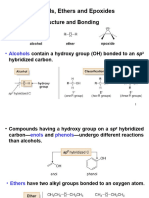
You might also like
- Schaum's Easy Outline of Organic Chemistry, Second EditionFrom EverandSchaum's Easy Outline of Organic Chemistry, Second EditionRating: 3.5 out of 5 stars3.5/5 (2)
- Organic Chemistry,: Substitution Reactions of Carbonyl Compounds at TheDocument29 pagesOrganic Chemistry,: Substitution Reactions of Carbonyl Compounds at Thesungyeon heoNo ratings yet
- (Topic 2) Carbonyl Chemistry 2 2022Document31 pages(Topic 2) Carbonyl Chemistry 2 2022VirginiaNo ratings yet
- Chapter 22 NotesDocument35 pagesChapter 22 NotesTiffany YehNo ratings yet
- Formation of C-C-bonds by Base Catalysed Condensation 2022Document39 pagesFormation of C-C-bonds by Base Catalysed Condensation 2022Thabiso GwijiNo ratings yet
- Carbonyl Alpha-Substitution ReactionsDocument64 pagesCarbonyl Alpha-Substitution Reactions張湧浩No ratings yet
- Things To Remember Only Alc Phe 2022-23Document17 pagesThings To Remember Only Alc Phe 2022-23poornaNo ratings yet
- 16H Carbonyl PDFDocument60 pages16H Carbonyl PDFJose Erick Ortega ValenciaNo ratings yet
- Formation of C-C Bonds by Acid-Catalyzed CondensationsDocument21 pagesFormation of C-C Bonds by Acid-Catalyzed CondensationsThabiso GwijiNo ratings yet
- Lesson 8 Aldehyds, ketonesDocument54 pagesLesson 8 Aldehyds, ketonesIris BallajNo ratings yet
- Wiley's Solomons, Fryhle Snyder Organic Chemistry -720-742Document23 pagesWiley's Solomons, Fryhle Snyder Organic Chemistry -720-742vishalgowni8888No ratings yet
- Aldol EnolateDocument36 pagesAldol Enolatescarllee rogerNo ratings yet
- Ethers, Epoxides, and Sulfides: Organic Chemistry, 7Document46 pagesEthers, Epoxides, and Sulfides: Organic Chemistry, 7Mon Jake Caoile PavicoNo ratings yet
- Ethers, Epoxides, and Sulfides: Organic Chemistry, 7Document34 pagesEthers, Epoxides, and Sulfides: Organic Chemistry, 7Elahine Yuliheht Orozco OcampoNo ratings yet
- Chapter 9 Aldehydes and KetonesDocument36 pagesChapter 9 Aldehydes and KetonesRoshan GillNo ratings yet
- Structure of Aldehydes and KetonesDocument42 pagesStructure of Aldehydes and KetonesPaul Jhon EugenioNo ratings yet
- Chapter FourDocument35 pagesChapter FourAsad MohNo ratings yet
- CH (8) L209 PDFDocument6 pagesCH (8) L209 PDFbebsybiswezNo ratings yet
- Maximizing Enolate Reactivity Through Solvent and Counterion SelectionDocument14 pagesMaximizing Enolate Reactivity Through Solvent and Counterion SelectionVirendra Singh RajputNo ratings yet
- ch20 Aldehydes and Ketones (Revised)Document40 pagesch20 Aldehydes and Ketones (Revised)Spry CylinderNo ratings yet
- Chapter 19. Aldehydes and Ketones: Nucleophilic Addition ReactionsDocument98 pagesChapter 19. Aldehydes and Ketones: Nucleophilic Addition Reactions張湧浩No ratings yet
- Unit 7 Carboxylic Acids Phenols AminesDocument55 pagesUnit 7 Carboxylic Acids Phenols AminesTZ YNo ratings yet
- Aldehyde & Ketone ReactionsDocument21 pagesAldehyde & Ketone ReactionsAinsssNo ratings yet
- Functional Groups Organic Vs Inorganic CompoundsDocument5 pagesFunctional Groups Organic Vs Inorganic CompoundsNo OneNo ratings yet
- BTG Alcoholes Prep 21 1Document26 pagesBTG Alcoholes Prep 21 1LaLo MaldonadoNo ratings yet
- 241 Chem CH-1: AlcoholsDocument21 pages241 Chem CH-1: AlcoholsAlanoud AllaNo ratings yet
- Carbonyl Group Properties and Organic CompoundsDocument18 pagesCarbonyl Group Properties and Organic CompoundsDr Said HassanNo ratings yet
- Organic Chemistry Lesson on Aldehydes and KetonesDocument4 pagesOrganic Chemistry Lesson on Aldehydes and KetonesMARY JANE ANGELICA SEVANo ratings yet
- Organic CompoundsDocument18 pagesOrganic CompoundsAnaya noorNo ratings yet
- Chapter 3Document28 pagesChapter 3c4.arsyadNo ratings yet
- Aldehydes and Ketones II Aldol Reactions: Enolate AnionsDocument9 pagesAldehydes and Ketones II Aldol Reactions: Enolate AnionsCyrene MBolañosNo ratings yet
- Alcohols, Phenols and Ethers - Important Notes For NEET ChemistryDocument12 pagesAlcohols, Phenols and Ethers - Important Notes For NEET Chemistryridha100% (1)
- 19 Enolates EnaminesDocument59 pages19 Enolates EnaminesFlowerNo ratings yet
- Carbonyl ReactionDocument34 pagesCarbonyl Reactionmichot feleguNo ratings yet
- Chapter 8 - FDocument33 pagesChapter 8 - Fcsmmvyxb4bNo ratings yet
- Chemistry II (Organic) Heteroaromatic Chemistry Lectures 2 & 3Document25 pagesChemistry II (Organic) Heteroaromatic Chemistry Lectures 2 & 3Subhabrata MabhaiNo ratings yet
- Alcohols, Phenols and Ethers 2Document167 pagesAlcohols, Phenols and Ethers 2Omkar Singh ShekhawatNo ratings yet
- Chapter Four PowerpointDocument109 pagesChapter Four PowerpointthanaNo ratings yet
- Organic Chemistry,: Alcohols, Ethers, EpoxidesDocument69 pagesOrganic Chemistry,: Alcohols, Ethers, EpoxidesilhamfaturachmanagusNo ratings yet
- Substitution Reactions of Aromatic CompoundsDocument14 pagesSubstitution Reactions of Aromatic CompoundsRavi Kiran KoduriNo ratings yet
- Organic Reactions PPT Alcohols.pptDocument43 pagesOrganic Reactions PPT Alcohols.pptsmithsashay74No ratings yet
- Carbon compounds guideDocument3 pagesCarbon compounds guideKrishna Prasanth rNo ratings yet
- لقطة شاشة 2023-05-27 في 6.17.55 مDocument34 pagesلقطة شاشة 2023-05-27 في 6.17.55 م9c5vtrtqsqNo ratings yet
- Qoiii I Class1Document25 pagesQoiii I Class1valentina viveros riosNo ratings yet
- Enols and EnolatesDocument36 pagesEnols and EnolatesManuel Curitol PiutrinNo ratings yet
- 02 - Enol - Enolates-Alpha Reaction-Std-Maret 2021Document147 pages02 - Enol - Enolates-Alpha Reaction-Std-Maret 2021Fauzia SavitriNo ratings yet
- Enolate Chemistry: 1. Some BasicsDocument20 pagesEnolate Chemistry: 1. Some BasicsIpsita TripathyNo ratings yet
- Alcohols, Phenols and EthersDocument51 pagesAlcohols, Phenols and EthersFatehNo ratings yet
- Dev GDocument22 pagesDev GDeeksha GangwarNo ratings yet
- Lectures 19-22 (LB) Alcohols-Phenols-EthersDocument61 pagesLectures 19-22 (LB) Alcohols-Phenols-Ethersvintu pvNo ratings yet
- Chapter 21 Ald KetonesDocument82 pagesChapter 21 Ald KetonesAndra Ch123No ratings yet
- Alkene Structure and ReactivityDocument33 pagesAlkene Structure and ReactivityДана ЧилибаеваNo ratings yet
- Aldehydes & KetonesDocument61 pagesAldehydes & KetonesfirehywotNo ratings yet
- Chapter 11: Alcohols, Ethers, and EpoxidesDocument27 pagesChapter 11: Alcohols, Ethers, and EpoxidesSaeed SabryNo ratings yet
- Aldehydes and Ketones IIDocument46 pagesAldehydes and Ketones IImidohemaNo ratings yet
- 19 Enolates Enamines-2Document59 pages19 Enolates Enamines-2ronNo ratings yet
- Unit 12 Aldehydes and Ketones UST TemplateDocument26 pagesUnit 12 Aldehydes and Ketones UST TemplateDaniel BalubalNo ratings yet
- Ketones and Aldehydes2022 Copy 1Document56 pagesKetones and Aldehydes2022 Copy 1Camille SolanaNo ratings yet
- Homologous Series Table AnswersDocument2 pagesHomologous Series Table AnswersAli r24No ratings yet
- 122 CH 5 Alcohols Thiolsand Ethers WDocument73 pages122 CH 5 Alcohols Thiolsand Ethers WEvelyn MushangweNo ratings yet
- RectangleDocument6 pagesRectanglesamocamo 123No ratings yet
- FTC Graph Practice (Student Version)Document1 pageFTC Graph Practice (Student Version)samocamo 123No ratings yet
- Universal Basic Income PowerpointDocument12 pagesUniversal Basic Income Powerpointsamocamo 123No ratings yet
- Honors Literature II: Grammar RulesDocument2 pagesHonors Literature II: Grammar Rulessamocamo 123No ratings yet
- FTC Graph Practice (Student Version)Document1 pageFTC Graph Practice (Student Version)samocamo 123No ratings yet
- UBI Pro Op Ed RADocument2 pagesUBI Pro Op Ed RAsamocamo 123No ratings yet
- Works Cited - 1584460790 - 5e70f3f61b7476.98210742Document1 pageWorks Cited - 1584460790 - 5e70f3f61b7476.98210742samocamo 123No ratings yet
- Congressional WebquestDocument8 pagesCongressional Webquestsamocamo 123No ratings yet
- Cherokee High School: Units of Study-"Big Ideas" Classroom ExpectationsDocument10 pagesCherokee High School: Units of Study-"Big Ideas" Classroom Expectationssamocamo 123No ratings yet
- 2019 EngDocument2 pages2019 EngnothardNo ratings yet
- Stocking Final AP Gov RubricDocument1 pageStocking Final AP Gov Rubricsamocamo 123No ratings yet
- Program of Work - Earl PinkettDocument9 pagesProgram of Work - Earl Pinkettsamocamo 123No ratings yet
- STUDENT Remote Learning Schedule S2 SY20-21 (1-19 To 1-22) - CorrectedDocument1 pageSTUDENT Remote Learning Schedule S2 SY20-21 (1-19 To 1-22) - Correctedsamocamo 123No ratings yet
- CHS School Letter1130Document1 pageCHS School Letter1130samocamo 123No ratings yet
- Work, Energy & Power: AP Physics CDocument53 pagesWork, Energy & Power: AP Physics Csamocamo 123No ratings yet
- PythagoreanismDocument6 pagesPythagoreanismsamocamo 123No ratings yet
- DILADocument1,369 pagesDILALilly Hoo LeeNo ratings yet
- Nicotin ADocument12 pagesNicotin ACristina OzarciucNo ratings yet
- Pengaruh Perebusan Terhadap Kandungan Asam Lemak Dan Kolesterol Kerang Pokea (BatissaDocument7 pagesPengaruh Perebusan Terhadap Kandungan Asam Lemak Dan Kolesterol Kerang Pokea (BatissaMesyaSeoKyuYoonwonitedNo ratings yet
- Nucleic Acids: DNA and RNA EssentialsDocument8 pagesNucleic Acids: DNA and RNA EssentialsAmmutha SokayahNo ratings yet
- Chemistry 2 Year Ch-08 & 10 Marks.50 Timing: 1:30 HourDocument2 pagesChemistry 2 Year Ch-08 & 10 Marks.50 Timing: 1:30 HourMusaddiq AzizNo ratings yet
- Chapter Three Amino Acids and Peptides: Mary K. Campbell Shawn O. FarrellDocument23 pagesChapter Three Amino Acids and Peptides: Mary K. Campbell Shawn O. FarrellsaddamixoNo ratings yet
- AntidoteDocument2 pagesAntidoteZeeia MendozaNo ratings yet
- Microbiology Basic and Clinical Principles 1st Edition Mckay Test BankDocument17 pagesMicrobiology Basic and Clinical Principles 1st Edition Mckay Test Bankjamesmartinezstzejixmcg100% (11)
- PWRSLMDocument3 pagesPWRSLMGANESH MURUGANNo ratings yet
- Aromaticity (Organic Chemistry)Document12 pagesAromaticity (Organic Chemistry)noneNo ratings yet
- FR 06 2021 06-53-24ROs Plastic Recyclers - FinalDocument14 pagesFR 06 2021 06-53-24ROs Plastic Recyclers - FinalLakshay UniplarNo ratings yet
- Carbamate Poisoning - MSDDocument2 pagesCarbamate Poisoning - MSDAswin ANo ratings yet
- Cobb 500 Broiler Performance & Nutrition SupplementDocument6 pagesCobb 500 Broiler Performance & Nutrition SupplementUmar Iqbal100% (1)
- Chapter 14 BiomoleculesDocument11 pagesChapter 14 BiomoleculesJaanvi SisodiaNo ratings yet
- WORKSHEET 4.4 Types of CarbohydratesDocument3 pagesWORKSHEET 4.4 Types of Carbohydratesmira bonzay100% (1)
- Biological Molecules Mcqs 2020Document3 pagesBiological Molecules Mcqs 2020PakistanWaqarMughalNo ratings yet
- Carbon Compounds and Their PropertiesDocument23 pagesCarbon Compounds and Their PropertiesAngie Kong Su MeiNo ratings yet
- Function of Each GearsDocument6 pagesFunction of Each GearsRacco Roy0% (1)
- Contractubex - US20100247689A1Document14 pagesContractubex - US20100247689A1hana.hovorkovaNo ratings yet
- Tugas Kimia Organik 2 Jane Nizar RahmanDocument11 pagesTugas Kimia Organik 2 Jane Nizar Rahmanjane nizar rahmanNo ratings yet
- Haloalkanes and Haloarenes ClassificationDocument23 pagesHaloalkanes and Haloarenes ClassificationQwertyNo ratings yet
- Citric Acid 1Document10 pagesCitric Acid 1Tawakal MuzaffarNo ratings yet
- S10 Q3 Enhanced Hybrid Module 4 Week 4 FinalDocument16 pagesS10 Q3 Enhanced Hybrid Module 4 Week 4 FinalBriannah loiuse P. AdalidNo ratings yet
- 13 Proteins and Nucleic AcidsDocument27 pages13 Proteins and Nucleic AcidsJerik ChristofferNo ratings yet
- Lewis Mander, Hung-Wen Liu - Comprehensive Natural Products II - Chemistry and Biology - Cofactors. Volume 7-Elsevier Science (2010)Document754 pagesLewis Mander, Hung-Wen Liu - Comprehensive Natural Products II - Chemistry and Biology - Cofactors. Volume 7-Elsevier Science (2010)Marcio BragaNo ratings yet
- Dyes BookDocument93 pagesDyes BookSebastian OchoaNo ratings yet
- Hydrocarbon and Alkyl Halide-1Document10 pagesHydrocarbon and Alkyl Halide-1Aarya Vardhan ShandilyaNo ratings yet
- Ethylhexylglycerin Safety ToxicologyDocument17 pagesEthylhexylglycerin Safety Toxicologybfh83730No ratings yet
- MLS 114 Lecture #2Document99 pagesMLS 114 Lecture #2NjeodoNo ratings yet
- 20 Common Amino Acids v2 PDFDocument1 page20 Common Amino Acids v2 PDFvinhson65-1No ratings yet