You might also like
- Pressure, Heat and Temperature - Physics for Kids - 5th Grade | Children's Physics BooksFrom EverandPressure, Heat and Temperature - Physics for Kids - 5th Grade | Children's Physics BooksNo ratings yet
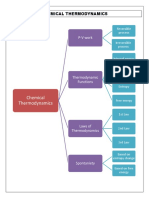
- Mind Map Physical ChemDocument1 pageMind Map Physical ChemhusnaNo ratings yet
- Feynman Lectures Simplified 2B: Magnetism & ElectrodynamicsFrom EverandFeynman Lectures Simplified 2B: Magnetism & ElectrodynamicsNo ratings yet
- Temperature MeasurementDocument21 pagesTemperature MeasurementDINESH KUMAR DRAVIDAMANI100% (2)
- Class 11 Chemistry Revision Notes ThermodynamicsDocument11 pagesClass 11 Chemistry Revision Notes ThermodynamicsAivenNo ratings yet
- Sheet - 01 - Thermodynamics - 2Document85 pagesSheet - 01 - Thermodynamics - 2Novack GamingNo ratings yet
- Nicu NCPDocument2 pagesNicu NCPYette Polillo Conde100% (1)
- DCGUPTA TermodinâmicaDocument210 pagesDCGUPTA TermodinâmicaLaerte Pereira100% (2)
- Calculation of Heat Transfer Coefficients in Agitated VesselsDocument4 pagesCalculation of Heat Transfer Coefficients in Agitated Vesselsdesignselva100% (1)
- Thermodynamic Revision 2021 Part 1Document70 pagesThermodynamic Revision 2021 Part 1Rawda AliNo ratings yet
- 0 Intro ThermodynamicsDocument16 pages0 Intro ThermodynamicsmoazamaakbarNo ratings yet
- 131.11b Heat Equations of Change Part 2Document20 pages131.11b Heat Equations of Change Part 2Jelor GallegoNo ratings yet
- ThermodynamicsDocument16 pagesThermodynamicsSibisi SinethembaNo ratings yet
- Thermodynamics Module 1Document12 pagesThermodynamics Module 1Kirtan KumarNo ratings yet
- End SEM SyllabusDocument45 pagesEnd SEM SyllabusShreyans KothariNo ratings yet
- Chemical Engineering ThermodynamicsDocument8 pagesChemical Engineering ThermodynamicsP P DNo ratings yet
- Momentum Transfer-Compressible Flow (Isentropic, Adiabatic, Isothermal)Document21 pagesMomentum Transfer-Compressible Flow (Isentropic, Adiabatic, Isothermal)Sedrick LopezNo ratings yet
- Analysis of Non-Structural DomainDocument13 pagesAnalysis of Non-Structural DomainPejalan KakiNo ratings yet
- Thermodynamics - Chapter 2Document19 pagesThermodynamics - Chapter 2Jana OsamaNo ratings yet
- Chem101 Ho7Document6 pagesChem101 Ho7nairbatnabamNo ratings yet
- Thermodynamic Potentials Phase Tran. Low Temp MaterialDocument26 pagesThermodynamic Potentials Phase Tran. Low Temp Materialsrinu reddyNo ratings yet
- 2251 SLOTcarnotDocument9 pages2251 SLOTcarnotFrancis JosephNo ratings yet
- MMAN2700ThermoProblemSheet7Solutions - Entropy 2nd LawDocument8 pagesMMAN2700ThermoProblemSheet7Solutions - Entropy 2nd Lawgrandw9524No ratings yet
- Module4 - (2) Conservation of Energy (Work, Work Flow & Heat)Document13 pagesModule4 - (2) Conservation of Energy (Work, Work Flow & Heat)John Dalton ValenciaNo ratings yet
- Lec3 - Final - Revised Stat MechDocument16 pagesLec3 - Final - Revised Stat MechnokosamNo ratings yet
- Dimensions and Units Measures of Amount or SizeDocument5 pagesDimensions and Units Measures of Amount or SizennbNo ratings yet
- Lect 5 Chemical EquilbibriumDocument18 pagesLect 5 Chemical EquilbibriumOmar YousefNo ratings yet
- PHY103E EEE SS Thermodynamics 2Document29 pagesPHY103E EEE SS Thermodynamics 2Robiul Islam RobinNo ratings yet
- CH-103-9th and 10th LectureDocument14 pagesCH-103-9th and 10th LectureDhruv DhirawaniNo ratings yet
- Physics FormulaeDocument6 pagesPhysics FormulaeAsh VinNo ratings yet
- Conduction & ConvectionDocument28 pagesConduction & ConvectionNirmal JayanthNo ratings yet
- 2 Slide Deck DST Heat Transfer Fundamentals 2023 PAO IIDocument22 pages2 Slide Deck DST Heat Transfer Fundamentals 2023 PAO IIAngel BlacioNo ratings yet
- Lesson 1a Overview of 3 Modes of Heat Transfer and Conduction Plane and Walls in SeiresDocument9 pagesLesson 1a Overview of 3 Modes of Heat Transfer and Conduction Plane and Walls in SeiresJAYSON VERANONo ratings yet
- Lesson 8 Generalized Thermal Resistance NetworkDocument24 pagesLesson 8 Generalized Thermal Resistance Networksurya kiranNo ratings yet
- CH2105 - ThermodynamicsDocument12 pagesCH2105 - ThermodynamicsJohnNo ratings yet
- TFS I CH5 Lecture NotesDocument16 pagesTFS I CH5 Lecture Notes高誠蔚No ratings yet
- Entropy and The Second Law of ThermodynamicsDocument6 pagesEntropy and The Second Law of Thermodynamicskhandaker raiyanNo ratings yet
- Apni Kaksha ThermodynamicsDocument27 pagesApni Kaksha ThermodynamicsVcNo ratings yet
- Process Engineering Thermodynamics: Dr. Dharmendra Kumar Bal Assistant Professor (SR.) ScaleDocument50 pagesProcess Engineering Thermodynamics: Dr. Dharmendra Kumar Bal Assistant Professor (SR.) ScaleAABID SHAIKNo ratings yet
- Transient Conduction Analysis The Lumped Capacitance Method: Class #6Document15 pagesTransient Conduction Analysis The Lumped Capacitance Method: Class #6Valentina Mejia GallonNo ratings yet
- Lecture Notes 2 ThermodynamicsDocument5 pagesLecture Notes 2 ThermodynamicsAndrewNo ratings yet
- Lecture 5 - 2nd Law of Thermodynamics - Entropy - 2022 PDFDocument18 pagesLecture 5 - 2nd Law of Thermodynamics - Entropy - 2022 PDFJey BlaQNo ratings yet
- FM IntroductionDocument14 pagesFM IntroductionJayden PangilinanNo ratings yet
- 2021-물리화학-Chapter 2 - 1st law of thermodynamicsDocument30 pages2021-물리화학-Chapter 2 - 1st law of thermodynamics박희원No ratings yet
- ThermoDynamics ProcessDocument2 pagesThermoDynamics ProcessSTUDY BEASTNo ratings yet
- ThermodynamicsDocument65 pagesThermodynamicsEdith EatonNo ratings yet
- CHE2040 Heat Transfer Notes Typed 2014Document302 pagesCHE2040 Heat Transfer Notes Typed 2014EdwinaNo ratings yet
- ASE2101 HT Transfer & Thermo - 3 - 2Document30 pagesASE2101 HT Transfer & Thermo - 3 - 22200851No ratings yet
- Lesson 1 ThermodynamicsDocument40 pagesLesson 1 ThermodynamicsRex OabelNo ratings yet
- Introduction To Thermodynamics UpdatedDocument21 pagesIntroduction To Thermodynamics UpdatedLia HamadaNo ratings yet
- Potential & Kinetic Energy and Flow WorkDocument10 pagesPotential & Kinetic Energy and Flow WorkCarla Francheska CalmaNo ratings yet
- JEE Advanced 2023 Revision Notes For Thermodynamics - Free PDF DownloadDocument18 pagesJEE Advanced 2023 Revision Notes For Thermodynamics - Free PDF DownloadKamini SrivastavNo ratings yet
- THERMODYNAMICS-3 Kca NotesDocument23 pagesTHERMODYNAMICS-3 Kca NotesAbraham ChackoNo ratings yet
- Chapter 4 - 32044472 - 2024 - 03 - 26 - 17 - 48Document7 pagesChapter 4 - 32044472 - 2024 - 03 - 26 - 17 - 48samyakpande1008No ratings yet
- Exam 3 Cheat Sheet: Heat TransferDocument2 pagesExam 3 Cheat Sheet: Heat TransferShyam PolacondaNo ratings yet
- Thermo ChemistryDocument8 pagesThermo ChemistryANGELA JOIE BAIZASNo ratings yet
- Diesel CycleDocument1 pageDiesel CycleGladys Ruth PaypaNo ratings yet
- Chbe 6300 Graduate Kinetics and Reactor Design: Carsten Sievers 8/20/2020Document27 pagesChbe 6300 Graduate Kinetics and Reactor Design: Carsten Sievers 8/20/2020AnnNo ratings yet
- Chemical ThermodynamicsDocument20 pagesChemical ThermodynamicsShantanu KadamNo ratings yet
- Dimensional Analysis Similarity Lesson2 Dimensional Parameters HandoutDocument11 pagesDimensional Analysis Similarity Lesson2 Dimensional Parameters HandoutRizqi RamadhanNo ratings yet
- asset-v1-DelftX+TP102x+3T2016+type@asset+block@Formula Sheet ATPDocument12 pagesasset-v1-DelftX+TP102x+3T2016+type@asset+block@Formula Sheet ATPkennethmsorianoNo ratings yet
- Applications of Differential EquationDocument8 pagesApplications of Differential EquationMubeen JavedNo ratings yet
- 1D SS Heat ConductionDocument65 pages1D SS Heat ConductionAbdullah aminNo ratings yet
- Monceda Mareinelle Chem110b Journal No. 1Document5 pagesMonceda Mareinelle Chem110b Journal No. 1Mar MoncedaNo ratings yet
- W07 The Properties of SolutionsDocument43 pagesW07 The Properties of SolutionsMar MoncedaNo ratings yet
- W02 First Law of ThermodynamicsDocument30 pagesW02 First Law of ThermodynamicsMar MoncedaNo ratings yet
- Instrumental Analysis - UV - VS - AAS - CTLE - FINAL - ReyesDocument57 pagesInstrumental Analysis - UV - VS - AAS - CTLE - FINAL - ReyesMar MoncedaNo ratings yet
- WittigDocument4 pagesWittigMar MoncedaNo ratings yet
- Thermodynamic CyclesDocument13 pagesThermodynamic CyclesAshish KumarNo ratings yet
- Wetted - Wall Column PDFDocument8 pagesWetted - Wall Column PDFSaurab Devanandan33% (3)
- Water, PH and BuffersDocument9 pagesWater, PH and BuffersKivo Al'jahirNo ratings yet
- Improving Calibration and Measurement Capability (CMC) of Psychrometer CalibrationDocument7 pagesImproving Calibration and Measurement Capability (CMC) of Psychrometer CalibrationMiguel Angel Pacahuala CristobalNo ratings yet
- Lab Report 3 Calorimetry IntroductionDocument3 pagesLab Report 3 Calorimetry IntroductionJullifer TubaNo ratings yet
- Chapter 7: Entropy 7.1: Entropy A Thermodynamic PropertyDocument25 pagesChapter 7: Entropy 7.1: Entropy A Thermodynamic PropertyMoisesNo ratings yet
- Le Chatelier Equilibrium Worksheet QuestionsDocument35 pagesLe Chatelier Equilibrium Worksheet QuestionsJaya Chitra Degala RamaluNo ratings yet
- Construction and Building Materials: Mohammad Tahersima, Paul TikalskyDocument12 pagesConstruction and Building Materials: Mohammad Tahersima, Paul TikalskyalejandroNo ratings yet
- Thermal Physics PDFDocument87 pagesThermal Physics PDFPriyanshu SharmaNo ratings yet
- Thermodynamics - Chapter 4Document11 pagesThermodynamics - Chapter 4LiyanaNo ratings yet
- Describe The Difference in The Temperature Profiles For Counter-Flow and Parallel Flow Heat Exchangers. Counter Flow Heat ExchangersDocument3 pagesDescribe The Difference in The Temperature Profiles For Counter-Flow and Parallel Flow Heat Exchangers. Counter Flow Heat Exchangersbyun baekNo ratings yet
- The Second Law of Thermodynamics (Continued) (Lecture 05) : - Prashant Uday ManoharDocument28 pagesThe Second Law of Thermodynamics (Continued) (Lecture 05) : - Prashant Uday ManoharHRIDAY MAHESHWARINo ratings yet
- tb13 PDFDocument22 pagestb13 PDFMavis VermillionNo ratings yet
- Homework 3Document5 pagesHomework 3delru1990safariNo ratings yet
- 1 - Molecular FluxDocument44 pages1 - Molecular FluxJason RoyNo ratings yet
- Manual AD 101Document2 pagesManual AD 101Edvard ŽerovnikNo ratings yet
- NPTEL Phase II - Mechanical Engineering - Refrigeration and Air ConditioningDocument6 pagesNPTEL Phase II - Mechanical Engineering - Refrigeration and Air ConditioningADITYANo ratings yet
- First Law of Thermodynamics: Law of Conservation of Energy: Du DQ + DWDocument60 pagesFirst Law of Thermodynamics: Law of Conservation of Energy: Du DQ + DWAnge1196100% (1)
- Lecture 4 - Free EnergyDocument16 pagesLecture 4 - Free EnergyyudhiprasetyoNo ratings yet
- The Second Law of ThermodynamicsDocument37 pagesThe Second Law of ThermodynamicsGODNo ratings yet
- ASTM01 Thermal Measurements The Foundation of Fire Standards GritzoDocument198 pagesASTM01 Thermal Measurements The Foundation of Fire Standards GritzoEleni AsimakopoulouNo ratings yet
- Detailed Design Condenser Shell and TubeDocument18 pagesDetailed Design Condenser Shell and TubeVin Fiyanthi TyaswarieNo ratings yet
- HeattransferconductionconvectionradiationguidednotesDocument4 pagesHeattransferconductionconvectionradiationguidednotesapi-289120259100% (3)
- Lab Report 1Document14 pagesLab Report 1api-355836337No ratings yet
- Bff3242 Heat Transfer 21516Document9 pagesBff3242 Heat Transfer 21516nadiyaxx0% (1)
- SCES3083 Topic6 Temperature and HeatDocument22 pagesSCES3083 Topic6 Temperature and Heat胡佳玲No ratings yet