You might also like
- Agosto Salgado Et Al 2023 Management of Advanced Thyroid Cancer Overview Advances and OpportunitiesDocument10 pagesAgosto Salgado Et Al 2023 Management of Advanced Thyroid Cancer Overview Advances and OpportunitiesNicole Antoine-Feill BarónNo ratings yet
- Ceftaroline Jesus VallejosDocument6 pagesCeftaroline Jesus VallejosDiego VasquezNo ratings yet
- Molecular Pathology of Breast CancerDocument42 pagesMolecular Pathology of Breast CancerleartaNo ratings yet
- Caracteristicas Moleculares Del Ca Endometrial 2020Document12 pagesCaracteristicas Moleculares Del Ca Endometrial 2020Jairo Lino BNo ratings yet
- Targeted Therapies To Improve CFTR Function in CysDocument17 pagesTargeted Therapies To Improve CFTR Function in CysAnthony Bravo CortezNo ratings yet
- Critical Reviews in Oncology / HematologyDocument14 pagesCritical Reviews in Oncology / HematologyMIGUEL MORENONo ratings yet
- Biomedicines 12 00704 v2Document17 pagesBiomedicines 12 00704 v2jamel-shamsNo ratings yet
- Muta GenesisDocument8 pagesMuta GenesisOphy FirmansyahNo ratings yet
- RCCM 201910-1943SODocument16 pagesRCCM 201910-1943SOanu deepNo ratings yet
- BF00744670Document10 pagesBF00744670Carolina RibeiroNo ratings yet
- Clinmed 17 1 84Document2 pagesClinmed 17 1 84little luluNo ratings yet
- No Longer Well Differentiated Diagnostic Criteria and Clin 2023 Surgical PaDocument12 pagesNo Longer Well Differentiated Diagnostic Criteria and Clin 2023 Surgical ParubenmacaNo ratings yet
- 大法2 PDFDocument22 pages大法2 PDFTianliang GuoNo ratings yet
- Recent Advances in Histopathology 23 (2015) (PDF) (UnitedVRG) PDFDocument192 pagesRecent Advances in Histopathology 23 (2015) (PDF) (UnitedVRG) PDFsargonNo ratings yet
- This Is Your Thyroid On Drugs Targetable Mutations An 2023 Surgical PatholoDocument17 pagesThis Is Your Thyroid On Drugs Targetable Mutations An 2023 Surgical PatholorubenmacaNo ratings yet
- Next Gen CARDocument11 pagesNext Gen CARJuhi VermaNo ratings yet
- How To Cite This Thesis: University of JohannesburgDocument63 pagesHow To Cite This Thesis: University of JohannesburgKaroline MinskNo ratings yet
- Evaluation of C3435T MDR1 Gene Polymorphism in Adult Patient With Acute Lymphoblastic LeukemiaDocument4 pagesEvaluation of C3435T MDR1 Gene Polymorphism in Adult Patient With Acute Lymphoblastic Leukemiaali99No ratings yet
- Phase Angle and Impedance Ratio Two Specular Ways To Analyze Body CompositionDocument5 pagesPhase Angle and Impedance Ratio Two Specular Ways To Analyze Body CompositionPaul HartingNo ratings yet
- Pooled Analysis of Fluorouracil-Based Adjuvant Therapy For Stage II and III Colon Cancer: Who Benefits and by How Much?Document10 pagesPooled Analysis of Fluorouracil-Based Adjuvant Therapy For Stage II and III Colon Cancer: Who Benefits and by How Much?asyaNo ratings yet
- Cohen 2019Document12 pagesCohen 2019Sanjay NavaleNo ratings yet
- PDL1Document12 pagesPDL1Arlen ElisaNo ratings yet
- CAR-T TherapyDocument14 pagesCAR-T TherapyrameshaachariarNo ratings yet
- 4th Edition World Health Organization Classification For Thyroid Tumors, Asian PerspectivesDocument24 pages4th Edition World Health Organization Classification For Thyroid Tumors, Asian PerspectivesbeosroNo ratings yet
- Artículo ArtritisDocument11 pagesArtículo ArtritisAndrea Munive AzcárateNo ratings yet
- Second-Line Chemotherapy in Metastatic Docetaxel-Resistant Prostate Cancer: A ReviewDocument10 pagesSecond-Line Chemotherapy in Metastatic Docetaxel-Resistant Prostate Cancer: A ReviewHector Javier BurgosNo ratings yet
- Molecular Oncology - 2015 - Jensen - Establishment and Characterization of Models of Chemotherapy Resistance in ColorectalDocument17 pagesMolecular Oncology - 2015 - Jensen - Establishment and Characterization of Models of Chemotherapy Resistance in ColorectalAliza JafriNo ratings yet
- Poorly Differentiated and Undifferentiated Thyroid CarcinomasDocument12 pagesPoorly Differentiated and Undifferentiated Thyroid CarcinomasVlak FooNo ratings yet
- Critical Reviews in Oncology / Hematology: ReviewDocument17 pagesCritical Reviews in Oncology / Hematology: ReviewentannabilakasdyNo ratings yet
- Paper 1 - PD1 Blockade Enhances ICAM1-Directed CAR T Therapeutic Efficacy in Advanced Thyroid CancerDocument14 pagesPaper 1 - PD1 Blockade Enhances ICAM1-Directed CAR T Therapeutic Efficacy in Advanced Thyroid CancerChauPhuongNo ratings yet
- Review Article Esophageal CancerDocument9 pagesReview Article Esophageal CancerMuhadis AhmadNo ratings yet
- 21 - Mark e Berry Guidelines Staging and TreatmentDocument9 pages21 - Mark e Berry Guidelines Staging and TreatmentNatalindah Jokiem Woecandra T. D.No ratings yet
- Ffi, I: About ApplyDocument3 pagesFfi, I: About Applywaled badawyNo ratings yet
- CancerDocument11 pagesCancerzohra benouadahNo ratings yet
- CT Findings of Chemotherapy-Induced Toxicity What Radiologists Need To Know About The Clinical and Radiologic Manifestations of Chemotherapy ToxicityDocument16 pagesCT Findings of Chemotherapy-Induced Toxicity What Radiologists Need To Know About The Clinical and Radiologic Manifestations of Chemotherapy ToxicityZai Rojas OrtizNo ratings yet
- Heterodimerization of Cholecystokinin 1 Nad Cholecystokinin 2 Receptor in Gallbladder CancerDocument10 pagesHeterodimerization of Cholecystokinin 1 Nad Cholecystokinin 2 Receptor in Gallbladder CancerJaya Kunal DubeyNo ratings yet
- Pharmacogenomics: New Challenges For Thai Anesthesiologists: J Med Assoc Thai 2017 100 (Suppl. 7) : S250-S258Document9 pagesPharmacogenomics: New Challenges For Thai Anesthesiologists: J Med Assoc Thai 2017 100 (Suppl. 7) : S250-S258Ronald André Buleje HinostrozaNo ratings yet
- NutrientsDocument14 pagesNutrientsSteve GannabanNo ratings yet
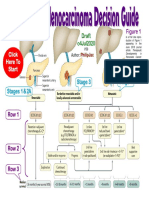
- Draft O4jul2020: Stages 1 & 2A Stage 3 Stage 4Document9 pagesDraft O4jul2020: Stages 1 & 2A Stage 3 Stage 4WemhoffpNo ratings yet
- Adrenal Histoplasmosis A Therapeutic Restoration of - 2022 - AACE Clinical CaseDocument2 pagesAdrenal Histoplasmosis A Therapeutic Restoration of - 2022 - AACE Clinical CaseMohammed Shuaib AhmedNo ratings yet
- Estresse OxidativoDocument1 pageEstresse OxidativoRafaelaNo ratings yet
- Radioactive Iodine Remnant Ablation: The Beta-Knife Completion ThyroidectomyDocument8 pagesRadioactive Iodine Remnant Ablation: The Beta-Knife Completion Thyroidectomynfp.panjaitanNo ratings yet
- Jadikan Dafpus 5Document8 pagesJadikan Dafpus 5trifamonika23No ratings yet
- The Evolution of Car T-Cell Therapy: Ethics and EfficiencyDocument8 pagesThe Evolution of Car T-Cell Therapy: Ethics and EfficiencyIJAR JOURNALNo ratings yet
- PA Curriculum MatrixDocument127 pagesPA Curriculum MatrixMichelle UwakkNo ratings yet
- Ajrccm 163 1 16310aDocument3 pagesAjrccm 163 1 16310amisulica2010No ratings yet
- EBRT in DTCDocument7 pagesEBRT in DTCElena FlorentinaNo ratings yet
- TNBCDocument13 pagesTNBCSilvi Zakiyatul IlmiyahNo ratings yet
- 1 s2.0 S2376060523000081 MainDocument4 pages1 s2.0 S2376060523000081 MainGeorge DragosNo ratings yet
- Critical Reviews in Oncology / HematologyDocument6 pagesCritical Reviews in Oncology / HematologyasdffdsaNo ratings yet
- 9 Randomized Phase III Evaluation of Cisplatin PlusDocument6 pages9 Randomized Phase III Evaluation of Cisplatin Plustrifamonika23No ratings yet
- Cost Effectiveness of Dapagliflozinmetformin VersuDocument2 pagesCost Effectiveness of Dapagliflozinmetformin VersuENY RUSMIYATINo ratings yet
- IJMLR311801Document5 pagesIJMLR311801sandeep raiNo ratings yet
- 02-1.WHO 2022-PitNETDocument21 pages02-1.WHO 2022-PitNETJihuhn YuNo ratings yet
- Calon2015 2Document13 pagesCalon2015 2Javier Cerda InfanteNo ratings yet
- Marrow UpdatesDocument9 pagesMarrow UpdatesVirat KohliNo ratings yet
- Breast Cancer Therapeutics and Biomarkers: Past, Present, and Future ApproachesDocument19 pagesBreast Cancer Therapeutics and Biomarkers: Past, Present, and Future ApproachesDavide RadiceNo ratings yet
- Ymj 47 243Document6 pagesYmj 47 243Marcia MejíaNo ratings yet
- EPOC, Lancet 2015Document10 pagesEPOC, Lancet 2015Sebastian BurgosNo ratings yet
- Rhumatologie-Pediatrique FRDocument4 pagesRhumatologie-Pediatrique FRCaity YoungNo ratings yet
- Hypoxic Ischaemic Encephalopathy HIE AsphyxiaDocument6 pagesHypoxic Ischaemic Encephalopathy HIE AsphyxiaCaity YoungNo ratings yet
- Afoem Sample Paper Stage B Written Exam Paper 7Document46 pagesAfoem Sample Paper Stage B Written Exam Paper 7Caity YoungNo ratings yet
- A I - E M: Drenal Nsufficiency Mergency AnagementDocument12 pagesA I - E M: Drenal Nsufficiency Mergency AnagementCaity YoungNo ratings yet
- Fluid Calculator DKADocument3 pagesFluid Calculator DKACaity YoungNo ratings yet
- Afoem Sample Paper Stage B Written Exam Paper 7Document43 pagesAfoem Sample Paper Stage B Written Exam Paper 7Caity YoungNo ratings yet
- 2014 A Health Professionals Guide To Using The ChartsDocument14 pages2014 A Health Professionals Guide To Using The ChartsCaity YoungNo ratings yet
- ToF ScriptDocument6 pagesToF ScriptCaity YoungNo ratings yet
- SSHADESS HandoutDocument1 pageSSHADESS HandoutCaity YoungNo ratings yet
- Reference Ranges For Urinalysis - Microbiology: Test Range Units DateDocument1 pageReference Ranges For Urinalysis - Microbiology: Test Range Units DateCaity YoungNo ratings yet
- Genetic Basis For Congenital Heart Disease: Revisited: CirculationDocument59 pagesGenetic Basis For Congenital Heart Disease: Revisited: CirculationCaity YoungNo ratings yet
- 2 - McDonald - Examples - of - Unhelpful - ThoughtsDocument2 pages2 - McDonald - Examples - of - Unhelpful - ThoughtsCaity YoungNo ratings yet
- Part 2: Male Reproductive System: Normal Physiology and StructureDocument30 pagesPart 2: Male Reproductive System: Normal Physiology and StructureCaity YoungNo ratings yet
- 4-5 GlomerularDiseasesDocument21 pages4-5 GlomerularDiseasesCaity YoungNo ratings yet
- IsbelDocument7 pagesIsbelCaity YoungNo ratings yet
- Sore Throat: Navigating The Differential DiagnosisDocument7 pagesSore Throat: Navigating The Differential DiagnosisCaity YoungNo ratings yet
- Jones Et - Al.1994Document6 pagesJones Et - Al.1994Sukanya MajumderNo ratings yet
- Controlador DanfossDocument2 pagesControlador Danfossfrank.marcondes2416No ratings yet
- VC AndrewsDocument3 pagesVC AndrewsLesa O'Leary100% (1)
- ATS2017 ProspectusDocument13 pagesATS2017 ProspectusGiri WakshanNo ratings yet
- Em FlexicokingDocument8 pagesEm FlexicokingHenry Saenz0% (1)
- MOS - Steel StructureDocument15 pagesMOS - Steel StructuredennisNo ratings yet
- Process Industry Practices Insulation: PIP INEG2000 Guidelines For Use of Insulation PracticesDocument15 pagesProcess Industry Practices Insulation: PIP INEG2000 Guidelines For Use of Insulation PracticesZubair RaoofNo ratings yet
- Rab Sikda Optima 2016Document20 pagesRab Sikda Optima 2016Julius Chatry UniwalyNo ratings yet
- A Process Reference Model For Claims Management in Construction Supply Chains The Contractors PerspectiveDocument20 pagesA Process Reference Model For Claims Management in Construction Supply Chains The Contractors Perspectivejadal khanNo ratings yet
- Scrum Handbook: Scrum Training Institute PressDocument66 pagesScrum Handbook: Scrum Training Institute PressFranky RiveroNo ratings yet
- Attachment 1 Fiber Data SheetDocument2 pagesAttachment 1 Fiber Data SheetflavioovNo ratings yet
- Reference by John BatchelorDocument1 pageReference by John Batchelorapi-276994844No ratings yet
- Lakh Only) Being The Amount Covered Under The Aforesaid Dishonoured Cheque, and So AlsoDocument2 pagesLakh Only) Being The Amount Covered Under The Aforesaid Dishonoured Cheque, and So AlsoShivam MishraNo ratings yet
- Gmail - ICICI BANK I PROCESS HIRING FOR BACKEND - OPERATION PDFDocument2 pagesGmail - ICICI BANK I PROCESS HIRING FOR BACKEND - OPERATION PDFDeepankar ChoudhuryNo ratings yet
- Uh 60 ManualDocument241 pagesUh 60 ManualAnonymous ddjwf1dqpNo ratings yet
- Energy-Roles-In-Ecosystems-Notes-7 12bDocument10 pagesEnergy-Roles-In-Ecosystems-Notes-7 12bapi-218158367No ratings yet
- Parker HPD Product Bulletin (HY28-2673-01)Document162 pagesParker HPD Product Bulletin (HY28-2673-01)helden50229881No ratings yet
- CL200 PLCDocument158 pagesCL200 PLCJavierRuizThorrensNo ratings yet
- Forces L2 Measuring Forces WSDocument4 pagesForces L2 Measuring Forces WSAarav KapoorNo ratings yet
- Reproduction in PlantsDocument12 pagesReproduction in PlantsAnand Philip PrasadNo ratings yet
- SahanaDocument1 pageSahanamurthyarun1993No ratings yet
- AppcDocument71 pagesAppcTomy lee youngNo ratings yet
- Research Methods in Developmental PsychologyDocument9 pagesResearch Methods in Developmental PsychologyHugoNo ratings yet
- Mangement of Shipping CompaniesDocument20 pagesMangement of Shipping CompaniesSatyam MishraNo ratings yet
- Effect of Plant Growth RegulatorsDocument17 pagesEffect of Plant Growth RegulatorsSharmilla AshokhanNo ratings yet
- Cocaine in Blood of Coca ChewersDocument10 pagesCocaine in Blood of Coca ChewersKarl-GeorgNo ratings yet
- Contigency Plan On Class SuspensionDocument4 pagesContigency Plan On Class SuspensionAnjaneth Balingit-PerezNo ratings yet
- Atom SDDocument5 pagesAtom SDatomsa shiferaNo ratings yet
- Wins Salvacion Es 2021Document16 pagesWins Salvacion Es 2021MURILLO, FRANK JOMARI C.No ratings yet
- Bathinda - Wikipedia, The Free EncyclopediaDocument4 pagesBathinda - Wikipedia, The Free EncyclopediaBhuwan GargNo ratings yet
- ADHD is Awesome: A Guide to (Mostly) Thriving with ADHDFrom EverandADHD is Awesome: A Guide to (Mostly) Thriving with ADHDRating: 5 out of 5 stars5/5 (3)
- The Age of Magical Overthinking: Notes on Modern IrrationalityFrom EverandThe Age of Magical Overthinking: Notes on Modern IrrationalityRating: 4 out of 5 stars4/5 (30)
- Think This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeFrom EverandThink This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeRating: 2 out of 5 stars2/5 (1)
- Love Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)From EverandLove Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Rating: 3 out of 5 stars3/5 (1)
- LIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionFrom EverandLIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionRating: 4 out of 5 stars4/5 (404)
- The Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsFrom EverandThe Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsRating: 4 out of 5 stars4/5 (4)
- Summary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisFrom EverandSummary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisRating: 4.5 out of 5 stars4.5/5 (42)
- Raising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsFrom EverandRaising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsRating: 5 out of 5 stars5/5 (1)
- By the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsFrom EverandBy the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsNo ratings yet
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedFrom EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedRating: 5 out of 5 stars5/5 (81)
- Raising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsFrom EverandRaising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsRating: 4.5 out of 5 stars4.5/5 (170)
- The Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaFrom EverandThe Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaRating: 4.5 out of 5 stars4.5/5 (266)
- Dark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.From EverandDark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.Rating: 4.5 out of 5 stars4.5/5 (110)
- Why We Die: The New Science of Aging and the Quest for ImmortalityFrom EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityRating: 4 out of 5 stars4/5 (5)
- Summary: Limitless: Upgrade Your Brain, Learn Anything Faster, and Unlock Your Exceptional Life By Jim Kwik: Key Takeaways, Summary and AnalysisFrom EverandSummary: Limitless: Upgrade Your Brain, Learn Anything Faster, and Unlock Your Exceptional Life By Jim Kwik: Key Takeaways, Summary and AnalysisRating: 5 out of 5 stars5/5 (8)
- Empath: The Survival Guide For Highly Sensitive People: Protect Yourself From Narcissists & Toxic Relationships. Discover How to Stop Absorbing Other People's PainFrom EverandEmpath: The Survival Guide For Highly Sensitive People: Protect Yourself From Narcissists & Toxic Relationships. Discover How to Stop Absorbing Other People's PainRating: 4 out of 5 stars4/5 (95)
- The Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeFrom EverandThe Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeRating: 4.5 out of 5 stars4.5/5 (253)
- Mindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessFrom EverandMindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessRating: 4.5 out of 5 stars4.5/5 (328)
- Summary: Thinking, Fast and Slow: by Daniel Kahneman: Key Takeaways, Summary & Analysis IncludedFrom EverandSummary: Thinking, Fast and Slow: by Daniel Kahneman: Key Takeaways, Summary & Analysis IncludedRating: 4 out of 5 stars4/5 (61)
- The Obesity Code: Unlocking the Secrets of Weight LossFrom EverandThe Obesity Code: Unlocking the Secrets of Weight LossRating: 4 out of 5 stars4/5 (6)
- The Marshmallow Test: Mastering Self-ControlFrom EverandThe Marshmallow Test: Mastering Self-ControlRating: 4.5 out of 5 stars4.5/5 (59)
- Manipulation: The Ultimate Guide To Influence People with Persuasion, Mind Control and NLP With Highly Effective Manipulation TechniquesFrom EverandManipulation: The Ultimate Guide To Influence People with Persuasion, Mind Control and NLP With Highly Effective Manipulation TechniquesRating: 4.5 out of 5 stars4.5/5 (1412)
- Cult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryFrom EverandCult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryRating: 4 out of 5 stars4/5 (45)