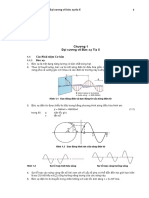
You might also like
- Làm Sao Giải 64 Rubik Với Những Công Thức Đơn GiảnFrom EverandLàm Sao Giải 64 Rubik Với Những Công Thức Đơn GiảnRating: 5 out of 5 stars5/5 (1)
- Bốn khám phá Căn bản Đặc biệt quan trọng cho Hóa học: Four basic Discoveries Especially Important for ChemistryFrom EverandBốn khám phá Căn bản Đặc biệt quan trọng cho Hóa học: Four basic Discoveries Especially Important for ChemistryNo ratings yet
- Chuong 5. Kỹ Thuật Phân Lập Và Tinh ChếDocument93 pagesChuong 5. Kỹ Thuật Phân Lập Và Tinh ChếNguyễn BìnhNo ratings yet
- Quang Xúc Tác Tio2Document49 pagesQuang Xúc Tác Tio2Nga Võ100% (2)
- Bài 1+2 Uv-VisDocument48 pagesBài 1+2 Uv-VisPhong Vũ BảoNo ratings yet
- Chương 6. Phương Pháp Phân Tích Thể TíchDocument8 pagesChương 6. Phương Pháp Phân Tích Thể TíchMinh MinhNo ratings yet
- Cours UV-VISDocument57 pagesCours UV-VISDungNo ratings yet
- Chuyên đề Tinh thể lỏng (hiepkhachquay)Document46 pagesChuyên đề Tinh thể lỏng (hiepkhachquay)hiepkhachquay100% (6)
- Bai Giang PTHC Chuong 2Document177 pagesBai Giang PTHC Chuong 2Xanh XanhNo ratings yet
- 3.Chương 3. Phương Pháp Khối LượngDocument27 pages3.Chương 3. Phương Pháp Khối LượngAnh Quoc LeNo ratings yet
- HOÁ KEO & CÁC HIỆN TƯỢNG BỀ MẶT-2010-2011Document122 pagesHOÁ KEO & CÁC HIỆN TƯỢNG BỀ MẶT-2010-2011Đức Biên100% (2)
- Chương 6 - LTDocument29 pagesChương 6 - LTNguyen Van TaiNo ratings yet
- Bài Giảng 4 Phương Pháp Phân Tích Thể TíchDocument51 pagesBài Giảng 4 Phương Pháp Phân Tích Thể TíchVũ HậuNo ratings yet
- Slide Phan Tich Bang Cong CuDocument333 pagesSlide Phan Tich Bang Cong CuPhan Minh QuyếtNo ratings yet
- Chiết Xuất SPE (Thầy Đức)Document70 pagesChiết Xuất SPE (Thầy Đức)KhanhNguyen0% (1)
- Pho X 2016 GT + BT PDFDocument47 pagesPho X 2016 GT + BT PDFTôn SevenNo ratings yet
- Oxy Hoa Nang Cao.Document11 pagesOxy Hoa Nang Cao.Winter's CryNo ratings yet
- Khoi PhoDocument34 pagesKhoi Phomilundethuong1990100% (4)
- Chủ đề 2- ĐIỆN TRƯỜNG-CƯỜNG ĐỘ ĐIỆN TRƯỜNG - 882021Document21 pagesChủ đề 2- ĐIỆN TRƯỜNG-CƯỜNG ĐỘ ĐIỆN TRƯỜNG - 882021Trang NgânNo ratings yet
- 314753596 Hệ Thống Sắc Ký Lỏng Hiệu Năng CaoDocument16 pages314753596 Hệ Thống Sắc Ký Lỏng Hiệu Năng CaoThái Ngọc Khánh ĐoanNo ratings yet
- SẮC KÝ 1Document22 pagesSẮC KÝ 1Nguyên Trân Nguyễn PhúcNo ratings yet
- Xác định thế ZetaDocument8 pagesXác định thế ZetaHung DaoNo ratings yet
- Phương Pháp Phổ Khối LượngDocument3 pagesPhương Pháp Phổ Khối LượngVu DungNo ratings yet
- TÀI LIỆU HÓA 12 ÔN TN THPTQG PDFDocument150 pagesTÀI LIỆU HÓA 12 ÔN TN THPTQG PDFNgânNo ratings yet
- Bai Giang LCMS 2012Document25 pagesBai Giang LCMS 2012tungduoc_tnNo ratings yet
- Hấp Phụ: -Hấp phụ vật lý có thể tạo nhiều lớp -Hấp phụ hóa học chỉ xảy ra ở lớp thứ nhấtDocument59 pagesHấp Phụ: -Hấp phụ vật lý có thể tạo nhiều lớp -Hấp phụ hóa học chỉ xảy ra ở lớp thứ nhấtmèo béoNo ratings yet
- PHỔ-cơ sở lí thuyết phương pháp phổ Raman và kĩ thuật thực nghiệmDocument6 pagesPHỔ-cơ sở lí thuyết phương pháp phổ Raman và kĩ thuật thực nghiệmVy ThanhNo ratings yet
- HPLCDocument28 pagesHPLCtranthuhachau211033% (3)
- Chuyên Đề Phức Chất PDFDocument75 pagesChuyên Đề Phức Chất PDFThanh TùngNo ratings yet
- Tom Tat Cac Bao Cao Khoa HocDocument639 pagesTom Tat Cac Bao Cao Khoa Hochoa1405No ratings yet
- Tặng Trọn Bộ eBook ĐỘC QUYỀN Cập Nhật 12.5.2021Document11 pagesTặng Trọn Bộ eBook ĐỘC QUYỀN Cập Nhật 12.5.2021katieNo ratings yet
- BÁO CÁO VỀ MÁY UVDocument32 pagesBÁO CÁO VỀ MÁY UVtrunghieuda08hhaNo ratings yet
- Bài Giảng Chuẩn Độ Điện ThếDocument53 pagesBài Giảng Chuẩn Độ Điện ThếQuynh Tram PhanNo ratings yet
- Hoa Hoc Xanh Trong Tong Hop Huu Co Tap 1 Xuc Tac Xanh Va Dung Moi Xanh Phan Thanh Son NamDocument485 pagesHoa Hoc Xanh Trong Tong Hop Huu Co Tap 1 Xuc Tac Xanh Va Dung Moi Xanh Phan Thanh Son Namjonathan kanshanNo ratings yet
- SO SÁNH NHIỆT ĐỘ SÔIDocument6 pagesSO SÁNH NHIỆT ĐỘ SÔIchjtonbaovtpNo ratings yet
- Sac Ky LongDocument22 pagesSac Ky LongTái TuầnNo ratings yet
- Chuyen de Phuc ChatDocument75 pagesChuyen de Phuc ChatTiến MinhNo ratings yet
- xử lý mẫu Phạm luậnDocument98 pagesxử lý mẫu Phạm luậnQuốcLong100% (6)
- Tổng Quan Về Oxit Titan Và Cơ Chế Xúc Tác QuangDocument13 pagesTổng Quan Về Oxit Titan Và Cơ Chế Xúc Tác QuangKỳ Trầm90% (10)
- Thí nghiệm sinh học đại cương - Bài 1: Kính hiển viDocument5 pagesThí nghiệm sinh học đại cương - Bài 1: Kính hiển viHeo AnNo ratings yet
- Dai Cuong Hoa Huu CoDocument2 pagesDai Cuong Hoa Huu CoHà Lê ThanhNo ratings yet
- 15 Hoa Dai Cuong - D-01 PDFDocument275 pages15 Hoa Dai Cuong - D-01 PDFTrần Công Lâm100% (2)
- tiểu luận hóaDocument22 pagestiểu luận hóaThanh HùngNo ratings yet
- Electroanalytical Methods Ch1Document23 pagesElectroanalytical Methods Ch1Lợi TrườngNo ratings yet
- Pin Điện Hóa...Document7 pagesPin Điện Hóa...DHHH UDCNTTNo ratings yet
- PhuongPhapDienHoa DuocDocument6 pagesPhuongPhapDienHoa DuocTâm LêNo ratings yet
- Đề 4 - Chuyển Động Tròn ĐềuDocument12 pagesĐề 4 - Chuyển Động Tròn ĐềuNguyễn Thanh HằngNo ratings yet
- Phuong Phap Quang PhoDocument67 pagesPhuong Phap Quang PhoKesy BachaNo ratings yet
- Pho Nguyen TuDocument295 pagesPho Nguyen TuLê Phạm Phương NamNo ratings yet
- Protein Docking Bai GiangDocument54 pagesProtein Docking Bai GiangMến Nguyễn HồngNo ratings yet
- AutoDock4.2.6 UserGuide-vneseDocument51 pagesAutoDock4.2.6 UserGuide-vnesethanhquangNo ratings yet
- Cong-Nghe-Nano Nhom5 - (Cuuduongthancong - Com)Document36 pagesCong-Nghe-Nano Nhom5 - (Cuuduongthancong - Com)Việt QuốcNo ratings yet
- Basicprotocol - QCuongDocument7 pagesBasicprotocol - QCuongQuốc CườngNo ratings yet
- 239 242Document7 pages239 242TÚ Trương AnhNo ratings yet
- Tao Nói PhétDocument5 pagesTao Nói Phét7loveninhbinh1408No ratings yet
- Tìm Hiểu Về Cae Và FemDocument19 pagesTìm Hiểu Về Cae Và FemTwOsEvEn SnAkENo ratings yet
- BaiTapNhom AT-TS 20202 EE3127Document13 pagesBaiTapNhom AT-TS 20202 EE3127Phạm Hoàng ĐôngNo ratings yet
- Overview of Application of Surface Response MethodDocument8 pagesOverview of Application of Surface Response Methodquangminh03051999No ratings yet
- Asm - Filenall - Nhóm 4Document34 pagesAsm - Filenall - Nhóm 4Tran Thanh Truyen (FPL DN K17)No ratings yet
- CSM DistributedMPCvieDocument10 pagesCSM DistributedMPCvieHuu Phuoc TranNo ratings yet