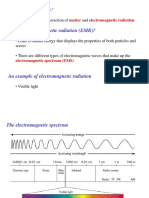
You might also like
- What is Charge? – The Redefinition of Atom - Energy to Matter ConversionFrom EverandWhat is Charge? – The Redefinition of Atom - Energy to Matter ConversionNo ratings yet
- UV-Visible Spectroscopy: Dr. V. KrishnakumarDocument32 pagesUV-Visible Spectroscopy: Dr. V. KrishnakumarRafael HenriqueNo ratings yet
- UV Visible Spectroscopy 2014Document48 pagesUV Visible Spectroscopy 2014endangNo ratings yet
- UV Spectroscopy 2016Document87 pagesUV Spectroscopy 2016M Mudassar AslamNo ratings yet
- Lecture 9 - Chapter 7-9-28-05Document78 pagesLecture 9 - Chapter 7-9-28-05a3d4sNo ratings yet
- UV SpectrosDocument81 pagesUV Spectrosnoon noonNo ratings yet
- Various Tools and Techniques Used For Structural ElucidationDocument28 pagesVarious Tools and Techniques Used For Structural ElucidationAvinashNo ratings yet
- Ultraviolet Spectroscopy SEM-4, CC-8 PART-1, PPT-2Document7 pagesUltraviolet Spectroscopy SEM-4, CC-8 PART-1, PPT-2Me DevilNo ratings yet
- Advanced Lecture2 (07-03-2021)Document23 pagesAdvanced Lecture2 (07-03-2021)Ziad HassanNo ratings yet
- Chapter 5 Organic Spectroscopy.Document118 pagesChapter 5 Organic Spectroscopy.Dr. Dhondiba Vishwanath60% (5)
- UV AnalysisDocument7 pagesUV AnalysiskchetiNo ratings yet
- Theory of Ultraviolet SpectrosDocument49 pagesTheory of Ultraviolet Spectrosanusha jrNo ratings yet
- UV Visible Spectroscopy ECEpptDocument61 pagesUV Visible Spectroscopy ECEpptBhavya TanneruNo ratings yet
- Analysis ReviewerDocument7 pagesAnalysis ReviewerjaguarmaeNo ratings yet
- Lecture 07 - OM - EM PDFDocument33 pagesLecture 07 - OM - EM PDFHemant KumarNo ratings yet
- Uv Visible SpectrosDocument14 pagesUv Visible SpectrosDevanshi JadaunNo ratings yet
- Uv Visible SpectrosDocument14 pagesUv Visible SpectrosDevanshi JadaunNo ratings yet
- UV - Vis Spectros PDFDocument25 pagesUV - Vis Spectros PDFNelson BarriosNo ratings yet
- Unit 2: CH8491 - Instrumental Methods of Analysis 2019-2020Document24 pagesUnit 2: CH8491 - Instrumental Methods of Analysis 2019-2020Akshay UdayNo ratings yet
- Optical Material and SensorsDocument28 pagesOptical Material and SensorsdinasharanaNo ratings yet
- UV Visible Spectroscopy EceDocument48 pagesUV Visible Spectroscopy EceRavi KumarNo ratings yet
- S.6 Atomic Structur Notes 1 Revision Past PapersDocument12 pagesS.6 Atomic Structur Notes 1 Revision Past PapersMaama PhionaNo ratings yet
- UV - Visible SpectrophotometryDocument171 pagesUV - Visible SpectrophotometryGudisa KufoNo ratings yet
- 546 5686798651313311232164897987lijdcoihsdiuhgfieygDocument24 pages546 5686798651313311232164897987lijdcoihsdiuhgfieygędën hãzárdNo ratings yet
- Excess Carriers and Junction Concepts: Unit IIIDocument48 pagesExcess Carriers and Junction Concepts: Unit IIIThamizharasanNo ratings yet
- Solar ElectricityDocument40 pagesSolar Electricitysonu kumarNo ratings yet
- SemiconductorDocument6 pagesSemiconductordksingh369No ratings yet
- IR and UV SpectroDocument69 pagesIR and UV SpectroSk KumarNo ratings yet
- Chemistry Unit-5Document12 pagesChemistry Unit-5santanu janaNo ratings yet
- Uv Visible Spectroscopy: by Nandesh V. PingaleDocument38 pagesUv Visible Spectroscopy: by Nandesh V. PingaleMohammed Adil ShareefNo ratings yet
- UV-visible SpectrosDocument61 pagesUV-visible SpectrosMuhammad BilalNo ratings yet
- SL Chapter 2 ReviewDocument27 pagesSL Chapter 2 ReviewakikoNo ratings yet
- Spectroscopy: Electromagnetic RadiationDocument19 pagesSpectroscopy: Electromagnetic RadiationPriyanka SharmaNo ratings yet
- UV-Visible Spectroscopy PPT B.TechDocument46 pagesUV-Visible Spectroscopy PPT B.TechGunnika SharmaNo ratings yet
- Electron Configurations-Transition, IonizationDocument33 pagesElectron Configurations-Transition, IonizationRayan BotanyNo ratings yet
- Mössbauer EffectDocument13 pagesMössbauer EffectPrasad RavichandranNo ratings yet
- Semi Conductor NotesDocument8 pagesSemi Conductor Notesjohhnysins1978No ratings yet
- AtomsDocument23 pagesAtomsRaj PatilNo ratings yet
- Chap 3.3 Physics of Semiconductors: PhononDocument9 pagesChap 3.3 Physics of Semiconductors: PhononHassan AzouzNo ratings yet
- Quantum Physic For SpectrumDocument2 pagesQuantum Physic For Spectrumswetha chandrasekaranNo ratings yet
- SBC 470 Principles of Organic SpectroscoDocument107 pagesSBC 470 Principles of Organic SpectroscoVaittianathan MahavapillaiNo ratings yet
- SPECTROSDocument48 pagesSPECTROSyt HehkkeNo ratings yet
- Charge Coupled Devices TechnologyDocument40 pagesCharge Coupled Devices Technologyapi-3733788100% (3)
- General Chemistry: More Than Two Electrons (With Opposite Spin) ."Document5 pagesGeneral Chemistry: More Than Two Electrons (With Opposite Spin) ."Marc Vincent CastilloNo ratings yet
- Solid State Chemistry-4: Dr. Shaista Ali CHEM-3207Document45 pagesSolid State Chemistry-4: Dr. Shaista Ali CHEM-3207Muhammad Shehzad HaiderNo ratings yet
- Radiography l2 NotesDocument63 pagesRadiography l2 NotesaasattiNo ratings yet
- Files 3-Lecture Notes CHEM-303 UV SpectrosDocument35 pagesFiles 3-Lecture Notes CHEM-303 UV SpectrosKrishna Moorthy G100% (1)
- Electronic SpectrosDocument82 pagesElectronic SpectrosEub EuNo ratings yet
- Uv Visiblespectroscopyppt 170925144657Document58 pagesUv Visiblespectroscopyppt 170925144657meenu sruthi priyaNo ratings yet
- UV-Visible Spectrophotometric Method and Validation of Organic CompoundsDocument4 pagesUV-Visible Spectrophotometric Method and Validation of Organic Compoundsianatul khafidlahNo ratings yet
- Class Lecture Atomic Model Conductor SemiconductorDocument8 pagesClass Lecture Atomic Model Conductor SemiconductorRajaul Morshad SaikotNo ratings yet
- Uv-Visible Spectroscopy: Presented By: Gabriel Engonga and Flora DikeDocument61 pagesUv-Visible Spectroscopy: Presented By: Gabriel Engonga and Flora DikeGabriel EngongaNo ratings yet
- Spectroscopy of Organic CompoundsDocument36 pagesSpectroscopy of Organic Compoundsnandhini_lgc0% (1)
- Report SpectrosDocument4 pagesReport SpectrosFff FffNo ratings yet
- Emags and Circ1Document14 pagesEmags and Circ1Compl3x CSGONo ratings yet
- Atomic Spectra and Flame TestsDocument37 pagesAtomic Spectra and Flame TestsMouli MishraNo ratings yet
- Ch6 PhotonicDocument11 pagesCh6 PhotonicZeyad AymanNo ratings yet
- Elementary Particles: The Commonwealth and International LibraryFrom EverandElementary Particles: The Commonwealth and International LibraryNo ratings yet
- Quantum and Nuclear Questions and AnswersDocument43 pagesQuantum and Nuclear Questions and AnswersemilyNo ratings yet
- Fieldspec Fieldguide TG - Rev4 - WebDocument136 pagesFieldspec Fieldguide TG - Rev4 - WebHugo Poma FernándezNo ratings yet
- ScintillationDetectors PDFDocument10 pagesScintillationDetectors PDFphanthanhhungNo ratings yet
- Chapter 3c X Ray DiffractionDocument57 pagesChapter 3c X Ray DiffractionarulmuruguNo ratings yet
- National Executive Management Seminar On Surface Finishing by Radiation Curing TechnologyDocument133 pagesNational Executive Management Seminar On Surface Finishing by Radiation Curing Technologysalih benliNo ratings yet
- Unit 20 Medical Physics Techniques CompleteDocument7 pagesUnit 20 Medical Physics Techniques CompleteSum Wing Samuel SoNo ratings yet
- Objective: Activity1. Let's Make Waves! What Happens When Waves Pass By?Document4 pagesObjective: Activity1. Let's Make Waves! What Happens When Waves Pass By?Nina Angela Cate100% (1)
- Proof of LENRDocument126 pagesProof of LENRAnonymous ZKt114No ratings yet
- Photoelectric EffectDocument3 pagesPhotoelectric EffectBIALIGYNo ratings yet
- Point and Line SourceDocument41 pagesPoint and Line SourceNomanNo ratings yet
- Holography (Unit 1)Document19 pagesHolography (Unit 1)Prince JunejaNo ratings yet
- Antenas RVRDocument140 pagesAntenas RVReleazarNo ratings yet
- Prism OpticsDocument32 pagesPrism OpticsFredNo ratings yet
- Theoretical Modeling of Tm-Doped Silica Fiber LasersDocument9 pagesTheoretical Modeling of Tm-Doped Silica Fiber Lasersteektak1No ratings yet
- 7 LensesDocument34 pages7 LensesAananth BalachandranNo ratings yet
- EPAPER solar-FloTHERMDocument19 pagesEPAPER solar-FloTHERMmukeshNo ratings yet
- An Assignment On Ultraviolet and Visible SpectrometerDocument12 pagesAn Assignment On Ultraviolet and Visible SpectrometerSonnet100% (1)
- Sample Data Reflection & RefractionDocument11 pagesSample Data Reflection & RefractionIssa GrantNo ratings yet
- Antenna Ece 8th Sem MakautDocument128 pagesAntenna Ece 8th Sem MakautRitan DasNo ratings yet
- QUESTIONS AND PROBLEMS (Anachem - Topic 1)Document4 pagesQUESTIONS AND PROBLEMS (Anachem - Topic 1)Michaella CabanaNo ratings yet
- 9-Dental Radiation PhysicsDocument27 pages9-Dental Radiation PhysicsAbdullah El-ZaaboutNo ratings yet
- 22.01 Problem Set 4 Homework Solutions: Q M) Q E M)Document5 pages22.01 Problem Set 4 Homework Solutions: Q M) Q E M)Aurélie MaillardNo ratings yet
- Wave GuideDocument12 pagesWave GuideSudhir KumarNo ratings yet
- Service Manual: Color Television Chassis No. SN-012Document28 pagesService Manual: Color Television Chassis No. SN-012inglarkinNo ratings yet
- Refraction, Lenses TestDocument5 pagesRefraction, Lenses TestAndrew Nicholas100% (1)
- IR Bruce GilbertDocument14 pagesIR Bruce Gilbertkane likNo ratings yet
- LQ Grade 7Document2 pagesLQ Grade 7Irene MartinNo ratings yet
- Raman Scattering - Lecture 10Document11 pagesRaman Scattering - Lecture 10Omar AlshekhliNo ratings yet
- Second Periodic TestDocument7 pagesSecond Periodic TestJennifer O. Catubig100% (1)
- Double Slit ExperimentDocument31 pagesDouble Slit ExperimentNorvemer SalcedaNo ratings yet