You might also like
- SLE Concept MapDocument1 pageSLE Concept Mapadrienne cervantesNo ratings yet
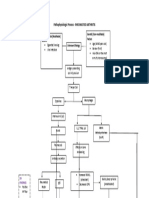
- Acute Lymphoblastic Leukemia Pathophysiology: Precipitating Factors: Etiology: Predisposing FactorsDocument3 pagesAcute Lymphoblastic Leukemia Pathophysiology: Precipitating Factors: Etiology: Predisposing FactorsromelynNo ratings yet
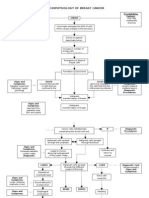
- Pathophysiology of Breast Cancer: Unkno Predisposing Factors: Precipitating FactorsDocument3 pagesPathophysiology of Breast Cancer: Unkno Predisposing Factors: Precipitating FactorsKevin Ercia100% (1)
- Physiology of LeptospirosisDocument1 pagePhysiology of LeptospirosisYum C80% (5)
- Pathophysiology Dengue Hemorrhagic FeverDocument1 pagePathophysiology Dengue Hemorrhagic Fevershiramu50% (4)
- Pathophysiology AML DiagramDocument4 pagesPathophysiology AML DiagramKlerra Hope60% (5)
- GENERAL-PATHOPHYSIOLOGY-OF-LEPTOSPIROSIS-1 (AutoRecovered)Document14 pagesGENERAL-PATHOPHYSIOLOGY-OF-LEPTOSPIROSIS-1 (AutoRecovered)Lemuel GuevarraNo ratings yet
- 6th Central Pay Commission Salary CalculatorDocument15 pages6th Central Pay Commission Salary Calculatorrakhonde100% (436)
- ABMLI Sample Questions 000Document7 pagesABMLI Sample Questions 000samy100% (1)
- Pathophysiology Diagram: Breast Cancer: Predisposing Factors: Precipitating FactorsDocument2 pagesPathophysiology Diagram: Breast Cancer: Predisposing Factors: Precipitating FactorsMarinill Soliman100% (1)
- Systemic Lupus Erythematosus Actual PathophysiologyDocument1 pageSystemic Lupus Erythematosus Actual PathophysiologyGhan Maria100% (2)
- SLE PathophysiologyDocument3 pagesSLE PathophysiologyRanela Kwinkee Pastor Salazar100% (7)
- Guillain Barre Syndrome PathophysiologyDocument4 pagesGuillain Barre Syndrome Pathophysiologykathy100% (13)
- Multiple Sclerosis PathoDocument1 pageMultiple Sclerosis PathoAleli DoNo ratings yet
- Dengue PoathoDocument6 pagesDengue PoathoCleobebs Agustin100% (2)
- Systemic Lupus ErythematosusDocument80 pagesSystemic Lupus ErythematosusLarissa Miguel Severa100% (2)
- Congestive Heart Failure Schematic DiagramDocument1 pageCongestive Heart Failure Schematic DiagramCyrus De Asis100% (1)
- Lymphomas: Varun. M M-Pharm (Pharmacology) IyriisemDocument32 pagesLymphomas: Varun. M M-Pharm (Pharmacology) IyriisemPintoo Pintu100% (2)
- Pa Tho Physiology of Meningioma (Edited Version)Document2 pagesPa Tho Physiology of Meningioma (Edited Version)Niño Villamarin75% (8)
- Placenta Previa PathophysiologyDocument1 pagePlacenta Previa Pathophysiologykathy85% (20)
- CHNDocument59 pagesCHNkathy100% (1)
- CP On Pre-EclampsiaDocument152 pagesCP On Pre-Eclampsiakathy100% (2)
- Guillain Barre Syndrome PathophysiologyDocument4 pagesGuillain Barre Syndrome Pathophysiologykathy100% (13)
- Rheumatology Notes From DR Osama LecturesDocument76 pagesRheumatology Notes From DR Osama LecturesMohamed ElAyadiNo ratings yet
- Euroline Ana Profile 3 (Igg) Test InstructionDocument16 pagesEuroline Ana Profile 3 (Igg) Test Instructionkholoud mohamedNo ratings yet
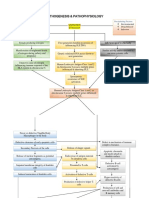
- Systemic Lupus Erythematosus PathophysiologyDocument8 pagesSystemic Lupus Erythematosus PathophysiologySharmaineTaguitagOmli100% (1)
- Sle Concept Map Part 1Document1 pageSle Concept Map Part 1Vane UcatNo ratings yet
- Pathophysiology of SLEDocument16 pagesPathophysiology of SLESeff CausapinNo ratings yet
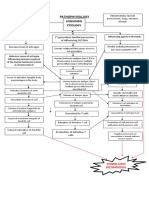
- Systemic Lupus Erythematosus (SLE) : Genetic Factors Environmental FactorsDocument5 pagesSystemic Lupus Erythematosus (SLE) : Genetic Factors Environmental Factorsjoyrena ochondraNo ratings yet
- Pa Tho Physiology Part 1Document1 pagePa Tho Physiology Part 1anonymous89ify100% (2)
- Med 1.11 - SleDocument5 pagesMed 1.11 - SleZazaNo ratings yet
- Case Study Systemic Lupus ErythematosusDocument54 pagesCase Study Systemic Lupus ErythematosusCharmaine Paran100% (3)
- Systemic Lupus Erythematosus PathophysiologyDocument8 pagesSystemic Lupus Erythematosus PathophysiologyAnonymous OU6w8lX9No ratings yet
- SLE PathophysiologyDocument3 pagesSLE PathophysiologyyasiraNo ratings yet
- Systemic Lupus ErythematosusDocument64 pagesSystemic Lupus Erythematosusganga2424100% (1)
- Systemic Lupus Erythematosus (SLE) Is A Multiorgan System Autoimmune DiseaseDocument9 pagesSystemic Lupus Erythematosus (SLE) Is A Multiorgan System Autoimmune Diseasecoyre100% (2)
- Systemic Lupous Erythematosus (SLE)Document46 pagesSystemic Lupous Erythematosus (SLE)Power La Victoria Floro100% (1)
- Cancer Pathophysiology FinalDocument3 pagesCancer Pathophysiology FinalAngelique Ramos Pascua100% (1)
- Systemic Lupus ErythematosusDocument25 pagesSystemic Lupus ErythematosusdaliaNo ratings yet
- Pathophysiology of Rheumatoid ArthritisDocument1 pagePathophysiology of Rheumatoid ArthritisGerardeanne ReposarNo ratings yet
- Dengue Hemorrhagic Fever PathophysiologyDocument4 pagesDengue Hemorrhagic Fever PathophysiologyKirk Espanol BigstoneNo ratings yet
- DengueDocument4 pagesDengueKathleen DimacaliNo ratings yet
- PathoConceptMap AIDSDocument3 pagesPathoConceptMap AIDSKristen Babauta50% (2)
- Systemic Lupus ErythematosusDocument2 pagesSystemic Lupus ErythematosusErnest caneteNo ratings yet
- Systemic Lupus Erythematosus Methods and Protocols - 1st EditionDocument268 pagesSystemic Lupus Erythematosus Methods and Protocols - 1st EditionMuhammad Noor Bumiputra100% (1)
- Lupus, SLEDocument2 pagesLupus, SLEFrances Anne Pasiliao100% (2)
- Bronchial Asthma PathophysiologyDocument1 pageBronchial Asthma PathophysiologyElisa Kerr100% (2)
- Pathophysiology of Acute Kidney InjuryDocument4 pagesPathophysiology of Acute Kidney InjuryJane Arian Berzabal0% (1)
- Pathophysiology of Diabetes Mellitus Type 1Document3 pagesPathophysiology of Diabetes Mellitus Type 1CajRofuli100% (2)
- Pathophysiology of AMLDocument1 pagePathophysiology of AMLjake251996100% (1)
- Pathophysiology - Rheumatoid ArthritisDocument1 pagePathophysiology - Rheumatoid ArthritisAngel FiloteoNo ratings yet
- Sle FinalDocument41 pagesSle FinalAsniah Hadjiadatu Abdullah100% (1)
- Pathophysiology of Acute Myelogenous Leukemia Fab m4Document3 pagesPathophysiology of Acute Myelogenous Leukemia Fab m4KristaMaeC.Lazo100% (2)
- Pa Tho Physiology and NCP Lung CancerDocument2 pagesPa Tho Physiology and NCP Lung CancerAlleizarg EuqorNo ratings yet
- Acute Lymphoblastic Leukemia Pathophysiology DiagramDocument3 pagesAcute Lymphoblastic Leukemia Pathophysiology DiagrammonishaNo ratings yet
- Dengue PathophysiologyDocument1 pageDengue PathophysiologyRafael Miguel Alon Protacio50% (2)
- HIV With PathophysiologyDocument2 pagesHIV With PathophysiologyAC, MDNo ratings yet
- Gonorrhea and Syphylis Pa Tho PhysiologyDocument3 pagesGonorrhea and Syphylis Pa Tho Physiologyjapz yeah100% (2)
- Pathophysiology (Chronic Renal Failure)Document3 pagesPathophysiology (Chronic Renal Failure)marshmalou86% (7)
- SLE Patogenesis & Patofisiologi PDFDocument8 pagesSLE Patogenesis & Patofisiologi PDFRIZQI IRFANSYAH100% (1)
- Systemic Lupus Erythematous: Precipitating Factors: Environmental Drug-Induced InfectionDocument10 pagesSystemic Lupus Erythematous: Precipitating Factors: Environmental Drug-Induced Infectioninah krizia lagueNo ratings yet
- Pa Tho Physiology Sle, CompDocument5 pagesPa Tho Physiology Sle, CompHassan Bj MarabongNo ratings yet
- Valmed PathoDocument1 pageValmed PathoJez RarangNo ratings yet
- 7-Aging and The Immune System - DR - TaghaviDocument42 pages7-Aging and The Immune System - DR - TaghavihumairashowkatNo ratings yet
- Immunology-Serology: Autoimmunity: Compiled By: J.T Cayetano UST-MT 2019Document5 pagesImmunology-Serology: Autoimmunity: Compiled By: J.T Cayetano UST-MT 2019Joshua Ty CayetanoNo ratings yet
- Pertaining To Extracellular Fluid Such As Plasma and Lymph. The Term Humoral Immunity Is Used To Denote Antibody Mediated Immune ResponsesDocument4 pagesPertaining To Extracellular Fluid Such As Plasma and Lymph. The Term Humoral Immunity Is Used To Denote Antibody Mediated Immune ResponsesZhon CabitacNo ratings yet
- Imun AwalDocument13 pagesImun AwalAdy RAstafaraNo ratings yet
- SCIDDocument9 pagesSCIDgarimaupadhyay20002No ratings yet
- Abruptio Placenta PathophysiologyDocument4 pagesAbruptio Placenta Pathophysiologyjamie carpioNo ratings yet
- DIC PathophysiologyDocument1 pageDIC Pathophysiologykathy100% (1)
- ARF PathophysiologyDocument2 pagesARF Pathophysiologykathy100% (9)
- CP On OsteomyelitisDocument147 pagesCP On Osteomyelitiskathy50% (2)
- CP-Guillain Barre SyndromeDocument35 pagesCP-Guillain Barre Syndromekathy60% (5)
- Liver CirrhosisDocument76 pagesLiver Cirrhosiskathy100% (2)
- THESIS in NURSINGDocument47 pagesTHESIS in NURSINGkathy85% (40)
- CP Placenta PreviaDocument95 pagesCP Placenta Previakathy60% (5)
- CP On Breast CancerDocument100 pagesCP On Breast Cancerkathy50% (2)
- CVADocument116 pagesCVAkathy100% (1)
- CP On Calculous CholelithiasisDocument102 pagesCP On Calculous Cholelithiasiskathy100% (3)
- Rheum-Terms and MCQsDocument26 pagesRheum-Terms and MCQsnickNo ratings yet
- NeuroscienceDocument234 pagesNeuroscienceDrAvik Chakraborty100% (1)
- Significantly Higher Frequencies of Presence of Serum Autoantibodies in Chinese Patients With Oral Lichen PlanusDocument7 pagesSignificantly Higher Frequencies of Presence of Serum Autoantibodies in Chinese Patients With Oral Lichen PlanusBimalKrishnaNo ratings yet
- Inova Catalog 2013Document56 pagesInova Catalog 2013wangxuliang100% (1)
- Diagnosis Banding SleDocument3 pagesDiagnosis Banding SleDwi ApriliziaNo ratings yet
- Anca Related PDFDocument3 pagesAnca Related PDFGREENCROSS PALDINo ratings yet
- Autoantibodies: Prof Sami Salman, FRCP, MRCP, DMR, Ces, MB CHBDocument40 pagesAutoantibodies: Prof Sami Salman, FRCP, MRCP, DMR, Ces, MB CHBamereNo ratings yet
- The JKL Medical DictionaryDocument186 pagesThe JKL Medical Dictionarymoekyaw7171No ratings yet
- Pocket Medicine, Fifth EditionDocument41 pagesPocket Medicine, Fifth Editionalaa100% (1)
- Rheumatology Questions and AnswersDocument31 pagesRheumatology Questions and Answerslittle lulu100% (1)
- M1001E04-Company Catalog-191115 - ImTec TestDocument42 pagesM1001E04-Company Catalog-191115 - ImTec TestТатьяна Исаева100% (1)
- Systemic Lupus Erythematosus in Dogs and CatsDocument7 pagesSystemic Lupus Erythematosus in Dogs and CatsJireh SlatNo ratings yet
- EUROIMMUNDocument66 pagesEUROIMMUNOskar Z ChávezNo ratings yet
- Prometric Book 2nd EditionDocument207 pagesPrometric Book 2nd Editionsajitha100% (2)
- Antihbc Igm ArcDocument6 pagesAntihbc Igm Arctesteste testeNo ratings yet
- Diabetologist Rate ListDocument28 pagesDiabetologist Rate Listirajput596_327930641No ratings yet
- Ds AQP4 CBA FA - 1128 - D - UK - A02Document2 pagesDs AQP4 CBA FA - 1128 - D - UK - A02nbiolab6659No ratings yet
- AssDocument5 pagesAssAlona Argie PianoNo ratings yet
- Patho Immune System PathologyDocument7 pagesPatho Immune System PathologyCoy Nuñez100% (1)
- Ana & Its InterpretationDocument48 pagesAna & Its InterpretationSiddharth DashNo ratings yet
- The Ai/Rheum Knowledge-Based Computer Consultant System in RheumatologyDocument8 pagesThe Ai/Rheum Knowledge-Based Computer Consultant System in RheumatologyMugundhan RamamoorthyNo ratings yet
- Clinical Significance of Antinuclear Antibody Staining Patterns and Associated Autoantibodies - UpToDateDocument13 pagesClinical Significance of Antinuclear Antibody Staining Patterns and Associated Autoantibodies - UpToDateMelanie NgNo ratings yet
- Dengue Fever Triggering Systemic Lupus Erythematosus and Lupus Nephritis A Case ReportDocument6 pagesDengue Fever Triggering Systemic Lupus Erythematosus and Lupus Nephritis A Case ReportputiridhaNo ratings yet
- Ana Detect Elisa: User S ManualDocument15 pagesAna Detect Elisa: User S ManualEdvin Bogdan GuguiNo ratings yet
- Isoniazid Induced Lupus Presenting As Oral Mucosal Ulcers With PancytopeniaDocument3 pagesIsoniazid Induced Lupus Presenting As Oral Mucosal Ulcers With PancytopeniaPutri YingNo ratings yet
- Test Description Revid TesDocument4 pagesTest Description Revid Tesjoe andarestaNo ratings yet
- Laboratory Testing For Cryoglobulins: Test of The MonthDocument3 pagesLaboratory Testing For Cryoglobulins: Test of The MonthDAWOODNo ratings yet