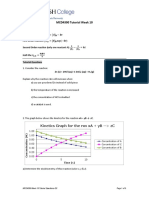
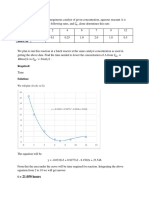
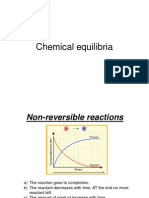
You might also like
- CHP 16 PDFDocument28 pagesCHP 16 PDFomarNo ratings yet
- Chemistry File 4Document4 pagesChemistry File 4Pawan Kumar100% (1)
- Rate Law Worksheet PDFDocument3 pagesRate Law Worksheet PDFJunghoon Lee100% (1)
- Kinetics Practice ProblemsDocument4 pagesKinetics Practice ProblemsLuke MeredithNo ratings yet
- bài tập rateDocument2 pagesbài tập rateMys Genie100% (1)
- Question Score A Chapter 1Document14 pagesQuestion Score A Chapter 1Dee -AdilaNo ratings yet
- MCD4390 Week 10 Tutorial QuestionsDocument5 pagesMCD4390 Week 10 Tutorial QuestionsGabbar100% (1)
- Spotlight - Crux (2023-24) - Day-1 - PPT - Chemistry (Sol.)Document6 pagesSpotlight - Crux (2023-24) - Day-1 - PPT - Chemistry (Sol.)Parth SonawaneNo ratings yet
- KineticsDocument13 pagesKineticsmnfazliNo ratings yet
- Hints & Solutions: Mno I MN I I Naso Naso NaiDocument7 pagesHints & Solutions: Mno I MN I I Naso Naso NaiAYUSH SHARMANo ratings yet
- Chapter 5 KineticsDocument69 pagesChapter 5 KineticsexpertwritersNo ratings yet
- Topic 16 Past Paper Questions 2011Document15 pagesTopic 16 Past Paper Questions 2011nadia sykesNo ratings yet
- Tugas Teknik Reaksi Kimia Lanjut EXAMPLE 10.2 & PROBLEM 10.3Document13 pagesTugas Teknik Reaksi Kimia Lanjut EXAMPLE 10.2 & PROBLEM 10.3Andre Fahriz Perdana HarahapNo ratings yet
- Ans-Sol JEEMain-2022 Phase-2 25-07-2022 M Chemistry FINALDocument7 pagesAns-Sol JEEMain-2022 Phase-2 25-07-2022 M Chemistry FINALChirayu SharmaNo ratings yet
- CHEMISTRY-19-11 - 11th (J)Document9 pagesCHEMISTRY-19-11 - 11th (J)Raju SinghNo ratings yet
- MATHEMATICS-19-11-11th (PQRS)Document9 pagesMATHEMATICS-19-11-11th (PQRS)Raju SinghNo ratings yet
- Rates of Reaction Suroviec Spring 2014Document43 pagesRates of Reaction Suroviec Spring 2014enesffsNo ratings yet
- 12 - Kinetics 2-7Document2 pages12 - Kinetics 2-7vikyappleNo ratings yet
- (C-4 A-1) SolutionDocument4 pages(C-4 A-1) SolutionSachin DedhiaNo ratings yet
- 2021 Chem Skills SolutionsDocument10 pages2021 Chem Skills SolutionsVictor GuanNo ratings yet
- Kinetics Practice QuestionDocument6 pagesKinetics Practice Question장채윤No ratings yet
- Rate of Reaction Part 2 XI MIPA 12Document10 pagesRate of Reaction Part 2 XI MIPA 12Shofwa AnnisaNo ratings yet
- Rates Review Questions ANSWERSDocument3 pagesRates Review Questions ANSWERSnadia sykesNo ratings yet
- Kinetics Revision Worksheet 2 (Solutions)Document8 pagesKinetics Revision Worksheet 2 (Solutions)Lee Jun HuiNo ratings yet
- Kinetics Note 2023Document127 pagesKinetics Note 2023oliviaadams637No ratings yet
- Practice Final CHE1112Document13 pagesPractice Final CHE1112dancer88838No ratings yet
- Hints & Solutions: Section - I (Chemistry)Document7 pagesHints & Solutions: Section - I (Chemistry)Ashish RanjanNo ratings yet
- Solutions - Revision Book Chemistry PDFDocument89 pagesSolutions - Revision Book Chemistry PDFJatin GoyalNo ratings yet
- MS of Chemistry XI 23-24. FinalDocument11 pagesMS of Chemistry XI 23-24. Finalanshulchauhan94595No ratings yet
- Chemical Kinetics NumDocument3 pagesChemical Kinetics NumVANESSA JOHNS CLASS XINo ratings yet
- Chemcal Kinetics (Tutorial Questions)Document3 pagesChemcal Kinetics (Tutorial Questions)renNo ratings yet
- L STPM 2019 SEM 1 MOCK ANSDocument2 pagesL STPM 2019 SEM 1 MOCK ANSm-7319562No ratings yet
- Answers: Rate K (H) (NO) Must Determine A and BDocument4 pagesAnswers: Rate K (H) (NO) Must Determine A and BWahyu YusupNo ratings yet
- JEE Main (Memory Based) - 24-01-2023 - Chemistry - Shift 1 PDFDocument3 pagesJEE Main (Memory Based) - 24-01-2023 - Chemistry - Shift 1 PDFKUNAL RAWATNo ratings yet
- CH 14 Kinetics Part1 WebDocument39 pagesCH 14 Kinetics Part1 Webblue educationNo ratings yet
- Chemistry Advanced Level Problem Solving (ALPS-7) - SolutionDocument9 pagesChemistry Advanced Level Problem Solving (ALPS-7) - SolutionSwapnil MandalNo ratings yet
- SM CH 2 Prin Davis 3Document39 pagesSM CH 2 Prin Davis 3Tophie CrunchNo ratings yet
- MS Xi Chem Hy Set 1Document5 pagesMS Xi Chem Hy Set 1pandeyrajesh156No ratings yet
- EquilibriumDocument46 pagesEquilibriumRoesma NarulitaNo ratings yet
- Chemisstry FormulaDocument11 pagesChemisstry FormulaSharifah RenahNo ratings yet
- Jee Main 24 Jan 2023 Shift 1 Chemistry Memory Based Paper SolutionDocument9 pagesJee Main 24 Jan 2023 Shift 1 Chemistry Memory Based Paper SolutionThe Daily PleasureNo ratings yet
- Solutions To Mock IIT Advanced Test - 1/JEE-2018/Paper - 2: (Chemistry)Document10 pagesSolutions To Mock IIT Advanced Test - 1/JEE-2018/Paper - 2: (Chemistry)rajeshNo ratings yet
- Key KineticsDocument3 pagesKey Kineticsnonononoway100% (1)
- Che - Jee Main (Jan) - 2023 - 24-01-2023 - Maths - QuestionsDocument7 pagesChe - Jee Main (Jan) - 2023 - 24-01-2023 - Maths - QuestionsAditya Dev SinghNo ratings yet
- 2019 NYJC H2 Chem P1 P2 P3 P4 AnswersDocument44 pages2019 NYJC H2 Chem P1 P2 P3 P4 Answersthe.volleyball.guyNo ratings yet
- Wa0163.Document5 pagesWa0163.Saish ShindeNo ratings yet
- Chapter 1 Reaction KineticsDocument8 pagesChapter 1 Reaction KineticsDinesh RamaNo ratings yet
- Chem Marking SchemeDocument8 pagesChem Marking SchemeForzen flamesNo ratings yet
- Chm271 - Tutorial 5 - Chemical KineticsDocument6 pagesChm271 - Tutorial 5 - Chemical Kineticsfiefy zmrNo ratings yet
- Che - Jee Main (Jan) - 2023 - 24-01-2023 - F.N (Maths) Memory Based QuestionsDocument7 pagesChe - Jee Main (Jan) - 2023 - 24-01-2023 - F.N (Maths) Memory Based QuestionsTaaha BaigNo ratings yet
- By Einar Helland Berger (Own Work) (CC BY SA 2.5 ), Via Wikimedia CommonsDocument49 pagesBy Einar Helland Berger (Own Work) (CC BY SA 2.5 ), Via Wikimedia CommonsHoàng Anh DbbyNo ratings yet
- Kinetics (Gjjkkkgty)Document5 pagesKinetics (Gjjkkkgty)Chrysler Kane DepnagNo ratings yet
- Ms Xi Chem Hy Set 1Document5 pagesMs Xi Chem Hy Set 1harshsingh95555No ratings yet
- Chap0-Dong Hoa HocDocument24 pagesChap0-Dong Hoa HocVan Nguyen Phuong NganNo ratings yet
- Assignment-1 CRE by Shailendra SirDocument5 pagesAssignment-1 CRE by Shailendra Sirgyandeep.rs.che23No ratings yet
- Subject: Chemistry Electrochemistry: Decreases PH of Solution (D) Electrolysis of CusoDocument28 pagesSubject: Chemistry Electrochemistry: Decreases PH of Solution (D) Electrolysis of CusoQwertyNo ratings yet
- Lakshya Jee Air (2025) Chemical Kinetics: Single Correct Questions 1. 4Document4 pagesLakshya Jee Air (2025) Chemical Kinetics: Single Correct Questions 1. 4Meet ShahNo ratings yet
- Part - I: Subjective Questions: Mole ConceptDocument23 pagesPart - I: Subjective Questions: Mole ConceptSubham RajNo ratings yet
- Topic 16 Past PapersDocument9 pagesTopic 16 Past PapersMahmoud Sameh-Abdel-LateefNo ratings yet
- Answers to Selected Problems in Multivariable Calculus with Linear Algebra and SeriesFrom EverandAnswers to Selected Problems in Multivariable Calculus with Linear Algebra and SeriesRating: 1.5 out of 5 stars1.5/5 (2)
- Focus 7Document2 pagesFocus 7Selma MeloNo ratings yet
- Chem Principles 7e ISM Focus 05 Even FINALDocument62 pagesChem Principles 7e ISM Focus 05 Even FINALSelma MeloNo ratings yet
- Chem Principles 7e ISM Focus 10 Even FINALDocument20 pagesChem Principles 7e ISM Focus 10 Even FINALSelma MeloNo ratings yet
- Atkins Chemical Principles Focus2 SolDocument44 pagesAtkins Chemical Principles Focus2 Sol김은우No ratings yet
- Chapter 6 IM Chang 11eDocument8 pagesChapter 6 IM Chang 11eSelma MeloNo ratings yet
- Chapter 2 IM Chang 11eDocument5 pagesChapter 2 IM Chang 11eSelma MeloNo ratings yet
- Chapter 5 IM Chang 11eDocument11 pagesChapter 5 IM Chang 11eSelma MeloNo ratings yet
- Chapter 3 IM Chang 11eDocument7 pagesChapter 3 IM Chang 11eSelma MeloNo ratings yet
- Chapter 4 IM Chang 11eDocument6 pagesChapter 4 IM Chang 11eSelma MeloNo ratings yet
- Chapter 1 IM Chang 11eDocument6 pagesChapter 1 IM Chang 11eSelma MeloNo ratings yet
- Chapter 17 IM Chang 11eDocument4 pagesChapter 17 IM Chang 11eSelma MeloNo ratings yet
- Chapter 8 IM Chang 11eDocument6 pagesChapter 8 IM Chang 11eSelma MeloNo ratings yet
- Wa0001.Document15 pagesWa0001.Thrivikram ArepalliNo ratings yet
- Ib PPT 6 HL PDFDocument38 pagesIb PPT 6 HL PDFzarna nirmal rawalNo ratings yet
- Chemistry Past Paper Ch4.1Document13 pagesChemistry Past Paper Ch4.1Raymond ChanNo ratings yet
- Science - Temperature of Sodium Thiosulphate and Rate of ReactionDocument4 pagesScience - Temperature of Sodium Thiosulphate and Rate of ReactionSmartPurdyNo ratings yet
- How Does Surface Area Affect The Rate of Reaction?Document8 pagesHow Does Surface Area Affect The Rate of Reaction?Monica Paris SisourathNo ratings yet
- Activation Energy2Document27 pagesActivation Energy2Chen ShyanNo ratings yet
- Catalysis PDFDocument104 pagesCatalysis PDFMandla Rebirth0% (1)
- Bourne 2003Document38 pagesBourne 2003Gopal KasatNo ratings yet
- Naoh +CH Ooo C H CH Ooona+C H OH: Results and DiscussionDocument2 pagesNaoh +CH Ooo C H CH Ooona+C H OH: Results and DiscussionJoseph Bien Mercado OdiñaNo ratings yet
- Formative C Grade 8Document9 pagesFormative C Grade 8TarshihiNo ratings yet
- 1 ChemicalKinetics (RPP&CHW)Document6 pages1 ChemicalKinetics (RPP&CHW)Mainak ChakrabortyNo ratings yet
- J. Electrochem. Soc. 1954 Belle 339 42Document4 pagesJ. Electrochem. Soc. 1954 Belle 339 42Sutanwi LahiriNo ratings yet
- Arihant 41 Years ChemistryDocument497 pagesArihant 41 Years Chemistrydevinder9100% (1)
- Chemical EquilibriaDocument15 pagesChemical EquilibriaiceggNo ratings yet
- Chem Ia Exemplar2Document12 pagesChem Ia Exemplar2Archit GargNo ratings yet
- 2 CreDocument3 pages2 CreDamien MarleyNo ratings yet
- Name:Saiful Islam Bin Ahmad HusniDocument11 pagesName:Saiful Islam Bin Ahmad Husninike7No ratings yet
- Chemical KineticsDocument28 pagesChemical KineticsPaul Michael CunananNo ratings yet
- Sintesis Sol GelDocument40 pagesSintesis Sol GelcarlosNo ratings yet
- Tutorial (Kinetics in Gas Phase) With AnswersDocument9 pagesTutorial (Kinetics in Gas Phase) With AnswershusnaNo ratings yet
- Laminar Flow Reactor ProblemDocument6 pagesLaminar Flow Reactor ProblemAileen Banua Añonuevo100% (1)
- Chemical Kinetics-Assignment PDFDocument37 pagesChemical Kinetics-Assignment PDFggk201367% (3)
- Organic Process Research & Development (1997), 1 (2), 97-105Document9 pagesOrganic Process Research & Development (1997), 1 (2), 97-105prashantNo ratings yet
- Expt 1-Factors Affecting Reaction RateDocument25 pagesExpt 1-Factors Affecting Reaction Ratetwinkledreampoppies100% (5)
- Chemistry Project Topics Class 12Document11 pagesChemistry Project Topics Class 12Mari MuthuNo ratings yet
- Cc2 Lec EnzymesDocument25 pagesCc2 Lec EnzymesJunea SeeNo ratings yet
- The Effects of Surface Area On The Rate of A ReactionDocument16 pagesThe Effects of Surface Area On The Rate of A ReactionNick SchlobohmNo ratings yet
- Chemical Kinetics 13thDocument32 pagesChemical Kinetics 13thRaju SinghNo ratings yet
- 1aa3 2012 Test1 SolutionsDocument23 pages1aa3 2012 Test1 SolutionsDavidNo ratings yet
- Determination of The Rate Equation For A ReactionDocument6 pagesDetermination of The Rate Equation For A ReactionFanilo RazafindralamboNo ratings yet