You might also like
- Gas Laws Worksheet Answer KeyDocument4 pagesGas Laws Worksheet Answer KeyJovenil Bacatan50% (2)
- Manufacturing Considerations in Liquids: - Water Condensate ...................... - Steam .................Document12 pagesManufacturing Considerations in Liquids: - Water Condensate ...................... - Steam .................lola&losa farhanNo ratings yet
- Lamarsh Solutions Ch-2 HalukDocument6 pagesLamarsh Solutions Ch-2 HalukMohamed Ahmed Khodeery50% (2)
- ChemDocument11 pagesChemBrigham John Ricaña Gisala100% (1)
- 10.1 Science Notebook (Answer Key)Document5 pages10.1 Science Notebook (Answer Key)Black arab GaladimaNo ratings yet
- Heat Transfer in Internal Combustion EnginesDocument7 pagesHeat Transfer in Internal Combustion EnginesMishelNo ratings yet
- Presentation About Wooden Toys-1Document30 pagesPresentation About Wooden Toys-1Usama Jahangir KhanNo ratings yet
- Problems in Metallurgical Thermodynamics and Kinetics: International Series on Materials Science and TechnologyFrom EverandProblems in Metallurgical Thermodynamics and Kinetics: International Series on Materials Science and TechnologyRating: 4 out of 5 stars4/5 (5)
- User Guide PDFDocument43 pagesUser Guide PDFJay MaradiyaNo ratings yet
- Introduction to Applied Thermodynamics: The Commonwealth and International Library: Mechanical Engineering DivisionFrom EverandIntroduction to Applied Thermodynamics: The Commonwealth and International Library: Mechanical Engineering DivisionRating: 2.5 out of 5 stars2.5/5 (3)
- Reactor Energy BalanceDocument31 pagesReactor Energy Balancemans_01No ratings yet
- EXP 12 Molar Mass of A Volatile LiquidDocument8 pagesEXP 12 Molar Mass of A Volatile LiquidMau TenNo ratings yet
- Advanced Engineering Thermodynamics: Thermodynamics and Fluid Mechanics SeriesFrom EverandAdvanced Engineering Thermodynamics: Thermodynamics and Fluid Mechanics SeriesRating: 4 out of 5 stars4/5 (9)
- Sarhad University of Science Project ReportDocument33 pagesSarhad University of Science Project ReportDanial NaeemNo ratings yet
- Properties of FluidDocument10 pagesProperties of FluidBrian CedricNo ratings yet
- Processes and Process VariablesDocument13 pagesProcesses and Process Variablesmamat88100% (1)
- Lecture 1 - ThermodynamicsDocument73 pagesLecture 1 - ThermodynamicsNgọc ĐàoNo ratings yet
- Self Evaluation For Class ViiDocument1 pageSelf Evaluation For Class ViistudieslbwNo ratings yet
- Mass Transfer I Online Education 1. Week DuzelDocument20 pagesMass Transfer I Online Education 1. Week DuzeljormungandNo ratings yet
- COMEDK Important Topics and Revised SyllabusDocument49 pagesCOMEDK Important Topics and Revised SyllabusVishal VermaNo ratings yet
- Physical AND Chemical: Mathema TicsDocument1 pagePhysical AND Chemical: Mathema TicsJenny LlanesNo ratings yet
- Process Variables and Ideal Gases: Cheme 101 Module 2 Part 1Document13 pagesProcess Variables and Ideal Gases: Cheme 101 Module 2 Part 1AcademicBMNo ratings yet
- Review For Physics Test 2Document2 pagesReview For Physics Test 2Walt BeckNo ratings yet
- Mass Balance Draft CHE 514N FORM-1-4-Calculation Sheets TemplateDocument8 pagesMass Balance Draft CHE 514N FORM-1-4-Calculation Sheets TemplateLouie G NavaltaNo ratings yet
- Formula collection-Thermodynamics-ver2020HT-1Document22 pagesFormula collection-Thermodynamics-ver2020HT-1Timmy NgoNo ratings yet
- Stem 6 Lesson 4 Final TermDocument44 pagesStem 6 Lesson 4 Final TermA - CAYAGA, Kirby, C 12 - HermonNo ratings yet
- Chemistry - Investigation On Structure & BondingDocument8 pagesChemistry - Investigation On Structure & BondingNej SnekNo ratings yet
- Lecture 5 & 6Document45 pagesLecture 5 & 6Abdulla DoskiNo ratings yet
- Chemistry Lecture NotesDocument3 pagesChemistry Lecture Notes21-54339No ratings yet
- (L-1) - (JEE 3.0) - States of Matter - 13th May.Document50 pages(L-1) - (JEE 3.0) - States of Matter - 13th May.Mohith VenkateshNo ratings yet
- CH 7 SlidesDocument15 pagesCH 7 SlidesMohamed SalahNo ratings yet
- BMMA4833 - Lecture 3Document23 pagesBMMA4833 - Lecture 3khairil adzhaNo ratings yet
- TD-3 - Ideal Gas LawDocument15 pagesTD-3 - Ideal Gas LawAhmed HassanNo ratings yet
- Chem Reviewer MidtermsDocument4 pagesChem Reviewer MidtermsJayrick James AriscoNo ratings yet
- Unit 1 - IntroductionDocument43 pagesUnit 1 - IntroductionIamzura Abdullah100% (1)
- Meen204 Module-01Document6 pagesMeen204 Module-01Genesis MedelNo ratings yet
- Chem 145Document5 pagesChem 145Omar QasimNo ratings yet
- Poster Chapter 2.2 & 3Document1 pagePoster Chapter 2.2 & 3Riyad RahmanNo ratings yet
- Entropy: Thermodynamics: An Engineering ApproachDocument45 pagesEntropy: Thermodynamics: An Engineering ApproachAdolfo Carlos Almendares ÁlvarezNo ratings yet
- Q2 - G11 - Investigating Reaction Rates (Observing - Collision Theory - Energy Flow - The Rate Law)Document123 pagesQ2 - G11 - Investigating Reaction Rates (Observing - Collision Theory - Energy Flow - The Rate Law)ecyel.trNo ratings yet
- Exergy Analysis in Industry: Dr. Khoa Ta DangDocument55 pagesExergy Analysis in Industry: Dr. Khoa Ta DangMinh Hoàng NguyễnNo ratings yet
- Media - Kinetic Theory of GasesDocument36 pagesMedia - Kinetic Theory of GasesYumi LuvenaNo ratings yet
- Liquid Solution Thermodynamic's Properties: Kesetimbangan Fasa (TK201411)Document20 pagesLiquid Solution Thermodynamic's Properties: Kesetimbangan Fasa (TK201411)Wiwi NorzahraNo ratings yet
- Lecture 01 Fundamentals of Thermodynamics 1aDocument28 pagesLecture 01 Fundamentals of Thermodynamics 1aIsmael Torres-PizarroNo ratings yet
- TD-1 - IntroDocument21 pagesTD-1 - IntroAhmed HassanNo ratings yet
- Specific gravity-PPARLAB - EX1Document6 pagesSpecific gravity-PPARLAB - EX1Apple Angeline BalbalosaNo ratings yet
- MCL241 L6 Combustion Flame Temperature Speed TypesDocument20 pagesMCL241 L6 Combustion Flame Temperature Speed TypesAditi SamdarshiniNo ratings yet
- Introduction To Physical PharmacyDocument21 pagesIntroduction To Physical PharmacyKaren GuzmanNo ratings yet
- ESO 201A: Thermodynamics: EntropyDocument44 pagesESO 201A: Thermodynamics: EntropyamanNo ratings yet
- Spontaneity Entropy and Free EnergyDocument79 pagesSpontaneity Entropy and Free Energyserene sanctuaryNo ratings yet
- DF WorksheetsDocument45 pagesDF WorksheetstestNo ratings yet
- 12 Handout 12Document3 pages12 Handout 12James TanNo ratings yet
- Mass Transfer Slides - CHE304 - Chapter 26Document51 pagesMass Transfer Slides - CHE304 - Chapter 26RehabNo ratings yet
- Moles, Density, and ConcentrationDocument12 pagesMoles, Density, and ConcentrationSterlingNo ratings yet
- CHD225 Chemical Reaction Engineering-I: Lecture-2Document17 pagesCHD225 Chemical Reaction Engineering-I: Lecture-2aayush4995No ratings yet
- Combustion - Part 1 - WatermarkedDocument30 pagesCombustion - Part 1 - Watermarked84105851No ratings yet
- G11week5 6 Lectures in Gen Chem 1 Week 5 and 6Document2 pagesG11week5 6 Lectures in Gen Chem 1 Week 5 and 6FMangonon, Jarrah Mae B.No ratings yet
- Group 7 FixDocument30 pagesGroup 7 Fixyuni fitriaNo ratings yet
- Created by James Riddle With Guidance From Dr. Matthew T. BalhoffDocument2 pagesCreated by James Riddle With Guidance From Dr. Matthew T. BalhoffChrisNo ratings yet
- Thermodynamics and Phase BehaviorDocument2 pagesThermodynamics and Phase Behaviorsaurabh pandeyNo ratings yet
- Fluids Review Supplemental Notes - 023609Document18 pagesFluids Review Supplemental Notes - 023609VICTORIA OMEGA JARAMILLANo ratings yet
- EXP 1 - Physics Finals PDFDocument10 pagesEXP 1 - Physics Finals PDFPearl ArcamoNo ratings yet
- Physics 2 - Lecture 1Document69 pagesPhysics 2 - Lecture 1Abonin, Carl Ivan D.L.No ratings yet
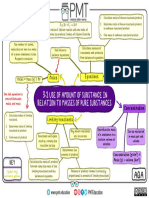
- 3.2. Use of Amount of Substance in Relation To Masses of Pure SubstancesDocument1 page3.2. Use of Amount of Substance in Relation To Masses of Pure Substancesameliarazaq12No ratings yet
- 109 41 FINAL FIZIK T4 DLP - Edited-6-35Document30 pages109 41 FINAL FIZIK T4 DLP - Edited-6-35Jasrul JamalNo ratings yet
- Energy Entropy and The Flow of Nature Thomas F Sherman Full ChapterDocument67 pagesEnergy Entropy and The Flow of Nature Thomas F Sherman Full Chapterdenise.sweeney729100% (7)
- Chemical Measurements: Why Do We Need To Study This Chapter?Document10 pagesChemical Measurements: Why Do We Need To Study This Chapter?SkygazerNo ratings yet
- Bussiness Plan of Dish WashDocument2 pagesBussiness Plan of Dish WashUsama Jahangir KhanNo ratings yet
- Entropy: T DS QDocument12 pagesEntropy: T DS QUsama Jahangir KhanNo ratings yet
- DO Good Have Good: Target CommunitiesDocument1 pageDO Good Have Good: Target CommunitiesUsama Jahangir KhanNo ratings yet
- Assignment Of: Thermodynamics: Group MembersDocument12 pagesAssignment Of: Thermodynamics: Group MembersUsama Jahangir KhanNo ratings yet
- 2020 AliExpress Black Friday Leaked Secret List (Super Discount)Document144 pages2020 AliExpress Black Friday Leaked Secret List (Super Discount)Usama Jahangir KhanNo ratings yet
- Ayesha AMBREEN Chen19111020Document6 pagesAyesha AMBREEN Chen19111020Usama Jahangir KhanNo ratings yet
- 1.1 Thermodynamics: Part A: Topics (All From Reference 1)Document7 pages1.1 Thermodynamics: Part A: Topics (All From Reference 1)Usama Jahangir KhanNo ratings yet
- Isolation of Cellulose Fibers From Banana Leaves (24pgs)Document24 pagesIsolation of Cellulose Fibers From Banana Leaves (24pgs)VŨ NGUYỄN TRẦNNo ratings yet
- Angelita Subdivision, Taliptip, Bulakan, Bulacan Angelita Subdivision, Taliptip, Bulakan, BulacanDocument49 pagesAngelita Subdivision, Taliptip, Bulakan, Bulacan Angelita Subdivision, Taliptip, Bulakan, BulacanDian LegaspiNo ratings yet
- Reading Sentence CompletionDocument3 pagesReading Sentence CompletionFahim AhmedNo ratings yet
- Modified Starches For The Food IndustryDocument5 pagesModified Starches For The Food IndustryMiguelArceMonroyNo ratings yet
- Grahams Law of Diffusion LabDocument3 pagesGrahams Law of Diffusion LabJhun Cards0% (1)
- Technical Paper - FormatDocument5 pagesTechnical Paper - FormatjohnjabarajNo ratings yet
- 55194HMPPSplashzoneWhitepaper.5d8cbbf884bccDocument3 pages55194HMPPSplashzoneWhitepaper.5d8cbbf884bccWan Ah-LunNo ratings yet
- HSG T.anh Cap Huyen 2020 2021 AN LAO 1Document9 pagesHSG T.anh Cap Huyen 2020 2021 AN LAO 1Chi ĐậuNo ratings yet
- T Mech PPR CatalogueDocument17 pagesT Mech PPR CatalogueDani DaniNo ratings yet
- Welcome: Didier Zimmermann, Eit Rawmaterials Central CLC Patrick Bourcet, Metz MétropoleDocument16 pagesWelcome: Didier Zimmermann, Eit Rawmaterials Central CLC Patrick Bourcet, Metz Métropolemangin philippeNo ratings yet
- Chapter 1 - DP FlowDocument8 pagesChapter 1 - DP FlowAmitabhaNo ratings yet
- Energy Conversion and ManagementDocument10 pagesEnergy Conversion and ManagementRamón CevallosNo ratings yet
- Kendriya Vidyalaya Sanghathan, Ahemdabad Region Schedule For Conduct of Test / Examination For The Session 2020-21 Class Xii Subject: ChemistryDocument1 pageKendriya Vidyalaya Sanghathan, Ahemdabad Region Schedule For Conduct of Test / Examination For The Session 2020-21 Class Xii Subject: ChemistryKamal AnandNo ratings yet
- Parker Ammonia-Manual00 DDocument37 pagesParker Ammonia-Manual00 DGöktug AhunbayNo ratings yet
- Larson - Ton KartaDocument9 pagesLarson - Ton KartaJelena GraovčevićNo ratings yet
- Astronomy Masters - Midterm TestDocument7 pagesAstronomy Masters - Midterm TestE. F.No ratings yet
- Daun Teh - KafeinDocument10 pagesDaun Teh - KafeinAila YumekoNo ratings yet
- High-Energy Cathode Materials (Li Mno Limo) For Lithium-Ion BatteriesDocument13 pagesHigh-Energy Cathode Materials (Li Mno Limo) For Lithium-Ion BatteriesEYERUSALEM TADESSENo ratings yet
- Practice Science Questions - Physics Velocity and AccelerationDocument5 pagesPractice Science Questions - Physics Velocity and Acceleration김재명No ratings yet
- Amorphous PolymerDocument14 pagesAmorphous PolymerWahyu SulistyoNo ratings yet
- Calorimetry Lab Methods and Materials-3Document3 pagesCalorimetry Lab Methods and Materials-3Hermela GhebremichaelNo ratings yet
- CV - AshwinDocument3 pagesCV - AshwinAngelo PallancaNo ratings yet
- Physical Sciences. Gsa.: The UniverseDocument13 pagesPhysical Sciences. Gsa.: The UniverseMirza ArslanNo ratings yet