You might also like
- Modern School For SaxophoneDocument23 pagesModern School For SaxophoneAllen Demiter65% (23)
- Haematopathology 3:: Leucocytosis/LeucopeniaDocument113 pagesHaematopathology 3:: Leucocytosis/LeucopeniaarwaNo ratings yet
- Chronic Lymphoproliferative DisordersDocument108 pagesChronic Lymphoproliferative DisordersSiti NurrazanNo ratings yet
- Disorders of White Blood Cells and Lymphoid TissuesDocument30 pagesDisorders of White Blood Cells and Lymphoid Tissuesammar amerNo ratings yet
- Eurocode 3: Design of Steel Structures "ReadyDocument26 pagesEurocode 3: Design of Steel Structures "Readywazydotnet80% (10)
- Segregation in CastingDocument17 pagesSegregation in CastingAsmaa Smsm Abdallh78% (9)
- Acute Leukaemias Lecture-1Document39 pagesAcute Leukaemias Lecture-1MarvellousNo ratings yet
- Hematology Review 2021-2Document142 pagesHematology Review 2021-2Maram AbdullahNo ratings yet
- Hydraulic Mining ExcavatorDocument8 pagesHydraulic Mining Excavatorasditia_07100% (1)
- Non-Hodgkin'S Lymphoma: Oliveros Francis!!!!!!!!!!!!!!!!!Document48 pagesNon-Hodgkin'S Lymphoma: Oliveros Francis!!!!!!!!!!!!!!!!!francis00090No ratings yet
- 20 Lymphoid and Plasma Cell NeoplasmsDocument12 pages20 Lymphoid and Plasma Cell NeoplasmsDaphne Hernaez100% (1)
- Lymphoid NeoplasmsDocument52 pagesLymphoid NeoplasmsAmalia Riska GNo ratings yet
- Acute Lymphoblastic Leukemia (ALL)Document14 pagesAcute Lymphoblastic Leukemia (ALL)Med PhuongNo ratings yet
- Acute Lymphoblastic LeukemiaDocument27 pagesAcute Lymphoblastic LeukemiaMahalakshmi PalanisamiNo ratings yet
- Acute Leukemia HandoutDocument10 pagesAcute Leukemia Handoutnonie jacobNo ratings yet
- Acute Lymphocytic Leukemia: MR: Mohammed Mahmoud AlhajDocument14 pagesAcute Lymphocytic Leukemia: MR: Mohammed Mahmoud Alhajحسن محمدNo ratings yet
- Acute Leukemia: Thirunavukkarasu MurugappanDocument22 pagesAcute Leukemia: Thirunavukkarasu MurugappanFelix Allen100% (1)
- Hematology 1 LeukemiaDocument140 pagesHematology 1 LeukemiamaryantoinetteriveraNo ratings yet
- T NK CellDocument117 pagesT NK CellPpds MtatasuhartaNo ratings yet
- Molecular Basis of Acute LeukemiaDocument31 pagesMolecular Basis of Acute LeukemiaVivek SharmaNo ratings yet
- Hematopathlogy 2Document87 pagesHematopathlogy 2evansmando12No ratings yet
- ' / 5 Introduction of LeukemiasaDocument37 pages' / 5 Introduction of LeukemiasashaikhaboausaibaNo ratings yet
- LNs HNDocument190 pagesLNs HNNinna Isabel VictorioNo ratings yet
- Precursor B and T CellDocument8 pagesPrecursor B and T CellKita kitaNo ratings yet
- Back Up Slide ALL SkingDocument20 pagesBack Up Slide ALL SkingFebry BieluciousNo ratings yet
- Paediatric Acute Lymphoblastic LeukemiaDocument49 pagesPaediatric Acute Lymphoblastic LeukemiaKishoreChandraKoradaNo ratings yet
- Chapter 13 Neoplastic Proliferations of White CellsDocument16 pagesChapter 13 Neoplastic Proliferations of White CellsOmar100% (1)
- PATHO LEC WBC Lymph Nodes Spleen Thymus Part1 CompressedDocument84 pagesPATHO LEC WBC Lymph Nodes Spleen Thymus Part1 CompressedAngelo HinonNo ratings yet
- Acute LeukaemiaDocument39 pagesAcute LeukaemiaAbby LiewNo ratings yet
- HaematologyDocument25 pagesHaematologyMenziPhiwokuhleSukatiNo ratings yet
- Acute Lymphoblastic LeukaemiaDocument9 pagesAcute Lymphoblastic LeukaemiaAdams ZarawuNo ratings yet
- Leukemia: Defintion: Leukemias Are Diseases in Which Localised or Generalised Proliferation orDocument12 pagesLeukemia: Defintion: Leukemias Are Diseases in Which Localised or Generalised Proliferation orsharon victoria mendezNo ratings yet
- Fiedacan Maala Acute Precursor B Cell Lymphoblastic Leukemia Diagnostic Evaluation and Flow Cytometric AnalysisDocument28 pagesFiedacan Maala Acute Precursor B Cell Lymphoblastic Leukemia Diagnostic Evaluation and Flow Cytometric AnalysisGia UrdaiaNo ratings yet
- Acute Monocytic Leukemia and Acute Lmphocytic Leukemia: OccurrenceDocument12 pagesAcute Monocytic Leukemia and Acute Lmphocytic Leukemia: OccurrenceAngelo Jude CobachaNo ratings yet
- Chapter 13 - Diseases of White Blood Cells, Lymph Nodes, Spleen, and ThymusDocument10 pagesChapter 13 - Diseases of White Blood Cells, Lymph Nodes, Spleen, and ThymusAgnieszka WisniewskaNo ratings yet
- Blockxiv Neoplasms Lymphoid 2006Document54 pagesBlockxiv Neoplasms Lymphoid 2006Ryo RyozNo ratings yet
- Luekemia by Dr. WongelDocument63 pagesLuekemia by Dr. Wongelmogesie1995No ratings yet
- Acute Leukaemia-Update: DR Niranjan N. RathodDocument89 pagesAcute Leukaemia-Update: DR Niranjan N. RathodratanNo ratings yet
- 6.3. LymphomaDocument41 pages6.3. LymphomaAkli JahNo ratings yet
- (Romanian Journal of Internal Medicine) Acute Lymphocytic Leukemia in Adults. Pathologic Features and PrognosisDocument6 pages(Romanian Journal of Internal Medicine) Acute Lymphocytic Leukemia in Adults. Pathologic Features and PrognosisPepeeNo ratings yet
- Acute Lymphoblastic LeukemiaDocument80 pagesAcute Lymphoblastic LeukemiaGizo YitayewNo ratings yet
- 4b TUMOR JAR RetikuloendotelialAAADocument98 pages4b TUMOR JAR RetikuloendotelialAAARyo RyozNo ratings yet
- LeukemiaDocument91 pagesLeukemiaShadin SNo ratings yet
- Non - Hodgkin LymphomaDocument25 pagesNon - Hodgkin LymphomaRo RyNo ratings yet
- Leukaemia: Definition: Leukemia Is A Malignant Disease of The Hematopoietic System (Blood Forming Cells)Document16 pagesLeukaemia: Definition: Leukemia Is A Malignant Disease of The Hematopoietic System (Blood Forming Cells)Arnab Ghosh100% (1)
- Overview of LymphomaDocument42 pagesOverview of Lymphomaadamu mohammadNo ratings yet
- ALL Ferri 2019 PDFDocument5 pagesALL Ferri 2019 PDFMia FernandezNo ratings yet
- The LeukemiasDocument52 pagesThe Leukemiasمصطفي خندقاوي100% (1)
- Chronic Lymphocytic Leukemia: Dr. Tjatur Winarsanto SPPD Rs Ciremai Cirebon 2012Document17 pagesChronic Lymphocytic Leukemia: Dr. Tjatur Winarsanto SPPD Rs Ciremai Cirebon 2012Mohammad Fadel SatriansyahNo ratings yet
- Lymph Node CytologyDocument39 pagesLymph Node Cytologykamranghani641No ratings yet
- Week 05. Acute LeukemiasDocument26 pagesWeek 05. Acute LeukemiasAshley ArnoldNo ratings yet
- AML, CML, ALL, CLL, HemophiliaDocument7 pagesAML, CML, ALL, CLL, HemophiliaJamara Kyla Dela CruzNo ratings yet
- Pa Tho Physiology of LymphomasDocument17 pagesPa Tho Physiology of LymphomasKent MasadoNo ratings yet
- ALL - Curs (Engl)Document10 pagesALL - Curs (Engl)Andreea TudurachiNo ratings yet
- 1 Acute LeukemiaDocument14 pages1 Acute Leukemiaسمير هزاعNo ratings yet
- Week 15-Mature-Lymphoid-Neoplasms-SCDocument65 pagesWeek 15-Mature-Lymphoid-Neoplasms-SCKyle CollladoNo ratings yet
- Acute Lymphoblastic LeukemiaDocument5 pagesAcute Lymphoblastic LeukemiavnykumalasariNo ratings yet
- LeukemiaDocument51 pagesLeukemiaKailash KhatriNo ratings yet
- Acute Lymphoblastic Leukemia - LecturioDocument17 pagesAcute Lymphoblastic Leukemia - LecturioCornel PopaNo ratings yet
- Chronic Myeloid Leukemia.2023aDocument21 pagesChronic Myeloid Leukemia.2023aFavourNo ratings yet
- Acute Leukemia: Yetty Movieta Nency Pediatric Hemato-OncologistDocument17 pagesAcute Leukemia: Yetty Movieta Nency Pediatric Hemato-OncologistalivanabilafarinisaNo ratings yet
- Chronic Lymphocytic LeukemiaFrom EverandChronic Lymphocytic LeukemiaMichael HallekNo ratings yet
- Lab Session 4Document15 pagesLab Session 4Ro RyNo ratings yet
- Introduction To Bacteriology II - 29012024Document13 pagesIntroduction To Bacteriology II - 29012024Ro RyNo ratings yet
- Lab Session 2Document21 pagesLab Session 2Ro RyNo ratings yet
- Coagulation TestingDocument29 pagesCoagulation TestingRo RyNo ratings yet
- The Monospot TestDocument9 pagesThe Monospot TestRo RyNo ratings yet
- POLYCYTHEMIA VERA PresentationDocument12 pagesPOLYCYTHEMIA VERA PresentationRo RyNo ratings yet
- Urine Microscopic Examination Jun2021Document2 pagesUrine Microscopic Examination Jun2021Ro RyNo ratings yet
- ApplicationGUIDELINES BookletDocument52 pagesApplicationGUIDELINES BookletRo RyNo ratings yet
- in 30 MinutesDocument5 pagesin 30 MinutesCésar DiazNo ratings yet
- Introduction To Atomistic Simulation Through Density Functional TheoryDocument21 pagesIntroduction To Atomistic Simulation Through Density Functional TheoryTarang AgrawalNo ratings yet
- Colorfastness of Zippers To Light: Standard Test Method ForDocument2 pagesColorfastness of Zippers To Light: Standard Test Method ForShaker QaidiNo ratings yet
- Focus Edition From GC: Phosphate Bonded Investments For C&B TechniquesDocument35 pagesFocus Edition From GC: Phosphate Bonded Investments For C&B TechniquesAlexis De Jesus FernandezNo ratings yet
- Data Sheet Eldar Void SpinnerDocument1 pageData Sheet Eldar Void SpinnerAlex PolleyNo ratings yet
- BLP#1 - Assessment of Community Initiative (3 Files Merged)Document10 pagesBLP#1 - Assessment of Community Initiative (3 Files Merged)John Gladhimer CanlasNo ratings yet
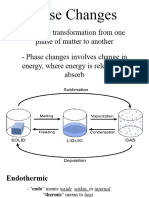
- General Chemistry 2 Q1 Lesson 5 Endothermic and Exotheric Reaction and Heating and Cooling CurveDocument19 pagesGeneral Chemistry 2 Q1 Lesson 5 Endothermic and Exotheric Reaction and Heating and Cooling CurveJolo Allexice R. PinedaNo ratings yet
- Airport & Harbour Engg-AssignmentDocument3 pagesAirport & Harbour Engg-AssignmentAshok Kumar RajanavarNo ratings yet
- Genomic Tools For Crop ImprovementDocument41 pagesGenomic Tools For Crop ImprovementNeeru RedhuNo ratings yet
- Chemistry Investigatory Project (R)Document23 pagesChemistry Investigatory Project (R)BhagyashreeNo ratings yet
- Form Expense ClaimDocument2 pagesForm Expense Claimviedelamonde_3868443No ratings yet
- Steel Design Fourth Edition William T Segui Solution Manual 1Document11 pagesSteel Design Fourth Edition William T Segui Solution Manual 1RazaNo ratings yet
- Mathematics BQP 2022Document43 pagesMathematics BQP 2022muhammadmansuri815No ratings yet
- 1916 South American Championship Squads - WikipediaDocument6 pages1916 South American Championship Squads - WikipediaCristian VillamayorNo ratings yet
- 1 Bacterial DeseaseDocument108 pages1 Bacterial DeseasechachaNo ratings yet
- S4 HANALicensing Model External V19Document28 pagesS4 HANALicensing Model External V19Edir JuniorNo ratings yet
- Siemens Rapidlab 248, 348, 840, 845, 850, 855, 860, 865: Reagents & ControlsDocument2 pagesSiemens Rapidlab 248, 348, 840, 845, 850, 855, 860, 865: Reagents & ControlsJuan Carlos CrespoNo ratings yet
- Modern and Nonlinear OpticsDocument181 pagesModern and Nonlinear Opticssoma_venuNo ratings yet
- Chapter 4 - Basic ProbabilityDocument37 pagesChapter 4 - Basic Probabilitynadya shafirahNo ratings yet
- Business-Communication Solved MCQs (Set-3)Document8 pagesBusiness-Communication Solved MCQs (Set-3)Pavan Sai Krishna KottiNo ratings yet
- Haier in India Building Presence in A Mass Market Beyond ChinaDocument14 pagesHaier in India Building Presence in A Mass Market Beyond ChinaGaurav Sharma100% (1)
- Cam 18 Test 3 ListeningDocument6 pagesCam 18 Test 3 ListeningKhắc Trung NguyễnNo ratings yet
- The Kicker TranscriptionDocument4 pagesThe Kicker TranscriptionmilesNo ratings yet
- E-Versuri Ro - Rihana - UmbrelaDocument2 pagesE-Versuri Ro - Rihana - Umbrelaanon-821253100% (1)
- HSCC SRH 0705 PDFDocument1 pageHSCC SRH 0705 PDFBhawna KapoorNo ratings yet
- Importance of Communications 05sept2023Document14 pagesImportance of Communications 05sept2023Sajib BhattacharyaNo ratings yet