You might also like
- Lecture Outline: Organic ChemistryDocument21 pagesLecture Outline: Organic Chemistrysungyeon heoNo ratings yet
- Ch07 LectureDocument87 pagesCh07 Lectureshantalle gabrielNo ratings yet
- Chapter 11 - AlkynesDocument48 pagesChapter 11 - AlkynesTempuraNo ratings yet
- Alcohol Ether and ExpoksideDocument64 pagesAlcohol Ether and ExpoksideAhmadBadruzzamanShuib100% (1)
- Ch03 - Basics3Document57 pagesCh03 - Basics3Saguvij FopoNo ratings yet
- Atropisomerism PDFDocument22 pagesAtropisomerism PDFAnil KumarNo ratings yet
- L9 Sympathomimetics Lytics REVISED 2017 PDFDocument49 pagesL9 Sympathomimetics Lytics REVISED 2017 PDFVea AngelesNo ratings yet
- Organic Chemistry, Second Edition Janice Gorzynski Smith, ch1Document40 pagesOrganic Chemistry, Second Edition Janice Gorzynski Smith, ch1sungyeon heoNo ratings yet
- Organic Chemistry,: Alcohols, Ethers, EpoxidesDocument69 pagesOrganic Chemistry,: Alcohols, Ethers, EpoxidesilhamfaturachmanagusNo ratings yet
- ELIMINATION REACTIONS: AN OVERVIEWDocument19 pagesELIMINATION REACTIONS: AN OVERVIEWSyuhadah NoordinNo ratings yet
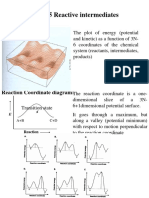
- Chap. 5 Reactive Intermediates: Energy SurfaceDocument20 pagesChap. 5 Reactive Intermediates: Energy SurfaceAnil KumarNo ratings yet
- Racemic Mixtures and Optical Activity DeterminationDocument184 pagesRacemic Mixtures and Optical Activity DeterminationBowo Aank ApriantoNo ratings yet
- Fizika I - Električna struja u tečnostima i gasovimaDocument2 pagesFizika I - Električna struja u tečnostima i gasovimaYuki AYukiNo ratings yet
- 2007Document9 pages2007Anil KumarNo ratings yet
- Organic Chemistry,: Structure & BondingDocument74 pagesOrganic Chemistry,: Structure & BondingilhamfaturachmanagusNo ratings yet
- Chapter 11 - Reactions of Alcohol (Compatibility Mode)Document13 pagesChapter 11 - Reactions of Alcohol (Compatibility Mode)Billy HoNo ratings yet
- Static StereochemistryDocument29 pagesStatic StereochemistryAsmitaNo ratings yet
- Reactions of Alcohols: Organic Chemistry, 7Document53 pagesReactions of Alcohols: Organic Chemistry, 7haha_le12100% (1)
- Practice Problems On Addition Reactions To Alkenes With AnswersDocument4 pagesPractice Problems On Addition Reactions To Alkenes With AnswersSangetha ChelladoraiNo ratings yet
- Chapter02 Smith3e PPTDocument40 pagesChapter02 Smith3e PPTLuisa PalomoNo ratings yet
- QOI0809 AlkenesDocument30 pagesQOI0809 Alkenesmtucker17No ratings yet
- Structure and Synthesis of Alcohols: Organic Chemistry, 7Document52 pagesStructure and Synthesis of Alcohols: Organic Chemistry, 7haha_le12100% (1)
- Sn12, E12 - Exam 2 Practice Problems and KeysDocument40 pagesSn12, E12 - Exam 2 Practice Problems and KeysNguyễn Khánh PhươngNo ratings yet
- Chapter04 Smith3e PPTDocument71 pagesChapter04 Smith3e PPTLuisa Palomo100% (1)
- Organic Chemistry, Second Edition Janice Gorzynski Smith, ch2Document16 pagesOrganic Chemistry, Second Edition Janice Gorzynski Smith, ch2sungyeon heoNo ratings yet
- Amino KiselineDocument13 pagesAmino KiselineGagy FocalSoundNo ratings yet
- Stereochemistry by Qari Abdullah SiddiqueDocument48 pagesStereochemistry by Qari Abdullah SiddiqueABDULLAH0% (1)
- 18 Organic ChemistryDocument26 pages18 Organic ChemistryRachel GreenNo ratings yet
- Module 2.3 NGP PDFDocument3 pagesModule 2.3 NGP PDFIshaan ChaturvediNo ratings yet
- Ch1b Ps3 Key SerDocument7 pagesCh1b Ps3 Key SerRichard ZhuNo ratings yet
- Organic Chemistry Midterm 1 Dir+eff++keyDocument1 pageOrganic Chemistry Midterm 1 Dir+eff++keyNorma Leticia RamosNo ratings yet
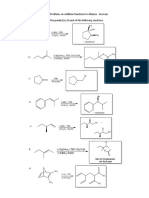
- Course Title: Organic Chemistry-I Course Code: Chm-553, Chm-507 Semester: MSC 1, Bs 5Document18 pagesCourse Title: Organic Chemistry-I Course Code: Chm-553, Chm-507 Semester: MSC 1, Bs 5Mian Naveed AhmedNo ratings yet
- SN1 Vs SN2 ReactionsDocument23 pagesSN1 Vs SN2 Reactionssamnas100No ratings yet
- Chapter 11Document30 pagesChapter 11kanilkadianNo ratings yet
- Aromaticity With Huckle's RuleDocument7 pagesAromaticity With Huckle's RuleSk ZNo ratings yet
- Alkyl Halides and Nucleophilic SubstitutionDocument53 pagesAlkyl Halides and Nucleophilic SubstitutionRaja DanishNo ratings yet
- Neighbouring Group Participation or NGP inDocument4 pagesNeighbouring Group Participation or NGP inbharatbhushansankhya100% (1)
- CHEM F111: General Chemistry II-Semester Lecture 35 (12-04-2019Document20 pagesCHEM F111: General Chemistry II-Semester Lecture 35 (12-04-2019Rachit ShahNo ratings yet
- Chapter 12 Lecture PDFDocument156 pagesChapter 12 Lecture PDFjoseph changNo ratings yet
- Org Chem Sem 3 Paper 2Document15 pagesOrg Chem Sem 3 Paper 2Rohit DeshmukhNo ratings yet
- Organic Chemistry For USTH Students Lecture 2: Electrophilic Addition To C CDocument107 pagesOrganic Chemistry For USTH Students Lecture 2: Electrophilic Addition To C CminhminhNo ratings yet
- Essential Organic Chemistry: Aromaticity: Reactions of Benzene and Substituted BenzenesDocument75 pagesEssential Organic Chemistry: Aromaticity: Reactions of Benzene and Substituted BenzeneschurvaloooNo ratings yet
- McMurry Chapter 2: Polar Covalent Bonds and Acid-Base ConceptsDocument44 pagesMcMurry Chapter 2: Polar Covalent Bonds and Acid-Base ConceptsKirilKocevskiNo ratings yet
- Organic Chemistry - Chapter 15 Benzene & Aromatic CompoundsDocument9 pagesOrganic Chemistry - Chapter 15 Benzene & Aromatic CompoundsSairille ManejaNo ratings yet
- SAR of FluoroquinolonesDocument5 pagesSAR of FluoroquinolonesTanjila IslamNo ratings yet
- Heterocyclic compounds overviewDocument116 pagesHeterocyclic compounds overviewLetin Shrivastav100% (1)
- Enol Dan EnolatDocument40 pagesEnol Dan EnolatRiyan KateeNo ratings yet
- The Structure and Reactions of Aromatic CompoundsDocument25 pagesThe Structure and Reactions of Aromatic CompoundsElizabeth Vivar100% (1)
- Carbonyl Compounds: Aldehydes and KetonesDocument58 pagesCarbonyl Compounds: Aldehydes and KetonesNur Aliyah Abdul RazakNo ratings yet
- Chemistry 108B Exam #1 reagentsDocument1 pageChemistry 108B Exam #1 reagentsNorma Leticia RamosNo ratings yet
- Benzene and Derivatives Members GroupDocument57 pagesBenzene and Derivatives Members GroupHaris KhanNo ratings yet
- Vollhardt 6e Lecture PowerPoints - Chapter 11Document58 pagesVollhardt 6e Lecture PowerPoints - Chapter 11superfr3shmNo ratings yet
- Aromaticity CompleteDocument104 pagesAromaticity Completewahidalwahdi100% (1)
- Sharpless Asymmetric EpoxidationDocument19 pagesSharpless Asymmetric Epoxidationjaanabhenchod100% (2)
- Carbonylalphacarbonreactions_Lecture2revFMGLDocument64 pagesCarbonylalphacarbonreactions_Lecture2revFMGLLinearNo ratings yet
- Enols and EnolatesDocument44 pagesEnols and Enolatessamocamo 123No ratings yet
- Reactive Intermediates: Carbanions and Their ReactivityDocument59 pagesReactive Intermediates: Carbanions and Their Reactivitydagne momNo ratings yet
- Aldehydes and Ketones IIDocument46 pagesAldehydes and Ketones IImidohemaNo ratings yet
- Carbonyl Alpha-Substitution ReactionsDocument64 pagesCarbonyl Alpha-Substitution Reactions張湧浩No ratings yet
- 10장Document37 pages10장sungyeon heoNo ratings yet
- Lecture Outline: Organic ChemistryDocument30 pagesLecture Outline: Organic Chemistrysungyeon heoNo ratings yet
- Lecture Outline: Organic ChemistryDocument25 pagesLecture Outline: Organic Chemistrysungyeon heoNo ratings yet
- Organic Chemistry,: Janice Gorzynski SmithDocument44 pagesOrganic Chemistry,: Janice Gorzynski Smithsungyeon heoNo ratings yet
- Alcohols Ethers and Epoxides Alcohols, Ethers, and Epoxides: Organic ChemistryDocument40 pagesAlcohols Ethers and Epoxides Alcohols, Ethers, and Epoxides: Organic Chemistrysungyeon heoNo ratings yet
- The Claisen Condensation: O CH CH Coch CH ODocument46 pagesThe Claisen Condensation: O CH CH Coch CH Osungyeon heoNo ratings yet
- Organic Chemistry,: Carboxylic Acids and The Acidity of The O-H BondDocument26 pagesOrganic Chemistry,: Carboxylic Acids and The Acidity of The O-H Bondsungyeon heoNo ratings yet
- Organic Chemistry,: Aldehydes and Ketones-Nucleophilic AdditionDocument37 pagesOrganic Chemistry,: Aldehydes and Ketones-Nucleophilic Additionsungyeon heoNo ratings yet
- 20장Document40 pages20장sungyeon heoNo ratings yet
- Herman Emil Fischer 1852 - 1919 Nobel Prize 1902 Sugars, Esters and PurinesDocument44 pagesHerman Emil Fischer 1852 - 1919 Nobel Prize 1902 Sugars, Esters and Purinessungyeon heoNo ratings yet
- The Aldol Condensation: Versatile Synthesis of β-Hydroxy Carbonyl CompoundsDocument38 pagesThe Aldol Condensation: Versatile Synthesis of β-Hydroxy Carbonyl Compoundssungyeon heoNo ratings yet
- Organic Chemistry, Second Edition Janice Gorzynski Smith, ch1Document40 pagesOrganic Chemistry, Second Edition Janice Gorzynski Smith, ch1sungyeon heoNo ratings yet
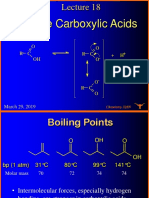
- Carboxylic Acid Lecture: Properties, Reactions and SpectroscopyDocument34 pagesCarboxylic Acid Lecture: Properties, Reactions and Spectroscopysungyeon heoNo ratings yet
- Still More Carbonyl Chemistry: March 26, 2019Document35 pagesStill More Carbonyl Chemistry: March 26, 2019sungyeon heoNo ratings yet
- Organic Chemistry, Second Edition Janice Gorzynski Smith, ch2Document16 pagesOrganic Chemistry, Second Edition Janice Gorzynski Smith, ch2sungyeon heoNo ratings yet
- Organic Chemistry, Second Edition Janice Gorzynski Smith, ch3Document29 pagesOrganic Chemistry, Second Edition Janice Gorzynski Smith, ch3sungyeon heoNo ratings yet
- Ohm's LawDocument79 pagesOhm's Laweugene rellamaNo ratings yet
- IFM - SM7000 - Debitmetre - Sortie AnalogiqueDocument4 pagesIFM - SM7000 - Debitmetre - Sortie Analogiquenapnap5No ratings yet
- Automatic Bottle Washing MachineDocument14 pagesAutomatic Bottle Washing Machineheisnoble100% (1)
- Inorganic ChemistryDocument3 pagesInorganic ChemistryLong ENo ratings yet
- VEML6075: Vishay SemiconductorsDocument11 pagesVEML6075: Vishay Semiconductorsyaumil akbarNo ratings yet
- Gratings & Cycle RacksDocument31 pagesGratings & Cycle Rackswawan setiawanNo ratings yet
- Complete Solar Energy Masterclass For Elec - Engineer Ahmed MahdyDocument159 pagesComplete Solar Energy Masterclass For Elec - Engineer Ahmed Mahdyae6037100No ratings yet
- Tehnicni Program SESSION 2018Document34 pagesTehnicni Program SESSION 2018kenlavie2No ratings yet
- Chapter 8: Blood Rheology: Christina KolyvaDocument24 pagesChapter 8: Blood Rheology: Christina Kolyvaclacsik bikin asikNo ratings yet
- EEE20006 Finalassessment2023Document5 pagesEEE20006 Finalassessment2023anam shafiqueNo ratings yet
- QSD 1976Document10 pagesQSD 1976Felipe ChagasNo ratings yet
- Phase Diagram - : Dr. Aneela Wakeel 02-01-2018Document42 pagesPhase Diagram - : Dr. Aneela Wakeel 02-01-2018Hassan KhanNo ratings yet
- MMPDS-11 Chapter 9 Guidelines for Presentation of DataDocument5 pagesMMPDS-11 Chapter 9 Guidelines for Presentation of Databananth1No ratings yet
- Spur Gears Component GeneratorDocument4 pagesSpur Gears Component GeneratorRodrigo NavarreteNo ratings yet
- Lesson Plan 6 Electrical Installation and Maintenance I. ObjectivesDocument6 pagesLesson Plan 6 Electrical Installation and Maintenance I. Objectivescecille mañacapNo ratings yet
- Welding Distortion Control in Thin Metal Plates by Altering Heat Input Through Weld ParametersDocument7 pagesWelding Distortion Control in Thin Metal Plates by Altering Heat Input Through Weld ParametersGanesh R NairNo ratings yet
- SuspensionDocument72 pagesSuspensionUkash sukarmanNo ratings yet
- PDS Amshield Rev 04Document4 pagesPDS Amshield Rev 04Praveen Sreekumaran NairNo ratings yet
- Review Analytic GeometryDocument10 pagesReview Analytic GeometryGerard GuangcoNo ratings yet
- Offer For 48V 700AH Li-Ion Battery PDFDocument1 pageOffer For 48V 700AH Li-Ion Battery PDFJosé MoralesNo ratings yet
- GR 9, Chem P-1Document23 pagesGR 9, Chem P-1kashif mohammedNo ratings yet
- Direct drive disperser guideDocument2 pagesDirect drive disperser guideAlvaro Nerviani AltieriNo ratings yet
- Form 3 Angles subtended at arc circumference are equalDocument2 pagesForm 3 Angles subtended at arc circumference are equalRita Lau50% (4)
- Inj. Pump Calibration Data: 1. Test ConditionsDocument1 pageInj. Pump Calibration Data: 1. Test ConditionsMiguel RojasNo ratings yet
- Nomenclature For Wire Leads Used As Conductors in Electron TubesDocument3 pagesNomenclature For Wire Leads Used As Conductors in Electron TubesCasey RybackNo ratings yet
- Metering SatamDocument3 pagesMetering SatamYohanes AzzisNo ratings yet
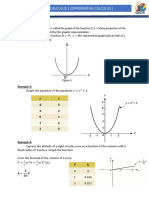
- Em 111-Calculus 1 (Differential Calculus) : Graph of A FunctionDocument3 pagesEm 111-Calculus 1 (Differential Calculus) : Graph of A FunctionJohn Philip NadalNo ratings yet
- MF4202 Additive ManufacturingDocument159 pagesMF4202 Additive ManufacturingOptimMart InternationaleNo ratings yet
- ZXONE 8300&8500&8700 Acceptance Test Guide - R1.7Document152 pagesZXONE 8300&8500&8700 Acceptance Test Guide - R1.7Felipe Ignacio Lopez GordilloNo ratings yet
- Vibrapac Maint PDFDocument211 pagesVibrapac Maint PDFJosé C. Canche DavidNo ratings yet