You might also like
- FITZEE Test Papers and Soln PDFDocument911 pagesFITZEE Test Papers and Soln PDFKushagra Srivastava100% (3)
- Physics Learning MaterialDocument7 pagesPhysics Learning Materialasdfghjkl0118No ratings yet
- Tugas - 1 - PSPK Firstiando YudaDocument10 pagesTugas - 1 - PSPK Firstiando YudaBrayonoFlo100% (1)
- Answers to Selected Problems in Multivariable Calculus with Linear Algebra and SeriesFrom EverandAnswers to Selected Problems in Multivariable Calculus with Linear Algebra and SeriesRating: 1.5 out of 5 stars1.5/5 (2)
- Physical Properties of RocksDocument10 pagesPhysical Properties of RocksZaeem HassanNo ratings yet
- Binder 1Document100 pagesBinder 1HemanthNo ratings yet
- RESERVOIR GEOMECHANICS COUPLING - EMI (Autoguardado)Document49 pagesRESERVOIR GEOMECHANICS COUPLING - EMI (Autoguardado)VP LeonelNo ratings yet
- Flotation Equipment and ProcessesDocument78 pagesFlotation Equipment and ProcessesANo ratings yet
- Transport Phenomena - Monash University Final - Exam - 2010 - SolutionDocument15 pagesTransport Phenomena - Monash University Final - Exam - 2010 - SolutionKunal BhardwajNo ratings yet
- Test Practice Exam Questions and Answers Solow ModelDocument5 pagesTest Practice Exam Questions and Answers Solow ModelFiranbek BgNo ratings yet
- MMSB Exercise SolutionsDocument49 pagesMMSB Exercise SolutionsYolanda Winarny Eviphanie HutabaratNo ratings yet
- Parallel Reaction NetworkDocument3 pagesParallel Reaction NetworkMahnoor AbbasNo ratings yet
- NomuraDocument2 pagesNomuraAnil NayakNo ratings yet
- ParallRxn PDFDocument1 pageParallRxn PDFAbhinav RajeshNo ratings yet
- ParallRxn PDFDocument1 pageParallRxn PDFWizra QamarNo ratings yet
- ParallRxn PDFDocument1 pageParallRxn PDFAbhinav RajeshNo ratings yet
- Sample & Practice Tutorial Assignment On UNIT IIDocument9 pagesSample & Practice Tutorial Assignment On UNIT IIFF02 Aniket BarhateNo ratings yet
- DO Sag Equation DerivationDocument4 pagesDO Sag Equation DerivationSnowy AshesNo ratings yet
- Method of DerivativeDocument4 pagesMethod of DerivativeSOHAM CHATTERJEENo ratings yet
- Difference Between Social and Natural ScienceDocument5 pagesDifference Between Social and Natural ScienceBilal Ahmad AliNo ratings yet
- 21.8 (A) Lindermann-Hinshelwood Mechanism: (Slow, RDS) + +Document18 pages21.8 (A) Lindermann-Hinshelwood Mechanism: (Slow, RDS) + +cmc107No ratings yet
- Neet Weekend Test: PhysicsDocument21 pagesNeet Weekend Test: PhysicsTHARUN THANGELLANo ratings yet
- NPTEL Online Course: Control Engineering: Assignment 1Document4 pagesNPTEL Online Course: Control Engineering: Assignment 1udayNo ratings yet
- Homework Report: Modelling of Homogenous Isothermal Tubular ReactorDocument10 pagesHomework Report: Modelling of Homogenous Isothermal Tubular ReactorSarah BessadiNo ratings yet
- AMATH 350 Assignment 5 Winter 2017: - Late Submissions Will Not Be AcceptedDocument2 pagesAMATH 350 Assignment 5 Winter 2017: - Late Submissions Will Not Be Acceptedthomas94josephNo ratings yet
- Matematika Diskrit: (n+1) (2a+nd) 2Document4 pagesMatematika Diskrit: (n+1) (2a+nd) 2arif sptNo ratings yet
- sm2 037 PDFDocument1 pagesm2 037 PDFNilton MafraNo ratings yet
- Problems and Solutions: The American Mathematical MonthlyDocument9 pagesProblems and Solutions: The American Mathematical MonthlyKhokon GayenNo ratings yet
- Chemical KineticsDocument20 pagesChemical Kineticsvijaylakshmi0727No ratings yet
- hw4 SolnDocument8 pageshw4 SolnMd Abid AfridiNo ratings yet
- Neet-Jee KineticsDocument16 pagesNeet-Jee KineticsSudheerkhan MuhammedNo ratings yet
- Chapter 14. Enzyme KineticsDocument44 pagesChapter 14. Enzyme KineticsKalai VaniNo ratings yet
- Kinetics Formula SheetDocument1 pageKinetics Formula SheetdfasdfNo ratings yet
- K-Gamma and K-Beta FunctionDocument5 pagesK-Gamma and K-Beta FunctionketashiNo ratings yet
- One-Dimensional and Steady-State Conduction: DT DX DT DR Ka X 1 2Document4 pagesOne-Dimensional and Steady-State Conduction: DT DX DT DR Ka X 1 2Green HiderNo ratings yet
- Reglas de IntegraciónDocument1 pageReglas de IntegraciónAndres RangelNo ratings yet
- Ans-Sol JEEMain-2022 Phase-2 25-07-2022 M Physics FinalDocument9 pagesAns-Sol JEEMain-2022 Phase-2 25-07-2022 M Physics FinalRajkumar SharmaNo ratings yet
- Ans&Sol JEE (Main) - 2022 (Phase-2) (25-07-2022) Morning (PCM) 0Document26 pagesAns&Sol JEE (Main) - 2022 (Phase-2) (25-07-2022) Morning (PCM) 0Aditya BadeNo ratings yet
- Nodia and Company: Gate Solved Paper Electronics & Communication 2005Document39 pagesNodia and Company: Gate Solved Paper Electronics & Communication 2005Tanmaya Tapaswini TripathyNo ratings yet
- Date PDFDocument25 pagesDate PDFSiddharth RaviNo ratings yet
- 2015Document10 pages2015ZaNo ratings yet
- Chemical-Kinetics-Ppt XIIDocument41 pagesChemical-Kinetics-Ppt XIIAnonymous RuslwNZZlNo ratings yet
- PHY1B Derivatives GaelanDocument81 pagesPHY1B Derivatives GaelanTshegofatso KgopaNo ratings yet
- Node 58 - 86Document15 pagesNode 58 - 86AliMubarakNo ratings yet
- Steady StateDocument30 pagesSteady Statesus023No ratings yet
- 028 CSTR Startup PDFDocument3 pages028 CSTR Startup PDFAnalytics ClubNo ratings yet
- Comp2024 Btest-11 MathematicsDocument16 pagesComp2024 Btest-11 MathematicsTtttNo ratings yet
- A × B C × B) C, When The Answer Is A Scalar. The Other Possibility A × (B C) Does Not MakeDocument8 pagesA × B C × B) C, When The Answer Is A Scalar. The Other Possibility A × (B C) Does Not MakeNatkhatKhoopdiNo ratings yet
- Week 5 - Theory - Unsteady State and Multiple ReactionsDocument39 pagesWeek 5 - Theory - Unsteady State and Multiple ReactionsJules ArseneNo ratings yet
- Rumus Dan Persamaan Yang Dapat Digunakan (Soal Farmakokinetika)Document1 pageRumus Dan Persamaan Yang Dapat Digunakan (Soal Farmakokinetika)asriNo ratings yet
- Emulation of Analog Controllers: 2.1: Control Design Via Time-Domain EmulationDocument47 pagesEmulation of Analog Controllers: 2.1: Control Design Via Time-Domain EmulationLêNhậtMinhNo ratings yet
- Part-02 Reaction Kinetics (KINETIKA KIMIA, 15 DES 2020)Document24 pagesPart-02 Reaction Kinetics (KINETIKA KIMIA, 15 DES 2020)Anisa NurulNo ratings yet
- Differential CalculusDocument4 pagesDifferential CalculusBILONG, KARYLLE KEITH, O.No ratings yet
- 108 Quiz1Document1 page108 Quiz1沈智恩No ratings yet
- Laplace COEP Updated PDFDocument9 pagesLaplace COEP Updated PDFJayashree MisalNo ratings yet
- Chemical Kinetics: Click Togoto The Table of ContentsDocument39 pagesChemical Kinetics: Click Togoto The Table of ContentsRivaldi Di CaprioNo ratings yet
- Ec 2003Document37 pagesEc 2003SRINIVASA RAONo ratings yet
- Definite & Indefinite IntegrationDocument24 pagesDefinite & Indefinite IntegrationSatyabrata NayakNo ratings yet
- M Ch-05 Sequences and SeriesDocument4 pagesM Ch-05 Sequences and Seriesmysoftinfo.incNo ratings yet
- Solving Real Business Cycle Models by Solving Systems of First Order ConditionsDocument18 pagesSolving Real Business Cycle Models by Solving Systems of First Order ConditionsBrunéNo ratings yet
- Capacitance - JEE Main 2021 July Chapter-Wise - MathonGoDocument5 pagesCapacitance - JEE Main 2021 July Chapter-Wise - MathonGoAyush GuptaNo ratings yet
- PHL BS With Affine DivsDocument8 pagesPHL BS With Affine DivsRaphaël FromEverNo ratings yet
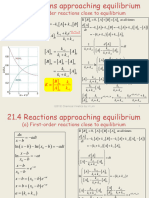
- (A) First-Order Reactions Close To Equilibrium: Da Ka K B DT K Ke A A K KDocument18 pages(A) First-Order Reactions Close To Equilibrium: Da Ka K B DT K Ke A A K Kcmc107No ratings yet
- Pragathi Full Test 5 Question Paper PDFDocument20 pagesPragathi Full Test 5 Question Paper PDFAkash A100% (2)
- 1035purl IPC TYS 2023Document11 pages1035purl IPC TYS 2023shiv lionNo ratings yet
- Equilibrium - Structure - Change: Thermodynamics Quantum TheoryDocument20 pagesEquilibrium - Structure - Change: Thermodynamics Quantum Theorycmc107No ratings yet
- Manho LimDocument25 pagesManho Limcmc107No ratings yet
- 4 +Proline-Catalyzed+AsymmetriDocument8 pages4 +Proline-Catalyzed+Asymmetricmc107No ratings yet
- 8 +Peptide+SynthesisDocument7 pages8 +Peptide+Synthesiscmc107No ratings yet
- 5 +crystallization +purificatiDocument11 pages5 +crystallization +purificaticmc107No ratings yet
- 2, United States Patent: Hiermeier Et atDocument11 pages2, United States Patent: Hiermeier Et atjackNo ratings yet
- 1 s2.0 S0013935121019435 MainDocument13 pages1 s2.0 S0013935121019435 MainEsmeralda SouzaNo ratings yet
- PLANCK's Constant by Photocell-Neha TDocument4 pagesPLANCK's Constant by Photocell-Neha TKittu100% (2)
- Physics IAL Mechanics Topical Paper Unit 1Document11 pagesPhysics IAL Mechanics Topical Paper Unit 1Nagham BarakatNo ratings yet
- Performance Comparative Analysis of Monocrystalline and Polycrystalline Single Diode Solar Panel Models Using The Five Parameters MethodDocument6 pagesPerformance Comparative Analysis of Monocrystalline and Polycrystalline Single Diode Solar Panel Models Using The Five Parameters MethodAlif Mu'tashimNo ratings yet
- 2-Ethylhexanol: Material Safety Data SheetDocument13 pages2-Ethylhexanol: Material Safety Data SheetOmar Eduardo Davalillo MarínNo ratings yet
- Corrosion Control Solution For Gas Treating AminesDocument3 pagesCorrosion Control Solution For Gas Treating AminesSalman AbuzuhairaNo ratings yet
- Amorphous Metal TransformersDocument17 pagesAmorphous Metal TransformersAnjeri PhelestusNo ratings yet
- Organic Photo ChemistryDocument21 pagesOrganic Photo ChemistryAmal Cherian KNo ratings yet
- 15MEC312 L5 1D Heat Conduction EquationDocument12 pages15MEC312 L5 1D Heat Conduction EquationVenkatesh VenkatNo ratings yet
- 18 silberberg8eISMChapter18 9eDocument68 pages18 silberberg8eISMChapter18 9efgb9qfb7x6No ratings yet
- GOFUN2017 ParticleSimulations SlidesDocument44 pagesGOFUN2017 ParticleSimulations SlidesAnonymous dDTRvVNo ratings yet
- Rockstrom Et Al 2009 - Planetary BoundariesDocument33 pagesRockstrom Et Al 2009 - Planetary Boundariestemp tempNo ratings yet
- FINAL EXAM MEC420 Feb 2018 - SUPPDocument7 pagesFINAL EXAM MEC420 Feb 2018 - SUPPfaezahjalalNo ratings yet
- The Stop Motion ProjectDocument3 pagesThe Stop Motion ProjectChanceNo ratings yet
- Lesson 1 Forces and MotionDocument4 pagesLesson 1 Forces and MotionW-d DomNo ratings yet
- Ads or PtionDocument53 pagesAds or PtionYamini KandregulaNo ratings yet
- (L1) Electrostatics-1 9th AprDocument33 pages(L1) Electrostatics-1 9th Aprkhushi singhNo ratings yet
- Chapter2-Campuran Pada Tingkat MolekulerDocument73 pagesChapter2-Campuran Pada Tingkat MolekulerAnnisah MardiyyahNo ratings yet
- GV-BEKA Wind - Energy - LRDocument24 pagesGV-BEKA Wind - Energy - LRRoffepNo ratings yet
- Fiitjee: All India Test SeriesDocument24 pagesFiitjee: All India Test SeriesJyothi BrothersNo ratings yet
- 1.1 PowerofamagnetDocument20 pages1.1 PowerofamagnetEngelo CaroNo ratings yet
- 1674 CompositesDocument22 pages1674 CompositessuneethaNo ratings yet
- Final ResearchDocument14 pagesFinal ResearchAlthea Monique R LamesNo ratings yet