You might also like
- Introduction to Applied Thermodynamics: The Commonwealth and International Library: Mechanical Engineering DivisionFrom EverandIntroduction to Applied Thermodynamics: The Commonwealth and International Library: Mechanical Engineering DivisionRating: 2.5 out of 5 stars2.5/5 (3)
- Advanced Engineering Thermodynamics: Thermodynamics and Fluid Mechanics SeriesFrom EverandAdvanced Engineering Thermodynamics: Thermodynamics and Fluid Mechanics SeriesRating: 4 out of 5 stars4/5 (9)
- Properties of FluidDocument10 pagesProperties of FluidBrian CedricNo ratings yet
- Heat Transfer & Heat Exchangers GuideDocument73 pagesHeat Transfer & Heat Exchangers GuideNgọc ĐàoNo ratings yet
- Self Evaluation for Class ViiDocument1 pageSelf Evaluation for Class ViistudieslbwNo ratings yet
- Mass Transfer I Online Education 1. Week DuzelDocument20 pagesMass Transfer I Online Education 1. Week DuzeljormungandNo ratings yet
- COMEDK Important Topics and Revised SyllabusDocument49 pagesCOMEDK Important Topics and Revised SyllabusVishal VermaNo ratings yet
- Physical AND Chemical: Mathema TicsDocument1 pagePhysical AND Chemical: Mathema TicsJenny LlanesNo ratings yet
- Process Variables and Ideal Gases: Cheme 101 Module 2 Part 1Document13 pagesProcess Variables and Ideal Gases: Cheme 101 Module 2 Part 1AcademicBMNo ratings yet
- Review For Physics Test 2Document2 pagesReview For Physics Test 2Walt BeckNo ratings yet
- Mass Balance Draft CHE 514N FORM-1-4-Calculation Sheets TemplateDocument8 pagesMass Balance Draft CHE 514N FORM-1-4-Calculation Sheets TemplateLouie G NavaltaNo ratings yet
- Formula collection-Thermodynamics-ver2020HT-1Document22 pagesFormula collection-Thermodynamics-ver2020HT-1Timmy NgoNo ratings yet
- Stem 6 Lesson 4 Final TermDocument44 pagesStem 6 Lesson 4 Final TermA - CAYAGA, Kirby, C 12 - HermonNo ratings yet
- ThermodynamicsDocument67 pagesThermodynamicsHimanshu RaiNo ratings yet
- 10.1 Science Notebook (Answer Key)Document5 pages10.1 Science Notebook (Answer Key)Black arab GaladimaNo ratings yet
- Chemistry - Investigation On Structure & BondingDocument8 pagesChemistry - Investigation On Structure & BondingNej SnekNo ratings yet
- Lecture 5 & 6Document45 pagesLecture 5 & 6Abdulla DoskiNo ratings yet
- Chemistry Lecture NotesDocument3 pagesChemistry Lecture Notes21-54339No ratings yet
- EXP 12 Molar Mass of A Volatile LiquidDocument8 pagesEXP 12 Molar Mass of A Volatile LiquidMau TenNo ratings yet
- (L-1) - (JEE 3.0) - States of Matter - 13th May.Document50 pages(L-1) - (JEE 3.0) - States of Matter - 13th May.Mohith VenkateshNo ratings yet
- CH 7 SlidesDocument15 pagesCH 7 SlidesMohamed SalahNo ratings yet
- BMMA4833 - Lecture 3Document23 pagesBMMA4833 - Lecture 3khairil adzhaNo ratings yet
- TD-3 - Ideal Gas LawDocument15 pagesTD-3 - Ideal Gas LawAhmed HassanNo ratings yet
- Chem Reviewer MidtermsDocument4 pagesChem Reviewer MidtermsJayrick James AriscoNo ratings yet
- Unit 1 - IntroductionDocument43 pagesUnit 1 - IntroductionIamzura Abdullah100% (1)
- Chem 145Document5 pagesChem 145Omar QasimNo ratings yet
- Module-1 ATF - IC Engine - Fundamental of IC EngineDocument50 pagesModule-1 ATF - IC Engine - Fundamental of IC EngineNandepu Sravan KumarNo ratings yet
- Poster Chapter 2.2 & 3Document1 pagePoster Chapter 2.2 & 3Riyad RahmanNo ratings yet
- Entropy: Thermodynamics: An Engineering ApproachDocument45 pagesEntropy: Thermodynamics: An Engineering ApproachAdolfo Carlos Almendares ÁlvarezNo ratings yet
- Q2 - G11 - Investigating Reaction Rates (Observing - Collision Theory - Energy Flow - The Rate Law)Document123 pagesQ2 - G11 - Investigating Reaction Rates (Observing - Collision Theory - Energy Flow - The Rate Law)ecyel.trNo ratings yet
- Exergy Analysis in Industry: Dr. Khoa Ta DangDocument55 pagesExergy Analysis in Industry: Dr. Khoa Ta DangMinh Hoàng NguyễnNo ratings yet
- Media - Kinetic Theory of GasesDocument36 pagesMedia - Kinetic Theory of GasesYumi LuvenaNo ratings yet
- Liquid Solution Thermodynamic's Properties: Kesetimbangan Fasa (TK201411)Document20 pagesLiquid Solution Thermodynamic's Properties: Kesetimbangan Fasa (TK201411)Wiwi NorzahraNo ratings yet
- Lecture 01 Fundamentals of Thermodynamics 1aDocument28 pagesLecture 01 Fundamentals of Thermodynamics 1aIsmael Torres-PizarroNo ratings yet
- TD-1 - IntroDocument21 pagesTD-1 - IntroAhmed HassanNo ratings yet
- Specific gravity-PPARLAB - EX1Document6 pagesSpecific gravity-PPARLAB - EX1Apple Angeline BalbalosaNo ratings yet
- MCL241 L6 Combustion Flame Temperature Speed TypesDocument20 pagesMCL241 L6 Combustion Flame Temperature Speed TypesAditi SamdarshiniNo ratings yet
- Introduction To Physical PharmacyDocument21 pagesIntroduction To Physical PharmacyKaren GuzmanNo ratings yet
- ESO 201A: Thermodynamics: EntropyDocument44 pagesESO 201A: Thermodynamics: EntropyamanNo ratings yet
- Spontaneity Entropy and Free EnergyDocument79 pagesSpontaneity Entropy and Free Energyserene sanctuaryNo ratings yet
- DF WorksheetsDocument45 pagesDF WorksheetstestNo ratings yet
- Mass Transfer Slides - CHE304 - Chapter 26Document51 pagesMass Transfer Slides - CHE304 - Chapter 26RehabNo ratings yet
- Moles, Density, and ConcentrationDocument12 pagesMoles, Density, and ConcentrationSterlingNo ratings yet
- CHD225 Chemical Reaction Engineering-I: Lecture-2Document17 pagesCHD225 Chemical Reaction Engineering-I: Lecture-2aayush4995No ratings yet
- Graduate Seminars on Chemical Reaction EngineeringDocument31 pagesGraduate Seminars on Chemical Reaction Engineeringmans_01No ratings yet
- Introduction to Combustion ConceptsDocument30 pagesIntroduction to Combustion Concepts84105851No ratings yet
- G11week5 6 Lectures in Gen Chem 1 Week 5 and 6Document2 pagesG11week5 6 Lectures in Gen Chem 1 Week 5 and 6FMangonon, Jarrah Mae B.No ratings yet
- Group 7 FixDocument30 pagesGroup 7 Fixyuni fitriaNo ratings yet
- MT 21006 TP Unit 3 Lec 3 and 4 06 and 09 Oct 2023 CombinedDocument19 pagesMT 21006 TP Unit 3 Lec 3 and 4 06 and 09 Oct 2023 CombinedRasika MalodeNo ratings yet
- EXP 1 - Physics Finals PDFDocument10 pagesEXP 1 - Physics Finals PDFPearl ArcamoNo ratings yet
- Physics 2 - Lecture 1Document69 pagesPhysics 2 - Lecture 1Abonin, Carl Ivan D.L.No ratings yet
- Processes and Process VariablesDocument13 pagesProcesses and Process Variablesmamat88100% (1)
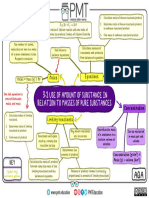
- 3.2. Use of Amount of Substance in Relation To Masses of Pure SubstancesDocument1 page3.2. Use of Amount of Substance in Relation To Masses of Pure Substancesameliarazaq12No ratings yet
- 109 41 FINAL FIZIK T4 DLP - Edited-6-35Document30 pages109 41 FINAL FIZIK T4 DLP - Edited-6-35Jasrul JamalNo ratings yet
- Download Energy Entropy And The Flow Of Nature Thomas F Sherman full chapterDocument67 pagesDownload Energy Entropy And The Flow Of Nature Thomas F Sherman full chapterdenise.sweeney729100% (5)
- Chemical Measurements: Why Do We Need To Study This Chapter?Document10 pagesChemical Measurements: Why Do We Need To Study This Chapter?SkygazerNo ratings yet
- Thermodynamics Fundamental Property RelationsDocument36 pagesThermodynamics Fundamental Property RelationsnNo ratings yet
- Chem 154 Physical Chemistry II: 1 The States of GasesDocument15 pagesChem 154 Physical Chemistry II: 1 The States of GasesPatrick BonayonNo ratings yet
- Problems in Metallurgical Thermodynamics and Kinetics: International Series on Materials Science and TechnologyFrom EverandProblems in Metallurgical Thermodynamics and Kinetics: International Series on Materials Science and TechnologyRating: 4 out of 5 stars4/5 (5)
- DO Good Have Good: Target CommunitiesDocument1 pageDO Good Have Good: Target CommunitiesUsama Jahangir KhanNo ratings yet
- New Microsoft Word DocumentDocument1 pageNew Microsoft Word DocumentUsama Jahangir KhanNo ratings yet
- Assignment Of: Thermodynamics: Group MembersDocument12 pagesAssignment Of: Thermodynamics: Group MembersUsama Jahangir KhanNo ratings yet
- 1.1 Thermodynamics: Part A: Topics (All From Reference 1)Document7 pages1.1 Thermodynamics: Part A: Topics (All From Reference 1)Usama Jahangir KhanNo ratings yet
- Ict Huss201101093Document1 pageIct Huss201101093Usama Jahangir KhanNo ratings yet
- Entropy: T DS QDocument12 pagesEntropy: T DS QUsama Jahangir KhanNo ratings yet
- Presentation About Wooden Toys-1Document30 pagesPresentation About Wooden Toys-1Usama Jahangir KhanNo ratings yet
- Bussiness Plan of Dish WashDocument2 pagesBussiness Plan of Dish WashUsama Jahangir KhanNo ratings yet
- Group#6 Presentation of Linear AlgebraDocument12 pagesGroup#6 Presentation of Linear AlgebraUsama Jahangir KhanNo ratings yet
- Bussiness PlanDocument4 pagesBussiness PlanUsama Jahangir KhanNo ratings yet
- Chen19111005 M JaishDocument20 pagesChen19111005 M JaishUsama Jahangir KhanNo ratings yet
- 1.1 Thermodynamics: Part A: Topics (All From Reference 1)Document7 pages1.1 Thermodynamics: Part A: Topics (All From Reference 1)Usama Jahangir KhanNo ratings yet
- Assignment Of: Thermodynamics: Group MembersDocument12 pagesAssignment Of: Thermodynamics: Group MembersUsama Jahangir KhanNo ratings yet
- 2020 AliExpress Black Friday Leaked Secret List (Super Discount)Document144 pages2020 AliExpress Black Friday Leaked Secret List (Super Discount)Usama Jahangir KhanNo ratings yet
- Ayesha AMBREEN Chen19111020Document6 pagesAyesha AMBREEN Chen19111020Usama Jahangir KhanNo ratings yet
- Ict Huss201101093Document1 pageIct Huss201101093Usama Jahangir KhanNo ratings yet
- Descriptive ParagraphDocument2 pagesDescriptive ParagraphUsama Jahangir KhanNo ratings yet
- Energy and Chemical Change: Chemistry: The Molecular Nature of Matter, 7EDocument47 pagesEnergy and Chemical Change: Chemistry: The Molecular Nature of Matter, 7EpopoojiNo ratings yet
- SCI1 ProbSet1Document2 pagesSCI1 ProbSet1Mikaella MonteronaNo ratings yet
- Test Bank For Chemistry Atoms First 1st Edition BurdgeDocument16 pagesTest Bank For Chemistry Atoms First 1st Edition Burdgeermintrudeletitia5lsyNo ratings yet
- Capítulo 6Document85 pagesCapítulo 6Lorena VivasNo ratings yet
- Fundamentals of Energy Conversion Engineering: August 2019Document40 pagesFundamentals of Energy Conversion Engineering: August 2019M Sohaib100% (1)
- Calculus of Thermodynamics HandoutDocument8 pagesCalculus of Thermodynamics HandoutMaria Charlene Caraos TapiaNo ratings yet
- BT8301 Iq PDFDocument21 pagesBT8301 Iq PDFPraveen yadhavNo ratings yet
- Thermodynamics Chapter 2 IntroductionDocument44 pagesThermodynamics Chapter 2 IntroductionChella YuanharNo ratings yet
- Key Thermodynamics Terms and DefinitionsDocument1 pageKey Thermodynamics Terms and DefinitionsBhushanNo ratings yet
- Thermodynamics & KineticsDocument113 pagesThermodynamics & KineticsAMMASI A SHARAN0% (1)
- Moran Safiro Solucionario Inglés (5 Edicion)Document847 pagesMoran Safiro Solucionario Inglés (5 Edicion)Raul75% (4)
- ME-131 Thermodynamics - I: BS Mechanical EngineeringDocument44 pagesME-131 Thermodynamics - I: BS Mechanical EngineeringHahshskakagaNo ratings yet
- Maxwell's Thermodynamical RelationsDocument19 pagesMaxwell's Thermodynamical RelationssNo ratings yet
- JR Chemistry - Chapter Wise Important Questions - Part 1Document21 pagesJR Chemistry - Chapter Wise Important Questions - Part 1githa80% (352)
- Chemical energetics: The branch of chemistry which deals with absorption of energy or evolution of energy in chemical reactionDocument68 pagesChemical energetics: The branch of chemistry which deals with absorption of energy or evolution of energy in chemical reactionDipu RokayaNo ratings yet
- Unit 1 FMM Session 1Document31 pagesUnit 1 FMM Session 1Palanivel Rajan A RNo ratings yet
- Principles of Engineering Thermodynamics 2nd Ed - E. M. Goodger (Macmillan, 1984) PDFDocument257 pagesPrinciples of Engineering Thermodynamics 2nd Ed - E. M. Goodger (Macmillan, 1984) PDFbikalNo ratings yet
- Applied Thermodynamics FOR Second Year / Third Semester Eee DeptDocument30 pagesApplied Thermodynamics FOR Second Year / Third Semester Eee DeptMuthuvel M100% (2)
- ME 1100 Thermodynamics Class NotesDocument88 pagesME 1100 Thermodynamics Class NotesVishalNo ratings yet
- Continuity EquationDocument9 pagesContinuity EquationEucharia100% (1)
- Test Bank For Chemistry Principles and Reactions 7th Edition William L Masterton DownloadDocument21 pagesTest Bank For Chemistry Principles and Reactions 7th Edition William L Masterton Downloadliterarysyncline63wjwpNo ratings yet
- Book 1 Thermodynamics Mechanical Engineering PDFDocument108 pagesBook 1 Thermodynamics Mechanical Engineering PDFoperationmanagerNo ratings yet
- Peter Atkins - Four Laws That Drive The Universe (2007)Document142 pagesPeter Atkins - Four Laws That Drive The Universe (2007)QuidamVigo VicusNo ratings yet
- THSTMDocument85 pagesTHSTMPatrick SibandaNo ratings yet
- DPP Class11 Chemistry Thermodynamics SolutionsDocument13 pagesDPP Class11 Chemistry Thermodynamics Solutionsajmerkaur947No ratings yet
- Fluid Mechanics With Lab Flumec1: School of Engineering and ArchitectureDocument134 pagesFluid Mechanics With Lab Flumec1: School of Engineering and ArchitectureSamantha SamanthaNo ratings yet
- Lecture 04 Oneside PDFDocument9 pagesLecture 04 Oneside PDFMrsriyansyahNo ratings yet
- Thermodynamics: Unit 6Document32 pagesThermodynamics: Unit 6rekha_1234No ratings yet
- Engineering ThermodynamicsDocument30 pagesEngineering ThermodynamicsAddisu DagneNo ratings yet
- Compressible Flow Usyd - EduDocument61 pagesCompressible Flow Usyd - EduMaueeMalicdemNo ratings yet