC 4WG 8marcianocalvi4611100% (2)
- Classical Encryption TechniqueDocument18 pagesClassical Encryption TechniquetalebmuhsinNo ratings yet
- Market Challengers StrategiesDocument19 pagesMarket Challengers Strategiestobbob007100% (20)
- Bubble SortDocument6 pagesBubble SortRollin RevieNo ratings yet
- A Dessertation Report Submitted in Partial Fulfillment of Requirements For The Award of The Degree ofDocument65 pagesA Dessertation Report Submitted in Partial Fulfillment of Requirements For The Award of The Degree ofMadhavpokale100% (1)
- Avanquest Perfect Image V.12 User GuideDocument174 pagesAvanquest Perfect Image V.12 User GuideShafiq-UR-Rehman Lodhi100% (1)
- ATA212001Document3 pagesATA212001Tarek DeghedyNo ratings yet
- 10 Problem For The Topic 9 & 10 Hicao GroupDocument4 pages10 Problem For The Topic 9 & 10 Hicao GroupArvin ArmojallasNo ratings yet
- The Impact of Social Networking Sites To The Academic Performance of The College Students of Lyceum of The Philippines - LagunaDocument15 pagesThe Impact of Social Networking Sites To The Academic Performance of The College Students of Lyceum of The Philippines - LagunaAasvogel Felodese Carnivora64% (14)
- Full Download Human Biology 11th Edition Starr Solutions ManualDocument35 pagesFull Download Human Biology 11th Edition Starr Solutions Manualsheathe.zebrinny.53vubg100% (41)