You might also like
- Ap Chemistry Course and Exam Description PDFDocument177 pagesAp Chemistry Course and Exam Description PDFKristine HallNo ratings yet
- Philippine LiteratureDocument82 pagesPhilippine LiteratureLovelene Chrizze100% (5)
- Monster Visual Discrimination CardsDocument9 pagesMonster Visual Discrimination CardsLanaNo ratings yet
- Determination of Dissolved Oxygen Using Winkler MethodDocument5 pagesDetermination of Dissolved Oxygen Using Winkler MethodSyamil Amir Hamzah50% (2)
- 2010-12 600 800 Rush Switchback RMK Service Manual PDFDocument430 pages2010-12 600 800 Rush Switchback RMK Service Manual PDFBrianCook73% (11)
- Experiment No. 3 Determination of Acetic Acid Content in VinegarDocument14 pagesExperiment No. 3 Determination of Acetic Acid Content in VinegarClandy CoNo ratings yet
- RDR 6 Quantitative Determination of Oxalate by Permanganate TitrationDocument5 pagesRDR 6 Quantitative Determination of Oxalate by Permanganate TitrationAlyssa Bautista100% (2)
- Experiment No. 4: Standardization of Sodium Thiosulphate Solution With A Standard Potassium Dichromate SolutionDocument20 pagesExperiment No. 4: Standardization of Sodium Thiosulphate Solution With A Standard Potassium Dichromate Solutionshiam50% (2)
- Exp 2 Iodometric TitrationDocument2 pagesExp 2 Iodometric TitrationDeep Dave100% (3)
- Lab Report Prepare and Standardize A 0.1 M NaOH SolutionsDocument8 pagesLab Report Prepare and Standardize A 0.1 M NaOH Solutionsrodneyperu100% (2)
- Volumetric AnalysisDocument66 pagesVolumetric AnalysisAvan100% (1)
- Experiment 5 - Oxidation-Reduction Titration IodimetryDocument3 pagesExperiment 5 - Oxidation-Reduction Titration IodimetryAlma Pabilane75% (12)
- Experiment 1: Separation and Identification of CationsDocument6 pagesExperiment 1: Separation and Identification of CationsJoseph Pelaelo100% (1)
- KHP LabDocument5 pagesKHP LabSantino MusaNo ratings yet
- Oxidation Reduction Titration Lab ReportDocument9 pagesOxidation Reduction Titration Lab ReportTanebi0% (3)
- LAB REPORT - Determination of Concentration Acetic Acid in VinegarDocument12 pagesLAB REPORT - Determination of Concentration Acetic Acid in Vinegarhisham100% (3)
- 2 The Laws of Universe and Life PROTEGIDO PDFDocument72 pages2 The Laws of Universe and Life PROTEGIDO PDFFransiscus Tobias100% (1)
- Permanganate TitrationDocument6 pagesPermanganate Titrationxavier bourret sicotte83% (6)
- Chem2 Ch13 Skoog Lab Volumetric Analysis TitrationDocument40 pagesChem2 Ch13 Skoog Lab Volumetric Analysis TitrationBochibo Snatch100% (1)
- Laboratory Plan 1:: Standardization of Sodium Hydroxide (Naoh) With Potassium Hydrogen Phthalate (KHP)Document4 pagesLaboratory Plan 1:: Standardization of Sodium Hydroxide (Naoh) With Potassium Hydrogen Phthalate (KHP)Alliza Kaye CasullaNo ratings yet
- Aspirin TitrationDocument3 pagesAspirin TitrationBiancaTardecillaNo ratings yet
- 06 and 07 Standardization of NaOH and Acid Base TitrationDocument16 pages06 and 07 Standardization of NaOH and Acid Base TitrationTyler Hardy80% (5)
- Determination of Copper (II) Concentration by Colorimetric MethodDocument5 pagesDetermination of Copper (II) Concentration by Colorimetric MethodBulawan LaNeref Oremse100% (2)
- Best Way To LearnDocument3 pagesBest Way To LearnRAMIZKHAN124No ratings yet
- Aldol Condensation Reaction Lab ReportDocument4 pagesAldol Condensation Reaction Lab ReportAvril Watson100% (3)
- Determination of Iron With 1,10-PhenanthrolineDocument5 pagesDetermination of Iron With 1,10-PhenanthrolineOmSilence26510% (1)
- Weak Acid Strong Base Titration LabDocument8 pagesWeak Acid Strong Base Titration Labapi-265089380100% (1)
- Journal ListDocument715 pagesJournal ListKannamma DorairajNo ratings yet
- Experiment 9: SPECTROPHOTOMETRIC DETERMINATION OF IRON IN AQUEOUS SOLUTIONS AS A COMPLEX OF 1,10-PHENANTHROLINEDocument2 pagesExperiment 9: SPECTROPHOTOMETRIC DETERMINATION OF IRON IN AQUEOUS SOLUTIONS AS A COMPLEX OF 1,10-PHENANTHROLINEJane Cuerquis100% (5)
- Determination of Total Alkalinity of Water SampleDocument6 pagesDetermination of Total Alkalinity of Water Sampleshaherbano Awan75% (8)
- Spectrophotometric Determination of IronDocument3 pagesSpectrophotometric Determination of IronDozdi93% (14)
- Determination of Nitrate in Drinking Water by UVDocument8 pagesDetermination of Nitrate in Drinking Water by UVRaja Gokhul50% (2)
- Class Opening Preparations Status ReportDocument3 pagesClass Opening Preparations Status ReportMaria Theresa Buscato86% (7)
- The Formula of Epsom Salt Lab ReportDocument3 pagesThe Formula of Epsom Salt Lab ReportAnthony Bui0% (2)
- Neutralization Titration ExperimentDocument10 pagesNeutralization Titration ExperimentJohn Dy100% (1)
- Determination of Acetic Acid in VinegarDocument15 pagesDetermination of Acetic Acid in VinegarSiti Syuhadah75% (4)
- Chapter 2: Chemicals, Apparatus, and Unit Operations of Analytical ChemistryDocument28 pagesChapter 2: Chemicals, Apparatus, and Unit Operations of Analytical Chemistryangela100% (2)
- 2 Determination of Na2CO3 and NaHCO3 in A Mixture by HCLDocument2 pages2 Determination of Na2CO3 and NaHCO3 in A Mixture by HCLShyamapada Shit100% (2)
- (PDF) Biochemistry and Molecular Biology of Plants: Book DetailsDocument1 page(PDF) Biochemistry and Molecular Biology of Plants: Book DetailsArchana PatraNo ratings yet
- Determination of Iron in WaterDocument5 pagesDetermination of Iron in WaterOmSilence2651No ratings yet
- DETERMINING CONCENTRATION OF ACETIC ACIDDocument20 pagesDETERMINING CONCENTRATION OF ACETIC ACIDamiraaikharah100% (1)
- Neutralization Titrations in A Aqueous SolutionsDocument4 pagesNeutralization Titrations in A Aqueous SolutionsUgur ASİT100% (2)
- Experiment 7 - Spectrophotometric DeterminationDocument5 pagesExperiment 7 - Spectrophotometric DeterminationAlma Pabilane96% (24)
- Standardization NaOHDocument5 pagesStandardization NaOHfrancenoelleNo ratings yet
- Titration Curves of Strong and Weak Acids and BasesDocument3 pagesTitration Curves of Strong and Weak Acids and BasesMatthew Runyon50% (2)
- Determining Sodium Hydroxide Concentration Using Acid-Base TitrationDocument5 pagesDetermining Sodium Hydroxide Concentration Using Acid-Base Titrationmo100% (2)
- Preparation and Standardisation of Base and Acid SolutionDocument11 pagesPreparation and Standardisation of Base and Acid Solutionنور حنانيNo ratings yet
- Ppotentiometric Titration of Benzoic Acid With 0.1M Sodium HydroxideDocument23 pagesPpotentiometric Titration of Benzoic Acid With 0.1M Sodium HydroxideCristine ConcepcionNo ratings yet
- Chemistry Lab ReportDocument4 pagesChemistry Lab Reportahlam23a0% (2)
- Determination of Acetic Acid in Vinegar LabDocument6 pagesDetermination of Acetic Acid in Vinegar LabTishko0% (1)
- Determinate of The Concentration of Acetic Acid in VinegarDocument22 pagesDeterminate of The Concentration of Acetic Acid in VinegarSYahira HAzwaniNo ratings yet
- Solution Preparation and StandardizationDocument3 pagesSolution Preparation and StandardizationIris Charmaine Olaso50% (4)
- Experiment 2: Title:Preparation of Solutions. ObjectivesDocument4 pagesExperiment 2: Title:Preparation of Solutions. ObjectivesOromay EliasNo ratings yet
- Volumetric Analysis of A Carbonate-Bicarbonate MixtureDocument8 pagesVolumetric Analysis of A Carbonate-Bicarbonate MixtureDanielle FloridaNo ratings yet
- Determination of The Concentration of Acetic Acid in VinegarDocument24 pagesDetermination of The Concentration of Acetic Acid in VinegarNadia Kama69% (13)
- Preparation of Butyl Acetate PDFDocument6 pagesPreparation of Butyl Acetate PDFjoiya100133% (3)
- Experiment 3 Redox Titration Percent Purity AnalysisDocument5 pagesExperiment 3 Redox Titration Percent Purity AnalysisnanaNo ratings yet
- LAB Report 3Document8 pagesLAB Report 3Phương Vân0% (1)
- Acid Base TitrationDocument19 pagesAcid Base TitrationPredrag DjurdjevicNo ratings yet
- Adsorption Lab ManualDocument6 pagesAdsorption Lab ManualSatyamGuptaNo ratings yet
- Precipitation Titration MethodsDocument3 pagesPrecipitation Titration MethodsBanana SenpaiNo ratings yet
- Lab Report (Spectroscopy)Document6 pagesLab Report (Spectroscopy)Levison Kasenga100% (2)
- Standardization of Sodium HydroxideDocument7 pagesStandardization of Sodium HydroxideSerenity0% (1)
- TLC Separation of Amino AcidsDocument5 pagesTLC Separation of Amino Acidshmtlion0% (5)
- 2-14 Determination of The Dissociation Constant of Weak AcidsDocument3 pages2-14 Determination of The Dissociation Constant of Weak Acidsdbroncos78087100% (6)
- TitrationDocument31 pagesTitrationAtul BansalNo ratings yet
- Practical Analytical 1 ,,chemistryDocument45 pagesPractical Analytical 1 ,,chemistryFadlin AdimNo ratings yet
- Analytical Chemistry Report 3Document6 pagesAnalytical Chemistry Report 3sciencetopic4No ratings yet
- 1221chemistry E Manual IDocument26 pages1221chemistry E Manual Iangel zoeNo ratings yet
- GaneshaDocument39 pagesGaneshaSudarshan TrichurNo ratings yet
- 7.1.answer Key - Track and FieldDocument8 pages7.1.answer Key - Track and FieldNarayanRajNo ratings yet
- Ohms Law ProblemsDocument2 pagesOhms Law ProblemsNarayanRajNo ratings yet
- 12 - Chapter 5 PDFDocument32 pages12 - Chapter 5 PDFA S HARSHANA ANo ratings yet
- Molarity and Molality ANSWERSDocument3 pagesMolarity and Molality ANSWERSNarayanRajNo ratings yet
- Brunton Paul Commentaries Posthumous Vol 1 OCRDocument368 pagesBrunton Paul Commentaries Posthumous Vol 1 OCRDSSidgiddiNo ratings yet
- C) Weight of The Calcium Oxalate Precipitate X G Mass of C O Mass of Cac O X 100 GF Weight of Precipitate / Weight of Sample X GF Z % Z / Theoretical Yield X 100 % YieldDocument1 pageC) Weight of The Calcium Oxalate Precipitate X G Mass of C O Mass of Cac O X 100 GF Weight of Precipitate / Weight of Sample X GF Z % Z / Theoretical Yield X 100 % YieldNarayanRajNo ratings yet
- Business EnvironmentDocument12 pagesBusiness EnvironmentAbhinav GuptaNo ratings yet
- Ek Pardesi Mera Dil Le Gaya Lyrics English Translation - Lyrics GemDocument1 pageEk Pardesi Mera Dil Le Gaya Lyrics English Translation - Lyrics Gemmahsa.molaiepanahNo ratings yet
- Act 1&2 and SAQ No - LawDocument4 pagesAct 1&2 and SAQ No - LawBududut BurnikNo ratings yet
- Types of NumbersDocument11 pagesTypes of NumbersbrajanosmaniNo ratings yet
- BOQ - Hearts & Arrows Office 04sep2023Document15 pagesBOQ - Hearts & Arrows Office 04sep2023ChristianNo ratings yet
- GKInvest Market ReviewDocument66 pagesGKInvest Market ReviewjhonxracNo ratings yet
- DNA Affirmative - MSDI 2015Document146 pagesDNA Affirmative - MSDI 2015Michael TangNo ratings yet
- CHEMICAL ANALYSIS OF WATER SAMPLEDocument5 pagesCHEMICAL ANALYSIS OF WATER SAMPLEAiron Fuentes EresNo ratings yet
- Technical Description: BoilerDocument151 pagesTechnical Description: BoilerÍcaro VianaNo ratings yet
- Smart EE June 2022 - PackageDocument19 pagesSmart EE June 2022 - PackageMercy FeNo ratings yet
- Kennedy 1945 Bibliography of Indonesian Peoples and CulturesDocument12 pagesKennedy 1945 Bibliography of Indonesian Peoples and CulturesJennifer Williams NourseNo ratings yet
- The Interview: P F T IDocument14 pagesThe Interview: P F T IkkkkccccNo ratings yet
- Internal Resistance and Matching in Voltage SourceDocument8 pagesInternal Resistance and Matching in Voltage SourceAsif Rasheed Rajput100% (1)
- 99th Indian Science Congress (Bhubaneshwar)Document95 pages99th Indian Science Congress (Bhubaneshwar)Aadarsh DasNo ratings yet
- Winkens Et Al 2009Document8 pagesWinkens Et Al 2009Marta SanchezNo ratings yet
- Strategic Flexibility: The Evolving Paradigm of Strategic ManagementDocument3 pagesStrategic Flexibility: The Evolving Paradigm of Strategic Managementnanthini kanasanNo ratings yet
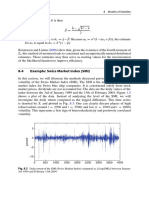
- 8.4 Example: Swiss Market Index (SMI) : 188 8 Models of VolatilityDocument3 pages8.4 Example: Swiss Market Index (SMI) : 188 8 Models of VolatilityNickesh ShahNo ratings yet
- Lineapelle: Leather & Non-LeatherDocument16 pagesLineapelle: Leather & Non-LeatherShikha BhartiNo ratings yet
- JTP Brochure - 2Document6 pagesJTP Brochure - 2YAKOVNo ratings yet
- Sample QuestionsDocument70 pagesSample QuestionsBushra MaryamNo ratings yet
- OMR Sheet Has Been Attached at The End: EST Erial ODocument8 pagesOMR Sheet Has Been Attached at The End: EST Erial OSangeeta BansalNo ratings yet
- Electrical Design Report For Mixed Use 2B+G+7Hotel BuildingDocument20 pagesElectrical Design Report For Mixed Use 2B+G+7Hotel BuildingEyerusNo ratings yet
- Earth Life Science Module 9 Second Quarter 1Document25 pagesEarth Life Science Module 9 Second Quarter 1Milo CatNo ratings yet
- Admission Procedure For International StudentsDocument8 pagesAdmission Procedure For International StudentsAndreea Anghel-DissanayakaNo ratings yet