Professional Documents
Culture Documents
Farmakologia Mutschler
Uploaded by
Krzysiek BoguckiCopyright
Available Formats
Share this document
Did you find this document useful?
Is this content inappropriate?
Report this DocumentCopyright:
Available Formats
Farmakologia Mutschler
Uploaded by
Krzysiek BoguckiCopyright:
Available Formats
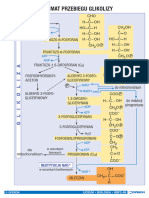
Biotransformacja 25
Tabela A 2.4-1. Drogi biotransformacji leków, reakcje I fazy
Reakcja Wzór Przykłady substratów
utlenianie
O
utlenianie alkoholi R CH2OH R
alkohol benzylowy,
R COOH
i aldehydów pirydoksyna
H
utlenianie łańcuchów barbiturany
alifatycznych OH
Farmakokinetyka
R CH3 + R COOH
R CH3
oksydacyjna H efedryna,
N-dealkilacja O lidokaina,
R1 N R1 NH2 + R2 metamfetamina
CH2R2 H
oksydacyjna histamina,
deaminacja O meskalina, A2
R NH2 R + NH3 noradrenalina
H
oksydacyjna R2 O R2 kodeina,
O-dealkilacja meskalina,
+ papaweryna
R1 O R1 H HO
utlenianie pochodne aniliny
aminy NH2 NH N O
aromatycznej OH
R R R
N-oksydacja R1
imipramina
R1 R3
+
N R3 N
-
R2 R2 O
S-oksydacja fenotiazyny
R1 R1 R1 O
S S O S
R2 R2 R2 O
desulfatacja R1 R1 tiobarbiturany,
paration
C S C O
R2 R2
R1 R3 R1 R3
P P
R2 S R2 O
epoksydacja aldryna,
R1 R2 karbamazepina
R1 R2
O
hydroksylacja R chloropromazyna,
R
substancji fenotiazyna,
aromatycznych propranolol
HO
MUTSCHLER-2009.indd Sek1:25 2010-03-18 20:07:42
Parametry farmakokinetyczne 53
stężenie w osoczu
stężenie w osoczu
Farmakokinetyka
10 20 30 40 50
godziny czas (t)
Ryc. A 2.6-12. Przebieg stężenia leku w osoczu po wielokrot- Ryc. A 2.6-13. Podwyższanie się stężenia leku w osoczu i osią-
nym podaniu doustnym leku o krótkim okresie półtrwania eli- ganie stanu równowagi stężeń przy wielokrotnym podaniu leku
minacji (t1/2 = 3 godziny) i długiej przerwie między podaniami (kumulacja).
A2
(τ = 24 godziny).
my stan równowagi (pseudo steady state). Średnie interakcje farmakokinetyczne, ustala – poprzez zmia-
stężenie Css,av wynosi: ny współczynnika oczyszczania – stężenie w stanie
stacjonarnym. Stan ten zostaje osiągnięty po mniej
AUC F·D więcej 5 okresach półtrwania.
Css,av = = (21)
τ CL · τ Na ryc. A 2.6-14 pokazano przebieg stężenia
w osoczu po czterokrotnym podaniu w ciągu doby
Z równania wynika, że – jak wspomniano wyżej dwóch różnych dawek leku z różnicą w odstępie po-
– przy wielokrotnym stosowaniu leku jego stężenie dawania w ciągu dnia i podczas nocy.
zależy od współczynnika oczyszczania.
Pominąwszy właściwości fizykochemiczne kse- W przypadku stosowania dużych dawek wieczorem
nobiotyku, wiele czynników osobniczych, takich jak przekroczone zostaje minimalne stężenie toksyczne,
choroby narządów uczestniczących w eliminacji lub natomiast przy obu sposobach dawkowania minimal-
też jednoczesne podawanie substancji wywołujących ne stężenia terapeutyczne nie zostają osiągnięte rano.
minimalne
stężenie
stężenie w osoczu
toksyczne
minimalne
stężenie
terapeutyczne
800 1400 1600 2000 000 400 800 1200 1600 2000 000 400
godzina
Ryc. A 2.6-14. Przebieg stężenia w osoczu po czterokrotnym podaniu dwóch różnych dawek leku w ciągu doby (według Rowlanda).
MUTSCHLER-2009.indd Sek1:53 2010-03-18 20:07:59
58 Farmakokinetyka w sytuacjach szczególnych
2.7. Farmakokinetyka w sytuacjach szczególnych
2.7.1. Zmiany farmakokinetyki wanych głównie w wątrobie. Na przykład w mar-
w stanach patologicznych skości wątroby metabolizm diazepamu, triamterenu
i lidokainy przebiega bardzo powoli.
Kinetyka większości leków jest zbadana głównie
u zdrowych ochotników, ponieważ dobrze nadają się Wydalanie z moczem w zależności od czynności
oni – jako względnie homogenna grupa – do ustalenia nerek. Jeśli lek jest wydalany głównie lub wy-
charakterystycznych dla danego leku lub jego posta- łącznie z moczem, czynność nerek, którą można
ci specyficznych parametrów farmakokinetycznych, ocenić za pomocą współczynnika oczyszczania
takich jak biodostępność, bądź następstw interakcji kreatyniny, wpływa na szybkość wydalania: wraz
z innymi lekami. W porównaniu z tym dane dotyczą- ze zmniejszającym się współczynnikiem oczysz-
ce farmakokinetyki u chorych są znacznie bardziej czania kreatyniny obniża się szybkość wydalania
skąpe, mimo że leki są przeznaczone do leczenia ksenobiotyków. Należy przy tym wziąć pod uwagę,
osób chorych. Dotychczas badano przede wszystkim że oprócz stanów patologicznych dla współczyn-
wpływ niewydolności krążenia oraz chorób wątroby nika oczyszczania kreatyniny istotny jest również
i nerek na farmakokinetykę leków. wiek. U niemowląt nerki nie osiągnęły jeszcze peł-
nej sprawności czynnościowej: szybkość przesą-
Zaburzenia wchłaniania. Na wchłanianie leków ma czania kłębuszkowego, która w przybliżeniu odpo-
wpływ przepływ krwi przez przewód pokarmowy. wiada współczynnikowi oczyszczania kreatyniny,
Jest on upośledzony przy zastoju w dużym krążeniu, wynosi u noworodków tylko ok. 10 ml/min (wartość
np. w niewydolności serca. Zmniejszona biodostęp- prawidłowa u dorosłych 120 ml/min). Oznacza to,
ność u chorych z niewyrównaną niewydolnością ser- że wydalanie przez nerki w pierwszych tygodniach
ca została opisana np. dla prokainamidu, chinidyny życia przebiega znacznie wolniej niż u zdrowych
i hydrochlorotiazydu. dorosłych. W wieku podeszłym współczynnik
oczyszczania kreatyniny ponownie ulega zmniej-
Zmiany dystrybucji. Zmniejszenie przepływu przez szeniu i powyżej 70 roku życia jest on często wy-
narządy obwodowe w następstwie niewydolności raźnie mniejszy. Jeśli współczynnik oczyszczania
serca może również wpływać na dystrybucję leków. kreatyniny w następstwie ciężkiej niewydolności
W takich przypadkach zmniejsza się na przykład ob- nerek jest znacznie ograniczony, w przypadku le-
jętość dystrybucji lidokainy, prokainamidu i chinidy- ków eliminowanych z moczem z powodu spowol-
ny. Utrata białek lub zaburzenia ich syntezy zmienia- nionej szybkości eliminacji należy:
ją wiązanie z białkami. Wykazano więc, że u chorych
■ zmniejszyć stosowane dawki lub
z zespołem nerczycowym związana część fenytoiny
ulega zmniejszeniu z 90 do 80%. Przy postępującej ■ zwiększyć okresy między podaniami.
niewydolności nerek możliwe jest wypieranie leków
z wiązania z białkami w następstwie retencji związ- Dostosowanie dawkowania do czynności nerek
ków podlegających wydalaniu z moczem. Ponadto jest obecnie przeprowadzane często zgodnie z wy-
obserwuje się zmiany jakościowe wiązania z białka- tycznymi podanymi przez Dettlego. Za pomocą dia-
mi wyróżniające się odmienną pojemnością i powi- gramu oznaczony zostaje współczynnik korekcji Q,
nowactwem wiązania. Dodatkowo ograniczone może przez który przemnożona zostaje zazwyczaj stosowa-
zostać wiązanie z tkankami, wskutek czego dochodzi na dawka lub przez który podzielony zostaje okres
do zmniejszenia objętości dystrybucji. między podaniami (zob. ryc. A 2.7-1). Podstawowym
W chorobach wątroby prawdopodobne jest wystę- warunkiem jest przy tym znajomość eliminowanej
powanie podobnych zaburzeń z powodu zmniejsze- ilości danej substancji Q0, którą można odczytać
nia syntezy białek. z odpowiednich tabel (zob. tab. A 2.7-1).
W nerczycach, które charakteryzują się dużą utratą
Wpływ na metabolizm. Jak opisano wyżej, elimi- białka drogą nerkową, szybkość wydalania z moczem
nacja związków lipofilnych przebiega głównie przez może ulec zwiększeniu. W takim przypadku w celu
przemiany oksydacyjne, a powstałe metabolity są na- utrzymania skuteczności stężeń w osoczu należy albo
stępnie sprzęgane, dlatego choroby wątroby mogą zwiększyć wielkość poszczególnych dawek, albo skró-
zmniejszać szybkość eliminacji leków metabolizo- cić okres między podaniami.
MUTSCHLER-2009.indd Sek1:58 2010-03-18 20:08:54
Farmakokinetyka w sytuacjach szczególnych 59
Tabela A 2.7-1. Wartości Q0 dla różnych leków
wartość kreatyniny (mg/100 ml)
8 4 3 2 1,5 1 0,75
1,0 Lek Q0
2 acetylodigoksyna 0,3
0,8 3 kwas acetylosalicylowy 1
Q'
0,6 amitryptylina 1
na
stężenie w osoczu
sy
digok ampicylina 0,12
Farmakokinetyka
0,4 1 2 karbenicylina 0,1
Q0
chloramfenikol 0,83
0,2
cymetydyna 0,25
klindamycyna 0,8
0 20 40 60 80 100
klirens kreatyniny (ml/min) digitoksyna 0,9
digoksyna 0,33
A2
fizjologiczny klirens kreatyniny
doksycyklina 0,9
Ryc. A 2.7-1. Wykres do wyznaczania współczynnika korekcji
gentamycyna 0,03
Q’, za pomocą którego można obliczyć indywidualnie dopaso-
waną do czynności nerek dawkę podtrzymującą (lub przerwy lidokaina 0,92
między podaniami) dla leków wydalanych z moczem (według
metylodigoksyna 0,5
Dettlego).
W celu wyznaczenia wartości Q’ należy odnaleźć na osi rzędnych minocyklina 0,9
po lewej stronie diagramu wartość odczytaną z tab. A 2.7-1 i od
tego punktu poprowadzić prostą 1 do prawego górnego rogu morfina 0,9
diagramu. Następnie sporządza się prostą 2, ciągnąc ją prosto- penicylina G 0,05
padle do góry z punktu na osi odciętych wyznaczonego przez
wartość klirensu kreatyniny pacjenta. Czynnik korekcji Q’ odnaj- prazosyna 0,9
duje się, prowadząc od punktu przecięcia prostych 1 i 2 prostą 3
prostopadłą do osi rzędnych po lewej stronie diagramu. rifampicyna 0,8
streptomycyna 0,04
2.7.2. Kinetyka u osób sulfatemoksazol 0,8
w podeszłym wieku trimetoprim 0,5
U osób w podeszłym wieku zazwyczaj nie występują
większe zaburzenia wchłaniania. Z powodu zmienio- (obniżanie się współczynnika oczyszczania kre-
nej czynności motorycznej żołądka szybkość wchłania- atyniny). Leki wydalane głównie drogą nerkową
nia może jednak ulegać nieznacznemu spowolnieniu. są wraz z postępującym wiekiem coraz wolniej eli-
minowane, np. okres półtrwania diacetolu, głów-
Znaczniejsze zmiany zachodzą w dystrybucji. Mogą nego metabolitu leku β-adrenolitycznego – acebu-
się one opierać na zmniejszeniu objętości całkowitej tololu, zwiększa się z 12 godz. u młodych ochotni-
wody organizmu lub zwiększeniu objętości tkanki ków do 17,5 godz. u osób w wieku powyżej 65 lat.
tłuszczowej, zmniejszonej syntezie albumin osoczo- Digoksyna jest u osób w podeszłym wieku również
wych, a także zwiększonej syntezie kwaśnej α1-gli- wolniej wydalana.
koproteiny. Wszystkie te czynniki mogą wpływać Jeśli do zależnego od wieku zmniejszenia współczyn-
na objętość dystrybucji. nika oczyszczenia kreatyniny dołączą się patologiczne
Możliwe zmiany biotransformacji u osób w pode- zmiany czynności nerek (np. w następstwie przewle-
szłym wieku opisano już w rozdziale A 2.4.7. kłego zapalenia kłębuszków nerkowych) i jeśli nie zo-
Zwracano także uwagę na zmniejszającą się czę- stanie określony odpowiedni sposób dawkowania, ry-
sto w podeszłym wieku sprawność czynności nerek zyko kumulacji substancji czynnej znacznie wzrasta.
MUTSCHLER-2009.indd Sek1:59 2010-03-18 20:08:54
74 Działanie farmakologiczne przez receptory
A agonista
A
receptor
białko we γ e białko
efektoro Gα fektoro
we kanały jonowe,
GTP β kinazy PI3,
fosfolipazy,
cyklazy adenylanowe,
kinazy receptorowe,
kinazy MAP
GTP GTP GTP GTP
Gαi Gαs Gαq Gα12,13
kanały jonowe, cyklazy fosfolipazy Rho
cyklazy adenylanowe, adenylanowe (cytoszkielet)
fosfolipazy
A
B 1
receptor
γ
α
łko e β
bia torow GDP
fe ek
4 2
γ
β
receptor
+ GTP
α
A
GDP
receptor
GDP
A
α
łko we
β
biaktoro
efe
β GTP
bia o α
efektołk
rowe γ białko we
receptor efektoro
3 A
Ryc. A 3.1-11. A – różnorodność dróg transdukcji sygnału szlakiem receptorów związanych z białkiem G. Aktywacja receptora zwią-
zanego z białkiem G przez agonistę powoduje dysocjację heterotrimerycznego białka G do podjednostek Gα i Gβ po wymianie
związanego do podjednostki α GDP na GTP. Receptory związane z białkiem G asocjują najczęściej z określoną podjednostką Gα (z
których 4 najważniejsze przedstawiono na rycinie). Poszczególne podjednostki Gα aktywują lub hamują różne systemy efektorowe,
m.in. kanały jonowe, fosfolipazy, cyklazy adenylanowe i białka wiążące GTP, np. Rho, które reguluje strukturę cytoszkieletu. Kinazy
PI3 – kinazy 3-fosfatydyloinozytolowe; Pi – fosforan nieorganiczny.
B – cykl aktywacji i dezaktywacji heterotrimerycznego białka G. Po związaniu agonistów do receptora 7-transbłonowego (1) dzię-
ki wymianie GDP na GTP (2) dochodzi do dysocjacji białka G (3), co powoduje aktywację białek efektorowych, które pośredniczą
w wysyłaniu odpowiedzi komórkowej (wazokonstrykcja, sekrecja, namnażanie komórek). Dzięki wewnętrznej aktywności GTP-azy
podjednostka Gα powoduje rozszczepienie GTP na GDP (4). Białko G asocjuje z powrotem do heterotrimeru, agonista dyfunduje
od receptora i przechodzi ponownie do stanu nieaktywnego.
MUTSCHLER-2009.indd Sek1:74 2010-03-18 20:09:08
Podstawy anatomiczne i fizjologiczne 705
7. Nerki i drogi moczowe,
gospodarka wodno-elektrolitowa
i kwasowo-zasadowa
7.1. Podstawy anatomiczne i fizjologiczne
Nerki pełnią podstawową funkcję w utrzymaniu ho- węglanów (diwęglanów), a tym samym utrzymanie
meostazy całego organizmu, biorą bowiem udział fizjologicznej wartości pH (izohydrii),
w takich procesach, jak:
■ (długotrwała) regulacja ciśnienia krwi poprzez
■ wydzielanie produktów przemiany materii (m.in. układ renina-angiotensyna-aldosteron,
związków znajdujących się prawidłowo w moczu,
■ odtwarzanie białka osoczowego (np. albumin, lizo-
np. mocznika, kwasu moczowego i kreatyny), a tak-
zymu, białka wiążącego retinol) i hormonów pep-
że substancji obcych (ksenobiotyków) i ich metabo-
tydowych (np. insuliny, parathormonu).
litów; jednocześnie odpowiadają za zatrzymywanie
komórek krwi i białek osoczowych oraz odzyski-
Nerki syntetyzują ponadto ważne hormony (kalcy-
wanie z moczu w wyniku resorpcji kanalikowej
triol, erytropoetynę) oraz są narządem wykonaw-
ważnych substratów (np. glukozy, aminokwasów),
czym hormonów pozanerkowych (np. wazopresyny,
■ kontrola gospodarki wodno-elektrolitowej po- aldosteronu, kalcytoniny, peptydów natriuretycz-
przez regulowane wydzielanie wody i elektrolitów nych, parathormonu). Może w nich także zachodzić
(np. jonów Na+, K+, Ca2+, Mg2+, Cl– i fosforanów), glukoneogeneza.
a tym samym utrzymywanie stałej objętości pły-
nu zewnątrzkomórkowego (izowolemii), stężenia
osmotycznego (izotonii) i równowagi jonowej (izo- 7.1.1. Anatomia makroskopowa
jonii),
nerki
■ regulowanie gospodarki kwasowo-zasadowej po-
przez wydzielanie protonów bądź retencję wodoro-
Nerki są narządem parzystym, przypominającym
kształtem fasolę, położonym w grzbietowej części
jamy brzusznej poniżej przepony i po obu stronach
kręgosłupa. Brzeg wypukły skierowany jest bocznie,
warstwa na wklęsłym brzegu dośrodkowym znajduje się wnę-
warstwa rdzeniowa
korowa nerki (kora) nerki (rdzeń) ka nerki (hilus), z której odchodzą naczynia, nerwy
torebka brodawka
i moczowód. Nerki i drogi moczowe
włóknista nerkowa U osoby dorosłej długość przekroju podłużnego
miedniczka nerki wynosi 10–12 cm, długość przekroju poprzecz-
nerkowa nego 5–7 cm, a masa 100–200 g.
tętnica
nerkowa Powierzchnię nerki chroni mocna torebka włókni-
kielich
nerkowy żyła sta. W obrazie mikroskopowym przekroju podłużne-
nerkowa go (zob. ryc. B 7.1-1) wyróżnia się:
tkanka moczowód
tłuszczowa ■ korę nerki (cortex renalis),
■ rdzeń nerki (medulla renalis), B7
■ miedniczkę nerkową (pelvis renalis) z moczowo-
dem.
słupy nerkowe Zewnętrzna warstwa korowa nerki jest jasna i drobno-
ziarnista, wewnętrzna warstwa rdzeniowa jest ciem-
Ryc. B 7.1-1. Przekrój podłużny nerki. niejsza i podłużnie prążkowana. Wnikając w rdzeń,
MUTSCHLER-2009.indd Sek1:705 2010-03-18 20:11:00
706 Podstawy anatomiczne i fizjologiczne
kora tworzy słupy nerkowe dzielące rdzeń na 8–17
piramid, które podstawą przylegają do kory, a wierz- część kręta kłębuszek nerkowy tętniczka doprowadzająca
chołkami zbiegają się w centrum nerki. Wierzcho- tętniczka odprowadzająca
łek każdej piramidy, nazywany brodawką nerkową,
uchodzi do woreczkowatego kielicha nerkowego.
część
Wychwytują one gotowy mocz (mocz ostateczny) kręta
i odprowadzają go do zbiornika w miedniczce ner- kanalik
kowej. proksymalny
(kręty)
Zaopatrywanie nerki w krew następuje przez odcho- część prosta
dzące od aorty tętnice nerkowe (arteria renali), które
po dotarciu do wnęki rozgałęziają się na tętnice mię- kanalik
dzypłatowe (arteriae interlobares). Te między pira- dystalny
midami biegną ku korze nerki i u podstawy piramid pętla Henlego
tworzą tętnice łukowate (arteriae arcuatae), od któ-
rych odchodzą tętnice międzypłacikowe (arteriae kanalik zbiorczy
interlobulares). Od tych ostatnich, w regularnych
odstępach, odgałęziają się zaopatrujące poszczególne
ciałka nerkowe (zob. poniżej) tętniczki zaopatrują- odcinek
przejściowy
ce (vasa afferentia), które najpierw tworzą (pierw-
szą) sieć kapilarną, a następnie łączą się ponownie
w naczynia odprowadzające (vasa efferentia). W tym
miejscu powstaje drugi, okołokanalikowy system ka- Ryc. B 7.1-2. Schemat budowy nefronu.
pilarny, którego zadaniem jest zaopatrywanie w krew
kanalików i kanalika zbiorczego (zob. poniżej).
Układ żylny zbudowany analogicznie do układu Ściany kapilar kłębuszków nerkowych zbudowane są
tętniczego odprowadza krew żylną do żyły nerkowej z dwóch warstw: śródbłonka składającego się z komórek
(vena renalis). posiadających liczne „okienka” oraz błony podstawnej,
w której zgromadzone są ujemnie naładowane glikozami-
noglikany (tzw. bariera anionowa). Na zewnątrz od błony
podstawnej odchodzą natomiast wypustki stopowate tzw.
podocytów, tj. komórek nabłonkowych torebki Bowmana.
7.1.2. Anatomia mikroskopowa nerki Wypustki te tworzą wokół kapilar warstwę z licznymi szpa-
Podstawowym elementem strukturalnym nerek
jest nefron (zob. ryc. B 7.1-2). Każda nerka ludzka
zawiera około 1 miliona tych jednostek morfologicz-
nych i funkcjonalnych. Nefron składa się z: kanalik dystalny
komórki plamki
■ ciałek nerkowych, gęstej
komórki tętniczka
■ aparatu kanalikowego. nabłonkowe odprowa-
dzająca
Ciałko nerkowe. Ciałko nerkowe (zob. ryc. B 7.1-3) tętniczka
doprowa-
składa się z kłębka naczyń włosowatych i otaczającej dzająca
go torebki Bowmana. Blaszka wewnętrzna torebki
Bowmana okrywa kłębuszek nerkowy, blaszka ze-
wnętrzna okrywa torebkę od zewnątrz i przechodzi
w ścianę kanalika proksymalnego. Jak wspomniano,
torebka
krew tętnicza dostaje się do kłębuszka nerkowego naczynia włosowate Bowmana
naczyniami doprowadzającymi (vasa afferentia), kłębka nerkowego
a opuszcza go przez naczynia odprowadzające (vasa kanalik proksymalny
efferentia). Obydwa położone blisko siebie naczynia (kręty)
tworzą biegun naczyniowy ciałka nerkowego, na-
przeciw którego, na początku aparatu kanalikowego, Ryc. B 7.1-3. Ciałko nerkowe z komórkami plamki gęstej (macula
znajduje się biegun moczowy. densa) (według Kristicia). Tętniczka doprowadzająca jest otwarta.
MUTSCHLER-2009.indd Sek1:706 2010-03-18 20:11:01
Podstawy anatomiczne i fizjologiczne 707
rami filtracyjnymi i stanowią ważny element błony filtra- łęzienia naczyń doprowadzających do drugiej sieci
cyjnej naczynia włosowatego kłębuszka nerkowego. Cien- kapilarnej, która oplata aparat kanalikowy znajdujący
ka silnie ujemnie naładowana błonka rozciągnięta między
się w korze i zewnętrznej strefie rdzenia nerki. We-
sąsiadującymi ze sobą wypustkami stopowatymi zamyka
„okienka” w śródbłonku. Torebkę Bowmana od światła wnętrzna warstwa rdzenia jest natomiast zaopatrywa-
kapilary oddzielają więc trzy warstwy, tworzące wspólnie na przez długie odcinki naczyń prostych (vasa recti),
tzw. barierę filtracyjną o łącznej powierzchni około 1 m2. które odchodzą od tętnic łukowatych (arteriae arcua-
Między poszczególnymi kapilarami, a w większej ilości tae) i naczyń doprowadzających najniżej położonych
przy biegunie naczyniowym, zgromadzone są komórki
kłębuszków nerkowych.
mezangialne tworzące dla kłębuszka rodzaj rusztowania.
Dzięki zdolności do fagocytozy mogą one usuwać złogi
gromadzące się w obszarze filtracyjnym, a zawartość akty-
ny i miozyny decyduje o ich kurczliwości. 7.1.3. Ukrwienie nerek
Wszystkie ciałka nerkowe położone są w obszarze
Łączna ilość krwi przepływającej przez obie nerki do-
warstwy korowej, przy czym te leżące bliżej po-
rosłego człowieka wynosi 1,2 l/min lub 1700 1/dzień,
wierzchni mają kłębuszki korowe, a te powyżej gra-
co odpowiada 20–25% pojemności minutowej ser-
nicy korowo-rdzeniowej kłębuszki przyrdzeniowe.
ca w pozycji spoczynkowej. Na korę nerki przypada
około 90% tej całości, na rdzeń zaledwie 10%. Nerki
Aparat kanalikowy. W przypadku aparatu kanaliko-
należą tym samym do najlepiej ukrwionych narzą-
wego istotny jest stosunek niezwykle długiego ukła-
dów, co jest konieczne do uzyskania wystarczającej
du kanalikowego do jego przekroju. W układzie ka-
ilości przesączu kłębuszkowego oraz stanowi waru-
nalikowym rozróżnia się następujące odcinki:
nek efektywnie pełnionej funkcji oczyszczającej.
■ kanaliki proksymalne z częścią krętą (pars convo- Pomiary przepływu naczyniowego u ludzi prze-
luta s. contorta) i częścią prostą (pars recta), prowadza się przez pośrednie oznaczenie tzw. kwasu
paraaminohippurowego (klirens PAH). PAH jest sub-
■ odcinek przejściowy (część cienka pętli Henlego),
stancją, która po jednorazowym przejściu przez nerkę
■ kanaliki dystalne z częścią krętą (pars convoluta jest prawie całkowicie eliminowana z krwi. Dlatego
s. contorta) i częścią prostą (pars recta), wartość klirensu (CLPAH) jest identyczna z przepły-
wem nerkowym osocza (RPF):
■ kanalik zbiorczy.
CLPAH = RPF
Część prosta kanalika proksymalnego i dystalnego
oraz odcinek przejściowy określane są wspólnie jako
pętla Henlego. Kręte odcinki kanalików leżą w części
korowej nerki, odcinki proste przeważnie bliżej czę- 7.1.4. Przesączanie kłębuszkowe,
ści rdzeniowej. powstawanie moczu
Na niewielkim odcinku kanalik dystalny styka pierwotnego
się z tętniczką doprowadzającą (vas efferens) oraz
z połączonym z nim ciałkiem nerkowym. Obszar na- Z osocza przepływającej krwi odsącza się w kłębusz-
błonka w miejscu zetknięcia się zawiera wiele jąder kach nerkowych prawie bezbiałkowy ultrafiltrat, Nerki i drogi moczowe
i nazywany jest plamką gęstą (macula densa) (zob. mocz pierwotny. Możliwość przejścia cząsteczek
ryc. B 7.1-3). Komórki leżące w obrębie plamki wraz z naczyń włosowatych do wnętrza torebki Bowmana
z komórkami sąsiadującymi z naczyniem doprowa- zmniejsza się stopniowo, począwszy od cząsteczek
dzającym tworzą aparat przykłębuszkowy. wielkości 1,5 nm. Przy cząsteczkach większych niż
Po kontakcie z ciałkiem nerkowym część krę- 4 nm przepuszczalność zanika całkowicie (tzw. filtr
ta (pars convoluta) kanalika dystalnego przechodzi kłębuszkowy). Ograniczona filtracja dotyczy rów-
w kanalik zbiorczy, który zbiera i odprowadza mocz nież cząsteczek o masie od 5000 Da do 50 000 Da.
z brodawek nerkowych. Każdy kanalik zbiorczy Przez nieuszkodzony filtr zatrzymywane są prawie B7
ma liczne dopływy z sąsiednich nefronów. Kanaliki całkowicie także najmniejsze białka osocza, albu-
zbiorcze łączą się pod ostrym kątem w większe ka- miny, ponieważ ich masa cząsteczkowa przekracza
naliki, które w wierzchołku piramidy, brodawce ner- 70 000 Da.
kowej, uchodzą do kielicha nerkowego. Przeciętna Filtracja kłębuszkowa zależy jednak nie tyl-
długość kanalika zbiorczego wynosi 22 mm. ko od szerokości szczelin filtracyjnych, ale przede
Zarówno kanaliki proksymalne i dystalne, jak i ka- wszystkim od efektywnego ciśnienia filtracyjnego
nalik zbiorczy są zaopatrywane w krew przez odga- (Peff) panującego w kłębuszkach nerkowych oraz
MUTSCHLER-2009.indd Sek1:707 2010-03-18 20:11:01
Inhibitory topoizomerazy 967
12.3. Inhibitory topoizomerazy
Istotnym warunkiem zahamowania podziału komórki obie nici DNA, przerywa je tymczasowo i umożliwia
(mitozy) jest umieszczenie inhibitora w DNA jądra rotację opisaną na ryc. B 12.3-1. W końcu miejsca
komórkowego. Podczas mitozy istnieje możliwość przerwania zostają zamknięte i enzym oddysocjowu-
syntezy DNA na nowo bez dużego nakładu energii je od DNA.
za pośrednictwem enzymów, które mogą przejściowo Hamowanie aktywności enzymu przez cytostatyki
przerywać nici DNA, aby następnie po skutecznej re- (zwłaszcza blokada jego dysocjacji od DNA) prowa-
plikacji ponownie je zespolić. Enzymy te, tak zwane dzi do tego, że ustabilizowany kompleks składający
topoizomerazy, są uważane za odwracalne nukleazy, się z topoizomerazy i DNA powoduje trwałe prze-
które wiążą się kowalencyjnie z fosforową grupą rwanie nici i związaną z tym śmierć komórki. W wie-
DNA, rozszczepiają wiązanie estrowe i w końcu po- lu nowotworach aktywność topoizomeraz jest zwięk-
nownie je łączą (zob. ryc. B 12.3-1). szona, dlatego inhibitory topoizomerazy wykazują
Rozróżnia się dwie postacie: topoizomeraza I pewną wybiórczość w odniesieniu do określonego
przerywa tylko jedną nić podwójnej helisy DNA, rodzaju nowotworów. Negatywnym zjawiskiem jest
co umożliwia wolną rotację przeciwnej nici i powsta- fakt, że inhibitory, zwłaszcza topoizomerazy II, sprzy-
nie wiązania fosfodiestrowego stanowiące warunek jają rozwojowi wtórnych nowotworów i wtórnych
replikacji. Topoizomeraza II wiąże kowalencyjnie białaczek.
nowotworów złośliwych
A B
Chemioterapia
topoizomeraza I (rozszczepienie jednej nici)
umożliwia wolną rotację i powstanie
3' grup estrów kwasu fosforowego
5'
B 12
topoizomeraza II (rozszczepienie podwójnej nici)
3'
5'
polimeraza DNA
3'
nowo zsyntetyzowany DNA
5'
Ryc. B 12.3-1. Zasada działania topoizomeraz. (A) W przypadku replikacji przez polimerazę DNA konieczna byłaby rotacja zrepli-
kowanego DNA z nakładem energetycznym w kierunku wskazanym przez strzałkę. (B) Dlatego korzystniejsze jest rozszczepienie
przez topoizomerazy.
MUTSCHLER-2009.indd Sek1:967 2010-03-18 20:09:59
968 Inhibitory topoizomerazy
12.3.1. Inhibitory topoizomerazy I rami i induktorami CYP3A. Około 22% dawki jest eli-
minowane przez nerki w postaci niezmienionej. Okres
półtrwania jest zależny od statusu metabolicznego.
Topotekan jest pochodną alkaloidu kamptotecyny.
U pacjentów bez zaburzeń metabolicznych wynosi
Jego okres półtrwania wynosi 2–3 godziny. Związek
około 6 godzin.
jest wydalany z moczem jako niezmieniony lub
Irinotekan okazał się skuteczny u pacjentów z za-
po przekształceniu pierścienia laktonowego w pro-
awansowanym rakiem okrężnicy i odbytnicy w połą-
cesie zależnej od pH hydrolizy. Interakcje tego leku
czeniu z fluorouracylem lub w monoterapii u chorych
na etapie biotransformacji nie są znane.
niereagujących na fluorouracyl.
Lek jest dawkowany w ilości 1,5 mg/m2 p.c. dzien-
Dawkowanie wynosi 125 mg/m2 w ciągu 90 minut
nie dożylnie we wlewie trwającym 30 minut przez
1 × tydzień przez 4 tygodnie. Charakterystycznym
5 kolejnych dni w cyklach co 3 tygodnie. Najczęściej
działaniem niepożądanym irinotekanu jest zależna
stosuje się 4 cykle terapeutyczne.
od dawki biegunka pojawiająca się z opóźnieniem
Topotekan jest przeznaczony do leczenia pacjen-
(u 20% chorych po pięciu dniach). Oprócz tego opi-
tek z przerzutowym rakiem jajnika po niepowodzeniu
sywano wystąpienie ostrego zespołu cholinergiczne-
pierwotnej lub kolejnej terapii. Prowadzone są pró-
go charakteryzującego się biegunką, bólami brzucha,
by stosowania tej substancji w postępującym raku
zapaleniem spojówek, nieżytem nosa, potliwością,
oskrzela. Najpoważniejszym działaniem niepożąda-
miozą, łzawieniem i zwiększonym napływem śliny.
nym jest zapalenie błony śluzowej jelita.
Wymienione objawy pojawiają się w ciągu pierwszych
24 godzin i można je wyleczyć za pomocą atropiny.
Irinotekan jest kolejną pochodną kamptotecyny.
Jest on przekształcany przez karboksyesterazy do me-
tabolitów SN-38, których działanie hamujące aktyw-
ność topoizomerazy I jest silniejsze od działania sub- 12.3.2. Inhibitory topoizomerazy II
stancji macierzystej. SN-38 ulegają następnie gluku-
ronizacji za pomocą glukuronozylotransferazy UGT Etopozyd jest częściowo syntetyczną podstawioną
1A1. U pacjentów z genetycznym uszkodzeniem ak- pochodną podofilotoksyny (sama podofilotoksyna
tywności tego enzymu toksyczność irinotekanu może jest związkiem hamującym mitozę, a nie inhibito-
być zwiększona. Jest to szczególnie niebezpieczne rem topoizomerazy i stosowana jest w miejscowym
u osób z jednoczesnym defektem dehydrogenazy leczeniu kłykcin kończystych). Etopozyd ulega de-
dihydropirymidyny, odpowiedzialnej za metabolizm metylacji w pozycji 3′ przez enzym CYP3A4. Okres
fluorouracylu, w przypadku jednoczesnego stosowa- półtrwania wynosi 8 godzin. Oprócz etopozydu
nia irinotekanu i fluorouracylu. Poza tym następuje w handlu jest dostępny również fosforan etopozydu,
oksydacyjny rozkład poprzez otwarcie zewnętrznego który jest biotransformowany do etopozydu za po-
pierścienia piperydynowego przez enzymy z rodziny mocą fosfataz zasadowych. Należy unikać jedno-
CYP3A. Dlatego należy wziąć pod uwagę możliwość czesnego podawania fosforanu etopozydu i inhibi-
wystąpienia interakcji z innymi substratami, inhibito- torów fosfataz.
R1 R2 R3
R3 kamptotecyna H H H
N HO C2H5
O CH3
R2 N
N O topotekan H OH
CH2 CH3
R1
O O
O N
irinotekan C2H5 H
N
SN-38 C2H5 H OH
MUTSCHLER-2009.indd Sek1:968 2010-03-18 20:09:59
Inhibitory mitozy 969
Etopozyd (albo difosforan etopozydu) jest stoso-
wany w wielu chorobach nowotworowych (np. raku
OH
jądra, raku oskrzela, w raku drobnokomórkowym H
H3C O OH
płuca, w ostrej białaczce monocytowej i mielomono-
cytowej, w chłoniakach). O
O O
Dawkowanie wynosi 100–120 mg/m2 p.c./dzień H H
dożylnie w powolnym wlewie (zbyt szybkie poda- O
O
wanie może wywołać spadek tętniczego ciśnienia O
krwi) przez 3–5 kolejnych dni. Do działań niepożą- O H
danych leku, oprócz hipotensji, należy supresja szpi-
ku. Cyklosporyna prowadzi do zwiększenia stężenia
H3CO OCH3
etopozydu w osoczu.
OH
Inhibitory obu topoizomeraz działają synergistycz-
nie między sobą lub łącznie z lekami alkilującymi,
zwłaszcza pochodnymi platyny, i dlatego są stosowa- etopozyd
ne w terapii skojarzonej w wielu chorobach nowo-
tworowych.
nowotworów złośliwych
Chemioterapia
12.4. Inhibitory mitozy
Hamowanie cyklu komórkowego jest możliwe po- uzyskane windezyna i winorelbina, hamują podział
przez blokadę mitozy. Dochodzi do tego w wyniku komórek na etapie metafazy. Alkaloidy barwinka blo-
uszkodzenia budowy aparatu mitotycznego, któ- kują również syntezę DNA i RNA.
B 12
ry jest niezbędny do tzw. wędrówki chromosomów Mimo bliskiego pokrewieństwa chemicznego sub-
do przeciwległych biegunów komórki w procesie mi- stancje te wykazują znaczne różnice w zakresie dzia-
tozy. Leki mogą: łania, farmakokinetyki i toksyczności.
Biologiczny okres półtrwania winblastyny i win-
■ hamować budowę wrzeciona kariokinetycznego
dezyny wynosi około 24 godz., winkrystyny 85 godz.,
(np. kolchicyna, alkaloidy barwinka i ich pochodne)
winorelbiny 40 godz. Siarczan winblastyny ulega
lub
deacylacji w wątrobie, a winkrystyna jest w 80%
■ blokować mikrotubule wrzeciona kariokinetycznego. eliminowana z żółcią. Biodostępność winorelbi-
ny wynosi 40% i jest ona metabolizowana przez
Punktem uchwytu w obu przypadkach jest podgrupa
CYP3A4.
β-tubuliny, jednakże miejsca wiązania wymienionych
Głównymi wskazaniami do stosowania winblastyny
leków są różne.
są ziarnica złośliwa i inne złośliwe nowotwory układu
Kolchicyna. Najdłużej znaną substancją hamującą mitozę chłonnego, ponadto mięsak Kaposiego towarzyszący
jest alkaloid zimowita jesiennego kolchicyna, która w proce- AIDS. Bardziej toksyczna winkrystyna jest stoso-
sie podziału jądra w stadium metafazy uniemożliwia tworze- wana zwłaszcza w ostrej białaczce limfoblastycznej
nie wrzeciona podziałowego, co powoduje powstanie jąder i szpikowej, w zaostrzeniu blastycznym przewlekłej
poliploidalnych. Jej mały zakres terapeutyczny wyklucza białaczki szpikowej, w chłoniakach, czerniaku złośli-
jednak zastosowanie tej substancji jako cytostatyku.
wym, raku piersi, w nowotworach OUN. Windezyna
jest stosowana w ostrej białaczce limfoblastycznej
i szpikowej, zaostrzeniu blastycznym przewlekłej
12.4.1. Alkaloidy barwinka (Vinca) białaczki szpikowej, czerniaku złośliwym, raku płu-
ca. Winorelbina znalazła zastosowanie w niedrobno-
Winblastyna i winkrystyna – dwa alkaloidy z Catha- komórkowym raku płuca, w raku piersi z przerzutami
ranthus roseus, podobnie jak częściowo syntetycznie albo innych narządów.
MUTSCHLER-2009.indd Sek1:969 2010-03-18 20:10:00
1149
Skorowidz
– działanie 658 adrenokortykotropowy hormon 397
A – interakcje 658 adrenoreceptory 65, 355
– jako odtrutka 1122 – agoniści 356
ABC transportery 12, 40–42 – wskazania 659 – antagoniści 356
– jelito 40 acetylokoenzym A 556 – blokery 356
– miejsca ekspresji 41 N-acetylotransferaza 115 – hamowanie 69
– nerki 41 ACTH zob. adrenokortykotropowy – kinetyka 357
– substraty 40 hormon – presynaptyczne 355
– zasada działania 41 Actinomycetes 971 – profil działania 357, 358
abciksimab 537 acyklowir 862–863 adsorbenty 1056
– działania niepożądane 538 – ciąża 102 adynamia 408, 416, 451, 1133
– powinowactwo do receptorów 537 – dawkowanie 912 aerozole 7, 16, 284
– specyficzność 538 – działania niepożądane 912 Aesculus hippocastanum 594
ACAT-2 555 – działanie 909 agonista 65–68
acebutolol 368–371 – interakcje 912 – kompetycyjny 67
– profil działania 369 – kinetyka 909 – niekompetycyjny 67
ACE inhibitory 605 – wskazania 757, 912 – pełny i częściowy 67
– dawkowanie 574, 606 acytretyna 767, 771–773 – receptorów adrenergicznych 356
– działania niepożądane 574 adenina 909, 959 – receptorów dopaminergicznych 328–
– fetopatie 103 adenozylometionina 29, 30 330, 389
– hiperkaliemia 606 adenozyna 622 ajmalina 317, 618–620
– interakcje 575, 606 – dezaminacja 622 akamprosat 1074
– kaszel 510 – działania niepożądane 622 akarboza 436, 443
– mechanizm działania 574 – indukcja snu 209 akonityna 1045, 1081
– mieszanie 581 – interakcje 622 – mechanizm działania 1081
– niewydolność serca 606 – przeciwwskazania 622 – występowanie 1081
– odma angioneurotyczna 603 – receptory 71, 651 – zatrucie 1081
– przeciwwskazania 574 – szybka biotransformacja – – leczenie 1082
aceklofenak 258–259 – wychwyt przez erytrocyty 622 – – objawy 1081
acemetacyna 254–255 – wzmożona produkcja 709 akroleina 961, 963, 1107
acetaldehyd 810 adenozynotrifosforan zob. ATP akseroftol zob. witamina A
acetanilid 93 adenozyny deaminaza 118, 958, 959 aktynomycyny 971–973
acetazolamid 737, 753 adenowirusy 118, 697, 905 – daktynomycyna 971
aceton ADH zob. wazopresyna – dawka 971
– w moczu 444 adiuretyna zob. wazopresyna – działania niepożądane 971
– zapach 431 adrenalina 176, 184, 293 aktywator plazminogenu 531, 532,
acetylacja 33, 115 – alergia 294 546–547
acetylator 115, 579 – dawkowanie 293 – alteplaza 547
acetylocholina 311–314, 353 – działania niepożądane 360 – reteplaza 547
– miejsce wiązania 75 – działanie 293, 359 – tenekteplaza 547
– rozkład 312 – interakcje 341 aktywność wewnętrzna 3, 67, 85
– siła działania 85 – glikoliza 359 aktywowany węgiel zob. carbo medi-
– uwalnianie 311 – lipoliza 359 cinalis
– wpływ na błony 75 – przeciwwskazania 360 ALA zob. δ-aminolewulinowy kwas
– zatrucie 180 – receptory 356–357 ALAD zob. dehydrataza ALA
acetylocholinoesteraza 365 – rozkład 355–356 alanina 836–839, 1009
– inhibitory 37, 200 – synteza 353–354 ALAS zob. syntaza ALA
– reaktywacja 1052 – uwalnianie 353 albendazol 112, 941–942
acetylocysteina 635–637 – wskazania 359 albumina 21, 60, 329, 525
– dawkowanie 1122 – wychwyt zwrotny 355 – filtracja 707
– działania niepożądane 658 – zatrucie 293 – spadek stężenia 21
MUTSCHLER-2009.indd Sek1:1149 2010-03-18 20:12:19
1150 Skorowidz
– wzrost stężenia 21 – działanie teratogenne 99 – przeciwwskazania 898
albuminuria 822, 1118 – uzależnienie 104 amid kwasu nikotynowego zob. wita-
aldehyd glutarowy 818, 822 alkuronium 315, 317 mina PP
– działanie 822 allele 114–116 amifostyna 965
aldesleukina 986 alletryna 786 amikacyna 830, 857
– dawkowanie 1009 Allium cepa 139 amilorid 661
– działania niepożądane 1009 allodynia 218, 219 – interakcje 126, 611
– działanie 1009 alloksantyna 288 aminofenazon 112, 260
aldoketoreduktazy 978 allopregnandiol 469, 470 aminoglikozydy 56, 102, 856
aldofosfamid 961, 962 allopurinol 270–271, 959 – dawkowanie 857–858
aldosteron 305, 446–452 – biodostępność 44 – działania niepożądane 858
– biosynteza 446 – ciąża 102 – interakcje 859
– działanie 447 – dawkowanie 288 – kinetyka 858
– hiperaldosteronizm 451 – działania niepożądane 95, 288 – mechanizm działania 856
– uwalnianie 449 – interakcje 288, 546, 1019 – wskazania 856
– znaczenie 449 – mechanizm działania 288 aminoglutetymid 450
aldosteronu antagoniści 567, 603, 606 – reakcje skórne 97 aminokwasy 10, 518
– dawkowanie 606 allosteryczne działanie 69, 150, 320, – dezaminacja 493, 810
– eplerenon 606 922 – piroliza 1103
– spironolakton 606 almotriptan 266 – resorpcja 671
– wskazania 606 – kinetyka 267 – transporter 10, 912
aldryna 25, 1093 aloesu emodyna 691 δ-aminolewulinowy kwas 1060
alemtuzumab 983, 1021 alopecja 954 amiodaron 46, 621
alendronian 418 alprazolam 27, 190, 193 – dawkowanie 621
– dawkowanie 419, 420 alprenolol 27, 35, 369 – działania niepożądane 621
alergeny 92, 94 alprostadil 505 – interakcje 546, 621
– blokada 640 – dawkowanie 506 amisulprid 164, 165, 173
– ekstrakty 777 – wskazania 506 amitryptylina 177–180
– iniekcja 645 alteplaza 547, 549 – dawkowanie 180
– karencja 645 Alzheimera choroba 199, 200, 335, – działania niepożądane 178
– kontakt 96 592, 1035 – przeciwwskazania 180
– mechanizm działania 494 Amanita muscaria 1089 – substrat CYP 27
– odczulanie 645 – pantherina 1089 amlodypina 570
– uwrażliwienie 640 – phalloides 1087 – substrat CYP3A4 27
alergia 92 – verna 1087 amoidyna 769
– celiakia 96 – virosa 1087 amnezja 323, 1133
– definicja 92 amanityna 1108 amoksycylina 93, 680, 844, 846
– krzyżowa 91, 94 – mechanizm działania 1087 amoniak 702, 980, 1023, 1083, 1108
– predyspozycje 92 amantadyna 102, 338, 342–345 amorolfina 898
alergiczna astma oskrzelowa 95 – dawkowanie 345 cAMP 14, 73
– reakcja 92 – działania niepożądane 342 AMPA 223
– – czynniki ryzyka 98 – przeciwwskazania 345 ampicylina 842, 843
– – patomechanizm 95 amatoksyna 1087 amprenawir 923–925
– – typy 95 ambroksol 658 amrinon 115
– – unikanie 98 – dawkowanie 659 amsakryna 971, 973
alfa-4-integryna 1022 – działania niepożądane 658 amyloid 199, 431, 1133
alfakalcydol 422, 803 – działanie 658 amyloidoza 1133
alfentanyl 241, 303, 307–309 amebiaza 456, 859, 876 amylopektyna 528, 671
– CYP3A4 27 ameby 929 amyloza 671
– maksymalne działanie 307 amezynium 364, 586 anafilaksja 95
– właściwości 308 amfebutamon 1084 anakinra 280, 281
alfuzosyna 367, 740 amfetamina 104, – dawkowanie 281
alginowy kwas 677 – dawkowanie 197 – działania niepożądane 281
alkaloidy barwinka 13, 954, 955 – działania niepożądane 184, 197 – wskazania 281
– sporyszu 102–104, 364–366, 491, – przeciwwskazania 197 analeptyki 216
503 amfoterycyna B 102, 107, 317, 897 – doksapram 216
alkohol 99 – dawka 898 analgetyki 217
– ciąża 102 – interakcje 611 analgezja 229, 232, 297, 1133
MUTSCHLER-2009.indd Sek1:1150 2010-03-18 20:12:19
Skorowidz 1151
anandamid 203 – – 5-HT3 350 atowakwon 112, 902, 930–933, 936
anastomoza 1133 – – leukotrienowego 649–650 ATP 12, 219
anastrozol 977 – – muskarynowego 351, 652–653 atrakurium 316, 317
Ancylostoma duodenale 940 – – NK1 350 atropina 381, 383, 755
androgeny 391 – – opioidowego 692 – dawkowanie 381
– ciąża 102 – – progesteronu 479 – kinetyka 382
– działanie 465 – wapnia 268–269, 569–573 – zatrucia 383
– transport 389 – witaminy K 543–546 AUC 43
– w lecznictwie 465 antagonizm chemiczny 69–70 Auerbacha splot 160
– znaczenie fizjologiczne 449 – fizjologiczny 69 AUMC 43
androstandion 464 – funkcjonalny 69 azatiopryna 277, 282, 1018–1019
androstanolon 464 antracykliny 971–973 – dawkowanie 1018
androstendion 464, 469, 977 antropozofia 138 – działania niepożądane 1019
anemia 519, 921, 1133 antyandrogeny 102, 104, 467, 978– – przeciwwskazania 1019
– aplastyczna 332 979 azelastyna 495, 758
– hemolityczna 374 antyarytmiki 619–620 azotany 612, 626–628
anergia 92 antybakteryjne leki 661 – dawkowanie 628
anestetyk dożylny 303–309 antybiogram 661, 831, 879 – działania niepożądane 628
– wziewny 300–303 antybiotyki cytostatyczne 971–974 – interakcje 628
anestezja 309 – β-laktamowe 93, 836–841 – kinetyka 628
– całkowita dożylna 309 – polipeptydowe 878 – mechanizm działania 626
– neuroleptanelgezja 310 antydepresanty 81, 1040 – NO 627
– neuroleptanestezja 310 antydiuretyczny hormon zob. wazo- – przeciwwskazania 628
– zrównoważona 309 presyna – tolerancja 627
aneuryna zob. witamina B1 antydiureza 713, 714 azotu podtlenek 301–302
angina pectoris zob. dusznica bolesna antyestrogeny 471–475 aztreonam 839, 852
angiogeneza 951 antygeny 94, 992 azytromycyna 862
angiografia 632, 1028, 1032 antygestageny 475–479
angioneuropatie 589 antykoagulanty 538–546
angiostatyna 951 antymetabolity 957–959
angiotensyna 449, 576
angiotensynogen 449, 576
antymon 815, 1040, 1054, 1108
antytoksyna 880, 1006, 1100
B
anilina 1080, 1108 apomorfina 348 bacytracyna 93, 837, 878
aniony 42, 515, 525 apoptoza 81, 604, 947–951 badanie kliniczne 127, 130–132
anoksja 322, 1133 aprotynina 510, 548 bakampicylina 846
anoreksja 801 arachidonowy kwas 94, 99, 221 baklofen 311, 320
ansamycyny 887 arekaidyna 379 bakterie
– dawkowanie 888 arekolina 379 – błona cytoplazmatyczna 818, 826
– działania niepożądane 888 arginaza 1065 – budowa ściany komórkowej 836
– interakcje 888 arginina 401, 539, 627 – flora fizjologiczna 436, 691
– kinetyka 887 arsen 22, 288, 979 – Gram-dodatnie 841, 842
– mechanizm działania 887 askorbinowy kwas zob. witamina C – Gram-ujemne 841, 842
– oporność 887 asparaginaza 954, 979–980 – koniugacja 829
– przeciwwskazania 888 astma oskrzelowa 639–654 – toksyczność 841
– wskazania 888 – glukokortykosteroidy 647 – transdukcja 829
antagonista 72, 74–78 – – wziewne 647 – zakażenia szpitalne 830–831
– aldosteronu 567, 606–607, 613 – – systemowe 649 bakterioliza 839
– kwasu foliowego 872–876, 957– – leczenie 644 bambuterol 651–652
958 – lukasty 649 – biodostępność 651
– opioidów 692 – monoklonalne przeciwciała 645 – metabolizm 652
– receptora ADP 536–537 – parasympatykolityki 652 balastowe substancje 688, 694
– – adrenergicznego 356 – patofizjologia 639 bar 103
– – androgenowego 466–467 – schemat leczenia 654 – zatrucie 1009
– – AT1 567, 577–578 – β2-sympatykomimetyki 650 barbiturany 303–305
– – glikoproteinowego Ilb/IIIa 535, – teofilina 651 – dawkowanie 305
537–538 ataranalgezja 309, 310 – działania niepożądane 305
– – H1 181 atenolol 369, 371, 372 – N-metylowane 303
– – H2 675–676 atorwastatyna 27, 559, 560 – tiobarbiturany 304
MUTSCHLER-2009.indd Sek1:1151 2010-03-18 20:12:19
1152 Skorowidz
Batemana równanie 51 beta-talasemia 521 blastopatie 100
Bcl-2 950, 951 bezsenność 173, 210–211 blastula 100
BCNU zob. karmustyna białko C 470, 530 bleomycyna 971, 973
BCRP 14, 40 – G 72, 355, 493, 947 bloker 18, 63
BDNF 226, 227 – S 470, 530 – aldosteronu 567, 606–607, 613
beksaroten 773, 980 – wiążące penicylinę 839, 840 – H+/K+-ATP-azy 673
benfotiamina 444, 807, 808 bibrokatol 756 – kanałów wapniowych 569–573, 630
benperidol 164, 165, 167 biegunki, leczenie 693–694 – – If 630
benserazyd 22, 338–340, 343 bifenyl 1070 – MAO 176, 341
bentonit 1050 bifonazol 786, 892, 894 – promieniowania 790
benzalkonium 823, 825 bilirubina 41, 526, 701 – receptora ADP 536–537
benzamidy 163, 171–173 – hiperbilirubinemia 41, 473, 1137 – – α-adrenergicznego 1107
benzimidazol 673, 674, 942–943 – wypieranie 109 – – β-adrenergicznego 368–373, 607,
benzbromaron 288, 289 – żółtaczka jąder podstawnych mózgu 629
benzodiazepiny 190–194 103 – – androgenów 466–467
– antagoniści 191 bimodalna krzywa 84 – – AT1 606
– ciąża 102, 104 bioaktywacja 36–38 – – glikoproteinowego Ilb/IIIa 537–
– dawkowanie 192–194 biodostępność 43–44 538
– działanie 191 bioinaktywacja 36–38 – – H1 181
– kinetyka 191 biologiczna wartość graniczna 217, – – H2 675–676
– receptory 191 1041–1042 – – 5-HT 350
benzoesowy kwas 30–32, 775 biologiczny rytm 123 – – leukotrienowego 649–650
benzoilu nadtlenek 774–775 biorównoważność 44 – – muskarynowego 351, 652–653
benzokaina 294–295 biorytm 123–124 – – NK1 350
benzomorfan 241 biosynteza 63, 82, 247, 354, 404 – – opioidowego 692
benzopiren 1101 – hormony płciowe 463–464, 468– BLT receptor 508
benzydyna 1103, 1104 469 BMI 205, 207, 444
benzylopenicylina 843, 845 – zaburzenia 62 BMP 80
benzylowy alkohol 25 biotransformacja 5, 24–40 borelioza 835, 1134
benzyna 1110 – bioaktywacja 36 Borrelia burgdorferi 835, 845, 881,
beryl 1106 – bioinaktywacja 36 1004
– zatrucie 1110 – efekt pierwszego przejścia 34 borny kwas 1110
betaksolol 369, 371, 752 – hamowanie aktywności enzymów bortezomib 985
beta-laktamowe antybiotyki 82, 93, 35 botulinowa toksyna 1039, 1081, 1100
777, 836–852 – indukcja białek 33 botulizm 1100
– cefalosporyny 848 – interakcje 109 bosentan 35, 583–584
– – dawkowanie 851 – reakcje I fazy 24 ból, droga czuciowa 217–224
– – działania niepożądane 851 – reakcje II fazy 29 – mediatory 219
– – działanie 838 – wpływ płci 39 – rodzaje 217–219
– – grupy 848 – – wieku 38 BPH zob. hiperplazja prostaty
– – kinetyka 851 biotyna zob. witamina H BPO zob. benzoilu nadtlenek
– karbacefemy 848 – niedobór 813 bradykardia 166, 293, 383, 616, 1107
– karbapenemy 851 – zastosowanie 813 – zatokowa 614
– – działania niepożądane 852 – znaczenie 813 bradykinina 95, 220, 509, 783
– – przeciwwskazania 852 biperiden 345, 346, 1053 bradyfrenia 337
– monobaktamy 852 bisakodyl 691–692 Broki ośrodek mowy 151
– oporność 840 bisfosfoniany 112, 418–421 brom 29, 774, 815, 1051
– penicyliny 841 – dawkowanie 420 – trądzik 774
– – dawkowanie 844 – działania niepożądane 420 bromazepam 190, 193
– – działania niepożądane 844 – działanie 420 bromheksyna 658, 659
– – grupy 845 – interakcje 421 bromki, zatrucie 1110
– reakcje alergiczne 841 – przeciwwskazania 421 bromokryptyna 341, 398
– toksyczność 841 – skuteczność 420 bromperidol 165, 167
betametazon 454, 782 – właściwości 420 broncholityczne leki 362
betanechol 378–379 – wskazania 420 brotizolam 212, 213, 215
– dawkowanie 378 bisoprolol 369, 371, 582, 607 brucyna 1110
– działania niepożądane 379 biwalirudyna 543 brywudyna 910, 914
– przeciwwskazania 379 bizmutu sole 108, 679 budezonid 648–649, 685
MUTSCHLER-2009.indd Sek1:1152 2010-03-18 20:12:20
Skorowidz 1153
budowa nerwu 155 cefaleksyna 850 chlamydie 756, 821, 834–835
– synapsy 156 cefalosporynazy 840 chlor 29, 126, 149, 191, 1111
budypina 345–346 cefalosporyny 93, 103, 830–835, 838, chloralu wodzian 211, 214, 1111
– dawkowanie 345 851 chlorambucyl 32, 962, 963
– działania niepożądane 345, 346 cefamycyny 848, 851 chloramfenikol 102, 104, 439, 756,
– interakcje 346 cefazolina 849 829, 865–866
bufotenina 204 cefepim 849 – dawkowanie 865
bulbus oculi 743 cefiksim 850 – działania niepożądane 865–866
– olfactorius 153 cefotaksym 851, 880 – działanie 865
bulimia 189, 349, 1085 cefotiam 849 – interakcje 866
bupiwakaina 294, 296 ceftazydym 849 – kinetyka 865
bupranolol 368–370 ceftibuten 850 – oporność 865
buprenorfina 240 ceftriakson 880–882 – przeciwwskazania 866
bupropion 27, 1084 cefuroksym 850 chloramina 820
Burkitta chłoniak 947 celekoksyb 27, 256–258 chlorany 1080
buserelina 460, 975 – dawkowanie 258 chlordiazepoksyd 191–193
buspiron 27, 194–195, 502 – działania niepożądane 256 chlorfenoksamina 496
– dawkowanie 195 – interakcje 254 chlorfenotan 103
– działania niepożądane 195 – przeciwwskazania 257 chlorheksydyna 818, 825–826
– przeciwwskazania 195 – wskazania 256 chlorkowe kanały 76, 191, 304, 943
busulfan 963 celiprolol 13, 111, 369, 371 chlorochina 102, 830, 930–937
butoksykarboksym 1095 ceramidy 762 chloroform 296, 513, 1069
butyrofenony 163, 167, 170 cetroreliks 460–462 chlorprotiksen 164, 165, 167, 169
butyrylocholinoesteraza 376 cetyryzyna 495, 497, 498 chlortalidon 731, 732
cGMP 73, 80, 594 chmiel 215, 473
cheilitis 773 cholangiografia 1027
chelatowe kompleksy 869, 1031–1032 cholecystografia 1027
C chemioterapeutyki 57, 826–833, 864
chemioterapia 891–903
cholecystokinina 205, 417, 664–670
cholegrafia 1028
Cmax 44, 112, 124 chemokiny 493, 1008, 1012 cholera 693, 817
Ca2+-ATP-aza 10, 802 – działania niepożądane 1012 – szczepionka 1001
Ca2+ jony 73, 186, 226, 321, 718, – klasy 1012 – uzupełnianie elektrolitów 693
724 – receptory 1012 cholesterol 9, 132, 437, 445–446, 459,
– kanał 77, 325 chemotaksja 245, 271, 507, 990 526, 555–562
– kinaza białkowa C 73 chemotaksyny 94 – inhibitory wchłaniania 561
– receptor 415 chenodeoksycholowy kwas 703, 704 – metabolizm 556
– resorpcja 710 – dawkowanie 704 – transport zwrotny 557
– wymiennik 599 – działania niepożądane 704 cholestyramina 546, 561–562
– zapotrzebowanie 718 – przeciwwskazania 704 – dawkowanie 562
CAD 950 chenodiol zob. chenodeoksycholowy – działania niepożądane 562
CAM zob. kalmodulina kwas cholinergiczny odruch 299
cAMP 73, 245, 355 chiasma opticum 208, 746 cholinoesteraza nieswoista 376
Candida albicans 785, 827, 899 chimeryczne przeciwciała 280, 281, cholinolityk 22, 380, 679
Cannabis indica 203 687, 983, 1019 cholowy kwas 669
carbo medicinalis 694, 1049–1051, chinidyna 108, 383, 611, 618–619 chondroblasty 122
1107 – działanie 619 chondrocyty 122, 388, 399
CCNU zob. lomustyna – interakcje 619–620 chondroityna 274, 542, 1090
CCS system 623 chinina 619, 931–934 choroba Alzheimera 199–200
CD2 766, 771 – działania niepożądane 934 – polekowa 95
CD20 280, 281 – działanie 934 – posurowicza 95
CD28 766 chinolina 824, 939 – reumatyczna 249, 253–256, 269–
CD52 983 chinolinol 824 285
CD95 81, 95, 949 chinolizydynowe alkaloidy 1082 – wieńcowa 565–566, 622–633
CD11a 770 – cytyzyna 1082 – wrzodowa 672–680
CD4 limfocyty 95, 640, 683, 908 – sparteina 1082 – zwyrodnieniowa stawów 249
CD8 limfocyty 95, 655 chinolony 694, 869–871 chrom 814, 1054, 1064, 1106
cefadroksyl 850 chinon 544, 804, 973, 1069 chylomikron 557
cefaklor 850 chiralne substancje 60, 61, 89, 129 ciąża 100–104
MUTSCHLER-2009.indd Sek1:1153 2010-03-18 20:12:20
1154 Skorowidz
cilazapril 573, 576 depolaryzacja 76–78, 146, 318
cisplatyna 124, 964 D dépôt 14, 397, 434, 471
– dawkowanie 965 DAG zob. diacyloglicerol depresja 123, 175
– działania niepożądane 965 dakarbazyna 965–966 – oddechowa 229
citalopram 182–183, 263 daklizumab 1016, 1020, 1021 dermatitis 791, 811
cisapryd 696, 697, 865 daktynomycyna 971 dermatomyositis 273
CKI 946 danaweryna 385 dermatozy 764, 766, 774, 789
clearance 13, 39, 45 danazol 478 desensytyzacja 65
Clostridium botulinum 1006, 1100 dantrolen 166, 300, 318–319 desfluran 300, 302
CO 1053, 1077–1078 dantron 767 desloratadyna 495, 498
CO2, ciśnienie parcjalne 514, 525, dapson 115, 116, 935 desmopresyna 16, 402, 403, 534, 738
552, 636 daptomycyna 877–878 desmosomy 762
CoA zob. koenzym A darunawir 923–924, 927 detajmium 618–620
colitis ulcerosa, leczenie 682–686 daunorubicyna 971, 972 detergenty 672, 819, 1068
COMT 338 dawka jednorazowa 83 determinanty, antygenowe 92, 992
– inhibitor 340–341 – letalna 83 dealkilacja 28, 360
compliance 83, 125, 582 – nasycająca 621 dezaminacja 355, 810
COX zob. cyklooksygenaza – podtrzymująca 83 dezipramina 177, 178
CSE inhibitor 81 – progowa 84 dezorientacja 963, 1139
cukrzyca 429–434 dawkowanie leku 83–84 dezoksymetazon 780, 782
– leczenie 434–444 dazatynib 984–985 dezoksyrybonukleinowy kwas (DNA)
cyjanokobalamina zob. witamina B12 DDT 1091–1093 522
cyjanowodór 1042, 1047, 1053, debryzochina, polimorfizm 115 dezynfekcja 817–822
1078–1079 deeskalacja 654 DHEA zob. dehydroepiandrosteron
cykliny 946 defekacja 668, 1135 449, 464
cyklofosfamid 27, 277, 435, 955, – odruch 692 diacetylohydrazyna 885, 887
961–963 – zaburzony mechanizm 688 diacyloglicerol 73, 355
cyklooksygenaza (COX) 221, 245, deferipron 1055 dializa 416, 999
248, 256 deferoksamina 520, 1053–1055 diastereomery 89
– inhibitory 256 – dawkowanie 1055 diazepam 27, 45, 190–193
cyklopentolat 755 – działania niepożądane 1055 – biotransformacja 193
cyklopiroks 901, 903 defosforylacja 359, 674, 675, 1035 diazoksyd 429
cykloseryna 855, 889 degranulacja 94, 95, 495, 640 didanozyna 112
cyklosporyna 57, 277, 279, 685, 770, dehalogenacja 29, 1069 dideoksyinozyna 919
1014 dehydrataza ALA 1060 dieldryna 1093
cymetydyna 109, 546, 675–676 dehydroaskorbinowy kwas 813 dienogest 477–479
– dawkowanie 676 dehydrocholowy kwas 703 dietylokarbamazyna 941, 943
– interakcje 676 dehydroepiandrosteron 11, 449, 464 difenhydramina 214, 349, 495
cynaryzyna 590–592 dehydrogenaza glukozo-6-fosforano- difenole 689, 691–692
CYP zob. cytochrom P-450 wa (G6PD), niedobór 116 digitoksygenina 608, 609
cyprofloksacyna 756, 834, 880 dekarboksylacja 29, 125 digitoksyna 608–611
cyproheptadyna 495, 503 dekarboksylazy inhibitor 22, 125, – dawkowanie 610
cyproteronu octan 467, 978 339–340 – działania niepożądane 611
– dawkowanie 978 dekontaminacja 859, 1061 – interakcje 611
– działania niepożądane 978 deksametazon 349, 350, 454, 940 – zatrucie 611
cytarabina 901, 958–960 deksibuprofen 251, 256 digoksyna 573, 607–611
– dawkowanie 960 deksketoprofen 251 dihydralazyna 579–580, 582–583
– interakcje 960 dekspantenol 758, 812 dihydroekwilina 471
cytochrom P-450 (CYP) 26–28, 464, dekstran 520, 526–528 dihydroergokryptyna 342, 365
536 dekstrometorfan 27, 243, 244 dihydroergotamina 365, 585, 594
cytostatyk 102, 104, 124, 954–956 delirium tremens 1073, 1074 dihydroergotoksyna 365, 593
cytrulina 553, 627 demencja 199, 1135 dihydrokodeina 235, 241, 243
cytrynowego kwasu cykl 432, 769, – alkoholowa 1073 dihydrokortyzol 448
808, 1098 – naczyniowa 566 dihydroksyaceton 791
cytrynowy kwas 525, 695, 723 dendryt 143, 148 dijodotyrozyna 396
czerwień wzrokowa 800 dendrytyczne komórki 122, 512, 766 diklofenak 250, 255
czynniki krzepnięcia 108, 402, 470, depersonalizacja 162 dikloksacylina 842, 843, 845
529–539 depigmentacja 593, 791 dikumarol 112, 543, 1098
MUTSCHLER-2009.indd Sek1:1154 2010-03-18 20:12:20
Skorowidz 1155
diltiazem 342, 569, 573, 621 dyfteryt 880 – – zaburzenia 450
dioksygenaza 24, 26 dynorfina 205 – resorpcja 668
dioksyny 103, 1070 dyskineza 165–166, 345, 347, 703 – roztwory 352
dioptryczny aparat 743, 747, 749 dyslipoproteinemia 433 – transportery 81
dipirydamol 535, 538, 622, 630 dysmelia 952 – uzupełnianie 349, 352
disacharydazy 671 dysocjacji stała 65, 66 elektromechaniczne sprzężenie 312–313
distygminy bromek 380 dyspepsja 282, 351, 442 elektrony 190, 733, 876
disulfiram 93, 102, 1074 dysplazja 660, 917, 948, 953 – łańcuch reakcji 27
diterpenowe alkaloidy 1081 dysregulacja ortostatyczna 585 – promieniowanie 1066
ditranol 766, 767–768 dyssomnia 210 elektroforeza 525, 555, 950
diuretyk 81, 102, 126, 416, 451, 605, dystonia 188, 342 eletriptan 266, 267
728 dystrybucja 5, 18–19 eliminacja 5, 6, 13
dizopiramid 1056 działanie addytywne 87 – okres półtrwania 46
DMPS 1052–1054 – biologiczne 3, 1010 – szybkość 24
– dawkowanie 1052 – niepożądane 92 embriogeneza 100
– działania niepożądane 1054 – placebo 132 embriopatia 100, 952
– odtrutka 1053 – szkodliwe 3 EMEA 131
DNA zob. dezoksyrybonukleinowy – teratogenne 37, 92 emetyna 348, 658
kwas dziurawiec 185 emodyny 691
dna moczanowa, leczenie 285–289 emulsje 7, 765
dobesylan wapnia 594 enalapril 573, 575, 606
dobutamina 587, 611, 1048 enalaprilat 574
docetaksel 27, 955, 970–971 E enancjomery 60, 89
– dawkowanie 971 ecstasy 307 endocytoza 120, 404, 522
dokozaheksaenowy kwas 806 Echinacea purpurea 1012 endolimfa 19, 107
doksapram 216 echinokandyny 899 endorfiny 205
doksazosyna 367, 368, 741 echogeniczność 1034 endostatyna 951, 952
doksepina 177–180, 186 ekonazol 786, 892, 894 enfluran 302
doksorubicyna 955, 972 EDRF 627 enfuwirtyd 904, 908, 925–926
doksorubicynol 972 EEG zob. elektroencefalografia – dawkowanie 926
doksycyklina 835, 860–862 EET zob. epoksyeikozatrienowe kwasy – działania niepożądane 926
doksylamina 214, 496, 498 efalizumab 767, 770–771, 1021 – oporność 925
dokuzan sodu 692 efedryna 362, 364 enoksacyna 110, 342, 869–972
dolasetron 350, 351 efekt allosteryczny 69 – dawkowanie 870
domperidon 265, 350, 696, 697 – cytotoksyczny 36 – oporność 871
donepezil 200–201 – genotoksyczny 36 – wskazania 971
L-dopa 339, 1036 – kuraropodobny 107, 302 enoksaparyna 541
dopamina 161, 166, 360 – pierwszego przejścia 9, 14, 16, enoksymon 612
– niedobór 336 34–35 entakapon 338, 340, 343
– synteza 353 – placebo 132 entaktyna 953
dornaza 661 – poantybiotykowy 828 enterotoksyna 1100
dorzolamid 737, 753 – pułapowy 233 enzymy 4
dostępność biologiczna 43–44 EGF zob. naskórkowy czynnik wzro- – hamowanie 35
down-regulation 65 stowy – indukcja 34
DRESS zespół 95, 97, 98 EGFR zob. receptor naskórkowego – mikrosomalne 26
dronabinol 242–243 czynnika wzrostowego – rozpuszczalne 24
dronedaron 477 egzema 776 – utleniające 28
droperidol 310 eikozanoidy 504–509 eozynopenia 451
drospirenon 478 ejakulacja 358 epilepsja zob. padaczka
drug targeting 22 EKG zob. elektrokardiogram epinefryna zob. adrenalina
Dubina-Johnsona zespół 41 ekonazol 786, 892 epirubicyna 971, 972–973
duloksetyna 181–182, 262 eksemestan 977, 978 epirubicynol 972
dusznica bolesna 623 elastaza 517, 655 eplerenon 606–607, 734
– Prinzmetala 624 elastyna 644, 762 epoetyna 521–522
– przedzawałowa 624 elektroencefalografia 321 epoksydy 26, 29, 247, 507
– schemat leczenia 625 elektrokardiogram 180, 244, 492, 611 epoksyeikozatrienowe kwasy 553
dydrogesteron 476, 478 elektrolity 525–526 EPSP zob. pobudzający potencjał post-
dyfuzja 9–12 – gospodarka 349, 350, 403, 445 synaptyczny
MUTSCHLER-2009.indd Sek1:1155 2010-03-18 20:12:20
1156 Skorowidz
Epsteina-Barr wirus 846, 912 farmakodynamika 4, 62–90 fluor 815
eradykacja Helicobacter pylori 673 farmakogenetyka 4, 114–117 fluorki 421, 815
erekcja zob. wzwód farmakokinetyka 4, 5–61 fluorochinolony 834, 869, 872
ergokalcyferol 802–803 farmakologia 3, 4 fluorouracyl 546, 959, 987, 1036
ergokornina 364–365 farmakon 3 flupentiksol 165, 169
ergokryptyna 364–365 Fas-receptor 949 flupirtyna 261
ergokrystyna 364–365 fawizm 116 flurbiprofen 251
ergometryna 366, 491 faza badań klinicznych 127 fluspirylen 165, 171
ergotamina 365–366 – farmaceutyczna 5 flutamid 978
erlotynib 984 – refrakcji 147 fluwastatyna 546, 560
erytrocyty 20, 444, 511–513 febuprol 703 fluwoksamina 183, 186
erytromycyna 104, 675, 697, 862– feksofenadyna 495, 498 folikulotropowy hormon 394, 459–
865 felbamat 325, 332 463, 480
– dawkowanie 865 felodypina 329, 570–571 foliowy kwas zob. witamina B12
– działania niepożądane 865 felypresyna 293 folitropina 462, 480, 974, 975
– interakcje 865 feminizacja 103, 479, 975 fomepizol 1052, 1071, 1072, 1075
erytropoetyna 388, 513, 521–522 fenazon 247, 252, 260 formaldehyd 523, 821
erytroza 808 fenobarbital 214, 325, 330 formoterol 363, 651
Escherichia coli 693, 979, 1089 fenoksybenzamina 366, 582 forskolina 82
estradiol 464, 468–473 fenol 31, 818, 819 fosamprenawir 923–924, 927
– dawkowanie 473 fenoterol 362, 491, 651 fosfolipaza A2 94, 222, 223
estramustyna 975 fenotiazyna 167–169 – C 73, 186, 355, 493
estriol 468, 471, 487 fenprokumon 544–545, 676 fosfomycyna 854
estrogeny 102, 459, 468–469 fentanyl 241, 303, 307–308 fosfor 140
– dawkowanie 473 fentikonazol 892 – radioaktywny 982
– działania niepożądane 473 fenylefryna 360, 755 fosforany 416, 570, 677, 718
– działanie 470–473 fenylobutazon 109, 260–261 fosgen 1047, 1076–1077
– interakcje 473 fenytoina 109, 111, 326, 329–330 foskarnet 905, 910, 913, 915–916
– przeciwwskazania 473 ferrihemoglobina 1079 fozynopril 573–575, 606
– środki terapeutyczne 471 ferrytyna 515 frowatriptan 266, 267
estron 468–471 ferukarbotran 1032 fruktoza 442, 671
etakrydyna 824 fetogeneza 100 FSH zob. folikulotropowy hormon
etakrynowy kwas 107, 733 fetopatie 101, 103 fulwestrant 475, 976, 977
etambutol 884, 885, 888 fibraty 534–535 fumarowy kwas 766, 769–770
etanercept 280–281, 687 fibroblasty 122, 279, 644, 951 fungicydy 1061
etanol 1072 fibronektyna 644 furosemid 416, 582, 605, 733–735,
– odtrutka 1052, 1053 fibryna 531, 951 1048
– zatrucie 1075 fibrynogen 531, 534, 566 fuzydynowy kwas 783
eter 290, 302 fibrynolityki 546
– halogenowany 302 fibrynoliza 530, 592
– znieczulenie 298, 299 Ficka prawo 10
etydronian 418–421 filtry UV 790 G
etylefryna 360–361 finasteryd 466, 740, 741
etoform 294 fitofarmaceutyki 137, 741 GABA 71, 75, 185, 215, 330
etomidat 303, 305 fizostygmina 380, 1053 gabapentyna 263, 331–332, 334
etopozyd 955, 968–969 flawiwirusy 698, 905 gadobutrol 1032
etosuksymid 326, 328, 333–334 flekainid 618 gadodiamid 1032
etylowy alkohol zob. etanol flucytozyna 102, 826, 899, 901 gadolin 1031–1032
eugenol 822–823 fludarabina 958 gadoksetowy kwas 1032
evidence-based medicine 136 fludrokortyzon 456 gadopentetowy kwas 1031, 1032
ewerolimus 1014–1017 flufenazyna 164, 168 galaktoza 671, 1034
ezetymib 561 flukonazol 896, 903 galaktozydaza 1023
flumazenil 309, 1053 galantamina 200–201
flunaryzyna 591, 592 gallopamil 572, 573, 621–622
F flunitrazepam 213
flukonazol 896, 903
galsulfaza 1024–1025
gamety 930
famcyklowir 912–914 fluokortolon 454 gametocyty 930, 934, 936
famotydyna 675–676 fluoksetyna 183, 186 – gametocytobójcze środki 932
MUTSCHLER-2009.indd Sek1:1156 2010-03-18 20:12:20
Skorowidz 1157
gametogeneza 100 glukoneogeneza 399, 405, 427 – interakcje 542
gamma promienie 1035 glukozamina 284, 539 – przeciwwskazania 542
gamolenowy kwas 806 glukoza 116, 123, 202, 352, 369, 428 – standardowa 439
gancyklowir 102, 852, 909, 914–915 glukuronian 29, 191 – wskazania 541
– dawkowanie 914 glukuronidacja 31, 37, 170, 612 heparynoidy 542
– działania niepożądane 914 glukuronidy 14, 309, 609, 798 hepatitis A 698, 700
– oporność 914 glutaminian 223, 808 – B 700
– przeciwwskazania 915 glutation 32, 116, 260, 824, 1070 – C 700
garbniki 694, 783 Golgiego aparat 424, 909 – D 702
gastryna 394, 416, 504, 664–667 gonadoliberyna 394, 474, 490, 975 – G 702
gaz rozweselający 296, 299, 301–302 gonadotropina kosmówkowa 396, hepatocyt 260, 405, 559, 923, 989
gazy 73, 299, 1034 459 hepatosplenomegalia 1024
gemcytabina 958 gorączka reumatyczna 270 hepcydyna 515
gemeprost 490, 505, 506 goserelina 460, 975 herbicydy 1042, 1098–1099
genetyczne defekty 31, 41, 114, 115, granisetron 350, 502, 503 heroina 234, 237, 239, 241
118 granulocyty 495, 507, 516, 517 herpes, wirus 785, 904–906
genotoksyczność 36–37, 691, 1101 grupy krwi 514, 527, 994 – leczenie 912–915
genotyp 114, 260, 700, 701, 945 gryzeofulwina 102, 892, 901, 903 Herxheimera odczyn 92
geny 73, 75 grzyby trujące 1087 heteroreceptory 150, 378, 495
– aktywacja 71, 948 guanina 286, 961, 1106 hioscyjamina 89, 1109
– amplifikacja 947, 948 hipercholesterolemia 118, 556, 558,
– ekspresja 64, 65, 71, 81 561, 704
– mutacja 75, 948 hiperchylomikronemia 558
– regulacja 34, 35, 62, 71 H hiperforyna 185
– skaczące 829 hiperglikemia 395, 401, 427–433,
– transkrypcja 34, 71, 77 Haemophilus ducreyi 882 436
– translokacja 856, 947 – influenzae 655, 661, 846 hiperkalcemia 107, 416, 419–421
– uszkodzenia 1066 Hagemana czynnik 509, 529, 532 hiperkaliemia 450, 574, 606, 611,
genisteina 473 haloperidol 167–171, 182, 326, 351 720–721
genitalia 882, 905 halotan 300, 302, 303 hiperpigmentacja 473, 593, 791
gentamycyna 135, 756, 830, 860 halucynacje 162, 420, 611, 676, 953 hiperplazja prostaty 468, 740
geriatryczne środki 815 halucynogen 203 hipertermia złośliwa, leczenie 117,
gestageny 102–104, 126, 459, 467 Hanscha analiza 88 299
GH zob. somatotropina H1-antyhistaminiki 150, 346, 1048 hipertonia 733
glibenklamid 438–439 H2-antyhistaminiki 150 hiperwolemia 451, 511, 527, 726
glikol 106, 1075 hapten 92, 96, 992 hiperycyna 185
– zatrucie 1074–1075 haszysz 203, 204 hipnoanalgetyki 217, 232
glikopeptydy 826, 852–854 HCG zob. gonadotropina kosmówkowa hipoglikemia 109, 373, 429
– dawkowanie 853–854 heksachlorofen 822–823 hipokalcemia 415, 605
– działania niepożądane 854 heksanol 1068 hipokaliemia 107, 346, 610, 719
– działanie 854 heksetydyna 824 hipoksantyna 286, 288–289, 958–959
– interakcje 854 heksobarbital 60 hipoksja 301, 432, 521, 601, 1049
– oporność 854 Helicobacter pylori 672–674, 679 hipotonia, leczenie 364, 457, 585–
– przeciwwskazania 854 – eradykacja 673, 680 586
glikoproteina P 12–13, 109, 573 – szczepionka 680 hippurowy kwas 32, 775
glikozydy antrachinowe 691 hem 513 hirudyna 542–543
– nasercowe 81, 107, 126, 416, 607– hemoglobina 117, 433, 513 histamina 95, 99, 123, 493–499
611 – budowa 513 – działanie 494
glikwidon 438 – wiązanie ditlenku węgla 514 – leki przeciwhistaminowe 495
glimepiryd 438–439 – wiązanie tlenu 513 – receptory 493, 495
glin 126, 407, 520 hemostaza 528–532 – synteza 493
– wodorotlenek 17, 416, 677 hepadnawirusy 698 – znaczenie 495
glinidy 425, 438, 440 heparyna 539–542 histydyna 493, 722
glitazony 70, 440–444 – dawkowanie 541 HIV 88, 699, 785, 827, 908–910
globulina 21, 389, 406, 465, 525 – do stosowania miejscowego 542 homeopatia 138–140
glukagon 373, 391, 424, 427–428, – drobnocząsteczkowa 540 hormon, definicja 387
1053 – działania niepożądane 541 – luteinizujący 396, 459, 469, 480
glukokortykosteroidy 352, 454 – fondaparinuks 541 5-HT zob. serotoniny transporter
MUTSCHLER-2009.indd Sek1:1157 2010-03-18 20:12:20
1158 Skorowidz
hydralazyna 579–580 inhibina 80, 393, 459, 480 irinotekan 116, 968, 987
hydrochinon 534, 544, 791, 973, inhibitor ACE 107, 573, 605 ischemia 631
1069 – agregacji trombocytów 631 isradypina 570–571
hydrochlorotiazyd 58, 731–732 – anhydrazy węglanowej 737, 753 itrakonazol 110, 865, 892, 895–897
hydrokodon 235, 244 – aromatazy 977–978 iwermektyna 941–943
hydrokortyzon 446–448, 457, 777, – cholinoesterazy 379 – dawkowanie 943
782 – COMT 337, 340 – działanie 943
hydroksychlorochina 283, 285 – cyklooksygenazy 256, 507, 535 – wskazania 943
hydroksykarbamid 980 – dekarboksylazy 22, 337, 33 izocyjanian 964, 1077
hydroksytryptamina 500 – epoksydazy skwalenowej 897 izoenzym cytochromu P-450 27
hydroksytryptofan 150, 500, 813 – fosfodiesterazy 82, 611 izofluran 300, 302
hydroksyzyna 194 – fuzji 925 izoniazyd 82, 104, 115, 810–811
hydromorfon 235, 239 – α-glukozydazy 436 izonikotynowy kwas 885–887
hydrożele 465, 765 – gyrazy 82, 102, 869–872 izoprenalina 617, 783
hymekromon 385, 703 – hemopolimerazy 932 izopropanol 822–823, 1056
– kinaz 984 izoprostan 247
– krzepnięcia krwi 530 izosorbid 612
– β-laktamaz 847 izotonia 8, 705
I – lipoksygenazy 509 izotopy radioaktywne 982–983
ibandronian 419, 420 – MAO 87, 341 izotretynoina 771–774
ibrytumomab 982 – mitozy 969–971 – działania niepożądane 772
ibuprofen 251, 254–256, 792 – monoaminooksydazy 81, 107, 176, – działanie 771
ibutylid 620–621 184 – interakcje 774
idarubicyna 971, 972 – odwrotnej transkryptazy 919–922 – leki 772
idoksurydyna 915 – pompy protonowej 81, 135, 275, – przeciwwskazania 772
iduronowy kwas, niedobór 1025 673
idursulfaza 1024–1025 – proteasomów 985
– proteazy HIV 13, 922–925
IFN zob. interferony
ifosfamid 962, 963 – 5α-reduktazy 467, 740 J
iloprost 507, 583–584, 590 – reduktazy HMG-CoA 81, 104, 559 Jacksona napady 323, 329
imatynib 585, 984 – syntezy kwasu nukleinowego 935 JAK zob. Janusowe kinazy
imidazol 373, 786 – topoizomerazy 967–969 Janusowe kinazy 78, 80, 1016
imidazolina 360 – trombiny 542 jaskra 750
imipenem 839, 852 – wchłaniania cholesterolu 561 – leczenie 170, 737, 751–755
imipramina 177–179, 195 – wychwytu zwrotnego noradrenali- jasmolon 1093
immunoglobuliny zob. przeciwciała ny/serotoniny 176–178, 181 jelita drażliwego zespół, leczenie 688
immunologiczna choroba 118, 321 inodylatatory 612 – unerwienie autonomiczne 160
– kompleksy 96, 271, 995 inozyna 622, 1013 jod 814, 820, 901, 1027–1030
– niedobór 907 insektycydy 1091–1097 – odjodowanie 1030
– odpowiedź 95, 517, 687, 787, 986, insulina 424–427, 429–432, 434–435 – radioaktywny 982
989 – działanie 426 jodki 1051, 1053, 1063, 1071
– reakcja krzyżowa 270 – gęstość receptorów 427 jodofory 820
– supresja 896 – uwalnianie 425 jodyna 820
immunomodulatory 278, 786, 1008– – zastosowanie 427 jonowe kanały 63, 73–76, 376, 619
1013 integraza 829, 905, 910, 917 – pompy 146
immunoprecypitacja 525 integryna 537, 1022 jopamidol 1028
indapamid 732 interakcje leków 106–112 jopromid 1028
indinawir 112, 922–924 – unikanie 113 jotrolan 1029
indometacyna 102, 254–255, 429 interferony 699–702, 986, 990, 1009
indukcja enzymatyczna 34–35, 39–40, interleukiny 221, 986, 996, 1009
55, 110 interneurony 160, 225, 227
infliksimab 686, 767 intrawazacja 953 K
infuzyjne płyny 106, 303, 721 inwersja chiralna 60, 61 kabergolina 342, 398
– rodzaje 723 ipekakuana 1050 kadm 815, 1041, 1054, 1062–1063
INH zob. izoniazyd ipratropium bromek 384, 617, 653 – leczenie 1063
inhalacja 363, 434, 646, 902, 1067 IPSP 149 – zatrucie 1062
inhibicja enzymatyczna 125, 819, iralukast 650 kalcyferol zob. witamina D
922, 989 irbesartan 577–578, 606 kalcypotriol 767–769, 804
MUTSCHLER-2009.indd Sek1:1158 2010-03-18 20:12:20
Skorowidz 1159
kalcytonina 391, 417–418 – transport 11, 40, 42 klokortolonu piwalan 778
kalendarz szczepień 1001–1004 – wymiennik 600, 725 klometiazol 1073–1074
kalidyna 509 Kaschina i Becka zespół 815 klomifen 474–475, 488
kalikreina 509–510, 991 kawa 111, 195 klomipramina 177–179, 195
kalikreinogen 509 – działanie diuretyczne 737 klonazepam 193, 331, 335
kalmodulina 75, 357, 553 – nadużywanie 196, 673 klonidyna 107, 373–375, 582, 752
kalorii dostarczanie 205, 442 keloid 764, 1066 – dawkowanie 374
kanał chlorkowy 76, 191, 212, 309 keratomalacja 801 – działanie 373
– jonowy 63, 71–79 keratoza 787–789 – wskazania 374
– potasowy 73, 75, 76 keratyna 22, 763, 791, 800 klopidogrel 535–537, 631
– sodowy 63, 75, 76, 78 keratynocyty 508, 761, 766, 770 klotrimazol 892, 894, 939
– wapniowy 63, 73, 75, 76, 109 ketamina 238, 303, 306–307 klozapina 166, 169–172
kanamycyna 756, 856, 860 – dawkowanie 307 koagulopatie 532, 1067
kandydiozy 784, 826, 892 – działania niepożądane 307 kodeina 108, 221, 226–227, 230–231
kancerogenność 366, 485, 783, 876, – wskazania 306 koenzym A 768
1043 ketokonazol 108, 111, 675, 895–897 kofeina 192–193, 343
kanrenon 734–736 – dawkowanie 894 kokaina 101, 282, 348–349
– dawkowanie 736 – działania niepożądane 897 koksyby 246–248
– kinetyka 735 – działanie 896 kolagen 273, 286, 422, 447
– wskazania 735 – interakcje 897 kolagenozy 270, 273, 574
Kaposiego mięsak 917, 969, 986 ketokwasy 807 kolchicyna 102, 287, 955, 969, 1082
kapsułki 7, 15, 214, 628, 685 ketolidy 862 kolistyna 878
kaptopril 573–575, 593 ketonowe ciała 427, 432, 444 komorowe arytmie 612
karbachol 378–379, 753 ketonuria 431 komory serca 596
karbamazepina 326, 334, 473 ketoprofen 251 – ciśnienie 597
karbapenemy 831, 851–853 ketotifen 495, 497, 650 – mięsień 598
– dawkowanie 852 kinazy białkowe 73 – przegroda 596
– działania niepożądane 852 – 3-fosfatydyloinozytolu 73 – rytm 598
– przeciwwskazania 852 – inhibitory 82 – skurcze dodatkowe 615
karbidopa 338–340 – Janusowa 80 – tachykardie 616
karbimazol 410–411 – MAP 79 – zaburzenia rytmu 360, 491, 589,
karbocysteina 658–659 – serynowo-treoninowa 78, 80 603
karboplatyna 964–965 – tyrozynowa 77, 78, 80 kompartment 6, 19, 44, 47–52
karbutamid 438–439, 1040 kinetyka nieliniowa 55–56 koniina 1045, 1082
karcynogeneza chemiczna 1101–1106 kininy 509–510 koniugacja 24, 829, 1104
kardiogenny wstrząs 586–588 kladrybina 958 kontaktowa alergia 97
– leczenie 588–589 klarytromycyna 675, 680, 832, 862– – wyprysk 776
karcynogeny 37, 947 865 kontrastowe środki 102, 410, 493,
– chemiczne 34, 953, 1101–1103 klaustrofobia 188 1027–1035
– definicja 1101 klawulanowy kwas 846, 885 kontrolowane badania 133, 138
– pierwotne 1101 klemastyna 497 kortykosteron 446, 447, 449
– wtórne 1101 klindamycyna 104, 775, 834, 866– kortykotropina 389, 394, 396, 397
karcynoid 1011 867, 937 kortyzol 106, 445–448
kardiomiocyty 77, 122, 553, 604 – dawkowanie 866 – niedobór 400
kardiomiopatia 491, 577, 602 – działania niepożądane 866 – zespół Cushinga 451
kardiotoksyczność 859, 972–973, – działanie 866 kortyzon 447, 452, 453, 457
1015 – interakcje 867 kotrimoksazol 98, 835, 876
karencja 645, 694 kliniczne badania 127, 130–132 kozłek lekarski 215
karmustyna 964 – farmakologia 4 krążenie jelitowo-żołądkowe 23
β-karoten 798–801 – toksykologia 1043 – płucne 35
karwedilol 369, 372, 607 kliochinol 824 – wątrobowo-jelitowe 22–23, 29
katalaza 819, 885, 886 klirens 59, 187, 278, 407, 584, 1056 krezol 822–823
katapleksja 196, 198–199 klobazam 190, 331, 334, 335 kromoglikanowy kwas 647
katarakta 747 klobetazolu propionian 777, 781 krople do oczu 750
katatonia 162 klobutinol 243–244 kryptokoki 895
katecholaminy 125, 355, 447, 611 klodronian 418–420 krzemiany 677, 1106
– synteza 353–354 klofazymina 890–891 ksylometazolina 361
kationy 42, 525 klofibrat 38 kumulacja 53–54
MUTSCHLER-2009.indd Sek1:1159 2010-03-18 20:12:20
1160 Skorowidz
kurara 313, 317 laronidaza 1024–1025 – żółciopędny 703
kwas acetylosalicylowy 253–254, latanoprost 505, 754 lekozależność 104–105
329, 535, 547 laudanozyna 318 Lennoxa-Gastauta zespół 324
– γ-aminomasłowy 191 leiszmanioza 929, 938–939 leptyna 205
– 5-aminosalicylowy 684–685 leflunomid 278 lerkanidypina 570–571
– arachidonowy 493, 504 lek 3 letrozol 955, 977–978
– askorbinowy 397, 813–814 – α-adrenolityczny 356 leukopenia 272, 326, 412
– asparaginowy 434, 979 – β-adrenolityczny 83, 268, 356, leukotrieny 223, 504, 507–509
– azelainowy 774–775 368–372 lewamizol 1008
– chenodeoksycholowy 703–704 – α-adrenomimetyczny 293, 361, 366 lewocetyryzyna 495
– etakrynowy 107, 733 – β-adrenomimetyczny 362–363 lewodopa 339–343
– foliowy 523–524 – antykoncepcyjny 107, 126, 441, lewofloksacyna 756, 880
– fusydowy 867 482–487 lewokabastyna 498, 758
– gadopentetowy 1031 – antyretrowirusowy 909, 980 lewometadon 236, 239
– glukuronowy 29–31 – cholinolityczny 299, 679 lewonorgestrel 477–479, 483–484,
– glutaminowy 185, 223 – dermatologiczny 129, 765 488
– hialuronowy 284, 1090 – hamujący łaknienie 205–207 lewopromazyna 167
– klawulanowy 838, 846 – immunosupresyjny 102, 277 lewotyroksyna 400, 404–405, 407
– kromoglikanowy 647 – interakcje 106 Leydiga komórki 389, 459, 460, 463
– linolenowy 806–807 – moczopędny 102, 126, 568–569 LH zob. hormon luteinizujący
– linolowy 783, 806 – nasenny 208–210 liberyna 393
– α-liponowy 444, 814 – neutralizujący kwas solny 126 libido 166, 332, 374, 398
– mlekowy 748 – nootropowy 201–202 lidokaina 296, 620
– nikotynowy 563–564 – opioidowy 217, 230–237, 239 limbiczny układ 152
– oleinowy 806 – podawanie 6–9 limfa 671, 726, 953
– pantotenowy 811–812 – przeciwarytmiczny 576, 613, 888 limfatyczne naczynia 465, 554, 726
– salicylowy 253, 767 – przeciwastmatyczny 123, 646 – węzły 517, 882
– siarkowy 31, 259, 465, 609 – przeciwbólowy 120, 217–290 limfocyty 93, 96, 271, 517–518
– tiaprofenowy 251 – przeciwdepresyjny 176–186 limfokiny 271, 986
– tioktanowy 814 – przeciwdrgawkowy 321–335 lindan 1092, 1093
– traneksamowy 548 – przeciwgruźliczy 885–889 linezolid 829, 831
– ursodeoksycholowy 702–704 – przeciwhistaminowy 104, 124, 214, linkozamidy 855, 864, 866–867
– walproinowy 104, 269, 329 349, 495–500 linolenowy kwas 806–807
kwinapril 606 – przeciwkaszlowy 243–244 lipaza 207, 359, 427, 539, 556
– przeciwnadciśnieniowy 102, 124, lipidy 14, 44, 108, 144
125, 564, 568 lipoliza 196, 358, 368, 399, 432
– przeciwobrzękowy 594 liponamid 807, 808, 814
L – przeciwpadaczkowy 321–335 liponowy kwas 444, 814
lacidypina 570, 571 – przeciwparkinsonowski 335–348 lipooksygenaza 507–509, 642, 684
laktacja 481 – przeciwpasożytniczy 786 lipoproteiny 554, 555, 558
β-laktamazy 830, 834, 840–841, 847 – przeciwpierwotniakowy 875, 938 liposomy 118, 765, 790
– lokalizacja 841 – przeciwreumatyczny 102, 104, 126, listerie 862, 1100
– reakcje alergiczne 841 284, 285 litu sole 186–187
– toksyczność 841 – przeciwświądowy 783 lizergid 204
β-laktamowe antybiotyki 82, 777, – przeciwwirusowy 757 lizozym 517, 989, 990
829 – przeciwwymiotny 348–353 lizyna 425, 434, 537, 548
laktitol 690, 702 – przeciwzakażeniowy 126 lizynopril 269, 573–575, 606
laktotropowy hormon 397 – przeciwzapalny niesteroidowy 247– – dawkowanie 574, 575
laktoza 671, 690 253 – działania niepożądane 574
laktuloza 102, 690, 702 – przeczyszczający 102, 688 – interakcje 574–576
laminina 951, 953 – psychostymulujący 195–199 – kinetyka 574
lamiwudyna 699, 700, 919 – psychotropowy 161–207 – przeciwwskazania 574
lamotrygina 324, 326, 330 – roślinny 137–138 lodoksamid 758
lanatozyd 609 – rozszerzający oskrzela 650–653 lomustyna 964
Langerhansa komórki 761, 787, 992 – – źrenicę 755 loperamid 693–694
– wyspy 388–391, 424 – sympatolityczny 373 lopinawir 923, 927
lanosterol 897 – trankwilizujący 189 loratadyna 495, 498, 650
lapis 774 – wykrztuśny 658–659 lorazepam 194, 325, 330–332
MUTSCHLER-2009.indd Sek1:1160 2010-03-18 20:12:20
Skorowidz 1161
lornoksykam 256 melanocyty 205, 397, 754, 761 metyloceluloza 106, 750
lowastatyna 442, 559 melanosomy 791 metylodigoksyna 608–610
losartan 577 melanoza 1064 metylodopa 374–375
LSD 104, 203, 501 melatonina 152, 209, 210, 388 metylofenidat 197
LTH zob. prolaktyna melanotropina 391, 396, 397 metyloparaben 823
lukasty 649 melfalan 962, 963, 986 mewalonowy kwas 559, 893
lumefantryna 112, 930–935 meloksykam 252, 256 mezlocylina 687, 847
lumirakoksyb 256–258 melperon 170 MHC 94, 96, 641, 766, 992, 995–996
lumirodopsyna 800 memantyna 69, 201 mianseryna 181, 186
lomustyna 964 menachinon 533 mibefradil 109
lutropina 462, 480, 974, 975 menadion 533 midazolam 309
lynestrenol 478, 483 meningocele 101 midodryna 390–391
meningokoki 832, 845, 1001, 1004 mięśnie, zwiotczenie 209, 298, 304,
menotropina 462 310
menstruacja 481, 678 mifepriston 479, 506
Ł mepiwakaina 294, 296 miglitol 436
łzy sztuczne 413, 760 merkaptoimidazol 410–412 migrena, leczenie 231, 249, 263–265
merkaptopuryna 958–959 mikcja 499, 619, 739–740
meropenem 852–853 mikobakterie 891
merozoity 930–931 mikonazol 892, 895, 903
M mesalazyna 509, 684 mikoplazmy 862
MAC 300, 301 meskalina 203 mikroalbuminuria 583
magnez 421, 491–492, 525, 622, 719 mesna 963 mikroangiopatia 433
– hipermagnezemia 721 mesuksymid 325, 333 mikrokrążenie 246, 447, 528, 592,
– hipomagnezemia 721 metabolizm 6, 16, 24, 35 792
– preparaty 722 metadon 236–237, 240 mikropigułka 482, 486
– związki 677 metale 515, 521, 862, 1059 mikroskładniki odżywcze 795–815
makroangiopatia 433 – ciężkie 40, 70, 283, 820–821 mikrosomy 26, 29
makroblast 513 metaloidy 1040, 1059–1066 mikrotubule 901, 942, 969–970
makrofagi 94, 223, 517 metaloproteazy 788 milrynon 612
makrolidy 82, 284, 675, 862–865 metamfetamina 197, 341 mineralokortykosteroidy 720
– dawkowanie 865 metamizol 252, 260–262 minocyklina 860–862
– działania niepożądane 865 – dawkowanie 260 minoksydyl 102, 579–580
– działanie 864 – działania niepożądane 260 miotyki 378, 753
– interakcje 865 metanol, zatrucie 1052 mioza 234, 748, 968
– oporność 864 metergolina 398 mirtazapina 181
malaria 817, 929–930 metformina 437, 441–443 mitochondrium 144, 149, 355, 425
malonowy kwas 775, 1075 methemoglobina 1057, 1080–1081 mitoksantron 971, 973
maltotrioza 671 metionina 260, 523, 697, 1036 mitomycyna 971, 973–974
maltoza 671 metoda galenowa 8 mitoza 79, 901, 945, 955, 967–970
mangan 815, 1031 metoheksital 214, 303–305 miwakurium chlorek 316, 318
mannitol 593, 737, 755 – dawkowanie 305 mizolastyna 498
MAO zob. monoaminooksydaza – działania niepożądane 305 mizoprostol 126, 255, 506, 678
marihuana 203–204 metoklopramid 350, 503, 696 mocznik 102, 526, 705
mastocytoza 495 metoksalen 768, 769 modafinil 198
mebendazol 102, 941–942 metoksypsoralen 769 model dwóch konformacji receptora
mebeweryna 385 metoksytyramina 338 66–67
medrogeston 476, 479, 488 metoprolol 269, 369, 607 – farmakokinetyczny 47–51
medroksyprogesteronu octan 476, metotreksat 277–285 – płynno-mozaikowy 9
479, 483, 978 metronidazol 104, 876–877 modelowanie molekularne 128–129
mefenytoina, polimorfizm 115 – dawkowanie 877 modulatory receptora estrogenowego
meflochina 102, 930, 931 – działania niepożądane 877 422, 473
mefruzyd 732 – interakcje 877 moklobemid 184, 186
megakariocyty 512, 518, 1010 – kinetyka 876 moksonidyna 373–374
megalocyty 522 – przeciwwskazania 877 molibden 814
megestrolu octan 476, 478, 978 metycylina 839–840 molsidomina 102, 629
Meissnera splot 160 metyksen 345–346 mometazon 447, 777
meksyletyna 618, 620 metylacja 33 monitorowanie leczenia 567
MUTSCHLER-2009.indd Sek1:1161 2010-03-18 20:12:21
1162 Skorowidz
– stężenia leku 56–57 nebiwolol 369, 372, 607 nikorandyl 631
monoaminooksydaza 28, 176 necynowe kwasy 1085 nikotyna 353, 1083–1084
– inhibitor 81, 107, 112, 176, 178, nedokromil 499, 643, 647, 758 nikotynamid 810–811
184 nefazodon 185 nikotynowy kwas 435, 563–564, 811
monoaminy 161, 164, 176–178, 242 nefopam 244 nimodypina 570, 572
monobaktamy 838, 852–853 nefrokalcynoza 678 nisoldypina 570, 572
monocyty 512, 517, 682, 991 nefron 390, 602, 706–707, 709, 729 nitraty 82, 87, 595, 626–629
monooksygenazy 24, 28, 886, 1069, nekroliza 97–98 nitrazepam 212, 213
1073 nekroza 81, 766, 792, 886, 981, 1066 nitrendypina 570, 572, 582
montelukast 509, 649, 654 neomycyna 97, 702, 856–857, 859 nitrobenzen 1080
– dawkowanie 649 neostygmina 317, 380, 1053 nitrogliceryna 35, 582, 628
– działania niepożądane 649 neowaskularyzacja 758, 983 nitroprusydek sodu 579–580
morfina 230, 240, 632 nerki 705–716 nitrowe związki zob. azotany
motylina 697, 865 – hormony 389 nizatydyna 675–676
mukopolisacharydy 284, 286, 658 – nadciśnienie 565 NLPZ 126, 246–248, 253–259
mukowiscydoza, leczenie 661 – niedokrwistość 521 nocyceptory 217, 219–224, 500, 501
mupirocyna 879 – współczynnik oczyszczania 45 nocyceptyna 229
mureina 836–839 – wydalanie 41–42, 110 nootropowe leki 201–202, 593
muscymol 1089 – zapalenie 59 noradrenalina 159–161, 176–178,
muskaryna 377–379 nerwowy układ 101, 107, 143–160 358–360
mydriatyki 755–756 – leki 161–207 – inhibitory 181–184
mykofenolan mofetilu 1019 – przewodzenie 145 – metabolizm 353–355
– typy synaps 148 nordazepam 194
– zakończenia 81 norepinefryna zob. noradrenalina
netylmycyna 856, 860 noretysteron 478, 479, 484
N neuralgia 262–263, 275, 326, 785 norfloksacyna 756
N2O zob. azotu podtlenek neuraminidaza 88, 129, 910 norgestimat 484
nabumeton 258 – inhibitory 911 norgestrel 478
N-acetylotransferazy 33, 115 neurastenia 188 norketamina 306
nadchlorany 410, 412, 1080 neuroborelioza 881 normoblasty 513
nadciśnienie, leczenie 564–568 neurodegeneracyjne choroby 121, normocyty 513, 522
nadmanganian potasu 819, 1065 335 norpetydyna 239
nadolol 372 neuroleptanalgezja 298, 310 nortryptylina 180, 185
NADP+ 28, 116, 523, 811 neuroleptanestezja 310 nos, zatkany 166, 178
NADPH 28, 116, 534, 1080 neuroleptyki 163–169, 188 noskapina 238, 244
nadtlenek azotynu 628 – atypowe 169, 172–173 NO syntaza 1018
– benzoilu 774–775 – o przedłużonym działaniu 174 nukleinowe kwasy 465, 805, 819,
– wodoru 38, 819, 1069 neuron 81–82, 122, 143 869–877
nafarelina 460, 461 – budowa 144 nukleotydy 71–73, 120, 278, 522
nafazolina 361 neuropatia 262, 433, 808, 921, 970 – analogi 912
nalokson 237–239, 242, 308, 1052 neuropeptyd 150, 223, 266, 604, 642 nukleozydy 699, 702, 905, 909
naltrekson 237 neuroprotekcyjne substancje 201 – analogi 912
nałóg 104–105 neuroprzekaźnik 65, 71, 149–150 nykturia 416, 451, 604, 742
NANC włókna 160, 642, 664 neurotoksyna 1089 nystatyna 757, 898–899, 903
naproksen 251, 255, 256, 269 neurotransmiter 81, 148, 627, 1091
narkoanalgetyk 217 newirapina 97, 922, 927
niacyna 796–798, 810
narkolepsja 161, 196–198, 307
narkotyna 238 nicergolina 202 O
narkoza 132, 155, 208, 296, 616, niedociśnienie, leczenie 585–586 obidoksym 1096
1049 niewydolność serca 565–566, 601– objętość dystrybucji 44
naskórkowy czynnik wzrostowy 77, 613 obstrukcja 639, 646
407, 974 – ostra 613–614 ochratoksyna A 1105, 1106
NAT zob. N-acetylotransferazy – przewlekła 613 odma 643, 655
natalizumab 1021–1022 nifedypina 124, 269, 569–573, 611 odtrutki 1047, 1049, 1051
natamycyna 757, 898–899, 939 nikardypina 570 – leczenie 1052–1056
nateglinid 440 nitrendypina 570 ofloksacyna 756
natriuretyki 438, 728 nikiel 97, 1065, 1106 okres półtrwania 46
natriureza 392, 416 niklozamid 940 oksacylina 842, 843, 845
MUTSCHLER-2009.indd Sek1:1162 2010-03-18 20:12:21
Skorowidz 1163
oksaliplatyna 964–965 – potyliczna 323 – podawanie 847–848
oksazepam 190–194 paklitaksel 129, 631, 970–971 – zakres działania 842
okskarbazepina 187–188 paliatywne leczenie, nowotwory 461, pentachlorofenol 1070
oksprenolol 370 462, 954–955 pentamidyna 900, 936–939
oksybutynina 385, 742 palifermina 792, 986 pentazocyna 233, 236, 241
oksygenaza 24 paliwizumab 660, 917, 1021 pentobarbital 305
oksykamy 246, 256 palmitynowy kwas 1034 pentoksyweryna 243, 244
– pochodne 97 palonosetron 350, 502, 503 pentostatyna 958–959
oksykodon 233, 239, 263 pankreatyna 695 pepsyna 665, 667, 671, 678, 695
oksykonazol 892, 985 pankuronium bromek 315, 317 pepsynogen 665, 667, 672, 679
oksylofryna 360–361 pantoprazol 673, 675, 680 peptydoglikan 836, 839, 854
oksymetazolina 361 pantotenowy kwas 811–812 peptydowe hormony 388, 392–394,
oksytocyna 391, 393, 401–403, 489– papaweryna 238, 384–385, 594 396, 417
491 paracetamol 252, 259–262 perazyna 165, 167, 168
oktenidyna 825 – dawkowanie 259 perfenazyna 167–168, 349, 351
oktreotyd 395, 401 – zatrucia 259 pergolid 342, 344
olanzapina 163, 170, 172, 188 parametry farmakokinetyczne 6, 43– perindopril 573–575
oleander, zatrucie 1045 57 permetryna 786, 1092, 1094
olejki eteryczne 284, 658, 728, 1081 paraokson 37, 1095, 1096 peroksydaza 404, 410, 411, 507, 815
oligodendrocyty 122, 144–145 parasympatykolityki 380, 384, 617, persorpcja 9, 11
oligonukleotydy antysensowne 118– 652, 679, 755 perystaltyka 126, 157, 265, 359, 401
122 parasympatykomimetyki 378–380, pestycydy 1039, 1041, 1044
oligopeptydowe hormony 393 751, 753 petydyna 236, 239–240, 307
oligospermia 283, 684 parathormon 414–418, 421 pierwiastki śladowe 526, 814–815,
oliguria 463, 502, 612 paration 1095 1059
olmesartan 577–578, 606 parekoksyb 256–258 pigmentacja 391, 397
olsalazyna 509, 684 parenteralne podanie 7, 8, 17, 233 – zmiany 593, 764, 791
ołów 22, 815, 1040 parestezja 263, 279, 366 pikosiarczan sodu 691–692
omalizumab 645–646, 653, 1021 parikalcytol 416–417 pilokarpina 85, 378–379, 753, 755
omeprazol 673–675, 680 Parkinsona zespół 107, 157, 165–166, pimekrolimus 782, 1014–1016
ondansetron 502, 503 335–337 pimozyd 167, 171, 352
onkogeny 117, 948, 952 – leczenie 125, 337 pindolol 368–370
opiaty zob. opioidy paroksetyna 182–183 pinocytoza 9, 11
opioidy 233–237, 693 paromomycyna 856–857, 859 pioglitazon 440–441
opium 238, 384, 1082 PBP 839–840, 845, 853 piperacylina 835, 844, 847, 860, 880
opsyna 749, 800 Pearla wskaźnik 485, 486 piracetam 202
orcyprenalina 362, 363, 617, 1048 pegaptanib 759 pirazynamid 828, 884–886, 890
orlistat 207 peginterferon 701 – dawkowanie 886
orotowy kwas 815 pektyna 694 – działania niepożądane 886
ortostatyczna regulacja 166, 173 pelagra 811 – działanie 886
oseltamiwir 904, 910–911 pemetreksed 957–958 – przeciwwskazania 886
osmodiuretyki 881 penam 838 pirenzepina 384, 679
osmolarność 390, 713–715, 718, 758, penbutolol 368, 370 pirogeny 244, 245
1027 pencyklowir 909–910, 912–914 piroksykam 251, 256, 284
osmotyczne ciśnienie 390, 402, 525, penicylamina 283–284, 1053–1055, pirolizydynowe alkaloidy 1085, 1106
689 1062 pirydoksyna zob. witamina B6
osteoporoza 276, 329, 415, 418 – dawkowanie 284 pirydostygminy bromek 380
– leczenie 418–422 – przeciwwskazania 284 pirymetamina 102, 104, 866, 873,
ototoksyczność 833, 854, 858, 860, penicyliny 284, 817, 828–830, 834– 935–937
954, 965 848 placebo 132
owulacja 166, 190, 460, 462, 475, 480 – acyloaminowe 847 platyna 731, 954, 964–965
ozon 819, 1075–1076 – benzylowa 845 plazmid 829, 840, 856, 861, 865
– dawkowanie 844 plazmina 82, 510, 531–532
– doustne 845 plazminogen 82, 531–532
– działania niepożądane 844 pneumokoki 657, 834, 842, 845, 880,
P – oporne na penicylinazę 845 1004
Paciniego ciałka 762 – o poszerzonym zakresie działania pobudzający potencjał postsynaptycz-
padaczka 173, 216, 269, 321, 324 846 ny 149
MUTSCHLER-2009.indd Sek1:1163 2010-03-18 20:12:21
1164 Skorowidz
podanie leku, drogi 7–8 prokinetyki 350, 681 psychodysleptyki 204
podofilotoksyna 905 prolaktyna 163, 391, 396–398 psylocybina 203–204
podtlenek azotu 296, 301–302 – hamowanie wydzielania 394, 398 psylocyna 203–204
polietylenoglikol 690, 700 prolek 38, 43, 110 PTH zob. parathormon
poliklonalne przeciwciała 993, 1014, prometazyna 168, 349, 351, 496 puryna 279, 526, 875, 909, 958–960
1019–1022 propafenon 618–620 PUVA 766–769, 774
polimerazy 698–700 propionowy kwas 246, 251, 690 pyrantel 940–941
polimorfizm genetyczny 27, 114, 117 propiweryna 385, 742 pyretroidy 1091–1094
polimorfizm CYP3A4 115 propofol 305–306 pyretrozyna 1093
– CYP2C19 115 propranolol 268–269, 368–373 pyritinol 202
– CYP2D6 115 propyfenazon 247, 252, 260
– N-acetylotransferazy 115 propylotiouracyl 407, 410–411
– pseudocholinoesterazy 116 prostacyklina 493, 504–507
polimyksyna B 102, 878 prostaglandyny 102, 220–226, 247– R
polipeptyd C 424 249, 504 rabeprazol 673, 675
pompa jonowa 146–147 – działanie 504–505 racemat 60, 89, 237, 255, 296
porfiria 486 – metabolizm 504 radioaktywne izotopy 982
poryny 830 – pochodne 490 radionuklidy 1035
posakonazol 895–896 – synteza 505 radiofarmaceutyki 1035–1036
postać galenowa 9, 44, 233, 777 prostamidy 754 raloksyfen 472–474, 487
potas 718 protamina 434 ramipril 573–575, 606
– preparaty 721–722 prostanoidy 504 randomizacja 134, 136, 139
– zaburzenia gospodarki 719–721, 725 – receptory 504 ranelinowy kwas 421
potencjał spoczynkowy 145 prostata 107, 269, 504, 739–740 ranibizumab 759
– czynnościowy 146 – leczenie 740–741 ranitydyna 675–676
pralidoksym 1096 – rak 461–462 rapamycyna 949, 1016
pramipeksol 342, 344 proteasomy 985 rasagilina 341, 343
pranlukast 650 – inhibitory 985 Raynauda choroba 268, 273, 285, 589
prawastatyna 559–560 proteaza 82, 449, 509, 910 reakcja I fazy 24–29
prazepam 192 – inhibitor 88, 135, 510, 910, 922– – II fazy 23, 29–33
prazykwantel 104, 939–941, 943 927 – alergiczna 92–98
prednikarbat 777, 781 proteinuria 117, 420, 433, 604 – anafilaktyczna 93, 95
prednizolon 37, 452–453 proteoliza 404–405, 427, 546 – cytotoksyczna 93
prednizon 37, 452–453 protrombina 254, 525, 529–531, 534 – poprzetoczeniowa 96
pregnandiol 469, 470 protyrelina zob. tyreoliberyna – pseudoalergiczna 99
pregnenolon 464 prowitaminy 795, 798, 801 reboksetyna 183–184
premedykacja 165, 298–299 przeciwciała 92–96, 983–984, 993– rebound efekt 373
preparat złożony 125–127 997 receptor 64–81
preprohormon 388, 393 – budowa 993–994 – agoniści/antagoniści 65
priapizm 185, 506, 595 – klasy 994 – błonowy 71
prilokaina 294, 296 – monoklonalne 645, 993 – farmakologiczny 64
primachina 116 – powstawanie 993 – jonotropowy 73–78
primidon 197, 325, 327, 329–331 – reakcje nadwrażliwości 93 – naskórkowego czynnika wzrostowe-
proakceleryna 530 – właściwości 994 go 949, 983–984
probenecid 110, 253, 258, 289, 912 przełom nadciśnieniowy 580, 582 – podtypy 64
probiotyki 686 – nadnerczowy 457 – wewnątrzkomórkowy 70
procyklidyna 345, 346 – tarczycowy 410, 413 – zmiany funkcji 65
progesteron 445–446, 468–470, 475– przesączanie kłębuszkowe 41–42 – związany z białkiem G 71
476, 479–481 przewodzenie pobudzenia 145, 148 – – z enzymami 78–81
program szczepień 998–1000 przyzwyczajenie 104 redoks układ 813
proguanil 880, 882–883 pseudoalergiczne reakcje 99, 1030 redukcja 28–29
prohormon 37, 930, 932–933, 935– pseudocholinoesteraza 376 refrakcja 147, 599
937 – polimorfizm 116 regulacja hormonalna 389–391
proinsulina 424, 431 psoraleny 766, 768–769 – presynaptyczna 150
prokaina 294–296, 815 – przeciwwskazania 769 REM faza 208–214
prokarbazyna 965–966 – stosowanie 769 remifentanyl 303, 307–309
prokaspazy 950 psychoanaleptyki 195 remisja 175, 186, 273, 276, 285
MUTSCHLER-2009.indd Sek1:1164 2010-03-18 20:12:21
Skorowidz 1165
renina 358, 449, 451, 552, 568 – mechanizm działania 1016
rentgenowskie środki kontrastowe S sitosterol 562, 741
1027–1030 sacharoza 436, 442, 671, 678 sitosterolemia 555, 561
repaglinid 440 safrol 1105–1106 Sjögrena zespół 270, 272–273, 285,
repelenty 936 sakwinawir 112, 908, 922–923, 925, 379
replikacja 118–120, 702, 871, 904– 927 sklerodermia 584
907 salazosulfapirydyna 26, 282–283, 685 skopolamina 299, 349, 351, 381, 384,
reproterol 363, 651 salbutamol 363, 651 755
resorpcja 5, 14–18, 671 salicylany 109, 246, 250, 254, 289 skrobia 526–528, 671, 1033
– hamowanie 690–692 salmeterol 363, 651, 653 – hydroksyetylowana 493, 528
restenoza 631 saluretyki 126, 187, 253, 289, 569 skuteczność leku 3
reteplaza 547 saponiny 658, 1087 skwalen 557, 893, 897
retikulocyty 513, 701, 1012 sarkoidoza 400, 455, 638, 721 SNRI 178, 181, 195, 206
retikulum endoplazmatyczne 73, 357, sarkoplazmatyczne retikulum 77, 117, solanina 1085
579, 669, 934 299, 311, 355 solny kwas 420, 424, 664, 672, 677,
– sarkoplazmatyczne 77, 600 SARS 905 1066
retinol zob. witamina A sartany 135, 576–578, 606 – leki neutralizujące 126
retinopatia 283, 433, 566, 751 satelitarne komórki 145 – wydzielanie 665
retinowy kwas 70, 771, 798, 799, 980 schistosomy 939 somatorelina 394
retrowirusy 108, 118–119, 869, 905, schizofrenie 162–163 somatostatyna 394–395, 399, 667
917 – leczenie 163 – analogi 395, 401
retynoidy 774 sedacja 99, 229, 232, 234, 241, 298 somatotropina 391, 394, 396, 398–
reumatyczne choroby, leczenie 269– sekrecja kanalikowa 41–42, 55 401
285 sekretolityki 661 – antagoniści 399
rewaskularyzacja 590, 632 selegilina 338, 341, 343, 347 sorbitol 433, 690, 703, 737, 1050
Reye’a zespół 254 selektyna 279, 1022 sotalol 368, 370, 620–621
rezerpina 102, 164, 374–375, 568 selen 786, 814, 815, 1059, 1064– sól glauberska 690
rezerwa receptorowa 64–65 1065 – gorzka 690
rezorcyna 774 sen 208 sparteina 1082
rifampicyna 102, 828, 885, 887–888, – fazy 208 – polimorfizm 115
890 – leki 211–215 spazmolityki 69, 384
riketsje 845, 860, 865 – zaburzenia 210 – neurotropowe 380–383, 742
Ringera roztwór 526 sennozyd 691 spektynomycyna 882
risperidon 163–165, 173–174 sensor 147, 291, 683 spiramycyna 862–865, 937
ritonawir 741, 922–927 sepsa 543, 627 spirapril 573, 575
rituksimab 982, 983, 987 SERCA 600 spironolakton 102, 104, 457, 606–607
riwastygmina 200–201 serotonina 174, 176–178, 220, 500– sprzęganie 24, 29–33
RNA wirus 697, 700, 702, 905 503 srebro 815
– leki 916–917 – inhibitory wychwytu zwrotnego SSRI 176, 178, 182–183, 195
rodopsyna 749, 800 181– 184, 195 stała szybkości eliminacji 46
rofekoksyb 256, 257 – transporter 82, 117 stan padaczkowy 324
roksytromycyna 675, 862–864 sertindol 165 – równowagi 6, 19, 46, 51
rokuronium bromek 315, 317 sertralina 182–183, 185 status epilepticus zob. stan padacz-
ropiwakaina 295–296 setrony zob. antagoniści 5-HT3 kowy
rosiglitazon 440–441 sewofluran 300, 302 statyny 44, 102, 104, 559–563, 633
rotawirusy 997, 1005 Sheehana zespół – dawkowanie 559
równanie Batemana 51 siarczan baru 1027, 1051 – działania niepożądane 559
rtęć 602, 815, 821, 1039, 1040 siarka 851, 1065 – przeciwwskazania 561
Ruffiniego ciałka 762 siarkowodór 1053, 1079 stawudyna 917, 919–921, 927
rutyna 594 sibutramina 206 – działania niepożądane 921
rybawiryna 671, 866 sildenafil 14, 27 stearynowy kwas 806
ryboflawina zob. witamina B2 silikony 1051 stenoza 615, 624
rybosom 120, 424, 642, 745, 855–856 siła działania leku 3 stereoizomery 89, 242, 317, 759
ryboza 808, 811, 916, 922 simwastatyna 110, 559–561 steroidowe hormony 70, 388, 464–
rycyna 1086 sirolimus 135, 631, 1014–1017 465, 469
rycynowy olej 690, 1069 – dawkowanie 1017 – analogi 388
rytm dobowy 123 – działania niepożądane 1017 sterylizacja 294, 527, 817, 823
MUTSCHLER-2009.indd Sek1:1165 2010-03-18 20:12:21
1166 Skorowidz
Stevensa-Johnsona zespół 97, 244, synapsa 144, 387, 668, 800, 1091 tenipozyd 27
278, 330, 922, 1054 – adrenergiczna 150, 355 tenofowir 904, 909, 919–920, 922,
stężenie minimalne terapeutyczne 47, – charakterystyka 149 927
53, 83 – ingerencja farmakologiczna 150 tenoksykam 256
– toksyczne 47, 53, 56 – przewodzenie bodźca 148 tenzydy 106, 1050, 1068
streptograminy 129, 832, 855, 864, synaptobrewina 319 teobromina 195–196, 737
868–869 synchronizacja 955 teofilina 104, 196, 650, 653, 656, 869
– dawkowanie 868 synergizm 87, 832 terapeutyczny reżim 340, 890, 987
– działania niepożądane 868 – hiperaddycyjny 125 terapia antysensowna 120
– interakcje 868 syntaza ALA 1060 – genowa 118–119
– przeciwwskazania 869 system terapeutyczny przezskórny 17, – za pomocą komórek pnia 121–122
streptokinaza 532, 546–547, 549, 792 135, 241 teratogenność 36, 952
streptokoki 862, 880 szczepienia 905, 997–1007 teratogeny 101–102
streptomycyna 102, 817, 828, 829, szok 1067 terazosyna 367–368, 741
856–859, 885 – anafilaktyczny 255, 851, 1033 terbinafina 900, 903
– dawkowanie 859 – kardiogenny 373 terbutalina 363, 651–652
– działania niepożądane 859 – septyczny 1089 terfenadyna 110, 352, 495, 499, 865,
– przeciwwskazania 859 śladowe pierwiastki 526, 814–815, 897
stres 162, 174, 188, 210, 448 1059, 1065 terlipresyna 402–403, 703
– mechaniczny 627 śluz 126, 299, 331, 374 termoreceptory 244
– oksydacyjny 34 śluzowaty obrzęk 409 termoregulacja 167, 244–245, 337,
strofantyna 8 śluzowe błony 15–16 511, 1073
strychnina 313, 320, 1049, 1085– terpeny 1081
1086, 1098 teryzydon 837, 855, 885, 889
substancja P 160, 220, 222–224, 226, testosteron 391, 417, 459–460, 463–
642 T 469
sufentanyl 241, 303, 307–309 tabletki 7, 15, 18 tetracykliny 82, 102–104, 860–862
sukcynylocholina 114 tachyarytmia 499, 610, 652 – dawkowanie 861
sukralfat 678 tachykardia 166, 196–198, 610, 652 – działania niepożądane 862
suksametonium 106, 293, 313, 316, takalcytol 768–769 – przeciwwskazania 862
318 takrolimus 683, 685, 782, 897, 1014– tetrahydrokortyzol 448
sulbaktam 846–847 1017 tetrakaina 294, 295, 756
sulfadiazyna 821, 872–873 takryna 200 tetramizol 1013
sulfametoksazol 872–876 taksany 955, 970–971 tetrazepam 320
sulfasalazyna zob. sulfazosulfapiry- tal 815, 1040, 1063 tetrodotoksyna 78
dyna talidomid 100, 102, 214–215, 810, tetryzolina 361, 758
sulfazosulfapirydyna 277, 282–283, 891, 952 tiabendazol 941–942
509, 684 talinolol 369, 371 tiagabina 325, 326, 328, 330–331
sulfonamidy 30, 33, 82, 95–98, 102– tamoksyfen 473, 955, 974, 976–978 tiamazol 410–411, 413
104, 872–875 tamol 783 tiamina zob. witamina B1
– dawkowanie 875 tamsulosyna 367–368, 741 tiapryd 166, 345–346
– działania niepożądane 875 tasiemiec 939–940, 942 tiazydy 102, 731–738
– interakcje 875 taurolidyna 821 tiklopidyna 537, 631
– przeciwwskazania 875 tauryna 669, 821 tilidyna 237, 242
– wskazania 874 tautomeria 256 tiludronian 418–420
sulpiryd 171–173, 350–351 tautomery 89 tiobarbiturany 303–305
sulproston 490, 505–506 tazaroten 766, 771 tiocyjanian 1079
sultiam 328, 330, 333 tazobaktam 846–847, 880 tioeter 28
suramina 937–938 tegaserod 688 tioguanina 958–959
surfaktant 660 teikoplanina 852–854 tiokonazol 892, 895
sygnału transdukcja 6, 64–65, 67, 71 teleangiektazje 782–783, 788 tioktanowy kwas 814
sympatolityki 356 telitromycyna 862–865 tiopental 214, 303–305, 1049
sympatomimetyki 362 telmisartan 577–578, 606 tiorydazyna 167–168
α-sympatykomimetyki 16, 150, 293, telomeraza 951 tiotepa 963
356, 360–361 telomery 951 tiotropium bromek 381, 384, 653,
β-sympatykomimetyki 103, 356, temazepam 193–194, 212–213, 215 656–657
362–363 temoporfina 981 tiouracyl, pochodne 410–411
symport 10 temozolomid 965–966 tirofiban 538
MUTSCHLER-2009.indd Sek1:1166 2010-03-18 20:12:21
Skorowidz 1167
tiuksetan ibrytumomabu 982, 1021 tretynoina 771–775, 791, 980 tyreotropina 390, 394, 396, 405–409
TIVA 309 triamcynolon 454, 777 tyroksyna 389–391, 396, 404–411,
tlenek azotu 160, 224, 858 triamteren 126, 611, 728, 730, 735– 1009
– etylenu 818, 823–824, 1105 736 tyrozyna 291, 354, 388, 391–392, 404
TNF 81, 95, 221–223, 245, 279–281 – dawkowanie 736 tyrozynemia 1023, 1025
tobramycyna 756, 856, 860 – działania niepożądane 736 tytoń 353, 486, 559, 605, 1083–1085
tokoferol zob. witamina E – interakcje 736 tyzanidyna 319
tokolityki 491–492 triazolam 212, 215
tokoliza 362, 491–492 triheksyfenidyl 345–346
toksoidy 997, 1001–1002, 1006 trijodotyronina 390–391, 396, 404–
toksoplazmoza 101, 826, 874, 918, 406, 409 U
937 triklosan 823 ubikwityna 985
toksyczność 106, 107, 127, 841, 1043, trimetazydyna 631 uniporter 10
1052 trimetoprim 102, 104, 830, 873–876, uran 1066
– reprodukcyjna 100 890 urapidil 367–368, 502
– z pobudzenia 201 – dawkowanie 876 urografia 1027–1029
toksyferyna 317 – przeciwwskazania 876 urokinaza 531, 546, 548, 549
toksykologia 3–4, 1037 trimipramina 178–179, 186 ursodiol 704
toksyna botulinowa 319–320, 1039, triptany 135, 264–268, 502 urykostatyki 288
1081, 1100 triptorelina 460–461, 975 urykozuryki 288
tolbutamid 109, 438–439, 1040 Tris 724 utlenianie 24–28, 35, 37
tolerancja 87, 105, 129–131, 237–238, trisomia 21 100 UV-A 768, 788–790
298 trombina 509, 529–531, 534–541, UV-B 767, 788–790
– na azotany 627–628 813 uzależnienie od leków 104–105
tolonium chlorek 1052, 1053, 1076, – inhibitory 542–543 – opiatów 239, 374
1080 trombocytopenia 96, 282, 329, 412,
tolterodyna 382, 384, 742 439, 518
toluidyna 1103 trombocytoza 451, 521
topiramat 268–269, 324, 328, 332, trombocyty 108, 122, 220, 253, 518 W
334 tromboksan A2 247–249, 253, 507 waldekoksyb 256–258
topoizomeraza 116, 871, 956 trombolityki 546–548 walproinian 324–325
– inhibitory 967–969 trombopenia 699 walsartan 577–578, 606
topotekan 968 trombopoetyna 512, 518, 1010, 1011 wankomycyna 102, 829–831, 852–
torasemid 733 trombostenina 530, 532 854
toremifen 976–977 trombozy 631 – dawkowanie 853
torsade de pointes 499, 620, 722 tropikamid 382, 384, 755 – działania niepożądane 854
tramadol 232, 237, 242, 263 tropisetron 350–351, 502–503 – interakcje 854
tramazolina 361 trucizna 3, 10, 103, 137, 1039 – przeciwdziałania 854
trandolapril 573, 575 tryptofan 150, 184, 388, 429, 500, wapń, antagoniści 569–572, 630
trankwilizatory 161, 189 811 – hiperkalcemia 721
transacylacja 856 TSH zob. tyreotropina – hipokalcemia 721
transducyna 800 TTS 17, 241, 472 – jony 718–719
transfer genów 118–119, 424 tuberkulinowa próba 280 – preparaty 722
transferyna 515–516, 520, 525 tuberkulostatyki 883 – stężenie w osoczu 525
transkobalamina 522, 525 tubokuraryna 99, 315, 317–318 – usunięcie jonów 539
transkortyna 389, 448, 525 tubulina 287, 969–970 warfaryna 544–545, 584, 675, 897
transkrypcja 34, 62, 71–72, 77–78, 80 tyfus 694 wazopresyna 390–391, 393, 401
– czynniki 221, 225, 282 tymol 822–823, 901 – analogi 402–403
– odwrotna 119 tymolol 370, 751 – działania 401
translokacja 70–71, 80, 769, 855–856, tymopentyna 423 – mechanizm działania 402
947 tymopoetyna 423 wchłanianie 5–18
transplantacja 583, 656, 698, 770, tymozyny 423 – bariery 9
962, 1020 tyramina 112, 184, 264, 362, 364 – kanalikowe zwrotne 41–42
transport czynny 10–11 tyreoglobulina 396, 404, 405, 410– – leków 14
tranylcypromina 184, 186 412 – mechanizmy 9
trastuzumab 983–984 tyreokalcytonina 391, 417–418 Wegenera ziarniniak 270, 273–274,
trazodon 185–186 tyreoliberyna 393–394 285
treosulfan 963 tyreostatyki 102–104, 409–413 wekuronium bromek 315, 317
MUTSCHLER-2009.indd Sek1:1167 2010-03-18 20:12:21
1168 Skorowidz
wenlafaksyna 176–177, 181–182, – ekstrakcji 45 – Parkinsona 165–166, 335–348
262–263 – oczyszczania 45–46 – piekących stóp 796, 1147
werapamil 569–573, 611, 621–622 – powinowactwa 21 – Reye’a 254
wersenian sodowo-wapniowy 1053 wstrząs, leczenie 586–587 – Sjögrena 273, 285
werteporfina 759 – anafilaktyczny 96 – Stevensa-Johnsona 95, 97
węgiel aktywowany 694, 1049–1050 wydalanie 5–6, 9, 21, 40–43 zileuton 509
węglowodory halogenowane 303, wymiotnica 1050 ziprazidon 173–174
1069–1071 WZW, leczenie 697–702 zjawisko pierwszej dawki 828
wiązanie z białkami 18, 20–21 – szczepienia 998–1000 złoto 282, 285, 815, 1054, 1962
wigabatryna 325–327, 330–331 wzwód, zaburzenia 594–595 Zollingera-Ellisona zespół 674
Wilmsa guz 971 zolmitriptan 27
winblastyna 955, 969–970 zolpidem 212–214
winkrystyna 969–970, 987 zonisamid 325–327, 330
witamina A 705, 771, 796–800
– B1 796–798, 807–809
Z zopiklon 212–215
zotepina 171–172
– B2 809–810 zaleplon 212–214 związek chiralny 60–61
– B6 810–811 zanamiwir 904, 907, 910–911 – fosforoorganiczny 379, 1052–1053
– B12 522–523 zaparcie, leczenie 688–693 zwiotczenie mięśni 80, 209, 298, 304
– C 813–814 zatrucia 1059–1106 zydowudyna 888, 915–917, 919–921
– D 415, 801–804 – alkaloidami sporyszu 366 zygota 100, 101, 931
– E 796–797, 804–805 – glikozydami nasercowymi 611
– H 812–813 – kolchicyną 288
– K 805
– PP (amid kwasu nikotynowego)
– kwasem acetylosalicylowym 254
– litem 187
Ż
810–811 – opioidami 238 żelatyny 526–528
wlew kroplowy 54 – tabela 1107 żelazo 511
włókno NANC 160, 642–643 – żelazem 520 – metabolizm 514–515
woda 19 zespół bezdechu sennego 210 – niedobór 519
– gospodarka 717 – Cushinga 451 – preparaty 519
– resorpcja 689 – jelitowy 688 – zapotrzebowanie 516
– wydalanie 689 – lęku uogólnionego 188 – zatrucie 520
wodzian chloralu 214 – Lennoxa-Gastauta 324 żółtaczka 276, 487, 1087
wskaźnik terapeutyczny 86 – Lyella 97 – cholestatyczna 467
– masy ciała 205 – metaboliczny 430 – hemolityczna 514
– Pearla 485–486 – nadnerczowo-płciowy 450 – jąder podstawnych 103, 109, 875
– Quicka 110, 545 – otępienny 199, 203 żywice jonowymienne 18, 407, 561–
współczynnik dyfuzji 10 – padaczki skroniowej 322 562
MUTSCHLER-2009.indd Sek1:1168 2010-03-18 20:12:21
FARMAKOLOGIA OGÓLNA
MUTSCHLER-2009.indd 1 2010-01-07 22:10:36
MUTSCHLER-2009.indd 2 2010-01-07 22:10:40
3
1. Definicje
Definicje
Substancje czynne są to substancje wywołujące ■ nauka o działaniu leków na zdrowe i chore orga-
w żywym organizmie działanie biologiczne. Mianem nizmy,
działania biologicznego określa się całokształt zmian
albo też ujmowana znacznie szerzej jako:
wywołanych przez substancję czynną w układzie bio-
logicznym. ■ nauka o wzajemnym oddziaływaniu między związ- A1
Substancje lecznicze są to substancje czynne kami chemicznymi i układami biologicznymi.
mogące służyć do zapobiegania, łagodzenia, lecze-
nia lub rozpoznawania chorób (pojęcie substancji Obie te definicje mogą nie być w pełni zadowalają-
leczniczej ma więc dające się oszacować działanie ce, ponieważ są albo niedostateczne, albo zbyt sze-
– w przeciwieństwie do substancji czynnej). Przez rokie. Niemniej jednak obie znajdują się pomiędzy
pojęcie lek rozumie się przeznaczone do stosowa- możliwymi do wyznaczenia granicami pojęcia far-
nia u ludzi lub zwierząt określone postacie środków makologia.
leczniczych (angielskie określenie drug jest iden- Granice między farmakologią i pokrewnymi na-
tyczne z pojęciem lek, nie odpowiada natomiast ukami, takimi jak fizjologia, patofizjologia, mikro-
niemieckiemu pojęciu Droge, które oznacza środek biologia, biochemia, biofizyka, biofarmacja, są płyn-
odurzający). ne, ponieważ nie tylko metody, którymi te dziedziny
Trucizna (substancja szkodliwa) jest substancją się posługują, są w dużej mierze identyczne, lecz
czynną wywołującą szkodliwe działania. W przypad- także pokrywają się stosowane i badane substancje.
ku wielu substancji, zwłaszcza substancji leczniczych, Przykładowo działaniem hormonów zajmują się far-
o tym, czy wywołane zostanie działanie korzystne makolodzy w tym samym stopniu co fizjolodzy, en-
czy też szkodliwe, decyduje dawka, natomiast tru- dokrynolodzy czy biochemicy.
cizna rozumiana w ścisłym sensie tego słowa działa Do specyficznych zadań farmakologii i toksyko-
tylko szkodliwie. logii realizowanych po części z innymi dyscyplinami
Siła działania jest mierzona dawką lub stężeniem należą:
niezbędnym do uzyskania określonego efektu: im
■ rozwój i badanie nowych środków leczniczych
mniejsza dawka (lub stężenie), tym większa siła efektu.
w celu wdrożenia nowych zasad leczenia;
Aktywność wewnętrzna (aktywność działania)
określa procent efektu maksymalnego uzyskiwany ■ udoskonalenie znanych już leków przez zmiany
w danym układzie biologicznym przez substancję strukturalne lub optymalizację stosowania, przy
(pobudzającą ten układ). czym szczególnie ważne jest tu zbadanie zależno-
Skuteczność – podobnie jak środek leczniczy lub ści pomiędzy budową chemiczną a działaniem far-
lek – jest pojęciem dającym się ocenić (klinicznie). makologicznym;
Oznacza uzyskiwane za pomocą leku wyleczenie,
■ ustalenie farmakokinetyki
poprawę, złagodzenie lub zapobieganie określonej
chorobie. ■ poszukiwanie możliwości zapobiegania zatruciom
Pojęcie farmakon jest w mowie potocznej uży- i ich zwalczania.
wane równoznacznie z określeniami środek leczni-
czy lub lek. W piśmiennictwie fachowym ma ono Dalszy podział farmakologii jest następstwem róż-
jednak charakter uogólniony i użyte w odniesie- norodnych zadań przypadających na poszczególne
niu do substancji czynnej biologicznie nie ocenia, jej działy.
czy jej terapeutyczne zastosowanie jest możliwe (ce-
lowe) czy też nie. W farmakologii ogólnej, na podstawie wyników ba-
dań farmakokinetycznych i farmakodynamicznych,
Wynika stąd, że farmakologia – zależnie od interpre- wprowadzono ogólnie obowiązujące zasady i stwo-
tacji samego słowa farmakon – może być definiowa- rzono teoretyczne podstawy innych dziedzin farma-
na albo w sposób bardzo ograniczony jako: kologii.
MUTSCHLER-2009.indd 3 2010-01-07 22:10:40
4 Definicje
Farmakokinetyka zajmuje się zmianami stężeń zagadnień farmakokinetycznych i farmakodynamicz-
leku w zależności od czasu: gdzie i jak szybko jest nych na poziomie molekularnym.
wchłaniana substancja lecznicza, jak zostaje ona Do zadań farmakologii klinicznej należy badanie
rozmieszczona w organizmie, jak enzymy organizmu nowych lub niedawno wprowadzonych do handlu le-
zmieniają budowę jej cząsteczki, gdzie, w jaki sposób ków i ich oddziaływania na ludzi. Zajmuje się ona
i jak szybko jest on wydalany. działaniem leków w najszerszym znaczeniu (np. rów-
nież farmakogenetyką, farmakoepidemiologią), do-
Farmakodynamika jest nauką o działaniu leku starcza więc przesłanek do racjonalnej farmakoterapii
w miejscu docelowym: gdzie, jak i dlaczego dochodzi oraz stanowi łącznik między farmakologią doświad-
do pojawienia się efektu farmakologicznego? czalną a medycyną kliniczną.
Coraz ważniejszą gałęzią farmakologii jest farma- Toksykologia jest nauką o szkodliwych właści-
kologia molekularna zajmująca się wyjaśnianiem wościach związków chemicznych u ludzi i zwierząt
(różne zagadnienia toksykologiczne – zob. część C).
MUTSCHLER-2009.indd 4 2010-01-07 22:10:40
Farmakokinetyka 5
2. Farmakokinetyka
Działanie leku jest wynikiem licznych, często bardzo Do fazy farmakokinetycznej należą takie procesy,
złożonych procesów, jakim podlega on w organizmie. jak:
U podłoża tego działania leży łańcuch reakcji, które
Farmakokinetyka
■ wchłanianie (resorpcja),
można podzielić na trzy fazy:
■ dystrybucja (rozmieszczenie),
■ farmaceutyczną,
■ eliminacja.
■ farmakokinetyczną,
■ farmakodynamiczną. Przez wchłanianie rozumie się pobranie leku przez
organizm, a przez pojęcie dystrybucja – przemiesz-
Faza farmaceutyczna, w przypadku najczęściej sto- czanie z krwi do tkanek. Eliminacja oznacza wszyst- A2
sowanych stałych postaci leku, obejmuje rozpad po- kie procesy prowadzące do zmniejszenia stężenia leku
staci leku i uwolnienie substancji leczniczej. Dlatego w organizmie, czyli biotransformację i wydalanie.
też jej przebieg zależy przede wszystkim od właści- Przy rozważaniach farmakokinetycznych orga-
wości galenowych leku. nizm jest rozpatrywany jako układ otwarty lub
Faza farmaceutyczna
podanie
rozpad postaci leku,
uwolnienie substancji leczniczej
Faza farmakokinetyczna
wchłanianie
biotransformacja
magazynowanie dystrybucja
wydalanie
miejsce działania
(receptory)
Faza farmakodynamiczna
efekt
farmakologiczny
skuteczność
(działanie kliniczne) działanie toksyczne
Ryc. A 2-1. Procesy zachodzące w organizmie po doustnym podaniu leku.
MUTSCHLER-2009.indd 5 2010-01-07 22:10:40
6 Farmakokinetyka
płynny, ponieważ istnieje stała wymiana substancji
i energii z otoczeniem. Jeśli bilans między podażą pobieranie metabolizm eliminacja
a wydalaniem zostaje wyrównany, mówi się o sta-
nie równowagi (steady-state). Ten dynamiczny stan
równowagi jest przedkładany ponad wszystkie inne,
a w razie jakichkolwiek zmian organizm stara się
go przywrócić.
Fazy farmaceutyczna i farmakokinetyczna określa-
ją razem stosunek między podaną dawką a stęże-
niem leku osiągniętym w organizmie. Fazę farma-
kokinetyczną odnosi się często do określonych rejo-
nów (tzw. kompartmentów) w organizmie (np. wą-
troby albo mózgu). W takim przypadku farmakoki-
netykę dzieli się w każdym kompartmencie na trzy
kompartment fizjologiczny
procesy:
■ pobieranie substancji leczniczej,
lek produkt przemiany materii
■ metabolizm (często w sensie przemiany materii),
■ końcową eliminację substancji wyjściowej albo
Ryc. A 2-2. Procesy fazy farmakokinetycznej. Pobieranie do
produktu przemiany materii z kompartmentu.
poszczególnych kompartmentów organizmu może zachodzić
Procesy te zostały schematycznie przedstawione pasywnie (drogą dyfuzji, zgodnie z własnościami biofizycznymi)
na ryc. A 2-2 na przykładzie wątroby. albo, jak przedstawiono na rycinie, za pomocą transporterów (to
samo dotyczy eliminacji).
Do fazy farmakodynamicznej zalicza się oddzia-
ływanie leku z receptorami oraz związane z tym
tj. parametrów farmaceutycznych i farmakokinetycz-
procesy powiązania receptora z efektorem (trans-
nych. Co za tym idzie badania farmakokinetyczne
dukcja sygnału), które powodują efekt farmakolo-
służą do:
giczny. Z opisanego przebiegu działania leku wy-
nika, że zależy on nie tylko od jego właściwości ■ ustalenia zasad wchłaniania,
farmakodynamicznych, ale również (i to w dużej
■ określenia uzyskanych stężeń leku w organizmie
mierze) od:
i ich przebiegu w czasie,
■ postaci leku i użytych substancji pomocniczych,
■ wyjaśnienia dróg biotransformacji i sposobów wy-
■ sposobu i miejsca podania, dalania oraz
■ stopnia i szybkości wchłaniania, ■ porównania różnych postaci i preparatów zawiera-
jących tę samą substancję czynną (badania biodo-
■ dystrybucji,
stępności i biorównoważności).
■ wiązania i rozmieszczenia w tkankach,
Opierając się na tych danych, można ustalić racjo-
■ biotransformacji (metabolizmu),
nalny sposób dawkowania (wielkość pojedynczych
■ drogi oraz szybkości wydalania, dawek, odstępy w podaniach).
2.1. Podawanie
Lek może być podawany albo na powierzchnię cia- ■ fizykalnych i chemicznych właściwości środka
ła, tj. skórę lub błony śluzowe, albo też za pomocą leczniczego,
różnych urządzeń wprowadzany (wstrzykiwany) ■ pożądanego czasu wystąpienia i utrzymywania się
do wnętrza ciała (strzykawki, pistolety ciśnieniowe). działania,
Miejsce i sposób podania oraz postać leku (zob. tab. ■ miejsca, w którym lek powinien działać,
A 2.1-1) zależą od: ■ stanu chorego.
MUTSCHLER-2009.indd 6 2010-01-07 22:10:41
Farmakokinetyka 7
Jeżeli działanie ma wystąpić szybko, konieczne gdy działanie leku powinno się ograniczać do miejsca
jest wybranie takiego sposobu podania, przy którym podania i w miarę możliwości nie dotyczyć całego
ze względu na ominięcie wchłaniania okres utajenia organizmu.
(latencja) pomiędzy podaniem a wystąpieniem dzia- Jeśli dąży się do uzyskania działania ogólnoustro-
łania jest krótki (wstrzyknięcie donaczyniowe). jowego, należy albo wstrzyknąć lek bezpośrednio
Jeśli natomiast dąży się do opóźnionego, utrzy- do krwi krążącej, albo też zastosować go w postaci
mującego się dłużej działania, najczęściej możliwe wchłanialnej.
są tylko takie sposoby podania, przy których lek dzia- Nierzadko wybór sposobu podania musi uwzględ-
ła dopiero po wchłonięciu. niać stan lub wiek chorego. Chorym nieprzytomnym
Docelowe (miejscowe) podanie na powierzchnię lub nie wolno podawać doustnie żadnych leków, ponie-
Farmakokinetyka
w określone miejsce ciała jest wskazane wówczas, waż w razie braku odruchu połykania istnieje nie-
Tabela A 2.1-1. Sposoby podawania leku (zmodyfikowane wg Schelera)
Miejsce podania Sposób podania Postać leku (przykłady)
1. Podanie na skórę lub błonę śluzową A2
na skórę powierzchniowo roztwory, zawiesiny, emulsje, pianki,
maści, pasty, plastry
na błony śluzowe
do jamy ustnej i języka podpoliczkowo, najęzykowo, tabletki, pastylki, drażetki,
podjęzykowo roztwory do płukania
do żołądka i jelit doustnie, dojelitowo tabletki, drażetki, kapsułki,
roztwory, zawiesiny, emulsje
do odbytnicy doodbytniczo czopki, kapsułki doodbytnicze, maści
do nosa donosowo krople, maści, żele, spreje
na nabłonek oskrzeli i pęcherzyków płucnych dopłucnie, aerozole, mgły
przez inhalacje
do spojówek dospojówkowo krople, maści i roztwory oczne
do dróg moczowo-płciowych dopochwowo globulki dopochwowe,
docewkowo maści, pręciki
2. Podanie wewnętrzne, parenteralne
z pominięciem wchłaniania
do serca dosercowo roztwór do wstrzyknięć
do tętnicy dotętniczo roztwór do wstrzyknięć, roztwór do wlewu
do żyły dożylnie roztwór do wstrzyknięć, roztwór do wlewu
do lędźwi dolędźwiowo roztwór do wstrzyknięć, roztwór do wlewu
do płynu mózgowo-rdzeniowego intratekalnie roztwór do wstrzyknięć, roztwór do wlewu
z włączeniem procesu wchłaniania
do skóry śródskórnie roztwór do wstrzyknięć
pod skórę podskórnie roztwór do wstrzyknięć, implanty
w mięsień domięśniowo roztwór do wstrzyknięć
do jamy brzusznej dootrzewnowo roztwór do wstrzyknięć, roztwór do wlewu
MUTSCHLER-2009.indd 7 2010-01-07 22:10:41
8 Farmakokinetyka
bezpieczeństwo zachłyśnięcia. U chorych z zaburze- ■ środek leczniczy bardzo szybko dociera do miejsca
niami żołądkowymi lub upośledzoną motoryką jelit działania.
także nie jest wskazane doustne podawanie leków.
Z drugiej strony trzeba starać się unikać wstrzykiwa- Ten sposób podawania jest więc wskazany przede
nia leków u chorych lękliwych lub u dzieci. wszystkim w przypadkach, w których szczególnie
W przypadku wielu substancji leczniczych, dzięki dużą rolę odgrywa czynnik czasu, np. w sytuacjach
określonym metodom galenowym, udaje się uzyskać nagłych oraz przy dożylnym znieczuleniu ogólnym.
postacie leków nadające się do wszystkich sposobów
podania. Trudno rozpuszczalne substancje można W porównaniu z innymi sposobami podawania
przekształcić w postacie rozpuszczalne i w związku do czynników niekorzystnych, oprócz większego na-
z tym nadające się do wstrzyknięć, np. przez dodanie kładu pracy i nieprzyjemnych doznań chorego (lęk
odpowiedniego rozpuszczalnika albo jeśli posiadają przed zastrzykiem), należy też zwiększone zagrożenie
one ugrupowania zjonizowane, uzyskanie odpowied- (np. niebezpieczeństwo utraty kończyny przy przy-
nich soli. Substancje wrażliwe na kwas solny można padkowym pozażylnym podaniu cytostatyku, zbyt
pokryć otoczką kwasoodporną i w ten sposób uzy- duże stężenie leku w miejscu działania przy zbyt
skać postać nadającą się do stosowania doustnego. szybkim wstrzyknięciu, zakażenie drobnoustrojami).
W przypadku niektórych substancji leczniczych licz- W przypadku leków o małej rozpiętości terapeu-
ba przeznaczonych do podawania postaci jest jednak tycznej szybkie wstrzyknięcie dożylne (bolus) może
ograniczona. Na przykład peptydy muszą być poda- prowadzić do poważnych działań niepożądanych
wane parenteralnie albo w niektórych przypadkach (np. zaburzeń rytmu serca po podaniu glikozydów
donosowo, ponieważ po podaniu doustnym ulegają nasercowych czy depresji czynności oddechowej
szybkiemu zniszczeniu przez proteazy. po opioidach).
W przypadku wstrzyknięć domięśniowych lub pod-
skórnych należy ściśle przestrzegać izohydrii i izo-
2.1.1. Drogi i sposoby podawania osmii podawanych roztworów, w przeciwnym razie
mogą wystąpić zjawiska nietolerancji miejscowej, ta-
Podanie miejscowe. Do przykładów podawania kie jak ból, a w szczególnych okolicznościach nawet
miejscowego, oprócz leczenia zewnętrznego chorób martwica.
skóry, można zaliczyć stosowanie leków rozkurcza-
jących oskrzela w postaci aerozoli, wstrzykiwanie Podanie doustne. Leki są najczęściej podawane
leków miejscowo znieczulających do tkanek, wpro- doustnie, ponieważ niezbędne dla tej drogi postacie
wadzanie roztworów cytostatyków do pęcherza mo- leków są względnie łatwe do wytworzenia, a ponad-
czowego. Korzyściom wiążącym się ze stosowaniem to chorzy najchętniej wybierają właśnie ten sposób
zazwyczaj znacznie mniejszych dawek, dzięki czemu podania. Zła wchłanialność środków leczniczych
możemy mieć do czynienia z brakiem lub niewiel- z przewodu pokarmowego (np. strofantyny) albo
kim tylko działaniem ogólnym, trzeba przeciwstawić działanie drażniące błonę śluzową żołądka mogą jed-
znacznie większe zagrożenie rozwoju alergii niż przy nak prowadzić do rezygnacji z podań doustnych lub
stosowaniu leków na skórę. – w drugim z wymienionych przypadków – zastoso-
wania postaci leku zaopatrzonych w otoczki odporne
Podanie parenteralne. Donaczyniowe (najczęściej na działanie soku żołądkowego.
dożylne) wstrzyknięcia lub wlewy charakteryzują się
tym, że: Podanie doodbytnicze. Ze względu na duże różni-
ce międzyosobnicze i zazwyczaj niewielki stopień
■ można je dokładnie dawkować, a ich biodostęp-
wchłaniania stosowanie doodbytnicze (zob. poniżej)
ność wynosi najczęściej 100% (tylko w wyjątko-
powinno zostać ograniczone do przypadków, w któ-
wych wypadkach dochodzi do absorpcji części leku
rych uzyskanie ściśle określonych działających stężeń
na sprzęcie infuzyjnym, a przez to do zmniejszenia
nie jest bezwzględnie konieczne lub nie występuje
biodostępności);
sytuacja niebezpieczna, zwłaszcza zagrażająca życiu.
■ dzięki szybkiemu rozcieńczeniu przez krew Dlatego nie należy stosować czopków zawierających
i jej dużą pojemność buforującą wymagania do- antybiotyki, natomiast uzasadnione jest doodbytnicze
tyczące izotonii i izohydrii płynu infuzyjnego podawanie środków przeciwbólowych lub przeciw-
są mniejsze niż przy podaniu domięśniowym lub gorączkowych u niemowląt i małych dzieci. Ponadto
podskórnym, przedkłada się doodbytnicze stosowanie leków u cho-
MUTSCHLER-2009.indd 8 2010-01-07 22:10:41
Farmakokinetyka 9
rych, u których występują skłonność do wymiotów uwzględnianie wielkości cząsteczek lub kryształów,
lub zaburzenia opróżniania żołądka (np. z migreną), użycie niewłaściwych, pogarszających wchłanianie
jeśli nie jest konieczne stosowanie pozajelitowe. środków pomocniczych, niedostatecznie szybki
rozpad postaci i zbyt wolne uwalnianie leku) upo-
Postać i biodostępność. W przypadku wszystkich śledzają biodostępność, podobnie jak nasilona bio-
postaci leku, po których podaniu zaangażowany transformacja podczas pierwszego przejścia przez
jest proces wchłaniania, występuje zagadnienie bio- przewód pokarmowy i wątrobę (tzw. efekt pierwsze-
dostępności. Wadliwe postacie galenowe (np. nie- go przejścia).
Farmakokinetyka
2.2. Wchłanianie
Przez pojęcie wchłaniania rozumie się przejście sub-
stancji z powierzchni ciała, do której należy również
błona śluzowa przewodu pokarmowego, albo ogra- A2
niczonych miejsc położonych wewnątrz organizmu glikoproteina płyn zewnątrz-
do krwi krążącej lub też układu limfatycznego, skąd komórkowy
następuje dystrybucja w całym organizmie. Lek może glikolipid
wykazywać działanie tylko wówczas, gdy dostanie się
w odpowiednich stężeniach do miejsca docelowego,
dlatego w przypadku kiedy nie jest on wstrzykiwany
donaczyniowo lub bezpośrednio w miejsce działania,
podstawowym warunkiem uzyskania efektów tera-
peutycznych jest zadowalające wchłanianie.
filamenty
cholesterol cytoszkieletu
2.2.1. Bariery przekraczane
intergralne
podczas wchłaniania białko błonowe
białko połączone z błoną
Właściwą barierę, która musi zostać pokonana pod-
czas wchłaniania, stanowi błona komórkowa, czyli cytoplazma
warstwa oddzielająca środowisko zewnętrzne od we-
wnętrznego. Wchłanianie – a także dystrybucja i wy- Ryc. A 2.2-1. Schematyczne przedstawienie budowy błony ko-
dalanie – nie jest możliwe bez transportu przez błony. mórkowej.
Obecny pogląd dotyczący budowy błon plazmatycz-
nych opiera się przede wszystkim na zaproponowa-
nym przez Lenarda i Singera modelu płynno-mozai-
kowym (zob. ryc. A 2.2-1).
2.2.2. Mechanizmy wchłaniania
Według tego modelu błona składa się z podwójnej i transportu
warstwy lipidowej, na której – oraz wewnątrz której
– jak wyspy znajdują się białka, tworzące swoistą Przenikanie substancji przez błonę może następować
mozaikę. Białka przenikające całą błonę tworzą pory jako:
w podwójnej warstwie lipidowej. Błonę należy sobie
■ (czysto) bierna dyfuzja (przejście),
przy tym wyobrazić nie jako twór statyczny, ale dy-
namiczny, tj. podlegający ciągłym zmianom. Zgodnie ■ ułatwiona (za pomocą nośnika) dyfuzja,
z powyższym do przenikania leków przez błonę służą
■ czynny transport (do lub na zewnątrz),
zasadniczo dwie różniące się znacznie struktury: war-
stwa lipidowa dobrze przepuszczalna dla substancji ■ pinocytoza, fagocytoza i persorpcja
lipofilnych i wypełnione wodą pory umożliwiające
penetrację substancjom hydrofilnym. (zob. ryc. A 2.2-2).
MUTSCHLER-2009.indd 9 2010-01-07 22:10:41
10 Farmakokinetyka
Dyfuzja. W przypadku biernej dyfuzji transport Ułatwiona dyfuzja. Dyfuzja (ułatwiona) zachodząca
leku jest – zgodnie z prawem Ficka – wprost pro- przy współudziale nośnika (carrier) jest, podobnie
porcjonalny do gradientu stężeń, powierzchni bło- jak prosta dyfuzja, procesem biernym. Napędzającą
ny, a także współczynnika rozdziału danego związku siłę stanowi gradient (różnica) stężeń pomiędzy róż-
oraz odwrotnie proporcjonalny do grubości błony. nymi kompartmentami, np. pomiędzy przestrzenią
Ponadto szybkość dyfuzji warunkowana jest przez zewnątrz- i wewnątrzkomórkową. Zależnie od rodza-
właściwy (specyficzny) dla danego związku współ- ju transportera wyróżnia się uniportery, które trans-
czynnik dyfuzji. portują tylko jeden rodzaj cząsteczek, symportery,
Czysta dyfuzja nie jest blokowana przez związki transportujące więcej rodzajów cząsteczek w tym
podobne ani też hamujące przemianę materii. Z iloś- samym kierunku, oraz antyportery, które umożliwia-
ciowego punktu widzenia przechodzenie związków ją przemieszczanie się różnych rodzajów cząsteczek
przez warstwy lipidowe dominuje w organizmie nad w przeciwnych kierunkach. Istnienie nośnika proce-
innymi procesami, dlatego rozpuszczalność w tłusz- su transportu charakteryzują: wysoka specyficzność
czach odgrywa podstawową rolę we wchłanianiu. strukturalna, wysycenie układu transportującego
Niektóre kwasy i zasady organiczne łatwiej dyfun- przy dużych stężeniach związku wiodących do oku-
dują przez błony lipidowe w stanie niezjonizowanym pacji wszystkich miejsc wiązania po jednej stronie
(bardziej lipofilnym), mówimy wówczas o dyfuzji błony, a także zdolność hamowania przez inhibitory.
niejonowej (non-ionic diffusion). Istniejące w obrębie Nie są one natomiast blokowane przez związki hamu-
błony pory mają pewne znaczenie tylko dla wchłania- jące przemianę materii.
nia źle rozpuszczalnych w tłuszczach nieelektrolitów
oraz w pełni zjonizowanych związków o względnie Transport czynny (aktywny). Transport czynny
niewielkiej masie cząsteczkowej. polega na przenoszeniu substancji w poprzek błony
przeciw gradientowi stężeń za pomocą specjalnych
białek transportowych, tzw. pomp lub ATPaz.
Najważniejszym czynnym transporterem jest Na+/
+
K -ATPaza występująca praktycznie we wszystkich
błonach komórkowych. Pozostałe pompy to Ca2+-
ATPaza i H+/K+-ATPaza. (Glikoproteina P, która
również działa jako ATPaza, zostanie omówiona
w punkcie A 2.2.3).
dyfuzja bierna
Ten wymagający zużycia energii proces jest kom-
petytywnie hamowany przez związki o zbliżonej bu-
dowie chemicznej oraz niekompetytywnie przez truci-
zny przemiany materii. Aminokwasy, rozliczne cukry,
a po części również rozpuszczalne w wodzie witami-
uniport
ny są przykładami związków wchłanianych na dro-
dze transportu czynnego. Z dużym prawdopodobień-
stwem można przyjąć istnienie aktywnego transportu
symport leków odznaczających się dużym podobieństwem
chemicznym do aminokwasów (np. lewodopy).
W przypadku czynnego transportu przyjmuje się ist-
antyport nienie transportu sprzężonego z jonami sodu, przy
czym i związek, i jony Na+ transportowane są w tym
samym kierunku (symport, kotransport). W proce-
sie tym powstaje ternarny kompleks między trans-
pierwotny transport porterem (substancją, nośnikiem) a jonami sodu.
ATP aktywny Wewnątrzkomórkowe stężenia jonów sodowych
dzięki pompie sodowej utrzymane są na niskim po-
ziomie, dlatego dochodzi do spadku stężenia tych jo-
substrat kosubstrat nów w środowisku wewnątrzkomórkowym. Podczas
przepływu jonów sodowych zgodnie z gradientem
Ryc. A 2.2-2. Możliwości przechodzenia substancji przez błonę. związek podlega jednocześnie transportowi przeciw
MUTSCHLER-2009.indd 10 2010-01-07 22:10:41
Farmakokinetyka 11
gradientowi. Niezbędna do tego transportu energia 2.2.3. Białka transportowe
jest tym samym dostarczana bezpośrednio przez pom- (dla substancji leczniczych)
pę sodową wskutek rozkładu ATP. Doświadczalnie
wykazano silną zależność miedzy transportem glu- Aby prawidłowo funkcjonować, organizm musi stale
kozy i aminokwasów oraz stężeniami jonów sodu. transportować liczne substancje przez błony biolo-
Wchłanianie glikozydów nasercowych również prze- giczne, np. składniki pokarmowe lub jony nieorga-
biega równolegle z wchłanianiem jonów sodowych. niczne. Ze względu na to nie dziwi duża ilość białek
transportowych występujących w organizmach zwie-
Transport czynny może przebiegać zarówno do we- rzęcych i ludzkich. Szacuje się, że około 7% wszyst-
wnątrz, jak i dzięki ATP-zależnym nośnikom z we- kich ludzkich genów koduje białka transportowe.
Farmakokinetyka
wnątrz na zewnątrz komórki. W tym ostatnim przy- W niniejszym rozdziale zajmiemy się przede wszyst-
padku transport czynny konkuruje często z bierną kim rolą białek transportowych w farmakokinetyce
dyfuzją do wnętrza komórki i zmienia w ten sposób leków. Szczególnie interesujący jest tzw. transport
całkowity (netto) transport przez błonę. wektorowy. Przez to pojęcie należy rozumieć kie-
rowany transport leków przez bariery komórkowe
Pinocytoza, fagocytoza, persorpcja. W czasie pi- (np. w wątrobie albo jelitach), które składają się
nocytozy drobne kropelki płynu, a podczas fagocytozy ze spolaryzowanych komórek. Według aktualnej wie-
cząsteczki stałe zostają przyjęte z przestrzeni poza- dzy za transport wektorowy leków odpowiedzialne A2
komórkowej (np. przewodu pokarmowego) wskutek są dwie rodziny białek, mianowicie białka transpor-
tego, że błona komórkowa ulega ugięciu i następnie towe typu SLC (Solute-Carrier-Family) oraz trans-
otacza zewnątrzkomórkowy materiał, wprowadzając portery ABC ( ATP-Binding-Cassette Family).
go w postaci pęcherzyka do wnętrza komórki. W cza-
sie persorpcji cząsteczki stałe, w niektórych warun- Białka transportowe typu SLC uczestniczą w trans-
kach nawet całe komórki, zostają przemieszczone porcie licznych kationowych, anionowych i niena-
międzykomórkowo, tj. pomiędzy komórkami nabłon- ładowanych substratów endogennych i ksenobioty-
ka wnikają do wnętrza organizmu. Mimo że te for- ków. Około 350 różnych białek podzielono na 47 ro-
my wchłaniania z punktu widzenia ilościowego mają dzin ze względu na ich sekwencję aminokwasową.
znaczenie tylko w przypadku nielicznych leków, trze- Mogą one transportować swoje substraty zarówno
ba im poświęcić uwagę ze względów patologicznych, do wewnątrz, jak i na zewnątrz komórki. Białka
np. w związku z rozwojem uogólnionych zakażeń SLC, które są najważniejsze dla transportu leków,
grzybiczych przy długotrwałym leczeniu antybioty- miejsca ich ekspresji oraz substraty zestawiono
kami, a także ze względu na choroby alergiczne. w tab. A 2.2-1.
Tabela A 2.2-1. Ważne transportery SLC (nośniki substancji rozpuszczalnych), miejsca ich ekspresji oraz substraty
Nazwa Nazwa Miejsce Substrat
genu białka ekspresji
SLC01A2 OATP1A2, OATP-A mózg, nerki, wątroba hormony tarczycy, feksofenadyna
SLC01B1 OATP1B1, OATP-C wątroba hormony tarczycy, prawastatyna,
rifampicyna, metotreksat, faloidyna
SLC01B3 OATP1B3, OATP8 wątroba jak SLC01B1 oraz digoksyna
SLC02B1 OATP2B1, OATP-B wątroba, łożysko, płuca, siarczan 3-estronu, siarczan dehydroepiandrosteronu,
nerki, serce, jelita glibenklamid, atorwastatyna, prawastatyna
SLC15A1 PEPT1 przewód pokarmowy, nerki kaptopril, enalapril, antybiotyki β-laktamowe, sulpiryd
SLC19A1 RFT wszędzie kwas foliowy, metotreksat
SLC19A2 ThTr1 wszędzie witamina B1
SLC19A3 ThTr2 wszędzie witamina B1
MUTSCHLER-2009.indd 11 2010-01-07 22:10:42
12 Farmakokinetyka
Białka transportowe typu ABC wykorzystują ener- filnych metabolitów leków powstających w ramach
gię z hydrolizy ATP, żeby transportować swoje substra- biotransformacji prowadzi do ścisłego współdziała-
ty wbrew gradientowi stężeń przez błony wewnątrz- nia między mechanizmami przemiany materii i trans-
komórkowe albo na zewnątrz komórki. Dotychczas portem.
zidentyfikowano 51 białek transportowych, które po-
dzielono na 7 rodzin (ABCA do ABCG). Wszystkie Szczególnie dobrze zbadanym przedstawicielem
transportery tego typu mają jedną lub dwie sekwen- białek transportowych ABC jest P-glikoproteina
cje aminokwasowe wiążące ATP, na których zacho- (ABCB1; P-gp; P-170, zob. ryc. A 2.2-3), którą od-
dzi hydroliza. Jeśli białko transportowe posiada tylko kryto podczas badań nad rozwojem oporności guzów
jedno miejsce wiążące ATP, muszą powstawać homo- na cytostatyki. Białko to może wypompowywać cy-
lub heterodimery umożliwiające transport. Ich zdol- tostatyki z wnętrza komórek guza, zmniejszając w ten
ność do transportowania na zewnątrz komórki hydro- sposób ich stężenie w miejscu docelowym. P-gp
C A
błona
apikalna
ATP ATP
NH2
HOOC
dyfuzja bierna
glikoproteina P
1 (pompa)
inhibitory
np. werapamil, cyklosporyna
substraty endogenne
Ryc. A 2.2-3. Zasada działania transporterów ABC na przykładzie glikoproteiny P. A) lokalizacja P-gp w enterocytach, B) lokalizacja
w błonie, C) struktura cząsteczkowa P-gp; 1 – dyfuzja bierna substratów P-gp do komórki, 2 – aktywny transport na zewnątrz przez
P-gp, 3 – inhibicja P-gp.
MUTSCHLER-2009.indd 12 2010-01-07 22:10:42
Farmakokinetyka 13
jest białkiem błonowym o masie cząsteczkowej wy- rozpuszczalnego w wodzie metabolitu przez błonę
noszącej 170 kD, kodowanym przez MDR1 (Multi- na zewnątrz komórki.
Drug-Resistance-Gen) i składającym się z dwóch
części. Każda część posiada 6 domen transmem- Zrozumienie opisanych procesów transportowych jest
branowych. W każdej połowie znajduje się miejsce szczególnie ważne dla farmakoterapii. W ten sposób, kie-
dy dwie substancje podane jednocześnie konkurują o ten
wiązania ATP, które dzięki hydrolizie ATP zapewnia sam transporter z ograniczonymi możliwościami trans-
energię potrzebną do transportu. W ten sposób we- portowymi, może dojść do zmiennych oddziaływań mię-
wnątrzkomórkowy substrat może być transportowa- dzy lekami. Dzięki temu odkryciu udało się na przykład
ny na zewnątrz (zob. ryc. A 2.2-3). zinterpretować długo niewyjaśnione interakcje glikozydu
nasercowego digoksyny, który nie ulega metabolizmo-
Farmakokinetyka
wi, a mimo to wchodzi w klinicznie znaczące interakcje
P-gp jest bardzo ważna dla farmakokinetyki wielu z innymi lekami (np. z chinidyną, cyklosporyną, propafe-
substancji (zob. tab. A 2.2-2), ponieważ występuje nie nonem, werapamilem). Poza tym niska i zmienna biodo-
tylko w komórkach guza, ale jest także ekspresjono- stępność (np. inhibitorów proteazy wirusa HIV, takich jak
wana fizjologicznie w wielu tkankach. Dlatego bierze indinawir, sakwinawir czy ritonawir) oraz niskie stężenie
udział w eliminacji licznych ksenobiotyków (clearan- tych substancji w OUN zostały wyjaśnione przez ekspresję
P-gp w przewodzie pokarmowym lub w komórkach bariery
ce, np. w wątrobie, jelicie, nerkach) albo chroni orga- krew-mózg. Ten aspekt ma znaczenie terapeutyczne, ponie-
nizm lub narząd przed wnikaniem obcych substancji waż OUN stanowi rezerwuar dla HIV. Hamując P-gp za po-
(funkcja barierowa, np. w jelicie albo mózgu). mocą specjalnych substancji (np. pochodnej cyklosporyny A2
PSC833), możliwe jest zarówno podwyższenie biodostęp-
P-gp często występuje w tych samych komórkach ności, jak i stężenia inhibitorów proteaz w OUN.
co enzym cytochrom P-450-3A4. Poza tym większość
substratów P-gp jest metabolizowana przez CYP3A4. Transportery ABC, które są najważniejsze dla trans-
Tym samym można zakładać, że oba białka są częś- portu leków, miejsca ich ekspresji oraz substraty ze-
cią większego mechanizmu, który zapewnia transport stawiono w tab. A 2.2-3.
Tabela A 2.2-2. A) miejsca ekspresji oraz B) substraty dla transportera ABC – glikoproteiny P (ABCB1)
A
Rodzaj
Narząd komórki Lokalizacja Transport do
wątroba hepatocyt błona kanalików żółci
jelita enterocyt błona apikalna światła przewodu pokarmowego
nerki komórki nabłonkowe cewki bliższej błona apikalna światła cewki
mózg komórki śródbłonka naczyń włosowatych błona luminalna krwi
B
Klasa substancji Substancje
β-adrenolityki celiprolol, talinolol
cytostatyki antracykliny, etopozyd, paklitaksel, alkaloidy barwinka
inhibitory proteaz HIV indinawir, nelfinawir, ritonawir, sakwinawir
różne digoksyna, erytromycyna, ranitydyna, terfenadyna, werapamil
MUTSCHLER-2009.indd 13 2010-01-07 22:10:42
14 Farmakokinetyka
Tabela A 2.2-3. Ważne transportery ABC, miejsca ich ekspresji oraz substraty
Nazwa Nazwa Miejsce Substraty
genu białka ekspresji
ABCB1 MDR1, P-gp zob. tab. A 2.2-2 zob. tab. A 2.2-2
ABCC1 MRP1 wszędzie leukotrieny C4, koniugaty glutationu,
glukuronidy, siarczany, metotreksat,
winkrystyna, sakwinawir
ABCC2 MRP2 przewód pokarmowy, leukotrieny C4, glukuronidy, koniugaty glutationu,
wątroba, nerki siarczany, karwedilol, indinawir, ritonawir,
prawastatyna
ABCC3 MRP3 przewód pokarmowy, jak ABCC2
wątroba, nerki
ABCC4 MRP4 wszędzie analogi nukleozydów, cGMP, cAMP, ADP,
6- merkaptopuryna, metotreksat
ABCC5 MRP5 wszędzie analogi nukleozydów, cGMP, cAMP,
6-merkaptopuryna, sildenafil (inhibitor)
ABCG2 BCRP wszędzie kamptotecyna, metotreksat, mitoksantron,
topotekan, imatynib
2.2.4. Wchłanianie leków substancja czynna (znajdująca się np. w obrębie
przewodu pokarmowego albo wewnątrzmięśnio-
wego magazynu = depot), warunkuje szybkość
Wchłanianie (resorpcja, absorption) zachodzi w przy-
wchłaniania. Jest ona uzależniona nie tylko od właś-
padku wielu leków drogą biernej dyfuzji. Szybkość
ciwości samej substancji (np. postać krystaliczna,
wchłaniania i wielkość wchłaniania (stosunek wchło-
wielkość cząsteczek), ale też od postaci leku (użyte
niętej części do podanej ilości) zależą od wielu czyn-
substancje pomocnicze, otoczki itp.). W przypadku
ników, z których za najważniejsze uznawane są:
substancji trudno rozpuszczalnych niekiedy pozo-
■ właściwości fizykochemiczne substancji leczni- stający do dyspozycji czas wchłaniania nie wystar-
czej, zwłaszcza jej rozpuszczalność, cza do całkowitego rozpuszczenia podanej ilości
leku. Poprzez silne rozdrobnienie (mikronizację)
■ wielkość oraz specyficzna powierzchnia cząste-
i co za tym idzie – zwiększenie specyficznej po-
czek,
wierzchni można uzyskać zwiększenie szybkości
■ postać leku, rozpuszczania. Silnie lipofilne substancje, np. wita-
mina A lub gryzeofulwina, niemal nierozpuszczalne
■ zastosowane substancje pomocnicze,
w wodzie, muszą przed wchłonięciem ulec solubi-
■ sposób i miejsce podania, lizacji. Taka solubilizacja może nastąpić w obrębie
jelita cienkiego, zwłaszcza za pomocą soli kwasów
■ czas kontaktu z powierzchnią wchłaniania,
żółciowych. Substancje o wysokim stopniu lipofilno-
■ wielkość powierzchni wchłaniania, ści, np. terbinafina, mogą być wychwytywane przez
układ limfatyczny wspólnie z lipidami (np. chole-
■ wartość pH w obrębie obszaru, w którym zachodzi
sterolem) w postaci chylomikronów i dlatego przy-
wchłanianie,
najmniej częściowo zniesiony jest efekt pierwszego
■ integralność błony przejścia przez wątrobę.
W wypadku jonów nieorganicznych zdolność
■ ukrwienie narządów, w których następuje wchła-
wchłaniania zmniejsza się wraz ze wzrostem stop-
nianie.
nia jonizacji i wielkością jonów. Przy organicznych
substancjach leczniczych stopień wchłaniania zale-
Substancja ulega wchłanianiu w postaci rozpusz- ży od współczynnika rozdziału (np. oktanol/woda):
czonej. Najczęściej szybkość, z jaką rozpuszcza się wchłanialność wzrasta początkowo ze zwiększają-
MUTSCHLER-2009.indd 14 2010-01-07 22:10:43
Farmakokinetyka 15
cym się współczynnikiem rozdziału aż do określo- z przewodu pokarmowego ma największe znaczenie.
nego maksimum, a następnie ulega zmniejszeniu. Następuje ono głównie w górnej części jelita cien-
Przyczyną leżącą u podłoża tych zjawisk jest to, kiego, wykazującego szczególnie dużą powierzchnię
że substancje o przeważającej hydrofilności źle prze- wchłaniania. Wbrew wcześniejszym poglądom żo-
chodzą przez lipofilne błony, a substancje silnie li- łądkowi jako miejscu wchłaniania przypada względ-
pofilne nie rozpuszczają się w dostatecznym stopniu nie niewielka rola, również w przypadku wchłaniania
w wodnym środowisku otaczającym powierzchnię słabych kwasów i substancji obojętnych z powodu
wchłaniania. Co za tym idzie – w jednostce czasu względnie niewielkiej powierzchni błony śluzowej
niedostateczna ilość substancji wchodzi w kontakt żołądka, a także często spotykanej niewielkiej roz-
z płaszczyzną wchłaniania. puszczalności słabych kwasów w kwaśnym środowi-
Farmakokinetyka
Kwaśne i zasadowe organiczne środki lecznicze sku. Może jednak dojść do przechodzenia substancji,
są wchłaniane głównie w niezjonizowanej, a więc zwłaszcza słabych zasad, z błony śluzowej do światła
lipofilnej postaci. Ponieważ stopień dysocjacji zale- żołądka.
ży z jednej strony od wartości pKa związku, z dru-
giej – od pH otaczającego środowiska, słabe kwasy Słabe zasady znajdują się w osoczu głównie w postaci
są lepiej wchłaniane w środowisku kwaśnym niż za- niezjonizowanej, dlatego mogą dyfundować z płynu ze-
wnątrzkomórkowego przez ścianę żołądka do jego światła.
sadowym, zasadowe przy wartości pH > 7. Sztucznie
wywołane zmiany wartości pH, np. przez podawanie
Po osiągnięciu światła żołądka ulegają jonizacji w wyniku
działania niskiego pH. W związku z tym niezjonizowana
A2
środków zobojętniających, mogą znacznie zmieniać część w ścianie żołądka jest mała. Dochodzi więc do dyfu-
stopień wchłaniania leków częściowo zdysocjowa- zji w kierunku światła żołądka nawet wówczas, gdy całko-
nych. Wchłanianie czwartorzędowych związków wite stężenie związku w soku żołądkowym (zjonizowane
oraz niezjonizowane) jest wyższe niż w osoczu.
amoniowych i innych w pełni zjonizowanych sub-
stancji następuje niezmiernie powoli i tylko w bar-
Czas trwania pasażu żołądkowego i co za tym idzie
dzo niewielkim stopniu. Po części są one wchłaniane
– czas pozostawania środka leczniczego w żołądku
w postaci par jonowych.
zależą od stanu wypełnienia i znajdujących się w tre-
Wchłonięty środek czynny jest szybko odtrans-
ści żołądkowej innych związków (szybkie opróżnie-
portowywany z krwią. Dzięki temu utrzymuje się
nie przy podawaniu leków na czczo, przyspieszone
gradient stężeń pomiędzy miejscem podania a krwią,
lub opóźnione przechodzenie przy jednoczesnym po-
co umożliwia dalsze wchłanianie. Dlatego w przypad-
daniu pokarmu).
ku substancji bardzo dobrze wchłanianych, np. etano-
lu, szybkość wchłaniania zależy od ukrwienia. Jeśli Tabletki o dużej objętości, źle się rozpadające opuszczają
jednak przepuszczalność przez błony jest ograniczo- żołądek tylko w wyniku silnych fal perystaltycznych, słu-
na, ona właśnie określa szybkość wchłaniania. żących do całkowitego opróżnienia żołądka z resztek po-
karmowych (tzw. fale wymiatające). Częste przyjmowanie
Wchłanianie przy podaniu podpoliczkowym lub pokarmów w ciągu dnia może więc przeszkadzać wchła-
nianiu substancji czynnej i opóźniać je aż do godzin noc-
podjęzykowym. Dobrze unaczyniona błona śluzo- nych. Przy podawaniu leku w wielu podzielonych dawkach
wa jamy ustnej i gardła (podanie podjęzykowe i pod- pojedynczych istnieje niebezpieczeństwo przedawkowania
policzkowe) stwarza dobre warunki wchłaniania dla w wyniku wchłonięcia całkowitej dawki dobowej podczas
substancji lipofilnych, niezjonizowanych. Korzystny nocnego powstrzymywania się od przyjmowania pokar-
jest brak wpływu działających w obrębie przewodu mów (zwielokrotnienie dawki).
pokarmowego enzymów trawiennych oraz to, że lek
nie musi natychmiast po wchłonięciu przejść przez Jeśli lek wpływa na motorykę żołądka lub wydziela-
filtr wątroby. Ze względu na stosunkowo niewielką nie soku żołądkowego, czas pozostawania w żołądku
powierzchnię wchłaniania podanie podpoliczkowe i jednocześnie kinetyka wchłaniania ulegają zmianie.
lub podjęzykowe stosuje się zwłaszcza w przypadku Etanol wywołuje przekrwienie i ma właściwości roz-
łatwo wchłanialnych substancji, które pozbawione puszczalnikowe, z tego względu przyspiesza wchła-
przy tym złego smaku. Ten sposób podania jest czę- nianie środków jednocześnie z nim podawanych.
sty przy zwalczaniu napadów dławicy sercowej nitro- Środki wrażliwe na kwas solny muszą być chro-
gliceryną podawaną w rozgryzalnych kapsułkach lub nione przez podawanie w postaciach powlekanych
w postaci aerozolu. otoczką niewrażliwą na kwas.
Jelito cienkie jest najważniejszym organem wchła-
Wchłanianie po podaniu doustnym. Jak już wspo- niania nie tylko pokarmów, ale również leków.
mniano, najczęściej stosowaną i najprostszą metodą Odpowiadająca za szybkie i w miarę możliwości
podania leku jest droga doustna, dlatego wchłanianie całkowite wchłanianie zwiększona powierzchnia po-
MUTSCHLER-2009.indd 15 2010-01-07 22:10:43
16 Farmakokinetyka
wstaje dzięki występowaniu fałdów błony śluzowej, niania mogą wystąpić działania ogólne, np. wzrost
kosmków, krypt oraz mikrokosmków. Wartość pH ciśnienia i odruchowa bradykardia po zastosowaniu
zmienia się od nieznacznie kwaśnej w dwunastnicy zawierających α-sympatykomimetyki kropli dono-
do lekko zasadowej w dalszych odcinkach jelita cien- sowych u niemowląt. Świadomie wykorzystuje się
kiego, dlatego dostateczne ilości zarówno słabych wchłanianie z błony śluzowej nosa, np. przy podawa-
kwasów, jak i słabych zasad występują w postaci niu roztworów desmopresyny w leczeniu moczówki
niezjonizowanej, a więc podlegającej wchłanianiu. prostej, a także analogów gonadoreliny w leczeniu
Z powodu długości jelita cienkiego czas przechodze- raka stercza. Po podaniu doustnym oligopeptydy ule-
nia przez nie treści pokarmowej (ok. 4 godz.) jest naj- gają zniszczeniu w przewodzie pokarmowym.
częściej wystarczający do wchłaniania podlegających
temu procesowi związków. Skrócenie czasu pasażu, Wchłanianie po podaniu do oka. Po podaniu na po-
np. po podaniu działających na jelito cienkie środ- wierzchnię oka, jeśli lek ma wniknąć w głąb, musi
ków przeczyszczających lub wskutek biegunki, może pokonać struktury zarówno lipofilne, jak i hydrofilne.
znacznie zmniejszyć stopień wchłaniania. Nabłonek i śródbłonek rogówki są barierami lipofil-
Przez długi czas uważano, że za intensywność nymi, natomiast przez podścielisko mogą dyfundować
wchłaniania w jelicie cienkim odpowiadają właś- tylko związki hydrofilne. W związku z tym warunki
ciwości fizykochemiczne leku. Obecnie wiadomo, do przenikania są szczególnie korzystne w przypadku
że wchłanianie składa się z kilku procesów cząstko- leku, który wykazuje jednocześnie właściwości lipo-
wych: pobrania do enterocytów (często przez białka i hydrofilne. Dotyczy to przede wszystkim słabych
transportowe), (ewentualnego) metabolizmu w ente- kwasów i zasad, które są częściowo niezjonizowa-
rocytach oraz końcowego transportu przez błonę ba- ne i w związku z tym rozpuszczalne w tłuszczach,
zolateralną (otaczającą naczynia krwionośne). Suma a częściowo występują w postaci zjonizowanej, czyli
tych trzech procesów określa intensywność wchła- rozpuszczalnej w wodzie.
niania.
Warunki wchłaniania w jelicie grubym odpowia- Wchłanianie z dróg oddechowych. Do wchłaniania
dają jakościowo i zasadniczo wchłanianiu w jelicie z płuc najlepiej nadają się substancje lotne. Jednak
cienkim, ale powierzchnia wchłaniania jest z powodu płuca ze swą olbrzymią powierzchnią pęcherzyków
braku kosmków znacznie mniejsza i dlatego wchła- płucnych wynoszącą 70–100 m2 zdolne są rów-
nialność jest niewielka. Niektóre leki, np. furosemid nież do wchłaniania roztworów i substancji stałych.
czy alkaloidy sporyszu, są bardzo słabo wchłaniane Aerozole, w przypadku których optymalne warunki
w jelicie grubym. wchłaniania występują przy wielkości cząsteczek
Substancje czynne podane drogą doustną po wchło- ok. l μm, służą przede wszystkim do leczenia miej-
nięciu trafiają z krążeniem wrotnym do wątroby. scowego w obrębie dróg oddechowych (np. leczenie
Tu przy odpowiednich właściwościach substancji dychawicy oskrzelowej). Podobnie jak przy podaniu
bardzo duża jej część może być metabolizowana. donosowym, należy się jednak liczyć z występowa-
niem działania ogólnego, jeśli zastosowane zostaną
Wchłanianie po podaniu doodbytniczym. Po po- środki ulegające wchłanianiu.
daniu doodbytniczym udaje się w dużej mierze unik-
nąć efektu pierwszego przejścia przez wątrobę, ponie- Wchłanianie przy podaniu na skórę. Wchłanianie
waż ilości leku wchłaniane w dolnych dwóch trzecich przez skórę, z fizjologicznego punktu widzenia nie-
odbytnicy trafiają bezpośrednio do żyły próżnej dol- spełniające żadnych zadań, zachodzi przez naskórek
nej, a nie do żyły wrotnej, jednak wchłaniane ilości (transepidermalnie) lub przezgruczołowo (transfoli-
są najczęściej znacznie mniejsze niż przy podaniu do- kularnie), jednak wchłanianie przez nieuszkodzoną
ustnym. Przy tej drodze podania stwierdza się bardzo skórę jest znacznie mniejsze niż przez błony śluzo-
znaczne różnice między- i wewnątrzosobnicze. we. Pozbawiona naczyń włosowatych warstwa zro-
gowaciala (stratum corneum) ze swoją bardzo małą
Wchłanianie po podaniu donosowym. Błona ślu- zawartością wody (ok. 10%), a dużą zawartością
zowa nosa, podobnie jak błona śluzowa jamy ustnej, apolarnych tłuszczów stanowi właściwą barierę dla
ma korzystne właściwości z punktu widzenia wchła- wchłaniania, ale zarazem zbiornik substancji czyn-
niania, nadaje się też do miejscowego, powierzchnio- nych. Największy stopień wchłaniania przy podaniu
wego stosowania środków zmniejszających jej obrzęk na skórę mają przede wszystkim substancje rozpusz-
przy katarze. W ostatnim z wymienionych przypad- czalne w tłuszczach, które jednak wykazują także pe-
ków należy jednak pamiętać, że w następstwie wchła- wien stopień rozpuszczalności w wodzie. Substancje
MUTSCHLER-2009.indd 16 2010-01-07 22:10:43
Farmakokinetyka 17
hydrofilne, a także tłuszcze i oleje są wchłaniane Szeroko rozpowszechnione przezskórne stosowanie leków
przez skórę tylko w niewielkim stopniu. wywalających przekrwienie w postaci wcieranych maści
i żeli w chorobach reumatycznych prowadzi tylko do nie-
Wiele czynników może wpływać na wchłanianie
znacznych dających się obiektywnie ocenić sukcesów tera-
przez skórę. Penetrację zastosowanych środków po- peutycznych.
prawia podwyższenie temperatury skóry przez ze-
wnętrzne źródła ciepła. W podobny sposób ułatwiają
Wchłanianie po podaniu parenteralnym. Jeżeli
wchłanianie bodźce powodujące przekrwienie, nie-
podaje się lek śródskórnie, w podskórną tkankę łącz-
które rozpuszczalniki, np. dimetylosulfotlenek, słu-
ną albo domięśniowo, szybkość wchłaniania zależy
żące jako promotory przenikalności, albo zwiększo-
w dużej mierze od gradientu stężeń i co za tym idzie
ne uwodnienie, np. preparaty zawierające mocznik.
Farmakokinetyka
– od ukrwienia tkanki. Ukrwienie mięśni szkieleto-
W obrębie zapalnych zmian skórnych wchłanianie
wych zależy natomiast od ich aktywności motorycz-
również ulega zwiększeniu. Mechaniczne, chemiczne
nej. Zazwyczaj związki wstrzyknięte w silnie una-
lub termiczne uszkodzenie naskórka, np. zranienie,
czynione mięśnie prążkowane są szybko wchłaniane,
odmrożenie, oparzenie, usuwa warstwę rogową, czyli
natomiast we wstrząsie wchłanianie ulega znacznemu
barierę utrudniającą wchłanianie.
osłabieniu i spowolnieniu.
Należy również zwrócić uwagę na zmieniającą się
W przypadku niektórych naczyń włosowatych
wchłanialność przez skórę w zależności od wieku.
U niemowląt warstwa rogowa nie jest w pełni wy-
wchłanianie jest ułatwione wskutek istnienia licz- A2
nych porów w śródbłonku. Takie włośnice, w których
kształcona, wchłanianie jest zatem większe. Należy
średnica porów wynosi ok. 3 μm, stanowią znacznie
także pamiętać o większym stosunku powierzchni
słabszą barierę niż lita warstwa śródbłonka, dlatego
ciała do masy u dzieci w porównaniu z dorosłymi.
mogą przez nią łatwo przechodzić również substancje
U osób w zaawansowanym wieku grubość warstwy
nierozpuszczalne w tłuszczach. Jest to możliwe nawet
rogowej również jest zmniejszona (skóra papierowa-
w przypadku związków o dużej masie cząsteczko-
ta), dlatego wchłanianiem rządzą podobne prawa.
wej (podskórne podania insuliny!), makrocząsteczki
Do niedawna skóra nie miała większego znaczenia dla nie są natomiast w stanie pokonać ściany naczyń wło-
wchłaniania leków o działaniu ogólnym, lecz w wyniku sowatych pozbawionych porów.
rozwoju odpowiednich postaci leków coraz częściej próbu-
je się wykorzystywać ją jako drogę podawania. Ze względu
na ograniczoną przenikalność skóry możliwe jest jedynie Modyfikowanie wchłaniania. W zależności od ro-
podawanie leków stosowanych w niewielkich dawkach dzaju pożądanego działania można próbować zwięk-
(dawki dobowe do 10 mg), ponadto logiczne jest podawa- szyć, zmniejszyć, przyspieszyć lub opóźnić wchłania-
nie tą drogą leków tylko wówczas, gdy stosowane substan- nie. W miarę możności całkowite (ilościowo) wchła-
cje charakteryzują się silnym efektem pierwszego przejścia
oraz krótkimi okresami półtrwania w osoczu.
nianie jest pożądane w przypadku leków, które mają
wykazywać działanie ogólnoustrojowe, dąży się na-
Tak zwane przezskórne systemy terapeutyczne (TTS, trans- tomiast do opóźnienia wchłaniania takich substancji,
dermal therapeutic system) naklejane na skórę w postaci dzięki którym przez powolne uwalnianie leku z ist-
plastrów są alternatywą dla niektórych środków stosowa-
nych doustnie. Ten sposób podania umożliwia uzyskanie niejącego depot (rezerwuaru) należy osiągnąć długo
przez relatywnie długi okres względnie równomiernego utrzymujące się działanie.
stężenia w osoczu, a także pozwala uniknąć przedsyste-
mowej eliminacji leku. Obecnie znajdują się w handlu tego Przyspieszenie wchłaniania, np. przez równoczesne po-
typu postacie leku zawierające np. nitroglicerynę, estradiol, danie hialuronidazy przy wstrzyknięciach podskórnych
gestagen oraz skojarzenie estrogen/gestagen, testosteron, lub domięśniowych, jest bardzo rzadko wykorzystywane
fentanyl i nikotynę, kolejne są przygotowywane. w lecznictwie. Szczególne znaczenie ma opóźnianie wchła-
Do stosowania w systemach przezskórnych nie nadają niania z preparatów depot. W przypadku wstrzykiwanych
się związki silnie alergizujące, a także drażniące miejsco- roztworów przedłużone wchłanianie i działanie można uzy-
wo. Składniki plastra mogą powodować działanie draż- skać przez:
niące, dlatego po co najmniej trzech dniach należy nakleić
nowy plaster na inne miejsce skóry. Ponadto jeszcze krótszy ■ rozpuszczanie lub zawieszanie substancji leczniczej
czas stosowania jest w niektórych przypadkach niezbędny w podłożu olejowym,
do uniknięcia rozwoju tolerancji. Próbuje się również wy- ■ dodanie zwiększających lepkość substancji wielkoczą-
tworzyć systemy ze sterowanym (pulsacyjnym) uwalnia- steczkowych opóźniających dyfuzję rozpuszczonego
niem substancji czynnej. środka leczniczego.
Ogólnoustrojowe działania niepożądane mogą wy- ■ adsorpcje środka leczniczego na odpowiedniej cząstecz-
ce nośnikowej, np. wodorotlenku glinu lub
stępować w razie stosowania na duże powierzchnie
skóry np. glukokortykosteroidów. ■ zastosowanie zawiesiny krystalicznej.
MUTSCHLER-2009.indd 17 2010-01-07 22:10:44
18 Farmakokinetyka
W przypadku tabletek lub drażetek uwalnianie związku W efektywnym leczeniu bólu towarzyszącego chorobom
czynnego udaje się opóźniać m.in. dzięki: nowotworowym pożądane jest przedłużone uwalnianie
morfiny, w przeciwnym wypadku szybka eliminacja leku
■ umieszczeniu środka leczniczego w trudno rozpuszczal-
uniemożliwia nieprzerwany sen nocny.
nej otoczce,
■ umieszczeniu środka leczniczego w tłuszczach lub wo- Wadą długotrwałego stosowania leków o przedłużonym
skach, uwalnianiu jest to, że pominięcie jednej dawki, np. przez
zapomnienie, wywiera znacznie większy wpływ na stęże-
■ związaniu leku z żywicą jonowymienną,
nie leku w osoczu niż w przypadku niepodania pojedynczej
■ zastosowaniu systemów osmotycznych. dawki prostej postaci leku stosowanej wielokrotnie w ciągu
dnia.
Jak ważne jest spowolnienie wchłaniania, pokazuje przy-
kład leczenia nadciśnienia za pomocą blokera kanałów Do niedawna przedłużone uwalnianie leku służyło wyłącz-
wapniowych typu dihydropirydyny. Po podaniu tabletki ni- nie uzyskaniu stałych stężeń w osoczu, natomiast obecnie
fedypiny, która szybko uwalnia substancję czynną, dochodzi dąży się do uzyskania systemów, które dzięki pulsacyjne-
do odruchowej tachykardii, która przynajmniej u pacjentów mu uwalnianiu substancji czynnej dopasowują się ściśle
z chorobą niedokrwienną serca może prowadzić do napa- do zmieniających się potrzeb organizmu. Przykładem może
dów dławicy piersiowej. Tego efektu można uniknąć dzięki być leczenie żeńskiej niepłodności za pomocą hormonu
spowolnieniu działania leku przez przygotowanie postaci uwalniającego gonadotropinę.
tabletki o przedłużonym uwalnianiu.
2.3. Dystrybucja
Dystrybucja jest określana jako odwracalny transport Przed uzyskaniem stanu równowagi dystrybucji roz-
leku z jednej części organizmu do innej. Przez pojęcie mieszczenie jest w dużym stopniu ustalane przez
równowagi dystrybucji rozumie się stan, w którym ukrwienie tkanek i narządów. W następstwie szyb-
stosunki stężeń w różnych częściach (tkankach orga- kiego przepływu krwi i zależnego od tego krótkiego
nizmu) pozostają stałe. przebywania w naczyniach włosowatych (ok. 2 s) po-
Kiedy lek dostanie się do układu krwionośnego, czątkowo część leku, która może dyfundować z krwi
jest on systemem naczyń krwionośnych transporto- do określonego narządu, jest tym większa im większe
wany dalej z prądem krwi. W następstwie różnicy stę- jest jego ukrwienie (zob. tab. A 2.3-1).
żeń między krwią i tkanką lek opuszcza łożysko na-
czyń krwionośnych, rozmieszczając się w całym or- Oznacza to, że narządy mające silnie rozwinięty układ
ganizmie. Jego przejście z krwi do tkanki i co za tym naczyń włosowatych w początkowej fazie dystrybucji
idzie – dystrybucja zależy, podobnie jak wchłanianie,
od wielu czynników (zmiennych). Ze strony organi-
zmu na dystrybucję substancji wpływają następujące Tabela A 2.3-1. Przeciętne ukrwienie u dorosłych (wg Thewsa,
czynniki: Mutschlera, Vaupela)
■ ukrwienie narządów i tkanek, Narząd Ukrwienie Masa
bezwzględne względne (pojemność (kg)
■ przepuszczalność błon, ml/min minutowa serca) %
■ różnice wartości pH między osoczem i tkanką. mózg 780 15 1,4
mięsień sercowy 250 5 0,3
Ze strony właściwości substancji ważne są przede
wszystkim: nerki 1200 23 0,3
■ wielkość cząsteczek, mięśnie szkieletowe 900 17 30
■ wiązanie z białkami osocza i białkami narządów skóra 400 8 4
oraz
wątroba 1500 28 1,5
■ rozpuszczalność i właściwości chemiczne.
MUTSCHLER-2009.indd 18 2010-01-07 22:10:44
Farmakokinetyka 19
całkowita woda organizmu
przestrzeń przestrzeń
wewnątrzkomórkowa zewnątrzkomórkowa
Farmakokinetyka
stałe składniki płyn wewnątrz- woda osocza przestrzeń płyn między-
komórkowe komórkowy śródmiąższowa komórkowy
Ryc. A 2.3-1. Kompartmenty organizmu.
A2
pobierają znacznie większe ilości leku niż źle ukrwio- Podane wyżej wartości odnoszą się wyłącznie
ne obszary. Na końcu procesu dystrybucji ustala się do osób dorosłych w średnim wieku. U niemowląt
stan równowagi niezależny od stopnia ukrwienia. udział płynów w masie ciała jest znacznie większy.
Przez pojęcie całkowita woda organizmu rozu-
mie się łączną ilość płynów występujących w ustro-
2.3.1. Kompartmenty ju. W zależności od posiadanych właściwości fizyko-
chemicznych, uwzględniając dystrybucję do różnych
Z czynnościowego punktu widzenia organizm moż- kompartmentów, można wyróżnić trzy typy środków
na podzielić na różne obszary, w których lek zostaje leczniczych, które podlegają rozmieszczeniu:
równomiernie rozmieszczony (kompartmenty):
■ wyłącznie w osoczu,
■ kompartment wewnątrzkomórkowy oraz
■ w osoczu i pozostałym obszarze pozakomórko-
■ kompartment zewnątrzkomórkowy (ryc. A 2.3-1). wym,
■ w przestrzeni zarówno poza-, jak i wewnątrzko-
Do kompartmentu wewnątrzkomórkowego (ok.
mórkowej.
75% masy ciała) należą płyn wewnątrzkomórkowy
i części stałe. Kompartment zewnątrzkomórkowy
Stężenie leku w osoczu stanowi istotną wartość, po-
(ok. 25% masy ciała) można podzielić na:
nieważ często koreluje ono ściśle z działaniem farma-
■ wodę zawartą w osoczu, kologicznym i może być dokładnie oznaczane za po-
mocą nowoczesnych metod analitycznych.
■ płyn śródmiąższowy,
Wielkocząsteczkowe (podane dożylnie) środki
■ płyn transkomórkowy. lecznicze, np. niektóre leki krwiozastępcze, nie mogą
opuścić układu krążenia. Na dystrybucję pozostałych
Woda zawarta w osoczu (ok. 4% masy ciała) obej- środków leczniczych pomiędzy osocze i płyn śród-
muje płyn wewnątrznaczyniowy, płyn śródmiąższowy tkankowy ma wpływ budowa naczyń włosowatych
(ok. 16–20% masy ciała), a także podlegający łatwo w danym obszarze lub narządzie. Wymiana następuje
procesom dyfuzji płyn międzykomórkowy oraz trud- szczególnie łatwo tam, gdzie śródbłonek włośniczko-
no dostępny dyfuzji płyn ścisłej tkanki łącznej skóry, wy posiada większe lub mniejsze pory (np. wątroba,
mięśni, chrząstki i kości. trzustka), albo też tam, gdzie nie występuje błona
podstawowa (np. wątroba). Przenikanie w mięśniach
Do płynu międzykomórkowego (ok. 1,5% masy także jest możliwe, ponieważ ich naczynia włosowa-
ciała) zalicza się płyn mózgowo-rdzeniowy, peri- te wykazują tzw. aktywność transcytotyczną. Łącznie
i endolimfę oraz płyny występujące w jamach ciała z wodną fazą osocza również rozpuszczone substan-
i narządach jamistych. cje zostają otoczone przez błonę i w utworzonych pę-
MUTSCHLER-2009.indd 19 2010-01-07 22:10:44
20 Farmakokinetyka
cherzykach mogą być transportowane z endotelium Rozmaite możliwości wiązania wyjaśniają zarazem,
do wnętrza tkanki mięśniowej. dlaczego wiązaniu z białkami podlegają tak różno-
Utrudnione jest natomiast przechodzenie z kapilar rodne substancje.
pozbawionych istniejących w endotelium i błonie pod- Wiązanie z białkami ksenobiotyków jest względnie
stawnej porów oraz dodatkowo otoczonych komór- niespecyficzne, zachodzi przede wszystkim w miej-
kami pomocniczymi. Kapilary mózgowe są np. ściśle scach odznaczających się dużym powinowactwem,
otoczone komórkami podporowymi gleju w obrębie których liczba jest względnie ograniczona.
splotu naczyniowego (plexus chorioideus), w którym
dochodzi do tworzenia płynu mózgowo-rdzeniowego.
Śródbłonek jest tu oddzielony od płynu mózgowo-rdze-
niowego dodatkową warstwą komórek. Następstwem
jest utrudnienie przenikania, mówi się więc o barierze
krew-mózg lub barierze krew-płyn mózgowo-rdze-
astrocyt
niowy. Związki lipofilne pokonują barierę z łatwoś-
cią, nierozpuszczalne w tłuszczach w bardzo małym
stopniu, o ile nie występuje czynny transport, jak
w przypadku aminokwasów. Podobnie jak w przy-
padku przewodu pokarmowego, również w barierze
mózgowo-rdzeniowej zidentyfikowano kilka układów
transportowych powodujących czynny zwrotny trans-
port leków i mogących na tej drodze przyczyniać się
do ochrony OUN przed substancjami obcymi (zob. ryc.
A 2.3-2). W przypadku procesów zapalnych, podobnie
jak w innych tkankach, przepuszczalność zwiększa śródbłonek
się tak, że związki, które w prawidłowych warun-
kach nie mogą przenikać przez barierę krew-mózg,
mogą wtargnąć do ośrodkowego układu nerwowego. nacz
włosoynie
wate
Przestrzeń wewnątrzkomórkowa jest oddzielona
od przestrzeni międzykomórkowej i osocza przez
lipofilne błony komórkowe. Dlatego też, wyłączając
związki podlegające czynnemu transportowi, do wnę-
trza komórki i jej organelli łatwo przechodzą tylko
związki lipofilne.
2.3.2. Wiązanie z białkami
krew
Ważnym czynnikiem wpływającym na dystrybucję
leku jest jego wiązanie z białkami, zwłaszcza:
OATP1A2
BCRP
■ osocza,
SLC
P-gp
ABC
ABC
■ tkanek,
gazy krwi/NO/CO
dyfuzja:
■ erytrocytów.
W wiązanie z białkami mogą być, zależnie od budo- komórka śródbłonka
wy chemicznej cząsteczki, zaangażowane wiązania:
SLC
■ jonowe,
tkanka mózgowa
■ wodorowe (tworzenie mostków) i
■ dipolowe (dipol-dipol) oraz
■ oddziaływania hydrofobowe. Ryc. A 2.3-2. Schemat budowy bariery krew-mózg.
MUTSCHLER-2009.indd 20 2010-01-07 22:10:44
Farmakokinetyka 21
W ludzkiej albuminie można wykazać istnie- cji czynnej. Natomiast zmiany zawartości albumin,
nie dwóch różnych miejsc wiązania (miejsca I i II). zwłaszcza występujące w różnych chorobach (zob.
Niektóre środki lecznicze wiążą się wybiórczo tylko tab. A 2.3-2) zmiany stężenia kwaśnej α1-glikopro-
z jednym z wymienionych miejsc (np. leki przeciw- teiny, mogą prowadzić do znacznych zmian w ilości
zakrzepowe typu kumaryn z miejscem wiązania I, wolnej postaci leku.
benzodiazepiny z miejscem wiązania II), natomiast Oprócz zależnych od budowy leku specyficz-
wszystkie inne z obydwoma miejscami. nych właściwości wiązanie z białkami jest też zależ-
W wiązaniu substancji zasadowych, np. proprano- ne od wartości pH osocza oraz wieku. Na przykład
lolu, lidokainy, dizopiramidu, petydyny lub trójpier- w kwasicy zmniejsza się ilość barbituranów wiąza-
ścieniowych leków antydepresyjnych, istotną rolę od- nych z białkami. U noworodków wiązanie z białkami
Farmakokinetyka
grywa również wiązanie z kwaśną α1-glikoproteiną. jest mniejsze niż u dorosłych. Ponieważ wydłużona
Dla substancji endogennych istnieje wiele specy- jest również eliminacja (zob. poniżej), często w na-
ficznych białek transportowych, należących do globu- stępstwie opóźnionego dojrzewania uczestniczących
lin (np. transkortyna dla kortyzolu). w nich narządów, noworodki są znacznie bardziej
Wiązanie z białkami jest odwracalne. Nieodwra- wrażliwe na środki lecznicze niż dorośli i starsze
calne (= kowalentne) połączenia, np. wskutek reakcji dzieci.
alkilujących cytostatyków z białkami, nie są zalicza-
ne do wiązań z białkami. Wiązanie z białkami ma następujący wpływ na siłę A2
Wiązanie z białkami jest tym silniejsze, im więk- i czas działania oraz eliminację leków: związana
szy jest współczynnik powinowactwa danego związku z białkami część leku nie może dyfundować, nie pod-
do białka. Ponieważ współczynniki powinowactwa lega też biotransformacji i wydalaniu. Ze względu
do różnych białek, np. białek osocza i białek tkanek, na to – pominąwszy wyjątki – tylko wolna postać leku
często różnią się między sobą, może to wpływać dostaje się do właściwego miejsca docelowego i wy-
na stałą równowagi dystrybucji: przesuwa się ona kazuje tam działanie. Związana część leku ma postać
w kierunku białek o większym współczynniku powi- zmagazynowaną, z której po obniżeniu stężenia po-
nowactwa (zob. ryc. A 2.3-3). staci wolnej (np. wskutek biotransformacji i wydala-
W następstwie ograniczonej liczby miejsc wią- nia) w celu przywrócenia stanu równowagi (w ciągu
żących stopień wiązania z białkami jest też determi- milisekund) uwolnione zostają cząsteczki leku.
nowany przez stężenie białek i substancji czynnej. Jeżeli we krwi znajduje się jednocześnie kilka
W przypadku większości leków skuteczne leczniczo leków, istnieje możliwość konkurencji (kompety-
stężenia są względnie niskie (< 10 μg/ml), dlatego cji) o miejsce wiązania i co za tym idzie – wzajem-
wiązanie z białkami w nieistotnym stopniu wpływa nego oddziaływania na siłę i czas działania, przede
na występujące zmiany (fluktuacje) stężeń substan- wszystkim wówczas, gdy związana część leku wy-
nosi > 80%. Zjawisko to jest jednak ograniczone
zwiększoną eliminacją środka po jego wyparciu z po-
Tabela A 2.3-2. Przyczyny zmniejszonego stężenia albuminy łączeń białkowych przez inny środek. Po ustaleniu
i podwyższonego stężenia kwaśnej α1-glikoproteiny (i odwrot- się nowego stanu równowagi całkowite stężenie leku
nie) jest mniejsze niż przed podaniem związku kompety-
cyjnego.
Albumina ↓ Kwaśna α1-glikoproteina ↑ Należy też wziąć pod uwagę, że leki mogą również
wypierać z połączeń białkowych związki endogenne,
niedożywienie reumatoidalne zapalenie stawów
np. bilirubinę lub glukokortykosteroidy, i w wyniku
marskość wątroby choroba Crohna tego zwiększać stężenie ich niezwiązanych części.
oparzenia ostry zawał serca
zespół nerczycowy oparzenia 2.3.3. Czynniki wpływające
niewydolność nerek choroby zakaźne na dystrybucję
nadczynność tarczycy otyłość
Wpływ cech rozpuszczalności środka leczniczego
Albumina ↑ Kwaśna α1-glikoproteina ↓
na dystrybucję objawia się m.in. tym, że silnie li-
niedoczynność tarczycy antykoncepcja doustna, pofilne związki gromadzą się w narządach o dużej
marskość wątroby zawartości tłuszczu, natomiast związki hydrofilne
na odwrót – w ogóle nie wchodzą do tkanki tłuszczo-
MUTSCHLER-2009.indd 21 2010-01-07 22:10:45
22 Farmakokinetyka
ry krew-mózg. Niemniej przy podaniach doustnych
wchłanianie również jest znacznie zmniejszone.
osocze tkanka/miejsce działania Działające obwodowe inhibitory dekarboksylazy,
np. karbidopa lub benserazyd, znacznie zwiększy-
ły możliwości leczenia lewodopą osób z chorobą
substancja związana substancja związana
z białkami z białkami Parkinsona. Przy podawaniu samej lewodopy ok.
90% leku ulega dekarboksylacji na obwodzie i tylko
10% dociera do właściwego miejsca działania w móz-
gu. W przypadku jednoczesnego podania inhibitora
substancja substancja dekarboksylazy dopamina tworzona jest niemal wy-
wolna wolna łącznie w ośrodkowym układzie nerwowym. Dzięki
temu można nie tylko zmniejszyć dawkę lewodopy
do jednej piątej czy nawet jednej dziesiątej, ale też
Ryc. A 2.3-3. Wpływ wiązania z białkami na dystrybucję leku. Po
i występowanie działań niepożądanych jest znacznie
uzyskaniu stanu równowagi dystrybucji stężenie niezwiązanej
(wolnej) części substancji jest takie samo w osoczu i tkankach. mniejsze.
Jednym ze szczególnie aktualnych aspektów wpływania
wej i nie przedostają się do wnętrza komórki, dlatego na dystrybucję jest tzw. drug targeting, tj. celowane dostar-
czanie substancji czynnej do pożądanego miejsca działania.
obserwowane są głównie zewnątrzkomórkowo. Działania niepożądane często występują właśnie dlatego,
Dystrybucja uwarunkowana szczególnym powi- że związki lecznicze poza zamierzonymi narządami i struk-
nowactwem do określonych składowych organizmu turami reagują również z innymi miejscami w organizmie.
może być przedstawiona na poniższym przykładzie:
tlenek węgla wskutek swego dużego powinowactwa Drug targeting wymaga odpowiedniego układu nośników,
który umożliwia wybiórczy transport do organu docelo-
do hemu jest związany niemal wyłącznie z hemoglo- wego i co za tym idzie – pożądaną selektywność działa-
biną i mioglobiną, z tego względu jego dystrybucja nia. Nośnikami mogą być zarówno endo-, jak i egzogenne
odpowiada rozmieszczeniu tych związków. Niektóre makrocząsteczki, a także komórki, np. erytrocyty. Jednym
pochodne akrydyny gromadzą się w zasadowych ze szczególnie interesujących przykładów jest kowalentne
strukturach tkanek, przede wszystkim w jądrach ko- wiązanie cytostatyków z (poli- i monoklonalnymi) przeciw-
ciałami przeciwnowotworowymi, jednak praktyczne osiąg-
mórkowych, izoniazyd natomiast w skórze. Arszenik nięcia uzyskiwane za pomocą takich systemów dotychczas
(trójtlenek arsenu) odkłada się przede wszystkim w większości rozczarowują.
w tkankach zawierających keratynę (skóra, paznok-
cie, włosy) w następstwie występującego w niej bo- Inną możliwością drug targeting jest wybiórcza enzyma-
gactwa grup SH. Jony strontu i ołowiu, które charak- tyczna bioaktywacja proleku zachodząca za pośrednictwem
enzymów występujących głównie w miejscu pożądanego
teryzują się podobnymi do jonów wapnia właściwoś- działania (np. w obrębie nowotworu).
ciami chemicznymi, rozmieszczają się podobnie jak
te ostatnie, tj. gromadzą się w kościach i narządach
Bez wpływania na dystrybucję również udaje się przy
bogatych w miąższ. Jod lub sole jodu są wychwyty-
wielu lekach uzyskać względnie wybiórcze działanie,
wane szybko za pomocą czynnego transportu przez
opiera się to w większości przypadków nie na gro-
tarczycę.
madzeniu się substancji czynnej w miejscu działania,
Z leczniczego punktu widzenia byłoby korzyst-
ale na różnicach we wrażliwości tkanek i narządów
ne, gdyby można było sterować dystrybucją, tj. móc
na lek.
zwiększać stężenie w miejscu działania w porówna-
niu ze stężeniami występującymi w pozostałych częś-
ciach organizmu, jednakże możliwości wpływania
na sposób dystrybucji są w większości przypadków 2.3.4. Szczególne zjawiska
niewielkie. związane z dystrybucją
W chemioterapii nowotworów złośliwych próbu-
je się osiągnąć bardziej celowane działanie poprzez Oprócz dotychczas opisanych procesów dystrybucji
wstrzyknięcia lub wlewy cytostatyków do tętnicy za- należy wziąć pod uwagę szczególne zjawiska zwią-
opatrującej guz. zane z dystrybucją zachodzące w obrębie przewodu
W przypadku cholinolityków (parasympatykoli- pokarmowego (zob. ryc. A 2.3-4).
tyków) przez stosowanie związków IV-rzędowych Związki, które są wydalane z żółcią do dwunast-
można osiągnąć wyłącznie działanie obwodowe, nicy, mogą ulegać częściowemu lub całkowitemu
ponieważ związki te nie są w stanie pokonać barie- wchłanianiu zwrotnemu (krążenie wątrobowo-jeli-
MUTSCHLER-2009.indd 22 2010-01-07 22:10:45
Farmakokinetyka 23
żołądek
Farmakokinetyka
jelito żółć
krążenie
jelitowo- L L -sprzężony L -sprzężony
-żołądkowe wydzielanie
hydroliza
L L
krążenie
jelitowo-
A2
-wątrobowe
L L L -sprzężony
reakcje II fazy
krew wątroba
Ryc. A 2.3-4. Krążenie jelitowo-wątrobowe i jelitowo-żołądkowe substancji czynnej (L).
towe) w dalej położonych odcinkach jelit. Na prze- ka również zawarte w ślinie środki lecznicze mogą
chodzenie substancji zasadowych z krwi do żołądka być po połknięciu częściowo ponownie wchłaniane
zwracano uwagę już uprzednio. Takie związki rów- w jelitach.
nież mogą być częściowo wchłaniane zwrotnie (krą- Łożysko jest względnie dobrze przenikalne zarów-
żenie jelitowo-żołądkowe) w jelicie cienkim. no dla lipofilnych, jak i hydrofilnych związków, po-
Istotne jest ponadto przechodzenie leków do śliny. nieważ jego błony posiadają liczne pory. Dlatego też
Ponieważ ślina niemal nie zawiera białek, występu- o barierze łożyskowej należy mówić bardzo ostroż-
jące w niej stężenia substancji obojętnych są równe nie. Prawie zawsze powinno się wychodzić z założe-
stężeniom substancji niezwiązanej z białkami w oso- nia, że leki, które przyjmuje matka, przechodzą rów-
czu. W przypadku słabych zasad lub kwasów są jed- nież do organizmu płodu.
nak możliwe różnice, ponieważ ślina w porównaniu Mleko matki wykazuje w porównaniu z osoczem
z osoczem wykazuje zazwyczaj niższe wartości pH. niższe pH i większą zawartość tłuszczu. Dlatego leki
Na podstawie wyżej podanych właściwości można o zasadowych i lipofilnych właściwościach osiągają
stwierdzić, że ślina nadaje się do badania stężeń duże stężenia w mleku matki. Szczególnie ważny
leku w organizmie. Korzystne jest to, że pobieranie jest w związku z tym fakt, że również etanol, niko-
śliny nie wymaga zabiegu inwazyjnego. Podobnie tyna, dioksyna oraz insektycydy (np. chlorofenotan)
jak przy wydalaniu z żółcią lub do światła żołąd- przechodzą do mleka matki.
MUTSCHLER-2009.indd 23 2010-01-07 22:10:46
24 Farmakokinetyka
2.4. Biotransformacja
Związki lipofilne są po filtracji kłębuszkowej w du- że mogą one uczestniczyć w metabolizmie substratów
żej mierze zwrotnie wchłaniane w kanalikach nerko- o różnorodnej budowie chemicznej. Układy enzyma-
wych, dlatego ich wydalanie z moczem może zacho- tyczne zaangażowane są nie tylko w biotransforma-
dzić wyłącznie bardzo powoli. O ile nie zostaną one cję ksenobiotyków, ale również w procesy przemia-
zmienione chemicznie, istnieje niebezpieczeństwo, ny materii związków endogennych (np. hormonów
że pozostaną w organizmie i będą gromadzone szcze- steroidowych, kwasów żółciowych, hemu). Oprócz
gólnie w tkance tłuszczowej. Nic więc dziwnego, enzymów występujących w narządach organizmu
że organizm posiada układy enzymatyczne mogące również flora bakteryjna jelit ma określony wpływ
przekształcić lipofilne ksenobiotyki w hydrofilne, ła- na biotransformację, zwłaszcza procesy redukcji i hy-
two usuwalne metabolity. Szybkość eliminacji związ- drolizy.
ków rozpuszczalnych w tłuszczach zależy więc w du- Na ryc. A 2.4-1 przedstawiono schematycznie
żej mierze od tego, jak szybko są one w organizmie ważniejsze procesy zachodzące w biotransformacji.
przekształcane (metabolizowane) w związki rozpusz-
czalne w wodzie.
Procesy przekształcania obcych substancji określa 2.4.1. Reakcje I fazy
się mianem biotransformacji. Zachodzi ona przede
wszystkim w wątrobie, a (zazwyczaj) w innych na- Reakcje I fazy są to procesy biotransformacji, w cza-
rządach (np. w jelitach, nerkach, płucach, śledzionie, sie których cząsteczka leku ulega zmianie w następ-
mięśniach prążkowanych, skórze, a nawet we krwi) stwie utlenienia, redukcji lub hydrolizy, natomiast
ma drugorzędny charakter. w wyniku II fazy zachodzi sprzęganie (koniugacja)
W przypadku powierzchniowo stosowanych środ- cząsteczki leku lub metabolitu uzyskanego w fa-
ków dermatologicznych znaczenie ma niekiedy meta- zie I z endogenną substancją. W wielu przypadkach
bolizm w skórze. Na przykład hydroliza C-21 estrów wskutek reakcji I fazy powstają warunki umożliwia-
glukokortykosteroidów nasila wiązanie z receptorami jące koniugację. Przykłady reakcji I fazy przedsta-
glukokortykosteroidowymi. Jednocześnie duża lipo- wiono w tab. A 2.4-1.
filność estrów ułatwia pokonanie trudno przenikalnej
zrogowaciałej bariery naskórkowej. W nowszym piśmiennictwie procesy czynnego trans-
portu przezbłonowego określane są jako reakcje fazy III.
Enzymy uczestniczące w biotransformacji, struktu-
ralnie związane z błonami siateczki endoplazmatycz-
nej (np. monoaminooksydazy, glukuronylotransfera-
zy), po części również zlokalizowane w mitochon- 2.4.1.1. Reakcje utleniania
driach oraz występujące obok związanych struk-
turalnie jako enzymy rozpuszczalne (np. esterazy, Szczególne znaczenie dla biotransformacji mają pro-
amidazy, sulfotransferazy), charakteryzują się często cesy utleniania, w których biorą udział oksygena-
niewielką specyficznością substratową. Oznacza to, zy, monooksygenazy, dioksygenazy. Oksydazy utle-
reakcje I fazy reakcje II fazy
lek metabolit I fazy metabolit II fazy
utlenianie, sprzęganie z:
redukcja, aktywnym kwasem
hydroliza glukuronowym, aktywnym
kwasem siarkowym,
aktywnym kwasem octowym,
aminokwasami i in.
Ryc. A 2.4-1. Najważniejsze procesy biotransformacji.
MUTSCHLER-2009.indd 24 2010-01-07 22:10:46
Farmakokinetyka 25
Tabela A 2.4-1. Drogi biotransformacji leków, reakcje I fazy
Reakcja Wzór Przykłady substratów
utlenianie
O
utlenianie alkoholi R CH2OH R
alkohol benzylowy,
R COOH
i aldehydów pirydoksyna
H
utlenianie łańcuchów barbiturany
alifatycznych OH
Farmakokinetyka
R CH3 + R COOH
R CH3
oksydacyjna H efedryna,
N-dealkilacja O lidokaina,
R1 N R1 NH2 + R2 metamfetamina
CH2R2 H
oksydacyjna histamina,
deaminacja O meskalina, A2
R NH2 R + NH3 noradrenalina
H
oksydacyjna R2 R2 kodeina,
O
O-dealkilacja meskalina,
+ papaweryna
R1 O R1 H HO
utlenianie pochodne aniliny
aminy NH2 NH N O
aromatycznej OH
R R R
N-oksydacja R1
imipramina
R1 R3
+
N R3 N
-
R2 R2 O
S-oksydacja fenotiazyny
R1 R1 R1 O
S S O S
R2 R2 R2 O
desulfatacja R1 R1 tiobarbiturany,
C S C O paration
R2 R2
R1 R3 R1 R3
P P
R2 S R2 O
epoksydacja aldryna,
R1 R2 karbamazepina
R1 R2
O
hydroksylacja R chloropromazyna,
R
substancji fenotiazyna,
aromatycznych propranolol
HO
MUTSCHLER-2009.indd 25 2010-01-07 22:10:47
26 Farmakokinetyka
Tabela A 2.4-1. Drogi biotransformacji leków, reakcje I fazy (kontynuacja)
Reakcja Wzór Przykłady substratów
redukcja O
aldehydów R R CH2OH wodzian chloralu
H
związków azowych R2 NH2 salazosulfapirydyna
N + R2
N R1
R1 H2N
grup nitrowych nitrazepam
NO2 NH2
R R
dehalogenacja R1 R3 R1 R3 halotan
C C
R2 Hal R2 H
hydroliza
estrów O kwas acetylosalicylowy,
R1 COOH + HO R2 kokaina,
R1 OR2 petydyna,
prokaina
amidów kwasowych O
prokainamid
R1 COOH + H2N R2
R1 NHR2
glikozydów glikozydy antranilowe,
H O R2 H O R2 glikozydy nasercowe
C C + HO R3
R1 O R3 R1 OH
epoksydów metabolit
R1 R3 R2 R3 karbamazepiny
R1 R4
R2 O R4
OH OH
inne
dekarboksylacja COOH H H histydyna,
+ CO2 lewodopa,
R1 R2 R1 R2 α-metylodopa
niają przez odłączenie wodoru lub elektronów. fragmenty siateczki endoplazmatycznej (frakcja mikroso-
Monooksygenazy wbudowują jeden atom tlenu malna). Związane w nich enzymy nazywa się enzymami
mikrosomalnymi. Należą do nich, oprócz monooksygenaz,
z cząsteczki tlenu w związek obcy, drugi atom zosta- również epoksydohydratazy i glukuronylotransferazy (zob.
je zredukowany z powstaniem wody. Dioksygenazy poniżej).
natomiast wprowadzają oba atomy cząsteczki tlenu
do ksenobiotyku.
Cytochrom P-450. Największe znacznie dla oksyda-
tywnej biotransformacji leków mają (mikrosomalne)
Mikrosomami nazywa się uzyskiwane podczas frakcjono- monooksygenazy zawierające hemoproteinę o typie
wanego wirowania homogenatów komórek wątrobowych cytochromu P-450. Istnieje wiele enzymów P-450
MUTSCHLER-2009.indd 26 2010-01-07 22:10:47
Farmakokinetyka 27
różniących się między sobą specyficznością wzglę- cie cienkim (w wierzchniej warstwie enterocytów)
dem substratu, ilością w danym miejscu ekspresji oraz może ograniczać biodostępność leków podawanych
innymi właściwościami (np. wrażliwością na induk- doustnie. Ilości poszczególnych enzymów CYP wy-
cję, polimorfizmem genetycznym). Izoenzymy cyto- kazują duże różnice międzyosobnicze. Uważa się,
chromu P-450 (CYP) uporządkowano zgodnie z na- że są za to odpowiedzialne dwa zjawiska: po pierw-
stępującymi zasadami: na podstawie sekwencji reszt sze, aktywność może być wyznaczona przez czynniki
aminokwasowych różne CYP podzielono na rodziny, wrodzone. Takie genetyczne polimorfizmy o istotnym
które charakteryzuje odpowiednia cyfra arabska (dla znaczeniu klinicznym zależnym od udziału określo-
metabolizmu związków obcych szczególnie ważne nych dróg metabolicznych w całkowitej eliminacji,
są rodziny 1-3). Rodziny posiadają liczne, oznaczo- a także rozpiętości terapeutycznej leku zostały opisa-
Farmakokinetyka
ne literami podrodziny. Poszczególni członkowie ne np. dla CYP2D6 i CYP2C19. Po drugie, występują
podrodziny zostali oznaczeni kolejną cyfrą arabską. również enzymy CYP, których aktywność niezależnie
Na przykład oznaczenie CYP1A2 jest związane od czynników wrodzonych regulowana jest przez in-
z drugim enzymem podrodziny A, rodziny 1. W tab. dukcję (m.in. CYP1A2, CYP2B6, CYP3A4).
A 2.4-2 zestawiono ważne dla metabolizmu leków
u ludzi enzymy CYP, ich cechy charakterystyczne Nazwa cytochrom P-450 pochodzi od jego właściwości po-
oraz niektóre substraty. Ilościowo najważniejszym legających na silnym pochłanianiu światła o długości fali
CYP jest CYP3A4, biorący udział w procesach utle-
450 nm po redukcji ditionianem sodu i wysyceniu tlenkiem A2
węgla (izoenzymy określane dawniej na podstawie pochła-
niania dużej liczby często stosowanych leków. niania światła jako cytochrom P-448 stanowią zgodnie
z obecnym nazewnictwem podrodzinę l A).
Największe ilości enzymów CYP występują w wą-
trobie. Metodami biochemicznymi oraz metodami Podczas utleniania pod wpływem monooksygenazy
biologii molekularnej udało się jednak wykazać wy- substrat (L-H) jest najpierw wiązany z trójwartościo-
stępowanie tych enzymów w wielu innych narządach wym żelazem cytochromu P-450. Następnie dochodzi
(np. w przewodzie pokarmowym, płucach, mózgu), do przeniesienia jednego elektronu za pomocą łań-
przy czym szczególnie ich występowanie w jeli- cucha reakcji (w którym wykazano udział NADPH
Tabela A 2.4-2. Ważne enzymy z rodziny cytochromu P-450 (CYP) oraz wybrane substraty
Enzym Substrat
CYP1A2 amitryptylina, klomipramina, klozapina, kofeina, estradiol, fluwoksamina, haloperidol, meksyletyna,
naproksen, olanapina, ondansetron, propranolol, ropiwakaina, takryna, teofilina, werapamil, zolmitryptan
CYP2A6 nikotyna
CYP2B6 bupropion, cyklofosfamid, efawirenz, ifosfamid, metadon
CYP2C8 cerywastatyna, paklitaksel, repaglinid
CYP2C9 amitryptylina, celekoksyb, diklofenak, fluoksetyna, fluwastatyna, glibenklamid, glipizyd, ibuprofen, irbesartan,
losartan, meloksykam, naproksen, fenprokumon, piroksykam, roziglitazon, tamoksyfen, tolbutamid, warfaryna
CYP2C19 amitryptylina, citalopram, klomipramina, cyklofosfamid, diazepam, heksobarbital, imipramina, indometacyna,
lanzoprazol, moklobemid, nelfinawir, omeprazol, pantoprazol, fenobarbital, fenytoina, prymidon,
progesteron, proguanil, propranolol, rabeprazol, tenipozyd, warfaryna
CYP2D6 alprenolol, amitryptylina, karwedilol, chloropromazyna, klomipramina, kodeina, dezypramina, dekstrometorfan,
flekainid, fluoksetyna, fluwoksamina, haloperidol, imipramina, metoprolol, meksyletyna, nortryptylina,
ondasetron, paroksetyna, perfenazyna, propafenon, risperidon, tamoksyfen, tiorydazyna, tiomolol, tramadol
CYP2E1 enfluran, etanol, halotan, izofluran, sewofluran, teofilina
CYP3A4 alprazolam, alfentanyl, amlodypina, astemizol, atorwastatyna, buspiron, klarytromycyna, chinidyna, cyklosporyna,
kofeina, dekstrometorfan, diazepam, diltiazem, docetaksel, domperidon, eplerenon, erytromycyna, estradiol, felodypina,
fentanyl, haloperidol, imatynib, indinawir, irinotekan, lerkanidypina, lidokaina, lowastatyna, metadon, midazolam,
nelfinawir, nifedypina, nisoldypina, nitrendypina, ondasetron, paklitaksel, progesteron, propranolol, ritonawir, sakwinawir,
sildenafil, simwastatyna, sirolimus, takrolimus, tamoksyfen, terfenadyna, testosteron, triazolam, werapamil,
winkrystyna, zolpidem
MUTSCHLER-2009.indd 27 2010-01-07 22:10:48
28 Farmakokinetyka
L H
(Fe3+) (Fe3+) L H
L OH e-
(Fe3+) L OH Cytochrom P-450 (Fe2+) L H
H2O O2
_ _
[(Fe3+) (O22-) L H] [(Fe2+) (O2. ) L H] [(Fe2+) (O2) L H] [(Fe3+)(O2. ) L H]
e-
Ryc. A 2.4-2. Mechanizm utleniania leku (L) przy współudziale cytochromu P-450.
i flawoproteidu) na żelazo cytochromu P-450 (przy wactwa do substratów FMO jest w porównaniu z en-
utlenieniu NADPH), w wyniku czego staje się ono zymami CYP znacznie mniejsza.
dwuwartościowe. Po dołączeniu tlenu cząsteczko-
wego i przyjęciu kolejnego elektronu dostarczonego Pozostałe enzymy utleniające. Innymi ważnymi en-
przez drugi łańcuch przenośnikowy powstały trzecio- zymami uczestniczącymi w procesach utleniania są:
rzędowy kompleks ulega rozpadowi do hydroksylo-
■ dehydrogenaza alkoholowa, utleniająca alkohole,
wanego substratu (zob. ryc. A 2.4-2).
zwłaszcza etanol, do odpowiednich aldehydów,
Podsumowując, uzyskuje się: ■ oksydaza aldehydowa, przekształcająca aldehydy
w kwasy,
L-H + O2 + NADPH + H+ ⇒ L-OH + H2O +NADP+.
■ monoaminooksydaza, która przede wszystkim ok-
Monooksydazy zawierające cytochrom katalizują
sydatywnie przekształca aminy biogenne (np. ka-
hydroksylację związków alifatycznych i aromatycz-
techolaminy).
nych, epoksydację podwójnych wiązań związków
oleinowych i aromatycznych, oksydatywną dealki-
lację związków N-, O- i S-alkilowych, oksydatywną
deaminację oraz utlenianie tioeterów i amin do sul-
2.4.1.2. Redukcja
fotlenków lub hydroksyloamin.
W porównaniu z utlenianiem procesy redukcji od-
Monooksygenazy flawinowe. Oprócz monooksy- grywają w biotransformacji tylko podrzędną rolę.
genaz zawierających cytochrom P-450 występują Związki karbonylowe przy współudziale dehydro-
również monooksygenazy zawierające flawoproteidy genazy alkoholowej albo cytoplazmatycznych aldo-
(FMO), które np. przekształcają drugorzędowe ami- keto-reduktaz mogą ulegać redukcji do alkoholi.
ny w hydroksyloaminy, a trzeciorzędowe w N-tlenki. W rozkładzie związków azowych do amin pierwszo-
Analogicznie do układu P-450 zidentyfikowano cały rzędowych poprzez hydrazowe związki pośrednie
szereg izoenzymów, różniących się powinowactwem uczestniczy prawdopodobnie wiele enzymów, m.in.
do substratów, lokalizacją i zasadami regulacji. należy brać pod uwagę udział reduktazy – NADPH
Ogólnie można stwierdzić, że specyficzność powino- cytochromu P-450. Nie jest też w pełni wyjaśniona
MUTSCHLER-2009.indd 28 2010-01-07 22:10:48
Farmakokinetyka 29
rola enzymów zaangażowanych w redukcję związ- 2.4.2. Reakcje II fazy
ków nitrowych do odpowiednich amin.
Z toksykologicznego punktu widzenia istotna jest re- W reakcjach sprzęgania, które zazwyczaj zachodzą
dukcyjna dehalogenacja alifatycznych związków za- przy udziale specyficznych transferaz, odróżnia się
wierających chlor, brom lub jod, np. tetrachlorku wę- te, w których wysokoenergetyczne związki endogenne
gla lub halotanu. są wiązane z alkoholowymi lub fenolowymi grupami
hydroksylowymi, grupami aminowymi, grupami sulf-
hydrylowymi i po części również grupami karboksy-
2.4.1.3. Biohydroliza lowymi, od tych, przy których po aktywacji ksenobio-
tyku następuje wiązanie z (nieaktywnym) związkiem
Farmakokinetyka
endogennym (do tych ostatnich należy sprzęganie
Ważnymi procesami biohydrolitycznymi są:
pewnych kwasów karboksylowych z aminokwasami.
■ rozpad estrów i amidów do kwasów i alkoholi lub Ważniejszymi reakcjami II fazy (zob. tab. A 2.4-3)
amin za pomocą esteraz (amidaz), jest sprzęganie z:
■ przekształcenie epoksydów do pokrewnych dioli ■ aktywowanym kwasem glukuronowym,
przez epoksydohydratazy (synonim epoksydohy-
■ aktywowanym siarczanem,
drolazy), A2
■ aminokwasami (zwłaszcza z glicyną),
■ hydroliza glikozydów przez glikozydazy,
■ oligopeptydami oraz tworzenie pochodnych kwasu
■ hydroliza glukuronianów przez glukuronidazy.
merkapturowego,
Estry i amidy są hydrolizowane przez te same enzymy, ■ aktywowanym kwasem octowym,
estry jednak znacznie szybciej niż amidy. Esterazy
■ S-adenozylometioniną.
występują zarówno wewnątrz-, jak i zewnątrzko-
mórkowo, są związane z mikrosomami i w postaci
Z wyjątkiem sprzęgania z kwasem octowym lub pro-
rozpuszczalnej. W metabolizmie związków obcych
cesów metylacji (zob. poniżej) w reakcjach tych zosta-
istotne są pseudocholinoesterazy i tzw. aliloesterazy
je zawsze wprowadzona do cząsteczki grupa kwaso-
rozszczepiające przede wszystkim alifatyczne estry
wa, która w następstwie tworzenia soli zdecydowanie
i amidy, a także aryloesterazy, do których wysokie
zwiększa hydrofilność i co za tym idzie – rozpuszczal-
powinowactwo wykazują estry i amidy z resztami
ność. Kwaśne koniugaty są szybko – również z udzia-
aromatycznymi.
łem procesów czynnych – wydalane z moczem lub
żółcią. Reakcje sprzęgania mają w związku z tym cha-
Enzymy hydrolityczne zawierają też bakterie flory
rakter dezaktywacji, tj. pozbawiania aktywności biolo-
jelitowej. Powodują one zwłaszcza rozszczepienie
gicznej, lub detoksykacji, ponieważ produkty sprzęga-
metabolitów fazy II w daleko położonych odcinkach
nia są w większości nieczynne biologicznie.
jelit. Jeśli uwolniona w ten sposób substancja czynna
W ostatnich latach odkryto jednak wiele powstają-
ulega wchłonięciu, występuje krążenie jelitowo-wą-
cych w fazie II czynnych metabolitów. Należą do nich
trobowe (zob. ryc. A 2.3-4).
np. hemiester hydroksylowanego, oszczędzającego
Epoksydohydratazy występujące w kompleksie
potas leku moczopędnego – triamterenu z kwasem
wieloenzymowym wspólnie z monooksygenazami
siarkowym oraz C-17 glukuroniany estrogenu i an-
odgrywają istotną rolę w metabolizmie epoksydów,
drogenów. 6-glukuronian morfiny działa przeciwbó-
z których powstają diole o grupach hydroksylowych
lowo tak jak związek macierzysty.
położonych obok siebie.
Z metabolitów II fazy mogą również powstać sub-
Oprócz opisywanej wyżej dehalogenacji, prze-
stancje czynne, które poprzez nieodwracalne wiąza-
biegającej z redukcją może też występować hydroli-
nia ze związkami endogennymi wywołują działania
tyczne rozszczepienie wiązania C-C1; produktem tej
toksyczne (zob. poniżej).
reakcji jest odpowiedni alkohol.
W niektórych przypadkach związki sprzężone
mogą ponadto ulegać hydrolizie do związków ma-
cierzystych. Sytuacja, którą przedstawiono wyżej,
2.4.1.4. Dekarboksylacja zdarza się często, kiedy sprzężone metabolity dostają
się z żółcią do jelit. Zjawisko to zachodzi natomiast
Do reakcji I fazy należy ponadto dekarboksylacja bardzo rzadko w przypadku sprzężonych metaboli-
występująca m.in. w przypadku aminokwasów. tów wydalanych z moczem.
MUTSCHLER-2009.indd 29 2010-01-07 22:10:48
30 Farmakokinetyka
Tabela A 2.4-3. Drogi biotransformacji leków, reakcje II fazy
Reakcja Wzór Przykłady substratów
sprzęganie
HOOC O O P P urydyna HOOC O O R
z aktywowanym alkohole,
kwasem glukuronowym aminy,
(UDP – kwas glukuronowy) HO OH H OR HO OH fenole,
OH OH
sulfonamidy
z aktywowanym R OH R O SO 3H aminy aromatyczne,
siarczanem fenole,
sulfonamidy
R NH2 R NH SO 3H
z glicyną kwas benzoesowy*,
O
kwas izonikotynowy*
R COOH + H2N COOH (jako metabolit
R NH COOH izoniazydu)
z glutaminą R COOH + H2N COOH R NH COOH kwas indolilooctowy*,
kwas fenylooctowy*
O
O NH2 O NH2
z aktywowanym izoniazyd,
O
kwasem octowym sulfonamidy
H2N R R
H3C NH
N-metylacja R1
metadon,
(z adenozylometioniną) R1 nikotynamid,
NH N CH3 noradrenalina
R2 R2
R R
+
N N
CH3
O-metylacja HO R O R katecholamina
(z adenozylometioniną) H3C
HO HO
* Sprzęganie zachodzi po aktywacji kwasu za pomocą ATP i koenzymu A.
2.4.2.1. Sprzęganie z aktywowanym estrów kwasu glukuronowego, poza rozszczepieniem
kwasem glukuronowym do substancji wyjściowej, może również następować
przmieszczenie reszty acylowej z grupy –OH na wę-
Z aktywowanym kwasem glukuronowym sprzęgane glu C-1 na grupy –OH na węglach C-2, C-3 lub C-4
są przede wszystkim te alkohole, które nie mogą ulec kwasu glukuronowego (migracja grupy acylowej).
szybkiemu utlenieniu, np. alkohole drugo- i trzecio-
rzędowe (zob. ryc. A 2.4-3). Fenole, kwasy karboksy- Kwas glukuronowy jest względnie silnym kwasem
lowe i aminy mogą być również sprzęgane z kwasem posiadającym dodatkowo alkoholowe grupy OH
glukuronowym. Eteroglukuroniany stanowią względ- i dlatego silnie hydrofilnym. Jest on przenoszony
nie stabilne związki, natomiast estry kwasu gluku- w postaci aktywowanego kwasu glukuronowego
ronowego są podatne na hydrolizę. W przypadku (UDP – kwas glukuronowy) na odpowiedni sub-
MUTSCHLER-2009.indd 30 2010-01-07 22:10:49
Farmakokinetyka 31
Cl
Cl aktywowany HOOC O O
kwas glukuronowy Cl
HOH2C Cl Cl
Cl HO OH
OH
trichloroetanol glukuronid trichloroetanolu
O O
Farmakokinetyka
OH
OH glicyna NH
O
kwas benzoesowy kwas hippurowy
O
OH
aktywowany siarczan
O S OH
A2
O
fenol ester fenylowy kwasu siarkowego
Ryc. A 2.4-3. Reakcje sprzęgania, w trakcie których powstają metabolity dobrze rozpuszczalne w wodzie, szybko wydalane z mo-
czem dzięki aktywnej sekrecji nerkowej.
strat przez związane z błonami glukuronylotransfe- sposób jak enzymy CYP. Wyraźne różnice w aktyw-
razy (urydyno-5′-difosforo-glukuronylo-transferazy ności między poszczególnymi enzymami, tak samo
– UGT) występujące przede wszystkim w wątrobie, jak w przypadku enzymów cytochromu P-450, mogą
a ponadto w nerkach i jelitach. Analogicznie do CYP- wynikać z działania czynników genetycznych lub być
450-zależnych monooksydaz występuje wiele izoen- indukowane.
zymów glukuronylotransferaz, różniących się specy-
ficznością powinowactwa do substratu oraz miejsca- Znane są dwie rodziny UGT obejmujące ogólnie 19 enzy-
mi ich ekspresji. Są one klasyfikowane w podobny mów – UGT1 i UGT2. Co ciekawe, wszystkich 9 enzymów
UGT1 jest kodowanych przez warianty splicingu tego sa-
mego genu. W ten sposób dzięki cięciu specyficznemu dla
Tabela A 2.4-4. Ważne glukuronozylotransferazy (UGT), wybra- poszczególnych tkanek (splicing alternatywny) z jednego
ne substraty i defekty genetyczne genu powstaje 9 różnych białek. UGT mają zdolność glu-
kuronidacji dużego zakresu różnorodnych strukturalnie
leków i endogennych substratów, a tym samym mają duże
UGT Substrat Zespół w przypadku znaczenie w procesie eliminacji. Zestawienie najważniej-
defektu genetycznego szych enzymów oraz ich substratów znajduje się w tab.
A 2.4-4.
1A1 bilirubina, zespół
etynyloestradiol, Criglera-Najjara,
SN 38 (metabolit zespół Gilberta
irynotekanu),
etopozyd 2.4.2.2. Sprzęganie z kwasem siarkowym
1A3 norbuprenorfina Z aktywowanym siarczanem (3’-fosfoadenozyno-
1A4 amitryptylina, imipramina 5’-fosfosiarczan, PAPS, zob. tab. A 2.4-3), przeno-
szonym przez cytozolowe sulfotransferazy (SULT)
1A6 naproksen, paracetamol
o różnej specyficzności, sprzęgane są przede wszyst-
1A9 propofol kim fenole. Dotychczas są znane trzy rodziny SULT
kodeina, ibuprofen,
(SULT1, 2 i 4), przy czym rodzina SULT1, a zwłasz-
2B7
ketoprofen, lorazepam, cza SULT1A1 odgrywa ważną rolę w rozkładzie le-
morfina, naproksen ków. Powstają hydrofilne półestry kwasu siarkowego,
które są wydalane z organizmu z moczem. W związ-
MUTSCHLER-2009.indd 31 2010-01-07 22:10:49
32 Farmakokinetyka
ku z tym należy wziąć pod uwagę fakt, że sulfono-
wanie, poza mniej toksycznymi, może również po-
wodować powstawanie reaktywnych, a tym samym O
niebezpiecznych, metabolitów. COOH
HN
HS O NH2
2.4.2.3. Sprzęganie z glicyną HN
COOH niebieski: kwas glutaminowy
czerwony: cysteina
Kwasy karboksylowe niepodlegające dalszym prze- zielony: glicyna
mianom oksydatywnym mogą ulegać sprzęganiu glutation
z glicyną. Należą tu podstawione przy atomie α-C
i aromatyczne kwasy karboksylowe, np. kwasy ben-
zoesowe. Klasyczne przykłady takich koniugatów
to tworzone z kwasów benzoesowych kwasy hippu-
rowe (zob. ryc. A 2.4-3) oraz powstające z kwasu sa- U ludzi występuje 20 izoenzymów S-transferazy glutationo-
licylowego kwasy salicylurowe. Reakcje są katalizo- wej (GST) o różnej specyficzności substratowej, które nale-
wane przez transacylazę. żą do dwóch podrodzin (GST mikrosomowe i cytozolowe).
Wśród siedmiu grup GST cytozolowych najważniejsze dla
metabolizmu leków są α-GST (GSTA1 i 2, indukowalne,
ich typowe substraty to busulfan i chlorambucyl), μ-GST
2.4.2.4. Powstawanie pochodnych (GSTM1 do 5, defekt genetyczny u 40% populacji, sub-
kwasu merkapturowego stratami są tiotepa, BCNU), π-GST (GSTP1) oraz θ-GST
(GSTT1 i 2, defekt genetyczny u 16% populacji).
Są to przebiegające wieloetapowo reakcje sprzęgania,
które po części zachodzą spontanicznie, ale są rów- Z utworzonych pierwotnych koniugatów z glutatio-
nież katalizowane przez glutationo-S-transferazy. nem zostają następnie odszczepione dwa aminokwa-
CYP450 bez udziału
Br Br
NADH + O2 enzymów kowalentne
O wiązanie
do makrocząsteczki
bromobenzol epoksyd
transferaza hydrataza
proces S-glutationowa epoksydowa
nieenzymatyczny
Br Br OH Br OH
OH S G OH
p-bromofenol odszczepienie kwasu –2H
glutaminowego i glicyny
acetylotransferaza
Br OH Br OH Br OH
– H2 O
S CysAc S Cys OH
pochodna kwasu 3,4-dihydroksy-
merkapturowego bromobenzol
Ryc. A 2.4-4. Powstawanie pochodnych kwasu merkapturowego z bromobenzolu obok innych reakcji biotransformacji. Reszty:
G –glutationylowa, Cys – cysteinylowa, Ac – acetylowa.
MUTSCHLER-2009.indd 32 2010-01-07 22:10:49
Farmakokinetyka 33
sy. Poprzez N-acetylację glicyny powstają jako pro- N-metylonikotynamidu z nikotynamidu. Powstające
dukty końcowe tzw. kwasy merkapturowe. na tej drodze czwartorzędowe zasady amoniowe
Sprzęganie z glutationem stanowi ważny proces są hydrofilne i mogą być czynnie wydalane. Metylacja
detoksykacji związków elektrofilnych. Wątpliwości fenolowych grup OH, jaka zachodzi np. w przypadku
nasuwa tu jednak silna reaktywność niektórych ko- katecholamin, stanowi raczej wyjątek niż regułę.
niugatów cysteiny, z których powstawać mogą tok- Za S-metylację odpowiedzialne są dwa różne enzy-
syczne produkty przemian. Dopiero znajdująca się my, mianowicie tiometylotransferaza i tiopurynome-
na końcu łańcucha przemian acetylacja oznacza tylotransferaza. Tiometylotransferaza jest enzymem
zmniejszenie reaktywności i toksyczności. błonowym, który katalizuje przede wszystkim mety-
Na ryc. A 2.4-4 przedstawiono oprócz innych pro- lację grup sulfhydrylowych łańcuchów alifatycznych.
Farmakokinetyka
cesów biochemicznych reakcje zachodzące podczas Substratami tego enzymu są przykładowo kaptopryl
przemiany bromobenzolu w odpowiednią pochodną i D-penicylamina. W odróżnieniu od tego tiopury-
kwasu merkapturowego. nometylotransferaza, będąca rozpuszczalną postacią
cytoplazmatyczną, metyluje grupy SH substancji
aromatycznych i heterocyklicznych (substratami
2.4.2.5. Acetylacja są np. 6-merkaptopuryna i azatiopryna). W przypad-
ku tego enzymu występuje genetyczny polimorfizm,
który może mieć poważne konsekwencje kliniczne. A2
Substancje posiadające grupy aminowe, które nie ule-
gają oksydacyjnym przemianom metabolicznym,
są często acetylowane za pomocą cytozolowych
N-acetylotransferaz. Dotychczas znane są dwa izoen- 2.4.3. Indukcja białek transportujących
zymy tego rodzaju: wszechobecny NAT1 oraz eks- i metabolizujących leki
presjonowany głównie w przewodzie pokarmowym
i w wątrobie NAT2.
Od dawna wiadomo, że niektóre leki oraz inne kseno-
Do ich substratów należą aminy aromatyczne
biotyki mogą wpływać na własne wydalanie. Należy
(np. anilina) i alkiloaminy, w których grupa amino-
przez to rozumieć, że np. lek przeciwpadaczkowy
wa znajduje się przy trzeciorzędowym atomie wę-
karbamazepina, po jej pierwszym podaniu, wyka-
gla. Acetylacja sulfonamidów i izoniazydu stanowi
zuje znacznie dłuższy biologiczny okres półtrwania
przykład tego typu procesów sprzęgania, w których
niż po powtarzalnej aplikacji. To oznacza, że ta sub-
na ogół dochodzi do zmniejszenia hydrofilności.
stancja może spowalniać własny metabolizm przez
Następstwem mogą być powikłania, np. wypadanie
nasiloną produkcję enzymów uczestniczących w bio-
kryształów (krystaluria), co może prowadzić do opi-
transformacji. Tego typu leki, które obecnie są dosyć
sywanych w przypadku sulfonamidów działań nie-
dobrze scharakteryzowane, nazywane są induktorami
pożądanych w obrębie dróg moczowych. Acetylacja
enzymatycznymi. Przez długi czas możliwości induk-
zmniejsza jednak skuteczność sulfonamidów, po-
cyjne przypisywano tylko enzymom zaangażowa-
nieważ niezbędna dla aktywności biologicznej gru-
nym w metabolizm. Dziś wiadomo, że poza tymi en-
pa aminowa zostaje zamaskowana grupą acetylową.
zymami indukcji ulegają także białka transportowe.
Poza sulfonamidami substratami dla NAT są także
Wspólna indukcja przez białka transportowe i enzy-
kwas aminosalicylowy, kofeina (substancja stosowa-
my metaboliczne prowadząca do wspólnej eliminacji
na do badania fenotypu acetylacji), klonazepam oraz
– a tym samym najczęściej również do eliminacji za-
izoniazyd.
trucia – wydaje się celowa.
Dla obu NAT opisano liczne warianty genetycz-
ne, przy czym tylko polimorfizmy NAT2 mają wpływ
Dawniej różne rodzaje indukcji nazywano według substan-
na status acetylacji i związane z nim działania niepo- cji leczniczych lub ksenobiotyków, dla których zostały one
żądane. pierwotnie opisane. W efekcie rozróżnia się induktory typu
fenobarbitalu, metylocholantrenu i rifampicyny. Jednak
przestano stosować tego typu podział, ponieważ odkryto
podobny podstawowy mechanizm działania wszystkich
2.4.2.6. Metylacja induktorów. Po pobraniu do komórki substancja induku-
jąca wiąże się do receptora wewnątrzkomórkowego, któ-
Wśród procesów biotransformacji metylacja wy- ry po połączeniu z drugim białkiem tworzy heterodimer.
Heterodimer wędruje do jądra komórkowego i wiąże się
stępuje względnie rzadko. Niekiedy stwierdza się z elementami regulatorowymi DNA. Dzięki temu specy-
N-metylację lub metylację nienasyconych związków ficznemu wiązaniu stymulowane są m.in. geny kodujące
heterocyklicznych. Przykładem może być tworzenie enzymy P-450 i białka transportowe.
MUTSCHLER-2009.indd 33 2010-01-07 22:10:50
34 Farmakokinetyka
Inna dobrze zbadana droga indukcji przebiega przez recep-
tory jądrowe należące do superrodziny receptorów steroi-
receptor Ah dowych (NR1, nuclear receptor family). Ze względu na to,
A h że początkowo receptory te były izolowane pod kątem
HSP 90 analogii do receptorów steroidowych i nie wiedziano nic
o ich potencjalnych ligandach, określano je jako „recepto-
HSP 90 ry sieroce” (orphan receptors). Z tej grupy receptorów dla
farmakokinetyki leków ważne są konstytutywny receptor
androstanu (CAR), receptor pregnanu X (PXR), receptor
A h farnezoidu X (FXR), receptory α i γ aktywowane przez pro-
liferatory peroksysomów (PPAR α i γ) oraz receptor witami-
rezeptor Ah ARNT ny D (VDR). Mechanizm indukcyjny dla tych wszystkich
typów receptorów jest identyczny: po związaniu substratu
ARNT
dochodzi do utworzenia heterodimeru z receptorem kwasu
9-cis-retinowego (RXR). Jak napisano wyżej dla receptora
Ah, utworzone heterodimery mogą się wiązać do określo-
nych struktur DNA i w ten sposób regulować transkrypcję
genów enzymów biorących udział w metabolizmie oraz
jądro białek transportowych. Najważniejsze białka, induktory
komórkowe
oraz geny docelowe zestawiono w tab. A 2.4-5.
XRE CYP1A1 Indukcja enzymatyczna powoduje następujące kon-
sekwencje dla terapii farmakologicznej:
Ryc. A 2.4-5. Molekularny mechanizm indukcji typu metylo- ■ W przypadku długotrwałego leczenia za pomocą
cholantrenowego; Ah – węglowodór aromatyczny, HSP – biał- induktorów enzymatycznych dochodzi do obniże-
ko szoku cieplnego, ARNT – jądrowy translokator kompleksu nia stężenia leku osiąganego w osoczu za pomocą
receptor Ah, XRE – czynnik wrażliwy na ksenobiotyk. dawki ustalonej na początku terapii.
Ten mechanizm opisano pierwotnie dla policyklicznych
■ Jeśli metabolity są mniej aktywne niż substancja
węglowodorów aromatycznych (polycyclic aromatic hy- wyjściowa, działanie słabnie, natomiast jeśli są bar-
drocarbons, PAH) (zob. ryc. A 2.4-5). dzie aktywne, działanie nasila się (zob. poniżej).
Scharakteryzowano również cytozolowy receptor dla wę- ■ Niektóre enzymy CYP (np. CYP1A1 oraz CYP1A2)
glowodorów arylohydroaromatycznych (aryl hydrocar- mogą prowadzić do bioaktywacji chemicznych
bons, Ah), który w stanie nieindukowalnym występuje karcynogenów, przez co indukcja może powodo-
jako dimer z białkiem szoku cieplnego (Hsp 90). Po zwią- wać zwiększenie ryzyka choroby nowotworowej.
zaniu PAH kompleks dysocjuje, a do receptora wiąże się
z kolei białko ARNT (Ah-receptor nuclear transloca-
Z tego względu induktory tych enzymów są szcze-
tor). Heterodimer składający się z receptora Ah i białka gólnie krytycznie badane w trakcie projektowania
ARNT może się wiązać do struktur XRE (xenobiotic re- leków.
sponsive element) DNA, które występują przykładowo
w promotorze flankującym 5’-koniec genu CYP1A1. ■ Stężenia endogennych substancji czynnych w oso-
To wiązanie prowadzi do zwiększenia transkrypcji i synte- czu mogą spaść poniżej wartości prawidłowych.
zy większej ilości białek.
■ Przy jednoczesnym stosowaniu innych leków ist-
Biorąc pod uwagę istnienie dużej liczby monooksygenaz nieje ryzyko występowania niebezpiecznych efek-
o różnej specyficzności substratowej, (zob. powyżej) zro- tów przemiany materii tych substancji: podczas po-
zumiałe jest, że poza fenobarbitalem i węglowodorami aro- dawania induktorów enzymatycznych mogą także
matycznymi istnieją także inne induktory enzymatyczne, spaść stężenia drugiego leku we krwi i jeśli daw-
które umożliwiają indukcję różnych izoenzymów.
kowanie zostanie zwiększone dla wyrównania stę-
W związku z tym należy wymienić dwa inne białka, które żenia leku, ewentualnie po odstawieniu induktora,
mają znaczenie dla konstytutywnej ekspresji białek meta- stężenie może wzrosnąć ponad wartość krytyczną.
bolizujących i transportujących leki. Chodzi tutaj o czynnik
transkrypcyjny NFE-2 (nuclear factor E2), który występuje
w cytozolu razem z kolejnym białkiem i ulega aktywacji
przy stresie oksydacyjnym przez uwalnianie z heterodime- 2.4.4. Efekt pierwszego przejścia
ru oraz reguluje szereg enzymów CYP i UGT. Drugim
białkiem jest czynnik transkrypcyjny HNF1α (hepatocyte
nuclear factor 1α), który występuje jako homodimer i tak- Krew żylna spływająca z przewodu pokarmowego
że wpływa na ekspresję enzymów CYP, UGT oraz trans- i zawarte w niej substancje dostają się do żyły wrotnej,
porterów. a z nią do wątroby. Zanim więc wchłonięty w żołądku
MUTSCHLER-2009.indd 34 2010-01-07 22:10:50
Farmakokinetyka 35
Tabela A 2.4-5. Mechanizmy indukcji enzymów metabolizujących z udziałem receptora (skróty – zob. tekst)
Receptor Miejsce ekspresji Regulowany gen Substancja indukująca
węglowodorów aromatycznych wątroba, nerki, język CYP1A1, CYP1A2, CYP1B1, β-naftoflawon,
(Ah-R) UGT1A1, UGT1A2 policykliczne
węglowodory aromatyczne
konstytutywny androstanu wątroba CYP2B6, CYP2C9, CYP2C19, fenobarbital, fenytoina
(CAR) CYP3A4, UGT1A1
farnezylowy X przewód pokarmowy, BSEP, UGT2B4 kwasy żółciowe
Farmakokinetyka
(FXR) wątroba, nerki
glukokortykosteroidów wszędzie CYP2C9, CYP2B6, CYP3A4, glukokortykosteroidy
CYP3A5, CAR, PXR
alfa aktywowany wątroba, serce, UGT1A9, UGT2B4 fibraty
przez proliferatory mięśnie, nerki
peroksysomów (PPARα)
pregnanu X przewód pokarmowy, CYP1A2, CYP2B6, CYP3A4, bosentan, karbamazepina,
(PXR) wątroba, limfocyty UGT1A1, UGT1A3, ABCB1, glukokortykosteroidy, fenobarbital, A2
ABCC2 rifampicyna, ritonawir i in.
witaminy D przewód pokarmowy CYP2B6, CYP2C9, CYP3A4, kalcytriol
SULT2A1
i jelicie cienkim lek dostanie się do prawego serca płciowych i morfiny. Indukcja enzymów przewodu
i stąd poprzez krążenie płucne do dużego krążenia, pokarmowego wywoływana np. przez rifampicynę
musi on najpierw przejść przez wątrobę. Dla działania może prowadzić do znacznego zwiększenia efektu
leku rozstrzygające znaczenie ma to, czy i jaka jego pierwszego przejścia. Tym samym określa go całość
część jest wychwytywana i metabolizowana w wątro- jelitowych i wątrobowych reakcji biotransformacji.
bie (zob. ryc. A 2.4-6). W związku z tymi zjawiskami
mówi się o tzw. efekcie pierwszego przejścia. Określa
on, jaka część leku podczas pierwszego przejścia 2.4.5. Hamowanie aktywności
ulega metabolizmowi lub zostaje zatrzymana w wą-
trobie. Związkami o dużym lub względnie dużym enzymatycznej
efekcie pierwszego przejścia są np. niektóre β-adre-
nolityki – propranolol i alprenolol, lek miejscowo Tak jak wiele leków całkowicie różniących się bu-
znieczulający i antyarytmiczny lidokaina, a przede dową chemiczną powoduje indukcję enzymatyczną,
wszystkim lek stosowany w chorobie niedokrwien- tak istnieją również liczne leki hamujące przebieg
nej serca – nitrogliceryna (triazotan glicerolu). Ten procesów transportowych biotransformacji i mogące
ostatni jest stosowany podjęzykowo nie tylko w celu w ten sposób wywołać wydłużenie i nasilenie dzia-
uzyskania szybkiego wystąpienia działania, ale także łania innych substancji. Do zahamowania aktywno-
ze względu na występujący przy podaniu doustnym ści enzymatycznej może dochodzić wówczas, gdy
efekt pierwszego przejścia. lek powoduje zmniejszenie syntezy lub zwiększenie
Oprócz przemian powodowanych przez enzy- rozkładu enzymów siateczki endoplazmatycznej.
my wątrobowe również metabolizm zachodzący Może też występować sytuacja, w której dwa leki
w świetle lub ścianie przewodu pokarmowego może lub więcej konkurują o miejsce wiązania z enzy-
być przyczyną efektu pierwszego przejścia. W jeli- mem i na tej drodze dochodzi do kompetycyjnego
tach występują – oprócz reakcji utleniania zachodzą- hamowania rozkładu jednego z nich. Szczególne
cych pod wpływem monooksygenaz zakaźnych od znaczenie kliniczne ma blokada enzymów działają-
CYP-450 (np. werapamilu lub cyklosporyny), – także cych w zakresie bliskim wysycenia. Jako przykład
reakcje koniugacji. Bardzo silną przedukładową eli- może posłużyć interakcja dikumarolu z inaktywacją
minację stwierdza się np. w przypadku hormonów fenytoiny.
MUTSCHLER-2009.indd 35 2010-01-07 22:10:51
36 Farmakokinetyka
światło przewodu pokarmowego
ściana wątroba
(żołądka) naczynia krwionośne
stała postać jelita
leku
stężenie
we krwi
rozpuszczanie
biotransformacja wychwyt,
biotransformacja
Ryc. A 2.4-6. Schematycznie przedstawiony efekt pierwszego przejścia.
2.4.6. Bioinaktywacja i bioaktywacja
lek
Jak opisano powyżej, metabolizm leków najczęściej
służy organizmowi do tego, żeby przeprowadzić po-
brane substancje w formę rozpuszczalną w wodzie,
która poza usprawnieniem usuwania przez nerki pro- naświetlanie biotransformacja
wadzi również najczęściej do obniżenia aktywności
farmakologicznej. Biotransformacja może więc pro-
wadzić do osłabienia działania lub całkowitej inakty- reaktywne metabolity
(elektrofile, reaktywne formy tlenu)
wacji (detoksykacji) albo też – choć rzadziej – do bio-
aktywacji i – jeśli czynny metabolit jest bardziej tok-
syczny niż związek macierzysty – do zatrucia. W tab.
A 2.4-6 zestawiono przykłady bioaktywacji i powsta- reakcja
wania toksycznych metabolitów. reakcja z kwasami peroksydacja z nukleofilnymi
nukleinowymi tłuszczów grupami
Procesom prowadzącym do wytworzenia się sub- białkowymi
stancji toksycznych (toksyfikacji) należy poświęcić
szczególną uwagę, ponieważ ich poznanie i zrozu-
mienie umożliwia przeprowadzenie odpowiednich
mutacje, hamowanie
zmian w cząsteczce leku, dzięki czemu nie dochodzi uszkodzenia aktywności
do powstawania jego toksycznych metabolitów. karcynogeneza,
teratogenność błon enzymatycznej
i transportu, alergie
Duża liczba efektów toksycznych jest wywołana przez tzw.
uszkodzenia chemiczne. Przez to pojęcie należy rozumieć
nieodwracalne zmiany w strukturze podstawowych skład- genotoksyczność cytotoksyczność
ników komórki, do których dochodzi w następstwie ich re-
akcji z substancją czynną. Zależnie od wpływu na DNA lub
inne makrocząsteczki rozróżnia się działania genotoksycz-
ne i cytotoksyczne (zob. ryc. A 2.4-7). Ryc. A 2.4-7. Następstwa zatrucia lekiem.
MUTSCHLER-2009.indd 36 2010-01-07 22:10:51
Farmakokinetyka 37
Tabela A 2.4-6. Bioaktywacja i biotoksykacja
H2N N NH2
NH NH NH bioaktywacja
CH(CH3) 2 N N
NH NH
Cl H3C CH3
Cl
cykloguanil
proguanil działa antymalarycznie
Farmakokinetyka
H3CO HO
bioaktywacja
O H O H
CH3 CH3
N N
HO HO
kodeina morfina
O O
CH3
CH2OH
CH3
CH2OH A2
O OH HO OH
bioaktywacja
CH3 H CH3 H
H H H H
O O
prednizolon
prednizon glukokortykosteroid
OC2H5 OC2H5
O P S biotoksykacja O P O
OC2H5 OC2H5
O2N O2N
paration paraokson
słaby inhibitor silny inhibitor
acetylocholinoesterazy acetylocholinoesterazy
H OH
N biotoksykacja N
CH3 CH3
O O
2-acetyloaminofluoren 2-(N-Hydroxy-acetyl)-aminofluoren
prekarcynogen karcynogen
OH
NH2
NH2
biotoksykacja
β-natyloamina α-hydroksy-β-natyloamina
karcynogen karcynogen
Do efektów genotoksycznych należą działania karcy- Toksyczne metabolity występują przede wszystkim
nogenne, mutagenne i teratogenne, a także przyspieszone wówczas, gdy w następstwie zbyt dużych dawek po-
starzenie jako następstwo narastania (kumulowania się)
błędów w procesach replikacji DNA. jemność (wydolność) reakcji biotransformacyjnych,
Do działań cytotoksycznych zalicza się uczulenia które zazwyczaj prowadzą do nietoksycznych pro-
na zmienione chemicznie białka, które działają jak anty- duktów przemiany (glukuronidacja, sulfatacja), staje
geny, a także śmierć komórek i martwicę w następstwie się niewystarczająca.
uszkodzenia błon lizosomów lub ważnych enzymów. Ważną przyczyną uszkodzeń chemicznych są za-
Najważniejsze organy uczestniczące w tworzeniu i wyda-
laniu toksycznych metabolitów – wątroba i nerki – są na- wierające elektrofilne rodniki produkty pośrednie po-
rażone na szczególnie duże ich stężenia i dlatego reakcje wstające w procesach utleniania i redukcji (zob. ryc.
toksyczne dotyczą najczęściej tych dwóch organów. A 2.4-7). Oprócz rodników występujących w związ-
MUTSCHLER-2009.indd 37 2010-01-07 22:10:52
38 Farmakokinetyka
Tabela A 2.4-7. Proleki
Prolek Postać czynna Cel stworzenia proleku
etylobursztynian erytromycyny erytromycyna poprawa smaku
półbursztynian metyloprednizolonu metyloprednizolon zwiększenie rozpuszczalności
(np. do wstrzyknięć i.v.)
aksetyl cefuroksymu cefuroksym
enalapryl enalaprylat
perindopryl perindoprylat zwiększenie wchłaniania
klofibrat kwas klofibrynowy
piwampicyna ampicylina
lewodopa dopamina przekroczenie bariery krew-mózg
dekaonian flufenazyny flufenazyna przedłużenie działania
omeprazol sulfenamid omeprazolu poprawa selektywności działania
cyklezonid izobutyryl cyklezonidu mniejsze układowe działania niepożądane
ku powstają też reaktywne formy tlenu (np. nadtlenek dążyć wówczas, gdy konieczne jest ułatwienie poda-
wodoru, rodniki hydroksylowe), które w przypadku wania lub poprawienie właściwości technologicz-
przeciążenia dróg inaktywujących (np. rozkładu nych, farmakokinetycznych, farmakodynamicznych
przez katalazę, dysmutazę nadtlenkową, witaminę lub toksykologicznych związku czynnego. Tak więc
C, witaminę E, glutation) nie mogą być dostatecznie synteza proleku jest celowa w przypadku substancji
szybko unieczynnione. czynnych obdarzonych nieprzyjemnym smakiem,
Drogą do uniknięcia tworzenia toksycznych meta- o niezadowalającej rozpuszczalności w wodzie przy
bolitów jest wprowadzanie do lecznictwa związków konieczności parenteralnego podawania leku, w przy-
leczniczych, które nie ulegają procesom oksydatywnej padku małej biodostępności przy stosowaniu doust-
biotransformacji lub ulegają im tylko w pomijalnych nym, silnego efektu pierwszego przejścia, krótkiego
ilościach. Synteza takich związków może przebiegać czasu działania, niedostatecznej dystrybucji do na-
w taki sposób, aby w miejscach cząsteczki prawidło- rządu docelowego, niezadowalającej selektywności
wo podlegającej biotransformacji wprowadzone zo- działania albo dużej toksyczności. W przypadku środ-
stały ugrupowania, które nie mogą zostać zmienione. ka, który mimo silnego powinowactwa do receptora,
Tego typu leki mają zazwyczaj długi okres półtrwania na skutek niedostatecznego wchłaniania, nie nadaje
w osoczu, co wiąże się z odpowiednimi korzyściami się do stosowania w lecznictwie, wchłanianie może
i wadami. zostać poprawione dzięki stworzeniu proleku.
Drugą możliwością zmniejszenia ryzyka związa-
nego z biotransformacją jest wprowadzenie do lecz- W tab. A 2.4-7 zestawiono przykłady proleków.
nictwa tzw. miękkich leków (soft-drugs), tj. związ- Na ogół przy tworzeniu proleków postępuje się w ten
ków czynnych, które są czynnymi metabolitami fazy sposób, że jedną z grup funkcyjnych występujących
I (np. oksazepam, metabolit wielu benzodiazepin) w substancji czynnej (np. grupę OH lub aminową)
albo ulegają nieoksydatywnej biotransformacji w po- wiąże się z odpowiednim związkiem (np. kwasem
żądanym miejscu, tzw. przewidzianym miejscu roz- karboksylowym), który następnie ulega odszczepie-
padu. Tak więc można np. syntetyzować leki, które niu w organizmie.
ulegają hydrolitycznemu rozkładowi do substancji
nieczynnych, niepodlegających dalej oksydatywnym
procesom metabolicznym.
2.4.7. Wpływ wieku
Proleki. Są to substancje, które same są nieczynne, na biotransformację
natomiast w wyniku procesów enzymatycznych lub
nieenzymatycznych w organizmie zostają przekształ- Wpływ wieku na biotransformację uwidacznia się
cone w postać czynną. Do uzyskania proleków można zwłaszcza u noworodków i osób w podeszłym wieku.
MUTSCHLER-2009.indd 38 2010-01-07 22:10:52
Farmakokinetyka 39
U noworodków i w większym stopniu u wcześnia- oczyszczania (tzw. high clearance drugs) są u osób
ków niektóre enzymy uczestniczące w biotransfor- w zaawansowanym wieku wydalane wolniej.
macji nie są w dostatecznym jeszcze stopniu aktyw- W podeszłym wieku wiązanie z białkami jest czę-
ne. Na przykład glukuronylotransferazy zaczynają się sto zmniejszone w następstwie obniżonego stężenia
tworzyć dopiero od chwili urodzenia, dlatego u no- albumin w osoczu krwi, w związku z czym część
worodków tylko w niewielkim stopniu mogą zacho- wolnego, dostępnego biotransformacji leku zwiększa
dzić procesy glukuronidacji. U dzieci w wieku 1–8 lat się, co oznacza przyspieszenie szybkości eliminacji.
szybkość biotransformacji jest natomiast większa niż
u dorosłych, co przypuszczalnie, przynajmniej częś- Dotychczas nie przeprowadzono systematycznych ba-
ciowo, może być związane z większą różnicą masy dań wpływu wieku na procesy transportowe. Dostępne
Farmakokinetyka
wątroby w stosunku do masy ciała. dane wskazują na zachowanie tej funkcji w starszym
wieku.
Dotychczas uważano, że reakcje zachodzące przy
współudziale CYP-450 przebiegają w podeszłym
wieku wolniej w związku z zależnym od wieku 2.4.8. Wpływ płci
zmniejszeniem aktywności enzymatycznej. Wyniki
na biotransformację
nowszych badań nie potwierdzają jednak tych opi-
nii; dowodzą natomiast – przynajmniej częściowo – i transport A2
że aktywność enzymatyczna jest utrzymywana nawet
w zaawansowanym wieku. Zdolność do indukcji en- Pojawia się coraz więcej dowodów na to, że kobiety
zymów I fazy i białek transportowych (np. CYP3A4 i mężczyźni różnią się pod względem ekspresji enzy-
i P-9P) również nie jest u chorych w zaawansowanym mów biorących udział w metabolizmie i białek trans-
wieku ograniczona. Należy wychodzić raczej z zało- portowych. Wykazano np., że CYP3A4, który meta-
żenia, że zmniejszony przepływ wątrobowy u osób bolizuje wiele leków, występuje w wątrobie kobiet
w podeszłym wieku (zależny od zmniejszającej się w prawie dwukrotnie większej ilości niż u mężczyzn.
pojemności minutowej) prowadzi do zmniejszenia Kliniczne konsekwencje nie są dotychczas znane,
współczynnika oczyszczania. Pogląd ten potwierdza- jednak liczne badania wskazują na istnienie zależnej
ją obserwacje, że związki o wysokim współczynniku od płci reakcji na terapię farmakologiczną.
MUTSCHLER-2009.indd 39 2010-01-07 22:10:53
40 Farmakokinetyka
2.5. Wydalanie
Wydalanie leku lub jego metabolitów prowadzi – po-
dobnie jak biotransformacja – do zmniejszenia stęże-
nia substancji czynnej w organizmie. Jest ono zależne jelito krew
od właściwości fizykochemicznych (masa cząstecz- enterocyt
kowa, wartość pKa, rozpuszczalność, ciśnienie par- ABC
UGT
cjalne) związku i zachodzi: MRP2
■ drogą jelitową (z kałem), ABC
P-gp
■ drogą wątrobową (z kałem), P-450
SLC
■ drogą nerkową (z moczem). ABC
MRP3
Wydalanie leków przez płuca albo przez skórę
SLC
i jej wytwory ma niewielkie znaczenie.
enterocyt
substancje metabolity
2.5.1. Wydalanie przez układ Ryc. 2.5-1. Pobieranie i eliminacja leków przez enterocyty.
pokarmowy Substancje są pobierane do enterocytów pasywnie lub przez
białka transportowe typu SLC. Są one następnie transportowane
do krwi w niezmienionej postaci albo są metabolizowane przez
Większość leków, zwłaszcza przeznaczonych do dłu- UDP-glukuronozylotransferazy (UGT). Utworzone metabolity
gotrwałego stosowania, podaje się doustnie. Dlatego mogą być transportowane do krwi albo ponownie wydzielane
w niektórych przypadkach powstaje możliwość, do światła jelita za pomocą transporterów ABC.
że substancja czynna pobrana przez enterocyty albo
jej metabolity, są ponownie transportowane do światła
jelita i wydalane ze stolcem. Ta ważna droga elimina- 2.5.2. Pobieranie leków przez wątrobę
cji przebiega z wykorzystaniem transporterów ABC
(m.in. P-gp, MRP2 albo BCRP) i może podlegać re-
i wydalanie wątrobowe
gulacji przez indukcję enzymatyczną. Na przykład
w ten sposób dzięki induktorowi PXR rifampicyna Po resorpcji jelitowej wszystkie leki transportowane
prowadzi do nasilonej jelitowej ekspresji CYP3A4, są żyłą wrotną do wątroby i przeważnie tam ulegają
P-gp oraz MRP2, dzięki czemu powstające w du- metabolizmowi. Wcześniej konieczny jest transfer
żych ilościach metabolity również mogą być szybciej ze strumienia krwi do hepatocytów. Zależy on przede
transportowane do światła jelita ( zob. ryc. A 2.5-1). wszystkim od właściwości biofizycznych leku. Ważną
W przypadku kilku innych leków, np. dla β-ad- rolę odgrywają również transportery położone bazo-
renolityków talinololu i karwedilolu, wykazano, lateralnie, które należą do rodziny SLC. Dzięki dużej
że po resorpcji są one ponownie wydzielane do świat- ilości ekspresjonowanych tam białek transportowych
ła jelita przez transportery ABC. Pod względem tok- do wątroby może zostać pobranych wiele różnych
sykologicznym ważne jest, że niektóre metale ciężkie substancji. Do tych transporterów należą m.in. trans-
są wydzielane dzięki przechodzeniu z krwi do światła portery anionów organicznych (OATP) i transporte-
jelita z kałem. ry kationów (OCT). Najlepiej zbadano pobieranie
Podsumowując, dzięki kombinacji reakcji bio- leków przez transporter anionów organicznych 1B1.
transformacji i procesów transportowych w przewo- Modyfikacje genetyczne albo jednocześnie podawa-
dzie pokarmowym organizm dysponuje komplekso- ne inhibitory tego białka, np. lek obniżający poziom
wym systemem eliminacji, który prowadzi do tego, tłuszczów w osoczu – gemfibrozyl, mogą wyraźnie
że w przypadku wielu ksenobiotyków tylko część po- zmniejszać pobieranie niektórych substancji przez
danej dawki trafia do żyły wrotnej. wątrobę, np. prawastatyny.
MUTSCHLER-2009.indd 40 2010-01-07 22:10:53
Farmakokinetyka 41
krew (strona
bazolateralna)
OATP1B1
OATP1B1
OATP1B1
SLC
SLC
SLC
ABC
BCRP
enzymII
Farmakokinetyka
en ABC fazy
faz zym
yI MRP2
ABC
P-gp
przewód żółciowy
hepatocyt wspólny (strona apikalna)
A2
substancje metabolity
Ryc. A 2.5-2. Pobieranie i eliminacja leków przez wątrobę.
Po pobraniu do wątroby przez białka transportowe 2.5.3. Wydalanie nerkowe
wiele leków jest metabolizowanych przez opisane
wcześniej systemy fazy I i II. Utworzone w ten spo-
Najważniejszym organem wydalniczym są nerki.
sób hydrofilne metabolity są z kolei transportowane
Szybkość i wielkość wydalania nerkowego są usta-
do żółci przez położone apikalnie białka transportowe
lane przez:
należące do rodziny ABC. Szczególnie ważne są tu-
taj ABCB1 (P-gp) i ABCC2 (MRP2). Ograniczenie ■ przesączenie kłębuszkowe,
działania tych białek, przez jednoczesne podanie in-
■ zwrotne wchłanianie kanalikowe
nych albo przez modyfikacje genetyczne, prowadzi
do tego, że metabolity powstałe w wątrobie nie mogą ■ sekrecję kanalikową.
być wydzielane do żółci, kumulują się w hepatocy-
tach, prowadząc w ten sposób do uszkodzenia wą- Przesączanie kłębuszkowe. Rozpuszczalność le-
troby. ków nie ma wpływu na przesączanie kłębuszkowe:
związki rozpuszczalne w tłuszczach są filtrowane
Defekt genetyczny w ABCC2 (MRP2) występuje w ze- równie dobrze jak rozpuszczalne w wodzie. Białka
spole Dubina-Johnsona. Prowadzi to do gromadzenia się nie przechodzą przez filtr kłębuszkowy, dlatego
sprzężonej bilirubiny. W celu jej usunięcia z hepatocytów,
dochodzi do nadekspresji białek transportowych położo- związane z nimi substancje nie podlegają przesą-
nych bazolateralnie (ABCC3, MRP3), które transportują czaniu kłębuszkowemu. W nerkach nie zachodzi
sprzężoną bilirubinę. Skutkiem tego jest hiperbilirubinemia dysocjacja kompleksu lek-białko, z tego względu
ze sprzężoną bilirubiną. dostępne filtracji stężenia wolnej substancji nie ule-
gają zmianie.
Z żółcią usuwane są przede wszystkim substancje, Szybkość filtracji zwiększa się przy zwiększe-
których masa cząsteczkowa wynosi ponad 500 Da lub niu ciśnienia w naczyniach włosowatych kłębuszka,
które osiągają tę masę w wyniku metabolizmu. (W zwiększeniu powierzchni filtracyjnej przez uaktyw-
przeciwieństwie do tego substancje o masie cząstecz- nienie pozostających w spoczynku kłębuszków ner-
kowej poniżej 300 Da są wydalane głównie z moczem). kowych i przy obniżeniu stężenia białek w osoczu,
a co za tym idzie – przy zmniejszeniu wiązania leków
Pobieranie i eliminację leków w wątrobie przedsta- z białkami. W przypadku hipoproteinemii, podob-
wia ryc. A 2.5-2. nie jak przy wypieraniu leku z połączeń białkowych
MUTSCHLER-2009.indd 41 2010-01-07 22:10:53
42 Farmakokinetyka
przez inny lek, znacznemu skróceniu może ulec czas
działania leków silnie wiązanych z białkiem.
mocz krew
Zwrotne wchłanianie kanalikowe. Zwrotne wchła-
nianie jest możliwe dzięki zwiększeniu stężenia leku α-Keto-
GL
w moczu w następstwie zwrotnego wchłaniania wody SLC SLC
w kanalikach nerkowych. Dla większości leków A– A– OAT1 A–
wchłanianie zwrotne jest biernym procesem prostej
dyfuzji, który zależy od rozpuszczalności leku, jego ABC
wartości pKa oraz wartości pH moczu. Leki rozpusz- A– MRP4
czalne w tłuszczach, które są dobrze wchłaniane
w przewodzie pokarmowym, z łatwością też prze- ABC SLC
chodzą przez nabłonek kanalików i są w dużej ilości A– A– A–
MRP2 OAT3
wchłaniane zwrotnie. Substancje hydrofilne, które
niemal nie są wchłaniane w jelitach, źle przechodzą
przez ścianę kanalika.
Słabe zasady (pKa 6–12) są w większym stopniu
SLC Kat+
wydalane przy obniżeniu wartości pH moczu, nato- Kat+ SLC Kat+ OCT2
miast słabe kwasy (pKa 3–7,5) są w większym stop- OCTN2 Na+ CAR
niu wydalane przy podwyższeniu wartości pH moczu
(przejście w rozpuszczalną w wodzie sól).
W zatruciach związkami zasadowymi, np. alkalo-
idami, można więc przez zakwaszenie moczu przy- SLC Kat+
spieszyć eliminację środka toksycznego, a w zatru- ABC Kat+ OCT2
Kat+ P-gp
ciach związkami kwaśnymi, np. barbituranami, moż-
na przyspieszyć tę eliminację przez alkalizację mo- kanalik proksymalny
czu.
W przypadku silnego kanalikowego wchłaniania
zwrotnego określonego środka można, dzięki zwięk-
szeniu wydalania moczu, nasilić jego eliminację.
A– lek anionowy Kat+ lek kationowy
Sekrecja kanalikowa. W przeciwieństwie do resorp-
cji zwrotnej, będącej procesem biernym, u podłoża Ryc. A 2.5-3. Procesy transportowe dla leków anionowych i ka-
sekrecji kanalikowej leży proces czynny. tionowych zachodzące w nerkach. Przeciwjony: Na+, karnityna
(CAR) i α-ketoglutaran (α-keto-GL).
Aby substancja mogła ulec sekrecji do światła kana-
lika, musi ona przejść ze strumienia krwi przez trans-
porter bazolateralny, a następnie, po stronie apikalnej,
ulec sekrecji z moczem również za pośrednictwem
białka transportowego. Opisane procesy są bardzo tem stężeń (wbrew gradientowi) wydalane do moczu.
dobrze scharakteryzowane (zob. ryc. A 2.5-3). Poszczególne substancje mogą wzajemnie konkuro-
wać o układ transportujący i kompetycyjnie hamować
Kationy organiczne pobierane są z krwi za pomocą trans- swoje wydalanie. Oprócz kwasów również organicz-
porterów SLC należących do rodziny OCT i wydzielane ne zasady mogą być czynnie wydalane przez układ
do moczu przez białka SLC, takie jak OCTN2 albo trans-
porter ABC P-gp. Aniony organiczne są transportowane nośnikowy występujący w komórkach kanalików.
z krwi do nabłonka kanalików za pomocą transporterów Obniżenie stężenia wolnej postaci leku w osoczu
anionów, takich jak OAT1 i OAT3, a stamtąd do moczu wskutek sekrecji kanalikowej zostaje (przynajmniej
za pomocą transporterów ABC, takich jak ABCC2 (MRP2) częściowo) wyrównane przez dysocjację kompleksu
i ABCC4 (MRP4), przy czym w ostatnim etapie biorą rów- lek-białko z wytworzeniem nowego stanu równowa-
nież udział białka SLC.
gi postać wolna/postać związana. Tak więc w przeci-
wieństwie do przesączania kłębuszkowego, któremu
Za pomocą zlokalizowanego w komórkach kanali- podlega tylko postać wolna, sekrecji kanalikowej
ka bliższego układu transportującego liczne kwasy pośrednio podlega również postać związana z biał-
organiczne, np. penicyliny, są niezgodnie z gradien- kami.
MUTSCHLER-2009.indd 42 2010-01-07 22:10:54
Farmakokinetyka 43
2.5.4. Wydalanie drogą oddechową ku jest mowa o czystych procesach dyfuzji biernej.
W porównaniu z wchłanianiem leków z dróg odde-
Wydalanie gazów – zwłaszcza po znieczuleniu ogól- chowych odwrócony jest kierunek gradientu stę-
nym – oraz związków lotnych następuje proporcjo- żeń. Wraz ze zmniejszającą się rozpuszczalnością
nalnie do gradientu stężenia lub ciśnienia pomiędzy we krwi zwiększa się wydalanie z powietrzem wy-
krwią a powietrzem oddechowym. W tym przypad- dychanym.
Farmakokinetyka
2.6. Parametry farmakokinetyczne;
podstawy obliczeń farmakokinetycznych
Opisane poniżej parametry farmakokinetyczne zosta- krzywej). Tę ostatnią wartość uzyskuje się, obliczając
ły uzyskane na podstawie oceny przebiegu stężeń le- stosunek iloczynu stężenia i czasu (C • E) do czasu t.
ków w czasie, a w odpowiednich przypadkach ich me- Stanowi ona podstawę do obliczenia tzw. MRT (mean A2
tabolitów we krwi, osoczu i surowicy, a także ilości residence time), tj. średniego czasu przebywania leku
leków i metabolitów odzyskanych z moczu. Płyny te w organizmie w postaci niezmienionej (tzw. średnie-
są łatwo dostępne, a stężenie we krwi odzwierciedla go czasu przetrwania, zob. poniżej).
przebieg procesów kinetycznych w ustroju. W celu
uzyskania krzywych przebiegu stężeń w czasie jako Dostępność biologiczna (biodostępność). Przez
odzwierciedlenia przebiegu różnorodnych proce- pojęcie dostępności biologicznej leku rozumie się
sów farmakokinetycznych niezbędne są wielokrotne rozmiar i szybkość, z jaką terapeutycznie skuteczna
oznaczenia stężeń leku we krwi, osoczu i surowicy. składowa (najczęściej niezmieniony lek, w przypad-
W zależności od znaczenia dla różnorodnych pro- ku proleku – czynny metabolit) została uwolniona
cesów farmakokinetycznych (wchłaniania, dystrybu- z postaci leku, wchłonięta i ostatecznie może osiąg-
cji, eliminacji) wyróżniono i opisano różne parametry nąć miejsce działania (zgodnie z tą definicją biodo-
farmakokinetyczne. stępność przy podaniu dożylnym wynosi 100%).
AUC (area under the curve). AUC oznacza pole W miejscu działania (np. na receptorach) stęże-
pod krzywą stężeń leku zależne od czasu (zazwyczaj nia związku czynnego w większości przypadków
krzywa stężeń leku w osoczu). Jest ono miarą obecnej nie mogą zostać zmierzone, dlatego pomocniczo
w organizmie ilości leku i może być względnie prosto określa się biodostępność za pomocą pomiarów stę-
wyliczone metodą trapezoidalną (zob. ryc. A 2.6-1), żeń leków w osoczu lub moczu.
w której z każdych dwóch punktów pomiaru i przy-
należnych im stężeń ustalona zostaje powierzchnia
odpowiednich trapezów:
(tn+1 – tn) · (cn + cn+1)
powierzchnia trapezu = (1) C2
2
C3
Suma wszystkich pól daje całkowitą trapezoidalną po-
C1
wierzchnię pod krzywą stężeń AUCtrap, która odpowiada C4
powierzchni pod krzywą stężeń do ostatniego mierzonego
punktu (stężenia), tj. AUCo-t(last). Dla całkowitego pola pod C5
stężenie w osoczu
krzywą stężeń AUCo-∞ można część powierzchni, niedają-
cą się ustalić na podstawie rzeczywistych punktów pomia-
rowych (AUCt(last)-∞), wyliczyć za pomocą ekstrapolacji. C6
Ten sposób postępowania uważany jest jednak za dopusz- C7
czalny tylko wówczas, gdy udział wyliczonej drogą ekstra-
polacji powierzchni nie przekracza 10, a maksymalnie 20% C0
powierzchni całkowitej.
t0 t1 t2 t3 t4 t5 t6 t7
czas
AUMC (area under the moment curve). AUMC
oznacza powierzchnię pod krzywą stężeń do tzw. Ryc. A 2.6-1. Oznaczanie powierzchni pod krzywą za pomocą
pierwszego momentu (tj. wyraźnej zmiany charakteru metody trapezoidalnej.
MUTSCHLER-2009.indd 43 2010-01-07 22:10:54
44 Farmakokinetyka
Ten sposób postępowania jest dopuszczalny przy wszyst- przy obu drogach podania. Miarę biodostępności
kich drogach podania, przy których lek z krwią dostaje uzyskuje się z poniższego równania:
się do właściwego miejsca działania (np. po podaniu do-
ustnym, doodbytniczym). Ustalenie biodostępności leku AUCx
zastosowanego miejscowo nie jest jednak przy tym spo- F= · 100 [%] (2)
AUCi.v.
sobie możliwe. Zastosowane miejscowo leki przenikają
najpierw do miejsca działania, a dopiero później dostają gdzie AUCx – pole pod krzywą stężeń przy dowolnej
się do krwiobiegu albo też – nie osiągnąwszy w ogóle drodze podania, AUCi.v. – pole pod krzywą przy po-
krwi – ulegają eliminacji. Dlatego stężenie w osoczu le-
daniu i.v.
ków stosowanych i działających miejscowo nie pozwa-
la na ocenę stężeń w miejscu działania. Biodostępność
jest problematycznym parametrem także wtedy, gdy Uzyskana w ten sposób wartość określana jest jako
struktura docelowa dla leku znajduje się w narządzie, któ- absolutna dostępność biologiczna.
ry po podaniu doustnym zostanie przez ten lek osiągnięty
przed krążeniem systemowym. Najlepiej zbadanym przed-
Jeżeli nie ma postaci leku przeznaczonej do podawa-
stawicielem są leki obniżające poziom lipidów we krwi
– statyny. Ich docelowy enzym, reduktaza HMG-CoA, nia dożylnego, można oznaczyć względną dostępność
znajduje się w hepatocytach, podczas gdy najważniej- biologiczną (RBA) danego preparatu w ten sposób,
szym działaniem niepożądanym jest zaburzenie działania że pole pod krzywą stężeń badanego preparatu odnosi
mięśni poprzecznie prążkowanych. W tym przypadku się do pola pod krzywą stężeń preparatu standardo-
wysoka biodostępność (mierzona na podstawie stężeń
wego:
w osoczu) prowadzi przede wszystkim do nasilonych
działań niepożądanych.
AUCx
F = AUC · 100 [%] (3)
standard
O ile dawniej przyjmowano, że – niezależnie od po-
staci galenowej – ta sama dawka substancji czynnej
Parametrami wyznaczającymi szybkość wchłaniania
wywołuje takie samo działanie, o tyle współcześnie
są:
wiadomo, że dostępność biologiczna i co za tym idzie
– również skuteczność danego leku stosowanego ■ maksymalne stężenie leku w osoczu (Cmax),
w postaci jednego preparatu handlowego może róż-
■ czas, jaki upływa między podaniem a uzyskaniem
nić się w porównaniu z innym i to w dużym zakresie.
maksymalnego stężenia (tmax).
Odnosi się to zwłaszcza do związków trudno roz-
puszczalnych. Przykładem leków, w przypadku któ-
Przy podaniu identycznych dawek tmax jest tym
rych występują bardzo duże różnice biodostępności
mniejszy, a Cmax tym większe, im większa jest szyb-
pomiędzy poszczególnymi preparatami handlowymi,
kość wchłaniania.
są: kwas acetylosalicylowy, allopurinol, digoksyna,
glibenklamid i tetracyklina. Ważnymi czynnikami
Biorównoważność. Dwa leki zawierające identycz-
wpływającymi na biodostępność są:
ną substancję czynną uważa się za biorównoważne,
■ szybkość i procent substancji czynnej uwalnianej tj. wywołujące takie samo działanie i mogące być sto-
z postaci leku, sowane zamiennie bez zagrożenia dla chorego, jeśli
nie różnią się one pod względem biodostępności lub
■ szybkość wchłaniania uwolnionej części leku,
różnice są niewielkie (< 20%). Oznacza to, że zależ-
■ wielkość efektu pierwszego przejścia. ne od czasu krzywe stężeń w osoczu w dużej mierze
pokrywają się, a występujące międzyosobnicze róż-
Pediatra Dost wykazał, że powierzchnia pod krzy- nice w stężeniach substancji czynnej są podobne.
wą stężeń leku we krwi w funkcji czasu odpowiada
ilości leku w organizmie i jest niezależna od szyb- Objętość dystrybucji. Przez pojecie objętości dys-
kości wchłaniania. Oznacza to, że przy tej samej trybucji (V) rozumie się wielkość (pojemność) prze-
dawce – zakładając całkowite wchłanianie do krwi strzeni, w której rozmieszcza się dany związek. Przy
– pola pod krzywą stężeń przy podaniu dożylnym założeniu, że cały organizm zachowuje się jak jed-
(i.v.) i np. doustnym są takie same. Wynika z tego, nolity obszar dystrybucji (model jednokompartmen-
że można ustalić stopień biodostępności leku przy towy, zob. poniżej), objętość dystrybucji po szybkim
dowolnej drodze podania. Lek powinien więc być dożylnym wstrzyknięciu (podanie w bolusie) odpo-
najpierw podany dożylnie w celu zapewnienia peł- wiada ilorazowi uzyskanemu z podzielenia podanej
nej biodostępności. W drugiej fazie taka sama daw- dawki przez (fikcyjne) stężenie początkowe C0:
ka powinna być podana dowolną drogą, np. doust-
D
nie. Następnie oblicza się pola pod krzywymi stężeń V= · 100 [1] (4)
C0
MUTSCHLER-2009.indd 44 2010-01-07 22:10:55
Farmakokinetyka 45
(Stężenie początkowe C0 oznacza stężenie w osoczu, CLorg = Q · E (7)
które wystąpiłoby, gdyby po wstrzyknięciu i.v. lek
został natychmiastowo równomiernie rozmieszczony przy czym
w całym organizmie).
Objętość dystrybucji może być identyczna z ob- E = (Ca – Cv)/Ca,
jętością osocza, objętością płynu pozakomórkowe- gdzie Ca – stężenie we krwi tętniczej,
go albo objętością całkowitej wody ustrojowej. Cv – stężenie we krwi żylnej.
Najczęściej jest to czysto fikcyjna wartość, która
czasem znacznie przekracza całkowitą objętość or- Wątrobowy współczynnik oczyszczania uzyskuje
ganizmu. Stanowi to wówczas wskazówkę, że okre- się zgodnie z (7)
Farmakokinetyka
ślony związek gromadzi się w niektórych narządach,
np. w tkance tłuszczowej. Jeśli przemnoży się stęże- CLH – QH · EH (8)
nie we krwi przez objętość dystrybucji, otrzymuje się
– jako wartość rzeczywistą – całkowitą ilość substan- gdzie QH – przepływ wątrobowy,
cji w organizmie. EH – wątrobowy współczynnik ekstrakcji.
Znaczenie kliniczne objętości dystrybucji polega
na tym, że wpływa ona w istotny sposób na wartość Odnośnie do wątrobowego współczynnika oczysz-
stężeń w osoczu. czania wśród eliminowanych substancji daje się wy- A2
różnić dwie podstawowe grupy leków:
Jeśli mimo całkowitej resorpcji i małego efektu
■ ograniczone przepływem (perfuzją),
pierwszego przejścia po podaniu dawki leku osią-
gane są tylko bardzo niewielkie stężenia osoczowe, ■ ograniczone pojemnością.
wskazuje to na fakt, że duża część dawki została
przetransportowana do głębokich kompartmentów, W przypadku pierwszej grupy, tzw. leków o dużym
np. do tkanki tłuszczowej. Ma to ogromne znaczenie współczynniku oczyszczania (high clearance drugs),
kliniczne, jako że takie kompartmenty mogą pełnić eliminacja zależy głównie od przepływu krwi przez
funkcję magazynującą – lek może być powoli wy- wątrobę. Współczynnik ekstrakcji przekracza tu 0,8,
dzielany z tkanki tłuszczowej do osocza przez dłu- tj. niemal cała ilość leku zostaje usunięta (wyeks-
gi czas – czego rezultatem jest znacznie wydłużone trahowana) z krwi podczas jej przepływu przez wą-
działanie. Ponadto w tym przypadku należy się liczyć trobę.
z długim okresem połowicznego zaniku. W przypadku drugiej grupy, tzw. leków o małym
współczynniku oczyszczania (low clearance drugs),
Współczynnik oczyszczania. Współczynnik oczysz- posiadających współczynnik ekstrakcji < 0,2, stop-
czania określa hipotetyczną objętość krwi (objętość niem ustalającym szybkość eliminacji jest przede
osocza), która w jednostce czasu zostaje uwolniona wszystkim pojemność enzymatyczna (wydolność en-
(oczyszczona) od leku. zymatyczna) wątroby.
Całkowity współczynnik oczyszczania (CL) ustala
się w ten sposób, że dzieli się dawkę (D) przez pole Przykładami leków należących do grupy pierwszej
pod krzywą stężeń (AUC): są: propranolol i lidokaina, przykładami leków, któ-
rych szybkość eliminacji jest ograniczona pojemnoś-
D
CL = (5) cią enzymatyczną są: diazepam i fenprokumon.
AUC
Jeśli związek jest eliminowany tylko przez jeden na-
Nerkowy współczynnik oczyszczania jest to, zgod-
rząd, to całkowity współczynnik oczyszczania rów-
nie z ogólną definicją współczynnika oczyszczania,
na się współczynnikowi oczyszczania tego narządu.
objętość krwi, która jest przez nerki całkowicie po-
W większości przypadków całkowity współczynnik
zbawiana danego związku w jednostce czasu. Można
oczyszczania składa się jednak z kilku współczynni-
go wyliczyć z następującego równania:
ków częściowych, z których ważniejszymi są: wątro-
bowy (CLH) i nerkowy (CLR).
Ae (∞) Ae (∞) · CL
CLR = = (9)
CL = CLR + CLH + CLX (6) AUC dawka
Narządowy współczynnik oczyszczania (CLorg) gdzie Ae (∞) (amount exereted) – ilość leku wyda-
uzyskuje się, mnożąc przepływ krwi przez ten narząd lanego z moczem w postaci niezmienionej w czasie
(Q) przez współczynnik ekstrakcji (E): do nieskończoności.
MUTSCHLER-2009.indd 45 2010-01-07 22:10:55
46 Farmakokinetyka
Współczynnik oczyszczania, zwłaszcza całkowity,
ma duże znaczenie w praktyce klinicznej, ponieważ 100
1h 10h 100h
jest on obok dawkowania podstawowym czynnikiem kwas acetylosalicylowy
determinującym wielkość średnich (average) stężeń
w osoczu krwi w stanie stacjonarnym (CSS,av) przy
współczynnik oczyszczania (l/h)
długotrwałym leczeniu. Zmniejszenie CL prowadzi
bezpośrednio do podwyższenia CSS,av, a co za tym 10 amiodaron
idzie – w przypadku leków o małej rozpiętości tera-
peutycznej do znacznego zwiększenia niebezpieczeń-
stwa przedawkowania. 1000h
1,0
Okres półtrwania eliminacji (okres półtrwania
w osoczu t1/2). Okres półtrwania eliminacji jest to czas,
w którym stężenie leku w osoczu ulega obniżeniu warfaryna digitoksyna
10000h
do połowy pierwotnej wartości. Można to uzyskać
z poniższego równania: 0,1
10 100 1000 10000
objętość dystrybucji (l)
ln2 0,693
t1/2 = = (10)
kel kel Ryc. A 2.6-2. Objętość dystrybucji i współczynnik oczyszczania
określają okres półtrwania (linie przerywane; zmodyf. na podst.
gdzie kel oznacza stałą szybkości eliminacji (zob. po-
Rowlanda i Tozera).
niżej).
Najczęściej jednak jest on wyznaczany graficznie
z krzywej stężeń leku w osoczu (zob. poniżej).
Mimo dużego znaczenia okresu półtrwania elimi-
Stężenia w osoczu po podaniu leku z powodu jed-
nacji dla leczenia nie można jednak pominąć faktu,
nocześnie zachodzącej dystrybucji i eliminacji zazwy-
że jest to tylko parametr pochodny zależny od obję-
czaj początkowo obniżają się znacznie szybciej niż
tości dystrybucji oraz współczynnika oczyszczania.
w okresie późniejszym, a eliminacja z różnych obsza-
Niska objętość dystrybucji i wysoki współczynnik
rów dystrybucji może przebiegać z różną szybkością,
oczyszczania prowadzą do krótkiego okresu półtrwa-
dlatego często wyznacza się wiele okresów półtrwania
nia substancji, natomiast lek o takiej samej objętości
eliminacji. Najczęściej jednak podawany jest najdłuż-
dystrybucji, ale o mniejszym współczynniku oczysz-
szy z nich tzw. okres półtrwania końcowej eliminacji.
czania posiada dłuższy okres półtrwania. Zależność
Od okresu półtrwania eliminacji należy ściśle odróżniać między współczynnikiem oczyszczania, objętością
okres półtrwania działania (biologiczny okres półtrwania), dystrybucji i okresem półtrwania przedstawia ryc.
tj. czas, w którym działanie leku zmniejsza się o połowę. A 2.6-2.
Okres półtrwania (końcowej) eliminacji jest ważnym Stała szybkości eliminacji. Jeśli za pomocą przed-
parametrem farmakokinetycznym. Według okresów stawienia graficznego określono wartość t1/2 w oso-
półtrwania leki można podzielić na krótko, średnio czu, można też obliczyć (całkowitą) stałą szybkości
długo i długo działające. Okres półtrwania elimina- eliminacji kel:
cji dostarcza ponadto podstaw do obliczenia sposo-
ln2
bu dawkowania przy wielokrotnym podawaniu leku, kel = (11)
t1/2
a więc w przypadku każdego długotrwałego leczenia
(zob. poniżej). Stałą szybkości eliminacji drogą nerkową kr uzyskuje
się przez oznaczenie wydalania związku z moczem;
Znaczenie kliniczne okresu półtrwania wynika metaboliczna stała szybkości eliminacji obliczana
z następujących faktów: przede wszystkim pozwala jest z równania:
na oszacowanie, kiedy substancja całkowicie opuści
organizm. Poza tym na podstawie okresu półtrwania kel = kel – kr (12)
można przewidzieć, kiedy po podaniu leku zostanie
osiągnięty stan równowagi (steady state). W tym sta- MRT (mean residence time, średni czas przetrwania).
nie dostarczana dawka odpowiada ilości wydalanej, MRT odpowiada średniemu czasowi przebywania
co prowadzi do stabilności stężenia leku w osoczu. niezmienionej cząsteczki leku w organizmie. Zawiera
MUTSCHLER-2009.indd 46 2010-01-07 22:10:55
Farmakokinetyka 47
w sobie wszystkie procesy kinetyczne, począwszy 2.6.1. Modele farmakokinetyczne
od zachodzącego in vivo uwolnienia, poprzez wchła-
nianie, dystrybucję, aż po eliminację. Wartość tę moż- Przez pojęcie modelu farmakokinetycznego rozumie
na obliczyć za pomocą następującego równania: się matematyczny opis wiernie oddający przebieg stę-
żenia w czasie w badanym systemie. W ścisłym zna-
AUMC czeniu każde równanie matematyczne, które opisuje
MRT = (13)
AUC stężenia leku w organizmie i zawiera w sobie zawsze
uproszczenie zachodzących złożonych procesów,
Minimalne stężenie terapeutyczne i minimalne
przedstawia model kinetyczny. Zazwyczaj jednak
stężenie toksyczne. Działanie leku występuje do-
przez pojecie modeli farmakokinetycznych rozumie
Farmakokinetyka
piero wówczas, gdy uzyska on określone stężenie
się tylko takie opisy matematyczne, przy których or-
we krwi i przekroczone zostanie stężenie progowe
ganizm jest podzielony na poszczególne przestrzenie
niezbędne do wywołania działania w miejscu docelo-
dystrybucji (kompartmenty). W obrębie pojedyncze-
wym (zob. ryc. A 2.6-3).
go obszaru dystrybucji (kompartmentu) stężenia da-
nego związku czynnego są identyczne. Przebiegające
To progowe stężenie w osoczu określa się jako mini-
procesy transportowe mogą być przejrzyście przed-
malne stężenie terapeutyczne lub minimalne stężenie
stawione za pomocą uproszczonych, blokowych dia-
efektywne (MEC). Z terapeutycznego punktu widze-
gramów (zob. poniżej). A2
nia górna granica stężeń w osoczu jest wyznaczana
przez maksymalne stężenie terapeutyczne odpowia-
O tak zwanym modelu jednokompartmentowym
dające zarazem minimalnemu stężeniu toksycznemu,
mówi się wówczas, gdy lek natychmiast po poda-
tj. stężeniu, przy którym występują pierwsze objawy
niu (dożylnym) zostaje równomiernie rozmieszczony
toksyczne. Zakres pomiędzy minimalnym stężeniem
w dostępnej przestrzeni dystrybucji (zob. ryc. A 2.6-4
terapeutycznym a minimalnym stężeniem toksycz-
A). Model jednokompartmentowy jest określony jako
nym określony jest jako zakres stężeń terapeutycz-
otwarty, jeśli możliwe jest zachodzenie procesów eli-
nych.
minacji.
Czas plateau. Jest to czas, w którym stężenia w oso-
Przy dwu- lub wielokompartmentowym modelu
czu przekraczają założone wartości, np. minimalne
(zob. ryc. A 2.6-4 B) dystrybucja leku do dostępnych
terapeutyczne stężenie związku czynnego.
przestrzeni zachodzi ze zróżnicowaną szybkością.
Rozróżnia się przy tym kompartment centralny, który
kinetycznie stanowi element transportujący, tj. krew,
i kompartmenty obwodowe. Jeśli wymiana substancji
pomiędzy kompartmentem obwodowym i centralnym
zachodzi bardzo wolno, mówi się o kompartmencie
głębokim.
minimalne stężenie toksyczne Kompartmenty farmakokinetyczne w większości
przypadków nie odpowiadają żadnym zdefiniowa-
nym anatomicznie obszarom dystrybucji w obrębie
zakres stężeń organizmu. Tym samym chodzi tu o uczestniczące
terapeutycznych wielkości. Z tego względu rozwijane są fizjologicznie
stężenie w osoczu
rzeczywiste modele, tzw. modele fizjologiczno-far-
minimalne makokinetyczne, w przypadku których brane są pod
stężenie uwagę parametry anatomiczne, fizjologiczne i fizy-
terapeutyczne kochemiczne. Tego typu model fizjologiczno-far-
makokinetyczny składa się z wielu występujących
po sobie lub obok siebie, wzajemnie powiązanych
kompartmentów (narządów, obszarów ciała) przed-
stawiających jednolite obszary dystrybucji, mogących
czas (t)
ewentualnie dodatkowo spełniać czynności elimina-
Ryc. A 2.6-3. Ustalanie zakresu stężeń terapeutycznych przez cyjne. Przy założeniu, że lek rozmieszcza się szybko
oznaczanie minimalnego stężenia terapeutycznego oraz mini- i równomiernie w każdym z kompartmentów, zależ-
malnego stężenia toksycznego. ny od czasu przebieg stężenia jest w poszczególnych
MUTSCHLER-2009.indd 47 2010-01-07 22:10:55
48 Farmakokinetyka
i.v.
C1
k10
i.v. k21
C1 k12
k10
C2
A B
A model jednokompartmentowy k10 stała szybkości eliminacji
B model dwukompartmentowy (czytać: k jeden-zero)
C1 kompartment centralny k12 stała transferu dla transportu z C1 do C2
C2 kompartment obwodowy k21 stała transferu dla transportu z C2 do C1
Ryc. A 2.6-4. Schemat blokowy przedstawiający modele farmakokinetyczne po wstrzyknięciu dożylnym.
kompartmentach wyznaczany przez narządowy prze- dC
vel = = kel · C (14)
pływ krwi oraz istniejący procent ekstrakcji leku. dt
Zaletą modeli fizjologiczno-farmakokinetycznych
gdzie: vel – szybkość eliminacji,
jest możliwość lepszego przenoszenia wyników ba-
kel – stała eliminacji,
dań na zwierzętach na człowieka w ramach badań no-
C – stężenie w osoczu w czasie t.
wych leków. Ponadto modele takie pozwalają na lep-
sze przewidywanie wpływu na kinetykę upośledzenia
Całkowanie daje funkcję eksponencjalną:
czynności narządów.
C = C0 · e–kel · t (15)
2.6.2. Kinetyka po wstrzyknięciu Na ryc. A 2.6-5 pokazano przykład takiej funkcji eks-
dożylnym (model ponencjalnej. Po zlogarytmowaniu otrzymuje się pro-
jednokompartmentowy) stą spełniającą równanie:
Po wstrzyknięciu dożylnym substancja dostaje się ln C = ln C0 – kel · t (16)
bezpośrednio do krwiobiegu, dlatego farmakokine-
tyka po jednorazowym podaniu i.v. jest najprostsza Nachylenie prostej jest miarą szybkości eliminacji:
do analizy. im większe nachylenie, tym szybciej zachodzi eli-
Jak długo stężenie substancji podlegającej eli- minacja. Z półlogarytmicznego wykresu pokazanego
minacji jest niewielkie w porównaniu ze stężeniem na ryc. A 2.6-5 można odczytać ważniejsze parametry
niezbędnym do wysycenia układu eliminującego, kinetyczne danego związku (zob. poniżej).
tak długo ilość eliminowanej w jednostce czasu sub-
stancji jest proporcjonalna do jej stężenia w osoczu,
a wydalany w jednostce czasu procent (nie ilość!) 2.6.3. Kinetyka po podaniu
jest stały (kinetyka I rzędu). Przy stosowaniu leków
dożylnym (model
zazwyczaj uzyskiwane są względnie małe stężenia
w osoczu, dlatego eliminacja zachodzi najczęściej dwukompartmentowy)
zgodnie z kinetyką I rzędu.
W przypadku zastosowania modelu jednokompart- Dystrybucja leku tylko w jednym kompartmencie
mentowego dla zmniejszającej się szybkości obniżania jest zjawiskiem stosunkowo rzadkim. Najczęściej lek
stężenia w osoczu otrzymuje się następujące równanie: rozmieszcza się w dwóch lub więcej kompartmen-
MUTSCHLER-2009.indd 48 2010-01-07 22:10:55
Farmakokinetyka 49
C log C
log C0
10 1
stężenie w osoczu
stężenie w osoczu
0,69
Farmakokinetyka
5
t1/2
czas (t) czas (t)
A B A2
A obraz liniowy, B obraz półlogarytmiczny, C0 fikcyjne stężenie początkowe
Ryc. A 2.6-5. Spadek stężenia leku w osoczu wskutek eliminacji po wstrzyknięciu dożylnym dla modelu jednokompartmentowego.
tach. Przy wstrzyknięciach dożylnych rozpoznaje się szybko się zmniejszają i dopiero po upływie pewnego
kinetykę, którą można opisać według modelu dwu- czasu układają się na mniej ostro przebiegającej krzy-
kompartmentowego, ponieważ przy wykresie półlo- wej. Na ryc. A 2.6-6 przedstawiono przebieg stężenia
garytmicznym wartości stężeń we krwi początkowo w osoczu po wstrzyknięciu i.v. przy istnieniu dwóch
C log C
10 1
log C1
stężenie w osoczu
stężenie w osoczu
0,69
log CZ
5
faza λ1
1
faza λZ
czas (t) czas (t)
A B
A obraz liniowy, B obraz półlogarytmiczny
Przerywana zielona prosta charakteryzuje fazę λ1, ciągła zielona prosta – fazę λZ
Ryc. A 2.6-6. Spadek stężenia leku w osoczu po wstrzyknięciu dożylnym dla modelu dwukompartmentowego.
MUTSCHLER-2009.indd 49 2010-01-07 22:10:56
50 Farmakokinetyka
obszarów dystrybucji (dwóch kompartmentów). Jako
równanie otrzymuje się w tym przypadku:
log C
C = C1 · e–λ1t + Cz · e–λz · t (17) 10
C1 i Cz są odcinkami osi rzędnych (zob. ryc. A 2.6-6),
stężenie w osoczu
C1 + Cz daje C0. λ1 i λz są tzw. stałymi hybrydyzacji.
Przez te pojęcia rozumie się stałe szybkości obejmu- 2
λZ
jące zarówno procesy dystrybucji, jak i eliminacji,
stąd określenie – hybrydy. λ1 charakteryzuje głównie 1
szybkość dystrybucji, λz – głównie szybkość elimina-
cji (zamiast obecnie powszechnie używanych symbo-
li λ1, λz itp. dawniej do oznaczania stałych hybrydy-
t1/2
zacji używano symboli α, β itp.).
Na ryc. A 2.6-7 przedstawiono graficznie wy-
znaczanie końcowego okresu półtrwania po nanie- 0,1
czas (t)
sieniu zmierzonych stężeń leku na skalę półloga-
rytmiczną. Ryc. A 2.6-7. Graficzne wyznaczanie t1/2 z fazy λZ.
2.6.4. Kinetyka przy jednorazowym powinny zostać opisane, musi zostać wprowadzony
podaniu doustnym kompartment startowy, który zawiera depot (rezer-
wuar) związku (zob. ryc. A 2.6-8).
Po podaniu doustnym, podobnie jak przy innych dro-
gach podania, w których zachodzi wchłanianie, prze- Model jednokompartmentowy z dodatkowym kom-
biegają bezpośrednio po sobie procesy: wchłaniania, partmentem startowym (zob. ryc. A 2.6-8 A) jest dla
dystrybucji i eliminacji. Przy konstruowaniu modelu celów praktycznych wystarczający wówczas, gdy
farmakokinetycznego, za pomocą którego procesy te dystrybucja w organizmie w porównaniu z wchłania-
podanie doustne kE1 k10
CE C1
podanie doustne kE1 k10
CE C1
k21
k12
C2
B
A model dwukompartmentowy CE kompartment startowy
B model trójkompartmentowy kE1 stała prędkości wchłaniania
Ryc. A 2.6-8. Schemat blokowy przedstawiający modele farmakokinetyczne po doustnym podaniu leku.
MUTSCHLER-2009.indd 50 2010-01-07 22:10:56
Farmakokinetyka 51
C log C
10 1
stężenie w osoczu
stężenie w osoczu
0,69
Farmakokinetyka
1
czas (t) czas (t)
A B A2
krzywa eliminacji równanie Batemana krzywa wchłaniania
A obraz liniowy, B obraz półlogarytmiczny
Ryc. A 2.6-9. Przebieg stężenia w osoczu po doustnym podaniu leku w przypadku występowania kompartmentu startowego i kom-
partmentu centralnego (równanie Batemana).
niem zachodzi szybko i w związku z tym stan równo- Wynikająca z tego krzywa wyznaczana jest przez
wagi między kompartmentami centralnym i obwodo- równanie:
wym ustala się bardzo szybko.
C0 · ki
C= · (e–kel · t – e–ki · t), (20)
ki – kel
Poniżej przedstawiono podstawowe obliczenia dla
tego modelu. Przy założeniu, że nie zachodzi jedno- zwane równaniem Batemana. W przypadku zasto-
cześnie eliminacja, dla szybkości zwiększania się stę- sowania wykresów półlogarytmicznych (zob. ryc. A
żenia leku we krwi stosuje się następujące równanie: 2.6-9) opadająca część krzywej przechodzi w prostą, któ-
ra przebiega równolegle do powoli zachodzących proce-
dC
vi = = ki (C0 – C) (18) sów częściowych, co zazwyczaj odpowiada eliminacji.
dt
Jak przedstawiono na ryc. A 2.6-10, na podstawie
gdzie: vi – szybkość wchłaniania, nachylenia prostych opadających części wykresów
ki – stała wchłaniania. można obliczyć stałą szybkości eliminacji, a przez
Całkowanie równania (18) przy założeniu, że w cza- ekstrapolację – teoretyczne stężenie leku w osoczu
sie t = 0 stężenie we krwi również wynosi 0, daje: w czasie t = 0.
Jeśli od stężeń zmierzonych w osoczu odejmie się
C = C0 (1 – e–ki · t) (19) wartości uzyskane przez ekstrapolację, uzyskuje się
krzywą wchłaniania. Ma ona, tak jak krzywa elimi-
W rzeczywistości jednak kinetyka całkowita ustala- nacji, najczęściej charakter ekspotencjalny, co ozna-
na na podstawie krzywych stężenia w osoczu wynika cza, że na wykresie półlogarytmicznym stanowi pro-
ze wszystkich procesów kinetycznych. stą (przerywana prosta na ryc. A 2.6-10). Z krzywej
wchłaniania można określić t1/2 wchłaniania.
Na ryc. A 2.6-9 przedstawiono wykresy w skali za- Jeśli model ma charakter dwukompartmentowy
równo liniowej, jak i półlogarytmicznej wyłącznie z dodatkowym kompartmentem startowym (ryc.
dla wchłaniania lub wyłącznie dla eliminacji, a także A 2.6-8 B), uzyskuje się przebieg krzywych przedsta-
jednocześnie dla wchłaniania i eliminacji. wiony na ryc. A 2.6-11.
MUTSCHLER-2009.indd 51 2010-01-07 22:10:57
52 Farmakokinetyka
W celu uzyskania sukcesu terapeutycznego koniecz-
ne jest zazwyczaj wielokrotne zastosowanie leku.
log C
Przy takim wielokrotnym podawaniu leku stężenie,
10 jakie dany związek osiąga w organizmie, zależy
od dawki, od odstępów między podaniami i czasu
półtrwania eliminacji. Jeśli czas półtrwania elimi-
stężenie w osoczu
nacji jest krótki w porównaniu z odstępami w poda-
5
waniu, związek jest niemal całkowicie eliminowany
w okresie między podaniami. Stężenie w osoczu
uzyskiwane po kolejnej dawce jest więc właściwie
równe stężeniu, jakie zostało uzyskane po dawce
poprzedzającej (zob. ryc. A 2.6-12). Jeśli okres pół-
trwania eliminacji jest mniej więcej taki sam lub na-
wet dłuższy niż odstępy między podaniami, to przy
1 końcu okresu między podaniami w organizmie znaj-
czas (t) dują się znaczne stężenia związku. Kolejna dawka
prowadzi do wyraźnie większych stężeń w osoczu
Ryc. A 2.6-10. Wykres przebiegu stężenia leku w osoczu po po- niż dawka poprzednia. Po następnej dawce stężenie
daniu doustnym w skali półlogarytmicznej służący do wyzna- w osoczu nadal wzrasta, jednocześnie rośnie stęże-
czenia kel. nie eliminowanego związku w jednostce czasu, tak
że w końcu ilość substancji eliminowanej pomiędzy
odstępami dorównuje ilości wchłanianej z poprzed-
2.6.5. Kinetyka przy wielokrotnym
niej dawki (zob. ryc. A 2.6-13).
podaniu
Stężenia w osoczu oscylują wówczas niemal między
Omówione w poprzednich podrozdziałach jednora- stałymi maksymalnymi (Css,max) i minimalnymi war-
zowe podanie leku jest raczej wyjątkiem niż regułą. tościami (Css,min); stan ten określany jest jako rzeko-
C log C
10 1
stężenie w osoczu
stężenie w osoczu
0,69
czas (t) czas (t)
A B
A obraz liniowy, B obraz półlogarytmiczny
Przerywana zielona prosta charakteryzuje fazę λ1, ciągła zielona prosta – fazę λZ
Ryc. A 2.6-11. Przebieg stężenia w osoczu po doustnym podaniu leku w przypadku występowania kompartmentów startowego,
centralnego i obwodowego.
MUTSCHLER-2009.indd 52 2010-01-07 22:10:57
Farmakokinetyka 53
stężenie w osoczu
stężenie w osoczu
Farmakokinetyka
10 20 30 40 50
godziny czas (t)
Ryc. A 2.6-12. Przebieg stężenia leku w osoczu po wielokrot- Ryc. A 2.6-13. Podwyższanie się stężenia leku w osoczu i osią-
nym podaniu doustnym leku o krótkim okresie półtrwania eli- ganie stanu równowagi stężeń przy wielokrotnym podaniu leku
minacji (t1/2 = 3 godziny) i długiej przerwie między podaniami (kumulacja).
A2
(τ = 24 godziny).
my stan równowagi (pseudo steady state). Średnie interakcje farmakokinetyczne, ustala – poprzez zmia-
stężenie Css,av wynosi: ny współczynnika oczyszczania – stężenie w stanie
stacjonarnym. Stan ten zostaje osiągnięty po mniej
AUC F·D więcej 5 okresach półtrwania.
Css,av = = (21)
τ τCL · τ Na ryc. A 2.6-14 pokazano przebieg stężenia
w osoczu po czterokrotnym podaniu w ciągu doby
Z równania wynika, że – jak wspomniano wyżej dwóch różnych dawek leku z różnicą w odstępie po-
– przy wielokrotnym stosowaniu leku jego stężenie dawania w ciągu dnia i podczas nocy.
zależy od współczynnika oczyszczania.
Pominąwszy właściwości fizykochemiczne kse- W przypadku stosowania dużych dawek wieczorem
nobiotyku wiele czynników osobniczych, takich jak przekroczone zostaje minimalne stężenie toksyczne,
choroby narządów uczestniczących w eliminacji lub natomiast przy obu sposobach dawkowania minimal-
też jednoczesne podawanie substancji wywołujących ne stężenia terapeutyczne nie zostają osiągnięte rano.
minimalne
stężenie
stężenie w osoczu
toksyczne
minimalne
stężenie
terapeutyczne
800 1400 1600 2000 000 400 800 1200 1600 2000 000 400
godzina
Ryc. A 2.6-14. Przebieg stężenia w osoczu po czterokrotnym podaniu dwóch różnych dawek leku w ciągu doby (wg Rowlanda).
MUTSCHLER-2009.indd 53 2010-01-07 22:10:58
54 Farmakokinetyka
Ten przykład wyraźnie uwidacznia trudności w pra- przepisanego leczenia, niekoniecznie jednak oznacza
widłowym dawkowaniu leków o wąskiej rozpiętości zwiększenie bezpieczeństwa leczenia.
terapeutycznej w celu uzyskania właściwych stężeń
w ciągu całej doby. Długotrwały wlew kroplowy. Określone, w dużej
mierze stabilne stężenie leku we krwi można uzyskać
Kumulacja. Opisane wyżej zwiększanie się stężenia stale, zastępując eliminowaną ilość leku powoli wle-
leku przy wielokrotnym podawaniu jest określane wanym kroplowo jego roztworem. Taki sposób poda-
mianem kumulacji. Zależy ona od względnych okre- wania jest korzystny zwłaszcza w przypadku leków
sów pomiędzy dawkami (stosunek okresu pomiędzy szybko eliminowanych (np. nitroprusydku sodu).
podaniami i okresu półtrwania eliminacji t1/2) W przypadku długotrwałego wlewu kropelkowego
dostarczanie leku odbywa się zgodnie z kinetyką 0
τ
ε= (22) rzędu, co oznacza, że w jednostce czasu podawana
t1/2
jest stała ilość leku. Natomiast eliminacja przebie-
i występuje przy ε < l, tj. wówczas gdy okres między ga zgodnie z kinetyką I rzędu. W efekcie krzywa
podaniami jest krótszy niż okres półtrwania elimina- (w uproszczeniu) stężenia substancji w osoczu prze-
cji. O ile więc okres pomiędzy podaniami jest odpo- biega tak, jak przedstawiono na ryc. A 2.6-15.
wiednio krótki, o tyle kumulacji może ulegać właści-
wie każdy związek. O substancjach kumulujących się Z rozpoczęciem wlewu stężenie przez pewien czas
w ścisłym znaczeniu tego słowa mówi się jednak tylko szybko narasta, następnie stopniowo przechodzi
wtedy, kiedy przy niewielu podaniach w ciągu doby w stabilne stężenie stanu stacjonarnego. To ostatnie
(1–2 razy) stężenie wzrasta. Do silnie kumulujących można obliczyć na podstawie równania:
się związków należą m.in. sole ołowiu, chlorofenotan,
fenobarbital, niektóre benzodiazepiny i digitoksyna. Css = szybkość wlewu/CL (23)
Względny okres między podaniami ma kliniczne Czas niezbędny do uzyskania stężeń stanu stacjonar-
znaczenie z jeszcze jednej przyczyny. Mianowicie im nego jest równy, podobnie jak w przypadku podawa-
większe jest ε, tym bardziej pominięcie jednej dawki nia przerywanego, około pięciu okresom półtrwania.
wpływa na stężenie leku w osoczu. Z tego względu Czas niezbędny do uzyskania działających stężeń
zmniejszenie częstotliwości przyjmowania z wie- można skrócić, jeśli wraz z początkiem wlewu jed-
lokrotnej na 1–2 razy dziennie, np. za pomocą pre- nocześnie dodatkowo zastosowana zostanie dawka
paratów o modyfikowanym uwalnianiu, umożliwia początkowa w postaci jednorazowego wstrzyknięcia
co prawda poprawę systematyczności przestrzegania (tzw. bolusa).
Css
stężenie w osoczu
0 1 2 3 4 5 6 0 1 2 3 4 5
t1/2
Ryc. A 2.6-15. Przebieg stężenia leku w osoczu w przypadku wlewu.
MUTSCHLER-2009.indd 54 2010-01-07 22:10:59
Farmakokinetyka 55
2.6.6. Kinetyka nieliniowa Tabela A 2.6-1. Przyczyny kinetyki nieliniowej (zmodyf. wg
Luddena)
W dotychczas opisywanych procesach kinetycznych
zakładano, że procesy te przebiegają zgodnie z kine- Proces Mechanizm Przykłady
tyką I rzędu – w dużej mierze niezależnie od zasto- wchłanianie wysycenie kwas askorbinowy,
sowanych dawek. Jeśli warunek ten jest spełniony, transporterów antybiotyki β-laktamowe,
ryboflawina
to występuje prawidłowo przebiegająca kinetyka li-
niowa. W przypadku zastosowania dużych dawek nie- efekt pierwszego wysycenie enzymów 5-fluorouracyl,
których substancji może się jednak zdarzyć, że ich ki- przejścia hydralazyna,
propranolol,
Farmakokinetyka
netyka przestaje przebiegać zgodnie z zasadami ki- werapamil
netyki I rzędu. Przyczyną takiej nieliniowej kinetyki
mogą być niemal wszystkie procesy kinetyczne, wiązanie z białkami ograniczona ceftraikson,
osocza pojemność dizopiramid,
a więc wchłanianie, dystrybucja i eliminacja, przy wiązania białek kwas walproinowy
czym odpowiedzialne za to mechanizmy w przypad- transportowych
ku poszczególnych procesów są zazwyczaj zbliżone wiązanie cyklosporyna
(zob. tab. A 2.6-1). z erytrocytami
U podłoża leży wysycenie enzymów lub białek
metabolizm wysycenie enzymów etanol, A2
transportowych (carriers), a także ograniczona po- fenytoina,
jemność wiązania białek transportowych: kwas salicylowy
niedobór kosubstratu paracetamol
■ W razie zastosowania dużych dawek część (pro-
hamowanie
cent) wchłaniania może ulec zmniejszeniu, je- przez produkt dikumarol
śli przy wchłanianiu z przewodu pokarmowego
autoindukcja karbamazepina,
uczestniczy białko transportowe. Do nieproporcjo- rifampicyna
nalnie dużego zwiększenia ilości leku w organizmie Wydalanie nerkowe
może natomiast dojść wówczas, gdy biodostępność
sekrecja wysycenie kwas p-aminohippurowy
jest ograniczana silnym efektem pierwszego przej- kanalikowa transporterów
ścia, a uczestniczące w tym enzymy pracują na gra-
nicy wysycenia. zwrotne wchłanianie wysycenie ryboflawina
kanalikowe transporterów
■ Przy ograniczonej pojemności wiązania z białkami
zwiększenie dawki powoduje wzrost wolnej czę-
ści substancji czynnej w osoczu. W wyniku tego
związek rozmieszcza się w tkankach w większych ■ Kolejną ważną przyczynę nieliniowości farmako-
ilościach, co oznacza wzrost objętości dystrybucji. kinetyki stanowi wysycenie białek transportowych
Jeśli eliminacja ogranicza się do wolnego leku, odpowiedzialnych za sekrecję lub zwrotne wchła-
czas półtrwania eliminacji skraca się, a całkowite nianie substancji czynnych w kanalikach nerko-
stężenie wzrasta w mniejszym stopniu, niż można wych.
tego oczekiwać w przypadku kinetyki liniowej.
Najbardziej znanym przykładem kinetyki nieliniowej
■ W przypadku wysycalności enzymu, w wyniku
jest eliminacja etanolu, która przebiega ze stałą szyb-
zwiększenia dawki zmniejsza się stała szybko-
kością, ponieważ metabolizujący enzym, dehydroge-
ści eliminacji, a po obniżeniu stężenia w osoczu
naza alkoholowa, zostaje wysycony już przy niewiel-
poniżej określonej wartości progowej ponownie
kich stężeniach etanolu we krwi.
wzrasta. Odzwierciedla się to jednak w przebiegu
W przypadku stosowania fenytoiny lub kwasu
stężenia w osoczu tylko wówczas, gdy odpowia-
salicylowego obserwuje się po zwiększeniu dawki
dająca za to reakcja jest główną drogą eliminacji
nieproporcjonalnie duży wzrost stężenia stanu rów-
(na związek pomiędzy wysyceniem aktywności
nowagi w następstwie wysycenia przez substrat en-
enzymu a efektem pierwszego przejścia zwrócono
zymów uczestniczących w metabolizmie.
uwagę uprzednio).
Odwrotna sytuacja występuje podczas indukcji
■ Indukcja lub hamowanie aktywności enzymów enzymatycznej: szybkość eliminacji zwiększa się,
metabolicznych prowadzi ponadto do odchyleń co prowadzi do kinetyki nieliniowej.
stężeń substancji czynnych od oczekiwanych war- Rozmiaru nieproporcjonalnych zmian stężenia
tości. leków w przypadku nieliniowej kinetyki nie da się
MUTSCHLER-2009.indd 55 2010-01-07 22:10:59
56 Farmakokinetyka
ściśle ocenić, dlatego precyzyjne dawkowanie ta- ■ przyspieszenie eliminacji alkaloidów w zatruciach
kich leków jest znacznie trudniejsze niż w przypadku za pomocą zakwaszania moczu.
środków o kinetyce liniowej.
2.6.8. Monitorowanie stężenia leku
2.6.7. Modyfikacja zależnych we krwi
od czasu zmian stężenia
leku w osoczu Pojęcie therapeutic drug monitoring (TDM) oznacza
pomiar stężenia leku w osoczu pacjenta w celu uzy-
Na zależne od czasu zmiany stężenia leku w osoczu skania danych na temat jego indywidualnej farmako-
– jednego z ważniejszych zagadnień w badaniach kinetyki, aby dzięki temu, w razie konieczności, od-
farmakokinetycznych – można zgodnie z równaniem powiednio dopasować dawkowanie. Oznaczanie stę-
(20) wpływać w różny sposób. Przede wszystkim na- żeń osoczowych w celu kontroli leczenia jest jednak
leży rozważyć, jaki wpływ na C0 będą miały zmiany sensowne tylko wówczas, gdy:
dawki. Mianowicie im większe C0, tym dłuższy czas
■ stosowany lek ma niewielką rozpiętość terapeu-
upływa do obniżenia stężeń w osoczu poniżej progu
tyczną, w związku z czym istnieje niebezpieczeń-
skutecznego działania. Jednak wydłużenie czasu dzia-
stwo zatrucia,
łania wyłącznie na podstawie zwiększenia dawek ma
sens jedynie w nielicznych przypadkach, ponieważ ■ występuje ścisły związek między stężeniami leku
jeśli nie występuje opóźnienie i wydłużenie wchła- i działaniem terapeutycznym,
niania (zob. poniżej), istnieje niebezpieczeństwo
przekroczenia minimalnych stężeń toksycznych.
Odpowiednim i szczególnie często stosowanym
sposobem postępowania, mającym na celu wydłu-
żenie działania leku, jest natomiast modyfikowanie
szybkości wchłaniania przez opóźnienie uwalniania
z postaci leku lub wydłużenie czasu rozpuszczenia Tabela A 2.6-2. Zakresy osoczowych stężeń terapeutycznych
związku czynnego. Wszystkie postacie depot oparte dla wybranych leków (wg Birchera)
są na zasadzie opóźnionego przechodzenia do organi-
zmu. Z idealnym preparatem o modyfikowanym (wy- Substancja Zakres terapeutyczny
dłużonym) uwalnianiu, które zapewnia (w przybliże- czynna (μg/ml)
niu) stałe stężenie we krwi przez dłuższy okres, ma się Leki przeciwpadaczkowe
do czynienia wówczas, gdy odpowiednimi metodami
galenowymi można po wchłonięciu dawki początko- karbamazepina
(monoterapia) 6-10
wej (inicjującej) doprowadzić do sytuacji, w której (leczenie skojarzone) 2-6
dalsze wchłanianie postaci czynnej jest w jednostce
fenobarbital 15-25
czasu równe ilości eliminowanej. Tego typu prepara- fenytoina 5-15
ty o modyfikowanym uwalnianiu jest jednak bardzo prymidon 5-10
trudno uzyskać. kwas walproinowy 5-100
Na czas działania leku można wpływać również Ksantyny
przez zmianę szybkości eliminacji. Stała szybko- teofilina 6-12
ści eliminacji jest wartością niezmienną dla danego
związku, dlatego należy przez modyfikację chemicz- Glikozydy nasercowe
ną leku zmienić albo biotransformację, albo szybkość digoksyna 0,0005-0,0025
digitoksyna 0,01-0,025
wydalania. Alternatywnie można próbować hamo-
wać wydalanie kanalikowe albo zwrotne wchłanianie Aminoglikozydy
leku przez jednoczesne zastosowanie innego związ- gentamycyna 4-12
ku. Znanymi, przedstawianymi już uprzednio przy- tobramycyna 4-12
kładami są: Immunosupresanty
■ opóźnienie wydalania penicylin przez probenecid cyklosporyna 4-12
lub inne związki zawierające grupy karboksylowe,
MUTSCHLER-2009.indd 56 2010-01-07 22:11:00
Farmakokinetyka 57
■ pożądane lub niepożądane działania występują do- 2.6.9. Farmakokinetyka
piero po dłuższym okresie utajenia, w określonych
■ należy się liczyć z bardzo znacznymi odchyleniami populacjach chorych
między stężeniami w osoczu pomimo stosowania
takich samych dawek, W klasycznej farmakokinetyce w celu obliczenia pa-
rametrów farmakokinetycznych (np. współczynnika
■ naturalny przebieg choroby podlega znacznym
oczyszczania, okresu półtrwania) uwzględnia się stę-
wahaniom, co uniemożliwia ocenę działania lecz-
żenia w osoczu pojedynczych ochotników. Średnie
niczego,
wartości i odchylenia standardowe tych parametrów
Farmakokinetyka
■ objawy przedawkowania są trudne do odróżnienia wskazują, z jaką zmiennością należy się liczyć w ob-
od objawów chorobowych (np. przy zaburzeniach rębie określonej populacji (np. grupy pacjentów z daną
rytmu serca). chorobą). Metoda ta nie jest jednak zadowalająca
w przypadku dwóch często występujących problemów.
Udowodnione zostały korzyści monitorowania Tak więc nie można ustalić oczekiwanego przebiegu
w przypadku leczenia środkami przeciwpadaczko- stężenia leku w osoczu u określonego chorego na pod-
wymi (szczególnie fenytoiną), solami litu, teofiliną, stawie nielicznych uzyskanych oznaczeń w początko-
niektórymi chemioterapeutykami (np. aminoglikozy- wej fazie leczenia. Niepełne dane, często występujące A2
dami, chloramfenikolem, flucytozyną) i cyklospory- w przypadku dużych badań klinicznych (sparse data
ną. Nie jest to tak jednoznaczne w przypadku leków situation) nie są również reprezentatywne dla przyję-
przeciwarytmicznych i glikozydów nasercowych. tych metod wyznaczania parametrów farmakokine-
W tab. A 2.6-2 zestawiono zakresy terapeutyczne tycznych. W celu rozwiązania obu tych problemów
stężeń we krwi leków wymagających monitorowa- niezbędne jest wzięcie pod uwagę w modelu farma-
nia. Monitorowanie może być ponadto celowe, jeśli kokinetycznym zmienności parametrów farmakokine-
podejrzewa się, że chory nie przestrzega zaleconego tycznych przy uwzględnieniu czynników fizjologicz-
stosowania/dawkowania leku, lub w przypadku inter- nych (np. czynności nerek, wieku, płci, rasy). Zadania
akcji leków. tego typu ocenia tzw. farmakokinetyka populacji, przy
czym najczęściej stosowana jest metoda non-MEM
Oceniając stężenia leku w surowicy, należy uwzględ- (non-linear mixed effect monitoring). Za pomocą tej
nić: dawkę i czas ostatniego podania, czas trwania metody możliwe jest przewidywanie na podstawie nie-
leczenia, a także masę ciała, wiek i choroby towarzy- licznych istniejących danych klinicznych przybliżone-
szące występujące u danego pacjenta. Monitorowanie go dalszego przebiegu stężeń w osoczu krwi u określo-
nie jest niezbędne, jeśli farmakodynamika leku nego chorego, a także wyliczenie dokładności takiego
(np. obniżenie stężeń glukozy we krwi przez insuli- pomiaru. Jeśli sytuacja kliniczna uniemożliwia częste
nę, zmniejszenie aktywności protrombiny, tj. zwięk- pobrania krwi (np. w onkologii dziecięcej) albo też
szenie wartości INR przez leki przeciwzakrzepowe, niezbędne jest dostosowanie dawkowania do osobni-
zwolnienie rytmu serca przez β-adrenolityki) może czego przebiegu stężeń we krwi, metoda ta ma duże
być dokładnie ustalona. znaczenie.
MUTSCHLER-2009.indd 57 2010-01-07 22:11:00
58 Farmakokinetyka
2.7. Farmakokinetyka w sytuacjach szczególnych
2.7.1. Zmiany farmakokinetyki skości wątroby metabolizm diazepamu, triamterenu
w stanach patologicznych i lidokainy przebiega bardzo powoli.
Kinetyka większości leków jest zbadana głównie Wydalanie z moczem w zależności od czynności
u zdrowych ochotników, ponieważ dobrze nadają się nerek. Jeśli lek jest wydalany głównie lub wyłącz-
oni – jako względnie homogenna grupa – do ustalenia nie z moczem, czynność nerek, którą można ocenić
charakterystycznych dla danego leku lub jego posta- za pomocą współczynnika oczyszczania kreatyniny,
ci specyficznych parametrów farmakokinetycznych, wpływa na szybkość wydalania: wraz ze zmniejsza-
takich jak biodostępność, bądź następstw interakcji jącym się współczynnikiem oczyszczania kreatyni-
z innymi lekami. W porównaniu z tym dane dotyczą- ny obniża się szybkość wydalania ksenobiotyków.
ce farmakokinetyki u chorych są znacznie bardziej Należy przy tym wziąć pod uwagę, że oprócz sta-
skąpe, mimo że leki są przeznaczone do leczenia nów patologicznych dla współczynnika oczyszczania
osób chorych. Dotychczas badano przede wszystkim kreatyniny istotny jest również wiek. U niemowląt
wpływ niewydolności krążenia oraz chorób wątroby nerki nie osiągnęły jeszcze pełnej sprawności czyn-
i nerek na farmakokinetykę leków. nościowej: szybkość przesączania kłębuszkowego,
która w przybliżeniu odpowiada współczynnikowi
Zaburzenia wchłaniania. Na wchłanianie leków oczyszczania kreatyniny, wynosi u noworodków tyl-
ma wpływ przepływ krwi przez przewód pokarmowy. ko ok. 10 ml/min (wartość prawidłowa u dorosłych
Jest on upośledzony przy zastoju w dużym krążeniu, 120 ml/min). Oznacza to, że wydalanie przez nerki
np. w niewydolności serca. Zmniejszona biodostęp- w pierwszych tygodniach życia przebiega znacznie
ność u chorych z niewyrównaną niewydolnością ser- wolniej niż u zdrowych dorosłych. W wieku pode-
ca została opisana np. dla prokainamidu, chinidyny szłym współczynnik oczyszczania kreatyniny po-
i hydrochlorotiazydu. nownie ulega zmniejszeniu i powyżej 70 roku życia
jest on często wyraźnie mniejszy. Jeśli współczynnik
Zmiany dystrybucji. Zmniejszenie przepływu przez oczyszczania kreatyniny w następstwie ciężkiej nie-
narządy obwodowe w następstwie niewydolności wydolności nerek jest znacznie ograniczony, w przy-
serca może również wpływać na dystrybucję leków. padku leków eliminowanych z moczem z powodu
W takich przypadkach zmniejsza się na przykład ob- spowolnionej szybkości eliminacji należy:
jętość dystrybucji lidokainy, prokainamidu i chinidy-
■ zmniejszyć stosowane dawki lub
ny. Utrata białek lub zaburzenia ich syntezy zmieniają
wiązanie z białkami. Wykazano więc, że u chorych ■ zwiększyć okresy między podaniami.
z zespołem nerczycowym związana część fenytoiny
ulega zmniejszeniu z 90 do 80%. Przy postępującej Dostosowanie dawkowania do czynności nerek
niewydolności nerek możliwe jest wypieranie leków jest obecnie przeprowadzane często zgodnie z wy-
z wiązania z białkami w następstwie retencji związ- tycznymi podanymi przez Dettlego. Za pomocą dia-
ków podlegających wydalaniu z moczem. Ponadto gramu oznaczony zostaje współczynnik korekcji Q,
obserwuje się zmiany jakościowe wiązania z białka- przez który przemnożona zostaje zazwyczaj stosowa-
mi wyróżniające się odmienną pojemnością i powi- na dawka lub przez który podzielony zostaje okres
nowactwem wiązania. Dodatkowo ograniczone może między podaniami (zob. ryc. A 2.7-1). Podstawowym
zostać wiązanie z tkankami, wskutek czego dochodzi warunkiem jest przy tym znajomość eliminowanej
do zmniejszenia objętości dystrybucji. ilości danej substancji Q0, którą można odczytać
W chorobach wątroby prawdopodobne jest wystę- z odpowiednich tabel (zob. tab. A 2.7-1).
powanie podobnych zaburzeń z powodu zmniejsze- W nerczycach, które charakteryzują się dużą utratą
nia syntezy białek. białka drogą nerekową, szybkość wydalania z mo-
czem może ulec zwiększeniu. W takim przypadku
Wpływ na metabolizm. Jak opisano wyżej, elimi- w celu utrzymania skuteczności stężeń w osoczu na-
nacja związków lipofilnych przebiega głównie przez leży albo zwiększyć wielkość poszczególnych dawek,
przemiany oksydacyjne, a powstałe metabolity są na- albo skrócić okres między podaniami.
stępnie sprzęgane, dlatego choroby wątroby mogą
zmniejszać szybkość eliminacji leków metabolizo-
wanych głównie w wątrobie. Na przykład w mar-
MUTSCHLER-2009.indd 58 2010-01-07 22:11:00
Farmakokinetyka 59
Tabela A 2.7-1. Wartości Q0 dla różnych leków
wartość kreatyniny (mg/100 ml)
8 4 3 2 1,5 1 0,75
1,0 Lek Q0
2 acetylodigoksyna 0,3
0,8 3 kwas acetylosalicylowy 1
Q'
0,6 amitryptylina 1
na
stężenie w osoczu
sy
digok ampicylina 0,12
Farmakokinetyka
0,4 1 2 karbenicylina 0,1
Q0
chloramfenikol 0,83
0,2
cymetydyna 0,25
klindamycyna 0,8
0 20 40 60 80 100
klirens kreatyniny (ml/min) digitoksyna 0,9
digoksyna 0,33
A2
fizjologiczny klirens kreatyniny
doksycyklina 0,9
Ryc. A 2.7-1. Wykres do wyznaczania współczynnika korekcji Q’,
gentamycyna 0,03
za pomocą którego można obliczyć indywidualnie dopasowaną
do czynności nerek dawkę podtrzymującą (lub przerwy między lidokaina 0,92
podaniami) dla leków wydalanych z moczem (wg Dettlego).
metylodigoksyna 0,5
W celu wyznaczenia wartości Q’ należy odnaleźć na osi rzędnych
po lewej stronie diagramu wartość odczytaną z tab. 2.7-1 i od minocyklina 0,9
tego punktu poprowadzić prostą 1 do prawego górnego rogu
diagramu. Następnie sporządza się prostą 2, ciągnąc ją prosto- morfina 0,9
padle do góry z punktu na osi odciętych wyznaczonego przez penicylina G 0,05
wartość klirensu kreatyniny pacjenta. Czynnik korekcji Q’ odnaj-
duje się, prowadząc od punktu przecięcia prostych 1 i 2 prostą 3 prazosyna 0,9
prostopadłą do osi rzędnych po lewej stronie diagramu.
rifampicyna 0,8
streptomycyna 0,04
2.7.2. Kinetyka u osób sulfatemoksazol 0,8
w podeszłym wieku trimetoprim 0,5
U osób w podeszłym wieku zazwyczaj nie występują
większe zaburzenia wchłaniania. Z powodu zmienio- nerek (obniżanie się współczynnika oczyszczania
nej czynności motorycznej żołądka szybkość wchła- kreatyniny). Leki wydalane głównie drogą nerko-
niania może jednak ulegać nieznacznemu spowolnie- wą są wraz z postępującym wiekiem coraz wolniej
niu. eliminowane, np. okres półtrwania diacetolu, głów-
nego metabolitu leku β-adrenolitycznego – acebu-
Znaczniejsze zmiany zachodzą w dystrybucji. Mogą tololu zwiększa się z 12 godz. u młodych ochotni-
się one opierać na zmniejszeniu objętości całkowitej ków do 17,5 godz. u osób w wieku powyżej 65 lat.
wody organizmu lub zwiększeniu objętości tkanki Digoksyna jest u osób w podeszłym wieku również
tłuszczowej, zmniejszonej syntezie albumin osoczo- wolniej wydalana.
wych, a także zwiększonej syntezie kwaśnej α1-gli- Jeśli do zależnego od wieku zmniejszenia współ-
koproteiny. Wszystkie te czynniki mogą wpływać czynnika oczyszczenia kreatyniny dołączą się pato-
na objętość dystrybucji. logiczne zmiany czynności nerek (np. w następstwie
Możliwe zmiany biotransformacji u osób w pode- przewlekłego zapalenia kłębuszków nerkowych)
szłym wieku opisano już w A 2.4.7. i jeśli nie zostanie określony odpowiedni sposób
Zwracano już także uwagę na zmniejszającą się dawkowania, ryzyko kumulacji substancji czynnej
często w podeszłym wieku sprawność czynności znacznie wzrasta.
MUTSCHLER-2009.indd 59 2010-01-07 22:11:00
60 Farmakokinetyka
2.8. Farmakokinetyka substancji chiralnych
Wiele leków o istotnym znaczeniu klinicznym znaj- iną. Dla L-tryptofanu wykazano blisko 100-krotnie
duje zastosowanie w postaci racemicznej, tj. składa silniejsze wiązanie z albuminami niż D-enancjome-
się z dwóch występujących w równych częściach ru. Enancjomery mogą również wykazywać różnice
enancjomerów. Oba enancjomery są – wyjąwszy w wiązaniu z białkami tkanek.
kierunek skręcenia wiązki spolaryzowanego światła
– fizykalnie identyczne, różnią się jednak trójwymia- Największe różnice w kinetyce enancjomerów stwier-
rowym układem przestrzennym. Wzajemne oddzia- dza się jednak w biotransformacji. Przez wiązanie
ływanie z – będącym również związkiem chiralnym racematów z czynnymi optycznie enzymami uczestni-
– endogennym białkiem (np. receptorem) pozwala czącymi w biotransformacji powstają diasteromerowe
oczekiwać znacznych różnic pomiędzy enancjo- kompleksy enzym-substrat, co powoduje, że oba enan-
merami. W przypadku farmakokinetyki można jed- cjomery mogą być metabolizowane w różnym stop-
nak oczekiwać różnic między enancjomerami tylko niu. Klinicznie zwracają uwagę takie następstwa jak
w takich procesach kinetycznych, w których uczest- w przypadku werapamilu, gdy po dożylnym podaniu
niczą chiralne makrocząsteczki endogenne, z którymi 10 mg uzyskuje się taki sam efekt (wydłużenie czasu
enancjomery ksenobiotyków tworzą diastereomery przewodzenia w sercu) jak po doustnym podaniu 80
różniące się właściwościami fizykochemicznymi. mg. Wyraźnie słabsze działanie werapamilu po poda-
W przypadku wchłaniania, przebiegającego naj- niu doustnym wyjaśnia fakt, że czynny S-enancjomer
częściej jako proces biernej dyfuzji, różnice między podlega większemu wątrobowemu efektowi pierwsze-
enancjomerami występują bardzo rzadko. Lewodopa go przejścia i osiąga znacznie mniejsze stężenia w oso-
i L-metotreksat podlegają procesowi czynnego trans- czu (zob. ryc. A 2.8-1) niż po podaniu dożylnym.
portu, dlatego jest zrozumiałe, że są one wchłaniane
szybciej i w większym procencie niż niepodlegające Następstwa stereospecyficznej biotransformacji
czynnemu transportowi postacie D. Nieco częściej niż są różne dla leków wolno i szybko eliminowanych.
w przypadku wchłaniania występują różnice w wią- W przypadku leków o niskim współczynniku oczysz-
zaniu enancjomerów z białkami. Tak więc wiąże się czenia, np. heksobarbitalu, stereoselektywny me-
z albuminami osocza przede wszystkim R-proprano- tabolizm prowadzi do obniżenia okresu półtrwania,
lol, natomiast S-propranolol z kwaśną α1-glikoprote- zwłaszcza enancjomeru preferencyjnie biotrans-
formowanego. Tak więc np. t1/2 R(–) heksobarbita-
lu jest krótszy niż S(+) enancjomeru. W przypadku
związków o szybkiej eliminacji, jak opisany wyżej
werapamil, stereoselektywna biotransformacja po-
woduje natomiast odmienną biodostępność obu enan-
log C cjomerów po podaniu doustnym.
Szczególną postać stereoselektywnego metabolizmu
przedstawia chiralna inwersja. Występuje ona m.in.
w przypadku niektórych niesteroidowych leków prze-
stężenie w osoczu
R-(+) ciwzapalnych z grupy pochodnych kwasów 2-ary-
lopropionowych. Na przykład R-ibuprofen zostaje
w organizmie w dużej mierze przekształcony w sil-
niej działający S-ibuprofen (zob. ryc. A 2.8-2).
S-(–)
Poza opisanymi wyżej odrębnościami w farmakoki-
netycznych i farmakodynamicznych właściwoścach
czas (t) obu enancjomerów racematu enancjomery mogą
wzajemnie wpływać na swoją kinetykę. Tego typu in-
Ryc. A 2.8-1. Stężenie w osoczu R (+)- i S (–)-werapamilu po do- terakcje enancjomer/enancjomer wykazano na przy-
ustnym podaniu racematu (wg Eichelbauma i wsp.). kład dla leku przeciwarytmicznego – propafenonu.
MUTSCHLER-2009.indd 60 2010-01-07 22:11:01
Farmakokinetyka 61
CH3 CH3 CH3
syntetaza
CH3 COOH acylo-CoA CH3 COSCoA hydrolaza CH3 COOH
H3C H3C H3C
(–)-(R)-Ibuprofen (–)-(R)-ibuprofeno-CoA
Farmakokinetyka
epimeraza
2-arylopropionylo-CoA
CH3 CH3 CH3
hydrolaza
CH3 COOH CH3 COSCoA CH3 COOH
H3C H3C H3C
A2
(+)-(S)-ibuprofen (+)-(S)-ibuprofeno-CoA
Ryc. A 2.8-2. Chiralna inwersja R-ibuprofenu.
MUTSCHLER-2009.indd 61 2010-01-07 22:11:01
62
3. Farmakodynamika
Farmakodynamika, jak wspomniano w definicji Amerykańska Agencja ds. Żywności i Leków
w rozdz. A 1, jest to nauka o wpływie leków na funk- (FDA) na początku 2007 roku zarejestrowała 1202
cje lub struktury biologiczne. Według tej definicji ba- leki, które wiązały się do około 630 struktur doce-
dania farmakodynamiczne mają na celu odpowiedzi lowych.
na pytania o: Substancje działające niespecyficznie charakte-
ryzują się:
■ sposób działania (profil działania, jakość działania,
działania zależne i niezależne od struktury che- ■ niespecyficznymi interakcjami z jednoznacznie zde-
micznej), finiowanymi strukturami,
■ mechanizm działania, ■ jedynie minimalną zmianą działania w wyniku
zmian struktury chemicznej, niemających charak-
■ miejsce działania,
teru zmian zasadniczych.
■ siłę działania (potency),
Jednak tylko nieliczne substancje należą do tego typu
■ intensywność działania (efficacy, maksymalny
leków. Przykładem są substancje przeczyszczające
efekt).
i moczopędne działające na zasadzie osmotycznej,
a także niektóre środki dezynfekcyjne.
Substancje działające specyficznie są to substancje
działające w małych dawkach lub stężeniach (naj-
Mechanizm działania. Mechanizm działania więk-
częściej w nano- lub mikromolarnym zakresie stężeń);
szości leków można zaklasyfikować do kilku typów,
ich efekt zależy ściśle od struktury chemicznej i w wy-
których przykłady przedstawiono w tab. A 3-1.
niku tego także od formy, wielkości i stereochemicznej
Leki działają między innymi przez:
orientacji cząsteczek, np. położenia grup funkcyjnych
i elektronów. Nieznaczne zmiany struktury chemicznej
■ interakcje z receptorami błonowymi (aktywacja lub
mogą istotnie wpływać na działanie farmakologiczne.
hamowanie receptora),
Związki z tym samym punktem uchwytu często mają
wspólne elementy strukturalne, tzw. grupy farmakofo- ■ otwieranie lub zamykanie kanałów jonowych zależ-
rowe, w odpowiednim ułożeniu przestrzennym (por. nych od potencjału bądź aktywowanych ligandami
np. parasympatykomimetyki działające bezpośrednio). (np. neuroprzekaźnikami lub substancjami egzo-
Działanie specyficzne polega również na tym, gennymi),
że związek łączy się z danymi strukturami w możli-
■ regulację transkrypcji genów przez receptory we-
wie selektywny sposób. Ze względu na to, że w przy-
wnątrzkomórkowe,
padku większości leków te wymagania spełnione
są tylko częściowo albo kiedy te same cząsteczki ■ wpływ na błonowe i wewnątrzkomórkowe systemy
docelowe (targets) są położone w różnych miejscach, transportowe (carrier, aktywne systemy przenośni-
poza pożądanym działaniem głównym należy się tak- kowe),
że często liczyć z efektami ubocznymi leku (działa-
■ hamowanie albo aktywację enzymów,
niami niepożądanymi).
Na poziomie molekularno-farmakologicznym spe- ■ zaburzenia biosyntezy w mikroorganizmach.
cyficzność wiązania polega na tym, że związek wy-
kazuje wystarczające powinowactwo do cząsteczki Inne mechanizmy działania, np. antymetabolitów,
docelowej oraz w efekcie wiązania posiada zdolność przeciwciał lub substancji działających na DNA, zo-
zwiększania lub hamowania jej aktywności. stały omówione w odrębnej części.
MUTSCHLER-2009.indd 62 2010-01-07 22:11:01
Farmakodynamika 63
Tabela A 3-1. Mechanizmy działania leków
Rodzaj mechanizmu Przykłady
Interakcja z receptorami błonowymi
Stymulacja receptora stymulacja receptorów adrenergicznych przez sympatykomimetyki
stymulacja receptorów muskarynowych przez parasympatykomimetyki
działające bezpośrednio
Blokada receptora hamowanie receptorów adrenergicznych przez α- lub β-blokery
blokada receptorów histaminowych H1 i H2 przez substancje antyhistaminowe
Wpływ na receptory jądrowe
Stymulacja receptorów jądrowych stymulacja receptorów hormonów tarczycy przez trójjodotyroninę
stymulacja PPARγ przez glitazon
Farmakodynamika
Hamowanie receptorów jądrowych hamowanie receptorów mineralokortykoidów przez elplerenon
Wpływ na kanały jonowe zależne od potencjału
Otwieranie kanałów jonowych zależnych od potencjału otwieranie kanałów potasowych przez aktywatory
Blokada kanałów jonowych zależnych od potencjału zamykanie kanałów sodowych przez środki miejscowo znieczulające
blokada kanałów wapniowych przez leki blokujące kanały wapniowe
Interakcje z przenośnikami (transporterami)
A3
Hamowanie aktywnych procesów transportujących hamowanie wychwytu zwrotnego monoamin przez leki przeciwdepresyjne
hamowanie magazynowania monoamin w pęcherzykach przez rezerpinę
Hamowanie transporterów hamowanie kotransportera Na+/K+/2CI– przez diuretyki typu furosemidu
hamowanie kotransportera Na+/2CI– przez tiazydy
Działanie na enzymy
Aktywacja enzymów aktywacja plazminy przez alteplazę
stymulacja cyklazy guanylanowej przez NO
Hamowanie enzymów hamowanie syntezy prostaglandyn przez niesteroidowe leki przeciwzapalne
hamowanie acetylocholinoesterazy przez pośrednio działające parasympatykomimetyki
hamowanie konwertazy angiotensyny przez jej inhibitory
Wpływ na biosyntezę mikroorganizmów
Hamowanie syntezy ściany komórkowej bakterii działanie bakteriobójcze antybiotyków β-laktamowych
Zaburzenia prawidłowej syntezy białek bakterii działanie bakteriostatyczne tetracyklin
Zaburzenia przemiany kwasu foliowego działanie bakteriostatyczne sulfonamidów
MUTSCHLER-2009.indd 63 2010-01-07 22:11:01
64 Farmakodynamika
3.1. Działanie farmakologiczne przez receptory
Przez pojęcie receptory farmakologiczne (farma- z zastosowaniem technik genetycznych udało się
koreceptory) rozumie się wewnątrzkomórkowe lub sklonować wiele receptorów i określić sekwencję
błonowe białka, które po przyłączeniu (endogennego, aminokwasów. Zintegrowano także izolowane recep-
fizjologicznego) liganda do specyficznego miejsca tory w sztucznych pęcherzykach lub błonach lipido-
wiążącego mogą wywołać transdukcję sygnału (akty- wych i wykazano, że nadal są one w pełni funkcjonal-
wowanie interakcji receptor-efektor). ne. Następnie udało się uzyskać syntezę receptorów
Zgodnie z tą definicją podstawowy wzór interakcji (ekspresję receptorów) po wprowadzeniu genów ko-
pomiędzy ligandem a receptorem można przedstawić dujących (transfekcji receptorów) do komórek, które
następująco: pierwotnie nie posiadają danego typu receptora.
L + R º [LR] → → E
L ligand
3.1.2. Podtypy receptorów
R receptor
E efekt
W enzymologii enzymy, które działają na ten sam
substrat, ale odróżniają się wartościami Km i Vmax,
Farmakoreceptor pełni więc podwójną funkcję:
określa się mianem izoenzymów. Analogicznie
■ rozpoznania sygnału przez zmiany działania pod w przypadku receptorów również można odróżnić
wpływem liganda i tworzenia kompleksu ligand- różne typy i podtypy. Praktycznie wykazano istnie-
-receptor, nie receptorów podobnych strukturalnie, ale odróż-
nialnych zarówno farmakologicznie, jak i metodami
■ przenoszenia sygnału (transdukcji sygnału) i wy-
molekularnymi dla wszystkich neuroprzekaźników,
woływania określonego efektu.
a także dla hormonów, witamin, czynników wzro-
stowych itp. Na przykład, noradrenalina oddziałuje
Liczba farmakoreceptorów, podobnie jak innych endo-
na receptory α- i β-adrenergiczne, które mogą być
gennych funkcjonalnych molekuł, jest ograniczona,
dalej dzielone na podtypy. Acetylocholina działa
dlatego wiązanie liganda ulega wysyceniu. Ponadto
na receptory nikotynowe i muskarynowe, wśród któ-
wiązanie liganda jest stereoselektywne i, w przeci-
rych także wyróżnia się podtypy wzajemnie ze sobą
wieństwie do reakcji enzymatycznej, przebiega od-
oddziałujące. Receptory dla glukokortykosteroidów,
wracalnie bez zmian chemicznych liganda.
estrogenu i progesteronu istnieją w (przynajmniej)
Dla farmakologów receptory mają szczególne znaczenie, dwóch podtypach (α i β), które w przypadku recep-
podobnie jak enzymy dla biochemików. Istnieją liczne po- torów glukokortykosteroidowych powstają w wyniku
dobieństwa między receptorami a enzymami. W enzymolo- różnego cięcia i wstawiania (splicing) kwasów nukle-
gii wprowadza się rozróżnienie całego kompleksu enzymu inowych. Inne podtypy receptorowe zostały omówio-
i jego aktywnego centrum, tzn. części cząsteczki związanej ne w odrębnej części.
ze zmianą działania substratu. Analogicznie można wy-
różnić cały kompleks receptora i miejsce wiążące – ligand Natura pracuje zatem, używając uniwersalnych
(binding site, recognition site). kluczy, tzn. ligandów fizjologicznych, do otwierania
specyficznych zamków, czyli podtypów receptora.
3.1.1. Izolacja receptorów,
badanie struktury, transfekcja 3.1.3. Rezerwa receptorowa
i ekspresja receptorów
Jeżeli tylko część receptorów musi być aktywowana
Ze względu na niską gęstość receptorów w tkance, w celu uzyskania maksymalnego efektu (transduk-
izolacja receptorów wydawała się przez długi czas cji sygnału), to mówi się o rezerwie receptorowej.
niemożliwa. Jednak dzięki zastosowaniu metod izo- Jest ona duża, kiedy liczba receptorów znacząco
lacji (m.in. solubilizacji białek receptorowych z nie- przewyższa liczbę efektorów. Rezerwa receptorowa
izotonicznymi detergentami, chromatografii powi- wzrasta najczęściej wraz z ilością procesów uczest-
nowactwa, chromatografii jonowymiennej), a także niczących w transdukcji sygnału. Jest ona najczęściej
MUTSCHLER-2009.indd 64 2010-01-07 22:11:01
Farmakodynamika 65
niewielka, jeżeli łańcuch reakcji wywoływany akty- 3.1.5. Zmiany funkcji receptora
wacją danego receptora składa się z niewielu elemen- wywołane stanami
tów wzmacniających sygnał. Rezerwa receptorowa
zwiększa się wraz ze wzrostem liczby elementów
chorobotwórczymi
wzmacniających sygnał.
W stanach patologicznych występują odchylenia
od prawidłowej czynności receptorów. Typowym
3.1.4. Desensytyzacja, regulacja przykładem choroby autoagresyjnej skierowanej
w górę i w dół na białka receptorowe jest miastenia rzekomoporaź-
na, w której powstają własne przeciwciała przeciw
(up- and down-regulation) receptorom nikotynowym w płytce nerwowo-mięś-
niowej. W wyniku związania się przeciwciał z recep-
Siła sygnału wywołanego uwolnieniem określonego
torami nikotynowymi nie są one wrażliwe na działa-
neuroprzekaźnika nie jest stała, lecz ulega osłabie-
nie neuroprzekaźnika. Efektem jest nużliwość mięś-
niu w stopniu zależnym od typu receptora: dochodzi
niowa.
do desensytyzacji, tzn. osłabienia wrażliwości syste-
Farmakodynamika
Także choroba Basedowa jest schorzeniem au-
mu.
toimmunologicznym, w którym wytwarzają się au-
Podłożem desensytyzacji może być wiele mechani-
toprzeciwciała przeciw receptorom tyreotropowym,
zmów. Tak więc aktywacja receptorów wewnątrzko-
które, w przeciwieństwie do ww. przykładów, mają
mórkowych powoduje też ich fosforylację. W wyniku
właściwości aktywujące receptor. Konsekwencją
tego wzrasta powinowactwo tych receptorów do białek
jest wzmożone wytwarzanie hormonu przez tarczy-
wewnątrzkomórkowych, mających hamujący wpływ
cę.
na transdukcję sygnału i w konsekwencji dochodzi
Można tu również wymienić zaburzenia tworze-
do osłabienia sygnału. Desensytyzacja zachodzi na- A3
nia receptorów dla LDL jako przyczynę rodzinnej
stępnie przez podwyższone wiązanie hamujących
hipercholesterolemii i defekt receptora antydiuretyny
białek G, a także przez osłabienie ekspresji genów
w moczówce prostej.
kodujących dany receptor lub przyspieszenie rozkła-
du kodującego go mRNA. Ponadto liczba receptorów
zmienia się zależnie od stanu funkcjonalnego całego
organizmu lub od stanu danego narządu. W konse- 3.1.6. Agoniści, antagoniści
kwencji utrzymującego się podwyższonego stężenia
liganda stymulującego obserwuje się zmniejszenie Podobnie jak endogenne (fizjologiczne) ligandy, rów-
liczby aktywnych receptorów poprzez ich internali- nież leki mogą wchodzić w interakcję z receptorami.
zację i nasilony rozkład (receptor down-regulation). Warunkiem koniecznym takiej interakcji jest – ana-
Jednym z praktycznych przykładów takiej regulacji logicznie do interakcji endogennego liganda z recep-
receptora w dół jest spadek liczby adrenoreceptorów torem – powstanie kompleksu substancja lecznicza-
β przy niewydolności serca. Zwiększenie liczby re- farmakoreceptor:
ceptorów (up-regulation, czyli regulacja receptorów
w górę) następuje przy ochronie receptorów przed P + R º [PR]
stymulacją, a tym samym zmniejszeniu ich zużycia,
np. przez podanie blokerów receptorów (antagoni- P substancja lecznicza
stów kompetycyjnych), przy odnerwieniu lub bra- R receptor
ku neuroprzekaźników. Substancje, które pośrednio
działają na określony system, mogą także zmieniać Czy i w jakim stopniu powstaje taki kompleks, zależy
liczby receptorów (heterologiczna regulacja w górę od powinowactwa substancji leczniczej do receptora:
lub w dół). Przykładem jest występujący po podaniu im większe powinowactwo, tym większa tendencja
estrogenu wzrost liczby receptorów dla oksytocyny do tworzenia kompleksu.
i spadek liczby receptorów dla estrogenu, a także Parametrem określającym powinowactwo jest stala
wzrost liczby receptorów β, np. w mięśniu sercowym powinowactwa KA. Odpowiada ona odwrotności stałej
przy podawaniu hormonów tarczycy. Te obserwacje dysocjacji KD, tzn. KA = 1/KD. Według prawa mas:
odpowiadają zmienionej wrażliwości tkanki na ok-
sytocynę i noradrenalinę (zob. indukcja enzymu przy [P] ⋅ [R] k2
KD = =
reakcji biotransformacji). [PR] k1
MUTSCHLER-2009.indd 65 2010-01-07 22:11:02
66 Farmakodynamika
gdzie [P] odpowiada stężeniu wolnego leku, [R]
stężeniu wolnych receptorów, [PR] stężeniu kom-
stan spoczynkowy (R) stan zaktywowany (R*)
pleksu receptor-lek, k1 to stała asocjacji i k2 stała
dysocjacji.
A
Istotne jest przy tym rozróżnienie:
■ substancji, które wiążą się z receptorem i go stymu-
lują, tzw. agonistów,
P + R → [PR] → → E,
oraz
■ substancji, które hamują lub całkiem blokują efekty B
aktywacji receptora, tzw. antagonistów.
Wiązanie lek-receptor. W przypadku wiązania sub-
stancji czynnej do receptora możliwe jest występo-
wanie wszystkich typów wiązań (np. jonowe, mostki
wodorowe, hydrofobowe wiązanie przez siły van der
Waalsa). Prawie zawsze różne typy wiązania wystę-
pują jednocześnie. Dla wstępnej fazy zbliżania się
substancji leczniczej i receptora, w przypadku sub- C
stancji ulegających jonizacji, podstawowe znaczenie
mają wiązania jonowe (zasadowe, kwasowe), ponie-
waż – w przeciwieństwie do innych typów wiązania
– charakteryzują się znacznym zasięgiem. Natomiast
wiązania dipol-dipol, mostki wodorowe i wiązania
hydrofobowe są przeważnie odpowiedzialne za na-
stępowe (odwracalne) utrwalenie wiązania.
D
Model dwóch konformacji receptora. Dotychczas
przy opisywaniu interakcji ligand-receptor nie wspo-
mniano, jakie zmiany fizykochemiczne zachodzą
w wyniku tego procesu. Zmiany te przedstawiono
w uproszczeniu na modelu dwóch stanów (zob. ryc.
A 3.1-1). Receptor występuje w dwóch stanach (kon-
formacjach): nieaktywnym (spoczynkowym) i aktyw-
nym (zaktywowanym). Obie konformacje występują
w dynamicznej równowadze, która w razie braku li-
ganda (endogennego lub egzogennego) przesunięta Ryc. A 3.1-1. Model dwóch stanów receptora.
jest zwykle całkowicie w kierunku stanu nieaktyw- A – brak liganda, receptory w stanie spoczynkowym; B – w obec-
nego. Jeżeli jednak receptory, także bez obecności ności pełnych agonistów równowaga przesuwa się zdecydowa-
liganda, mogą znaleźć się w stanie zaktywowanym, nie w kierunku stanu zaktywowanego; C – w obecności częścio-
mówi się o receptorach o aktywności konstytutywnej. wych agonistów/antagonistów równowaga przesuwa się słabiej
Modelom tym odpowiadają: w kierunku stanu zaktywowanego; D – w obecności (pełnego)
antagonisty następuje przesunięcie równowagi w kierunku sta-
■ substancje agonistyczne, które wiążą się z recepto- nu spoczynkowego.
rem preferencyjnie w stanie zaktywowanym i prze-
suwają dalej równowagę w stronę stanu zaktywo-
wanego;
MUTSCHLER-2009.indd 66 2010-01-07 22:11:02
Farmakodynamika 67
■ wiązanie się antagonistów (kompetycyjnych, zob. wewnętrzną. Najczęściej podaje się przy tym aktyw-
poniżej), zapobiegające aktywacji receptora przez ność wewnętrzną jako relatywną aktywność wewnętrz-
wiązanie się do receptora nieaktywowanego; ną α. Odpowiada ona ilorazowi efektu wywoływanego
przez dany ligand (EA) i maksymalnego efektu możli-
■ substancje o właściwościach odwrotnych agonistów
wego w danym układzie biologicznym (Em):
hamujące konstytutywną aktywność receptora.
EA
α∼
Em
3.1.6.1. Pełni i częściowi agoniści
Maksymalna relatywna aktywność wewnętrzna wy-
Zdolność związku do wywoływania działania po utwo- nika z równania EA/Em = l.
rzeniu kompleksu z receptorem nazywana jest aktyw- Agoniści o aktywności wewnętrznej równej l okre-
nością wewnętrzną (intrinsic activity, i.a.). Jest to mia- ślani są jako pełni agoniści, substancje o aktywności
ra maksymalnego działania, jakie może wywoływać wewnętrznej większej niż 0, a mniejszej niż l nazy-
substancja w określonym systemie biologicznym. wa się częściowymi agonistami. Substancje mające
Farmakodynamika
Agonistą nazywa się substancję, która wykazuje za- właściwości zarówno pełnych agonistów, jak i anta-
równo powinowactwo do receptora, jak i aktywność gonistów, słabiej od agonistów przesuwają równo-
I. Działanie agonisty
+ transdukcja efekt A3
sygnału
receptor agonista
II. Hamowanie kompetycyjne
brak transdukcji
+ sygnału brak efektu
receptor antagonista
kompetycyjny
III. Hamowanie niekompetycyjne
zmniejszenie zmniejszenie
+ + transdukcji efektu
sygnału
receptor agonista antagonista
niekompetycyjny
antagonista
niekompetycyjny
zmniejszenie
+ transdukcji zmniejszenie
sygnału efektu
receptor agonista
Ryc. A 3.1-2. Schemat przedstawiający interakcje lek-receptor (modyf. wg Ariensa).
MUTSCHLER-2009.indd 67 2010-01-07 22:11:02
68 Farmakodynamika
wagę ze stanu nieaktywnego w kierunku aktywnego do receptora, w przeciwieństwie natomiast do agoni-
(zob. ryc. A 3.1-1 C). Działanie takie decyduje o tym, stów nie są zdolni do wywołania efektu. Nie wykazu-
że częściowi agoniści mają dwojakie właściwości, tzn. ją aktywności wewnętrznej:
agonistyczne i antagonistyczne. Pomimo wysokich
stężeń pełnego agonisty, wywołujących silniejszy P + R º [PR] →
|| E
efekt niż wywołany przez określonego częściowego
agonistę, obecność tego ostatniego hamuje działanie Ponieważ agoniści i kompetycyjni antagoniści kon-
pełnych agonistów. Przy niskich stężeniach pełnego kurują o to samo miejsce rozpoznawcze (recognition
agonisty lub w razie jego braku, częściowy agonista site) – według prawa działania mas – zwiększenie stę-
wywołuje działanie aktywujące (agonistyczne). żenia jednego z nich wypiera drugą substancję z wią-
zania do receptora.
Na ryc. A 3.1-3 przedstawiono krzywe zależności stęże-
3.1.6.2. Antagoniści nie-efekt, przy 0 bez dodania kompetycyjnego antagonisty,
przy l i 2 z dodatkiem określonych ilości kompetycyjnego
Antagonistów można podzielić na następujące typy: antagonisty. Zanim agonista może wywołać efekt przy l i 2,
musi wyprzeć antagonistę z receptora, tzn. agonista musi
■ kompetycyjni (ryc. A 3.1-2, II), być podany w stężeniach wyższych niż w przypadku 0,
zanim pojawi się uchwytny efekt. Podobnie w celu osiąg-
■ niekompetycyjni (ryc. A 3.1-2, III), nięcia maksymalnego efektu konieczne są wyższe stężenia
agonistów.
■ funkcjonalni (ryc. A 3.1-6),
■ chemiczni (ryc. A 3.1-7). Istotną właściwością kompetycyjnych antagonistów
jest równoległe przesuwanie krzywej zależności
Antagoniści kompetycyjni. Substancje takie wiążą stężenia-efekt w prawo (zob. ryc. A 3.1-3). Stopień
się do receptorów w tym samym miejscu co agoniści. takiego równoległego przesunięcia krzywej agoni-
Kompetycyjni antagoniści wykazują powinowactwo stycznej na osi jest miarą powinowactwa antagonisty
EAB/Em EAB/Em
0 1 2
1,0 1,0 0
0,5 0,5
2
0
10-8 10-7 10-6 10-5 mol (A) 10-8 10-7 10-6 10-5 mol (A)
0: krzywa zależności stężenie-efekt A 0: krzywa zależności stężenie-efekt A
w przypadku nieobecności B. w przypadku nieobecności B’.
1 i 2: krzywa zależności stężenie-efekt A 1 i 2: krzywa zależności stężenie-efekt A
w przypadku obecności B; w przypadku 2 w przypadku obecności B’; w przypadku 2
przy trzy razy większym stężeniu B przy trzy razy większym stężeniu B’
niż w przypadku 1. niż w przypadku 1.
Ryc. A 3.1-3. Wpływ wzrastających stężeń kompetycyjnego an- Ryc. A 3.1-4. Wpływ wzrastających stężeń niekompetycyjne-
tagonisty (B) na krzywą stężenie-efekt agonisty (A) (wg Ariensa). go antagonisty (B’) na krzywą stężenie-efekt agonisty (A) (wg
Na osi rzędnych przedstawiono efekt EAB wywoływany przez A + Ariensa). Na osi rzędnych przedstawiono efekt EAB – wywoływa-
B w stosunku do maksymalnego efektu Em, a na osi odciętych ny przez A + B’ w stosunku do maksymalnego efektu Em, a na osi
– molarne stężenie A. odciętych – molarne stężenie A.
MUTSCHLER-2009.indd 68 2010-01-07 22:11:03
Farmakodynamika 69
w ten sposób, że nachylenie krzywej i maksymalny
EAB/Em efekt ulegają zmniejszeniu. Przy wyższych stężeniach
1,0 0
niekompetycyjnych antagonistów efekt agonisty zo-
1 staje całkowicie zablokowany. Mimo że receptory
mogą być całkowicie wysycone agonistą, efekt anta-
gonisty niekompetycyjnego nie zostaje przezwyciężo-
ny nawet przy wysokich stężeniach agonisty – w prze-
ciwieństwie do hamowania kompetycyjnego. W tym
2
więc przypadku prawo działania mas nie obowiązuje.
0,5
Typowym niekompetycyjnym antagonistą jest me-
mantyna.
Antagoniści mieszani kompetycyjni-niekompety-
cyjni. Poza antagonistami kompetycyjnymi i niekom-
petycyjnymi znane są także takie substancje, które
Farmakodynamika
10-8 10-7 10-6 10-5 M (A) w niskich stężeniach mają kompetycyjne, a w wyż-
szych niekompetycyjne działanie antagonistyczne
0: krzywa zależności stężenie-efekt A (zob. ryc. A 3.1-5). Oznacza to, że przy niskich stę-
w przypadku nieobecności C. żeniach krzywa stężenie-efekt agonisty ulega prze-
sunięciu w prawo, natomiast w stężeniach wyższych
1 i 2: krzywa zależności stężenie-efekt A
w przypadku obecności wzrastających stężeń C. nachylenia krzywej i maksymalny efekt ulegają osła-
bieniu.
Przykładem mogą tu być neurotropowo-miotropo-
Ryc. A 3.1-5. Wpływ wzrastających stężeń mieszanego kompe- wo działające spazmolityki. A3
tycyjno-niekompetycyjnego antagonisty (C) na krzywą stężenie-
-efekt agonisty (A) (wg Ariensa). Antagoniści funkcjonalni i fizjologiczni. Antago-
Na osi rzędnych przedstawiono efekt EAc wywoływany przez A + C nistami funkcjonalnymi nazywa się substancje hamu-
w stosunku do maksymalnego efektu Em, a na osi odciętych jące efekt innego agonisty poprzez działanie agoni-
– molarne stężenie A.
styczne na odmienne receptory. Przykładem może
być antagonistyczne działanie między substancjami
cholinergicznymi i histaminergicznymi a β-adrener-
do receptora: przy takich samych stężeniach silnie gicznymi na mięśnie oskrzeli (zob. ryc. A 3.1-6).
działający antagoniści o wysokim powinowactwie
wywołują znaczne przesunięcie równoległe, a słabo Antagoniści chemiczni. Przez pojęcie antagonisty
działające substancje – tylko nieznaczne. chemicznego rozumie się substancję, która reaguje
Typowym przykładem kompetycyjnych antago- chemicznie z substancją czynną i powoduje jej in-
nistów są blokery α- i β-adrenoceptorów, substancje aktywację w sposób niezależny od działania recep-
H1- i H2-antyhistaminowe lub antyandrogeny. torowego (zob. ryc. A 3.1-7). Ten typ antagonizmu
Antagoniści niekompetycyjni. Jak wynika z ryc.
A 3.1-2, niekompetycyjni antagoniści osłabiają dzia-
Antagonizm
łanie agonisty w różny sposób. Antagonista może funkcjonalny
nie działać na to samo białko receptorowe co agoni- (fizjologiczny)
sta, lecz oddziałuje w innym rejonie, tzw. działanie
allosteryczne (zob. ryc. A 3.1-2, III, góra). Działanie
hamujące wynika z tego, że warunki wiązania agoni-
agonista
sty do miejsca rozpoznawczego zmieniły się w sposób (A)
niekorzystny. Kolejnym typem jest hamowanie nie- efekt
kompetycyjne poprzez osłabianie efektów działania
agonisty już po jego połączeniu z receptorem (zob. agonista
ryc. A 3.1-2, III, dół). We wszystkich przypadkach (B)
krzywa zależności stężenie-efekt agonisty zmienia się
następująco (zob. ryc. A 3.1-4): efekty agonisty ule-
gają osłabieniu w zależności od stężenia antagonisty Ryc. A 3.1-6. Antagonizm funkcjonalny (fizjologiczny).
MUTSCHLER-2009.indd 69 2010-01-07 22:11:03
70 Farmakodynamika
3.1.7. Receptory
wewnątrzkomórkowe i błonowe
agonista brak
(np. heparyna) efektu
Badania nad lokalizacją receptorów wykazały istnie-
nie receptorów zarówno błonowych, jak i wewnątrz-
+ komórkowych.
antagonizm chemiczny
(np. protamina)
3.1.7.1. Receptory wewnątrzkomórkowe
nieaktywny produkt reakcji Do receptorów wewnątrzkomórkowych, będących
(kompleks heparyna/protamina) czynnikami transkrypcyjnymi, należą receptory:
■ hormonów steroidowych (glukokortykosteroidów,
Ryc. A 3.1-7. Antagonizm chemiczny.
mineralokortykosteroidów, androgenów, estroge-
nu, progesteronu, hormonu witaminy D),
ma znaczenie przede wszystkim przy leczeniu prze-
■ kwasu retinowego,
dawkowania leków i w przypadku zatruć (np. hamo-
wanie działania heparyny przez siarczan protaminy; ■ hormonów tarczycy.
zapobieganie zatruciu chlorkiem baru przez podanie
siarczanu sodu; detoksyfikujące działanie różnych Poza tym do tej grupy receptorów zalicza się także
substancji chelatujących przy zatruciu metalami receptory aktywowane przez proliferatory peroksyso-
ciężkimi). Istotą działania antagonizmu chemicznego mów (PPAR), będące cząsteczkami docelowymi dla
jest obniżanie stężenia substancji czynnej w biofazie. fibratów i glitazonów.
domena
dimeryzująca
domena LBD domena LBD
przyłączająca L przyłączająca L
kofaktory kofaktory
DNA- DNA-
BD BD
DNA
AC T T CCGGT AC T T CCGGT
TGAA GGCCA TGAA GGCCA
element
odpowiedzi
na DNA
Ryc. A 3.1-8. Schemat wewnątrzkomórkowego dimeru receptorowego z końcem aminowym (zielony), domeną wiążącą DNA (po-
marańczowy) i końcem karboksylowym (żółty). Poszczególne sekwencje aminokwasowe w domenach wiążących DNA i ligand są od-
powiedzialne za translokację. DNA-BD – domena wiążąca DNA; L – ligand; LBD – domena wiążąca ligand.
MUTSCHLER-2009.indd 70 2010-01-07 22:11:03
Farmakodynamika 71
Receptory wewnątrzkomórkowe występują w cyto- hormon-receptor wiąże się z czynnikiem transkryp-
plazmie (np. receptory dla glukokortykosteroidów) cyjnym, blokuje jego wiązanie do swojej promoto-
i w jądrze komórkowym (np. receptory dla androge- rowej sekwencji DNA. W wyniku tego nie dochodzi
nów i estrogenów). W cytoplazmie są one powiązane do odpowiednich transkrypcji, tzn. hormon hamuje
z białkami szoku cieplnego (hit shock protein – HSP) w tym przypadku ekspresję genów.
odpowiedzialnymi za ich odpowiednie ułożenie (skła-
danie). W przypadku tych ostatnich można wyróżnić Opisana regulacja genów przez receptory wewnątrzkomór-
trzy regiony (zob. ryc. A 3.1-8): kowe może być wyjaśniona na przykładzie glukokortyko-
steroidów. Ich przeciwzapalne działanie wynika przynaj-
■ domenę wiążącą DNA, odpowiedzialną za wiązanie mniej częściowo z hamowania ekspresji genów prozapal-
specyficznych sekwencji nukleotydowych w rejo- nych, takich jak interleukina-2 i cyklooksygenaza-2.
nie promotora DNA,
■ domenę efektorową (tzw. domenę transaktywującą),
w rejonie N-końca, regulującą ekspresję genów, 3.1.7.2. Receptory błonowe
■ domenę wiążącą ligand, w rejonie C-końca.
Farmakodynamika
Receptory błonowe można podzielić na:
Poza tymi trzema najważniejszymi domenami we- ■ receptory związane z białkami G,
wnątrzkomórkowe białka receptorowe posiadają
■ receptory jonotropowe (kanały jonowe zależne
jeszcze dwa ważne rejony: domenę odpowiedzialną
od napięcia i ligandów),
za translokację białka do jądra komórkowego oraz
domenę odpowiedzialną za dimeryzację, za pomocą ■ receptorowe kinazy białkowe (receptory związane
której dwa białka receptorowe asocjują, tworząc ho- z enzymami).
loproteinę, i dzięki temu nabywają zdolności wiąza- A3
nia sekwencji rozpoznawczych DNA.
3.1.7.2.1. Receptory związane z białkiem G
Poszczególne receptory odróżniają się zarówno pod
względem funkcji, jak i sekwencji białek. Największe
Receptory związane z białkiem G (GPCRs) stanowią
podobieństwo (homologia strukturalna) występu-
nie tylko największą grupę w obrębie rodziny recep-
je w rejonie domeny wiążącej DNA, najmniejsze –
torów błonowych w ludzkim genomie, ale także grupę
w domenie transaktywującej, odpowiedzialnej za ak-
o największej różnorodności. Przekazują do wnętrza
tywację transkrypcji genów.
komórki informacje o różnych bodźcach zewnątrzko-
Znaczne różnice domen transaktywujących umożliwiają mórkowych.
uzyskanie selektywnych przeciwciał dla różnych recepto- Określenie receptory związane z białkiem G odnosi
rów wewnątrzkomórkowych, dlatego ten region był nazy-
wany domeną immunogenną, zanim poznano jego funkcje. się do receptorów związanych z białkiem wykazują-
cym interakcje z nukleotydem guaninowym. Do tej
Ze względu na właściwości lipofilne, steroidy i reti- grupy należą bardzo istotne dla farmakoterapii recep-
noidy przechodzą przez błonę drogą dyfuzji prostej, tory wielu neuroprzekaźników, m.in.:
natomiast hormony tarczycy dzięki dyfuzji ułatwionej.
■ adenozyny,
Transdukcja sygnału. Efekty stymulacji receptorów ■ adrenergiczne,
wewnątrzkomórkowych (zob. ryc. A 3.1-9) wynika-
■ ATP-(P2Y),
ją początkowo z tego, że kompleks ligand-receptor
powstaje w wyniku wiązania się liganda do końca ■ dopaminowe,
C-karboksylowego. Następnie oddysocjowuje biał-
■ GABAB,
ko szoku cieplnego i domena wiążąca ligand może
się połączyć poprzez tzw. palce cynkowe (zinkfinger) ■ metabotropowe glutaminergiczne,
do DNA. Cztery cysteiny tworzące palcową strukturę
■ histaminowe,
poprzez tworzenie kompleksu z cynkiem są odpo-
wiedzialne za wiązanie się do specyficznej sekwencji ■ muskarynowe (M-cholinoreceptory),
DNA, tzw. element rozpoznawczy (hormon responsi-
■ opiatowe,
ve element – HRE) (np. dla estrogenu – ERE; glu-
kokortykosteroidu – GRE), i w konsekwencji za ak- ■ serotoninergiczne (z wyjątkiem receptorów 5-HT3,
tywację określonych genów. Jeżeli jednak kompleks zob. poniżej).
MUTSCHLER-2009.indd 71 2010-01-07 22:11:04
72 Farmakodynamika
ligand
HSP 90 HSP 90 2 HSP 90
hormon-
-receptor
czynnik
kompleks ligand- transkrypcyjny
-hormon-receptor
jądro komórkowe
transkrypcja
brak
transkrypcja wiązania
mRNA
mRNA brak transkrypcji
białko
rybosom rybosom
odpowiedź komórkowa
Ryc. A 3.1-9. Transdukcja sygnału receptorów wewnątrzkomórkowych. HSP – białko szoku cieplnego (szczegóły w tekście).
Bardzo dużo receptorów dla hormonów i mediatorów cyklazę (Gs), hamujące cyklazę (Gi) lub aktywujące
(np. hormonu antydiuretycznego, glukagonu, somato- fosfolipazę (Gq). We wszystkich przypadkach za in-
statyny, prostaglandyn) również należy do tej grupy. terakcję receptora z białkiem G jest odpowiedzialna
Na ryc. A 3.1-10 wskazano, że receptory związa- trzecia pętla wewnątrzkomórkowa. Decyduje ona też
ne z białkiem G posiadają siedem helikalnych domen o tym, z jakim typem białka G zachodzi interakcja.
przechodzących przez błonę komórkową (I-VII),
a także trzy pętle zewnątrzkomórkowe (ZP1–ZP3) Białka G tworzą rodzinę białek heterotrimerów, które
i wewnątrzkomórkowe (WP1–WP3). Z tego powodu składają się z podjednostki α oraz podjednostki β, γ.
są one również nazywane receptorami siedmioheli- Podjednostka α zawiera miejsce wiązania nukleoty-
kalnymi. du guaninowego (guanozynobifosforan – GDP lub
trifosforan – GTP), hydrofobowe podjednostki β, γ
Transdukcja sygnału. W przypadku receptorów łączą białko G z błoną komórkową. W stanie nie-
związanych z białkiem G związanie liganda z re- aktywowanym GDP jest związany z podjednostką
ceptorem powoduje zmianę konformacji białka re- α, a cały kompleks nie jest połączony z receptorami
ceptora, która za pośrednictwem białka G (białka błonowymi. W wyniku stymulacji odpowiednich re-
wiążącego nukleotyd guaninowy) wywołuje kaskadę ceptorów błonowych początkowo białko G łączy się
dalszych reakcji. Przy tym białko G może wpływać z tym receptorem i GDP zostaje zastąpiony przez
bezpośrednio na kanały jonowe lub aktywuje albo też GTP. Następnie oddzielają się od siebie podjednostki
hamuje powstawanie przekaźnika drugiego rzędu po- α i β, γ, a podjednostka α zawierająca GTP aktywuje
przez interakcję z enzymem (ryc. A 3.1-11) i wywołuje (w przypadku białka Gs) lub blokuje (w przypadku
dalsze reakcje. Ze względu na takie wielorakie funk- białka Gi) białko docelowe. Podobnie podjednostki
cje istnieje wiele różnych białek G, np. stymulujące β, γ mogą aktywować białka efektorowe (np. fosfo-
MUTSCHLER-2009.indd 72 2010-01-07 22:11:04
Farmakodynamika 73
przestrzeń
H2N zewnątrzkomórkowa
ligand
ZP1 ZP2 ZP3
NH2
błona komórkowa
błona komórkowa
I II III IV V VI VII
WP1 WP2 HOOC
I–VII domeny
transbłonowe COOH
Farmakodynamika
WP3
przestrzeń
wewnątrzkomórkowa
Ryc. A 3.1-10. Schemat receptora o siedmiu domenach przechodzących przez błonę komórkową połączonego z białkiem G.
ZP – pętla zewnątrzkomórkowa; WP – pętla wewnątrzkomórkowa
A3
lipazę C). Reakcja zostaje przerwana w momencie, Wtórne przekaźniki, powstające w wyniku reakcji
gdy podjednostka α po przyłączeniu GTP uzysku- enzymatycznych, np. cAMP, IP3 i DAG, wywołują
je właściwości GTP-azy i w wyniku tego dochodzi kolejne reakcje, m.in. aktywację kinaz białkowych,
do rozszczepienia GTP na GDP i nieorganiczną resztę a tym samym fosforylację białek i uwolnienie jonów
fosforanową (zob. ryc. A 3.1-11). Ten proces hydro- wapnia.
lizy jest przyspieszany przez tzw. białko GAP (GTP-
ase activating protein), jeden z enzymów należących Uruchomienie wtórnego przekaźnika umożliwia efek-
do rodziny białek RGS (regulators of G-protein sig- tywne wzmocnienie sygnału. Poza tym różne komór-
naling). Po rozszczepieniu GTP cały układ wraca ki docelowe, ze względu na wiązanie z receptorami
do początkowego stanu spoczynku. i enzymami zależnymi od wtórnych przekaźników,
różnie reagują na hormon albo przekaźnik.
Do enzymów podlegających regulacji przez białka G Dwa najpopularniejsze i najdłużej znane wtórne prze-
należą: kaźniki to cAMP oraz inozytolotrifosforan (IP3).
cAMP jest aktywatorem kinazy białkowej A (PKA), któ-
■ cyklaza adenylanowa, której stymulacja powoduje ra wykazuje liczne działania w procesie przemiany materii
tworzenie cyklicznego nukleotydu 3′,5′-adenozyno- i ekspresji genów.
monofosforanu (cAMP), IP3 działa poprzez aktywację kanałów receptorowych
IP3 w retikulum endoplazmatycznym, uwalniając Ca2+
■ fosfolipaza C, która poprzez rozszczepienie fosfa- z wewnątrzkomórkowych magazynów wapniowych, di-
tydyloinozytolo-(4,5)-bisfosforanu wytwarza dwa acyloglicerol jest aktywatorem zależnej od Ca2+ kinazy
przekaźniki drugiego rzędu inozytolo-1,4,5-trifo- białkowej C (PKC).
sforan (IP3) i l,2-diacyloglicerol (DAG), Kolejnymi przedstawicielami grupy wtórnych przekaź-
ników są cGMP oraz dwa gazy – NO i CO.
■ fosfodiesteraza VI, która rozszczepia cGMP, zaan-
gażowana w proces widzenia,
3.1.7.2.2. Receptory jonotropowe
■ kinaza 3-fosfatydyloinozytolu (kinaza PI3), która
(receptory związane
reguluje liczne funkcje błonowe za pośrednictwem
z kanałami jonowymi)
kinazy białkowej B (PKB) oraz
■ kanały potasowe i neuronalne kanały wapniowe, Kanały jonowe, w których powstawanie zaangażowa-
których aktywność regulują podjednostki β i γ. nych jest ponad 150 genów, należą do z największych
MUTSCHLER-2009.indd 73 2010-01-07 22:11:05
74 Farmakodynamika
A agonista
A
receptor
białko we γ e białko
efektoro Gα fektoro
we kanały jonowe,
GTP β kinazy PI3,
fosfolipazy,
cyklazy adenylanowe,
kinazy receptorowe,
kinazy MAP
GTP GTP GTP GTP
Gαi Gαs Gαq Gα12,13
kanały jonowe, cyklazy fosfolipazy Rho
cyklazy adenylanowe, adenylanowe (cytoszkielet)
fosfolipazy
A
B 1
receptor
γ
α
łko e β
bia torow GDP
fe ek
γ
4 2
β
receptor
GTP
α
+ P
A
GDP
receptor
GDP
A
α
łko we
β
biaktoro
efe
β GTP
bia o α
efektołk
rowe γ białko we
receptor efektoro
3 A
Ryc. A 3.1-11. A – różnorodność dróg transdukcji sygnału szlakiem receptorów związanych z białkiem G. Aktywacja receptora zwią-
zanego z białkiem G przez agonistę powoduje dysocjację heterotrimerycznego białka G do podjednostek Gα i Gβ po wymianie
związanego do podjednostki α GDP na GTP. Receptory związane z białkiem G asocjują najczęściej z określoną podjednostką Gα (z
których 4 najważniejsze przedstawiono na rycinie). Poszczególne podjednostki Gα aktywują lub hamują różne systemy efektorowe,
m.in. kanały jonowe, fosfolipazy, cyklazy adenylanowe i białka wiążące GTP, np. Rho, które reguluje strukturę cytoszkieletu. Kinazy
PI3 – kinazy 3-fosfatydyloinozytolowe; Pi – fosforan nieorganiczny.
B – cykl aktywacji i dezaktywacji heterotrimerycznego białka G. Po związaniu agonistów do receptora 7-transbłonowego (1) dzię-
ki wymianie GDP na GTP (2) dochodzi do dysocjacji białka G (3), co powoduje aktywację białek efektorowych, które pośredniczą
w wysyłaniu odpowiedzi komórkowej (wazokonstrykcja, sekrecja, namnażanie komórek). Dzięki wewnętrznej aktywności GTPazy
podjednostka Gα powoduje rozszczepienie GTP na GDP (4). Białko G asocjuje z powrotem do heterotrimeru, agonista dyfunduje
od receptora i przechodzi ponownie do stanu nieaktywnego.
MUTSCHLER-2009.indd 74 2010-01-07 22:11:05
Farmakodynamika 75
rodzin białek sygnałowych. Pod względem liczebności Siłą powodującą ruch jonów przez kanał jonowy
znajdują się na trzecim miejscu po receptorach związa- (do wnętrza komórki lub na zewnątrz) jest gradient
nych z białkiem G i kinazach białkowych. Odgrywają stężeń między wnętrzem komórki i przestrzenią ze-
one ważną rolę w wielu procesach biologicznych, wnątrzkomórkową. Skala przepływu jonów zależy
np. przy powstawaniu potencjałów czynnościowych, od liczby otwartych kanałów, czasu ich otwarcia
skurczów mięśni serca, szkieletowych i gładkich, i przepuszczalności dla danych jonów (tzw. prze-
transportu nabłonkowego, aktywacji komórek T albo wodność).
sekrecji insuliny. U ssaków ich geny są silnie konser- Jeżeli kanały są otwierane lub zamykane pod
wowane. Z drugiej strony mutacje tych genów odpo- wpływem ligandów, mówi się o kanałach zależnych
wiadają za liczne choroby, takie jak zespół długiego od ligandów (receptory jonotropowe). Jeżeli nato-
QT, mukowiscydoza, migrena, kongenitalna hiperin- miast do otwierania i zamykania kanałów dochodzi
sulinemia albo określone formy padaczki. w wyniku zmian potencjału błony komórkowej (hi-
Kanały jonowe są integralnymi, złożonymi z kil- perpolaryzacja lub hipopolaryzacja), mówi się o ka-
ku podjednostek, białkami błonowymi, tworzącymi nałach zależnych od potencjału (voltage dependent
kanały porowe, które mogą być otwierane lub zamy- ion channels, voltage operated ion channels).
Farmakodynamika
kane przez zmiany konformacyjne. Ze względu na ła-
twy dostęp do nich od strony zewnątrzkomórkowej
Wśród receptorów jonotropowych można wymienić
stanowią one preferowaną strukturę docelową dla
m.in. receptory:
leków. Podjednostka tworząca pory, która w najwęż-
szym miejscu posiada jedynie 1–2-krotność średnicy ■ ATP (P2X),
jonu, nazywana jest jednostką α, podczas gdy pod-
■ GABAA,
jednostki pomocnicze to β, γ itd. Z powodu różnej
budowy przestrzennej i lokalizacji ładunków elek- ■ jonotropowe receptory glutaminergiczne,
trycznych otwarte receptory jonotropowe umożliwia- A3
■ glicynowe,
ją przepływ tylko określonych jonów. Odpowiednio
więc do rodzaju jonów selektywnie przechodzących ■ serotoninergiczne 5-HT3,
przez dany kanał odróżnia się kanały sodowe, potaso-
■ nikotynowe (N-cholinoreceptory)
we, wapniowe i chlorowe.
■ K+ (regulowane przez ATP, aktywowane przez Ca2+/
kalmodulinę, regulowane przez białko Gi „GIRK”).
receptorowe kanały jonowe,
np. nikotynowy receptor acetylocholiny Interakcja ligand-receptor prowadzi w kanałach jo-
nowych regulowanych ligandem do podwyższenia
albo obniżenia prawdopodobieństwa otwarcia kanału
miejsce wiązania związana i w efekcie do nasilonej albo zmniejszonej wymiany
acetylocholiny acetylocholina
odpowiednich jonów. W ten sposób np. acetylocholi-
acetylocholina Na+
na lub nikotyna wiążą się do podjednostek α recepto-
ra nikotynowego, otwierając kanał i wywołując po-
wstawanie potencjału czynnościowego przez napływ
jonów sodu.
Na ryc. A 3.1-12 przedstawiono schematycznie
budowę receptorów nikotynowych (mięśniowych)
struktura konformacja konformacja jako przykład receptora jonotropowego. Składa się
pentamerowa zamknięta otwarta
on z dwóch podjednostek α i po jednej podjednostce
β, γ i δ, które tworzą razem kanał jonowy w błonie
Ryc. A 3.1-12. Struktura i sposób aktywacji nikotynowego re- komórkowej.
ceptora acetylocholiny. Ten kanał jonowy regulowany przez
Fizjologiczne ligandy działające na receptory jo-
ligandy jest pentamerem, który składa się np. z dwóch podjed-
notropowe nazywa się szybkimi neuroprzekaźnikami,
nostek α oraz jednej podjednostki β, γ, δ. Po związaniu dwóch
cząsteczek acetylocholiny do podjednostek α i spowodowanej ponieważ po połączeniu z receptorem ich aktywacja
tym zmiany konformacji dochodzi do otwarcia kanału. Napływ jest niemal natychmiastowa.
jonów Na+ zachodzący teraz zgodnie z gradientem stężeń zo-
staje ułatwiony przez ujemnie naładowane aminokwasy po we- Kanały jonowe regulowane napięciem. Kanały
wnętrznej stronie ujścia kanału. Preferowany napływ jonów Na+ jonowe bramkowane napięciem także mogą być re-
jest możliwy dzięki selektywnemu filtrowi w środku kanału. ceptorami dla leków, np. nifedypiny i werapamilu
MUTSCHLER-2009.indd 75 2010-01-07 22:11:05
76 Farmakodynamika
jako blokerów kanału wapniowego oraz lidokainy przez co dochodzi do przejściowego, selektywnego
jako blokera kanału sodowego. W przeciwieństwie napływu jonów. Podczas gdy aktywacja kanałów
do kanałów jonowych regulowanych ligandem ot- wapniowych i sodowych prowadzi do pobudzenia,
wieranie i zamykanie kanałów jonowych zależnych otwarcie kanałów potasowych i chlorkowych po-
od napięcia, jak już wspomniano, zachodzi przez woduje hiperpolaryzację błony komórkowej, przez
zmianę potencjału błonowego. Kanały jonowe co zmniejsza się pobudliwość i spada prawdopodo-
bramkowane napięciem znajdują się w komórkach bieństwo otwarcia się kanałów sodowych i wapnio-
pobudliwych, np. w neuronach i mięśniu sercowym, wych. Po szybkiej początkowej aktywacji kanałów
i mają zasadnicze znaczenie dla powstawania, prze- jonowych następuje powolniejsza faza inaktywacji,
kazywania i zakończenia pobudzenia. W większości która najczęściej kończy się jeszcze podczas fazy
przypadków są one otwierane przez depolaryzację, depolaryzacji.
A Kanały jonowe regulowane napięciem (np. kanał Na+)
I II III IV
por por por por przestrzeń
zewnątrzkomórkowa
+ + + +
+ + + +
przestrzeń
H2N P P wewnątrzkomórkowa
P P COOH
B Stany kanału jonowego regulowanego napięciem (np. kanał Na+)
Na+
zamknięty
kanał Na+
repolaryzacja depolaryzacja błony
lub hiperpolaryzacja
błony
nieaktywny otwarty
kanał Na+ kanał Na+
Na+
Na+
Ryc. A 3.1-13. A – struktura kanału Na+ regulowanego napięciem. Cząsteczka białkowa składa się z około 2000 aminokwasów po-
dzielonych na 4 powtarzalne domeny, z których każda zawiera 6 segmentów transbłonowych. Czwarty segment każdej z domen
posiada dużą zawartość dodatnio naładowanych aminokwasów, takich jak arginina i lizyna. Te segmenty wpływają na zmianę kon-
formacji kanału Na+, kiedy powstanie potencjał czynnościowy w miejscu błony komórkowej, gdzie znajduje się kanał Na+. Pętle
znajdujące się między segmentami transbłonowymi 5 i 6 stanowią wyściełającą część poru znajdującą się w błonie komórkowej.
Odcinki białka kanałowego leżące po stronie cytozolowej mogą ulegać fosforylacji przez wewnątrzkomórkowe kinazy białkowe,
co umożliwia regulację aktywności kanału.
B – rozróżnia się trzy podstawowe stany kanałów Na+ zależnych od napięcia. Po pojawieniu się potencjału czynnościowego kanał
Na+ otwiera się, przechodząc ze stanu spoczynkowego („zamkniętego”), jednak ulega inaktywacji po kilku milisekundach. Dopiero
po repolaryzacji błony komórkowej przez aktywację kanałów K+ zachodzi zmiana konformacji, która ponownie przeprowadza białko
kanałowe do aktywowalnego stanu wyjściowego („zamkniętego”).
MUTSCHLER-2009.indd 76 2010-01-07 22:11:06
Farmakodynamika 77
Poza kanałami otwieranymi przez depolaryzację ■ kanał Ca2+ (typu: L, N, T, P/Q),
istnieją także kanały jonowe bramkowane napię-
■ kanał K+ (Kv-, hERG, KCNQ, Kir).
ciem, które ulegają aktywacji przez hiperpolaryza-
cję błony komórkowej. Spełniają one ważną funkcję
Znaczenie tych kanałów można pokazać na przykła-
w komórkach wykazujących rytmiczną aktywność,
dzie komórki mięśnia sercowego. Napływ jonów Na+
np. w węzłach zatokowo-przedsionkowych i w nie-
do komórki mięśnia sercowego umożliwia szybką
których neuronach.
depolaryzację błony, która jest konieczna do otwarcia
kanału Ca2+ typu L bramkowanego napięciem. Jony
Przykłady kanałów jonowych bramkowanych napię-
wapnia napływające dzięki temu do komórki prowa-
ciem to:
dzą do uwalniania Ca2+ z retikulum sarkoplazmatycz-
■ kanał Na+, nego i umożliwiają inicjację skurczu kardiomiocytów.
Farmakodynamika
Receptor z aktywnością kinazy tyrozynowej
A C
EGF EGF EGF
EGF
kotwica błonowa Ras
A3
P P
P P GRB RAS
SOS RAF
aktywny
nie- ny MAPKK
P
w
akty MAPKK
P Thr
MAPK
MAPK
P Tyr
fosforylacja czynnika
transkrypcyjnego wzrost
różnicowanie
transkrypcja białko
ELK genów rozwój
P
jądro
komórkowe
Ryc. A 3.1-14. Transdukcja sygnału idącego od nabłonkowego czynnika wzrostu (EGF) za pośrednictwem receptora EGF. A – po sty-
mulacji receptora dochodzi (B) do dimeryzacji i autofosforylacji reszt tyrozynowych na cytozolowych domenach receptora. Zachodzi
rekrutacja białek adaptorowych, takich jak GRB i SOS, oraz wiązanie (C) do fosforylowanej reszty tyrozynowej receptora EGF.
Aktywowane białko SOS stymuluje małe białko RAS wiążące GTP, które z kolei aktywuje kinazę serynowo-treoninową RAF. Stymuluje
to z kolei kinazę kinazy białkowej aktywowanej przez mitogeny (MAPKK), która z kolei fosforyluje kinazę MAP na resztach tyrozy-
nowych i treoninowych. Zaktywowana MAPK ulega translokacji do jądra komórkowego i fosforyluje różne czynniki transkrypcyjne
(ELK i. in.). Umożliwia to transkrypcję genów stymulowanych czynnikami wzrostu. Ich translacja do białek prowadzi do proliferacji
komórki.
MUTSCHLER-2009.indd 77 2010-01-07 22:11:06
78 Farmakodynamika
Receptor z przyłączoną kinazą tyrozynową
receptor
A erytropoetyny
erytropoetyna
JAK P P JAK Tyr
r STAT
-Ty Tyr
JAK
-
Tyr ADP Tyr P
ATP STAT
ATP Tyr P
JAK
proliferacja
ADP regulacja i różnicowanie
transkrypcji genów komórek macierzystych
jądro erytrocytów
komórkowe
Ryc. A 3.1-15. Schemat receptora ze zasocjowaną kinazą tyrozynową (np. receptora erytropoetyny). A – po aktywacji receptora
przez erytropoetynę jest on fosforylowany na odcinkach cytozolowych przez kinazę tyrozynową JAK. B – JAK wiąże się do fosfo-
rylowanych domen receptora i może fosforylować białka sygnałowe, np. STAT. Aktywowane białka STAT penetrują błonę jądrową
i regulują transkrypcję genów, co w tym przypadku umożliwia proliferację i różnicowanie erytrocytów z komórek macierzystych.
Kanały K+ aktywowane depolaryzacją repolaryzują 3.1.7.2.3. Receptory powiązane
błonę komórkową i przez zmianę konformacji umoż- z enzymami
liwiają przeprowadzenie wcześniej nieaktywnych ka- (receptory metabotropowe)
nałów Na+ i Ca2+ w stan aktywowalny, dzięki czemu
możliwe jest znowu ich pobudzenie. Do tej grupy receptorów należą:
Przechodzenie jonów sodu przez kanały sodowe zależne ■ receptory wykazujące aktywność kinazy tyrozyno-
od napięcia można selektywnie zatrzymać już za pomo- wej,
cą 10–9–10–8 mol/l tetrodotoksyny. Jest ona produkowana
przez bakterie, m.in. Pseudomonas, i poprzez łańcuch po- ■ receptory związane z kinazą tyrozynową,
karmowy lub od symbiontów trafia do różnych narządów
organizmu, gromadząc się przede wszystkim w jajnikach ■ receptory wykazujące aktywność cyklazy guanyla-
i wątrobie licznych zwierząt, np. japońskiej ryby fugu nowej,
i kilku innych gatunków ryb z rodziny rozdymkowych
oraz kosterowatych, australijskiej mątwy oraz różnych ■ receptory związane z kinazą serynowo-treoninową,
gatunków żab.
■ receptory śmierci, które wywołują programowaną
śmierć komórki (apoptozę).
Transdukcja sygnału. W przypadku receptorów jono-
tropowych interakcja liganda z receptorem prowadzi
do zwiększenia lub zmniejszenia prawdopodobieństwa Receptory wykazujące aktywność kinazy tyrozy-
otwierania kanału, a w konsekwencji do nasilonej lub nowej (receptory kinazy tyrozynowej) charakteryzu-
osłabionej wymiany odpowiednich jonów. W ten spo- ją się tym, że posiadają tylko zewnątrzkomórkową
sób wiąże się np. acetylocholina lub nikotyna do pod- domenę wiążącą ligand, a części białka znajdujące
jednostki α receptorów nikotynowych, otwiera kanał się w cytozolu są domeną o właściwościach kinazy
i wywołuje potencjał czynnościowy poprzez napływ tyrozynowej. Białko to pełni więc zarówno funkcję
jonów sodowych do wnętrza komórki. receptora, jak i enzymu.
MUTSCHLER-2009.indd 78 2010-01-07 22:11:07
Farmakodynamika 79
ANP monotlenek
azotu
GCA
sGC
cGMP
GTP GTP
-
fosfo za 5
te ra IRAG
dies
Ca2+
GMP
PKG
receptor IP3
Farmakodynamika
P AG L
fosfataza IR
miozynowa
PKG
kanał K+ Ca2+
Ca2+ miozyna miofosfata ER
zyn za P
ow
P a
P
aktyna
PKG
P
A3
K+ Relaksacja komórek
mięśni gładkich
Ryc. A 3.1-16. Cyklaza guanylanowa (GCA) związana z błoną jako receptor dla przedsionkowego peptydu natriuretycznego (ANP)
i rozpuszczalna cyklaza guanylanowa (sGC) jako receptor dla NO regulują cytozolowe stężenie cGMP. Aktywacja kinazy białkowej G
(PKG) przez cGMP prowadzi trzema głównymi szlakami do relaksacji mięśni gładkich, np. naczyń: fosforylacja IRAG (substratu PKG
zasocjowanego z IP3) hamuje uwalnianie Ca2+ z retikulum endoplazmatycznego (ER), aktywacja fosfatazy miozynowej przez PKG po-
woduje defosforylację główek miozynowych i stymulację kanałów K+ aktywowanych Ca2+, przez co następuje repolaryzacja komórki
i obniża się prawdopodobieństwo otwarcia kanałów Ca2+ zależnych od napięcia.
W dalszą transdukcję sygnału zaangażowane są kinazy biał- stowych (np. czynnik wzrostowy epidermalny
kowe aktywowane mitogenem (MAP-kinazy). Ze względu lub fibroblastów). Receptory dla insuliny i IGF-1
na to, że regulują one wiele procesów komórkowych, takich są bardzo podobne: składają się z dwóch podjedno-
jak ekspresja genów, mitoza, różnicowanie oraz apoptoza,
mają ogromne znaczenie dla całego organizmu. Na przy- stek α i dwóch β powiązanych między sobą most-
kład ich działanie stymulujące proliferację jest decydujące kami siarczkowymi. W przeciwieństwie do nich
w transdukcji sygnału większości onkogenów. receptory czynników wzrostowych są pojedynczą
MAP-kinazy dzielą się na 4 grupy: kinazy regulowane cząsteczką białka (monomerem), ale po przyłą-
przez sygnał zewnątrzkomórkowy (ERK), kinazy JNK (kinaza czeniu liganda dochodzi do tworzenia dimerów,
fosforylująca N-terminalną część białka Jun), kinazy p38 oraz
szczególna forma ERK – ERK5. Kaskada sygnałowa ERK a następnie, tak jak w przypadku receptorów in-
jest stymulowana głównie przez czynniki wzrostu. JNK i ki- sulinowych oraz IGF-1, do autofosforylacji reszt
nazy p38 są aktywne w obecności czynników stresogennych, tyrozynowych w cytozolowym odcinku receptora.
np. uwalniania cytokin, promieniowania UV, szoku ciepl- Powoduje to tworzenie się miejsc przebywania bia-
nego lub osmotycznego. ERK5 jest natomiast aktywowana łek sygnałowych, które wiążą się z fosforylowany-
zarówno przez czynniki wzrostu, jak i czynniki stresogenne.
mi resztami tyrozynowymi receptora. W ten sposób
receptorowe kinazy tyrozynowe zostają włączone
Do tej grupy należy receptor dla insuliny, IGF-1, do kaskady sygnałowej Ras, która kieruje wzrostem
a także receptory dla różnych czynników wzro- komórki i proliferacją.
MUTSCHLER-2009.indd 79 2010-01-07 22:11:08
80 Farmakodynamika
Receptor związany z kinazą serynowo-treoninową
A TGF-β
R-I R-II
-β R-II
TGF
R-I P
P
ATP
P
ATP ADP P Smad2 Smad4
P
ADP
białko Smad2 P
P
proliferacja P
białko
różnicowanie
apoptoza transkrypcja
jądro
komórkowe geny regulowane przez TGF-β
Ryc. A 3.1-17. Model kompleksu receptorowego TGF-β regulowanego ligandem. Receptory z tej rodziny odgrywają ważną rolę we
wzroście i różnicowaniu komórek. A – TGF-β-zależna aktywacja receptora TGF-β I (R-I) dzięki fosforylacji przez receptor TGF-β II (R-II)
prowadzi do szybkiej fosforylacji Smad2. Nazwa białko Smad wywodzi się od genów kodujących, które po raz pierwszy zidentyfiko-
wano w badaniach genetycznych nad Drosophila i C. elegans jako sma (small body size). Połączenie obu nazw prowadzi do powstania
nazwy Smad. B – fosforylacja Smad2 po heterooligodimeryzacji ze Smad4 powoduje translokację kompleksu do jądra komórkowe-
go, gdzie, po powstaniu interakcji z kolejnymi czynnikami jądrowymi, działa ono jako czynnik transkrypcyjny.
Receptory związane z kinazą tyrozynową są biał- tory te są monomerycznym białkiem i posiadają
kami monomerycznymi z jedną domeną przechodzą- zewnątrzkomórkowe miejsce wiązania ligandów
cą przez błonę komórkową, które łączą się w dime- aktywujących i wewnątrzkomórkową domenę en-
ry po przyłączeniu się liganda. Receptory te jednak zymatyczną. Związanie liganda do receptora z ak-
nie posiadają własnej domeny kinazy tyrozynowej. tywnością cyklazy guanylanowej powoduje akty-
Należą do nich liczne receptory dla cytokin, a tak- wację domeny cyklazy guanylanowej. W efekcie
że receptory czynników wzrostowych, prolaktyny z guanozynotrifosforanu (GTP) powstaje cykliczny
i erytropoetyny. Po aktywacji i dimeryzacji przyłą- guanozynomonofosforan (cGMP), który jako prze-
czają się kinazy Janusowe (JAK) i fosforylują reszty kaźnik drugiego rzędu wywołuje kolejne reakcje,
tyrozynowe receptora. Efektem tego jest asocjacja np. zwiotczenie mięśni gładkich albo sekrecję chlor-
białek STAT (Signal Transducers and Activators of ku do światła jelita.
Transcription) z fosforylowanymi domenami recep-
torowymi. Zasocjowane białka STAT są następnie Ważnym przykładem receptorów związanych z ki-
dodatkowo fosforylowane przez kinazy JAK. Na ko- nazą serynowo-treoninową są receptory dla transfor-
niec fosforylowane białka STAT dimeryzują, ulegają mujących czynników wzrostowych β (transforming
translokacji do jądra komórkowego i tam aktywują growth factors β), które występują w dwóch typach,
specyficzne geny. TGFβR-I i TGFβR-II. Za ich pomocą rozwijają swo-
je działanie cytokina BMP2 (białko morfogenetyczne
kości 2) oraz aktywina i inhibina. Ogólnie istnieje
Do receptorów wykazujących aktywność cyklazy ponad 30 wielofunkcyjnych białek rodziny TGF-β,
guanylanowej (związanej z błoną komórkową) za- które indukują albo hamują proliferację i różnicowa-
licza się przede wszystkim receptory dla przedsion- nie komórek oraz regulują migrację i adhezję różnych
kowego peptydu natriuretycznego (ANP). Recep- rodzajów komórek.
MUTSCHLER-2009.indd 80 2010-01-07 22:11:08
Farmakodynamika 81
Dla receptorów transformujących czynników Receptory śmierci należące do rodziny receptorów
wzrostowych β obowiązuje następujący mecha- czynników nekrozy nowotworów (TNF) są zintegro-
nizm transdukcji sygnału: najpierw cytokina TGF-β wane z błoną większości komórek. Ważnymi przed-
wiąże się z TGFβ-R-II, następnie wiąże się stawicielami tej grupy są receptor TNF1 i CD95.
z TGFβ-R-I, tworząc heterodimer. Kolejnym kro- Wiązanie TNF do swojego receptora prowadzi do ho-
kiem jest transfosforylacja z TGFβ-R-II na TGFβ- motrimeryzacji i rekrutacji białka adaptorowego, któ-
R-I, co powoduje właściwe przekazanie sygnału. re asocjuje z tzw. domenami śmierci tych trzech pod-
Aktywowany kompleks receptorowy uruchamia jednostek. Powstały kompleks aktywuje kaskadę
ekspresję genów za pośrednictwem tzw. bia- kaspazową, która prowadzi do inaktywacji enzymów
łek Smad, które wędrują do jądra komórkowego i białek strukturalnych oraz do fragmentacji genomo-
w swojej aktywnej formie. wego DNA. Te procesy nazywane są programowaną
śmiercią komórki albo apoptozą.
Farmakodynamika
3.2. Efekty działania leków zależne od receptorów
3.2.1. Działanie leków 3.2.2. Działanie leków
na systemy transportowe na enzymy
(transportery)
Mechanizm działania wielu leków polega na hamo-
Transport małych cząstek organicznych albo jonów, waniu lub (rzadko) aktywowaniu enzymów. Podobnie A3
które są zbyt polarne, żeby samodzielnie pokonać jak w wypadku interakcji lek-receptor, dochodzi naj-
błonę, zachodzi często za pomocą cząstek transpor- pierw do tworzenia kompleksu lek-enzym i w wyni-
towych. Poza transporterami dla neurotransmiterów ku tego do blokady lub aktywacji enzymu, zależnie
w zakończeniach nerwowych (transportery specyficz- od typu substancji leczniczej.
ne dla neurotransporterów, np. dla noradrenaliny, se-
rotoniny, GABA), które służą do zwrotnego wychwytu
Hamowanie enzymów. Hamowanie enzymów wy-
wydzielanych transmiterów do neuronu presynaptycz-
wołane lekami może mieć charakter kompetycyjny
nego, i transporterami dla elektrolitów (np. symporte-
lub niekompetycyjny. O działaniu kompetycyjnym
rów Na+/K+/2Cl– i Na+/Cl–), które występują głównie
mówi się wówczas, gdy lek w sposób odwracalny
w komórkach nabłonkowych mających funkcję sekre-
konkuruje z substratem o miejsce wiązania do danego
tolityczną (m.in. w kanalikach nerkowych, nabłonku
enzymu. W wypadku niekompetycyjnego hamowania
oskrzelowym i błonie śluzowej jelita), występują także
lek reaguje nieodwracalnie z centrum aktywnym
transportery dla glukozy czy aminokwasów.
enzymu lub działa hamująco na efekty następujące
Punktami uchwytu dla takich leków, jak antyde-
już po połączeniu się substratu z enzymem, nie wpły-
presanty, diuretyki, glikozydy nasercowe i inhibito-
wając na sam proces wiązania.
ry pomp protonowych są zwłaszcza transportery dla
Najważniejszymi przykładami blokowania enzy-
neurotransmiterów i elektrolitów. Antydepresanty
mów przez środki lecznicze są:
hamują aktywny transport (zwrotny) noradrenaliny
i/lub serotoniny. Diuretyki charakteryzowane są jako ■ inhibitory monoaminooksydazy,
selektywne inhibitory transportu elektrolitów: silne
■ niesteroidowe leki przeciwzapalne hamujące cyklo-
diuretyki, takie jak furosemid, blokują symportery
oksygenazę,
Na+/K+/2Cl–, natomiast tiazydy blokują symporte-
ry Na+/Cl–. Glikozydy nasercowe hamują transport ■ leki hamujące tworzenie kwasu moczowego po-
jonów sodowych na zewnątrz komórki oraz trans- przez hamowanie oksydazy ksantyny,
port do wnętrza komórki jonów potasu przez blo-
■ pośrednie parasympatykomimetyki hamujące cho-
kadę pompy sodowo-potasowej (Na+/K+-ATPaza).
linoesterazę,
Inhibitory pomp protonowych stosowane w chorobie
wrzodowej zmniejszają produkcję kwasu solnego ■ inhibitory reduktazy hydroksy-metylo-glutaryloko-
w żołądku przez hamowanie pompy protonowo-po- enzymu-A (inhibitory reduktazy HMG-CoA, inhi-
tasowej (Na+/K+-ATPaza). bitory CSE, statyna),
MUTSCHLER-2009.indd 81 2010-01-07 22:11:09
82 Farmakodynamika
■ inhibitory fosfodiesterazy,
przestrzeń zewnątrzkomórkowa ■ inhibitory konwertazy angiotensyny (inhibitory
ACE),
■ inhibitory kinazy tyrozynowej.
Wewnątrzkomórkowe kinazy tyrozynowe bez własnej dome-
ny receptorowej (non-receptor-tyrosin-kinases) uczestniczą
COOH
NH2 w licznych procesach transdukcji sygnału i podobnie jak re-
przestrzeń wewnątrzkomórkowa ceptorowe kinazy tyrozynowe, regulują najważniejsze funk-
cje komórkowe, takie jak proliferacja, różnicowanie, apopto-
za i antyapoptoza oraz wzrost neuronów. Mutacje albo nad-
ekspresja tych enzymów uważane są za przyczyny różnych
neuron presynaptyczny chorób nowotworowych i łagodnych hiperplazji. Ponad 70%
znanych onkogenów stanowią zmutowane kinazy tyrozy-
nowe! Znaczenie kinaz tyrozynowych w innych chorobach
uwidacznia się np. przy procesach zapalnych i cukrzycy.
5-HT Wiele środków stosowanych w zakażeniach wykazu-
je swoje działanie również poprzez hamowanie enzy-
mów drobnoustrojów, np. inhibitory gyrazy poprzez
transporter
5-HT interakcję gyraza-DNA, azolowe środki przeciw-
Gi T 1D grzybicze poprzez blokadę metylazy lanosterolu lub
5-H
wirusostatyki przeciw HIV działające w wyniku in-
terakcji z wirusowymi polimerazami lub proteazami.
Aktywacja enzymów. Aktywacja enzymów jest naj-
5-HT2A,2C częściej wywoływana przez działanie na tzw. przekaź-
Gi niki 2 rzędu. Bezpośrednim aktywatorem cyklazy ade-
nylanowej jest forskolina, która jest używana jedynie
regulacja aktywności w badaniach eksperymentalnych i nie ma zastosowania
neuronalnej terapeutycznego. Nitraty aktywują poprzez NO cykla-
zę guanylanową. Działanie wielu czynników podczas
krzepnięcia krwi polega na tym, że zamieniają nie-
aktywne czynniki w aktywne proteazy. Fibrynolityki
zmieniają plazminogen w plazminę, również proteazę.
neuron postsynaptyczny
Ryc. A 3.2-1. Działanie transportera serotoniny (5-HT) oraz pre- 3.2.3. Działanie leków poprzez
i postsynaptycznych receptorów 5-HT. Transportery 5-HT wystę-
pują w błonie zakończeń aksonów neuronów serotoninergicznych wpływ na biosyntezę
i odpowiadają za wychwyt zwrotny neurotransmitera serotoniny w drobnoustrojach
do neuronu presynaptycznego po jego uwolnieniu poza komórkę.
Transporter 5-HT zawiera 12 segmentów transbłonowych. Istnieją Leki stosowane w zakażeniach wykazują swoje dzia-
również receptory 5-HT związane z białkiem G, których zadaniem łanie, jak wyczerpująco opisano w B 11.2, najczęś-
jest dalsze przekazywanie sygnału na błonę postsynaptyczną, ciej poprzez wpływ na biosyntezę w drobnoustro-
podczas gdy jednocześnie hamują egzocytozę neutransmiterów jach. Działanie antybiotyków β-laktamowych wyni-
na zakończeniach aksonów neuronów presynaptycznych. ka z zaburzeń syntezy ściany komórkowej bakterii.
Bakteriostatyczne działanie tetracyklin i makrolidów
jest z kolei efektem hamowania syntezy białek w ry-
bosomach bakterii. Sulfonamidy działają jako anty-
metabolity w procesie syntezy kwasu foliowego ko-
niecznym do wzrostu bakterii. Izoniazyd działający
przeciwgruźliczo jako antymetabolit kwasu nikoty-
nowego hamuje procesy przemiany materii w prąt-
kach gruźlicy.
MUTSCHLER-2009.indd 82 2010-01-07 22:11:09
Farmakodynamika 83
3.3. Dawkowanie i zależność działania leku
od dawki lub stężenia
3.3.1. Dawkowanie między dawkami jest istotnym parametrem reżimu
dawkowania. Z powodu tzw. compliance pacjentów
Jak wspomniano w części farmakokinetycznej, wa- – przez to pojęcie rozumie się przestrzeganie zaleceń
runkiem racjonalnej farmakoterapii jest dobór odpo- lekarzy przez pacjentów – pożądane jest dawkowa-
wiedniej dawki. Dawka taka powinna wywoływać nie raz dziennie. W przypadku substancji o krótkim
oczekiwane działanie terapeutyczne bez toksycznych czasie półtrwania, jeżeli nie istnieją formy retard
efektów niepożądanych wynikających z przedawko- (opóźnionego uwalniania), konieczne jest częstsze
wania. dawkowanie, aby osiągnąć stałe wystarczające stęże-
Efekt leku zależy od stężenia w miejscu działania nie we krwi.
– przy określonej dawce zależy od masy ciała (lub
Farmakodynamika
jego powierzchni) – dlatego należy to uwzględnić Należy jednak wziąć pod uwagę, że działanie leku może
w razie konieczności bardzo dokładnego dawkowa- być dłuższe niż jego czas półtrwania w osoczu. Na przykład
działanie leków β-adrenolitycznych jest 3–4 razy dłuższe
nia (np. przy lekach o niewielkim zakresie bezpie- niż czas półtrwania. Z tego powodu jest możliwe podawa-
czeństwa). W praktyce w celu uproszczenia wytycz- nie leków blokujących receptory β o krótkim t1/2 (kilka go-
nych dawkowania najczęściej zakłada się średnią dzin) tylko raz dziennie.
masę ciała – 70 kg.
W tabeli A 3.3-1 zebrano najważniejsze pojęcia Dawkowanie u niemowląt i małych dzieci. Dawko-
związane z dawkowaniem. wanie u niemowląt i małych dzieci wymaga szczegól-
nej ostrożności. Jak wspomniano w A 2.7, farmako- A3
Dawka początkowa, dawka podtrzymująca. W przy- kinetyka u noworodków i niemowląt często znacznie
padku leków o długim półokresie eliminacji, aby odbiega od obserwowanej u dorosłych. Eliminacja
szybko uzyskać minimalne stężenie terapeutyczne, jest opóźniona szczególnie w pierwszym roku życia.
stosuje się zwykle większą dawkę początkową (pri- Można jednak obliczyć odpowiednią dawkę dla tego
ming dose, loading dose), zależną od objętości dys- okresu życia, opierając się na dawkowaniu u dorosłych
trybucji, a następnie zmniejsza się dawkę do poziomu i biorąc pod uwagę powierzchnię ciała według wzoru:
dawki podtrzymującej. W ten sposób szybko osiąga
się poziom terapeutyczny we krwi. KO
NUdziecko = NDdorosły ⋅ ,
1,73
Odstępy między dawkami. Poza pojedynczą,
dzienną, początkową i podtrzymującą dawką odstęp KO – powierzchnia ciała dziecka (m2).
Tabela A 3.3-1. Nazewnictwo dawek
Nazwa Skrót Wyjaśnienie
Dawka jednorazowa ED typowa dawka wywołująca efekt terapeutyczny
Jednorazowa
dawka maksymalna EMD maksymalna dozwolona dawka
Dawka dzienna TD typowa dawka w ciągu 24 godzin
Maksymalna dawka dzienna TMD maksymalna dawka w ciągu 24 godzin
Dawka typowa ND ogólnie odpowiada dawce jednorazowej
Dawka letalna LD dawka śmiertelna
Dawka początkowa dawka stosowana na początku terapii do czasu osiągnięcia oczekiwanego
poziomu działania
Dawka podtrzymująca dawka stosowana po osiągnięciu oczekiwanego efektu (stężenia w narządzie),
służąca podtrzymaniu efektu terapeutycznego
MUTSCHLER-2009.indd 83 2010-01-07 22:11:09
84 Farmakodynamika
Powierzchnię ciała dziecka można obliczyć (wg 3.3.2.1. Zależność dawka-efekt w grupie
Wagnera) z następującego wzoru:
Badania zależności dawka-efekt prowadzi się w fa-
KO = 0,09 ⋅ W0,73 zach przedklinicznych i klinicznych badania nowych
leków (zob. A 10). Jest to praktykowane zarówno
W – masa ciała (kg). w przypadku ogólnych badań toksykologicznych,
jak i przy badaniu toksyczności narządowej. Wyniki
Dokładniejsze informacje można uzyskać z odpo- takich badań można przedstawić w postaci krzywej
wiednich tabel. częstości. Dawka leku, która wywołuje efekt u 50%
W przypadku wielu leków reguła obliczania daw- osobników, jest miarą jego aktywności i określana
ki na podstawie powierzchni ciała nie znajduje zasto- jest jako ED50.
sowania. Dotyczy to niektórych leków działających Symetryczna dystrybucja przedstawiona na ryc.
na OUN, np. dawki leków przeciwpadaczkowych ob- A 3.3-1 jest raczej wyjątkiem niż regułą. W prakty-
liczone w ten sposób są za małe. Kodeinę należy sto- ce zwykle stwierdza się dystrybucję asymetryczną.
sować, opierając się na masie ciała (tzn. 0,5 mg/kg). Jeżeli jednak przedstawi się dawki na skali logaryt-
W wielu przypadkach trudno jest ściśle ustalić micznej, ponownie otrzymuje się krzywą symetrycz-
dawkowanie u dziecka, jeżeli nie dysponuje się wy- ną (zob. ryc. A 3.3-2).
nikami odpowiednich badań.
Odchylenie standardowe σ jest miarą rozpiętości wartości
w stosunku do wartości średniej ED50 i jest zdefiniowa-
ne w ten sposób, że część krzywej od ED50 – σ do ED50
3.3.2. Zależność działania leku + σ zawiera 68% wszystkich wartości. Natomiast część
zawarta pomiędzy ED50 – 2σ a ED50 + 2σ zawiera 95,4%
od dawki (lub stężenia) wartości.
Badania zależności między dawką (lub stężeniem) W niektórych przypadkach krzywa zależności dawka-
a działaniem leku można wykonywać na dwa spo- -efekt wykazuje dwa piki. Mówi się wtedy o krzywej
soby: występowanie danego efektu w zależności bimodalnej. W tym przypadku nie występuje homo-
od dawki badane jest w grupie (tzw. zależność czę-
stości występowania efektu od dawki) lub bada się
siłę działania na jednym osobniku (zależność działa-
nie-dawka/stężenie w dosłownym znaczeniu). Wraz
ze wzrostem dawki/stężenia w pierwszym przypadku
140
obserwuje się wzrost liczby pacjentów, którzy wyka- 1000 osobników
zują obecność danego efektu, w drugim wzrost nasi-
120
lenia działania tego efektu, aż do osiągnięcia efektu
Liczba reagujących osobników
maksymalnego.
100
Jeżeli przedstawi się uzyskane w ten sposób wyniki
w układzie osi współrzędnych (na osi odciętych (x) 80
dawkę lub stężenie; na osi rzędnych (y) efekt) w spo-
sób graficzny, otrzymamy krzywe zależności działa- 60
nie-dawka/stężenie. Przy analizie takich krzywych
istotne są przede wszystkim: 40
■ dawka progowa, tzn. najmniejsza dawka, przy któ-
20
rej obserwuje się dane działanie,
■ maksymalne osiągalne działanie,
10 40 70 100 130 160 190
■ minimalna dawka wywołująca maksymalne działa- dawka (mg)
nie, ED50
■ nachylenie krzywej determinuje zakres dawek mię- Ryc. A 3.3-1. Wykres zależności między dawką leku i liczbą
dzy pojawieniem się danego działania a działaniem osobników wykazujących specyficzny efekt. Widoczny jest sy-
maksymalnym: przy małym nachyleniu zakres ten metryczny podział przez pionową linię przerywaną dzielącą dia-
jest duży, natomiast przy silnym nachyleniu jest on gram na dwie równe części – odpowiada to ED50. Jest to przy-
mały. kład prawidłowej statystycznej dystrybucji.
MUTSCHLER-2009.indd 84 2010-01-07 22:11:10
Farmakodynamika 85
3.3.2.2. Krzywa zależności dawka-efekt
u pojedynczego osobnika
100
80 Charakterystyka krzywej zależności dawka-efekt
Liczba reagujących
osobników (%)
u pojedynczego osobnika podlega zasadzie prawa
60
działania mas. Jeżeli przedstawi się dawkę na loga-
40 rytmicznej skali odciętych, a siłę efektu na osi rzęd-
20 nych, otrzyma się krzywą o kształcie litery S.
– 3σ – 2σ – 1σ + 1σ + 2σ + 3σ W przypadku agonistów wygląd krzywej pozwala
0 określić siłę działania, a także aktywność wewnętrz-
33,3 100 300
ną. Położenie na osi x jest miarą siły działania,
dawka (mg) a wielkość maksymalnego efektu odpowiada aktyw-
ED50 ności wewnętrznej.
Ryc. A 3.3-2. Krzywa zależności dawka-efekt grupy (krzywa Na ryc. A 3.3-3 przedstawiono krzywe zależności stężenie-
łączna). efekt różnych parasympatykomimetyków na izolowanym
Farmakodynamika
jelicie cienkim świnki morskiej. Patrząc na te krzywe, moż-
na zauważyć, że:
■ acetylocholina wywołuje silniejszy efekt, tzn. działa
genna populacja, ale dwie odrębne grupy o odmien- w mniejszych dawkach niż karbachol i pilokarpina,
nej wrażliwości. Krzywa zależności dawka-efekt
w wypadku takiej mieszanej populacji składa się ■ karbachol wywołuje taki sam efekt maksymalny jak ace-
tylocholina, jest on jednak osiągany w mniejszych daw-
z dwóch krzywych o kształcie litery S, nakładających kach,
się na siebie. Oznacza to, że w skład populacji wcho-
dzą dwie odrębne grupy i dla każdej można obliczyć ■ pilokarpina wywołuje słabszy efekt maksymalny, co ozna- A3
cza, że nawet w dużych stężeniach nie można osiągnąć
niezależne ED50. takiego samego maksymalnego efektu jak po acetylocho-
linie.
3.3.2.3. Wskaźniki
i wartości farmakologiczne
EA/Em
1,0 acetylocholina Z krzywych zależności dawka-efekt lub stężenie-
karbachol
-działanie można uzyskać ważne wartości farmakolo-
giczne, które opisano poniżej:
pilokarpina
ED50. Wielokrotnie już wspomniana wartość ED50
(dosis efektiva 50) jest dawką, przy której osiąga się
połowę (50%) efektu maksymalnego lub przy której
0,5
50% osobników wykazuje oczekiwany efekt (odpo-
wiednio – ED95 oznacza dawkę, która wywołuje 95%
efektu).
LD50. LD50 (dosis letalis 50) jest szczególnym przy-
padkiem ED50, przy której 50% badanych zwierząt
0 umiera.
10-8 10-7 10-6 10-5 10-4 10-3
M
Wartość pD2. Wartość pD2 określa siłę działania
Ryc. A 3.3-3. Krzywe zależności stężenie-efekt różnych para-
agonisty i odpowiada ujemnej wartości dziesiętnego
sympatykomimetyków na izolowanym jelicie cienkim świnki logarytmu stężenia danej substancji, które wywołuje
morskiej. Na osi odciętych przedstawiono stężenie molarne, maksymalny efekt.
a na osi rzędnych efekt wywołany przez lek EA w relacji do efektu
maksymalnego Em. Należy zwrócić uwagę na mniejszy maksy- Wartość pA2. Wartość pA2 jest parametrem powino-
malny efekt pilokarpiny. Szczegóły w tekście. wactwa antagonisty i odpowiada ujemnej wartości
MUTSCHLER-2009.indd 85 2010-01-07 22:11:10
86 Farmakodynamika
dziesiętnego logarytmu stężenia antagonisty, które W obu przypadkach obserwuje się taki sam wskaźnik tera-
dwukrotnie przesuwa krzywą zależności stężenie- peutyczny LD50/ED50 = 1000.
Na podstawie wyglądu tych krzywych można się jednak
-efekt agonisty w prawo.
łatwo przekonać, że właściwie bezpieczeństwo terapeu-
tyczne obu leków L1 i L2 nie jest jednakowe. Na przykład
Wskaźnik terapeutyczny (okno terapeutyczne). w przypadku l, w którym maksymalny efekt terapeutyczny
Wskaźnik terapeutyczny jest miarą bezpieczeństwa L1 osiąga się znacznie poniżej dawki letalnej: zakres bez-
dotyczącą zakresu pomiędzy dawką terapeutyczną pieczeństwa wynosi 20%, natomiast przy maksymalnie
efektywnej dawce L2 zwierzęta giną: brak jest odpowied-
a toksyczną. Lek jest tym bardziej bezpieczny, im
niego zakresu bezpieczeństwa.
większy jest to zakres. Zwykle podaje się go w formie Ze względu na możliwość występowania tego typu błę-
współczynnika (indeksu), jako stosunek LD50 do ED50: du niejednokrotnie proponuje się używanie zamiast warto-
ści LD50/ED50 innych współczynników, np. LD5/ED95 lub
LD25/ED50. Jeśli krzywa dawka-efekt i krzywa śmiertel-
LD50 ności danej substancji są równoległe (jak w przypadku 1)
Wskaźnik terapeutyczny =
ED50 możliwa jest właściwie lepsza ocena zakresu terapeutycz-
nego. W przypadku l otrzyma się przykładowo następujące
wartości: LD25/ED75 dla L1 ok. 500, a dla L2 ok. 50. W ten
Należy jednak wziąć po uwagę, że takie dane zakre-
sposób znaczne różnice zakresu terapeutycznego obu le-
su terapeutycznego mogą prowadzić do fałszywych ków stają się bardziej widoczne.
wniosków. Wykazuje to następujący przykład: na ryc. Jeżeli natomiast krzywa dawka-działanie i krzywa śmier-
A 3.3-4 przedstawiono dwie krzywe zależności daw- telności danej substancji nie są równoległe (jak w przypad-
ka-działanie (krzywe A i B) i dwie krzywe letalności ku 2), wtedy wartości LD25/ED75 lub LD5/ED95 mają bar-
dzo ograniczone zastosowanie. W przypadku 2: LD25/ED75
(krzywe C i D) dla dwóch leków L1 i L2.
dla L1 wynosi ok. 100, a dla L2 ok. 50, mimo że wskaźnik
terapeutyczny różni się tylko dwukrotnie. Wynika z tego,
W tym przykładzie należy wziąć po uwagę, że: że z przebiegu krzywych można otrzymać bardzo duże
różnice bezpieczeństwa terapeutycznego pomimo pozornie
■ w pierwszym przypadku krzywe A i C należą do L1, podobnego zakresu terapeutycznego.
a B i D do L2, Precyzyjne określenie bezpieczeństwa terapeutyczne-
go jest więc możliwe w wyniku analizy całego przebiegu
■ w drugim przypadku krzywe A i D należą do L1, krzywej dawka-działanie i krzywej dawka-śmiertelność,
a B i C do L2. a nie w wyniku obliczenia określonych ilorazów.
100 A B C D 100
+ +
śmiertelność (%)
działanie (%)
50 50
+ +
10-8 10-7 10-6 10-5 10-4 10-3
g/g m.c. zwierzęcia
Ryc. A 3.3-4. Graficzne przedstawienie różnych zakresów terapeutycznych dwóch leków o takich samych współczynnikach terapeu-
tycznych LD50/ED50 (zmodyfikowane wg Kuschinsky’ego i Lüllmanna). Na osi rzędnych (y) przedstawiono śmiertelność w %, a na osi
odciętych (x) dawkę w gramach na gram masy ciała zwierzęcia. ED75 i LD25 zaznaczono znakiem + na krzywych. Szczegóły w tekście.
MUTSCHLER-2009.indd 86 2010-01-07 22:11:11
Farmakodynamika 87
3.3.3. Synergizm uzyskać ten sam efekt. Powinno się przy tym odróż-
nić tolerancję farmakokinetyczną od farmakodyna-
O synergizmie mówi się wówczas, gdy zastosowa- micznej.
nie kombinacji dwóch lub więcej substancji wywołuje
efekt większy niż suma działań poszczególnych skład- Tolerancja farmakokinetyczna polega na osła-
ników. Jeżeli natomiast efekt ten jest jedynie sumą bianiu działania najczęściej w wyniku indukcji en-
tych działań, mówi się o działaniu addytywnym. zymów (przy prekursorach [pro-drugs], ewentualnie
Warunkiem koniecznym do otrzymania efektu także przy zmniejszonej biotransformacji). W przy-
synergistycznego jest działanie na różne receptory padku tolerancji farmakodynamicznej występuje
lub systemy efektorowe. Jednak przy ocenie takiego zmiana liczby lub wrażliwości receptorów albo też
efektu należy wziąć po uwagę, że jak opisano wyżej, sprzężenia receptor-efektor (zob. zmiany wrażliwo-
krzywe dawka-efekt nie mają najczęściej przebiegu ści). Ponieważ barbiturany są szczególnie silnymi
liniowego, lecz sigmoidalny. Jest jednak niedopusz- induktorami enzymów, a morfina wywołuje szcze-
czalne wnioskowanie, że występuje synergizm, jeże- gólnie silnie zjawisko tolerancji farmakodynamicz-
li obserwowane działanie nie jest w sposób liniowy nej, w pierwszym przypadku dochodzi do rozwoju
Farmakodynamika
skorelowane z dodatkiem pojedynczej dawki. Taki tolerancji typu barbituranów, a w drugim przypadku
wniosek jest uzasadniony jedynie wtedy, gdy ob- – typu morfiny.
serwowane efekty znajdują się w liniowym zakresie Natomiast w przypadku tachyfilaksji dochodzi
krzywej. W celu precyzyjnego określenia typu inter- niezwykle szybko do rozwoju tolerancji (minuty lub
akcji konieczna jest analiza krzywej dawka-efekt lub godziny), a po zaprzestaniu jej podawania relatywnie
stężenie-efekt. Można wtedy ocenić, czy występuje szybko można znowu wywołać efekt o prawidłowym
interakcja addytywna, synergistyczna (hiperaddy- nasileniu. Znanym przykładem może być tachyfi-
tywna) czy hipoaddytywna (= antagonistyczna). laksja po podaniu pośrednich sympatykomimetyków,
np. efedryny. Po szybko po sobie następujących po- A3
Przykładem synergizmu może być efekt obserwowany daniach efedryny zapasy noradrenaliny zmniejszają
po łącznym podaniu środka nasennego i diuretyku tiazy- się coraz bardziej, ponieważ mechanizmy odnawia-
dowego (zob. blokada nefronu) lub inhibitora MAO i leku
sympatykomimetycznego. nia jej zasobów nie nadążają za ich opróżnianiem.
W następstwie dochodzi do stopniowego osłabienia
działania. Po przerwaniu podawania efedryny system
3.3.4. Tolerancja i tachyfilaksja powraca jednak szybko do normy w wyniku odna-
wiania zapasów noradrenaliny.
O tolerancji mówi się wówczas, gdy przy ponownych Zjawisko tachyfilaksji obserwuje się też przy le-
podaniach danego leku należy zwiększać dawkę, aby czeniu nitratami (tzw. tolerancja na nitraty).
MUTSCHLER-2009.indd 87 2010-01-07 22:11:11
88 Farmakodynamika
3.4. Zależność między strukturą chemiczną leku
a jego działaniem
Od początku badań przyrodniczych ukierunkowa- są tym trudniejsze do sformułowania, im bardziej
nych farmakologicznie wiele grup badawczych zaj- skomplikowany jest system. Aby zmniejszyć liczbę
muje się relacją między strukturą chemiczną leku czynników wchodzących w interakcję, początko-
a działaniem farmakologicznym. Pomimo wielu nie- we badania przeprowadza się zwykle na możliwie
powodzeń w tej dziedzinie wysiłki w tym kierunku prostych systemach (takich jak błony komórkowe,
są jednak kontynuowane, a nawet zwiększone dzięki frakcja komórkowa, kultury komórkowe, izolowa-
zastosowaniu nowych technik, takich jak rentgenow- ne narządy), co jednak ogranicza zakres wniosko-
ska analiza struktury, wielowymiarowy jądrowy rezo- wania.
nans magnetyczny (MR) i metody oparte na technice Kolejnym ważnym problemem przy racjonalnym
komputerowej. Motywacją tych starań jest nadzieja, wdrażaniu nowych leków jest zbyt fragmentaryczna
że za pomocą odpowiednich programów kompute- wiedza o zależności struktura-toksyczność, będąca
rowych (Computer Aided Drug Design, Molecular niezwykle istotnym elementem zależności struktura-
Modeling) będzie można na podstawie struktury che- -działanie.
micznej danej substancji przewidzieć jej działanie lub Szczególnie obiecujące wydaje się poszukiwanie
obliczyć siłę i profil działania. Ułatwiłoby to z pew- zależności struktura-działanie, a tym samym zna-
nością poszukiwania i badania wdrożeniowe nowych lezienie najlepiej dopasowanej substancji czynnej,
substancji (zob. A 10). kiedy jest dokładnie znana trójwymiarowa struktu-
Problemy przy badaniach nad zależnością struktura ra docelowej cząsteczki oraz miejsce jej wiązania
chemiczna-działanie wynikają z tego, że wielu parame- do leku. W ten sposób, dzięki poznaniu struktury
trów istotnych dla badania leków nie można uwzględnić proteazy HIV oraz inhibitora neuraminidazy za po-
jednocześnie. Mowa tu o takich czynnikach, jak roz- mocą technik komputerowych, zaprojektowano in-
puszczalność, właściwości stereochemiczne czy roz- hibitory proteaz stosowane w leczeniu AIDS.
mieszczenie elektronów. Jest więc zrozumiałe, że jak
dotąd znaleziono jedynie ograniczoną liczbę ogólnie
obowiązujących zależności struktura-działanie. Wiele 3.4.1. Jakościowa i ilościowa
badań ogranicza się do tego, że porównują działanie
zależność struktura-działanie
substancji wyjściowej i jej pochodnych, wyciągając
wnioski o relacjach między strukturą i działaniem
Zależność struktura-działanie może mieć charakter
w obrębie wąskiej grupy leków.
jakościowy lub ilościowy.
Nie ma wątpliwości, że takie postępowanie jest pomocne Jakościowa zależność struktura-działanie. Służy
w badaniu nowych substancji leczniczych. Na przykład ona do wykrywania elementów struktury koniecznych
z naturalnej substancji kokainy otrzymano syntetyczną pro-
kainę przez systematyczne badania, których efekt jest kla- do działania leku i opisywania reguł, według któ-
syczną ilustracją relacji struktura chemiczna-działanie far- rych na podstawie struktury danej substancji można
makologiczne. Nie należy jednak zapominać, że w ten spo- zaklasyfikować ją do grupy związków biologicznie
sób otrzymane informacje o zależnościach między strukturą aktywnych. Znajomość jakościowej zależności struk-
chemiczną a efektem farmakologicznym mają ograniczone tura-działanie jest istotna dla racjonalnego badania
zastosowanie i zwykle po krótkim czasie stwierdza się
wiele wyjątków. Substancje nawet bardzo różniące się che- i wdrażania nowych leków.
micznie od prokainy, niezawierające azotu, mogą działać
miejscowo znieczulająco. Ilościowa zależność struktura-działanie. Badania
ilościowej zależności struktura-działanie mają na celu
In vivo za odmienne działanie leku mogą też być uzyskanie formuły matematycznej obrazującej relację
odpowiedzialne różnice w farmakokinetyce (różni- pomiędzy siłą efektu farmakologicznego a wielkoś-
ce we wchłanianiu, wiązaniu do białek, dystrybucji, cią określonych parametrów charakterystycznych dla
metabolizmie, wydalaniu). W konsekwencji stwier- substancji (tzw. analiza Hanscha). Wiele przykładów
dzenia dotyczące zależności struktura-działanie wskazuje, że działanie leku jest związane:
MUTSCHLER-2009.indd 88 2010-01-07 22:11:11
Farmakodynamika 89
■ ze współczynnikiem rozdziału (np. oktanol/woda), ctwo do receptora nikotynowego jest ok. 200 razy mniej-
sze. W przypadku acetylo-α-metylocholiny jest dokładnie
■ z parametrami elektronicznymi (stałe Hammeta), odwrotnie – powinowactwo do receptora nikotynowego
jest takie samo, natomiast do receptora muskarynowego
■ z właściwościami stereochemicznymi. jest ok. 50 razy mniejsze niż acetylocholiny.
Zarówno enancjomery, jak i diastereoizomery wykazują
Pomimo dostępności licznych równań i programów często, jak już wspomniano, odmienne właściwości far-
komputerowych służących do obliczania ilościowej makokinetyczne i farmakodynamiczne. Silniej działający
zależności struktura-działanie nie udało się bezpo- enancjomer jest nazywany eutomerem, a słabiej (lub nie-
średnio za ich pomocą otrzymać leków o optymal- działający w ogóle) diastomerem.
nych właściwościach. Wprawdzie jest możliwe, jak
opisano wyżej, aby zaczynając od substancji wyjścio- Odpowiednio do różnic farmakodynamicznych
wej o znanych właściwościach biologicznych, obli- obowiązują następujące reguły: im silniej działa
czyć efektywność substancji pochodnych (homologi, dana substancja, tym większe są zazwyczaj różni-
analogi z innymi podstawnikami). Dzięki temu liczba ce między izomerami. Inaczej mówiąc, duża siła
substancji, które muszą być zsyntetyzowane i prze- działania wymaga, aby lek pasował bardzo dobrze
Farmakodynamika
badane farmakologicznie, żeby otrzymać potencjalny do miejsca wiążącego receptora, a co za tym idzie
lek, została znacznie zmniejszona. – niewielkie zmiany geometrii odzwierciedlają się
wyraźnie w utracie powinowactwa. Zasada ta obo-
wiązuje jednak z pewnymi ograniczeniami – izo-
3.4.2. Zależność struktura-działanie mery muszą wykazywać różnice geometryczne
w przypadku izomerów w części molekuły istotnej dla jej działania farma-
kodynamicznego.
Izomery są chemicznymi cząsteczkami o takim sa- Może się więc zdarzyć, że różnice aktywności pomiędzy A3
mym wzorze sumarycznym, lecz o różnym prze- enancjomerami są małe lub nawet żadne, jeżeli asyme-
strzennym rozmieszczeniu atomów. Dalsza klasyfi- tryczne centrum znajduje się w części molekuły nieistotnej
kacja izomerów rozróżnia: dla jej działania, np. (+)-metylo-benzylat-choliny jest tak
samo aktywny jak jej enancjomer (–).
■ izomery strukturalne,
■ stereoizomery. Istotnym przykładem różnic działania enancjome-
Izomery strukturalne, np. izomery położeniowe lub tauto- rów są leki blokujące receptory β, które są repre-
mery, różnią się sposobem i rozmieszczeniem atomów. Są zentatywne dla wielu innych substancji leczni-
to więc zupełnie inne cząsteczki. czych. W przypadku leków blokujących receptory β
W wypadku stereoizomerów przeciwnie, sposób i sek- enancjomer S działa zazwyczaj 100 razy silniej niż
wencja rozmieszczenia atomów są takie same, różnice wy- ich enancjomer R.
stępują jednak w przestrzennej orientacji niektórych frag-
mentów cząsteczki. Jeżeli stereoizomery są odbiciem lu- W tym kontekście należy zaznaczyć, że wiele
strzanym, mówi się o enancjomerach: w przeciwnym razie leków stosowanych w lecznictwie występuje w po-
określa się je jako diastereoizomery. staci racematów, a nie czystych izomerów. Dlatego
Do substancji posiadających enancjomery należą sub- różne działanie enancjomerów ma duże teoretyczne
stancje mające centra chiralne (substancje chiralne). Do dia- i praktyczne znaczenie. Jeżeli enancjomer działa tak
stereoizomerów zaliczają się na przykład izomery cis/trans,
a także cząsteczki o dwóch centrach chiralnych. jak np. L-hioscyjamina, a inny jak D-hioscyjamina,
Enancjomery mają identyczne właściwości fizykoche- oznacza to, że w wypadku racematu (w tym przy-
miczne (z wyjątkiem skręcania światła spolaryzowanego), na- padku atropiny) 50% podawanej substancji to „nie-
tomiast diastereoizomery wykazują różnice fizykochemiczne. aktywne zanieczyszczenia” lub „balast izomerowy”.
Leki mające izomery strukturalne różnią się zazwyczaj wy- Jest to jednak trudne do uniknięcia, jeżeli – tak jak
raźnie właściwościami farmakologicznymi. Tak jest w przy- w przypadku L-hioscyjaminy – in vitro i/lub in vivo
padku izomerów strukturalnych estrów kwasu aminoben- dochodzi spontanicznie do racemizacji, tzn. z jedne-
zoesowego typu prokainy. W przypadku rozmieszczenia go (czystego) enancjomeru powstaje inny. W przy-
grup aminowych w pozycji para działają silniej miejscowo padku stabilnych enancjomerów „balast izomerowy”
znieczulająco, natomiast w układzie orto takie działanie
jest najsłabsze. Pochodna acetylocholiny, acetylo-β-mety- jest ewentualnie akceptowalny, jeżeli tylko „nie-
locholina, ma takie samo powinowactwo do receptorów aktywny” izomer nie ma działania niepożądanego,
muskarynowych jak acetylocholina, natomiast powinowa- np. toksycznego.
MUTSCHLER-2009.indd 89 2010-01-07 22:11:11
90 Farmakodynamika
3.5. Stosunek między farmakokinetyką
i farmakodynamiką
W przeszłości opisywano farmakokinetykę i farma-
A
kodynamikę leku zupełnie niezależnie. Nowa dyscy-
plina – modelowanie farmakokinetyczno-farmakody- Farmakokinetyka Farmakodynamika
namiczne (PK/PD modeling) zajmuje się łączeniem D
obu dyscyplin i w konsekwencji integruje zmiany
koncentracji leku z wywoływanym działaniem (zob.
ryc. A 3.5-1). k10
C model Emax efekt
W najprostszej sytuacji istnieje bezpośrednia za-
leżność między stężeniem w osoczu i obserwowanym
efektem. Często jednak, rozpatrując stosunek działa-
nia do stężenia, należy uwzględniać albo obwodowe
kompartmenty farmakokinetyczne, albo hipotetyczny B
kompartment efektu. Przebieg zmian stężenia w ta-
kim kompartmencie może być związany z efektem, Farmakokinetyka Farmakodynamika
np. poprzez model sigmoidalnego Emax (zob. ryc. D
A 3.5-2).
Jeżeli efekt pojawia się z wyraźnym czasowym k21
k10
opóźnieniem, niezwiązanym z dystrybucją farmako- C Cp model Emax efekt
kinetyczną, lecz np. polega na syntezie lub rozkładzie k12
endogennych substancji, uwzględnia się w modelu
farmakokinetycznym odpowiedni współczynnik (in-
direct response model).
C
Konsekwentne uwzględnianie zależności między far- Farmakokinetyka Farmakodynamika
makokinetyką i farmakodynamiką prowadzi do lep- D
szego zrozumienia działania leków. Szczególnie efekt
kompartment
k10 k12
C Cp model Emax efekt
Farmakokinetyka Farmakodynamika k10
dawka ⇒ stężenie – czas stężenie ⇒ efekt
efekt
stężenie
Ryc. A 3.5-2. Możliwości połączenia farmakokinetyki i farmako-
dynamiki przy modelowaniu PK/PD (A – relacja bezpośrednia, B
czas stężenie
– relacja pośrednia poprzez kompartment obwodowy, C – rela-
cja pośrednia poprzez kompartment efektorowy).
PK/PD
dawka ⇒ efekt – czas
efekt
w ramach badań i wdrażania nowych leków model
PK/PD może istotnie przyspieszyć ustalenie opty-
czas
malnej dawki. Kombinacja modeli PK/PD i kinetyki
populacji może również dawać podstawy do stoso-
Ryc. A 3.5-1. Zasady modelowania PK/PD. wania indywidualnego dawkowania.
MUTSCHLER-2009.indd 90 2010-01-07 22:11:11
Działania niepożądane leków 91
4. Działania niepożądane leków
Gdy zapewnia się, że dany lek nie powoduje żadnych efek- O tym, że działanie początkowo uważane za niepożą-
tów niepożądanych, pojawia się uzasadnione podejrzenie, dane może z czasem stać się działaniem podstawowym,
że jest on również pozbawiony działania podstawowego. świadczy przykład rozwoju leków moczopędnych oraz
G. Kuschinsky doustnych leków przeciwcukrzycowych pochodnych sul-
fonamidowych. Początkowo były one używane jako leki
bakteriostatyczne, jednak kiedy odkryto, że stosowanie sul-
fonamidów jako efekty uboczne powoduje wzrost wydzie-
lania moczu oraz obniżenie poziomu cukru, zaczęto syste-
W nielicznych tylko przypadkach możliwe jest usu-
matycznie syntetyzować kolejne pochodne tych związków,
nięcie przez lek stanu patologicznego bez równoczes- doprowadzając do uzyskania substancji, które miały silne
nego wpływu na inne funkcje organizmu. Podczas działanie moczopędne i przeciwcukrzycowe bez działania
stosowania prawie wszystkich leków należy liczyć bakteriostatycznego.
się bowiem z możliwością wystąpienia działań niepo-
żądanych, czyli działań występujących oprócz dzia- Potocznie przez sformułowanie działania uboczne
łania terapeutycznego. W zależności od rodzaju oraz rozumie się dzisiaj z reguły tylko niepożądane dzia-
Działania niepożądane
konkretnego przypadku działania leków mogą być łania leków. Z tego względu wyrażenia te są zwykle
pożądane lub niepożądane, nieszkodliwe lub groźne, używane zamiennie. Ponieważ niepożądane działania
zależne lub niezależne od dawki oraz przewidywalne leków wpływają silnie na terapię farmakologiczną,
bądź też nie. I tak na przykład antyproliferacyjne dzia- znajomość ich rodzaju oraz częstości występowania
łanie glukokortykosteroidów u pacjentów z łuszczycą jest niezbędna do określenia stosunku ryzyka zwią-
leków
będzie pożądane, a u pacjentów z wypryskiem endo- zanego z chorobą do ryzyka związanego z leczeniem,
gennym wręcz przeciwnie. Do nieszkodliwych dzia- a także przy ocenie stosunku korzyść-ryzyko. Ponie-
łań niepożądanych zalicza się na przykład przemija- waż rzadko występujące działania niepożądane mogą
jące nudności, podczas gdy wystąpienie groźnych dla zostać wykryte dopiero po dłuższym czasie od wpro-
życia efektów ubocznych, np. w postaci uszkodzenia wadzenia nowego leku na rynek, leki takie należy
A4
miąższu wątroby, wymagać może leczenia stacjonar- przepisywać z ostrożnością i uważnie obserwować
nego bądź też prowadzić może do przedłużenia te- ich działanie.
rapii, trwałych szkód zdrowotnych, a nawet śmierci. Lek może być uznany za niebudzący zastrzeżeń
Do działań niepożądanych możliwych do przewi- wyłącznie wówczas, gdy podczas jego prawidłowe-
dzenia należy między innymi szkodliwy wpływ cyto- go stosowania nie trzeba się liczyć z takimi działa-
statyków na tworzenie się leukocytów. Reakcje aler- niami niepożądanymi, które mogłyby w niemożliwy
giczne natomiast są (z wyjątkiem ich występowania do zaakceptowania sposób podwyższyć ryzyko tera-
u alergików) zasadniczo nieprzewidywalne. pii w odniesieniu do jej oczekiwanego celu.
MUTSCHLER-2009.indd 91 2010-01-07 22:11:11
92 Działania niepożądane leków
4.1. Działania niepożądane swoiste
dla leku i zależne od dawki
Tego rodzaju działania niepożądane leków charakte- du nerwowego, przez zaburzenia żołądkowo-jelito-
ryzują się tym, że dają się wyjaśnić za pomocą me- we, uszkodzenia miąższu wątroby i nerek, zmiany
chanizmu działania leku i dlatego są przewidywalne. w obrazie krwi, aż do działań teratogennych lub ra-
Ich siła i zakres są przy tym zależne od dawki. Jeśli kotwórczych.
lek zostanie użyty w zbyt dużej dawce (przedawko-
wany), to spowoduje wystąpienie działań niepożąda- Choroby polekowe. Przez to pojęcie rozumie się
nych u każdego człowieka. Ponieważ jednak ludzie takie stany chorobowe, które nie ustępują nawet
różnią się między sobą indywidualną tolerancją po odstawieniu leku. Należą do nich m.in. głuchota
na leki, istnieje zawsze możliwość, że dawka dobrze po długim używaniu streptomycyny lub dyskinezy
tolerowana przez większość pacjentów u niektórych po stosowaniu neuroleptyków.
spowodować może wystąpienie działań niepożąda-
nych. Istotną przyczyną tego rodzaju biologicznego Wtórne niepożądane działania leków. Jako wtór-
zróżnicowania są – podobnie jak w przypadku badań ne działania niepożądane leków określa się niepo-
przedklinicznych na zwierzętach – podłoże genetycz- żądane skutki wynikające z podstawowego działania
ne i czynniki środowiskowe oraz wynikające z nich danego leku. Dobrym przykładem są tu zaburzenia
różnice we wchłanianiu, dystrybucji i eliminacji (far- fizjologicznej flory bakteryjnej, lub zmiana czynnika
makokinetyce leku), a także w gęstości receptorów zakaźnego podczas leczenia antybiotykami o dużym
lub ich rozmieszczeniu. zakresie. Do tej kategorii działań niepożądanych za-
W związku ze znaczną liczbą stosowanych le- licza się również odczyn Herxheimera, który polega
ków działania niepożądane mogą występować pod na nagłym uwolnieniu do krwiobiegu dużych ilości
wieloma postaciami, od zaburzeń centralnego ukła- toksyn z bakterii zabitych przez antybiotyk.
4.2. Reakcje alergiczne na leki
Słowo „alergia” oznacza zmienioną odpowiedź organizmu tego procesu jest podłoże genetyczne, a oprócz tego
na określoną substancję (alergen). Odpowiedź ta może być ważną rolę odgrywają tu częstość i rodzaj kontaktu
zwiększona (hiperergia), osłabiona (hipoergia) bądź też z antygenem.
może w ogóle nie występować (anergia). Obecnie przyjęło
się, że słowo alergia używane jest wyłącznie w znaczeniu Większość leków to substancje o małych czą-
hiperergii. steczkach, które same w sobie nie mają żadnych
właściwości antygenowych. Aby uzyskać potencjał
Około 15–20% obserwowanych działań niepożąda- immunogenny, lek musi najpierw jako preantygen
nych leków związanych jest z reakcjami alergicznymi. (półantygen, hapten) połączyć się kowalencyjnie
Zwykle dotyczą one skórnych odczynów polekowych z endogennym związkiem wielkocząsteczkowym,
– najczęściej obserwuje się osutkę plamisto-grudko- z reguły białkowym, tworząc kompleks antygenowy
wą, która zalicza się do reakcji typu późnego (zob. (pełny antygen), przeciwko któremu wytwarzane
poniżej), a w dalszej kolejności pokrzywkę i obrzęk są przeciwciała. Przy czym swoistość przeciwciał
naczynioruchowy (reakcje typu natychmiastowego). skierowana jest przeciwko lekowi, a nie związkowi
W przeciwieństwie do charakterystycznych dla wielkocząsteczkowemu. Oznacza to, że to w obrębie
leku efektów niepożądanych omówionych w roz- leku lub jego części znajdują się determinanty antyge-
dziale 4.1 reakcje alergiczne są w dużej mierze nie- nowe, które są rozpoznawane przez swoiste przeciw-
zależne od dawki oraz niespecyficzne dla danej ciała. Wiele leków jest tzw. prohaptenami, co znaczy,
substancji. U ich podłoża zawsze leżą mechanizmy że jako takie nie są one immunologicznie aktywne,
immunologiczne. Warunkiem wystąpienia reakcji ale po przekształceniu w organizmie do reaktywnych
alergicznej jest uprzedni kontakt z antygenem, któ- metabolitów mogą w połączeniu z endogennymi biał-
ry określany jest mianem uwrażliwienia. Istotne dla kami tworzyć pełne antygeny.
MUTSCHLER-2009.indd 92 2010-01-07 22:11:12
Działania niepożądane leków 93
Tabela A 4.2-1. Ważne grupy leków, między którymi dochodzi do zjawiska krzyżowej reakcji alergicznej (wg Jägera)
Grupa chemiczna Związki, między którymi dochodzi
lub klasa związków do reakcji krzyżowej
Grupa paraaminowa acetanilid, barwniki azowe, kosmetyki, kwas paraaminobenzoesowy, fenotiazyna, p-fenylenodiamina,
wywoływacze fotograficzne, pochodne prokainy, sulfonamidy, pochodne sulfonylomocznika
Sulfonamidy związki hamujące anhydrazę węglanową (np. acetazolamid), niektóre diuretyki (tiazydy, furosemid),
disulfiram oraz (rzadko) środki miejscowo znieczulające z grupą p-aminofenylową,
pochodne sulfonylomocznika
Antybiotyki β-laktamowe amoksycylina, ampicylina, karbenicylina i inne półsyntetyczne pochodne
penicyliny, cefalosporyny
Antybiotyki zawierające bacytracyna, dihydrostreptomycyna, neomycyna, streptomycyna, rzadziej framycetyna, gentamycyna,
deoksystreptaminę kanamycyna i paromycyna (zawierają deoksystreptaminę w postaci glikonu)
Kwas prawie wszystkie niesteroidowe leki przeciwzaplane
acetylosalicylowy (u podstawy leży jednak reakcja pseudoalergiczna)
Grupa etylenoaminowa aminofilina, prometazyna
Grupa tiazynowa leki przeciwhistaminowe, różne pochodne fenotiazyny
Działania niepożądane
Grupa terpenowa substancje zapachowe (np. lawendowa, fiołkowa, goździkowa), wyroby gumowe, plastry,
substancje znajdujące szerokie zastosowanie w syropach oraz mazidłach
Reakcja przeciwciała z określoną determinantą nych jednak przypadkach, przy wielokrotnie powta-
leków
antygenową leku leży u podstaw antygenowości gru- rzanej ekspozycji na antygen dojść może do nadmier-
powej. Przez to pojęcie rozumie się zjawisko, kiedy nej odpowiedzi, która jest szkodliwa dla organizmu.
to różniące się pod względem chemicznym i farmako- Ze względu na rodzaj tej odpowiedzi reakcje nad-
logicznym leki mogą wywoływać takie same reakcje wrażliwości zależne od przeciwciał podzielić można A4
alergiczne, jeśli posiadają takie same determinanty na następujące typy:
antygenowe. Dlatego alergia na określoną substancję
■ typ I – (reakcje amafilaktyczne),
może być przyczyną wystąpienia reakcji alergicznej
wobec innej, podobnej strukturalnie substancji, nawet ■ typ II – (reakcje cytotoksyczne),
jeśli organizm nie miał z nią nigdy kontaktu. Zjawi-
■ typ III – (reakcje z udziałem kompleksów immuno-
sko to nazywa się alergią krzyżową (zob. tab. A 4-1).
logicznych).
Charakterystycznym jej przykładem jest antygeno-
wość grupowa, a w konsekwencji alergia krzyżowa Zob. ryc. A 4.2-1 oraz tab. A 4.2-2.
na związki w pozycji para pierścienia aromatyczne-
go z pierwszorzędową grupą aminową (np. prokaina,
Reakcje typu I. Reakcje nadwrażliwości typu I za-
kwas p-aminosalicylowy, sulfonamidy).
leżne są od przeciwciał klasy IgE, które znajdują się
W zależności od rodzaju mechanizmów leżących
w błonie mastocytów i granulocytów zasadochłon-
u podłoża reakcji alergicznych rozróżnia się reakcje
nych (zob. ryc. A 4.2-1). Występują one najczęściej
nadwrażliwości zależne od przeciwciał oraz od lim-
w czasie od kilku minut do jednej godziny po kontak-
focytów T.
cie z antygenem i z tego powodu nazywane są reak-
Podobne w przebiegu do reakcji alergicznych
cjami typu natychmiastowego.
są reakcje pseudoalergiczne, które jednakże nie należą
W przypadku istniejących predyspozycji po kon-
do reakcji o podłożu immunologicznym (zob. poniżej).
takcie z określonym antygenem (jad pszczół, pyłki,
truskawki, białko ryb, penicyliny i in.) organizm od-
4.2.1. Reakcje nadwrażliwości powiada, produkując szczególnie dużo przeciwciał
klasy IgE, co przy późniejszym kontakcie z tym sa-
zależne od przeciwciał mym antygenem może doprowadzić do rozwinięcia
Reakcje pomiędzy antygenem a przeciwciałem prze- się reakcji anafilaktycznej. Przeciwciała klasy IgE
biegają zwykle bez widocznych objawów. W określo- łączą się swoim odcinkiem Fc z receptorami IgEFc
MUTSCHLER-2009.indd 93 2010-01-07 22:11:12
94 Działania niepożądane leków
obecnymi na powierzchni mastocytów i granulocy- pazy A2 i doprowadzi w następstwie do uruchomie-
tów zasadochłonnych. Jeżeli dany antygen (alergen) nia kaskady kwasu arachidonowego, a w rezultacie
podczas kolejnej ekspozycji połączy się z dwoma do syntezy i uwalniania leukotrienów oraz cytokin.
miejscami wiązania na przeciwciałach klasy IgE Za pośrednictwem tych nowo powstałych media-
(mostkowanie), spowoduje to, poprzez napływ jo- torów oraz dzięki uwolnieniu już istniejących sub-
nów wapnia do wnętrza komórki, aktywację fosfoli- stancji z ziarnistości cytoplazmatycznych dochodzi
Typ I Typ II
antygen na powierzchni komórki
antygen receptor Fc
IgG
makrofag komórka
IgE docelowa
reakcja cytotoksyczna
receptor Fc przeciwciało
degranulacja mastocytów dopełniacz komórka
docelowa
liza za pośrednictwem
skurcz oskrzeli układu dopełniacza
pokrzywka ↑ przepuszczalności
naczyń
Typ III Typ IV
antygen
CD4+-
komórka T
CD -4
chemotaksyny, mediatory reakcji zapalnej
odkładanie się MHC-II TCR
aktywacja układu dopełniacza
kompleksów
immunologicznych komórka prezentująca
antygen (APC)
dopełniacz
reakcje limfokiny
przeciwciało zapalne
antygen
aktywowane makrofagi
śródbłonek błona podstawna
Ryc. A 4.2-1. Reakcje nadwrażliwości. Typ I to reakcja natychmiastowa zachodząca za pośrednictwem przeciwciał klasy IgE. Typ
II indukowany jest przez przeciwciała klasy IgG, co prowadzi do aktywacji układu dopełniacza oraz fagocytozy. Typ III to reakcja
zachodząca za pośrednictwem kompleksów immunologicznych z przeciwciałami klasy IgG przeciwko antygenom rozpuszczalnym,
a także związanym z macierzą pozakomórkową. Typ IV to komórkowa reakcja zapalna, która zachodzi za pośrednictwem limfocytów
Th i makrofagów lub też bezpośrednio z udziałem cytotoksycznych limfocytów T. TCR – receptor limfocytów T (T Cell Receptor).
MUTSCHLER-2009.indd 94 2010-01-07 22:11:12
Działania niepożądane leków 95
do wyzwolenia charakterystycznych reakcji wtór- mediatorów wywołują obrzęk i pokrzywkę (urtica-
nych. Bardzo aktywne mediatory (histamina, seroto- ria). W wielu przypadkach reakcja amafilaktyczna
nina, bradykinina, heparyna, leukotrieny) powodują ma charakter ograniczony (np. w astmie oskrzelo-
ciężkie zaburzenia czynnościowe (anafilaksję). Roz- wej czy katarze siennym), jednak u osób szczególnie
szerzenie naczyń i wzrost przepuszczalności naczyń predysponowanych może dojść do jej uogólnienia
włosowatych wskutek działania histaminy i innych (np. po iniekcji określonych leków czy użądleniu
Tabela A 4.2-2. Klasyfikacja patomechanizmów alergicznych reakcji na leki z przykładami. Możliwe jest nakładanie się różnych ty-
pów reakcji.
Zmodyfikowana klasyfikacja Uczestniczące w odpowiedzi Objawy kliniczne i obraz
wg Gella-Coombsa immunologicznej komórki, mediatory choroby z przykładami
(wg czasu reakcji) i mechanizmy efektorowe wywołujących je leków
Typ I
Reakcje typu natychmiastowego IgE; degranulacja komórek tucznych Pokrzywka, obrzęk, reakcja anafilaktyczna;
(od sekund do minut, ew. godziny) antybiotyki β-laktamowe, sulfonamidy,
pochodne pirazolonu, białka obcogatunkowe
Typ II
Działania niepożądane
Reakcje opóźnione IgG/IgM oraz FcR; Hemoliza, trombocytopenia, agranulocytoza;
(od minut do godzin, ew. dni) zniszczenie komórek za pośrednictwem FcR penicylina, sulfonamidy, metyldopa,
tiouracyl, chinina
Typ III*)
Reakcje opóźnione IgG/IgM, aktywacja układu dopełniacza; Zapalenia naczyń (ew. towarzyszące
leków
(od minut do godzin, ew. dni) odkładanie się kompleksów zapalenie stawów, błon surowiczych,
immunologicznych kłębuszków nerkowych)
Typ IV
Reakcje typu późnego A4
(od godzin do dni, ew. tygodnie)
Typ IVa**) Komórki Th1; Wyprysk kontaktowy; neomycyna,
IFN-γ, IL-2, TNF-α; chloramfenikol, bufeksamak, metale
aktywacja monocytów (nikiel, chrom).
Trwała osutka polekowa;
barbiturany, sulfonamidy, tetracykliny,
fenylbutazon, niesteroidowe
leki przeciwzapalne, paracetamol
Typ IVb Komórki Th2; Polekowa osutka plamisto-grudkowa,
IL-4, IL-5, IL-13; reakcja pęcherzowy rumień wielopostaciowy; penicyliny.
zapalna zależna od eozynofili Polekowy odczyn z eozynofilią
i objawami układowymi (DRESS);
leki przeciwpadaczkowe, allopurinol, sulfonamidy
Typ IVc Komórki T cytotoksyczne; perforyna/ Polekowa osutka plamisto-grudkowa,
granzym B, Fas-ligand (CD95); pęcherzowa i krostkowa, zespół Stevensa-
zniszczenie komórek (np. keratynocytów) -Johnsona (SJS), toksyczna nekroliza naskórka (TEN);
za pośrednictwem limfocytów CD4 lub CD8 allopurinol, przeciwbakteryjne sulfonamidy,
niesteroidowe leki przeciwzapalne pochodne oksykamu,
leki przeciwpadaczkowe (karbamazepina, lamotrygina,
fenytoina, fenobarbital), newirapina
Typ IVd Komórki T; IL-8, GM-CSF; Ostra uogólniona osutka krostkowa (AGEP);
mobilizacja antybiotyki β-laktamowe
i aktywacja neutrofili
*) Przykładem uogólnionej reakcji typu III jest choroba posurowicza, a miejscowych (zjawisko Arthusa) – płuco hodowcy drobiu, płuco farmera i celiakia.
**) Pierwowzorem reakcji typu IV jest odczyn tuberkulinowy.
MUTSCHLER-2009.indd 95 2010-01-07 22:11:13
96 Działania niepożądane leków
przez pszczoły lub osy). W efekcie dochodzi do sil- Przykładem uogólnionej reakcji typu III jest tak
nego spadku ciśnienia tętniczego krwi (wstrząs ana- zwana choroba posurowicza, która dawniej często
filaktyczny). Oprócz tego mogą wystąpić skurcze występowała po podaniu obcogatunkowej surowicy.
mięśni gładkich oskrzeli, które w krańcowych przy- Jeżeli kompleksy immunologiczne są względ-
padkach mogą doprowadzić do śmierci. nie duże, to odkładają się w miejscu powstawania
Reakcje typu I mogą w zasadzie zostać wywołane i wywołują tam miejscową reakcję zapalną (zjawisko
przez wszystkie leki, które tworzą antygeny w połącze- Arthusa). Przykładem miejscowych reakcji typu III
niu z cząsteczkami organizmu. Względnie często re- są alergiczne choroby płuc – płuco farmera (antyge-
akcje typu I obserwuje się w przypadku antybiotyków nem jest tu spleśniałe siano), płuco hodowcy ptaków
β-laktamowych, sulfonamidów i ich pochodnych. (antygenem są odchody gołębi i kur), a także celiakia
(alergenem jest gliadyna z ziaren zbóż).
Do leków, po których podaniu mogą wystąpić
Reakcje typu II. Reakcje nadwrażliwości typu II
reakcje typu III, należą na przykład penicyliny oraz
zachodzą po związaniu się przeciwciał klasy IgG
penicylamina.
lub IgM z antygenami obecnymi na powierzchni ko-
mórek. W wyniku tego dochodzi do ich opsonizacji
i aktywacji układu dopełniacza, co prowadzi do lizy 4.2.2. Reakcje nadwrażliwości
komórki (reakcja cytotoksyczna). W reakcjach typu
drugiego uczestniczą antygenowe struktury komó-
zależne od limfocytów T
rek krwi, na których powierzchni odkładają się leki
Tego typu reakcje, określane też jako nadwrażliwość
będące haptenami. W zależności od rodzaju zaan-
typu komórkowego, polegają na specyficznych re-
gażowanych komórek obserwuje się różnego typu
akcjach immunologicznych, które zachodzą za po-
zaburzenia. Na przykład w przypadku erytrocytów
średnictwem limfocytów T (reakcje nadwrażliwości
dochodzi do anemii hemolitycznej, a w przypadku
typu IV) i przebiegają bardzo podobnie do specyficz-
płytek krwi do trombocytopenii ze zwiększoną ten-
nej komórkowej odpowiedzi immunologicznej na pa-
dencją do krwawień. Kiedy uczestniczą w niej neu-
togeny. W trakcie pierwszego kontaktu z antygenem
trofile może dojść do agranulocytozy, a następnie
rozpoczyna się faza uwrażliwienia, która jest kli-
zaburzeń mechanizmów obrony przeciwbakteryjnej.
nicznie niema. Lek lub jego metabolit łączy się jako
Reakcje typu II z reguły występują po pewnym cza-
hapten z endogennym białkiem, tworząc kompleks
sie, to znaczy najwcześniej wiele godzin po podaniu
hapten-białko, który jest następnie wiązany na po-
leku.
wierzchni komórek reprezentujących antygen (APC)
Jeśli chodzi o leki, to reakcje typu II powodują
i zostaje rozpoznany przez limfocyty T. W wyni-
między innymi metyldopa, penicyliny, sulfonamidy,
ku interakcji z receptorem komórkowym obecnym
pochodne tiouracylu oraz chinina.
na powierzchni limfocytu T dochodzi do aktywacji
Innymi przykładami tego typu reakcji są reakcje niezgod- i różnicowania się limfocytów T, dzięki czemu po-
ności grupowej krwi po przetoczeniu (reakcje poprzetocze- wstają m.in. specyficzne względem tego antygenu
niowe), niezgodność w zakresie czynnika Rh oraz myasthe-
nia gravis. komórki pamięci. Niektóre leki (np. sulfametoksazol)
mogą w pewnych okolicznościach powodować tego
Reakcje typu III. Reakcje nadwrażliwości typu III typu reakcje bezpośrednio, to znaczy bez uprzed-
wywoływane są przez syntezę i odkładanie się kom- niego wiązania się z endogennym białkiem potrafią
pleksów immunologicznych, co prowadzi do reakcji słabo wiązać się z receptorem na błonie limfocytu T
zapalnej po aktywacji układu dopełniacza. i stymulować go w obecności białka MHC. Kiedy do-
Lokalizacja uszkodzeń zależna jest od wielkości chodzi do ponownego kontaktu z alergenem, komórki
powstających kompleksów antygen-przeciwciało. pamięci uwalniają cytokiny (faza uwalniania).
Jeżeli kompleksy te są względnie niewielkie, mogą Reakcje typu IV można podzielić ze względu
być przenoszone przez krążenie systemowe i wte- na uczestniczące w nich limfocyty T (Th1 lub Th2),
dy reakcja jest uogólniona. Mogą one odkładać się cytokiny, które są przez nie wydzielane (IFN-γ, IL-2
na przykład w kłębuszkach nerkowych, błonach su- bądź IL-4, IL-5), oraz komórki efektorowe (makrofa-
rowiczych, stawach oraz w śródbłonku naczyń, po- gi, granulocyty obojętnochłonne, limfocyty T) na re-
wodując w tych miejscach reakcje zapalne. Reakcje akcje typu IVa, IVb, IVc i IVd.
typu III po podaniu leków objawiać się mogą więc Określenie tego typu reakcji nadwrażliwością typu
jako zapalenie naczyń towarzyszące kłębuszkowemu późnego bierze się stąd, że zachodzą one po upływie
zapaleniu nerek, zapalenie błon surowiczych oraz za- dni, a maksymalne nasilenie osiągają czasem nawet
palenie stawów. dopiero po kilku tygodniach od zainicjowania.
MUTSCHLER-2009.indd 96 2010-01-07 22:11:13
Działania niepożądane leków 97
Jako przykład reakcji nadwrażliwości zależnej od limfocy- rakteryzują się występowaniem zmian rumieniowych
tów T może służyć reakcja tuberkulinowa, która zachodzi i pęcherzowych oraz martwicą skóry. Zmiany te obej-
po wstrzyknięciu białkowych antygenów prątka gruźlicy.
mują duże powierzchnie skóry i przypominać mogą
Do reakcji nadwrażliwości zależnych od limfocytów T
należą również reakcje odrzucania przeszczepu. Po trans- oparzenia drugiego stopnia. Zwykle zajęte są również
plantacji przeszczep będzie tym silniej i szybciej odrzucany, błony śluzowe (jamy ustnej, narządów płciowych,
im bardziej różnią się antygeny zgodności tkankowej daw- spojówki). Zmianom skórnym towarzyszą objawy
cy od antygenów biorcy. Antygeny zgodności tkankowej ogólne, takie jak gorączka, bóle stawów i kończyn,
(antygeny transplantacyjne, HLA) są obecne na wszystkich
a także osłabienie prowadzące niekiedy nawet do za-
komórkach organizmu posiadających jądro.
burzeń świadomości. W przypadku zespołu Steven-
sa-Johnsona zajęciu przez pęcherze ulega mniej niż
Alergie na leki, które zachodzą za pośrednictwem re-
10% powierzchni ciała, a w zespole Lyella więcej niż
akcji nadwrażliwości typu IV objawiają się głównie
30%. W przypadku tego ostatniego w 45% przypad-
zmianami skórnymi. Przykładami są tu wyprysk kon-
ków dochodzi do zgonu, głównie z powodu wtórnych
taktowy i często występująca osutka plamisto-grud-
zakażeń skóry. W mieszanych postaciach SJS-TEN
kowa.
dochodzi do zajęcia 10–30% powierzchni ciała.
Do reakcji nadwrażliwości typu IV należy naj-
prawdopodobniej również pęcherzowy rumień wie- Za przyczynę tych reakcji uważa się zależną od limfocytów
lopostaciowy (zespół Stevensa-Johnsona, toksyczna nadwrażliwość typu IVc, jednak dokładny patomechanizm
nekroliza naskórka) i polekowa osutka krostkowa, nie jest do końca wyjaśniony. Okres utajenia wynosi do 14
a także trwała osutka polekowa oraz polekowy od- dni od podania leku, co utrudnia ustalenie związku z kon-
Działania niepożądane
czyn z eozynofilią i objawami układowymi (zespół kretnym preparatem.
Substancje, które mogą wyzwalać tego typu reakcję,
DRESS – Drug Related Eosinophilia with Systemic to przede wszystkim allopurinol, sulfonamidy, niesteroido-
Symptoms). we leki przeciwzapalne – pochodne oksykamu, leki prze-
Do leków, które najczęściej powodują powstawa- ciwpadaczkowe (karbamazepina, lamotrygina, fenytoina
nie tego rodzaju zmian skórnych, należą leki przeciw- i fenobarbital), a także nienukleozydowy inhibitor odwrot-
infekcyjne (β-laktamy, sulfonamidy) oraz przeciwpa- nej transkryptazy – newirapina.
leków
W przypadku leczenia przewlekłego największe ryzyko
daczkowe leki z pierścieniem aromatycznym. występuje w trakcie pierwszych dwóch miesięcy terapii.
W przypadku alergicznego wyprysku kontakto-
Ostra uogólniona osutka krostkowa – AGEP (Acu-
wego faza uwrażliwiania trwa około 10–14 dni i z re-
te Generalized Exanthematous Pustulosis) jest ciężką
A4
guły przez dłuższy czas pozostaje trwała względem
reakcją skórną na lek, której towarzyszą gorączka,
danego antygenu. Każdy ponowny kontakt z nim
leukocytoza i eozynofilia. Ustępuje po odstawieniu
prowadzi najpóźniej w ciągu 48 godzin do powstania
wywołującego ją leku i w przeciwieństwie do SJS
stanu zapalnego. Mechanizm jest prawdopodobnie
czy TEN rzadko prowadzi do śmierci. Jej występo-
taki sam jak w przypadku reakcji tuberkulinowej – re-
wanie często pozostaje niepowiązane z faktem za-
akcja typu IVa. Alergenami kontaktowymi są zwykle
życia leku. W większości przypadków wywoływana
związki niskocząsteczkowe i lipofilne, które dobrze
jest przez antybiotyki β-laktamowe, ale można ją rów-
penetrują w głąb naskórka. Alergię kontaktową czę-
nież zaobserwować po zażyciu takich leków, jak ibu-
sto wywołują metale, takie jak chrom i nikiel. Alergia
profen, metamizol, paracetamol czy celekoksyb.
kontaktowa na leki występuje na przykład po miej-
scowym zastosowaniu preparatów zawierających
Trwała osutka polekowa objawia się pojedynczymi pla-
neomycynę, chloramfenikol czy bufeksamak. mami na skórze, które po wygojeniu przechodzą w trwa-
łe przebarwienia. W przypadku ponownego zastosowania
Polekowa osutka plamisto-grudkowa, z charakterystycz- wywołującego ją leku dochodzi do ponownego wystąpienia
nymi wykwitami skórnymi zaliczana jest do najczęstszych zmian w tych samych miejscach, niejednokrotnie bardziej
działań niepożądanych i może powstać po zastosowaniu nasilonych. Najczęściej wywołują ją barbiturany, sulfona-
wielu leków. Występuje 8–12 dni po rozpoczęciu terapii midy, tetracykliny, fenylbutazon, niesteroidowe leki prze-
i z reguły goi się bez komplikacji. U jej podłoża leży naj- ciwzapalne, a także paracetamol.
prawdopodobniej reakcja typu IVb.
Polekowy odczyn z eozynofilią i objawami układowy-
Pęcherzowy rumień wielopostaciowy występują- mi – DRESS (Drug Related Eosinophilia with Systemic
cy po podaniu leków obejmuje rzadko objawiające Symptoms) został opisany po raz pierwszy jako reakcja
nadwrażliwości po podaniu fenytoiny i innych leków
się, ale bardzo ciężkie w przebiegu reakcje skórne przeciwpadaczkowych. Dlatego początkowo był nazywa-
– zespół Stevensa-Johnsona (SJS) oraz toksyczną ny zespołem nadwrażliwości na leki przeciwpadaczkowe.
nekrolizę naskórka (TEN, zespół Lyella), które cha- W okresie 1–8 tygodni po podaniu leku dochodzi do wystą-
MUTSCHLER-2009.indd 97 2010-01-07 22:11:13
98 Działania niepożądane leków
Tabela A 4.2-3. Możliwe czynniki ryzyka wystąpienia skórnych jest ryzyko wystąpienia toksycznej nekrolizy naskór-
reakcji alergicznych ka po podaniu sulfonamidów bądź zespołu DRESS
po podaniu leków przeciwpadaczkowych. Podobnie
Czynniki ryzyka wpływ na wystąpienie reakcji alergicznych zwiększyć
Antygenowość grupowa/alergie krzyżowe mogą współistniejące zakażenia. Na przykład pacjenci
chorzy na AIDS mają 10–50 razy większe ryzyko wy-
Droga podania (przezskórna > doustna > dożylna) stąpienia skórnej reakcji alergicznej w trakcie terapii
Dawka (niższa > wyższa) kotrimoksazolem niż pacjenci bez AIDS.
Terapia przerywana
Czynniki farmakogenetyczne (typ metaboliczny) 4.2.3. Sposoby zapobiegania
oraz immunogenetyczne (typ HLA)
reakcjom alergicznym
Choroby towarzyszące, np. infekcje wirusowe, choroby
autoimmunologiczne, przewlekła białaczka limfatyczna
Siłę reakcji alergicznych oraz częstość ich występo-
wania można zmniejszyć dzięki przestrzeganiu nastę-
pienia plamisto-grudkowej wysypki oraz gorączki, powięk- pujących zasad:
szenia węzłów chłonnych, zajęcia narządów (nerek, wątro-
by) i eozynofilii. Śmiertelność wynosi ok. 10%. Do leków,
■ jasne określenie wskazań dla danego leku i w miarę
które mogą powodować tę reakcję, zaliczamy leki przeciw- możliwości prowadzenie monoterapii,
padaczkowe, które ze względu na zawarty w ich cząsteczce
pierścień aromatyczny mogą powodować nadwrażliwość
■ poszerzenie wywiadu o informacje dotyczące wcześ-
krzyżową. Zespół DRESS powodować mogą także allopu- niejszych reakcji alergicznych,
rinol i sulfonamidy.
■ unikanie terapii miejscowej lekami silnie alergizu-
jącymi (np. penicylinami),
Czynniki, które mogą doprowadzić do wystąpienia
■ uważna obserwacja pacjentów leczonych przewle-
ostrych skórnych reakcji alergicznych, ujęte są w tab.
kle – np. kontrola obrazu krwi i układu krzepnię-
A 4.2-3.
cia,
U pacjentów uwrażliwionych na związki spokrew-
nione chemicznie istnieje podwyższone ryzyko wystą- ■ informowanie pacjenta o możliwych objawach
pienia reakcji krzyżowej. W przypadku wielu substancji reakcji alergicznych (np. skórnych), również jeśli
decyduje o tym rodzaj metabolizmu – tak jak w przy- chodzi o leki bez recepty, aby mógł on je w razie
padku wolnych acetylatorów, u których podwyższone czego samodzielnie rozpoznać i odstawić lek.
MUTSCHLER-2009.indd 98 2010-01-07 22:11:13
Działania niepożądane leków 99
4.3. Reakcje pseudoalergiczne
Oprócz reakcji alergicznych na lek istnieją rów- przeciwzapalnych, która polega na hamowaniu
nież tzw. reakcje pseudoalergiczne, których mecha- syntezy prostaglandyn i wynikającej z tego zwięk-
nizm nie polega na odpowiedzi immunologicznej, szonej syntezie leukotrienów,
ale na bezpośrednim uwalnianiu przez daną substancję
■ reakcje anafilaktoidalne po podaniu środków kon-
mediatorów reakcji zapalnej, aktywacji układu dopeł-
trastowych,
niacza czy wpływaniu na kaskadę kwasu arachidono-
wego. W tego typu przypadkach nie jest konieczne ■ obrzęk naczyniowy po podaniu inhibitorów kon-
wcześniejsze uwrażliwienie na daną substancję – re- wertazy angiotensyny wskutek podwyższenia stę-
akcje te zachodzą również przy pierwszym podaniu żenia bradykininy,
leku. Do reakcji pseudoalergicznych należą np.:
■ spadek ciśnienia krwi i skurcz oskrzeli po iniek-
■ tzw. astma aspirynozależna po podaniu kwasu ace- cji tubokuraryny, wynikający z uwolnienia hista-
tylosalicylowego i innych niesteroidowych leków miny.
4.4. Wpływ na zdolność prowadzenia
Działania niepożądane
pojazdów mechanicznych
Co piąty wypadek drogowy w Polsce związany Niezależnie od wpływu na czas reakcji zdolność
jest z zażywaniem leków. do prowadzenia pojazdów może ulec pogorszeniu
leków
Wiele leków może wpływać na czas reakcji po- przez negatywny wpływ na zdolność widzenia po za-
przez swoje działanie sedatywne. Należą do nich stosowaniu leków okulistycznych (np. leków rozsze-
na przykład środki nasenne i uspokajające, leki psy- rzających źrenicę) lub przez zmiany metaboliczne
chotropowe, opioidowe leki przeciwbólowe, leki występujące na skutek błędów w dawkowaniu insuli- A4
przeciwhistaminowe, niektóre leki przeciwnadciśnie- ny u diabetyków.
niowe i przeciwpadaczkowe oraz środki zawierające Niezbędne jest udzielenie pacjentowi tego typu
alkohol. (Równoczesne spożycie alkoholu nasila z re- informacji przez lekarza lub aptekarza w przypadku
guły sedację po lekach). stosowania danego leku po raz pierwszy. Pacjent po-
Po krótkich zabiegach, przy których używa się winien być również świadomy tego, że należy kry-
środków znieczulających, upośledzenie zdolności tycznie oceniać swoje zdolności w zakresie zdolności
kierowania pojazdami może utrzymywać się bardzo do prowadzenia pojazdów lub podejmowania innych
długo, dlatego w takim przypadku należy unikać pro- niebezpiecznych czynności, np. obsługiwania ma-
wadzenia pojazdów przez 48 godzin. szyn.
MUTSCHLER-2009.indd 99 2010-01-07 22:11:14
100 Działania niepożądane leków
4.5. Działania niepożądane
w czasie rozwoju zarodkowego, płodowego
oraz w okresie poporodowym i karmienia piersią
Pomimo licznych ostrzeżeń kobiety w ciąży nadal za- pełnego rozdzielenia się komórek potomnych lub grupy ko-
żywają leki bez istotnych wskazań. Rodzi to niebez- mórek zygoty podczas wczesnego stadium rozwojowego.
Jeżeli zaburzenie to jest symetryczne, dochodzi do powsta-
pieczeństwo rozwinięcia się uszkodzeń płodu (tok- nia połączonych ze sobą identycznych organizmów (bliź-
syczność reprodukcyjna), ponieważ większość leków niąt syjamskich). Dla dokładniejszego ich nazwania dodaje
przenika z łatwością przez łożysko, a komórki za- się nazwę zrośniętej części ciała (np. thoracopagus = zroś-
rodka są szczególnie wrażliwe na działanie środków nięcie przez klatkę piersiową). Kiedy zaburzenie to rozwija
farmakologicznych (tzw. toksyczność selektywna). się niesymetrycznie, jeden zarodek rozwija się prawidłowo,
a drugi pozostaje w postaci szczątkowej.
Żaden lek nie może być uznany za w 100% bezpiecz-
ny. Z drugiej strony nie jest znany żaden czynnik, Embriopatie. Podczas embriogenezy w ściśle określo-
który powodowałby szkody w każdym przypadku. nych przedziałach czasowych dochodzi do rozwoju na-
Z istniejących badań wynika na przykład, że różycz- rządów z poszczególnych blastem. Rodzaj uszkodzeń
ka powoduje uszkodzenia płodu w 50% przypadków, zależy od momentu ekspozycji na czynnik szkodliwy.
a talidomid w 30%. Jeżeli w momencie narażenia dana blastema jest jeszcze
niezróżnicowana, a uszkodzenie nie jest letalne istnieje
W zależności od stadium rozwoju płodowego możliwość całkowitego przywrócenia stanu prawidłowego.
uszkodzenia mogą wystąpić w trakcie: Jeżeli czynnik szkodliwy zadziała w trakcie różnicowania
się blastemy powstaje typowa embriopatia w postaci izolo-
■ blastogenezy, wanej wady rozwojowej. Po zakończeniu różnicowania się
■ embriogenezy, czynniki szkodliwe nie mogą już powodować wad rozwo-
jowych. Okres, w którym zadziałanie czynnika uszkadzają-
■ fetogenezy. cego może spowodować rozwinięcie się wady rozwojowej,
nazywany jest okresem krytycznym (wrażliwym). Innymi
Blastopatie. Mianem blastopatii określa się zaburzenia słowy, konkretna wada rozwojowa powstać może wyłącz-
rozwojowe, które zachodzą podczas blastogenezy, czyli nie w określonym czasie, a jej rodzaj zależy w mniejszym
od momentu zapłodnienia do 15 dnia ciąży. Ciężkie uszko- stopniu od rodzaju czynnika szkodliwego, a w większym
dzenia w tym okresie prowadzą do obumarcia zarodka. od okresu rozwojowego zarodka. Największe ryzyko po-
Uszkodzenia mniejszego stopnia mogą ulec całkowitemu wstania wad rozwojowych istnieje pomiędzy 4. a 8. tygo-
cofnięciu, ponieważ do tego momentu rozwojowego słabo dniem ciąży. Na ryc. A 4.5-1 schematycznie przedstawiono
zróżnicowane komórki zarodka mają duże zdolności rege- fazy rozwoju, w których ludzki zarodek i płód są szczegól-
neracyjne. Blastopatie jako wady rozwojowe ujawniają się nie zagrożone.
często w postaci wad podwójnych, kiedy to dochodzi do nie-
Tabela A 4.5-1. Okresy rozwojowe płodu
Nazwa Okres Procesy biologiczne Zaburzenia rozwoju
Gametogeneza przed zapłodnieniem rozwój męskich aberracje chromosomalne
i żeńskich komórek płciowych (np. trisomia chromosomu 21)
Blastogeneza 0–15 dzień pierwszy podział zygoty, śmierć zarodka, podwójne wady rozwojowe
rozwój blastuli, symetryczne i asymetryczne
różnicowanie w embrioblast
i trofoblast
Embriogeneza 16–60 dzień tworzenie się narządów izolowane wady rozwojowe, np. rozszczepy,
i układów, różnicowanie się narządów, anomalie układu sercowo-naczyniowego,
przyłączenie się do krążenia matczynego, uszkodzenia wskutek zakażeń wirusowych,
utworzenie łożyska np. embriopatia różyczkowa
Fetogeneza 61 dzień – poród dalszy wzrost, zakończenie różnicowania się uszkodzenia wskutek infekcji,
narządów, dojrzewanie np. przez krętki, toksoplazmę,
choroba hemolityczna noworodków
MUTSCHLER-2009.indd 100 2010-01-07 22:11:14
Działania niepożądane leków 101
Stadium podziałów Okres rozwoju zarodka (w tygodniach) okres rozwoju płodu (w tygodniach) poród
zygoty, implantacji
zarodka i dwulistnej 3 4 5 6 7 8 12 16 20-36 38
tarczy zarodkowej oznacza najczęstsze miejsce działania czynnika teratogennego
OUN serce podniebienie mózg
serce oko oko ucho ucho
ręce
nogi zewnętrzne narządy płciowe
ośrodkowy układ nerwowy
serce
kończyny górne
oczy
kończyny dolne
zęby
zwykle niewrażliwy podniebienie
na substancje
teratogenne zewnętrzne narządy płciowe
Działania niepożądane
uszy
śmierć w okresie poważne anomalie morfologiczne defekty fizjologiczne i mniej poważne
przedporodowym anomalie morfologiczne
leków
Ryc. A 4.5-1. Schematyczne przedstawienie okresów rozwoju, podczas których zarodek, a następnie płód jest narażony na czynniki
teratogenne (wg Ariensa, Mutschlera, Simonisa). Czerwone pola oznaczają okresy szczególnego zagrożenia, żółte pola – okresy
mniejszej wrażliwości.
Do wad rozwojowych powstających na wczesnych eta- Szczególne znaczenie pośród infekcji pierwotniako-
A4
pach rozwoju zarodkowego należą dysrafie (rozszczepy). wych ma toksoplazmoza. Toxoplasma gondi jest szeroko
Należy do nich m.in. rozszczep kręgosłupa (spina bifida). rozpowszechnionym pierwotniakiem, który ma zdolność
Jeżeli dojdzie do przemieszczenia się na zewnątrz roz- zarażania wielu ptaków i ssaków, ale nie zawsze powoduje
szczepu opon rdzenia kręgowego, określa się to mianem u nich objawy choroby. Człowiek jest względnie odporny.
przepukliny oponowej (meningocele). Jeżeli przemieszcze- U dorosłych rzadko dochodzi do rozwinięcia się ciężkiej
niu ulegnie dodatkowo również rdzeń kręgowy, nazywa się postaci tego zakażenia. Jeżeli jednak dojdzie do zakażenia
to przepukliną oponowo-rdzeniową (meningomyelocele). matki we wczesnym okresie ciąży, to przez łożysko może
Wady rozwojowe powstające w późniejszym okresie dojść do zakażenia płodu, a następnie rozwinięcia się cięż-
dotyczą zwykle serca i naczyń. Podobnie jak rozszczepy, kich wad rozwojowych (wodogłowia, odkładania się soli
stanowią one około 40% ogółu wad rozwojowych. Inne ro- wapnia w mózgu i ślepoty jako oznaki przebytego zapale-
dzaje wad rozwojowych zestawiono w tabeli A 4.5-1. nia mózgu). Chociaż uszkodzenia płodu w przypadku tok-
Oprócz czynników genetycznych do przyczyn wystą- soplazmozy w późniejszym okresie ciąży występują częś-
pienia embriopatii zalicza się infekcje wirusowe, promie- ciej, to są one łagodniejsze.
niowanie jonizujące, cukrzycę, padaczkę oraz substancje Po wprowadzeniu do leczenia penicyliny rzadko obec-
chemiczne. Innym ważnym czynnikiem w tym okresie roz- nie występującą fetopatią jest mająca niegdyś istotne zna-
wojowym jest niedobór kwasu foliowego. czenie kiła wrodzona (lues congenita). Wywoływana przez
krętek blady (Treponema pallidum) prowadzi do wystąpie-
nia wielu zaburzeń ujawniających się po porodzie, takich
Fetopatie. W trakcie dojrzewania płodu rozwój narządów jak zmiany skórne, zapalenia chrząstek i kości, uszkodzenia
jest już zakończony. Dlatego w tym okresie czynniki uszka- zębów, uszkodzenia ucha wewnętrznego oraz wiele innych
dzające działają na struktury już zróżnicowane. W przypad- bardzo zróżnicowanych objawów.
ku tego rodzaju uszkodzeń oprócz zaburzeń hormonalnych
(niedoboru hormonów tarczycy) lub konfliktu w zakresie
czynnika Rh chodzi przede wszystkim o infekcje pierwot-
niakowe i bakteryjne. Płód reaguje na nie za pomocą pry- 4.5.1. Działanie teratogenne
mitywnych, mezenchymalnych reakcji obronnych (np. fa-
gocytozy zarazków), ponieważ dopiero pod koniec ciąży
pojawia się zdolność do wywoływania typowych objawów Działanie teratogenne, czyli szkodliwe dla płodu,
reakcji zapalnej. a w szczególności powodujące wady rozwojowe,
MUTSCHLER-2009.indd 101 2010-01-07 22:11:14
102 Działania niepożądane leków
należy do najgroźniejszych efektów niepożądanych go). Taka ocena może być jednak wyłącznie orien-
występujących po zastosowaniu leków. Dlatego przy tacyjna, ponieważ opiera się jedynie na obserwa-
stosowaniu leków w okresie ciąży należy bardzo cjach i nie można na jej podstawie określić stopnia
uważnie oszacować możliwe ryzyko. Jeśli chodzi prawdopodobieństwa. Żaden ze środków leczniczych
o potencjał teratogenny leków, istnieją zarówno iloś- nie może być uznany za absolutnie pewny.
ciowe, jak i jakościowe różnice pomiędzy gatunkami W celu zmniejszenia ryzyka wystąpienia wad wro-
i dlatego nie można bezkrytycznie przenosić na ludzi dzonych na skutek działania teratogennego w lecze-
wyników prób na zwierzętach. Na przykład u zwierząt niu kobiet we wczesnej ciąży należy:
wykazano teratogenne działanie kofeiny i penicyliny,
■ stosować leki wyłącznie w przypadku istotnych
podczas gdy u ludzi nigdy takiego nie stwierdzono.
wskazań, mając na uwadze ryzyko dla matki i dzie-
Częściowo można to tłumaczyć tym, że w ekspery-
cka,
mentach na zwierzętach używane są większe dawki,
których nie stosuje się u ludzi, bądź też tym, że istnie- ■ unikać stosowania leków niedawno wprowadzo-
ją różnice w biotransformacji. nych na rynek.
Do leków o udowodnionym działaniu teratogennym
u ludzi zalicza się m.in. cytostatyki, leki przeciwpa- Należy przy tym szczególnie podkreślić, że zdecy-
daczkowe, retinoidy, alkohol oraz wycofany ze sprze- dowanie większa liczba embriopatii wynika ze spoży-
daży środek nasenny – talidomid. Ponieważ substancje wania w ciąży alkoholu niż ze stosowania leków.
teratogenne nie prowadzą do wystąpienia wad rozwo-
jowych w każdym przypadku, można przyjąć, że dla
ich wystąpienia istotna jest obecność dodatkowych
czynników (np. różnice rasowe, wiek, styl życia). 4.5.2. Pozostałe działania
W celu określenia skali ryzyka wystąpienia wad niepożądane w okresie ciąży
wrodzonych środki lecznicze zostały podzielone
na określone grupy ryzyka. „Czerwona lista” rozróż- Oprócz działania teratogennego leki zastosowane
nia grupy od Gr 1 (brak ryzyka) do Gr 11 (ryzyko w ciąży mogą spowodować również inne działania
wystąpienia działania mutagennego/karcynogenne- niepożądane:
Tabela A 4.5-2. Niektóre substancje, które są przeciwwskazane w czasie ciąży (ryzyko działania teratogennego lub toksycznego na
płód, ryzyko poronienia)
Grupa leków/lek
Inhibitory konwertazy polimyksyna B Leki przeciwdnawe Alkaloidy sporyszu
angiotensyny pirymetamina allopurinol
rifampicyna (1. trymestr) kolchicyna Prostaglandyny
Anaboliki streptomycyna
sulfonamidy Hormony Środki kontrastowe
Leki przeciwcukrzycowe tetracykliny androgeny zawierające jod
pochodne biguanidu trimetoprim antyandrogeny
pochodne sulfonylomocznika wankomycyna estrogeny Retinoidy
(w dużych dawkach) (ogólnie)
Leki przeciwnadciśnieniowe Doustne leki przeciwzakrzepowe gestageny
antagoniści kanału wapnia (pochodne kumaryny) glukokortykosteroidy Tyreostatyki
minoksydyl (ogólnie)
rezerpina Leki przeciwreumatyczne Witamina A
chlorochina Leki immunosupresyjne (w dużych dawkach)
Leki przeciwinfekcyjne indometacyna
acyklowir związki złota Związki jodu Witamina D i pochodne
aminoglikozydy fenylbutazon (w dużych dawkach)
amfoterycyna B penicylamina i in. Środki przeczyszczające
(ogólnie) (oprócz laktulozy Leki działające na OUN
chloramfenikol Leki moczopędne i środków pęczniejących) alkohol
flucytozyna kanrenian potasu amantadyna
gancyklowir diuretyki pętlowe Leki hipolipemizujące przeciwbólowe opioidy
gryzeofulwina spironolakton inhibitory reduktazy barbiturany
inhibitory gyrazy tiazydy HMG-CoA (statyny) benzodiazepiny
mebendazol fibraty disulfiram
meflochina
pochodne nitroimidazolu Molsidomina Cytostatyki
MUTSCHLER-2009.indd 102 2010-01-07 22:11:15
Działania niepożądane leków 103
■ porażenie ośrodka oddechowego oraz objawy odsta- Tabela A 4.5-3. Substancje, dla których nie ma danych o szkod-
wienne po zastosowaniu analgetyków opioidowych, liwym wpływie na ciążę, pod warunkiem że są stosowane w
dawkach terapeutycznych
■ maskulinizację płodów żeńskich wywołaną stoso-
waniem androgenów lub gestagenów o działaniu
androgennym, Grupa leków/lek
■ feminizację płodów męskich wywołaną stosowa- Antagoniści receptora β1
niem gestagenów o działaniu antyandrogennym, Leki zobojętniające (z wyjątkiem preparatów zawierających jony sodu)
■ fetopatie wywołane stosowaniem inhibitorów kon- Leki przeciwinfekcyjne
wertazy angiotensyny, cefalosporyny
erytromycyna
■ uszkodzenie słuchu po zastosowaniu antybiotyków linkozamidy
penicyliny
aminoglikozydowych,
Preparaty żelaza
■ zaburzenia rozwoju zębów i uszkodzenie szkieletu
po zastosowaniu tetracyklin, Preparaty zawierające enzymy
■ niedoczynność tarczycy po zastosowaniu tyreosta- Hormony
tyków. insulina
hormony tarczycy
Zwiększone ryzyko poronienia oraz komplikacji Środki przeczyszczające
laktuloza
Działania niepożądane
okołoporodowych występuje po podaniu prostaglan- środki pęczniejące
dyn, alkaloidów sporyszu i środków znieczulenia
Metyldopa
ogólnego.
W tab. A 4.5-2 zestawione są niektóre substancje Paracetamol
przeciwwskazane w ciąży z powodu ryzyka wystą-
Środki kontrastowe zawierające bar
pienia działania uszkadzającego płód lub z powodu
leków
zwiększenia ryzyka poronienia lub komplikacji oko- Sukralfat
łoporodowych. β2-sympatykomimetyki
W tab. A 4.5-3 z kolei zostały wymienione niektó-
re leki, dla których nie są do tej pory znane żadne Kompleks witamin B A4
szkodliwe działania w ciąży.
cje silnie wiążące się z białkami poprzez wypieranie
4.5.3. Działania niepożądane po porodzie bilirubiny z jej wiązań z białkami oso-
występujące w okresie cza mogą powodować żółtaczkę jąder podstawnych
poporodowym oraz w czasie mózgu (kernicterus), czyli ciężkie uszkodzenia ko-
karmienia piersią mórek nerwowych wskutek odkładania się bilirubiny.
U wcześniaków, noworodków oraz niemowląt Leki w okresie karmienia piersią. Wiele leków,
w pierwszym trymestrze życia enzymy biorące udział w tym szczególnie lipofilne o właściwościach słabych
w biotransformacji nie są jeszcze w pełni aktywne. zasad, przechodzi do mleka matki. Dotyczy to także
U małych dzieci aktywność glukuronylotransfera- rozmaitych lipofilnych trucizn, takich jak chlorfeno-
zy jest niższa w porównaniu z osobami dorosłymi. tan, dioksyny czy nikotyna.
Oznacza to, że substancje, które w celu wydalenia W przypadku krótkotrwałego narażenia na niewiel-
z organizmu niemowlęcia muszą zostać wcześniej kie dawki u karmionego piersią dziecka nie należy się
sprzęgnięte z kwasem glukuronowym, są eliminowa- obawiać silnych działań niepożądanych. Jednak w ra-
ne bardzo powoli. Ten niedobór w zakresie enzymów zie konieczności dłuższego podawania, zwłaszcza le-
może być również powodem występowania ciężkich ków o węższym indeksie terapeutycznym, należy zde-
objawów toksycznych po podaniu u wcześniaków cydować o zmianie preparatu. Środki lecznicze zostały
i noworodków zbyt dużych dawek chloramfenikolu podzielone ze względu na stopień ryzyka ich stosowa-
bądź depresji oddechowej po podaniu morfiny. Rów- nia w okresie karmienia analogicznie jak w przypadku
nież sulfonamidy wywołują u wcześniaków i nowo- leków podawanych w ciąży.
rodków często działania niepożądane ze względu W tab. A 4.5-4 zestawiono leki, których nie po-
na niedojrzałość nerek i wątroby. Oprócz tego substan- winna stosować matka w okresie karmienia piersią.
MUTSCHLER-2009.indd 103 2010-01-07 22:11:15
104 Działania niepożądane leków
Tabela A 4.5-4. Leki, których nie powinno się stosować w okresie karmienia piersią
Grupa leków/lek
Inhibitory konwertazy nowobiocyna Inhibitory reduktazy HMG-CoA Alkaloidy sporyszu
angiotensyny prazykwantel (statyny)
pirymetamina Retinoidy (ogólnie)
Anaboliki rifampicyna Hormony
sulfonamidy androgeny Tyreostatyki
Leki przeciwpadaczkowe tetracykliny antyandrogeny
trimetoprim estrogeny Leki działające na OUN
Leki przeciwhistaminowe (w dużych dawkach) amantadyna
blokujące receptor H2 Doustne leki przeciwcukrzycowe gestageny przeciwbólowe opioidy
(pochodne kumaryny) glukokortykosteroidy barbiturany
Leki przeciwinfekcyjne (ogólnie) benzodiazepiny
aminoglikozydy Niesteroidowe leki sole litu
przeciwgrzybicze pochodne przeciwreumatyczne Leki immunosupresyjne neuroleptyki
azolowe (ogólnie) teofilina
chloramfenikol Atropina Związki jodu kwas walproinowy
klindamycyna
erytromycyna Leki moczopędne Środki przeczyszczające Cytostatyki
inhibitory gyrazy kanrenian potasu (z wyjątkiem środków
izoniazyd spironolakton pęczniejących i laktulozy)
metronidazol
pochodne nitroimidazolu Leki przeciwreumatyczne Lewodopa
4.6. Lekozależność
Przez pojęcie lekozależności (drug dependen- nięcia przykrych objawów towarzyszących brakowi
ce), zgodnie z propozycją Światowej Organizacji substancji.
Zdrowia (WHO), rozumie się wszelkie postacie nad- Definicja uzależnienia została wprowadzona
używania leków. Uzależnienie definiuje się przy ze względu na trudność określenia różnic pomiędzy
tym następująco: „Uzależnienie jest to psychiczny, przyzwyczajeniem a nałogiem. Ponieważ terminy te
a niekiedy fizyczny stan wynikający z interakcji są ciągle używane, zostaną opisane poniżej.
między żywym organizmem a substancją, charakte-
ryzujący się zmianami zachowania i innymi reak- Przyzwyczajenie (drug habituation) polega na chęci re-
gularnego przyjmowania określonej substancji po to, żeby
cjami, do których należy konieczność przyjmowa- doświadczyć przy tym stanu euforii (uzależnienie psychicz-
nia substancji w sposób ciągły lub okresowy, w celu ne). Nie występuje przy tym zależność fizyczna, co ozna-
doświadczenia jego wpływu na psychikę bądź unik- cza, że po odstawieniu substancji nie występują objawy
Tabela A 4.6-1. Substancje mogące prowadzić do uzależnienia
Rodzaj substancji Uzależnienie Uzależnienie Przyzwyczajenie
psychiczne fizyczne
Morfina +++ +++ ++
Barbiturany/alkohol ++ ++ +
Kokaina +++ (+) –
Amfetamina i pochodne ++ – +
Meskalina/LSD ++ – +
Konopie indyjskie (= haszysz) ++ – (+)
MUTSCHLER-2009.indd 104 2010-01-07 22:11:16
Działania niepożądane leków 105
odstawienne. Nie występuje również albo występuje w nie- W obrębie różnych grup substancji, które mogą pro-
wielkim stopniu, skłonność do zwiększania dawki. Jeżeli wadzić do powstawania lekozależności, w tab. A 4.6-1
w trakcie zażywania występują jakieś szkody, to dotyczą
wymieniono niektóre z nich wraz z opisem form uza-
one osoby zażywającej, a nie społeczeństwa.
leżnienia, jakie wywołują.
Przyzwyczajenie różni się od tolerancji. O rozwoju
Niebezpieczeństwa i problemy związane z nad-
tolerancji mówi się, kiedy w ciągu przyjmowania sub-
używaniem leków dla osoby uzależnionej i społe-
stancji w celu uzyskania tych samych efektów konieczne
czeństwa są powszechnie znane. Prawdopodobnie
jest zwiększenie przyjmowanej dawki.
nigdy nie uda się ich całkowicie pozbyć, czy roz-
Nałóg (addiction) oznacza „stan okresowego lub ciągłe-
wiązać. Dlatego zjawisko uzależnienia powinno być
go zatrucia, szkodliwy dla jednostki i/lub otoczenia, który
utrzymywane w rozsądnych granicach poprzez in-
spowodowany jest powtarzanym zażywaniem naturalnej
formowanie, unikanie niepotrzebnego wypisywania
bądź syntetycznej substancji”. Na to pojęcie składają się:
leków o potencjale uzależniającym oraz rozsądne
■ uzależnienie psychiczne, a najczęściej także fizyczna za- prawodawstwo.
leżność od działania środka,
■ silne pragnienie lub potrzeba (przymus) ciągłego zaży-
wania środka oraz posiadania go za wszelką cenę,
■ tendencja do zwiększania dawki.
Działania niepożądane
leków
A4
MUTSCHLER-2009.indd 105 2010-01-07 22:11:16
106 Interakcje leków
5. Interakcje leków
W przypadku jednoczesnego stosowania dwóch lub nie przyjmowanych leków. Dlatego znaczne ryzyko
wielu leków istnieje możliwość wzajemnego oddzia- jej wystąpienia występuje u pacjentów starszych,
ływania, co może prowadzić do nasilenia działania, cierpiących na wiele chorób, którzy w zasadzie stale
zmiany działania niepożądanego bądź toksyczności przyjmują sporo leków. Jak wykazano w badaniach,
albo też osłabienia lub nawet zahamowania oczeki- większość z tych pacjentów w ciągu dnia otrzymuje
wanych efektów. Wyrażenie interakcja nie oznacza od trzech do dziewięciu (albo nawet więcej) leków.
zatem, czy jest to działanie pozytywne czy negatyw- Później często trudno odróżnić działanie niepożądane
ne. Ponieważ z reguły interakcji nie można przewi- wynikające z samego działania leku od powstającego
dzieć na podstawie struktury chemicznej pojedynczej na skutek interakcji.
substancji leczniczej lub danej grupy leków, obecnie
przez pojęcie interakcje rozumie się najczęściej in- Ogólnie około 20% działania niepożądanego wy-
terakcje niepożądane. Przy stosowaniu paru leków nika z interakcji leków. Rzecz jasna, jest ona też przy-
należy uwzględniać ich właściwości farmakodyna- czyną wielu przypadków śmiertelnych. Dlatego ko-
miczne i farmakokinetyczne. Ponadto trzeba także nieczne jest ciągłe wskazywanie na zagrożenie, jakie
brać pod uwagę fakt, że na skutek indywidualnych niesie ze sobą. Ponieważ jednak nie każda opisana
różnic genetycznych interakcje pojawiają się w róż- interakcja ma także znaczenie terapeutyczne, należy
nym nasileniu. każdorazowo oszacować znaczenie kliniczne danej
Prawdopodobieństwo wystąpienia interakcji le- interakcji w porównaniu z ryzykiem, jakie ze sobą
ków rośnie ekspotencjalnie wraz z liczbą jednocześ- niesie.
5.1. Interakcje farmaceutyczne
Przez pojęcie interakcje farmaceutyczne rozumiemy ■ zmiana właściwości reologicznych maści zawie-
niezgodności chemiczne, fizyczne i fizyko-chemicz- rającej glikol polietylenowy na skutek dołączenia
ne, które najczęściej mają miejsce poza organizmem octanu kortyzolu,
i które prowadzą do negatywnych zmian danych
■ utworzenie związku kompleksowego aminogliko-
środków leczniczych. Często dochodzi do nieroz-
zydu i penicyliny przy ich jednoczesnym podaniu
poznanych (tzw. ukrytych) niezgodności, które nie
w tym samy roztworze infuzyjnym,
są następnie uwzględniane. Poniżej podane zostały
przykłady licznych niepożądanych reakcji lub nie- ■ utworzenie trudno rozpuszczalnych połączeń zawie-
skutecznych terapii, za które odpowiedzialne są in- rających kationy leków (np. kodeiny, prokainy) z ten-
terakcje farmaceutyczne: zydami anionowymi (np. siarczanem sodu),
■ kłaczkowanie z wytworzeniem wody (np. metylo- ■ powstanie nierozpuszczalnego osadu mieszaniny
celuloza) przez wysokie stężenia etanolu, tiopentalu z roztworem suksametonium.
5.2. Interakcje farmakodynamiczne
Interakcje farmakodynamiczne charakteryzują się żenia zwrotnego dochodzi do wzmocnienia lub osła-
tym, że przy jednoczesnym podaniu substancji czyn- bienia ich działania. Dobra znajomość właściwości
nych na skutek wspólnego oddziaływania na ten sam farmakodynamicznych stosowanych leków pozwala
receptor lub narząd wykonawczy bądź procesów sprzę- na wykorzystanie pozytywnych dla chorego interak-
MUTSCHLER-2009.indd 106 2010-01-07 22:11:16
Interakcje leków 107
cji, a na uniknięcie niepożądanych. Poniżej omówio- Niesteroidowe leki przeciwreumatyczne, na skutek
ne zostaną tylko takie interakcje, które teoretycznie zahamowania syntezy prostaglandyn, zmniejszają
są dobrze znane, niemniej w praktyce mogą się zda- przeciwnadciśnieniowe działanie inhibitorów enzy-
rzać na skutek przeoczenia. Liczne dalsze przykłady mu konwertującego (ACE-Is) czy diuretyków.
są opisane w poszczególnych rozdziałach.
Wzmożenie działania nefro- i ototoksycznego.
Nasilenie efektu osłabienia aktywności ośrodko- Antybiotyki z grupy aminoglikozydów, np. gentamy-
wego układu nerwowego. Typowe dla działania cyna, wzmagają działanie nefrotoksyczne cefalotyny.
wielu leków osłabienie aktywności ośrodkowego Podobnie działanie ototoksyczne antybiotyków z tej
układu nerwowego może zostać nasilone na skutek grupy jest nasilone w przypadku jednoczesnego sto-
ich wzajemnych kombinacji. Należy o tym pamiętać sowania leków moczopędnych działających na pętlę
szczególnie przy stosowaniu alkoholu, leków obni- nefronu (pętla Henlego), jak furosemid czy też kwas
żających ciśnienie, leków trankwilizujących, leków etakrynowy. Wzmożone działanie ototoksyczne diu-
przeciwdepresyjnych, neuroleptyków, opioidowych retyków pętowych spowodowane jest ich wpływem
środków przeciwbólowych, leków przeciwpadacz- na skład elektrolitów w endolimfie ucha wewnętrz-
kowych, leków przeciwhistaminowych, metyldopy, nego.
klonidyny czy rezerpiny.
Wzmożenie działania leków zwiotczających (ku-
Przeciwstawne oddziaływanie na stężenie cukru raropodobnych). Dla anestezjologa ważne są inter-
we krwi. Leki nieselektywnie blokujące receptory akcje stabilizujących (niedepolaryzujących) leków
β-adrenergiczne, np. propranolol, nasilają hipogli- zwiotczających z antybiotykami wykazującymi dzia-
kemizujące działanie insuliny. Mogą więc przedłu- łanie kuraropodobne, takimi jak antybiotyki amino-
żać reakcję hipoglikemiczną, maskując jednocześnie glikozydowe. Mogą one bowiem potęgować działa-
jej objawy. Glukokortykosteroidy i doustne środki nie leków zwiotczających.
antykoncepcyjne osłabiają spadek stężenia cukru
we krwi pod wpływem doustnych środków przeciw- Nasilenie toksyczności glikozydów nasercowych.
cukrzycowych i insuliny. Hiperkalcemia oraz hipokaliemia nasilają działanie
glikozydów nasercowych. Wynika z tego, że pacjen-
Interakcje leków
Przeciwstawny wpływ na ciśnienie tętnicze. Osoby towi otrzymującemu glikozydy nasercowe nie tylko
z nadciśnieniem tętniczym najczęściej przez wiele nie wolno wstrzykiwać płynów zawierających związ-
lat biorą leki hipotensyjne. Długoletnie stosowanie ki wapnia, ale należy również pamiętać, że powinien
leków zwiększa możliwość interakcji. Przy każ- być on szczególnie troskliwie obserwowany przy jed-
dym podaniu leków nasercowych lub krążeniowych noczesnym stosowaniu preparatów zwiększających
należy zwracać uwagę na ewentualne niepożądane wydalanie potasu. Dotyczy to leków przeczyszczają-
zmiany ciśnienia tętniczego, szczególnie jego spadek cych i saluretyków, które często stosowane są razem
do wartości, które mogą wywoływać stany podciś- z glikozydami nasercowymi, a także glukokortyko-
A5
nieniowe, groźne np. dla kierowców. Na szczególną steroidów. Także amfoterycyna B wzmagając utratę
uwagę zasługują nie tylko leki przeciwarytmiczne potasu, zwiększa toksyczność glikozydów naserco-
oraz leki działające na naczynia wieńcowe, ale także wych.
antagoniści receptorów α-adrenergicznych stosowani
w przeroście prostaty. Uwagę chorych z nadciśnie- Wzajemne oddziaływanie antycholinergiczne.
niem tętniczym należy zwracać na to, że alkohol ma W przypadku równoczesnego stosowania trójpier-
niekorzystny wpływ nie tylko na ich chorobę podsta- ścieniowych leków przeciwdepresyjnych z leka-
wową, ale w niektórych przypadkach także na nie- mi przeciwhistaminowymi H1 pierwszej genera-
kontrolowane spadki ciśnienia tętniczego. Również cji, środkami stosowanymi w chorobie Parkinsona
wiele leków psychotropowych wpływa na ciśnienie czy opoidowymi lekami przeciwbólowymi dochodzi
krwi. Trójpierścieniowe leki przeciwdepresyjne an- do nasilenia działania antycholinergicznego, a przez
tagonizują spadek ciśnienia tętniczego zachodzący to istnieje niebezpieczeństwo wystąpienia bezmoczu,
pod wpływem wielu leków, takich jak α-metyldopa, zaparć czy odblokowania kąta przesączania w napa-
rezerpina czy klonidyna. Jednoczesne podawanie dzie jaskry.
nieselektywnych inhibitorów monoaminooksydazy
oraz pośrednio działających leków sympatykomime- Zwiększona skłonność do krwawień. Przy stoso-
tycznych może powodować znaczne zmiany (zarów- waniu leków przeciwzakrzepowych typu dikumaro-
no wzrost, jak i spadek) ciśnienia tętniczego krwi. lu dochodzi do zwiększenia skłonności do krwawień
MUTSCHLER-2009.indd 107 2010-01-07 22:11:16
108 Interakcje leków
na skutek interakcji farmakodynamicznej z następu- ■ kwasem walprionowym ze względu na hamowanie
jącymi, stosowanymi jednocześnie lekami: agregacji trombocytów oraz zmniejszenie liczby
płytek krwi.
■ kwasem acetylosalicylowym w następstwie zahamo-
wania agregacji trombocytów oraz obniżenia synte- Wzajemne zniesienie działania. Przykładem tego
zy protrombiny (w dawkach większych niż 1,5 g), typu interakcji jest zahamowanie działania lewodopy
czy któregoś z agonistów dopaminergicznych przy
■ chinidyną na skutek zmniejszonego wytwarzania
równoczesnym stosowaniu któregoś z neurolepty-
czynników krzepnięcia związanych z syntezą wi-
ków, np. haloperidolu.
taminy K,
5.3. Interakcje farmakokinetyczne
Do interakcji typu farmakokinetycznego może dojść chłonną jelita. Spowodowane takim właśnie mecha-
w każdym momencie farmakokinetycznej fazy dzia- nizmem zmniejszenie wchłaniania zostało wykazane
łania leku, czyli w czasie wchłaniania, dystrybucji, m.in. w przypadku digoksyny. Odwrotne zjawisko
biotransformacji lub wydalania. W przeciwieństwie obserwowane jest w następstwie zwolnienia przesu-
do interakcji farmakodynamicznych możliwość prze- wania się treści jelitowej. Na przykład leki antycholi-
widzenia interakcji farmakokinetycznych jest trud- nergiczne mogą wzmagać wchłanianie innych leków.
niejsza, gdyż w niewielu tylko przypadkach proce- Nierozpuszczalne, a zatem i niewchłanialne związ-
sy farmakokinetyczne są swoiste dla danego leku. ki kompleksowe powstają w wyniku jednoczesnego
Należy oczywiście liczyć się stale z wystąpieniem stosowania bifosforanów czy tetracyklin oraz związ-
tego typu interakcji. Można je stwierdzić określając ków zawierających sole magnezu, wapnia lub żelaza.
zmiany w stężeniu danego środka leczniczego we krwi Stosowanie preparatów zawierających tetracykliny
czy osoczu przy podaniu wielokrotnym w porówna- jednocześnie z mlekiem, lekami zobojętniającymi albo
niu z podaniem pojedynczym. Także doświadczenia preparatami żelaza, tłumaczy niezadowalające działa-
in vitro z zastosowaniem enzymów rekombinowa- nie tego antybiotyku. Podobne zjawisko obserwuje się
nych, oczyszczonych preparatów mikrosomalnych także w przypadku inhibitorów gyrazy. Sucralfat (zob.
czy transferowanych komórek stwarzają możliwość str. 648) zmniejsza resorpcję digoksyny, a sole bizmu-
określenia interakcji farmakokinetycznych. tu zmniejszają biodostępność fenytoiny.
Stosowane w celu obniżenia stężenia lipidów
wymienniki anionowe, takie jak kolestyramina lub
5.3.1. Interakcje występujące kolestipol, mogą w następstwie tworzenia soli bądź
przy wchłanianiu leku adsorpcji zmniejszać lub całkowicie hamować
wchłanianie innych substancji czynnych. Zauważono
Interakcje w zakresie wchłaniania mogą być m.in. następujące interakcje: zmniejszenie lub nawet cał-
związane ze zmianą wartości pH spowodowaną przez kowite zniesienie działania hormonów tarczycy, te-
jeden z leków. Na przykład jednoczesne podanie leku tracyklin, kwasu cheno- czy ursodezoksycholowego
zobojętniającego (antacida) z lekiem o charakterze oraz wyraźne zmniejszenie wchłaniania soli żelaza.
kwasu lub zasady spowoduje zmianę wchłaniania Należy przyjąć jako zasadę, że przy stosowaniu wy-
związaną ze wzrostem pH w górnej części przewo- mienników anionowych przed upływem co najmniej
du pokarmowego. Tego rodzaju interakcja została jednej godziny nie należy podawać innych leków.
stwierdzona w przypadku ketokonazolu i niektórych Cytostatyki zmniejszają biodostępność wielu le-
leków działających na retrowirusy. Wpływ na wchła- ków na skutek uszkodzenia błony śluzowej jelita.
nianie jednego leku może mieć drugi z leków, prze- Antybiotyki o dużym zakresie działania mogą
dłużając lub skracając jego przesuwanie się w prze- w następstwie uszkodzenia flory jelitowej zakłócać
wodzie pokarmowym lub wiążąc go kompleksowo. krążenie jelitowo-wątrobowe substancji, które w wą-
Przyspieszając przesuwanie się treści jelita przez po- trobie podlegają sprzęganiu, a w jelitach są przez flo-
danie metoklopramidu, można znacznie ograniczyć rę jelitową ponownie uwalniane. Klinicznie ważne
wchłanianie trudno wchłanianych leków ze wzglę- są m.in. interakcje z lekami antykoncepcyjnymi (ist-
du na niedostatecznie długi kontakt z powierzchnią nieje niebezpieczeństwo niepożądanej ciąży).
MUTSCHLER-2009.indd 108 2010-01-07 22:11:16
Interakcje leków 109
5.3.2. Interakcje w zakresie żonej skłonności do krwawień. Podobny efekt został
transportu opisany także w odniesieniu do klofibratu.
Niebezpieczeństwo hipoglikemii po podaniu do-
ustnych leków przeciwcukrzycowych pochodnych
Tak jak to wcześnej opisano, transport aktywny od-
sulfonylomocznika, np. tolbutamidu, rośnie, gdy jed-
grywa wielokrotnie istotną rolę przy wchłanianiu
nocześnie podawany jest kwas acetylosalicylowy lub
środków leczniczych z przewodu pokarmowego,
fenylobutazon.
wchłanianiu przez komórki oraz ich dystrybucji
Salicylany mogą nasilać hepatotoksyczność meto-
do różnych kompartmentów. Zasadnicze znaczenie
treksatu i działanie niepożądane fenytoiny.
ma tutaj przezbłonowy transporter glikoproteina P.
Analogicznym zjawiskiem jest wypieranie biliru-
Tak jak w przypadku izoenzymu cytochromu P-450
biny z miejsc wiążących na albuminie przez salicyla-
(zob. poniżej), może dochodzić do interakcji między
ny i sulfonamidy, co łączy się z niebezpieczeństwem
różnymi lekami transportowanymi za pośrednictwem
wystąpienia tzw. kernicterus u noworodków, czyli
tego białka.
żółtaczki jąder podstawnych.
Istotnym klinicznie przykładem takiej interakcji
w zakresie glikoproteiny P jest kombinacja rifam-
picyny z digoksyną. Przy jednoczesnym doustnym
przyjęciu obydwu leków digoksyna osiąga stężenie 5.3.4. Interakcje
subterapeutyczne. Jest ona bowiem po początkowych w zakresie biotransformacji
wchłonięciu następnie wielokrotnie aktywnie trans-
portowana z powrotem do światła jelita na skutek Najważniejsze interakcje pomiędzy lekami związane
zachodzącej za pośrednictwem rifampicyny indukcji są z ich biotransformacją. W przypadku jednoczesne-
glikoproteiny P w dwunastnicy. go zastosowania leków metabolizowanych przez ten
sam enzym, głównie przez enzymy cytochromu P-
450, na skutek konkurowania o to samo miejsce wią-
5.3.3. Interakcje w zakresie żące na enzymie, może dochodzić do zahamowania
lub indukcji aktywności enzymu, a przez to do osła-
dystrybucji bienia lub nasilenia metabolizmu jednego lub obydwu
Interakcje leków
leków. W przeszłości interakcje o podłożu metabo-
W przypadku gdy we krwi znajduje się więcej leków,
licznym wykrywane były na ogół dopiero w później-
powstaje możliwość konkurencji o miejsca wiązania
szych fazach badań klinicznych lub po wprowadzeniu
na białkach osocza. Ta konkurencja o miejsca wiąza-
leków do handlu. W obecnych czasach nie powinno
nia na białkach jest zjawiskiem częstym, ale w prak-
dochodzić do takich sytuacji, na skutek pogłębionej
tyce ograniczonym do leków silnie wiążących się
wiedzy na temat interakcji w zakresie biotransforma-
z białkami, o stosunkowo małej objętości dystrybucji
cji uczestniczących w metabolizmie enzymów.
i małej rozpiętości terapeutycznej. Ten typ interakcji
zachodzi wówczas, gdy przynajmniej jeden z leków Doskonałym przykładem tego typu interakcji jest mibe- A5
stosowany jest w zakresie dawek kilkusetmiligramo- fradil, lek blokujący kanały wapniowe T, który tak silnie
hamował przemiany wielu jednocześnie podanych leków,
wych lub gramowych. Wprowadzanie do leczenia że doszło do licznych wypadków śmierci i oczywiście
substancji silnie działających, które wykazują dzia- do wycofania tego leku z obrotu.
łanie terapeutyczne już w dawkach kilkumiligramo-
wych, znacznie zmniejsza ryzyko tego typu inter- Interakcje w zakresie hamowania aktywności en-
akcji, ponieważ liczba pozostających do dyspozycji zymów. Do interakcji w zakresie hamowania ak-
miejsc wiązania na białkach jest dostateczna nawet tywności enzymów dochodzi wówczas, kiedy jeden
wówczas, gdy dwa jednocześnie podane leki konku- z dwóch podanych równocześnie leków hamuje roz-
rują o identyczne miejsca wiązania. kład drugiego. Takie opóźnienie biotransformacji
Leki przeciwreumatyczne pochodne fenylobutazo- może prowadzić do przedłużenia okresu półtrwania,
nu (fenylobutazon), a także inne niesteroidowe leki zwiększenia stężenia substancji leczniczej czy nasile-
przeciwzapalne mogą wypierać leki przeciwzakrzepo- nia działań niepożądanych.
we z wiązań białkowych. Dochodzi wówczas, do cza- Metabolizm fenytoiny lub tolbutamidu jest hamo-
su wytworzenia się nowego stanu równowagi (steady wany przez izoniazyd, chloramfenikol lub leki prze-
state), do przejściowego wzrostu stężenia wolnego ciwkrzepliwe. Może to prowadzić do wzrostu stę-
leku. To z kolei powoduje zwiększone wydalanie żenia fenytoiny w osoczu do wartości toksycznych.
leku przeciwzakrzepowego, ale też do silniejszego Do leków hamujących biotransformację oksydacyjną
hamowania syntezy protrombiny, a przez to do wzmo- innych substancji należą cymetydyna, będąca względ-
MUTSCHLER-2009.indd 109 2010-01-07 22:11:16
110 Interakcje leków
Tabela A 5.3-1. Interakcje w zakresie hamowania aktywności enzymów
Lek, którego rozkład Inhibitor Efekt
jest hamowany enzymatyczny
terfenadyna itrakonazol kumulacja terfenadyny, przedłużenie QT, tachyarytmia
simwastatyna, lowastatyna gemfibrozil nasilone niebezpieczeństwo mięśniaka
teofilina fluwoksamina nasilone niebezpieczeństwo toksycznego działania teofiliny
fenprokumon trójcykliczne związki przeciwdepresyjne nasilone niebezpieczeństwo krwawień
nie nieswoistym inhibitorem licznych enzymów sunkowo wysokiego 65-proc. efektu pierwszego przejścia.
z grupy cytochromu P-450, stosowane doustnie leki Przy całkowitej blokadzie CYP3A4 biodostępność wzra-
przeciwgrzybicze z grupy azoli, a także erytromycy- sta o dalsze 65%, co jest równoznaczne z jej 14-krotnym
wzrostem, wskazując na bardzo istotne znaczenie tej in-
na, która silnie blokuje izoenzym CYP3A4. terakcji.
Inhibitory gyrazy cyprofloksacyna i enoksacyna
na skutek blokady CYP1A2 opóźniają biotransfor-
Interakcje w zakresie indukcji enzymatycznej. Jak
macją teofiliny.
to wcześniej opisano, na skutek indukcji enzyma-
Niektóre przykłady istotnych interakcji tego typu
tycznej wzrasta ilość lub aktywność biorących udział
zostały podane w tab. A 5.3-1.
w biotransformacji enzymów w wątrobie lub innych
Następujący przykład wskazuje na istotne znaczenie wiel- tkankach. Następstwem tego jest nasilenie biotrans-
kości efektu pierwszego przejścia dla oceny klinicznej formacji przyjmowanego jednocześnie leku, a przez
zachodzącej interakcji (w zakresie hamowania aktywno- to zmniejszenie jego stężenia w osoczu i osłabienie
ści enzymów): biodostępność prawastatyny wynosi 18%; działania. W przypadku kiedy induktor enzyma-
zachodzący za pośrednictwem CYP3A4 efekt pierwsze- tyczny zostanie odstawiony, a dawka drugiego leku
go przejścia wynosi 12%. Blokada CYP3A4 zwiększa bio-
dostępność do 30%, czyli 1,7 razy. Efektywność interakcji nie ulegnie zmniejszeniu, następstwem już nieistnie-
jest w tym przypadku stosunkowo mała. Biodostępność jącej interakcji może być niebezpieczne przedawko-
simwastatyny wynosząca zaledwie 5% zależna jest od sto- wanie (zob. ryc. A 5.3-1).
W przypadku proleków, które działają dopiero
po uprzednim zmetabolizowaniu, indukcja enzyma-
stężenie tyczna prowadzi nie do zahamowania, lecz do nasile-
pochodnej nia siły działania.
kumaryny
krzepliwość
(wskaźnik Quicka)
0 5.3.5. Interakcje w zakresie
krwawienie
wydalania leków
25
50 Interakcje przy wydalaniu przez nerki mogą zacho-
dzić w następstwie zmian pH moczu lub być wyni-
75 kiem konkurencji o miejsca wiązania w systemach
transportu odpowiedzialnych za wydalanie lub ak-
100 % tywne wchłanianie zwrotne. Jak to wcześniej opisano,
związki obniżające pH moczu (np. kwasy) przyspie-
dzienne dawki pochodnej kumaryny szają wydalanie słabych zasad, które przy niższym pH
dzienne dawki fenobarbitalu podlegają silniejszej jonizacji. W podobnym stopniu
związki podwyższające pH moczu (np. wodorowę-
Ryc. A 5.3-1. Schematyczny obraz następstw indukcji enzyma- glan sodu) wzmagają wydalanie słabych kwasów.
tycznej i jej wygasania w wyniku zastosowania fenobarbitalu Konkurencja o miejsca wiązania w przypadku
jako drugiego leku u chorego leczonego pochodną kumaryny transportu czynnego jest odpowiedzialna za hamo-
(wg Kahla). wanie przez probenecid wydalania penicylin lub in-
MUTSCHLER-2009.indd 110 2010-01-07 22:11:16
Interakcje leków 111
Tab. A 5.3-2. Interakcje w zakresie indukcji enzymatycznej
Lek, którego działanie Induktor enzymatyczny Efekt
ulega modyfikacji
doustne środki przeciwkrzepliwe barbiturany, kapsazepina, fenytoina, zmniejszenie działania przeciwkrzepliwego
rifampicyna
doustne środki antykoncepcyjne barbiturany, kapsazepina, fenytoina, spadek aż do utraty właściwości antykoncepcyjnych
rifampicyna
glukokortykosteroidy barbiturany, kapsazepina, fenytoina, zmniejszenie działania glukokortykosteroidów
rifampicyna
glukokortykosteroidy fenobarbital, fenytoina zmniejszone działanie przeciwdrgawkowe,
zwiększone ryzyko ataku
teofilina karbamazepina, palenie zmniejszone działanie przeciwastmatyczne
nych leków będących kwasami (np. indometacyny). być popijane wystarczającą ilością płynów (przy-
Dawniej mechanizm ten był wykorzystywany celem najmniej ¼ litra), gdyż środek leczniczy jest wte-
podwyższenia stężenia penicyliny we krwi w przy- dy szybciej uwolniany z danej postaci leku, a przez
padkach względnej oporności na ten lek. Dzisiaj tego to o wiele lepiej resorbowany. Ogólnie przyjmuje się,
typu postępowanie jest wykorzystywane do zmniej- że powinno się stosować wodę bieżącą lub mineralną,
szenia neurotoksyczności cidofowiru. W podobny gdyż mleko, soki owocowe, kawa czy herbata mogą
sposób wpływają na siebie w procesie wydalania ka- wchodzić w interakcje z lekiem.
nalikowego sulfonamidy, pochodne sulfonylomoczni- Przykładem leków, których przyjmowanie po po-
ka oraz fenylobutazonu. siłku, w porównaniu z ich zażywaniem na pusty żo-
Rozszerzające naczynia krwionośne prostaglandy- łądek, opóźnia i zmniejsza wchłanianie, są leki prze-
ny, które zwiększają przepływ krwi przez nerki i fil- ciwgruźlicze rifampicyna i izoniazyd lub lek blokują-
Interakcje leków
trację kłębuszkową, będą w przypadku zahamowania cy receptory β-adrenergiczne, celiprolol. Tetracykliny
ich syntezy przez niesteroidowe leki przeciwreuma- i inhibitory gyrazy zażyte łącznie z mlekiem lub po-
tyczne, przedłużać proces wydalania soli litu z nastę- karmami zawierającymi jony wapnia, magnezu lub
powym wzrostem stężenia litu w osoczu. żelaza spowolniają wchłanianie na skutek powstawa-
nia, jak wspomniano powyżej, nierozpuszczalnych
związków chelatowych.
W przeciwieństwie do tego wzrasta np. stosunek
5.3.6. Interakcje pomiędzy lekami wchłaniania gryzeofulwiny podawanej łącznie z po-
A5
a produktami spożywczymi siłkiem bogatotłuszczowym. Także spironolakton,
fenytoina lub ketokonazol są wchłaniane szybciej
Przyjmowanie posiłków może wpływać zarówno i w większym stopniu przy ich przyjmowaniu razem
na szybkości wchłaniania, jak i na całkowitą ilość z pokarmem.
wchłoniętego środka leczniczego. Z reguły przyj- W tab. A 5.3-3 przedstawione są przykłady inter-
mowanie leków jednoczesne z posiłkiem przedłuża akcji leków z produktami spożywczymi.
czas ich pobytu w żołądku, co prowadzi do opóź- W przeciwieństwie do licznych danych opartych
nienia wchłonięcia, a co za tym idzie – rozpoczęcia na doświadczeniach na zwierzętach istnieje tylko nie-
działania. W przypadku wyraźnych właściwości lipo- wielka liczba obserwacji dotycząca wpływu składni-
filnych przyjmowanie leku jest wskazane w trakcie ków pożywienia na biotransformację leków u ludzi.
lub po tłustym posiłku (zob. poniżej), gdyż prowadzi Metabolizm leków jest przyspieszony przez induktory
to do poprawie resorpcji. Leki rozpuszczalne w wo- enzymatyczne w pożywieniu powstające np. w czasie
dzie są lepiej wchłaniane na czczo. Leki, których pieczenia mięsa na węglu drzewnym. W przypadku
składniki drażnią lub uszkadzają śluzówkę przewodu leków blokujących receptory β-adrenergiczne, takich
pokarmowego, powinny być przyjmowane w trakcie jak propranolol, stwierdzono, że jednoczesne spo-
lub bezpośrednio po posiłku bez względu na poten- żywanie posiłku osłabia tzw. efekt I przejścia, czyli
cjalne zmniejszenie ich resorpcji (np. niesteroidowe następstwa pierwszego przejścia leku wchłoniętego
leki przeciwreumatyczne). Poza tym leki powinny z przewodu pokarmowego przez wątrobę.
MUTSCHLER-2009.indd 111 2010-01-07 22:11:17
112 Interakcje leków
Tab. A 5.3-3. Interakcje pomiędzy lekami a pożywieniem
Lek Mechanizm interakcji Wielkość Wskazania
zmian
albendazol nasilona resorpcja przez tłuste jedzenie 4-krotny wzrost biodostępności polecane tłuste posiłki podczas terapii
artemeter/ nasilona resorpcja przez tłuste jedzenie przynajmniej podwojona przyjmować podczas posiłku
lumefantryna biodostępność obydwu leków
atowakwon nasilona resorpcja przez tłuste jedzenie 2-3-krotne zwiększenie biodostępności przyjmować podczas posiłku
po standardowym śniadaniu
bisfosfoniany tworzenie związków cholatowych tstotne zmniejszenie biodostępności przyjmować na czczo, co najmniej
pół godziny przed śniadaniem,
popijając czystą wodą
cuprofloksacyna tworzenie związków chylatowych 30-proc. zmniejszenie biodostępności należy przyjmować 2 godz. przed
narfloksacyna z produktami mlecznymi lub po spożyciu produktów
ofloksacyna mlecznych
didanozyna hydroliza na skutek uzależnionego od jedzenia zmniejszenie o 50% Cmax należy przyjmować 2 godz.
wydzielania kwasu i długiego pasażu i biodostępności przed lub po spożyciu posiłku
gancyklowir zwiększona resorpcja pod wpływem zwiększenie biodostępności o 20% należy przyjmować w trakcie posiłku
tłustego posiłku
indinawir zmniejszona resorpcja pod wpływem zmniejszenie Cmax o 60% należy przyjmować 2 godz. przed
tłustego posiłku lub po spożyciu posiłku
bądź w trakcie posiłku ubogotłuszczowego
izoniazyd posiłek zawierający dużo węglowodanów zmniejszenie Cmax o 20%, w przypadku posiłku zawierającego
zmniejsza szybkość i wielkość resorpcji a biodostępności o 20% dużo węglowodanów należy
przyjmować dopiero 1 godz. później
meflokina posiłek zwiększa szybkość zwiększenie Cmax o 50%, należy przyjmować bezpośrednio
i wielkość resorpcji a biodostępności o 30% po posiłku
D-penicylamina tworzenie związków chelatowych zmniejszenie biodostępności należy przyjmować 2 godz. przed
o 50% lub po spożyciu posiłku
sakwinawir dłuższe przebywanie w żołądku zwiększona biodostępność należy przyjmować w ciągu 2 godz.
zwiększa resorpcję po spożyciu posiłku
Pożywienie może zmniejszyć aktywność izoenzy- ny dowóz prekursorów katecholamin może doprowa-
mów cytochromu P-450. Na przykład składniki soku dzić do niebezpieczeństwa skrajnych wahań (wzro-
grejpfrutowego zmniejszają aktywność CYP3A4 w je- stów lub spadków) ciśnienia tętniczego.
litach. Powoduje to zmniejszenie przedwątrobowego Środki spożywcze bogate w azotyny mogą, np.
efektu pierwszego przejścia w odniesieniu do wielu łącznie z często stosowanym dawniej lekiem prze-
leków. W związku z powyższym jednoczesne picie ciwbólowym aminofenazonem, powodować powsta-
soku grejpfrutowego zwiększa biodostępność wielu wanie w żołądku rakotwórczych N-nitrozo-związ-
leków-substratów dla CYP3A4 (np. werapamilu lub ków. Właśnie z tego względu aminofenazon został
cyklosporyny. wycofany ze sprzedaży.
W przypadku stosowania nieselektywnych inhi- Zawartość w pokarmie znacznych ilości witaminy
bitorów monoaminooksydazy należy unikać pokar- K osłabia działanie leków przeciwzakrzepowych typu
mów bogatych w tyraminę, takich jak sery, ponieważ dikumarolu.
na skutek zahamowania rozkładu tyraminy zwiększo-
MUTSCHLER-2009.indd 112 2010-01-07 22:11:17
Interakcje leków 113
5.4. Unikanie interakcji
Za pomocą następujących środków można zmniejszyć ■ zmniejszenie dawki leku, kiedy przyjmowany jed-
(a nawet uniknąć) niebezpieczeństwo interakcji: nocześnie lek hamuje jego metabolizm, podnosząc
przez to jego stężenie we krwi,
■ unikanie zbędnych kombinacji,
■ regularna kontrola stężenia (stężeń) w osoczu leku
■ stała ocena wskazań dla każdego leku w przypad-
o małej rozpiętości terapeutycznej, w celu uniknię-
ku pacjentów (szczególnie osób starszych), którzy
cia toksycznych efektów na skutek zahamowania
jednocześnie biorą kilka leków,
aktywności enzymów lub niewystarczającego dzia-
■ rozpoznanie potencjalnych interakcji już przed po- łania przy jednoczesnym podaniu drugiego leku,
daniem leku pacjentowi, jeżeli tylko to możliwe,
■ wyjaśnienie pacjentowi możliwości zaistnienia
wybranie alternatywnego leku bez zagrożenia in-
ewentualnych interakcji i uczulenie go na wystą-
terakcji,
pienie możliwych symptomów takich wzajemnych
■ przesunięcie czasowe przyjmowania leku, który oddziaływań.
wykazuje interakcje w zakresie wchłaniania,
Interakcje leków
A5
MUTSCHLER-2009.indd 113 2010-01-07 22:11:18
114 Farmakogenetyka
6. Farmakogenetyka
Od dawna wiadomo, że zarówno u pojedynczych dynczych pacjentów. W przeprowadzonych następnie
pacjentów, jak i w określonych grupach populacji badaniach dotyczących dużych populacji określono
stwierdza się czasami inną, zwiększoną lub zmniej- częstość występowania defektu, tzn. odsetek osób
szoną, reakcję na dane leki aniżeli u reszty ludności. wolno metabolizujących, tzw. wolnych metaboliza-
Prowadzone ostatnio badania wskazują, że takie nie- rów (poor metabolizers; PM), w porównaniu z oso-
oczekiwane działania leków są często uwarunkowane bami szybko metabolizującymi, tzw. szybkimi meta-
czynnikami dziedzicznymi. Na podstawie tej wiedzy bolizerami (extensive metabolizers; EM). Przyczyny
powstała farmakogenetyka, która jest dziedziną doty- tych różnic udało się w ostatnich latach wyjaśnić
czącą genetycznie determinowanej zmienności dzia- metodami biologii molekularnej na poziomie DNA.
łania leków. Także badania rodzin pozwoliły na ustalenie poszcze-
gólnych sposobów dziedziczenia (recesywny lub do-
Po raz pierwszy w warunkach klinicznych zwrócono uwa- minujący).
gę na tego rodzaju zjawiska po wprowadzeniu do stoso-
wania leku zwiotczającego mięśnie – sukcynylocholiny.
U niektórych pacjentów po podaniu standardowych da- Znaczenie różnic farmakogenetycznych zależy
wek leku obserwowano istotnie przedłużony czas działa- od tego, jaką rozpiętością terapeutyczną cechuje się
nia, który można było przypisać genetycznie uwarunko- dana substancja czynna, czy dochodzi do wytworze-
wanemu defektowi enzymu rozkładającego – pseudocho- nia aktywnych metabolitów oraz w jakim zakresie
linoesterazy. dany enzym bierze udział w całkowitym metabolizo-
waniu leku. A zatem terapeutyczne konsekwencje do-
Takie farmakogenetycznie uwarunkowane różnice tyczące każdego leku muszą być oceniane w sposób
w działaniu występują często na podłożu farmako- indywidualny. W przypadku wielu substancji defekt
genetycznego polimorfizmu. Są one powodowane genetyczny prowadzi do gromadzenia się substancji
dziedziczonymi monogenowo, różniącymi się od sie- wyjściowej, a przez to przy powolnym metabolizo-
bie allelami tego samego genu, zajmującymi ten sam waniu do nasilenia działań niepożądanych. Czasami
locus, tzn. w takim samym miejscu w genomie u po- jednak, tak jak w przypadku leku przeciwbólowe-
szczególnych pacjentów występują różne sekwencje go kodeiny, następstwa są całkowicie odwrotne.
DNA (genotypy). W wyniku tego powstaje wiele feno- Kodeina jest prolekiem i do jej bioaktywacji z wy-
typów. Fenotyp jest zdefiniowany jako rozpoznawal- tworzeniem morfiny potrzebny jest CYP2D6 (zob.
na ekspresja genotypu, która objawia się na przykład poniżej). Z tego powodu po podaniu kodeiny efekt
różnicą polegającą na powolnym i szybkim metaboli- analgetyczny u wolnych metabolizerów jest mniejszy
zowaniu (zob. poniżej) w danej populacji pacjentów. niż u szybkich metabolizerów.
Farmakogenetyka zajmuje się badaniem: Za pomocą testów farmakogenetycznych określa się albo
genotyp, albo fenotyp pacjenta. W celu ustalenia genoty-
■ dziedzicznie uwarunkowanych różnic w działaniu pu pobiera się DNA z limfocytów z próbki krwi pełnej.
leków, a także ich przyczyn na poziomie moleku- Fenotyp można określić przez podanie substancji testowej,
larnym, która jest metabolizowana przez badany enzym. Aktywność
enzymu – a przez to fenotyp – określa się na podstawie sto-
■ znaczenia zmienionego kliniczne działania leków, sunku substancji wyjściowej do metabolitów.
■ testów umożliwiających identyfikację zagrożonych
Badania dostępnej obecnie informacji genetycznej,
pacjentów w miarę możliwości przed rozpoczę-
szczególnie tej związanej z polimorfizmem pojedyn-
ciem leczenia.
czych nukleotydów (single nucleotide polymorphism;
SNP), oraz rosnąca lista kandydatów posiadających
Jak podano we wstępie, punkt wyjściowy dla pierw- znaczenie funkcjonalne SNPs rozbudziły duże ocze-
szych badań farmakogenetycznych stanowiły nie- kiwania w kwestii zastosowania wiedzy farmako-
oczekiwane działania leków pojawiające się u poje- genetycznej w celu optymalizacji i indywidualizacji
MUTSCHLER-2009.indd 114 2010-01-07 22:11:18
Farkamogenetyka 115
terapii. Jednak do chwili obecnej uległy one tylko dualne różnice w aktywności CYP3A4 w wątrobie
częściowej realizacji. mogą wahać się o faktor 30. Jednakże polimorfizmy
w określonych regionach CYP3A4 są bardzo rzadkie
Polimorfizm dotyczący CYP2D6 (sparteiny-debry- i dlatego nie mogą być odpowiedzialne za widoczne
zochiny). U 5–10% ludności Europy stwierdza się zmiany. Możliwe, że są one związane z jeszcze nie-
genetyczny defekt dotyczący utlenienia sparteiny, al- określonymi polimorfizmami czynników transkryp-
kaloidu stosowanego w przeszłości jako lek antyaryt- cyjnych.
miczny, która zachodzi za pomocą CYP2D6, enzymu
biorącego udział w prawie 25% wszystkich reakcji Polimorfizm dotyczący N-acetylotransferazy. Po
biotransformacji środków leczniczych. Dokładne ba- wprowadzeniu do leczenia leku przeciwgruźliczego
dania wykazały, że prewalencja wolnych metabolize- izoniazydu opisano występowanie dużych różnic in-
rów wynosi wśród Europejczyków 7,7%, natomiast dywidualnych pod względem metabolizowania tej
wśród populacji orientalnych tylko 1–2%. substancji czynnej. Wskutek polimorfizmu dotyczą-
cego cytozolowej N-acetylotransferazy (NAT2) z 5
U osób tych występują różne warianty CYP2D6 (dotych- wariantami alleli izoniazyd ulega szybkiej inaktywa-
czas zidentyfikowano ponad 50), spośród których 5 wystę- cji na drodze acetylacji u mniej więcej połowy lud-
puje bardzo często. Zmutowane allele prowadzą do utraty
albo do ograniczenia czynności enzymu. Sposób dziedzi- ności Europy i u 70–90% Japończyków, Chińczyków
czenia jest autosomalny recesywny i dlatego fenotyp PM i Eskimosów, natomiast powolnej inaktywacji w po-
pojawia się tylko wtedy, kiedy pacjent ma dwa allele z de- zostałej populacji. A zatem pod względem genoty-
fektem. powym i fenotypowym można różnicować szybkich
Ostatnio zaobserwowano także zaskakującą duplikację i wolnych acetylatorów. W tym ostatnim przypadku
lub multiplikację genów kodujących CYP2D6, która pro-
wadzi do istotnego zwiększenia liczby enzymów. dochodzi do wzmożonego wytwarzania produktów
oksydacji izoniazydu, w wyniku czego zwiększa
Polimorfizm dotyczący CYP2D6 ma istotne znacze- się jego toksyczne działanie na wątrobę. W związ-
nie kliniczne, ponieważ występuje u dużego odsetka ku z powyższym u wolnych acetylatorów zażywa-
ludności Europy, a także związany jest z enzymem, jących izoniazyd zwiększa się istotnie możliwość
który metabolizuje liczne, często stosowane leki wystąpienia zapalenia wątroby (hepatitis). Ponadto
mające stosunkowo małą rozpiętość terapeutycz- u tej grupy osób z cząsteczki izoniazydu odszcze-
ną, w tym np. leki antyarytmiczne, neuroleptyki piana jest hydrazyna, która powoduje wzmożo-
czy opioidy. ną inaktywację pirydoksyny. Następstwem tego
jest zwiększenie zaburzeń w obrębie układu nerwo-
Polimorfizm dotyczący CYP2C19. Polimorfizm ten wego. W związku z tym powstające pod wpływem
Farmakokinetyka
został wykryty w wyniku tego, że u pojedynczych pa- izoniazydu neuropatie występują prawie wyłącznie
cjentów stwierdzono bardzo dużą wrażliwość na me- u osób z powolną acetylacją. W przeciwieństwie
fenytoinę, lek przeciwdrgawkowy niedostępny obecnie do tego osoby o szybkim i powolnym acetylowa-
w handlu. U 3–6% ludności Europy i u 13–23% orien- niu nie różnią się od siebie pod względem działania
talnych grup pacjentów wykrywa się defekt CYP2C19 przeciwgruźliczego izoniazydu.
o sposobie dziedziczenia autosomalnym recesywnym, Oprócz izoniazydu wiele innych leków stano-
który daje się wyjaśnić defektem dwóch alleli. Środki wi substrat dla NAT2, m.in. amrinon, klonazepam,
lecznicze, które są metabolizowane przez CYP2C19, kofeina, dapson, hydralazyna, metamizol i sulfona- A6
to m.in. barbiturany, diazepam i omeprazol. W przy- midy.
padku pacjentów z defektem enzymu CYP2C19, którzy
otrzymają normalną dawkę któregoś z leków podlega- Rzadziej występujące polimorfizmy. Dotyczą one
jących biotransformacji przez CYP2C19, może dojść m.in. metylotransferazy tiopurynowej (TPMT) oraz
do nieoczekiwanie wysokiego stężenia leku we krwi, dehydrogenazy dihydropirymidynowej (DPD). U pra-
a przez to do nasilenia działania niepożądanego. Kiedy wie co trzechsetnego pacjenta występuje homozygo-
przy zwolnionym metabolizmie dodatkowo zachodzi tyzm pod względem defektu TPMT. Metylotransferaza
interakcja prowadząca do zahamowania zmutowanego tiopurynowa bierze udział w metabolizowaniu mer-
CYP2C19, istnieje niebezpieczeństwo wystąpienia re- kaptopuryny i azatiopryny. W przypadku, kiedy
akcji toksycznej. nie dostosuje się w tej grupie pacjentów dawki obu
tych substancji (wystarczy 6–10% normalnej daw-
Polimorfizm dotyczący CYP3A4. CYP3A4 bierze ki!), dochodzi do znacznych działań niepożądanych,
udział w metabolizmie wszystkich leków. Indywi- m.in. mielosupresji i neurotoksyczności.
MUTSCHLER-2009.indd 115 2010-01-07 22:11:18
116 Farmakogenetyka
DPD katalizuje redukcję pirymidyn (np. 5-fluoroura- dochodzi do ciężkich niedokrwistości hemolitycz-
cylu. U pacjentów z ograniczoną aktywnością enzymu nych. Tego rodzaju hemolizę określa się jako fa-
(ok. 3% ludności) po podaniu typowych dawek mogą wizm, ponieważ może ona zostać wywołana również
wystąpić zagrażające życiu neutropenie. Oprócz tego przez surową zieloną fasolę (Vicia faba = bób) (zob.
opisane są także inne, rzadko występujące defekty ryc. A 6-1). Stwierdzono, że przyczyną tego dzia-
genetyczne innych cytostatyków. Dotyczy to m.in. łania niepożądanego jest niedobór dehydrogenazy
transferazy glukuronylowej difosforanu urydyny 1A1 glukozo-6-fosforanowej (G6PD). Enzym ten katali-
(UGT1A1), która katalizuje inaktywację aktywnego zuje powstawanie NADPH z jednoczesnym utlenie-
metabolitu związku hamującego topoizomerazę-I iri- niem glukozy. NADPH jest wykorzystywany do re-
notekanu i która jest odpowiedzialna za prawie 50% generacji zredukowanego glutationu (GSH). Przy
indywidualnych różnic przy biotransformacji tego występowaniu zmniejszonej ilości GSH następuje
leku. wzmożone tworzenie mostków dwusiarczkowych
Rzadko również pojawia się wymieniony powy- przez białka znajdujące się w błonie komórkowej
żej polimorfizm dotyczący pseudocholinoesterazy erytrocytów, co wywołuje niestabilność błony ko-
o sposobie dziedziczenia autosomalnym recesywnym mórkowej.
(częstość występowania allelu K z defektem wynosi Niedobór G6PD prowadzi do hemolizy, nie tylko
1,5%). kiedy zaburzone jest powstawanie GSH, ale także
kiedy występuje nadmierne zużycie glutationu spo-
Niedobór dehydrogenazy glukozo-6-fosforano- wodowane przez wyżej wymienione leki. Ponieważ
wej. Oprócz genetycznej zmienności enzymów roz- odpowiedni gen jest dziedziczony z chromosomem
kładających leki istnieją czynniki dziedziczne, które X, a w przypadku czynnościowo sprawnego genu
wpływają na działanie leków niezależnie od ich me- jest wytwarzana dostateczna ilość enzymu, działanie
tabolizmu. Na przykład po podaniu różnych leków niepożądane występuje wyłącznie u mężczyzn i u ko-
przeciw zimnicy, zwłaszcza primachiny i dapsonu, biet z homozygotyzmem.
u rdzennej, murzyńskiej ludności Afryki, u ludności
żyjącej w okolicy Morza Śródziemnego (Greków, Niedobór reduktazy methemoglobiny. W przy-
mieszkańców Sardynii), a także u Hindusów w 10% padku niedoboru tego enzymu dochodzi do wzrostu
2 GSH 2 NADP glukozo-6- P
reduktaza dehydrogenaza
glutationowa glukozo-6- P
GSSG 2 NADPH glukoniano-6- P
niedobór
hemoliza dehydrogenazy
glukozo-6- P
SH
SH
Ryc. A 6-1. Hemoliza wywołana niedoborem dehydrogenazy glukozo-6-fosforanowej. Niedobór ten prowadzi do zmniejszonego
wytwarzania NADPH i w wyniku tego do zwiększenia liczby mostków dwusiarczkowych w białkach błon komórkowych, co wywołuje
hemolizę; GSSG – utleniony glutation.
MUTSCHLER-2009.indd 116 2010-01-07 22:11:18
Farkamogenetyka 117
Tabela A 6-1. Kolejne przykłady polimorfizmów genowych. Podano pozycję aminokwasu lub zamiany zasady. Ins – insercja (wsta-
wienie), del – delecja (wypadnięcie)
Produkt genu Polimorfizm Kliniczny fenotyp
reduktaza 5,10- metylenote- C(677)T i A(1298)C T677 podwyższa toksyczność i oporność metotreksatu
trahydrofolianu
kompleks reduktazy szereg haplotyp różny efekt fenprokumonu
epoksydowej witaminy K
receptor β2-adrenergiczny Arg16Gly, Gln27Glu 16Gly zmniejsza odpowiedź na salbutamol
i Thr164Ile
receptor β1-adrenergiczny Arg389Gly Gly389 zmniejsza odpowiedź układu krążenia na antagonistów
receptorów β1-adrenergicznych
transporter dla serotoniny polimorfizm dłuższego zmienione działanie leków przciwdepresyjnych
lub krótszego promotora
receptor serotoninowy 2A His452Tyr Tyr452 zmniesjza działanie przeciwpsychotyczne klozapiny
(5-HT2A)
enzym konwertujący angiotensynę II Ins/del intron 16 homozygota del/del zmniejsza działanie
inhibitorów ACE przy proteinuriach
receptor czynnika wzrostu naskórka mutacja eksonu 18-21 w przypadku tej mutacji jest lepsza reakcja
(EGF-R) na gefitinib przy raku płuc
ERBB2=HER2 (nowy), onkogen EGF-R nadekspresja białek w przypadku nadekspresii ERBB2 terapia
za pomocą heceptyny przy raku sutka
białko XRCC1 Arg194TRP, Arg280His Gln399 prowadzi do oporności oksaliplatyny/5-fluorouracylu
i Arg399Gln
zawartości methemoglobiny we krwi wskutek nie- u osób predysponowanych ogromne zwiększenie stę-
dostatecznej redukcji methemoglobiny do hemo- żenia wapnia wewnątrz komórek, co prowadzi do sil-
globiny (zob. poniżej). W porównaniu ze stanem nych skurczów mięśni, a niekiedy do zagrażającej
Farmakokinetyka
prawidłowym proces ten może być zmniejszony życiu gorączki.
od dwudziestu do pięćdziesięciu razy. W lżejszych
formach – przede wszystkim u osób będących hete- Porfirie. Choroby te są spowodowane zaburzeniami
rozygotami – do methemoglobinemii dochodzi przy różnych enzymów biorących udział w biosyntezie
stosowaniu określonych leków, np. pochodnych hemu, w wyniku czego w narządach odkładają się
aniliny, chloramfenikolu, azotanów, sulfonamidów. produkty pośrednie syntezy hemu lub fałszywe pro-
Objawami methemoglobinemii są sinica i dusz- dukty jego syntezy. Niektóre leki, np. barbiturany,
ność. sulfonamidy, estrogeny, mogą przez indukcję synte- A6
tazy kwasu aminolewulinowego, głównego enzymu
Hipertermia złośliwa. To niebezpieczne powikłanie syntezy hemu, doprowadzić do powstania ostrych ob-
znieczulenia ogólnego jest spowodowane zmienioną jawów klinicznych. Dlatego ich stosowanie jest bez-
strukturą kanału wapniowego w siateczce sarkopla- względnie przeciwwskazane u tych osób.
zmatycznej (przy tzw. receptorze rianodynowym). Tabela A 6.1 zawiera przegląd polimorfizmów ge-
Otwarcie kanału spowodowane przez suksametonium nowych, które są odpowiedzialne za występowanie
lub różne środki znieczulające, np. halotan, wywołuje indywidualnych reakcji na leki.
MUTSCHLER-2009.indd 117 2010-01-07 22:11:18
118 Terapia genowa i stosowanie oligonukleotydów antysensownych
7. Terapia genowa,
stosowanie oligonukleotydów
antysensownych i komórek pnia
7.1. Terapia genowa
Mutacje genowe prowadzą do tego, że określone cjonalnej dla receptora interleukiny-2, której brak
białka nie są wytwarzane w ogóle (defekt homozygo- prowadzi do niedoboru odporności. Jednak u wielu
tyczny) lub powstają w niedostatecznej ilości (defekt leczonych w ten sposób pacjentów doszło do rozwo-
heterozygotyczny) bądź posiadają niewystarczającą ju leukemii związanej z zastosowanymi wektorami
aktywność. Przykładami defektów monogenowych genu – osłabionymi retrowirusami (zob. poniżej),
jako czynników wywołujących choroby są rodzinna za pomocą których wprowadzono prawidłowe geny
hipercholesterolemia – jedna z najczęstszych chorób do komórek. Zauważono przy tym, że retrowirus
uwarunkowych genetycznie (częstość występowania zawierający konstrukcję funkcjonalnego genu przy
1:500 w Europie Zachodniej), związana z mutacją re- integracji DNA z genomem aktywuje protoonkogen
ceptora LDL lub apolipoproteiny B100, mukowiscy- LMO2, który już wcześniej był łączony z białaczką
doza, hemofilia A, niedobór antytrypsyny α1, zespól u dzieci. Ze względu na poważne ryzyko w niektó-
kruchego chromosomu X lub wielokrotnie dziedziczo- rych krajach wprowadzono moratorium na leczenie
ne ataksje. za pomocą terapii genowej z zastosowaniem retro-
W przypadku chorób genetycznych często nie uda- wirusowych układów wektorowych.
je się zastąpić brakujących lub mających defekty bia- Dopiero w ostatnim czasie – ponownie za pomocą
łek za pomocą terapii substytucyjnej. Wyjątek stano- wprowadzenia genu przez retrowirusy – autologiczne
wi leczenie substytucyjne, polegające na uzupełnianiu komórki T tworzącego przerzuty czerniaka zostały
niedoboru czynnika VIII w hemofilii A czy deaminazy tak zaprogramowane, że utworzyły w błonach osocza
adenozyny w rzadko występującej chorobie immuno- namnażające się struktury, prezentujące białka ko-
logicznej SCID (Severe Combined Immunodeficiency mórek czerniaka, przeciw którym była w ten sposób
Disease; ciężki, złożony niedobór immunologiczny). wyzwalana reakcja immunologiczna. W przypadku
Problemy stwarza ograniczona dostępność, niedo- niektórych pacjentów uzyskano w ten sposób całko-
stateczna stabilność, a także trudność w dostarczaniu witą regresję nowotworu, a w przypadku innych po-
odpowiedniego białka w formie aktywnej w spo- nadroczny okres recesji.
sób wybiórczy do oczekiwanego miejsca działania, Strategie przyszłości przy dalszym rozwoju tej
np. do błony komórkowej lub do wnętrza komórki. technologii powinny kłaść nacisk na opracowanie
Stąd już blisko do korygowania defektów gene- nowych, zarówno bezpiecznych, jak i efektywnych
tycznych za pomocą somatycznej terapii genowej. sposobów transferu genów. Wcześniej wypróbo-
W tym przypadku próbuje się doprowadzić do wy- wane systemy wektorowe, takie jak adenowirusy,
zdrowienia poprzez trwałe wprowadzenie do geno- wirusy adenosatelitarne, liposomy, okazały się nie-
mu docelowej komórki prawidłowego genu koor- przydatne w terapii genowej ze względu na powsta-
dynującego syntezę białka. Somatyczna terapia ge- wanie reakcji immunologicznych u pacjentów bądź
nowa może być przez to alternatywą terapeutyczną ich małą skuteczność we wprowadzeniu genu do ko-
dla ciężkich, uwarunkowanych genetycznie chorób, mórek docelowych. Kiedy nawet uda się otrzymać
w których szanse na wyleczenie przy stosowaniu odpowiednie wektory dla genów, pozostanie jeszcze
konwencjonalnej terapii są niewielkie. Jednak w cią- najtrudniejszy problem do rozwiązania, który utrud-
gu ostatnich 10 lat problemy związane z tą techno- nia substytucję brakującego lub wadliwego genu
logią nie zostały rozwiązane, a możliwości zastoso- – mianowicie komórki manipulowane genetycznie,
wania przecenione. Pomimo olbrzymich starań udało które nie występowały do tej pory w organizmie,
się uzyskać utrzymujący się parę lat sukces jedynie będą rozpoznawane jako obce i eliminowane przez
w przypadku wymienionego powyżej związanego układ odpornościowy.
z chromosomem X niedoboru immunologicznego
SCID, poprzez pozaustrojowe leczenie komórek he- Retrowirusowe układy wektorowe. Mechanizm re-
matopoetycznych pnia za pomocą podjednostki funk- plikacji wspomnianych powyżej retrowirusów polega
MUTSCHLER-2009.indd 118 2010-01-07 22:11:19
Terapia genowa i stosowanie oligonukleotydów antysensownych 119
promotor –
sekwencja wzmacniająca
czynnik IX wektor z niedoborem
replikacyjnym
transfekcja
RNA zakażenie cytoplazma
odwrotna
jądro transkryptaza
komórkowe
env pol wirusowy RNA
gag
hybryda RNA-DNA
komórka pakująca
podwójny łańcuch DNA jądro
komórkowe
wydzielanie czynnik IX integracja
transkrypcja
komórka translacja DNA gospodarza
docelowa
Ryc. A 7.2-1. Mechanizm retrowirusowego transferu genu do somatycznej komórki na przykładzie IX. Retrowirusowe DNA zostaje
włączone do genomu komórki, będącej w trakcie podziału komórkowego (wg Vermy i Somii). Szczegóły w tekście.
na tym, że jednołańcuchowy RNA genomu wirusa zo- Zaleta używania retrowirusów polega na tym, że in-
staje na drodze odwrotnej transkrypcji przepisany (za tegracja DNA z genomem umożliwia długotrwałą eks-
pomocą enzymu odwrotnej transkryptazy) w postać presję genu. Natomiast wadą tej metody jest to, że re-
DNA, a następnie – ponownie enzymatycznie z udzia- trowirusy zakażają jedynie komórki w trakcie ich po-
łem enzymu integrazy – ulega włączeniu do genomu działu komórkowego. Poza tym nie zostały dotychczas
dzielącej się komórki i namnożeniu. rozwiązane w wystarczającym stopniu problemy zwią-
Aby zapobiec niekontrolowanemu rozprzestrze- zane z działaniem swoistym komórkowo. Duże zagro-
nianiu się stosowanych w terapii genowej retrowi- żenie stwarza także potencjalna mutagenność uwarun-
rusów w organizmie, muszą mieć one defekt repli- kowana zmianą DNA gospodarza (zob. powyżej).
kacyjny. Uzyskuje się to metodami inżynierii gene- W przeciwieństwie do somatycznej terapii geno-
tycznej przez eliminację określonych wirusowych wej – będąca teoretycznie efektywniejszym rodza-
genów potrzebnych do replikacji (np. gag, poi, env), jem terapii genowej – zarodkowa terapia genowa
co daje wystarczająco dużo miejsca do wbudowania z etycznego i prawnego punktu widzenia (prawo
Terapia genowa
DNA o znaczeniu leczniczym. Aby można było jed- o ochronie embrionów) nie zyska prawdopodobnie
nak namnażać wirusa z defektem replikacyjnym, tzw. żadnego znaczenia. W tym przypadku do dyspozy-
komórki pakujące muszą zawierać specjalne promo- cji (w Niemczech jeszcze zakazane) są nieszkodliwe
tory, które są transferowane razem z genem wiruso- techniki diagnostyki i selekcji preimplantacyjnej,
wym. W takich komórkach replikuje się manipulowa- umożliwiające uniknięcie defektu genetycznego przy
ne wirusy, które potem można stosować ex vivo lub in rodzinnej predyspozycji, stwierdzonej na podstawie
vivo (zob. ryc. A 7.1-1). przeprowadzonego wywiadu. A7
MUTSCHLER-2009.indd 119 2010-01-07 22:11:19
120 Terapia genowa i stosowanie oligonukleotydów antysensownych
7.2. Terapia antysensowna
Oprócz terapii genowej istnieją także inne możliwo- katalizowanemu przez endogenne enzymy. Z tego
ści wpływania na ekspresję genów. Zalicza się do nich powodu podjęto wiele prób uzyskania ich stabilności
stosowanie tzw. oligonukleotydów antysensownych. przez modyfikację chemiczną. Przykładem takiej mo-
Składają się one z krótkich sekwencji nukleotydów dyfikacji jest zastąpienie atomu tlenu w części fosfo-
(przeciętnie zawierających 15–25 zasad), zbudowa- ranowej oligonukleotydu grupą metylową (metylofo-
nych dokładnie komplementarnie do genu, którego sforan) lub atomem siarki (tiofosforan, oligomer S).
ekspresja ma zostać zahamowana (zob. ryc. A 7.2-1). Fomiwirsen jest pierwszym oligonukleotydem
Komplementarne oligonukleotydy mogą przykła- antysensownym wprowadzonym na rynek w USA,
dowo ulec hybrydyzacji z regulatorowymi sekwen- którego stosowanie jest wskazane przy zapaleniu
cjami DNA przy końcu 5’ danego genu i w ten sposób siatkówki wywołanym przez wirusa cytomegalii
oddziaływać na jego ekspresję. Inne możliwe zasto- człowieka (CMV). Ta postać zapalenia siatkówki
sowanie tych oligonukleotydów polega na ogranicze- występuje u 15–40% chorych na AIDS i może bardzo
niu translacji poprzez interakcje z mRNA. W obydwu szybko doprowadzić do ślepoty. Fomiwirsen jest sto-
przypadkach problem stanowi jednak to, że oligonu- sowany przeciwko występującej przy replikacji wiru-
kleotydy wprawdzie przechodzą na drodze endocyto- sa wczesnej odpowiedzi komórkowej, zapobiegając
zy przez błonę komórkową i następnie wiążą się z do- przez to postępowi choroby. Powinien być raz w ty-
celową strukturą, ale ulegają szybkiemu rozkładowi godniu wstrzykiwany do ciała szklistego.
ATA A C C
oligonukleotyd antysensowny
3'
3'
C GA CC T G T A
3' 3'
mRNA
aminokwasy
UG G
UG G
transkrypcja translacja
UAU
UUAU
rybosom
AU
G AC A
5'
5'
GCU G G AC
rozkład
GCU G
jądro
komórowe białko
5' 5'
Ryc. A 7.2-1. Zasada stosowania oligonukleotydów antysensownych w celu hamowania translacji genu (wg Askariego i McDonella).
MUTSCHLER-2009.indd 120 2010-01-07 22:11:19
Terapia genowa i stosowanie oligonukleotydów antysensownych 121
7.3. Terapia za pomocą komórek pnia
Komórki pnia są to komórki, które jeszcze nie uległy właściwości komórek pnia. Jednakże w określonych
zróżnicowaniu, a które mogą się w ograniczony spo- warunkach przy ich podziale powstają tzw. komórki
sób rozmnażać i które zachowały zdolność przekształ- progenitorowe, które są zdolne najpierw ulegać dal-
cania się w komórki somatyczne. Zgodnie z oczeki- szemu namnażaniu, a w końcu różnicowaniu w doj-
waniem występują one u zarodka i płodu, a i potem rzałe komórki. Już od wielu lat wprowadza się tera-
zostały stwierdzone także w większości narządów pie z zastosowaniem komórek pnia osób dorosłych,
osób dorosłych. Dlatego wyróżniamy komórki pnia np. przy przeszczepie szpiku kostnego w przypadku
embrionalne, osób dorosłych, a także pępowinowe białaczki. Jak do tej pory jedynie w doświadczeniach
(zob. ryc. A 7.3-1A i B). na zwierzętach wypróbowano zastosowanie komórek
pnia dorosłych osób w przypadku chorób mięśnia
Linie embrionalnych komórek pnia pochodzące sercowego czy neurodegeneracyjnych.
z wewnętrznych komórek blastocysty są pluripo-
tencjalne, tzn. mogą się różnicować na więcej niż Niezależnie od wyników badań z zastosowaniem ko-
200 różnych typów komórek organizmu człowieka. mórek pnia zarodka czy osób dorosłych coraz więk-
Warunkiem tego jest jednakże konieczność występo- sze uznanie znajduje koncepcja istnienia nowotworo-
wania czynników specyficznych dla danej tkanki. Bez wych komórek pnia, która może istotnie przyczynić
nich embrionalne komórki pnia dzielą się w sposób się do zrozumienia tworzenia się i rozwoju tkanki no-
ograniczony i zachowują swoją pluripotencjalność. wotworowej. Wykazano na przykład, że w przypadku
Do chwili obecnej nie wprowadzono żadnego po- białaczki czy nowotworu sutka tkanka nie składa się
stępowania medycznego z zastosowaniem embrio- z jednakowych komórek. Często, podobnie jak na-
nalnych komórek pnia. Związane jest to szczególnie rządy, składa się ona z różnorodnych komórek, po-
z faktem istnienia w wielu krajach moratorium na sto- siadających hierarchiczną strukturę. Podstawę guza
sowanie i produkcję komórek pnia, których pozyski- stanowią przy tym komórki pnia, które mogą tworzyć
wanie prowadzi do uszkodzenia ludzkiego zarodka. każdy typ komórki w tkance nowotworowej.
Komórki pnia osób dorosłych są niezbędne przy Odkrycie komórek pnia nowotworu stawia pod
odnawianiu wyspecjalizowanych komórek somatycz- znakiem zapytania konwencjonalną terapię przeciw-
nych. Tak jak w przypadku embrionalnych komórek nowotworową, gdyż prowadzi ona do uszkodzenia
pnia mogą one przy podziale komórkowym zachować każdej komórki, która się szybko dzieli. Komórki
Terapia genowa
zarodek zarodek zarodek zarodek zarodek dziecko dorosły
jednokomórkowy 3-dniowy 5–7-dniowy 4-tygodniowy 6-tygodniowy
rak
komórki pnia potworniak
embrionalne płodowe dorosłego
komórki pnia komórki pnia pluripotencjalne
totipotencjalne pluripotencjalne lub multipotencjalne A7
lub multipotencjalne
pępowinowe lub łożyskowe embrionalne
komórki pnia komórki pnia
pluripotencjalne pluripotencjalne
lub multipotencjalne
Ryc. A 7.3-1 A. Typy komórek pnia, które występują podczas rozwoju człowieka.
MUTSCHLER-2009.indd 121 2010-01-07 22:11:19
122 Terapia genowa i stosowanie oligonukleotydów antysensownych
Pluripotencjalne komórki pnia
Komórki pnia specyficzne dla tkanek ? ?
hematopoetyczne neuronalne mezenchymalne
komórki pnia komórki pnia komórki pnia
Komórki
progenitorowe
chondroblasty,
limfoidalne i mieloidalne fibroblasty,
komórki progenitorowe kardiobroblasty,
mioblasty,
osteoblasty,
preadipocyty
komórki prekursorowe komórki prekursorowe komórki prekursorowe
Zróżnicowane
komórki
adipocyty,
limfocyty T i B, chondrocyty,
komórki dendrytyczne, fibroblasty,
erytrocyty, granulocyty, astrocyty, kardiomiocyty,
monocyty, komórki NK, neurony, miocyty,
trombocyty oligodendrocyty osteocyty
Ryc. A 7.3-1 B. Możliwe drogi różnicowania pluripotencjalnych komórek pnia.
pnia jednak najczęściej dzielą się wolno i nie pod- należy zaliczyć oddziaływanie wybiórczo na komórki
legają tej strategii. Stanowi to jedno z możliwych pnia nowotworu. Nie jest to jednak obecnie możliwe
wytłumaczeń wysokiej częstości nawrotów przy ze względu na brak zrozumienia procesów kontroli
niektórych chorobach nowotworowych. Konieczne wzrostu komórek pnia. Nie istnieje także wyjaśnienie
są w związku z tym nowe formy terapii, do których rozwoju nowotworu z komórek pnia.
MUTSCHLER-2009.indd 122 2010-01-07 22:11:20
Chronofarmakologia (biorytmy w działaniu leków) 123
8. Chronofarmakologia
(biorytmy w działaniu leków)
Czynności wszystkich istot żywych – od jednoko- niewydolność serca, reumatoidalne zapalenie stawów,
mórkowych aż do ludzi – podlegają wpływom czasu, AIDS) mogą również wpływać na rytmy biologiczne,
co wyraża się w rytmach biologicznych. Można je wy- wywołując określone zaburzenia. Takie negatywne
kazać na poziomie wszystkich czynności organizmu. zmiany rytmów biologicznych stwierdza się również
Oprócz rytmów okołomiesięcznych (rytmy miesięcz- u osób pracujących na zmiany oraz po lotach między-
ne) i okołorocznych (rytmy roczne) występują prze- kontynentalnych (tzw. jet lag).
de wszystkim rytmy okołodobowe (rytmy dzień-noc, Nic więc dziwnego, że stwierdzono zależne od
rytmy 24-godzinne). Rytmy te są zdeterminowane pory dnia różnice w działaniu wielu różnych grup le-
genetycznie (tzw. zegary biologiczne lub wewnętrz- ków – mogą one wynikać z różnic zarówno w farma-
ne), jednakże mogą one podlegać wpływom czynni- kodynamice, jak i w farmakokinetyce. Poniżej przed-
ków środowiska zewnętrznego (wyznacznik czasowy). stawiono niektóre ważniejsze przykłady.
U ludzi i zwierząt największe znaczenie mają ryt-
my okołodobowe, których dotyczy także większość Leki znieczulające miejscowo i przeciwbólowe.
badań. Wykazano to np. w odniesieniu do tempera- Stężenie i wydzielanie endogennych substancji prze-
tury ciała, częstości skurczów serca, ciśnienia tętni- ciwbólowych (endorfin) podlega rytmom okołodobo-
czego krwi, przepływu narządowego krwi, czynności wym. To samo dotyczy progu bólowego i co jest z tym
płuc i nerek, a także stężenia neuroprzekaźników, związane – działania leków znieczulających miej-
hormonów, enzymów, elektrolitów i glukozy. scowo i przeciwbólowych. Na przykład ból zęba
Interesujące jest, że również zmiany chorobowe jest odczuwany wczesnym popołudniem nie tak do-
występują z niejednakową częstością w ciągu dnia tkliwie jak rano czy w nocy. Znoszące ból działanie
lub w trakcie roku. Zawały serca oraz nagła śmierć leków znieczulających miejscowo jest w tym czasie
sercowa występują najczęściej między godziną 8 a 12 wyraźnie dłuższe niż o innych porach doby (zob. ryc.
rano. Napady dychawicy oskrzelowej obserwuje się A 8-1).
bardzo często ok. godz. 4 nad ranem. Do najbardziej Objawy reumatoidalnego zapalenia stawów wyka-
znanych rytmów sezonowych należy depresja zimo- zują również wahania zależne od pory dnia. Chorzy
wa. Z drugiej strony stany chorobowe (np. depresje, skarżą się głównie rano na sztywność stawów i obrzę-
ki – w ciągu dnia dolegliwości te zmniejszają się sa-
moistnie. Na uwagę zasługuje fakt, że uszkadzające
śluzówkę żołądka działanie kwasu acetylosalicylo-
wego jest wyraźnie mniejsze przy jego podawaniu
efekt (AUC)
1000 wieczorem niż rano.
Glukokortykosteroidy. Jednym z najważniejszych
przykładów znaczenia chronofizjologii bądź chrono-
800
farmakologii jest wahające się w ciągu doby stężenie
kortyzolu we krwi. Działania niepożądane glukokor-
tykosteroidów można zmniejszyć przy dostosowaniu
Chronofarmakologia
600 ich podawania do rytmu dnia, to samo dotyczy fizjo-
logicznego sprzężenia zwrotnego ujemnego pomię-
dzy korą nadnerczy i przysadką.
400
5 8 11 14 17 Leki przeciwastmatyczne. Napady dychawicy
oskrzelowej występują, jak już wspomniano, szcze-
pora dnia (godz.) gólnie często w nocy. Jest to zrozumiałe, jeśli weź-
mie się pod uwagę badania chronobiologiczne.
Ryc. A 8-1. Rytm dobowy działania leku znieczulającego miej- W czasie spoczynku działanie histaminy i acetylo-
scowo – artikainy (wg Lemmera). choliny kurczące oskrzela jest szczególnie nasilone
A8
MUTSCHLER-2009.indd 123 2010-01-07 22:11:20
124 Chronofarmakologia (biorytmy w działaniu leków)
w następstwie fizjologicznego podwyższenia oporu no, że u pacjentek z rakiem jajników podawanie le-
dróg oddechowych w wyniku nieznacznej jedynie ków dostosowane do rytmu okołodobowego prawdo-
aktywności układu współczulnego. Wydzielanie kor- podobnie poprawia wyniki leczenia i przeżywalność,
tyzolu jest w tym czasie najmniejsze. Fakty te należy a jednocześnie zmniejsza działania niepożądane.
uwzględniać przy podejmowaniu racjonalnej farma- Nefrotoksyczność cisplatyny okazała się przy poda-
koterapii dychawicy oskrzelowej. waniu leku wieczorem wyraźnie mniejsza niż przy
Przy podawaniu teofiliny rano z reguły uzyskuje podawaniu rano.
się wyższe stężenia maksymalne w osoczu, jak rów-
nież krótszy czas potrzebny do uzyskania wartości
Cmax niż przy jej podawaniu wieczorem. Ze względu
na to, że w nocy konieczne są wyższe stężenia teofili-
ny, w celu uniknięcia upośledzenia funkcji układu od- 100
dechowego, wieczorem wskazane jest w wielu przy-
padkach stosowanie większych dawek leku. Wydaje 90
się również, że w nocy niezbędne jest podawanie
HR (uderzenia/min)
osobom chorym na astmę większych dawek leków 80
β2-adrenomimetycznych i antagonistów receptorów
muskarynowych. 70
Leki przeciwnadciśnieniowe. Zarówno u osób zdro- 60
wych, jak i u pacjentów z pierwotnym nadciśnieniem
w nocy dochodzi najczęściej do obniżenia ciśnienia 50
skurczowego i rozkurczowego, a także do zmniejsze-
nia częstości skurczów serca (zob. ryc. A 8-2). 190
Różne leki przeciwnadciśnieniowe (np. leki blo- 180
kujące receptory β-adrenergiczne) obniżają podwyż-
szone ciśnienie krwi przede wszystkim w czasie dnia, 170
co jest związane ze zwiększonym w tym okresie na- 160
RRskr. (mm Hg)
pięciem układu współczulnego. Diuretyki natomiast
działają w nocy tak samo silnie jak w dzień. 150
Podczas stosowania szybko działającej formy le- 140
ków blokujących kanały wapniowe – nifedypiny,
stwierdzono, podobnie jak w przypadku teofiliny, 130
wyższe stężenia we krwi po podaniu rano niż po za- 100
stosowaniu w nocy. Różnice takie nie występują przy
stosowaniu formy retard. Na ryc. A 8-2 przedstawio-
no 24-godzinny wpływ 2-krotnego podawania w cią- 120
gu dnia postaci o przedłużonym działaniu (retard) ni-
fedypiny na ciśnienie krwi i częstość skurczów serca 110
u chorych na nadciśnienie.
RRrozk. (mm Hg)
100
Leki przeciwhistaminowe (anty-H2). Również ta gru-
pa leków stanowi ważny przykład tego, że badania 90
chronofarmakologiczne mogą poprawiać skuteczność
farmakoterapii. Obserwacje okołodobowych zmian 80
wydzielania soku żołądkowego u osób zdrowych
i u pacjentów z chorobą wrzodową wykazały, że leki 7 11 15 19 23 3 7
przeciwhistaminowe (blokujące receptory histamino- pora dnia (godz.)
we H2), niezależnie od ich okresu półtrwania, mogą
być podawane tylko jeden raz wieczorem, a nie jak Ryc. A 8-2. Rytm okołodobowy częstości skurczów serca
dotychczas – wielokrotnie w ciągu dnia. (HR) i ciśnienia krwi (RR) u osób z nadciśnieniem tętniczym
przed (linia przerywana) oraz piątego dnia po przyjęciu (linia
Cytostatyki. Na podstawie wyników doświadczeń ciągła) nifedypiny retard 2 razy dziennie (rano i wieczorem)
na zwierzętach w badaniach klinicznych stwierdzo- (wg Lemmera).
MUTSCHLER-2009.indd 124 2010-01-07 22:11:20
Preparaty złożone 125
9. Preparaty złożone
Preparaty złożone
Preparat złożony powinno się wprowadzać do leczenie Przeciwko stałym preparatom złożonym przemawiają
tylko wówczas, gdy za pomocą pojedynczego leku (w następujące argumenty:
odpowiedniej dawce) nie można uzyskać pożądanego
■ liczba działań niepożądanych, szczególnie reakcji
efektu lub gdy zastosowanie preparatu składającego
się z dwóch leków pozwala uniknąć bądź zmniejszyć
alergicznych, najczęściej jest tym większa, im wię- A9
cej leków przyjmuje się jednocześnie,
działania niepożądane leku pierwotnego. Zadaniem
firm produkujących preparaty złożone jest wykaza- ■ tylko w wyjątkowych przypadkach składniki prepa-
nie, że każdy składnik deklarowany jako substancja ratu złożonego wykazują taką samą farmakokinety-
czynna istotnie uczestniczy w działaniu preparatu kę i czas półtrwania; wartości te mogą się ponadto
albo że preparat złożony wywołuje mniej działań nie- różnorodnie zmieniać w czasie leczenia w wyniku
pożądanych niż pierwotny składnik czynny. indukcji lub inhibicji enzymatycznej,
Istotnym argumentem przemawiającym za stały-
■ przebieg leczenia i ewentualne interakcje przy jed-
mi preparatami złożonymi, jeśli tylko składniki tego
noczesnym stosowaniu różnych leków stają się
preparatu z punktu widzenia farmakodynamicznego
mniej przejrzyste i zauważalne.
powinny być podawane razem, jest poprawa współ-
pracy pacjentów z lekarzem (compliance) i z tego
W przypadku wielu preparatów złożonych trudno
powodu lepsza realizacja programu leczenia. W ba-
mieć pewność, czy przez kombinację substancji lecz-
daniach dotyczących compliance stwierdzono, że na-
niczych można uzyskać istotną poprawę działania.
wet w warunkach klinicznych nie więcej niż połowa
Wielokrotnie zakładane potęgowanie działania (sy-
pacjentów przyjmuje leki zgodnie z zaleceniem leka-
nergizm hiperaddycyjny) występuje rzadko.
rza. U pacjentów leczonych ambulatoryjnie również
Poniżej podano wybrane przykłady racjonalnych
stwierdzono, że przestrzeganie przepisanego leczenia
i nieracjonalnych preparatów złożonych.
jest tym gorsze, im więcej leków zostało zaordyno-
wanych do jednoczesnego zażywania.
9.1. Racjonalne preparaty złożone
Leki przeciwparkinsonowe. Ostatnio stwierdza się powodu można podawać mniejsze dawki L-dopy,
zasadniczy postęp w leczeniu choroby Parkinsona zmniejszając jednocześnie działania niepożądane.
w następstwie wprowadzenia do lecznictwa lewodo-
py (L-dopy). Aby jednak uzyskać wymagany poziom Leki przeciwnadciśnieniowe. W leczeniu nadciś-
terapeutyczny w mózgu, konieczne było stosowanie nienia także często jest wskazane stosowanie terapii
dużych ilości L-dopy, ponieważ ok. 90% zastoso- skojarzonej, ponieważ skuteczność w leczeniu nad-
wanej dawki ulega dekarboksylacji w tkankach ob- ciśnienia za pomocą monoterapii można uzyskać
wodowych, a zatem tylko ok. 10% może przenikać tylko w mniej więcej 50% przypadków. Oznacza to,
do mózgu. Katecholaminy powstałe z dekarboksyla- że w przypadku pozostałych 50%, aby uzyskać po-
cji L-dopy na obwodzie wywołują poważne działania żądane obniżenie ciśnienia krwi, niezbędne jest jed-
niepożądane, szczególnie w przewodzie pokarmo- noczesne podawanie dwóch lub więcej leków prze-
wym i układzie krążenia. Istotną poprawę uzyska- ciwnadciśnieniowych o różnych punktach uchwytu
no dopiero przy jednoczesnym podawaniu L-dopy działania. Biorąc pod uwagę, że u chorych na nadciś-
z działającymi obwodowo inhibitorami dekarboksy- nienie często stwierdza się złą współpracę pacjenta
lazy dopa. Inhibitory te nie przenikają przez barierę z lekarzem – wysokie ciśnienie krwi nie powoduje
krew-mózg, nie działają zatem w ośrodkowym ukła- bowiem początkowo żadnych istotnych zaburzeń
dzie nerwowym. W tkankach obwodowych natomiast – uzasadnione jest stosowanie stałych preparatów
mniejsza ilość L-dopy ulega dekarboksylacji. Z tego złożonych.
MUTSCHLER-2009.indd 125 2010-01-07 22:11:20
126 Preparaty złożone
Diuretyki. W grupie leków moczopędnych tzw. diure- du na swoje właściwości osmotyczne wykazują
tyki pętlowe (saluretyki) zwiększają wydzielanie jonów działanie przeczyszczające, natomiast zawierające
sodu, potasu i chloru. Głównym niebezpieczeństwem glin – zapierające. Łączenie preparatów zawierają-
tego leczenia, zwłaszcza przy większych dawkach, cych związki magnezu i glinu może być uzasadnio-
jest hipokaliemia. Istnieją jednak diuretyki, które powo- ne w celu przeciwdziałania wpływowi pojedynczych
dują zatrzymywanie potasu w organizmie, dlatego ko- składników na perystaltykę jelit.
rzystne jest stosowanie u pacjentów ze zdrowymi ner-
kami równocześnie diuretyku wzmagającego wydzie- Hormonalne preparaty antykoncepcyjne. Jedno-
lanie potasu z lekiem moczopędnym oszczędzającym czesne podawanie estrogenów i gestagenów okazało
potas. Należy ponadto zwracać uwagę, aby farmako- się skuteczne jako doustna antykoncepcja hormonalna.
kinetyka obu stosowanych leków była podobna i żeby
odpowiednio dobrane dawki pojedynczych składników Leki przeciwzakażeniowe. Właściwe jest również
znosiły ich przeciwstawne efekty związane przejściem łączne stosowanie trimetoprimu z sulfonamidem,
potasu przez ścianę nefronu. Dotyczy to w szerokim np. z sulfametoksazolem. Przez równoczesne bloko-
zakresie łączenia triamterenu lub amiloridu z tiazyda- wanie przemiany kwasu foliowego w dwóch różnych
mi. Stosowanie takich leków łącznie nie musi się wią- miejscach uzyskuje się ograniczenie wytwarzania się
zać z koniecznością kontroli stężenia potasu we krwi. oporności.
Opóźnienie wytwarzania się oporności jest rów-
Leki neutralizujące kwas solny. Leki neutralizujące nież podstawą do łącznego stosowania dwóch lub
kwas solny w żołądku zawierające magnez ze wzglę- więcej leków w leczeniu gruźlicy lub zakażenia HIV.
9.2. Nieracjonalne preparaty złożone
Nienarkotyczne leki przeciwbólowe. W ostatnich Leki nasercowe. Nie należy stosować preparatów zawie-
latach liczba preparatów złożonych zaliczanych rających łącznie glikozydy nasercowe i inne leki wpły-
do nienarkotycznych leków przeciwbólowych ule- wające na czynność serca, ponieważ glikozydy naserco-
gła zmniejszeniu. Wiele z nich zostało uznanych za we charakteryzują się bardzo wąską rozpiętością tera-
nieracjonalne (nieuzasadnione). Na przykład dodatek peutyczną i w związku z tym w ich przypadku koniecz-
chininy jest nie tylko zbyteczny, ale również niewłaś- ne jest indywidualne dostosowanie leczenia. Ponadto
ciwy ze względu na możliwość działań niepożąda- glikozydy nasercowe mają szczególne właściwości
nych (zaburzenia czynności serca). Istnieją przesłan- farmakokinetyczne, np. długi czas półtrwania w osoczu.
ki wskazujące, że stosowanie preparatów złożonych
Leki przeciwzakażeniowe. Nie powinno się sto-
z kofeiną jest nieuzasadnione.
sować np. preparatów zawierających antybiotyki
Leki przeciwreumatyczne. Dotychczas często sto- łącznie z lekami przeciwkaszlowymi, ponieważ leki
sowane stałe preparaty złożone zawierające gluko- przeciwkaszlowe utrudniają wykrztuszanie śluzu,
kortykosteroidy i niesteroidowe leki przeciwzapalne a pacjenci nieznający składu preparatu mogą go po-
nie powinny być nadal stosowane w lecznictwie, traktować jako zwykły lek przeciwkaszlowy, nie bio-
ponieważ glukokortykosteroidy mogą być podawane rąc pod uwagę, że zawiera on antybiotyk.
jedynie przez krótki czas oraz w dawkach dostosowa- Problematyczne jest również stosowanie miejsco-
nych do stanu klinicznego pacjenta, natomiast nieste- we preparatów zawierających łącznie lek przeciwza-
roidowe leki przeciwzapalne muszą być stosowane każeniowy i glukokortykosteroidy, ponieważ stoso-
przez dłuższy czas. Ponadto glukokortykosteroidy, wanie tych preparatów w różnych chorobach skóry
które same nie działają wrzodogennie, zwiększają bez ustalenia właściwego rozpoznania (działają one
częstość występowania owrzodzeń w przewodzie po- zarówno przy wypryskach, jak i przy zakażeniach
karmowym związanych ze stosowaniem niesteroido- skóry) wywołuje częściej działania niepożądane niż
wych leków przeciwzapalnych. preparaty stosowane oddzielnie.
W przeciwieństwie do tego łączne podawanie Preparaty złożone powinny być zalecane tylko
niesteroidowych leków przeciwzapalnych (NLPZ) wtedy, gdy ich stosowanie jest w pełni uzasadnio-
z mizoprostolem może być racjonalne, ponieważ ne i kiedy wykazują wyraźne korzyści w porówna-
mizoprostol może zmniejszać częstość występowa- niu z pojedynczymi składniki. Należy podkreślić,
nia indukowanych przez NLPZ uszkodzeń śluzówki że w ostatnich latach coraz więcej nieracjonalnych
przewodu pokarmowego. preparatów wycofanych jest z rynku.
MUTSCHLER-2009.indd 126 2010-01-07 22:11:21
Poszukiwanie i badanie nowych leków 127
10. Poszukiwanie i badanie nowych leków
Celem poszukiwania nowych leków jest uzyskanie Przed zastosowaniem badanej substancji po raz
Poszukiwanie i badanie
lepszych możliwości terapeutycznych. Najbardziej pierwszy u człowieka niezbędna jest szczegółowa oce-
korzystna sytuacja istnieje wówczas, gdy nowy lek na jej różnych właściwości farmakologicznych (różno-
stwarza możliwość leczenia choroby dotychczas nie- rodnych działań) przez przetestowanie zarówno w do-
nowych leków
uleczalnej. Niejednokrotnie jednak musimy zadowa- świadczeniach in vitro, jak i na zwierzętach. W związ-
lać się tym, że nowy lek upraszcza lub ulepsza stoso- ku z tym wyróżnia się następujące etapy badań:
waną już terapię. ■ badania przedkliniczne (lub doświadczalne bada-
Wstępnym etapem tego poszukiwania jest synte- nia farmakologiczne i toksykologiczne),
za lub izolacja substancji potencjalnie czynnej, która
– po określeniu budowy i właściwości chemicznych – ■ badania kliniczne.
będzie poddawana badaniom przedstawionym na ryc. Wytyczne dotyczące badania leków są zgodne z prze-
A 10
A l0-1. Na każdym etapie trzeba liczyć się przy tym pisami służby zdrowia danego państwa; uwzględnia-
z koniecznością przerwania dalszych badań ze wzglę- ją one również wytyczne międzynarodowych towa-
du na niewystarczającą siłę działania, niekorzyst- rzystw fachowych.
ne działanie farmakologiczne lub na występowanie
efektów toksycznych. Ocenia się, że na 6000–10 000 Badanie.
przebadanych związków do lecznictwa przechodzi W Niemczech podstawy prawne związane z bada-
zaledwie jeden. niem środków leczniczych podlegają Ustawie
substancja badana
doświadczalna ocena toksyczności ostrej skryning
farmakologia
i toksykologia
ocena toksyczności farmakodynamika farmakokinetyka u zwierząt
przewlekłej
farmakologia
kliniczna faza I obserwacja porejestracyjna
faza II
faza III
dopuszczenie do lecznictwa
faza IV opracowanie
formy leku
Ryc. A 10-1. Schemat poszukiwania nowych leków. Po doświadczeniach in vitro i na zwierzętach (farmakologia doświadczalna)
przeprowadza się badania na ludziach (farmakologia kliniczna). Na początku wykonuje się opracowania galenowe (formy leku) oraz
przeprowadza badania toksyczności przewlekłej.
MUTSCHLER-2009.indd 127 2010-01-07 22:11:21
128 Poszukiwanie i badanie nowych leków
o środkach leczniczych z 1976 r., ostatnio zmienio- uwzględniają wytyczne europejskich oraz innych
nej w 2005 r., a w Polsce obowiązuje Prawo farma- urzędów zdrowia, a także międzynarodowych towa-
ceutyczne z 6 września 2001 r. Powyższe przepisy rzystw fachowych.
10.1. Badania przedkliniczne
W badaniach przedklinicznych substancje badane które odgrywają istotną rolę w patogenezie chorób.
podlegają przede wszystkim farmakologicznemu W związku z tym istotnie wzrosła ilość potencjalnych
skryningowi (screen – przesiewać), tzn. wielu róż- punktów uchwytu działania leków. Obecnie poznano
nym badaniom, które umożliwiają w ogólnym zary- ok. 1500 takich nowych punktów celowych (targets)
sie określenie profilu działania. W ostatnich latach i przypuszcza się, że liczba ta jeszcze istotnie wzroś-
coraz częściej wykorzystuje się przy tym analizy nie. Znaczenie zmian patobiochemicznych w choro-
biochemiczne i badania z zakresu biologii i farmako- bach ludzi bada się obecnie przy użyciu tzw. zwierząt
logii molekularnej, które umożliwiają szybszą i częś- knock-out lub transgenicznych. W przypadku zwie-
ciowo bardziej szczegółową charakterystykę profilu rząt knock-out zostają wyłączone docelowe geny,
działania testowanej substancji. Ponadto zwiększa a u zwierząt transgenicznych ludzki obcy gen zostaje
się częstość wczesnego rozpoznania niedostatecznie włączony do genomu zarodka i w ten sposób przy po-
aktywnych lub wywołujących działania niepożąda- dziale komórkowym jest przekazywany do komórek
ne substancji, które nie mogą być zaakceptowane potomnych.
(unikanie fałszywie pozytywnych ocen). Pozwala Od czasów coraz lepszego wyjaśnienia genomu
to na wyeliminowanie takich substancji z dalszych mikroorganizmów, co nastąpiło od połowy lat 90. XX
kosztownych badań już we wczesnej fazie ich oceny. wieku, stale rosną nadzieje na zidentyfikowanie no-
Oczekuje się, że szczególnie efektywne systemy te- wych, charakterystycznych jedynie dla różnych ga-
stowania (tzw. wysoko wydajny skryning) zmniejszą tunków zarazków, punktów uchwytu leków przciw-
częstość uzyskiwania fałszywie pozytywnych wyni- zakaźnych, które różnią się wystarczająco od struktur
ków, co wyeliminuje z dalszych badań początkowo ludzkiego organizmu.
bardzo obiecujące substancje. W ten sposób można
doprowadzić do przyspieszenia badań i zmniejszenia Wysoko wydajny skryning. Osiągnięcia biotech-
związanych z tym kosztów. nologiczne (genotechnika, fermentacja) pozwala-
Postęp w tej dziedzinie wynika w ostatnich latach ją za pomocą badań in vitro uzyskać łatwy dostęp
z różnorodnych nowych rozwiązań. Należą do nich do punktów celowych i w ten sposób osiągnąć no-
przede wszystkim: watorskie testowanie nowych substancji czynnych.
Postęp w zakresie wprowadzenia do laboratoriów
■ możliwość identyfikacji nowych punktów uchwy-
bardzo czułych technik pomiaru umożliwia miniatu-
tu działania leków (biologicznej struktury docelo-
ryzację odpowiednich badań, np. tzw. binding study
wej),
(wiązanie z ligandem). W ten sposób wprowadzono
■ wysoko wydajny skryning, obecnie do badań rutynowych 96 testów na jednym
przyrządzie z użyciem mikroszalek. Dalsza miniatu-
■ dostępność obszernej „biblioteki substancji biolo-
ryzacja pozwoli w najbliższej przyszłości na jeszcze
gicznie czynnych”,
większe przyspieszenie procesu badań (superwyso-
■ metody kombinatoryjnej chemii w połączeniu z au- ko wydajny skryning). Poza tym zmniejszy się ilość
tomatyczną syntezą związków. substancji niezbędnej do jednego testu, co pozwoli
na zbadanie wpływu potencjalnej substancji czynnej
Należy także pamiętać o postępach z zakresu mole- na liczne struktury docelowe, a przez to na proste
kularnego modelowania, które umożliwiają znaczne i szybkie określenie jej profilu działania.
zwiększenie wydajności przebiegu procesu poszuki-
wań nowych leków. Biblioteki substancji aktywnych i chemia kom-
binatoryjna. Dostępne obecnie liczne zestawienia
Punkty celowe. Zachodzące w ostatnich latach po- (tzw. biblioteki) substancji aktywnych stanowią waż-
stępy w biologii molekularnej, m.in. badania w za- ne uzupełnienia, które mogą być wykorzystywane
kresie genomu i proteomu, doprowadziły do pozna- w miarę potrzeby. Jednak niezbędnym warunkiem
nia większej liczby struktur w ludzkim organizmie, zidentyfikowania głównej struktury, przy jednoczes-
MUTSCHLER-2009.indd 128 2010-01-07 22:11:21
Poszukiwanie i badanie nowych leków 129
nym wykluczeniu niewłaściwych struktur, jest bardzo Pochodzenie środków biologicznie czynnych.
staranny dobór substancji do testu. Substancje czynne działające przeciwzakaźnie i cy-
Należy także pamiętać, że związki chemiczne tostatyczne znajdowane są często w naturze, gdzie
mogą być łączone standardowo i często automatycz- umożliwiają przeżycie danym gatunkom w porówna-
nie w różny sposób (chemia kombinatoryjna). W ten niu z gatunkami konkurencyjnymi. Jednakże wyeks-
sposób w ciągu kilku tygodni można stworzyć setki trahowanie aktywnej struktury może być procesem
tysięcy nowych związków, które mogą być dostar- kosztownym. Poza tym komercyjna stabilna synteza
czone do skryningu. bądź produkty optymalizowania często przewyższa-
Poszukiwanie i badanie
ją kompleksowe struktury produktów naturalnych.
Modelowanie molekularne. Ulepszone metody mo- Przykładem jest paklitaksel, wytwarzany w małych
delowania molekularnego, pozwalają na precyzyjne ilościach przez wolno rosnący cis pacyficzny, który
nowych leków
zaprojektowanie struktury chemicznej, która będzie znalazł zastosowanie jako cytostatyk dopiero po zna-
wywoływała optymalną interakcję między substancją lezieniu w cisie europejskim struktury wyjściowej
czynną a punktem docelowym. podlegającej częściowej syntezie. Kliniczne zastoso-
wanie cefalosporyny jako środka przeciwzakaźnego
Przykładem zakończonego sukcesem opracowania nowego stało się możliwe po odkryciu silnie działających
leku na drodze zidentyfikowania punktu docelowego i mo- pochodnych, a dla zastosowania naturalnej strepto-
delowania molekularnego jest odkrycie inhibitora neurami-
graminy były konieczne pochodne rozpuszczalne A 10
nidazy, mającej zastosowanie we wczesnej terapii wirusa
grypy. Łączy się on z aminokwasem w aktywnym centrum w wodzie.
ważnego dla rozprzestrzeniania się wirusa w drogach odde-
chowych enzymu neuraminidazy. Kompleksowe testy farmakologiczne. Związki,
które przy odpowiednim badaniu przesiewowym były
Na podstawie danych związanych ze strukturą obiecujące i przeszły zadowalająco badania na ostrą
czy za pomocą „metody in silico” nie jest jeszcze toksyczność, są następnie poddawane poszerzonym
możliwe dokładne przewidywanie właściwości far- badaniom farmakologicznym. W tym zakresie
makokinetycznych, które mają istotne znaczenie przeprowadza się zwłaszcza następujące badania:
w działaniu leku. Także przewidywanie efektów tok-
■ badanie działania głównego, a także jakościowy
sycznych z danych dotyczących struktury chemicznej
i ilościowy zakres tego działania,
jest możliwe tylko w przybliżeniu. A zatem w celu
określenia profilu działania ciągle jeszcze niezbędne ■ badania mające na celu wyjaśnienie punktu uchwy-
są doświadczenia na zwierzętach, które powinny być tu i mechanizmu działania,
wykonywane równocześnie z innymi badaniami uzu-
■ badania wpływu na czynność poszczególnych na-
pełniającymi (testy alternatywne, testy zastępcze).
rządów oraz określenie swoistości i narządowej
Przez badania uzupełniające rozumie się doświadczenia wybiórczości działania,
na systemach enzymatycznych, na izolowanych komór- ■ badanie tolerancji miejscowej i ogólnej,
kach lub hodowlach komórek, a także na narządach izolo-
wanych. Do metod tych należą np. badania tolerancji miej- ■ badanie toksyczności,
scowej na zalęgłych jajach kurzych (Hen-Egg-Test, Chorio-
Allantois-Membrane; HET-CAM) zamiast na rogówce oka ■ przy związkach chiralnych zbadanie różnorodnych
królika, badania fototoksyczności na kulturach komórek efektów obydwu enancjomerów.
keratynocytów bądź na modelach ludzkiej skóry (np. rekon-
struowany naskórek). Te ostatnie pozwalają także na ocenę
wchłaniania przezskórnego podawanych miejscowo leków Na podstawie uzyskanych wyników przeprowadza
dermatologicznych. Ustalenie metabolizmu środka lecz- się ponownie selekcję, a związki wybrane do ewen-
niczego, a także jego interakcji jest możliwe z zastosowa-
niem stabilnych transfekowanych genów danych enzymów tualnego stosowania u ludzi bada się na podostrą tok-
(komórki V79). Do badania działania mutagennego służy syczność po podawaniu podskórnym. Jeśli wyniki
tzw. test Amesa. Opracowuje się test embrionalnych ko- tych badań okażą się obiecujące, przeprowadza się
mórek pnia na mysiej linii komórkowej (EST) do badania badania farmakokinetyki u zwierząt. Jednocześnie
embriotoksyczności. Postęp w zakresie uwiarygodnienia ocenia się działania mutagenne i teratogenne, a na-
tych testów zwiększy z pewnością częstość ich stosowania.
Już teraz zabronione jest badanie na zwierzętach fitotok- stępnie toksyczność przewlekłą.
syczności różnych związków chemicznych i kosmetyków.
Jednakże testy uzupełniające nigdy nie będą mogły całko- Badania toksyczności podostrej, podprzewlekłej i przewle-
wicie zastąpić badań na zwierzętach. Na przykład dystry- kłej prowadzi się przez okres 2–4 tygodni lub – w zależno-
bucja czy wpływ leku na organizm możliwe są do zbadania ści od planowanego czasu podawania u ludzi – ponad 3–9
tylko na zwierzętach. (–12) miesięcy. Badania te przeprowadza się na 2 gatunkach
MUTSCHLER-2009.indd 129 2010-01-07 22:11:21
130 Poszukiwanie i badanie nowych leków
zwierząt (l gryzoń i l niegryzoń). Badane substancje poda- noznacznego odniesienia tych wyników do ludzi.
je się doustnie z pożywieniem lub przez sondę żołądkową Jednak wyniki te, szczególnie gdy dotyczą czynno-
albo ewentualnie inną drogą w zależności od przewidywa-
ści poszczególnych narządów, są znacznie bardziej
nej drogi podawania u ludzi. Ważne jest określenie tzw.
no-effect-levels, to znaczy największych dawek, po których wiarygodne niż to się ogólnie przyjmuje. Substancja,
nie występują żadne efekty biologiczne. A zatem po tych która przykładowo działa u zwierząt spazmolitycznie,
dawkach nie powinno być różnic w porównaniu z kontro- znosi również stany skurczowe u człowieka. Można
lą w ilości spożywanego pokarmu, masy ciała, wzrostu, też oczekiwać, że związek, który zwiększa częstość
obrazu krwi, a także czynności poszczególnych narządów.
akcji serca u zwierząt, będzie również przyspieszał
Ponadto powinno się zastosować dawki, które wywołują
efekty toksyczne lub śmierć. Wszystkie zwierzęta po za- czynność serca u człowieka. To samo dotyczy wy-
kończeniu badań są poddawane sekcji, a ich narządy podle- ników wysoko wydajnych badań skryningowych
gają ocenie makroskopowej i mikroskopowej. dotyczących wiązania z receptorami, enzymami lub
białkami kanałów jonowych itp. Znacznie trudniejsze
Badania kliniczne można rozpocząć tylko wówczas, jest przeniesienie wyników badań substancji, które
gdy wyniki badań przedklinicznych wykażą z dużym wpływają na procesy psychiczne.
prawdopodobieństwem, że badana substancja nadaje W zakresie oceny działań niepożądanych również
się do terapeutycznego lub diagnostycznego zastoso- nie można wyjaśnić wszystkich niepewności za po-
wania u człowieka oraz że wykazuje określoną wyż- mocą doświadczeń na zwierzętach i badań uzupeł-
szość w stosunku do innych podobnie działających niających. Mimo tych ograniczeń przeprowadzenie
leków. doświadczeń na zwierzętach jest niezbędnym warun-
W związku z tym powstaje często pytanie o możli- kiem do zastosowania określonej substancji badanej
wość przenoszenia wyników uzyskanych w doświad- u człowieka, ponieważ tylko w ten sposób można
czeniach na zwierzętach na ludzi. Jest oczywiste, zmniejszyć ryzyko, do zakresu możliwego do zaak-
że nie ma absolutnej zgodności i możliwości jed- ceptowania.
10.2. Badania kliniczne
Jak już wspomniano, badanie (testowanie) leku na lu- z tym osoby, które same nie mogą podejmować decyzji
dziach, dopuszczalne jest dopiero wówczas, gdy za- prawnych, np. dzieci, osoby chore psychiczne czy z zabu-
kładane korzyści dla danego człowieka lub dla społe- rzeniami pamięci, są szczególnie chronione. Badania in-
terwencyjne, tj. takie, które związane są z przyjęciem ba-
czeństwa przewyższają ryzyko związane z tym eks- danego związku, w przypadku terapii genowej można za-
perymentem. Najwyższe wymagania etyczne stawia cząć dopiero po uzyskaniu pozwolenia osób sprawujących
się przy badaniach na zdrowych ochotnikach, którzy opiekę. W przypadku innych związków leczniczych można
nie będą mieli żadnych korzyści z zastosowania da- rozpocząć wykonywanie badań 60 dni po zaakceptowa-
nego leku. Z tego powodu jest zrozumiałe, dlaczego niu wniosku, o ile wcześniej nie będzie sprzeciwu. Osoby
opiekujące się należy powiadamiać co do ciężkich działań
nie można zrezygnować z szerokich i całościowych niepożądanych (śmierć, inwalidztwo, wystąpienie koniecz-
badań przedklinicznych. ności lub przedłużenie leczenia stacjonarnego).
Zasadnicze wymagania etyczne dotyczące badań klinicz- Fazy badań klinicznych. W badaniach klinicznych
nych zawarte są w unowocześnionej Deklaracji Helsińskiej wyróżnia się zwyczajowo 4 fazy (I–IV).
Światowej Organizacji Zdrowia. Wymagania formalne
co do poprawy jakościowej leczenia i w ten sposób zwięk- Przez fazę I rozumie się pierwsze zastosowanie
szenia bezpieczeństwa postępowania terapeutycznego danej substancji u człowieka, co przeprowadza się
weszły w życie w 1992 r. jako wytyczne Good Clinical najczęściej u ludzi młodych i zdrowych. W wyjąt-
Practicefor Trials on Medical Products in the European kowych przypadkach można jednak, ze względów
Community (wytyczne GCP) oraz w dokumentach następ- etycznych lub naukowych, np. przy badaniu cytosta-
czych. Zawarte są tam m.in. zestawienia wzorów danych
badań przedklinicznych, zasady powoływania niezależnej tyków, zastosować dany lek po raz pierwszy u spe-
komisji etycznej przed rozpoczęciem badań, zgoda szcze- cjalnie wybranych pacjentów.
gółowo zaznajomionego z celem badań ochotnika, plan ba-
dań w wersji pisemnej, opis produkcji środka podlegającego W fazie I przeprowadza się następujące badania:
badaniu zgodnej ze standardami produkcji leków, wyczer-
pująca dokumentacja wyników, a także zarządzenia doty- ■ ocenia się tolerancję potencjalnej substancji aktyw-
cząca kwalifikacji lekarzy prowadzących badania. Zgodnie nej,
MUTSCHLER-2009.indd 130 2010-01-07 22:11:21
Poszukiwanie i badanie nowych leków 131
■ obserwuje się, czy u ludzi występują efekty far- W czasie badań klinicznych II i III fazy prowadzi
makodynamiczne, które stwierdzono w badaniach się rozpoczęte w czasie fazy I badania farmakokine-
przedklinicznych, tyczne. Uwzględnia się przy tym określone odrębno-
ści kinetyki u pacjentów z dużym stopniem ryzyka,
■ dokonuje się obserwacji występujących ewentual-
np. z upośledzoną czynnością wątroby lub nerek, jak
nie nieoczekiwanych efektów farmakodynamicz-
też interakcję z lekami, przede wszystkim tymi, które
nych, pozwalających na rozszerzenie wskazań
mogą być u tych pacjentów zastosowane.
leczniczych,
Poszukiwanie i badanie
■ przeprowadza się wstępne badania farmakokine- Dopuszczenie do obrotu. Po ukończeniu fazy III
tyczne u ludzi, wyniki określające jakość farmaceutyczną (ist-
nienie aktywnych fragmentów, środki pomocni-
nowych leków
■ ustala się propozycje dotyczące dawkowania w ce-
cze, zanieczyszczenia, metody produkcji i badania
lu dalszych badań klinicznych.
itd.), a także badań przedklinicznych i klinicznych
przedstawia się władzom służby zdrowia. Władze
Na podstawie wyników badań fazy I przystępuje się te analizują przedstawioną dokumentację i udziela-
do fazy II. W tej fazie przeprowadza się krótkie bada- ją (albo nie udzielają) zezwolenia na dopuszczenie
nia skuteczności działania i względnego bezpieczeń- nowego leku do sprzedaży. W Unii Europejskiej ist-
stwa na ograniczonej liczbie pacjentów ze schorze- nieją dwie formy dopuszczenia leku do obrotu: cen- A 10
niem, do którego leczenia substancja była przewidy- tralna poprzez odpowiednie instytucje w Londynie
wana. (European Medicines Evaluation Agency, EMEA)
W sumie w fazie II (badania eksploracyjne) bie- oraz zdecentralizowana, która opiera się na systemie
rze udział 100–500 pacjentów. Plan badań powinien wzajemnego uznawania miedzy państwami człon-
umożliwić ocenę intensywności działania, znaczenia kowskimi. Podanie dotyczące leku bardzo innowa-
klinicznego oraz ogólnych właściwości leku. Wyniki cyjnego, uzyskanego na drodze biotechnologicznej,
tych badań stanowią podstawę do planowania badań składa się do EMEA, podczas gdy leki zawierają-
fazy III. W fazie tej oprócz określenia zmian parame- ce znane środki lecznicze lub kombinację środ-
trów charakterystycznych dla danej choroby powinna ków leczniczych są dopuszczane do obrotu przede
być wyznaczona zależność efekt-dawka (najmniejsza wszystkim przez urzędy krajowe.
dawka działająca i największa dawka tolerowana),
a także określone dawkowanie do dalszych badań kli- W Unii Europejskiej pozwolenie na dany lek jest waż-
nicznych. Przy lekach przewidzianych do dłuższego ne początkowo 5 lat. W tym czasie przeprowadza się
stosowania przeprowadza się badania na wytwarzanie poszerzone badania skuteczności i bezpieczeństwa,
się tolerancji, kumulacji i interakcji z innymi lekami. zwracając szczególną uwagę na leki stosowane dłu-
Kontynuuje się rozpoczęte w badaniach przedkli- gotrwale. Każda nowa substancja lecznicza podlega
nicznych określenie stabilności, postaci farmaceu- w pierwszych 5 latach automatycznemu obowiązko-
tycznej i toksyczności leku, które, w miarę możliwo- wi przepisywania na receptę przez leczącego lekarza,
ści, powinny być w tym okresie zakończone. w wyniku czego zwiększa się bezpieczeństwo stoso-
W tym czasie ponownie należy zdecydować, wania leku w tej niezwykle krytycznej fazie. Na pod-
czy powinno się prowadzić dalsze badania dane- stawie obecnego stanu wiedzy po upłynięciu tego
go leku. W przypadku pozytywnej decyzji przystę- okresu można wystąpić z wnioskiem o przedłużenie
puje się do fazy III, która ma dostarczyć dowodu rejestru na kolejnych 5 lat lub zwolnienie z obowiąz-
(określonego biometrycznie) na aktywność i bez- ku przepisywania tego leku na receptę. Tak więc, po-
pieczeństwo nowej substancji. Próby niezbędne zwolenie na dany lek jest wznawiane co 5 lat.
do weryfikacji uzyskanych danych są przeprowadza- Odmowa rejestru następuje wówczas, gdy powsta-
ne na dużej liczbie pacjentów (próby losowe). Z tego ją wątpliwości co do skuteczności i bezpieczeństwa
powodu badania fazy III przeprowadza się w formie działania leku przy jego szerszym stosowaniu albo
badań wieloośrodkowych, tzn. wykonuje się badania gdy nie spełnia on określonych wymagań jakości far-
w różnych ośrodkach według takiego samego planu maceutycznej (czystość, zawartość substancji czyn-
z udziałem więcej niż 1000 pacjentów. Niezbędne nej i in.). Nie wymaga się jednak, aby lek był cał-
są przy tym badania porównawcze z innymi lekami kowicie pozbawiony działań niepożądanych. Należy
i z placebo (badania kontrolowane – zob. poniżej). natomiast wielokrotnie sprawdzać, czy działania nie-
W przypadku chorób przewlekłych pacjenci muszą pożądane pozostają w odpowiednim dopuszczalnym
być leczeni często do roku. Lekarze klinicyści mogą stosunku do skuteczności leku, zwłaszcza gdy istnie-
włączać do badań lekarzy nieklinicznych. ją inne możliwości lecznicze.
MUTSCHLER-2009.indd 131 2010-01-07 22:11:21
132 Poszukiwanie i badanie nowych leków
Po zarejestrowaniu należy wymagać od produ- kwasu acetylosalicylowego do hamowania agregacji
centa nadsyłania aktualnych sprawozdań dotyczą- płytek krwi lub późne objawy toksyczne.
cych bezpieczeństwa leku. Jeśli istnieje podejrzenie Większe znaczenie niż spontaniczne doniesienia
poważnego ryzyka (działania niepożądane, istnienie czy raporty mają celowane obserwacje stosowania
wzajemnych interakcji), wprowadza się tzw. plan (nie badania interwencyjne) z właściwą dokumenta-
stopniowego postępowania, w którym władze nad- cją wyników – mogą one ujawnić względnie wcześ-
zorcze wraz z producentem analizują wyniki cało- nie dowody na występowanie nieoczekiwanych
ściowe. Przy uzasadnionym podejrzeniu poważnego działań leku, niezależnie od tego, czy są one pożą-
ryzyka zezwolenie na dopuszczenie do obrotu zostaje dane czy niepożądane. Kontrola środka leczniczego
wycofane. Przez pojęcie poważnego ryzyka rozumie po jego zarejestrowaniu nosi nazwę inwigilacji far-
się takie, które zgodnie z dostępną wiedzą medyczną makologicznej. Monitorowanie preparatu podczas
przewyższa możliwą do zaakceptowania wielkość. jego stosowania służy również do ustalenia znaczenia
tego leku w relacji z innymi, biorąc szczególnie pod
Badania porejestracyjne. Rejestracja i dopuszcze- uwagę ryzyko jego stosowania.
nie do obrotu nie kończy badań nad działaniem leku. Od obserwacji efektów stosowania leku należy
Właściwą wiedzę na temat działania i bezpieczeń- odróżnić badania kliniczne, które przeprowadza się
stwa można bowiem uzyskać dopiero po bardziej po jego rejestracji (badania fazy IV). Są to badania
powszechnym i dłuższym jego stosowaniu w prak- długotrwałe lub interwencyjne często z udziałem wie-
tyce. W przypadku chorób przewlekłych dopiero lu tysięcy pacjentów, podczas których ustala się dalej
po rejestracji uzyskuje się dane dotyczące wpływu profil działania badanego leku. Ze względu na dużą
na długość i jakość życia (tzw. badania interwencyj- liczbę pacjentów można w ich trakcie wykryć rzadko
ne – zob. poniżej). Badania przedrejestracyjne często występujące działania niepożądane. Coraz większe
zmuszają do ograniczenia oceny wpływu leku na tzw. znaczenie mają badania porównawcze z innymi leka-
parametry zastępcze, np. obniżenia podwyższonego mi o ustalonym znaczeniu.
ciśnienia krwi lub zmniejszenia podwyższonego stę- W przypadku nowych wskazań ocenia się ponow-
żenia cholesterolu. Czasem dopiero po latach poja- nie bezpieczeństwo leku, co wymaga nowych badań
wiają się dodatkowe wskazania (np. wprowadzenie klinicznych II i III fazy.
10.3. Działanie placebo
Szczególną właściwością farmakoterapii u ludzi bóle głowy, pobudzenie, depresje) lub zaburzenia
jest to, że również podanie leku, który nie zawie- czynności przewodu pokarmowego. Szczególnie in-
ra żadnego środka leczniczego, czyli tzw. placebo, teresujący jest fakt, że efekty placebo są związane
prowadzi w całkiem dużym procencie (aż do 50%!) ze świadomością i nie występują nigdy u pacjentów
do uzyskania poprawy stanu zdrowia lub nawet wy- nieświadomych lub znajdujących się w narkozie.
leczenia. Zależy to od typu choroby, osobowości Przyczyna działania placebo jest tylko częściowo
pacjenta i sugestywnego wpływu lekarza. Działanie znana. Jakiś czas temu stwierdzono, że substancje
leków u pacjentów jest zatem związane w dużym endogenne, np. endorfiny, odgrywają istotną rolę
stopniu z działaniem sugestii. w działaniu placebo. U osób reagujących na działanie
Leki „pozorne” (placebo) mogą wywoływać placebo dochodzi do nasilenia ich wydzielania pod
jednak nie tylko efekty korzystne, ale także działa- wpływem jego podania. Związane jest z tym działa-
nia niepożądane. Po podaniu placebo obserwowano nie analgetyczne placebo – występujące przynajmniej
u 10–25% pacjentów zaburzenia czynności ośrod- u znacznej liczby pacjentów, zwłaszcza gdy poda się
kowego układu nerwowego (np. uczucie zmęczenia, dodatkowo antagonistę opioidów, np. nalokson.
MUTSCHLER-2009.indd 132 2010-01-07 22:11:21
Poszukiwanie i badanie nowych leków 133
10.4. Rodzaje badań leków
Odróżnia się następujące rodzaje badań leków: Takie badania przeprowadza się np. we wczesnym
okresie fazy II. W tym przypadku także podczas ob-
■ badania interwencyjne i nieinterwencyjne,
serwacji efektów stosowania leku nie ma grupy kon-
■ badania prospektywne i retrospektywne, trolnej.
Badania kontrolowane są etycznie uzasadnione
■ badania kontrolowane i niekontrolowane,
Poszukiwanie i badanie
i dozwolone jedynie wtedy, gdy nie ma możliwości
■ badania porównawcze między- i wewnątrzosobni- stosowania w tej grupie pacjentów zdecydowanie sku-
cze, teczniejszego leczenia. Jeżeli w III fazie badań okaże
nowych leków
się, że określone postępowanie lecznicze nie wyka-
■ badania otwarte i próby ślepe,
zuje wyraźnej przewagi skuteczności, nie powinno
■ badania poszukiwawcze i weryfikujące. się kontynuować tego postępowania w dalszych ba-
daniach. Próby długoterminowe (przewlekłe) powin-
Badania interwencyjne i nieinterwencyjne. W przy- ny być przerwane, gdy w ocenie okresowej zostanie
padku badań interwencyjnych dany lek zostaje pod- wykazana statystycznie znamienna wyższość okre-
dany testowi, w którym sposób jego stosowania ślonego schematu leczenia. Z tego wynika wyraźnie, A 10
jest ustalony z góry przez plan badania. Natomiast że stosowanie placebo w ciężkiej chorobie jest do-
w przypadku badań nieinterwencyjnych decyzje do- puszczalne tylko wtedy, kiedy w stosunku do danej
tyczące zastosowanej terapii i wybór leku następują choroby nie dysponuje się żadnym skutecznym le-
zgodnie ze zwykłą praktyką lekarską. Nie jest do- kiem.
zwolone w tym przypadku zastosowanie dodatko- Wiąże się z tym ostatnio także obowiązek produ-
wych metod diagnostycznych czy badań kontrolnych, centów, aby uczestnikom badań klinicznych, którzy
a analiza danych następuje jedynie za pomocą metod otrzymują nowy środek leczniczy, umożliwić jego
epidemiologicznych. przyjmowanie także po zakończeniu badań aż do mo-
mentu jego oficjalnej rejestracji.
Badania prospektywne i retrospektywne. W przy-
padku badań prospektywnych badania są dokładnie Grupy porównawcze. Istnieją różnorodne możli-
zaplanowane od początku aż do końca, a wszystkie wości przy ustalaniu grup kontrolnych. Na przykład
parametry podlegają ocenie. Przez odpowiedni dobór w dwóch lub więcej niezależnych grupach pacjentów
pacjentów zapewniona jest optymalna porównywal- prowadzi się ten sam sposób leczenia. W tym przy-
ność (np. podział według wieku, płci i in.). padku mówi się o grupach równoległych (paralel-
W badaniach retrospektywnych wykorzystuje się nych), a badania mają na celu międzyosobnicze po-
informacje dotyczące działania leku (oczekiwanego równanie działania leków.
lub niepożądanego) uzyskane z kart pacjentów lub Najważniejszą formą porównania wewnątrzosob-
z badań uzupełniających. Jest oczywiste, że znaczenie niczego jest badanie krzyżowe (cross-over). W tym
badań retrospektywnych jest niewątpliwie mniejsze przypadku początkowo pewna część badanej grupy
niż badań prospektywnych. Jednakże badania te są nie- otrzymuje badany preparat, druga zaś preparat po-
zbędne do oceny ryzyka występowania działań nie- równawczy. Po pewnej, wystarczająco długiej prze-
pożądanych oraz ewentualnych nowych zastosowań. rwie w pobieraniu leków – tzw. okres wymywania
Korzystna jest przy tym szybka dostępność wyników. (wash-out-time), który jest niezbędny, aby osiągnąć
w możliwie maksymalnym stopniu stan wyjściowy
Badania kontrolowane. Występowanie efektu place- – badanie kontynuuje się, wymieniając jednak grupy,
bo wskazuje wyraźnie, że w trakcie badania leku nale- tzn. obie grupy otrzymują wymiennie badane pre-
ży odróżnić istotne efekty farmakodynamiczne danej paraty. W ten sposób możliwe jest przeprowadzenie
substancji aktywnej od efektów od niej niezależnych. porównania wewnątrzosobniczego oraz porównania
Tego rodzaju badanie jest możliwe tylko za pomocą obu grup.
badań porównawczych, w których w identycznych Szczególne znaczenie ma fakt, że w trakcie pró-
warunkach porównuje się działanie nowego (poten- by krzyżowej obie grupy różnią się jedynie pod
cjalnego) leku na jednej grupie pacjentów z efektami względem sposobu leczenia. Natomiast inne różnice,
placebo i/lub znanego leku standardowego (badania np. w zakresie wieku, chorób towarzyszących, a prze-
kontrolowane). Gdy wszyscy pacjenci otrzymują ten de wszystkim stanu choroby, mogą być wyrównywa-
sam lek, wówczas są to badania niekontrolowane. ne. Można w ten sposób zminimalizować w znacz-
MUTSCHLER-2009.indd 133 2010-01-07 22:11:21
134 Poszukiwanie i badanie nowych leków
nym stopniu również błędy wynikające z przypadko- u ludzi. Metoda ta jest bezwzględnie zalecana szcze-
wego przyporządkowania pacjentów do grupy bada- gólnie w III fazie badań klinicznych. Jest oczywiste,
nej (randomizacji). że w nagłych wypadkach postępowanie to musi być
zweryfikowane.
Badania otwarte i próby ślepe. Przy badaniu otwar-
tym zarówno pacjent, jak i badający lekarz nie wie, Badania poszukiwawcze i weryfikujące. Badania
czy przeprowadzana jest próba z preparatem bada- leków można również podzielić ze względu na cel
nym, porównawczym czy z placebo. Próby otwarte badań. Badania poszukiwawcze (eksploracyjne) słu-
są przeprowadzane wówczas, gdy można określać je- żą stworzeniu określonej hipotezy, weryfikujące nato-
dynie obiektywne, niezależne (lub tylko w niewielkim miast – sprawdzeniu tej hipotezy. Pierwszy typ badań
stopniu) od psychiki pacjenta parametry (np. zmiany przeprowadzany jest w ramach II fazy badań kli-
przemiany materii). Jeśli działanie danego leku może nicznych, w czasie których poszukuje się dowodów
być oceniane tylko za pomocą kryteriów subiektyw- na działanie (aktywność) danego leku, natomiast dru-
nych lub takich, które podlegają silnym wpływom gi wiąże się z III fazą badań klinicznych, w czasie
czynników psychicznych, należy bezwzględnie prze- których określa się jego skuteczność i bezpieczeń-
prowadzić próbę ślepą. stwo stosowania. Oba typy tych badań różnią się
Przy pojedynczej próbie ślepej tylko lekarz, podstawowymi wymaganiami w zakresie planowa-
a nie pacjent, wie, czy podawany jest lek badany, nia i weryfikacji wyników. A zatem przy badaniach
czy preparat porównawczy. W takiej sytuacji nie moż- weryfikujących konieczne jest dokładne planowanie
na wykluczyć możliwości nieświadomego wpływu biometryczne z określeniem liczebności grupy bada-
lekarza na końcowy wynik. nej i kontrolnej, a także zapewnieniem określonego
W podwójnie ślepej próbie ani pacjent, ani lekarz randomizowanego doboru pacjentów do poszczegól-
nie wie, jaka substancja została zastosowana u pacjen- nych grup badawczych. Określenie niezbędnej liczby
ta. Podwójnie ślepa próba – szczególnie gdy zostanie pacjentów w grupie następuje na podstawie oceny
zastosowana technika próby krzyżowej – jest meto- klinicznie znaczącej różnicy między dwoma porów-
dą, która przy prawidłowym przeprowadzeniu próby nywanymi schematami leczenia, możliwym do zaak-
i właściwej ocenie statystycznej wyników umożliwia ceptowania prawdopodobieństwem błędu, uzyskanej
uzyskanie pewnych i obiektywnych danych dotyczą- w fazie III ocenie skuteczności, a także rozrzutu pa-
cych działania leku i jego efektów niepożądanych rametrów docelowych.
MUTSCHLER-2009.indd 134 2010-01-07 22:11:21
Poszukiwanie i badanie nowych leków 135
10.5. Pseudoinnowacje,
innowacje przełomowe i stopniowe
Nie każdy wprowadzony do terapii nowy środek razy. Inne stopniowe innowacje zostały osiągnięte dzięki
leczniczy oznacza przełom lub postęp w porównaniu metodom galenicznym (farmaceutyczno-technologicznym),
szczególnie poprzez wprowadzenie preparatów o przedłu-
z dostępnymi lekami. W tym przypadku mówi się
Poszukiwanie i badanie
żonym działaniu (retard).
o pseudoinnowacji. Należą tutaj przezskórne systemy terapeutyczne, które
W przeciwieństwie do tego zasadnicza poprawa te- w przypadku substancji o dużym efekcie I przejścia lub
rapii danymi środkami leczniczymi jest określana jako krótkim okresie półtrwania umożliwiają przez wystarcza-
nowych leków
tzw. przełomowa (skokowa) innowacja, a stopniowa jąco długi czas utrzymanie stosunkowo stałego i powtarzal-
nego stężenia we krwi. Jako przykłady można podać plastry
(krokowa) innowacja oznacza stopniowy postęp. z fentanylem lub estradiolem.
Długi czas działania umożliwiają także implanty. Obec-
Największym sukcesem w badaniach środków lecz- nie jest dostępny środek antykoncepcyjny zawierający ges-
niczych jest odkrycie leku w terapii choroby dotąd tagen o trzyletnim, pewnym czasie działania.
nieuleczalnej lub leku o całkowicie innym punkcie
uchwytu działania, co prowadzi do wyraźnej popra- Inne innowacyjne możliwości terapeutyczne to trwa- A 10
wy przewidywanej długości życia lub do uniknięcia łe połączenie środka leczniczego z produktem me-
działań niepożądanych. dycznym. (Przez pojęcie produktu medycznego rozu-
Przykładami takich nowych przełomowych in- mie się produkty trwałe, np. narzędzia, nici, cement
nowacji są antagoniści receptorów NMDA, sartany, do kości, a także maści, które nie wykazują żadnych
triptany, statyny, inhibitory pompy protonowej, inhi- działań farmakologicznych, lecz wyłącznie oddziału-
bitory kinazy tyrozynowej, czy środki przeciwwiru- ją fizycznie).
sowe w terapii AIDS.
Tak więc już od dłuższego czasu wykorzystywany jest za-
W ten sposób zydowudyna spowolniła po raz pierwszy wierający gentamycynę cement do kości, służący do moco-
progresję choroby prowadzącej w większości przypadków wania implantów. Korzystne byłoby jednak w tym przypad-
do szybkiej śmierci. Wirusy jednakże na drodze mutacji ku precyzyjne kierowanie uwalnianiem, tak aby osiągnąć
rozwijają oporność. Skuteczność działania leku można początkowo wysoki poziom leku do szybkiej inaktywacji
przedłużyć poprzez stosowanie preparatów złożonych bakterii. W przypadku stosowania cewników dożylnych
z wielu związków działających przeciw HIV o różnych me- i do pęcherza można zredukować częstość występowania
chanizmach działania. W preparatach złożonych stosuje się komplikacji związanych z infekcjami poprzez pokrycie
dodatkowo środki hamujące proteazę HIV, której działanie cewnika antybiotykiem (np. makrolidem oraz rifampicyną)
jest szczególnie ważne przy postępującej chorobie, co sta- lub środkiem dezynfekującym.
nowi także przełomową innowację. Poprzez pokrycie stentów do naczyń wieńcowych siroli-
musem lub paklitakselem udaje się zmniejszyć ryzyko reo-
Stopniowe innowacje udało się uzyskać w licznych gru- kluzji; w ten sposób w ostatnim czasie zmniejszono ryzyko
pach leków. Postępem terapeutycznym jest wprowadzenie komplikacji w przypadku stentów pokrytych w porównaniu
np. atypowych neuroleptyków, antagonistów receptorów z niepokrytymi.
β1-adrenergicznych, wolno działających antagonistów ka- Oczekuje się szybszego i lepszego umocowania implan-
nałów wapniowych, zasadowych difosforanów, penicylin tów w ortopedii i stomatologii poprzez zastosowanie specy-
o szerokim zakresie działania, czy nowych inhibitorów gy- ficznych czynników wzrostu.
MUTSCHLER-2009.indd 135 2010-01-07 22:11:21
136 Poszukiwanie i badanie nowych leków
10.6. Evidence-based medicine
Termin evidence-based medicine oznacza postępo- odgrywa przy tym kliniczne doświadczenie lekarza
wanie medyczne, w którym podejmuje się decyzje prowadzącego.
diagnostyczne i/lub terapeutyczne na podstawie sy- Zasady te można realizować za pomocą następują-
stematycznie gromadzonych i poddawanych ocenie cego algorytmu postępowania:
wyników badań naukowych. Główną ideą takiego
■ zdefiniowanie problemu (problem dotyczący pa-
sposobu postępowania jest osiągnięcie najlepszego
cjenta, konieczne lub możliwe do zastosowania
wyniku dla leczonego pacjenta i w miarę możliwości
środki łącznie z alternatywnymi, zdefiniowanie po-
oszczędzanie środków.
żądanego wyniku),
W odniesieniu do farmakoterapii oznacza to wybie-
ranie zarówno leku, jak i schematu terapeutycznego ■ przegląd piśmiennictwa w celu znalezienia najlep-
dla danego pacjenta w sposób sumienny i racjonalny, szej dokumentacji (zob. tab. A 10-1),
tzn. na podstawie najlepszych aktualnie dostępnych
■ ocena ich ważności (bliska prawdy) i znaczenia
danych naukowych pochodzących z badań podstawo-
(zastosowanie praktyczne),
wych i klinicznych. Przy tym widoczne są wyraźne
różnice w wartości np. wyników doświadczalnych ■ integracja (tzn. dostosowanie z uwzględnieniem
(doświadczenia in vitro lub na zwierzętach) i badań indywidualnych uwarunkowań i życzeń pacjenta),
klinicznych, w tym szczególnie badań potwierdzają-
■ ewaluacja (krytyczna ocena uzyskanego wyniku).
cych. Szczególną wartość (poziom oceny I) stanowią
sporządzone przez ekspertów i opublikowane w uzna- Obecnie nie można jeszcze rozstrzygnąć, czy za po-
nym czasopiśmie prace przeglądowe na temat danego mocą medycyny naukowo udokumentowanej będzie
środka leczniczego (zob. tab. A 10-1). można, oprócz lepszej terapii, osiągnąć również
Decyzje lekarzy nie powinny być jednak podej- oczekiwane obniżenie kosztów leczenia. Wydaje
mowane w sposób schematyczny wyłącznie na pod- się, że nie jest całkowicie wykluczony nawet dalszy
stawie tej pochodzącej z zewnątrz wiedzy. Znacznie wzrost kosztów ponoszonych przez służbę zdrowia
ważniejszą kwestią jest uwzględnienie indywidual- wskutek optymalizacji terapii i związanego z tym
nych uwarunkowań każdego pacjenta. Istotną rolę wzrostu oczekiwanej długości życia.
Tabela A 10-1. Ocena dokumentacji
I Przynajmniej jedna systematyczna praca przeglądowa na podstawie metodycznie pełnowartościowych badań klinicznych
II Przynajmniej jedna dostatecznie duża, metodycznie pełnowartościowa randomizowana próba kliniczna
III Metodycznie pełnowartościowe badania bez randomizacji
IV Więcej niż jedno metodycznie pełnowartościowe badanie niedoświadczalne
V Opinie uznanych autorytetów, komisji ekspertów, badania opisowe
(I = najbardziej wartościowa dokumentacja, V = najmniej wartościowa dokumentacja)
MUTSCHLER-2009.indd 136 2010-01-07 22:11:22
Poszukiwanie i badanie nowych leków 137
10.7. Suplement I: Leki roślinne (fitofarmaceutyki)
Przez fitoterapię rozumie się stosowanie roślin lub za ich działanie w wielu przypadkach nie są w ogóle
uzyskiwanych z nich preparatów (wyciągów, mace- lub są tylko częściowo poznane. Ponadto skład su-
racji, soków, herbatek, nalewek) do leczenia danych rowców roślinnych zależy od wielu różnych czyn-
stanów chorobowych. Fitofarmaceutykami są zgodnie ników i zmienia się mniej lub bardziej w zależności
z tym leki, które zawierają produkty roślinne tj. kom- od miejsca uprawy, warunków klimatycznych i in-
Poszukiwanie i badanie
pleksowe mieszanki substancji pochodzenia roślin- nych zmiennych czynników środowiska zewnętrz-
nego, mające w całości działać jako środek aktywny. nego. Co prawda, dzięki kontrolowaniu upraw i spe-
Według powyższej definicji do fitofarmaceutyków cjalnie wystandaryzowanym procedurom (uzyskując
nowych leków
nie zalicza się substancji wyizolowanych z roślin. tzw. specjalne wyciągi) można część tych czynników
wyeliminować, ale trudno jest uzyskać (jeśli w ogóle
Początek farmakoterapii wiąże się ze środkami leczniczymi jest to możliwe) identyczną jakość surowców. Wiele
uzyskiwanymi z roślin. Doświadczenia zdobywane przez po- preparatów roślinnych, zawierających tę samą rośli-
kolenia były zapisywane w zielnikach, a potem również w ofi-
cjalnych podręcznikach farmakoterapii. Dopiero w związku nę jako produkt wyjściowy, jest wytwarzanych przez
z opracowaniem i wprowadzeniem do praktyki laborato- firmy różnymi metodami, co dodatkowo utrudnia
ryjnej stale ulepszanych metod analityczno-chemicznych, ich ocenę. A 10
począwszy od pierwszych lat XIX w., podjęto próbę izola- W odniesieniu do wielu leków roślinnych nie
cji z roślin leczniczych związków w czystej postaci. Wiele ustalono jeszcze kryteriów skuteczności działania.
obecnie stosowanych leków zawiera takie substancje wyizo-
lowane z roślin lub odpowiadające im połączenia chemiczne. Ponieważ przepisom dotyczącym dopuszczania fito-
Takimi lekami są np. atropina uzyskana z pokrzyku, chinina farmaceutyków do obrotu przyznano, ze względów
z kory drzewa chinowego, digoksyna z naparstnicy, ergota- politycznych, szczególne warunki, nie wiadomo,
mina ze sporyszu, morfina z maku lekarskiego, penicylina ile ze znajdujących się na rynku fitoterapeutyków
z pleśni, rezerpina z Rauwolfia serpentina, salicylina (struk- jest rzeczywiście aktywnych. Bez uzyskania ofi-
tura podstawowa dla kwasu salicylowego) z kory wierzby,
teofilina z liści herbaty. W następnych latach wyizolowano, cjalnej zgody, a jedynie po zarejestrowaniu roślinne
a następnie zbadano farmakologicznie wiele nowych sub- środki lecznicze mogą być wprowadzone na rynek,
stancji z roślin. Istotną część leków z nowo wprowadzonych jednak bez żadnych wskazań (zob. rozdz. 10.8).
do lecznictwa uzyskano właśnie tą drogą. Natomiast w przypadku określonego działania i bez-
pieczeństwa stosowania leki roślinne, podobnie jak
Mimo że w wyniku wymienionych metod i technik środki pochodzenia chemicznego o danej strukturze
zastąpiono leki roślinne czystymi związkami ak- chemicznej, są dozwolone do leczenia określonych
tywnymi, preparaty roślinne nadal należą do często chorób.
stosowanych leków i to zarówno z przepisu lekarza, Należy jednakże ocenić pozytywnie, że w ostat-
jak i w ramach samoleczenia. Polska zajmuje dru- nich latach zwiększono wymagania, aby leki roślinne
gie po Niemczech (6,6% wszystkich leków) miejsce badać klinicznie według ogólnie uznanych kryteriów.
w Europie pod względem spożycia leków pochodze- Wiele leków roślinnych zostanie poddanych bada-
nia roślinnego. Wielu pacjentów przedkłada fitofarma- niom klinicznym metodą podwójnie ślepej próby.
ceutyki jako leki naturalne nad jakoby niebezpieczne Dotyczy to takich preparatów, jak wyciągi z kozłka
leki chemiczne. Nie bierze się przy tym pod uwagę, lekarskiego, miłorzębu dwuklapowego (Ginkgo bi-
że jeśli leki roślinne są farmakologicznie aktywne, loba), dziurawca, kasztanowca i głogu. Ich działanie
wynika to z działania zawartych w nich substan- jest opisane w odpowiednich rozdziałach. W związku
cji chemicznych, więc preparaty roślinne nie mogą z tym należy jeszcze raz podkreślić, że z wyżej poda-
być z góry uznawane za nieszkodliwe (bezpieczne). nych powodów wyniki badań dotyczą tylko określo-
Na przykład glikozydy roślinne lub alkaloidy należą nych preparatów, a nie wyciągów z danej rośliny.
do najsilniejszych znanych trucizn. Należy również Można się spodziewać, że w celu zwiększenia
pamiętać, że w wielu gatunkach roślin znajdują się bezpieczeństwa leków roślinnych będą przeprowa-
bardzo silne alergeny. dzane odpowiednie badania preparatów roślinnych
Innym istotnym problemem związanym z fitofar- i że zostaną ustalone jednolite kryteria ich oceny ja-
maceutykami jest to, że składniki odpowiedzialne kościowej.
MUTSCHLER-2009.indd 137 2010-01-07 22:11:22
138 Poszukiwanie i badanie nowych leków
10.8. Suplement II: Homeopatia
Choroba i leczenie są to procesy niematerialne 10.8.2. Potęgowanie
(Samuel Hahnemann)
Przy przygotowywaniu preparatów homeopatycz-
Przykładem tzw. szczególnych metod terapeutycz-
nych substancje wyjściowe są rozcieńczane przez
nych, do których zalicza się m.in. medycyna antro-
dodawanie rozcieńczonego etanolu (przy równo-
pozoficzna, jest homeopatia stworzona przez niemie-
czesnym wstrząsaniu) lub przez intensywne rozcie-
ckiego lekarza Samuela Hahnemanna (1755–1843).
ranie z laktozą, najczęściej w proporcji l:10 (dzie-
Szczególne metody terapeutyczne charakteryzują się
sięciokrotne potęgowanie, D) lub l:100 (stukrotne
w tym przypadku tym, że z jednej strony nie odpo-
potęgowanie, C). Zabiegi te stanowią według pojęć
wiadają współczesnym ogólnie przyjętym wyma-
przyrodniczych rozcieńczanie, natomiast według
ganiom naukowym, a z drugiej strony zastosowano
poglądów homeopatycznych potęgowanie. Oznacza
w stosunku do nich specjalne uregulowania prawne.
to, że przy potęgowaniu (rozcieńczaniu) siła dzia-
Leki mogą być rejestrowane bez uzyskania zgody.
łania leku homeopatycznego nie zmniejsza się,
Oznacza to, że nie podlegają one ogólnie przyjętym
ale może się zwiększać. Stopień potęgowania okre-
międzynarodowym i krajowym wytycznym rejestra-
śla się przez podanie liczby kolejnych etapów pro-
cyjnym i mogą być bez odpowiednich badań przyjęte
cedury potęgowania. Na przykład D30 powszech-
przez Ministerstwo Zdrowia, a następnie wprowa-
nie stosowane potęgowanie, oznacza rozcieńczenie
dzone do obrotu. Nie powinny być jednak łączone
1:1000 000 000 000 000 000 000 000 000 000 (1:1030).
z określonymi wskazaniami.
Krytycy stwierdzają, że przy takim potęgowa-
niu nie można się spodziewać, aby zachowała się
jeszcze wystarczająca ilość substancji aktywnej.
10.8.1. Zakres działania leku Przedstawiciele homeopatii uważają, że nie chodzi
i zasada podobieństwa tu o ilość leku, lecz o jego jakość. Niezależnie od tego
istnieją określone poszlaki wskazujące, że przy po-
Do podstawowych zasad homeopatii należy zakres tęgowaniu (rozcieńczaniu) zwiększa się dostępność
działania leku, tzn. zespół objawów, które występu- substancji aktywnej w stosunku do środowiska (roz-
ją u zdrowego (pozornie) człowieka po podaniu leku puszczalnika), co może wyjaśnić efekt potęgowania.
homeopatycznego, co stanowi podstawę do zastoso- Powyższy pogląd jest jednoznacznie sprzecz-
wania danego leku u chorego. Lek homeopatyczny ny z ogólnie akceptowanymi i uznanymi prawdami
najbardziej nadaje się (według poglądów homeopa- fizykochemicznymi. Na przykład struktura wody
tii) do leczenia tego chorego, którego obraz choroby i alkoholu nie jest na tyle stabilna, aby można było
jest najbardziej zbliżony do zakresu działania leku założyć eksponowanie cząsteczek substancji czyn-
– Similia similibus curentur – podobne leczy się po- nej, co by z kolei mogło tłumaczyć zjawisko potę-
dobnym. gowania – odwrotnie oba te związki ulegają stałemu
W odniesieniu do wielu leków homeopatycznych i szybkiemu przebudowywaniu. Poza tym trudno
nie można jednak określić charakterystycznego (ty- sobie wyobrazić, że w tym procesie następuje po-
powego) zakresu ich działania. W wielu bardzo sta- tęgowanie wybiórczo jednej substancji, natomiast
rannie przeprowadzonych badaniach kontrolowanych liczne inne związki zawsze obecne w wodzie desty-
wykazano, że w stosunku do licznych leków stoso- lowanej, mieszaninie wody i alkoholu lub czystej
wanych jako leki homeopatyczne nie można było laktozie nie ulegają wspólnemu potęgowaniu. Jeśli
wykazać różnic między grupą otrzymującą badany np. przygotujemy sól kuchenną (Natrium chloratum),
lek a grupą dostającą placebo. Wielokrotnie cytowa- ważny lek homeopatyczny w formie D20, to dlacze-
ne są wykonane przez samego Hahnemanna próby go nie następuje jednocześnie podobne potęgowanie
z korą drzewa chinowego. Stwierdził on mianowicie, chlorku potasu i magnezu, które zawsze znajdują się
że po zażyciu tej kory wystąpiły u niego dreszcze nawet w najczystszej soli kuchennej? Niezrozumiałe
i gorączka. Należy przyjąć, że prawdopodobnie była jest także, dlaczego w procesie potęgowania wzmo-
to niewłaściwa obserwacja. Zasada podobieństwa żeniu ulegają tylko pożądane efekty lecznicze,
nie została jak dotychczas skrupulatnie i naukowo a nie działania niepożądane, np. działanie rakotwór-
sprawdzona. cze związków arsenu.
MUTSCHLER-2009.indd 138 2010-01-07 22:11:22
Poszukiwanie i badanie nowych leków 139
10.8.3. Dowody działania W związku z powyższymi danymi należy dodać, że przy
lekach allopatycznych wymaga się oprócz właściwego ba-
dania działania również oceny zależności efekt-dawka oraz
Homeopatia jest uznawana przez zwolenników za przy zastosowaniu różnych form farmaceutycznych badań
skuteczną metodę leczenia i w związku z tym jest sto- porównawczych dostępności biologicznej i bioekwiwalen-
tności. W odniesieniu do leków homeopatycznych takich
sowana u wielu pacjentów. Z tego powodu w inte-
badań ani nie się planuje, ani się ich (systematycznie)
resie pacjentów leży uzyskanie, podobnie jak w od- nie przeprowadza.
niesieniu do innych tzw. leków allopatycznych (allos
Poszukiwanie i badanie
to po grecku inny), wiarygodnych danych dotyczą-
cych ich działania farmakologicznego i farmakotera- 10.8.4. Kiedy leki homeopatyczne,
peutycznego. Dane te należałoby uzyskać za pomocą również według poglądów
nowych leków
kontrolowanych i powtarzalnych badań, tzn. według lekarzy homeopatów,
jednoznacznych kryteriów naukowych. Jednakże wy-
nie są wskazane?
magania te mimo upływu ok. 200 lat nie zostały nie-
stety spełnione.
Według poglądów lekarzy homeopatów nie powinno
W związku z tym homeopaci często argumentują, że za- się stosować leków homeopatycznch, kiedy:
sadnicza różnica miedzy homeopatią a medycyną uniwer-
sytecką polega na różnicach w zakresie problematyki te-
■ jest możliwe leczenie przyczynowe (np. przy zaka- A 10
oretyczno-poznawczej. Ostatnio w dyskusjach na temat żeniach bakteryjnych),
homeopatii podkreśla się, że chodzi tu o (niejednoznacznie ■ można zastosować leczenie substytucyjne (np. u oso-
rozstrzygający) paradygmat. Jeśli nawet można podobnie
interpretować zagadnienie potęgowania, nie zmienia to fak- by chorej na cukrzycę – insulinę, a u chorych z nie-
tu, że na homeopatach ciąży obowiązek dostarczenia do- dokrwistością złośliwą - witaminę B12),
wodów na aktywność leków homeopatycznych. Działanie
lub aktywność można przecież wykazać niezależnie od pa- ■ występuje ostra choroba zagrażająca życiu (np. za-
radygmatu. Każdy, kto zajmował się poważnie homeopa- wał serca) lub
tią od strony medycyny uniwersyteckiej, musi oczekiwać,
że leki homeopatyczne zostaną przebadane według tych sa- ■ po zastosowaniu leczenia wyrównawczego (np. po
mych zasad co inne leki stosowane w medycynie uniwersy- próbie normalizacji homeostazy) nie można ocze-
teckiej. Oczywiście, przy badaniu działania leków homeo- kiwać żadnej dalszej poprawy (np. u wszystkich
patycznych wybór danego leku dla określonego pacjenta chorych z nowotworami złośliwymi).
następuje według kryteriów homeopatii.
Oznacza to, że homeopatia poniekąd automatycznie
i nieodwołalnie traci znaczenie wówczas, gdy medy-
Należy podkreślić, że trzeba stanowczo domagać się
cyna uniwersytecka dysponuje przyczynowo lub sub-
badania leków homeopatycznych z uwzględnieniem
stytucyjnie działającymi lekami.
zasad randomizacji i podwójnie ślepej próby.
Przegląd piśmiennictwa homeopatycznego wska-
zuje, że cytowane badania nie odpowiadają ogólnie
przyjętym kryteriom. Wiele badań zostało wykona- 10.8.5. Zastosowanie leków
nych niefachowo lub wykorzystano w nich wyniki homeopatycznych
negatywne albo też wyniki te okazały się niepowta-
rzalne. Wykonano np. badania z zastosowaniem po- Wybrane, oryginalnie cytowane z publikacji homeo-
dwójnie ślepej próby na pacjentach z nadciśnieniem patycznych przykłady zastosowania homeopatycz-
samoistnym, którzy otrzymywali lek przeciwnadciś- nych środków leczniczych pozostawia się do oceny
nieniowy (allopatyczny) lub – odpowiednio do obra- czytelnika.
zu chorobowego pacjenta – Aconitum D6, Arnica D4,
Barium carbonicum D6, Plumbum D6, Nux vomica Przeziębienie. Cebulę (Allium cepa) stosuje się przy
D4, Lachesis D12 bądź Cuprum D6. Leki homeopa- następujących objawach: silna, powodująca owrzo-
tyczne okazały się w tych badaniach całkowicie nie- dzenia wydzielina z nosa, ale łagodna wydzielina
skuteczne. W jednej próbie z zastosowaniem placebo z oczu, uczucie jakby krtań ulegała roztrzaskaniu lub
u pacjentów z reumatoidalnym zapaleniem stawów rozdarciu.
po 6 miesiącach leczenia homeopatycznego nie uzy-
skano żadnej statystycznie znamiennej różnicy mię- Oparzenia. Przy oparzeniach środkiem z wyboru aż
dzy grupą otrzymującą lek a grupą placebo. do momentu przyjścia lekarza jest kantarydyna. O ile
MUTSCHLER-2009.indd 139 2010-01-07 22:11:22
140 Poszukiwanie i badanie nowych leków
to możliwe, dzieciom należy podawać Cantharis D6 10.8.6. Działania niepożądane
(2 łyżeczki) co pół godziny.
Homeopatię uważa się za pozbawioną działań niepo-
Trudności w uczeniu się. Przy trudnościach w ucze-
żądanych. Jest to o tyle zdumiewające, że już Samuel
niu się stosuje się przede wszystkim fosfor, chlorek
Hahnemann w § 156 swego głównego dzieła
sodu i siarkę.
Organon napisał: „Rzadko zdarza się, aby lek ho-
meopatyczny nie wywoływał niepożądanych za-
Zaparcia. Opium D30 należy stosować przy następu-
burzeń”. Niskie potęgi leków homeopatycznych
jących objawach: brak parcia, grudki kałowe są okrą-
(Dl-D4) mogą wywoływać znaczne działania niepo-
głe, czarne, twarde i suche. Wydalony kał nie rozmię-
żądane lub toksyczne, jeśli zawierają np. silnie dzia-
ka. Brzuch jest wzdęty i bolesny.
łające alkaloidy (akonitynę, atropinę). Mogą rów-
nież działać rakotwórczo (np. arsen, wilczomlecz,
Pozostałe. Calcium phosphoricum D12 stosuje się
wawrzynek). Od homeopatii należałoby oczekiwać
zwłaszcza u pacjentów spożywających nadmierne
nie tylko dowodów działania, ale również dowodów
ilości zup. W przeciwieństwie do tego calcium car-
bezpieczeństwa stosowania.
bonicum stosuje się u niezręcznych, ociężałych, lecz
także u niesprawiających kłopotów wychowawczych
i pogodnych dzieci.
MUTSCHLER-2009.indd 140 2010-01-07 22:11:22
FARMAKOLOGIA
SZCZEGÓŁOWA
B
MUTSCHLER-2009.indd 141 2010-01-07 22:11:22
MUTSCHLER-2009.indd 142 2010-01-07 22:11:22
Układ nerwowy 143
1. Układ nerwowy
Układ nerwowy
1.1. Podstawy anatomiczne i fizjologiczne
Im bardziej zaawansowany rozwojowo jest żywy or- Inny sposób podziału układu nerwowego polega
ganizm, tym bardziej potrzebuje on – oprócz organów na wyróżnieniu: B1
zaopatrzenia (w pokarm) i wydalania – nadrzędnych,
■ autonomicznego (wegetatywnego),
sprawnych układów informacyjnych, koordynowania
i sterowania. Zapewniają to – w przypadku (wyżej ■ somatycznego układu nerwowego (kontrolowa-
rozwiniętego) organizmu zwierząt i człowieka – ukła- nego dowolnie).
dy nerwowy oraz hormonalny. U ludzi system nerwo-
wy, a szczególnie mózg, osiągnął stopień sprawności Oba układy składają się z części ośrodkowej i obwo-
znacznie przekraczający możliwości wszystkich in- dowej.
nych istot żywych.
Układ nerwowy służy:
1.1.1. Tkanka nerwowa
■ odbieraniu bodźców pochodzących ze środowiska
lub powstających wewnątrz ciała, Tkanka nerwowa – powstająca w rozwoju z ektoder-
my – jest zbudowana z komórek nerwowych (neu-
■ zamianie tych bodźców na impulsy nerwowe, ich
ronów) i komórek glejowych – ektodermalnej tkanki
dalszemu przekazywaniu i przetwarzaniu,
pełniącej funkcje podporowe i pomocnicze.
■ koordynowaniu i sterowaniu czynnościami ciała
dzięki impulsom przekazywanym z centrum na ob-
wód. 1.1.1.1. Komórka nerwowa
Ponadto w układzie nerwowym zachodzą wszelkie Komórka nerwowa (neuron) jest najmniejszą funk-
zjawiska duchowe i psychiczne. cjonalną jednostką układu nerwowego, która jest od-
powiedzialna za przyjmowanie informacji, jej prze-
Z anatomicznego i funkcjonalnego punktu widzenia twarzanie i przekazywanie, tzn. za specyficzne funk-
wyróżnia się: cje układu nerwowego. Poza ciałem komórki (zwa-
nym też somą lub perikarionem) posiada ona zwykle
■ ośrodkowy układ nerwowy (OUN) złożony z móz-
liczne wypustki. Wypustka służąca do przekazywania
gu i rdzenia kręgowego, które są przede wszystkim
informacji pod postacią potencjału czynnościowego
odpowiedzialne za sterowanie funkcjami ciała,
do innej komórki nerwowej nazywana jest neurytem
■ obwodowy układ (system) nerwowy, obejmują- lub aksonem. Pozostałe rozgałęziające się drzewia-
cy drogi przewodzące z obwodu do ośrodkowego sto wypustki, które odbierają impulsy od wcześniej
układu nerwowego (aferentne, wstępujące, dośrod- położonego ogniwa całego łańcucha połączeń neuro-
kowe, szlaki sensoryczne), włącznie z komórkami nalnych, nazywane są dendrytami (grec. dendron =
nerwowymi umiejscowionymi na obwodzie, oraz drzewo).
drogi z ośrodkowego układu nerwowego na obwód Typowa budowa neuronu przedstawiona jest sche-
(eferentne, zstępujące, odśrodkowe, motoryczne matycznie na ryc. B 1.1-1.
szlaki). (Drogi eferentne prowadzące do gruczo-
łów określane są jako sekrecyjne, wydzielnicze). Neuryt (akson) i włókno nerwowe. Neuryt składa się z ak-
soplazmy i otaczającej ją błony plazmatycznej (aksolema,
Do obwodowego układu nerwowego należy rów- błona aksoplazmatyczna). Długość aksonu może wahać się
nież autonomiczny układ trzewny, tj. unerwiający od kilku milimetrów do ponad jednego metra. Na końcu
przewód pokarmowy. akson jest rozgałęziony i tworzy zakończenia aksonalne.
MUTSCHLER-2009.indd 143 2010-01-07 22:11:22
144 Układ nerwowy
A B szczelina
mitochondrium synaptyczna
ciałka Nissla
synaptosomy
B synapsa synapsa
jądro komórkowe aparat Golgiego
pory jądrowe
jąderko
wzgórek dendryt
aksonu
neuryt jądro komórkowe plazmolemma
jąderko
akson siateczka
endoplazmatyczna
osłonka rdzenna C z rybosomami neuryt
jądro komórki
glejowej przewężenie
Ranviera akson
akson
przewężenie Ranviera
osłonka rdzenna
Ryc. B 1.1-1. Schemat neuronu (według Kretschmanna). A – Ciało neuronu (soma) z neurytem; B – obraz ciała neuronu w mikroskopii
elektronowej; C – (powiększony) fragment ryc. A.
Nazwa włókno nerwowe obejmuje akson z otoczką Nerwy. Większość włókien nerwowych przebiega w gru-
glejową (osłonką aksonalną). W obrębie OUN jest ona pach, które w ośrodkowym układzie nerwowym nazywa się
zbudowana z wypustek oligodendrocytów (zob. poniżej), wiązkami (fasciculi), a na obwodzie nerwami. Niezależnie
natomiast w przypadku włókien nerwów obwodowych ak- od tego, czy włókno nerwowe jest rdzenne czy bezrdzenne,
son otoczony jest tzw. komórkami Schwanna. Odróżnia się otoczone jest tkanką łączną, tzw. śródnerwiem (endoneu-
włókna nerwowe ubogie i bogate w lipidy, które określa- rium). Grupy włókien nerwowych otoczone są onerwiem
ne są odpowiednio jako bezrdzenne (bezmielinowe) oraz (perineurium) w cienkie wiązki, a grupy takich wiązek
rdzenne (mielinowe). W przypadku cienkich włókien bez- otoczone są z kolei przez nanerwie (epineurium) – tworząc
rdzennych aksony są otoczone tylko komórkami Schwan- nerwy. Wymienione otoczki stanowią barierę dla dyfuzji
na. Natomiast grubsze, rdzenne włókna nerwowe posiadają większości jonów, a także dla substancji czynnych.
otoczkę składającą się z licznych blaszek zawierających
lipidy i białka, które spiralnie otaczają akson. Substancja
tworząca te blaszki (lamele) nazywana jest mieliną, a cała
otoczka zwana jest otoczką mielinową lub rdzenną. W przy-
1.1.1.2. Neuroglej
padku obwodowych rdzennych włókien nerwowych otocz-
ka glejowa posiada w regularnych odstępach przerwy, tzw. Komórki glejowe pełnią ważne funkcje w układzie
przewężenia Ranviera. Mają one znaczenie dla szybkości nerwowym. Są to funkcje osłonowe, podporowe,
przewodzenia impulsów (zob. poniżej).
izolujące i metaboliczne, a także kontrolne w obrę-
MUTSCHLER-2009.indd 144 2010-01-07 22:11:22
Układ nerwowy 145
bie przestrzeni pozakomórkowej. W przeciwieństwie zycznymi czy chemicznymi, tj. do pobudzenia wraz
do komórek nerwowych, które (prawie w ogóle) z przenoszeniem na odległość tego pobudzenia i w ten
nie mogą się dzielić, komórki glejowe zachowują sposób oddziaływania na funkcje narządów. Oprócz
zdolność podziału podczas całego życia. komórek nerwowych służą do tego komórki czucio-
Układ nerwowy
we (komórki zmysłowe, receptory) i mięśniowe.
Komórki glejowe ośrodkowego układu nerwowego.
W mózgu i rdzeniu kręgowym neurony zajmują jedynie
niecałe 50% objętości. Przestrzeń pomiędzy nimi wypeł-
niają komórki glejowe. Do komórek glejowych OUN nale- 1.1.2.1. Potencjał spoczynkowy
żą astrocyty, oligodendrocyty i komórki ependymalne.
i potencjał czynnościowy
Astrocyty (neuroglej gwiaździsty, zwany niekiedy ma-
kroglejem – przyp. tłum.) – największe z komórek glejo-
wych posiadają liczne wypustki, za pomocą których łączą
Powstanie i przekazywanie pobudzenia we włóknach B1
się zarówno z komórkami nerwowymi, jak i naczyniami nerwowych i mięśniowych opiera się na procesach
włosowatymi. Wypustki astrocytów wchodzące w kontakt jonowych, rozgrywających się w otaczającej komór-
z naczyniami włosowatymi tworzą rozszerzenia w kształcie ki błonie komórkowej (teoria jonowa przewodzenia
stopy, które pokrywają ok. 80% ich powierzchni. Astrocyty pobudzenia). W związku z różnicą dystrybucji jonów
pełnią przede wszystkim funkcję tkanki podporowej. Two-
rzą one bariery między sąsiadującymi neuronami i w razie
pomiędzy wnętrzem włókien nerwowych a przestrze-
uszkodzenia tkanki nerwowej w pewnym zakresie zastępują nią zewnątrzkomórkową powstaje na granicy obu
ją, tworząc blizny. Ponadto astrocyty uczestniczą w utrzy- ośrodków różnica potencjału (napięcie) – nazywana
mywaniu stałego środowiska jonowego w przestrzeni ze- potencjałem błonowym.
wnątrzkomórkowej. Zmiany potencjału błonowego komórek nerwo-
Ponadto astrocyty oddzielają neurony od innych struk-
tur, np. naczyń (funkcja izolująca), tworząc na naczyniach
wych i mięśniowych tworzą podstawy do przewodze-
zachodzące na siebie warstwy błon. nia pobudzenia i w efekcie – do przekazywania syg-
nału i np. wywołania skurczu.
Oligodendrocyty (glej skąpowypustkowy przyp. tłum.)
są małe, a ich wypustki mniej liczne i krótsze niż w przy-
padku astrocytów. Tworzą one, jak wspomniano wcześniej, Potencjał spoczynkowy. Wnętrze pobudliwej
osłonki glejowe aksonów w ośrodkowym układzie nerwo- komórki wykazuje w stosunku do przestrzeni ze-
wym. Jako tzw. komórki satelitarne – liczne z nich znajdują wnętrznej negatywny potencjał o wartości od –60
się bezpośrednio przy komórkach nerwowych. do –100 mV – określany jako potencjał spoczynkowy
Ependyma (glej wyściółkowy – przyp. tłum.) jest poje- (błonowy potencjał spoczynkowy).
dynczą warstwą komórek o kształtach od sześciennego Przyczyną ujemnego potencjału wewnątrz komór-
do pryzmatycznego, która tworzy wykładzinę wewnętrz-
nych przestrzeni mózgu i rdzenia kręgowego. Szczególną
ki w stosunku do otoczenia są różnice w stężeniach
formą komórek wyściółkowych (ependymalnych) są ko- jonów w obu przestrzeniach (zob. tab. B 1.1-1), któ-
mórki nabłonkowe splotu naczyniówkowego (plexus cho- re utrzymywane są dzięki aktywnemu transportowi,
riodei). a szczególnie dzięki Na+/K+-ATP-azie.
Komórki glejowe obwodowego układu nerwowego. W wyniku tych różnic stężenia jony K+ mają ten-
Do komórek glejowych obwodowego układu nerwowego dencję do dyfundowania do przestrzeni zewnątrzko-
należą komórki Schwanna i komórki płaszcza. Komórki mórkowej – wykorzystując przy tym otwarte nawet
Schwanna tworzą, jak już wspomniano, osłonkę obwodo- w fazie potencjału spoczynkowego kanały potasowe,
wych aksonów. Komórki płaszcza (płaszczowe) otaczają
komórki nerwowe rdzenia i zwojów wegetatywnych. w szczególności przez kanały potasowe typu Kir peł-
Migroglej. Mianem mikrogleju (komórki mikroglejowe,
mezoglej, glej Hortegi) określa się aktywne fagocytarnie
komórki, które pochodzą z mezenchymy i wwędrowują za
pomocą ruchów ameboidalnych do ośrodkowego układu Tabela B 1.1-1. Wewnątrz- i zewnątrzkomórkowe stężenia jo-
nerwowego. Są one makrofagami układu nerwowego. nów. Wartości dotyczą komórki nerwowej i z niewielkimi odchy-
leniami także komórki mięśniowej (wg Traczyka)
1.1.2. Pobudzenie komórek Jon Stężenie zewnątrz- Stężenie wewnątrz-
nerwowych, przewodzenie komórkowe (mmol/l) komórkowe (mmol/l)
i przekazywanie pobudzenia Na+ 145 14,5
+
K 4 140
Istotną cechą organizmów żywych jest ich pobudli-
–
wość, tzn. zdolność określonych komórek do ujaw- Cl 115 4-20
niania swoistej reakcji na pobudzenie bodźcami fi-
MUTSCHLER-2009.indd 145 2010-01-07 22:11:23
146 Układ nerwowy
dem zewnętrznej strony błony komórkowej. Później
dochodzi po odbudowania dawnego (wyjściowego)
(mV) depolaryzacja potencjału spoczynkowego (repolaryzacja). Tego ro-
+40 (faza narastania) dzaju procesy de- oraz repolaryzacji, które jako krót-
+20
kotrwałe zmiany potencjału można rejestrować za po-
nadstrzał mocą elektrod, określa się jako potencjał czynnościo-
wy lub impuls nerwowy (zob. ryc. B 1.1-2). Polega on
repolaryzacja
na gwałtownych zmianach właściwości błony wywo-
-20 łanych działaniem bodźca: napięciowozależne kanały
sodowe zostają na krótko (< 1 ms) otwarte, w wyniku
-40 próg czego sód napływa pasywnie do aksoplazmy zgodnie
-60 następcza hiperpolaryzacja z gradientem stężeń. Skutkiem jest odwrócenie po-
laryzacji (tzw. nadstrzał lub overshoot; zob. ryc. B
-80 1.1-2). W wyniku bezpośrednio potem następujące-
faza reakcji go szybkiego zmniejszenia się przepuszczalności dla
bezwzględnej względnej
-100 jonów sodu oraz zwiększenia się przepuszczalności
0 1 2 3 4 5 6 dla jonów potasu – na skutek przejściowego otwarcia
(ms)
napięciowozależnych, zamkniętych w trakcie poten-
Ryc. B 1.1-2. Przebieg potencjału czynnościowego (wewnątrz- cjału spoczynkowego kanałów potasowych (Kv; v =
komórkowa rejestracja z pojedynczego włókna nerwowego). napięcie) – dochodzi do repolaryzacji. Kiedy tylko
potencjał błonowy osiągnie wartość ≥ –70 mV, do-
chodzi do ponownego zamknięcia Kv. Występująca
w przypadku niektórych komórek hiperpolaryzacja
niące funkcję prostownicze w kierunku do wnętrza
następcza, tj. ujawnienie się pod koniec potencjału
(ir = inward rectifier). Tego rodzaju wypływ potasu
czynnościowego potencjału błonowego mającego
na zewnętrz jest jednak hamowany istnieniem jo-
wartość bardziej ujemną niż wyjściowy potencjał
nów przeciwstawnych, w szczególności anionów
spoczynkowy, wiąże się z krótkotrwałym otwarciem
białczanowych, które nie mogą przedostawać się
kanałów potasowych aktywowanych jonami wap-
przez błonę komórkową. Wypływ na zewnętrz nie-
nia. Kiedy dojdzie do ich zamknięcia – co następuje
których dodatnio naładowanych ładunków prowa-
po znormalizowaniu się podwyższonego wewnątrz-
dzi w końcu do powstania elektrycznego potencjału
komórkowego stężenia wapnia, do czego dochodzi
błonowego zapobiegającego dalszej dyfuzji jonów
w czasie potencjału czynnościowego w wyniku ot-
K+ na zewnętrz. Z kolei jony sodowe właściwie
warcia napięciowozależnych kanałów wapniowych
w żaden sposób nie wpływają na potencjał spoczyn-
– potencjał błonowy powraca do swojej wartości
kowy, ponieważ niepobudzona błona komórkowa
wyjściowej.
jest praktycznie dla nich nieprzepuszczalna. Stąd też
potencjał spoczynkowy w zasadniczej mierze zwią-
Pompy jonowe. Prądy jonowe powstające podczas
zany jest z potencjałem dyfuzyjnym K+. W dalszej
potencjału czynnościowego są procesami transportu
kolejności wpływ na potencjał błonowy – skądinąd
biernego, które powstają w związku z istnieniem róż-
w mało zorganizowany sposób – ma chlorkowy po-
nicy ich stężeń między przestrzenią wewnątrz- i ze-
tencjał dyfuzyjny. (Chlorki obecne są w przestrzeni
wnątrzkomórkową. W trakcie pojedynczego poten-
wewnątrzkomórkowej w znacznie mniejszym stęże-
cjału czynnościowego tylko nieliczne jony sodowe
niu niż w zewnątrzkomórkowej).
wędrują do wnętrza, a jony potasowe na zewnątrz
neuronu, dlatego też wiele tysięcy potencjałów czyn-
Potencjał czynnościowy. Jeżeli pod wpływem dzia-
nościowych może być przewodzonych przez włókno
łania bodźca chemicznego lub fizykalnego dojdzie
nerwowe – zanim powstaną istotne zmiany stężeń tych
do zmiany błonowego potencjału spoczynkowego,
jonów. Jednak w celu zapewnienia stałej gotowości
tj. do przesunięcia jego wartości w kierunku bardziej
włókien nerwowych na pobudzenie (pobudliwość)
dodatnim (depolaryzacja), i zostanie przekroczona
– jony, które uległy przemieszczeniu do wewnątrz lub
określona wartość progowa (potencjał progowy),
na zewnątrz komórki, muszą być przetransportowane
– dochodzi do jego dalszego obniżenia, co przebiega
z powrotem. Ponieważ ten proces transportu jonów
już w błyskawiczny sposób – w trakcie bardzo krót-
zachodzi przeciw gradientowi ich stężeń, potrzebne
kiego czasu (< 0,1 ms). Przejściowo wnętrze komórki
są do tego aktywne systemy transportujące, tzw. pom-
nerwowej staje się nawet elektrycznie dodatnie wzglę-
py jonowe. System transportowy, który usuwa jony
MUTSCHLER-2009.indd 146 2010-01-07 22:11:23
Układ nerwowy 147
Na+ na zewnątrz, równocześnie transportuje jony K+ ■ nabłonek zmysłowy (rzęskowy) w uchu wewnętrz-
w przeciwnym kierunku. Jest to sodowo-potasowa- nym służący do odbioru fal dźwiękowych,
ATP-aza (pompa sodowo-potasowa), która uzysku-
■ receptory nacisku, bólu i temperatury skóry odbie-
je energię z rozszczepienia trifosforanu adenozyny
rające odpowiednie bodźce z zewnątrz,
Układ nerwowy
(ATP) i wymienia 3 jony Na+ na 2 K+. Tak więc
w wyniku jednego cyklu tej stale działającej pompy ■ baroreceptory w tętnicach rejestrujące ciśnienie
Na/K+ z komórki usunięty zostaje jeden ładunek do- krwi,
datni. Mówi się w tym przypadku o pompie elektro-
■ chemoreceptory kontrolujące stężenie gazów odde-
gennej.
chowych we krwi.
Faza refrakcji. Jeżeli włókno nerwowe będzie stymu-
lowane podczas trwania potencjału czynnościowego
Komórki czuciowe (sensoryczne) posiadają – po- B1
dobnie jak wszystkie komórki pobudliwe – potencjał
lub bezpośrednio po nim – nie dojdzie do pobudzenia.
spoczynkowy. Przy zadziałaniu odpowiedniego bodź-
Oczywiste jest, że włókno nerwowe nie jest zdolne
ca, tzn. w przypadku zmiany parametru chemicznego
w tym czasie do zareagowania na pobudzenie: przez
lub fizycznego, do którego odbioru dany receptor
krótki czas jest ono niewrażliwe na pobudzenie kolej-
jest wyspecjalizowany, dochodzi z reguły do obniże-
nym bodźcem. Stan ten, który trwa w przypadku ko-
nia potencjału błony, tzn. do depolaryzacji. Depola-
mórek nerwowych ok. 2 ms, jest określany jako faza
ryzacja trwa tak długo, jak długo działa dany bodziec,
bezwzględnej refrakcji (zob. ryc. B 1.1-2). Po nim
a jej wielkość jest zależna od siły bodźca (zob. ryc.
następuje dłuższy okres zmniejszonej pobudliwości,
B 1.1-3). Zmiany potencjału błonowego ograniczo-
tzw. względnej refrakcji. W tej fazie mogą wprawdzie
ne do receptywnej okolicy komórki sensorycznej,
powstawać potencjały czynnościowe, lecz do ich wy-
a więc nierozprzestrzeniające się na całą komórkę,
zwolenia konieczna jest silniejsza depolaryzacja niż
nazywa się potencjałem receptorowym (potencjałem
zazwyczaj.
generatorowym), a sama zamiana działającego bodź-
Stan braku lub obniżonej pobudliwości wynika
ca w potencjał receptorowy nazywa się transdukcją
z inaktywacji kanałów sodowych. Kanały sodowe,
bodźca (sygnału).
które w czasie fazy depolaryzacji ulegają krótkotrwa-
Poprzez potencjał receptorowy są wywoływane
łemu otwarciu, są ponownie błyskawicznie zamknię-
pobudzenia, które są przekazywane dalej wzdłuż
te i przez pewien czas nie mogą być otworzone. Pod-
aksonu; sam potencjał receptorowy ulega trans-
czas trwania fazy repolaryzacji kanały sodowe pozo-
formacji do rytmicznej serii pobudzeń (zob. ryc. B
stają wprawdzie nadal zamknięte, przechodzą jednak
1.1-3). Częstotliwość tych pobudzeń zależy od wiel-
w stan umożliwiający ich aktywację.
kości potencjału receptorowego. Czas trwania tej
Z fazy bezwzględnej refrakcji wynika maksymal-
serii pobudzeń zawiera informacje o czasie trwania
na częstotliwość, z jaką mogą być wywoływane po-
bodźca.
tencjały czynnościowe i przewodzone przez włókna
nerwowe. Na przykład w fazie refrakcji trwającej
2 ms maksymalna częstotliwość wynosi 500 s–1 (Hz).
W warunkach in vivo częstotliwość taka nigdy nie zo-
staje jednak osiągnięta.
bodziec
1.1.2.2. Wywoływanie pobudzenia
receptorów fizjologicznych potencjał
receptorowy
Bodźce działające na organizm od zewnątrz lub wy-
stępujące we wnętrzu ciała są rejestrowane przez (fi-
zjologiczne) receptory i następnie zamieniane na se-
rię pobudzeń. Służące do odbioru sygnałów (senso-
ry, czujniki) komórki zmysłowe (czuciowe) cechują potencjał
czynnościowy próg
się całkowicie odmienną budową i przeznaczone
są do specyficznego odbioru określonych rodzajów
bodźców. Jako przykłady można wymienić:
Ryc. B 1.1-3. Potencjały receptorowe i wyzwolone serie poten-
■ receptory siatkówki służące do odbioru energii cjałów czynnościowych w zależności od siły bodźca (według
świetlnej, Thewsa, Mutschlera, Vaupela).
MUTSCHLER-2009.indd 147 2010-01-07 22:11:24
148 Układ nerwowy
1.1.2.3. Przewodzenie pobudzenia dynczego potencjału czynnościowego, lecz jakościo-
przez nerwy wy i ilościowy kontekst przekazywanej informacji
i przekazywanie informacji musi być przenoszony w inny sposób. Rodzaj bodźca
(ból, światło, fale dźwiękowe, itd.) jest przekazywa-
ny w ten sposób, że pobudzeniu ulegają tylko pewne
Mechanizm przewodzenia pobudzenia. W wyni-
wrażliwe dla danego bodźca neurony, natomiast in-
ku różnicy potencjału pomiędzy pobudzonym i są-
tensywność bodźca – jak to już opisano – jest kodo-
siednim, jeszcze niepobudzonym miejscem wystę-
wana w częstotliwości impulsów.
pują lokalne zmiany potencjału (przepływ prądu
elektrotonicznego – przyp. tłum.), dzięki którym
pobudzenie rozprzestrzenia się z określoną prędkoś-
cią wzdłuż włókna nerwowego (zob. ryc. B 1.1-4). 1.1.2.4. Przenoszenie bodźca
W przypadku obwodowych neuronów mielinowych przez synapsy
przewodzenie pobudzenia ma charakter skokowy,
Przenoszenie (przekazywanie) bodźca z aksonu
tzn. od przewężenia do przewężenia, natomiast
do następnej komórki (neuronu, komórki mięśniowej
we wszystkich innych nerwach zachodzi w sposób
lub wydzielniczej) dokonuje się w synapsach. Sy-
ciągły. Sposób skokowy umożliwia dużą szybkość
napsy pełnią:
przewodzenia; wynosi ona do 120 m/s. W przeci-
wieństwie do nich w bezrdzennych (bezmielino- ■ funkcję wentylu, gdyż przenoszą pobudzenie za-
wych) wegetatywnych nerwach prędkość przewo- wsze tylko w jednym kierunku (od końca aksonu
dzenia osiąga wartość jedynie 0,5 do 15 m/s. do następnego w kolejności neuronu),
■ funkcje uczenia się i pamięci, gdyż w przypadku
Przewodzenie informacji przez nerwy. Przewodzo-
częstszego używania łatwiej przewodzą pobudze-
ne włóknami nerwowymi potencjały czynnościowe
nia niż wtedy, gdy są rzadko wykorzystywane (zja-
(impulsy nerwowe) służą – jak to już wcześniej wspo-
wisko plastyczności synaptycznej),
mniano – do przekazywania informacji w organizmie.
Ponieważ posiadają one taką samą amplitudę i postać ■ funkcje torowania i hamowania, ponieważ grupy
– informacja nie jest przenoszona za pomocą poje- synaps mogą łącznie nasilać proces pobudzenia lub
go osłabiać.
A
1.1.2.4.1. Typy synaps
kierunek przewodzenia
W zależności od umiejscowienia synaps rozróżnia
się następujące ich rodzaje (zob. ryc. B 1.1-6):
■ synapsy aksosomatyczne, łączące zakończenie
włókna nerwowego z ciałem komórki,
błona akson
■ synapsy aksodendrytyczne (zakończenie włókna
B nerwowego z okolicą dendrytów bliską ciału ko-
mórki)
■ aksoaksonalne (zakończenie na końcu neurytu).
Najczęściej spotykane są pierwsze dwa typy synaps.
W efekcie znaczna cześć powierzchni somy (ciała ko-
osłonka pobudzone niepobudzone
mielinowa przewężenie przewężenie mórki) oraz początkowych części dendrytów jest po-
kryta synapsami. Najczęściej na pojedynczym neuro-
Ryc. B 1.1-4. Przewodzenie pobudzenia w włóknach nerwowych nie znajduje się kilka tysięcy synaps.
warunkowane miejscowym przepływem słabych prądów (eletro-
tonicznych) na początku pobudzenia. Pobudzone okolice aksonu Jak to zostało uwidocznione na ryc. B 1.1-5 – włók-
zostały zaznaczone kolorem jasnoczerwonym. A – ciągłe przewo- no nerwowe na końcu ulega poszerzeniu do tzw. za-
dzenie w włóknach bezrdzennych; B – skokowe przewodzenie kończenia guziczkowego Helda-Auerbacha (kolbki
w włóknach rdzennych. Odcinek między kolejnymi przewężenia-
końcowej), które zawiera substancje przekaźnikowe
mi pokonywany jest przez linie prądu w sposób skokowy.
(neurotransmitery). Część błony kolbki synaptycz-
MUTSCHLER-2009.indd 148 2010-01-07 22:11:24
Układ nerwowy 149
ną znajduje się anatomiczna nieciągłość (przerwa)
A – szczelina synaptyczna (szerokość 10–20 nm),
będąca barierą dla przewodzenia pobudzenia, która
błona
postsynaptyczna może być pokonana jedynie za pomocą chemicz-
Układ nerwowy
nych neuroprzekaźników.
mitochondria pęcherzyk Do przekazania pobudzenia dochodzi wówczas,
gdy dojdzie do uwolnienia wystarczających ilości
soma substancji przekaźnikowej równocześnie w obrębie
kilku synaps (ok. 10) lub też w obrębie jednej synap-
sy aktywowanej wysoką częstotliwością przychodzą-
cych pobudzeń (zjawisko przestrzennego lub czaso-
wego sumowania). B1
błona
presynaptyczna 1.1.2.4.2. Synapsy pobudzające i hamujące
błona w układzie nerwowym
szczelina subsynaptyczna
synaptyczna W zależności od sposobu działania na następny neu-
ron odróżnia się synapsy pobudzające i hamujące.
Synapsy pobudzające sprzyjają wywołaniu potencja-
B łu czynnościowego, podczas gdy synapsy hamujące
synapsa synapsa wykazują odmienne działanie. Pomimo ich przeciw-
aksosomatyczna aksodendrytyczna stawnej funkcji procesy w obu typach synaps zacho-
dzą według podobnej zasady podstawowej.
Pobudzający potencjał postsynaptyczny (EPSP – exci-
tatory post-synaptic potential). Jeżeli pobudzenie przewo-
dzone przez akson dotrze do zakończenia presynaptyczne-
go, tj. do zakończenia guziczkowego Helda-Auerbacha,
dochodzi do uwolnienia neuroprzekaźnika z pęcherzyków
synaptycznych. Dyfunduje on przez szczelinę synaptyczną
do błony subsynaptycznej i tam wchodzi w kontakt z recep-
torami molekularnymi (farmakologicznymi), wywołując
w ten sposób – w przypadku synapsy pobudzającej – chwi-
lowy wzrost przepuszczalności dla małych kationów (Na+
synapsa
aksoaksonalna i K+). Głównym skutkiem wywołanego napływu jonów
Na+ jest krótkotrwała depolaryzacja nazywana postsynap-
tycznym potencjałem pobudzającym (EPSP).
Hamujący potencjał postsynaptyczny (IPSP – inhibitory
post-synaptic potential). Synapsy hamujące są z morfolo-
gicznego i funkcjonalnego punktu widzenia bardzo podob-
ne do synaps pobudzających. Uwalniany neuroprzekaźnik
wywołuje jednak w błonie subsynaptycznej krótkotrwałe
Ryc. B 1.1-5. Morfologiczna charakterystyka synaps. A – sche- podwyższenie potencjału błonowego – hiperpolaryzację.
matyczna budowa synapsy; B – klasyfikacja synaps na podsta- Te zmiany potencjału błony przeciwne do efektów EPSP
wie ich lokalizacji (według Thewsa, Mutschlera, Vaupela). są nazywane hamującym potencjałem postsynaptycznym
(IPSP).
Dochodzi do tego w ten sposób, że przekaźnik prowadzi
do silnego zwiększenia przepuszczalności dla jonów K+
nej w okolicy powierzchni styku określa się jako i Cl–. Na chwilowe podwyższenie potencjału błonowego
błonę presynaptyczną. Naprzeciw niej położona wpływa szczególnie nasilony napływ jonów chloru (prze-
jest nieco pogrubiona błona subsynaptyczna, nazy- sunięcie w kierunku wartości bardziej ujemnych) – co wy-
wana zwykle w farmakologii błoną postsynaptyczną wołuje powstanie IPSP. Każda hiperpolaryzacja oddala
– co nie jest prawidłowe z punktu widzenia fizjo- potencjał błonowy od progu pozwalającego na wywołanie
potencjału czynnościowego i tym samym zmniejsza pobud-
logii. (Fizjolog określa część błony komórkowej liwość neuronu, dlatego w trakcie IPSP jest znacznie trud-
leżącą poza strefą styku synaptycznego błoną post- niej wywołać potencjał czynnościowy niż podczas stanu
synaptyczną). Pomiędzy błoną pre- i postsynaptycz- spoczynkowego.
MUTSCHLER-2009.indd 149 2010-01-07 22:11:24
150 Układ nerwowy
1.1.2.4.3. Mechanizmy regulacji Przekaźnictwo synaptyczne może być modulowa-
presynaptycznej ne przez:
■ podawanie prekursorów neuroprzekaźników
Receptory dla neuroprzekaźników występują nie tyl- (np. tryptofanu lub hydroksytryptofanu),
ko w subsynaptycznej, ale również w części presy-
■ hamowanie magazynowania neuroprzekaźników
naptycznej błony komórkowej (zob. ryc. A 3.2-1).
w pęcherzykach (np. przez rezerpinę),
Wśród receptorów presynaptycznych rozróżnia się
autoreceptory, na które oddziałuje substancja prze- ■ uwalnianie neuroprzekaźników z pęcherzyków sy-
kaźnikowa uwalniania w tej samej synapsie, oraz he- naptycznych (np. przez pośrednio działające sym-
teroreceptory – pobudzane przez inne neuroprzekaź- patykomimetyki),
niki. Na przykład w obrębie synapsy adrenergicznej
■ zablokowanie uwalniania neuroprzekaźników
– uwalniającej noradrenalinę jako neuroprzekaźnik
do szczeliny synaptycznej (np. przez toksynę jadu
– znajdują się adrenergiczne receptory α2 będące au-
kiełbasianego),
toreceptorami oraz receptory cholinergiczne zalicza-
ne do heteroreceptorów. Pobudzenie autoreceptorów ■ hamowanie wychwytu zwrotnego uwolnionych
prowadzi albo do hamowania, albo rzadziej do zwięk- neuroprzekaźników ze szczeliny synaptycznej
szenia uwalniania neuroprzekaźnika – w wyniku (np. przez leki przeciwdepresyjne, kokainę lub he-
ujemnego lub dodatniego sprzężenia zwrotnego. Po- micholinę),
budzenie heteroreceptorów wywołuje – także w za-
■ stymulację autoreceptorów presynaptycznych (np.
leżności od typu receptora – hamowanie lub nasilenie
przez α2-sympatykomimetyki),
uwalniania neuroprzekaźnika.
■ blokadę heteroreceptorów presynaptycznych (np.
przez antagonistów receptora angiotensyny II),
1.1.2.4.4. Kotransmisja
■ pobudzenie receptorów postsynaptycznych (np.
przez bezpośrednio działające sympatykomimetyki
Liczne – jeżeli nie większość – zakończenia nerwowe
lub parasympatykomimetyki),
zawierają dwa lub więcej neuroprzekaźników, które
zwykle (ale nie zawsze) są uwalniane jednocześ- ■ hamowanie receptorów postsynaptycznych (np.
nie do szczeliny synaptycznej. Zjawisko to zwane przez blokery receptorów β-adrenergicznych, leki
jest kotransmisją. Należy tu wymienić kotransmi- antyhistaminowe H1 lub H2, antagonistów recepto-
sję noradrenaliny z trifosforanem adenozyny (ATP) ra angiotensyny II lub stabilizujące leki zwiotcza-
lub neuropeptydem Y albo równoczesne uwalnianie jące mięśnie),
acetylocholiny z naczynioruchowym (poli)peptydem
■ blokadę postsynaptycznych napięciowozależnych
jelitowym (VIP – vasoactive intestinal peptide) lub
kanałów jonowych (np. przez blokery kanałów
substancji P.
wapniowych lub leki znieczulające miejscowo),
Kotransmitery mogą modulować proces trans-
misji synaptycznej w różny sposób: albo wpływają ■ allosteryczny wpływ na działanie neuroprzekaźni-
w obrębie błony presynaptycznej w sposób dodatni ków (np. przez benzodiazepiny),
lub ujemny na syntezę bądź uwalnianie przekaźnika,
■ blokadę biotransformacji neuroprzekaźników
albo też błonę subsynaptyczną – zmieniając przez
(np. przez inhibitory monoaminooksydazy lub blo-
odpowiednie receptory działanie innych neuroprze-
kery acetylcholinesterazy),
kaźników.
■ działanie na postsynaptyczną kaskadę transdukcji
sygnału (np. przez inhibitory fosfodiesterazy).
1.1.2.5. Farmakologiczne wywieranie
wpływu na synaptyczne
przekaźnictwo pobudzenia
1.1.3. Anatomia mózgu
Działanie wielu grup leków wiąże się z ich wpływem
na synaptyczne przekaźnictwo pobudzenia. Poniżej – Mózg dorosłego mężczyzny waży średnio 1390 g,
dla lepszego zrozumienia sposobu ich działania – ze- dorosłej kobiety – 1250 g. Wypełnia on kostną pusz-
stawiono różne miejsca uchwytu takich działań w ob- kę czaszki – będąc otoczony oponami mózgowymi.
rębie synapsy. Z rozwojowego punktu widzenia wyróżnia się nastę-
MUTSCHLER-2009.indd 150 2010-01-07 22:11:25
Układ nerwowy 151
pujące części mózgu, które przedstawiono na ryc. B
1.1-6 na przekroju przez płaszczyznę środkową:
zakręt bruzda zakręt
■ kresomózgowie (telencephalon), przedśrodkowy środkowa zaśrodkowy
Układ nerwowy
■ międzymózgowie (diencephalon), płat czołowy płat ciemieniowy
■ śródmózgowie (mesencephalon),
■ tyłomózgowie (rhombencephalon) z móżdżkiem
zakręt
(cerebellum), mostem (pons) i rdzeniem przedłu- kątowy
żonym (medulla oblongata).
Kresomózgowie i międzymózgowie bywają określa- B1
płat skroniowy płat potyliczny
ne razem jako przodomózgowie (prosencephalon),
z kolei rdzeń przedłużony z mostem i śródmózgo- zakręt bruzda
wiem – jako pień mózgu. czołowy dolny boczna
Kresomózgowie. Kresomózgowie tworzą półkule Ryc. B 1.1-7. Widok boczny lewej półkuli mózgu (według
mózgowe i połączenia między nimi – drogi spoidło- Kahlego). Płaty mózgu zostały oznaczone różnymi kolorami.
we, z których najważniejszą jest ciało modzelowate
(corpus callosum). Obie półkule są oddzielone głębo-
ką szczeliną zwaną szczeliną podłużną mózgu (fissura ■ korową istotę szarą – określaną terminem kory móz-
longitudinalis). gu (cortex cerebri), która warstwą grubą na 1,5–
W każdej półkuli można wyróżnić dwie główne 5 mm otacza białą istotę rdzenną mózgu,
części: część podstawną z węchomózgowiem (rhie-
■ leżącą głębiej pośrednią i centralną istotę szarą,
nencephalon) i wyspą, oraz istotnie większą część
tzw. jądra podstawy (np. jądro ogoniaste – nucleus
– płaszcz (pallium) z różnymi płatami (lobi). Na po-
caudatus, jądro soczewkowate – nucleus lentifor-
wierzchni płaszcza mózgu występują liczne zakręty
mis, jądro migdałowate – nucleus amygdalae).
(gyri) tworzone przez bruzdy (sulci).
Tak jak cały ośrodkowy układ nerwowy – również
Na ryc. B 1.1-7 przedstawiono cztery płaty mózgu:
mózg zbudowany jest z istoty szarej i białej. W isto-
cie szarej zlokalizowane są ciała komórek nerwo- ■ płat czołowy (lobus frontalis),
wych, natomiast istota biała jest utworzona z włókien
■ płat ciemieniowy (lobus parietalis),
nerwowych. W przypadku istoty szarej można wy-
różnić: ■ płat skroniowy (lobus temporalis),
■ płat potyliczny (lobus occipitalis).
kresomózgowie Płat czołowy rozciąga się od bieguna czołowego do bruzdy
środkowej (sulcus centralis). W jego obrębie leżą głównie
okolice korowe odpowiedzialne za pierwszorzędowe funk-
cje ruchowe (w szczególności zakręt przedśrodkowy, gyrus
praecentralis). W części trójkątnej (pars triangularis) za-
krętu czołowego dolnego (gyrus frontalis inferior) znajduje
się najważniejsza okolica przełączeniowa dla funkcji mowy
(tzw. ośrodek mowy Broca).
Płat ciemieniowy jest położony z tyłu od bruzdy środ-
kowej (sulcus centralis). W obrębie najbardziej ku przo-
dowi położonej części – zakrętu przedśrodkowego (gyrus
międzymózgowie postcentralis) i sąsiadujących okolic kończą bieg somato-
sensoryczne drogi nerwowe. Te pola korowe odpowiedzial-
śródmózgowie ne są za przetwarzanie informacji sensorycznej (czuciowej)
móżdżek z obwodu i wnętrza ciała.
pień mózgu W obrębie płata skroniowego pod bruzdą boczną (sulcus
rdzeń lateralis) zlokalizowana jest pierwszorzędowa okolica słu-
przedłużony
chowa (kora słuchowa), z którą łączą się od tyłu i dołu okoli-
ce korowe odpowiedzialne za wyższe funkcje słuchowe.
Ryc. B 1.1-6. Przekrój mózgu w płaszczyźnie środkowej, granice Na przyśrodkowej powierzchni płata potylicznego prze-
między poszczególnymi częściami mózgu (według Waldeyera). biega bruzda ostrogowa (sulcus calcarinus). W sąsiadują-
MUTSCHLER-2009.indd 151 2010-01-07 22:11:25
152 Układ nerwowy
cej z bruzdą okolicy leży pierwszorzędowa okolica wzro- Śródmózgowie (mesencephalon). Śródmózgowie
kowa (kora wzrokowa), przechodząca w okolice położone jest najmniejszą częścią mózgu. Zawiera ono m.in.
na przyśrodkowych i bocznych powierzchniach półkul
miejsca przełączeniowe dla drogi wzrokowej i słu-
mózgowych, które służą przetwarzaniu informacji wzro-
kowej. chowej, należące do układu ruchowego komórki
zwojowe oraz jądra początkowe nerwów czaszko-
Należące do jąder mózgowia – jądro ogoniaste (nucle- wych.
us caudatus), jak również część jądra soczewkowate-
go (nucleus lentiformis) – skorupa (putamen), tworzą Móżdżek (cerebellum). Móżdżek leży w tylnym
razem prążkowie (corpus striatum), które jest ważną dole czaszkowym i jest oddzielony za pomocą na-
stacją przełączeniową w układzie ruchowym. Często miotu móżdżku (tentorium cerebelli) – skórzastej
razem z funkcjonalnie mu przyporządkowaną gałką błony – od płatów potylicznych mózgu. Wyróżnia
bladą (pallidum; zob. poniżej) jest ono określane mia- się nieparzystą część środkową – robak móżdżku
nem jąder podstawy (ryc. B 1.1.-8). (vermis cerebelli) i dwie półkule móżdżku. Na po-
wierzchni móżdżku znajdują się blisko leżące rów-
Międzymózgowie (diencephalon). Międzymózgo- nolegle przebiegające zwoje (folia cerebelli). Móż-
wie zawiera wzgórze (thalamus) – potężne nagroma- dżek jest połączony ze śródmózgowiem (mesen-
dzenie ciał komórek nerwowych. Pod tą częścią cephalon), pniem (pons) i rdzeniem przedłużonym
międzymózgowia leżą niskowzgórze (subthalamus) (medulla oblongata; zob. poniżej) przez konary
i podwzgórze (hypothalamus), a nad nią (w kierunku móżdżku (crura cerebelli).
grzbietowym) – nadwzgórze (epithalamus). Móżdżek jest odpowiedzialny za utrzymywanie
Wzgórze (thalamus; zob. ryc. B l.1-8) jest ważnym odpowiedniego napięcia mięśni szkieletowych i rów-
ośrodkiem przełączeniowym dla wstępujących (afe- nowagi, a także za koordynację ruchów.
rentnych) pobudzeń wstępujących włókien do kory
mózgowej. Obok są przekazywane zstępujące impul- Most (pons). Przez most przebiegają liczne wstępują-
sy z kory mózgowej do systemu ruchowego. ce i zstępujące drogi nerwowe. Jądra mostu są miej-
Niskowzgórze (subthalamus) zawiera jądra ukła- scami przełączeniowymi układu ruchowego.
du ruchowego, z którego największe znaczenie mają
jądro niskowzgórzowe (nucleus subthalamicus) i gał- Rdzeń przedłużony (medulla oblongata). Rdzeń
ka blada (globus pallidus). Gałka blada (określana przedłużony jest, jak to wynika z nazwy, przedłuże-
w skrócie łacińskim pallidum zamiast globus palli- niem rdzenia kręgowego, który łączy go z mostem.
dum) jest oddzielona włóknami torebki wewnętrz- W rdzeniu przedłużonym znajdują się ważne z ży-
nej (capsula interna) od otaczających struktur istoty ciowego punktu widzenia ośrodki kontroli oddycha-
szarej. Gałka blada ma połączenia do skorupy (pu- nia (ośrodek oddechowy) i krążenia (ośrodek naczy-
tamen), wzgórza (thalamus), jądra niskowzgórzowe- nioruchowy). Ponadto w obrębie rdzenia przedłużo-
go (nucleus subthalamicus) oraz do innych bardziej nego leżą ośrodki odruchowe związane z kaszlem,
z tyłu (doogonowo) leżących jąder. kichaniem, połykaniem i ssaniem, a także ośrodek
Podwzgórze (hypothalamus; zob. ryc. B 1.1-8) wymiotny.
tworzy najniższy poziom i dno międzymózgowia.
W jego obrębie leżą nadrzędne ośrodki autonomicz- Układ siatkowaty (formatio reticularis). W całym
nego układu nerwowego, które są odpowiedzialne pniu mózgu aż do międzymózgowia znajduje się –
za regulację ciśnienia krwi, oddychania i temperatu- oprócz wstępujących dróg czuciowych i zstępujących
ry. Poprzez podwzgórze dokonuje się również wpływ dróg ruchowych – sieć połączonych ze sobą neuro-
na regulację funkcji hormonalnych. Z pomocą lejka nów, zwanych układem siatkowatym. Uczestniczy on
z podwzgórzem łączy się przysadka mózgowa (hypo- w regulacji poziomu czuwania, modulacji informacji
physe). czuciowych, a także w sprawności funkcji wegeta-
Nadwzgórze (epithalamus) składa się z uzdecz- tywnych i hormonalnych.
ki (habenula) – miejsca przełączeniowego dla im-
pulsów drogi węchowej oraz z szyszynki (epiphyse, Układ limbiczny (rąbkowy). Określenie układ lim-
glandula pinealis). Ta ostatnia tworzy małe szyszko- biczny pochodzi od topograficznego ulokowania
wate ciało, które jest zlokalizowane na tylnej ścianie względem ciała modzelowatego (zob. powyżej),
trzeciej komory. Z szyszynki, połączonej przez pod- które otacza jak rąbek (limbus). Filogenetycznie
wzgórze z siatkówką, w zależności od oświetlenia, jest to starsza cześć mózgu, do której należą hipo-
a tym samym od pory dnia, uwalniana jest melato- kamp (hippocampus), sąsiadujący z nim zakręt przy-
nina. hipokampowy (gyrus parahippocampalis), zakręt zę-
MUTSCHLER-2009.indd 152 2010-01-07 22:11:25
Układ nerwowy 153
szczelina podłużna mózgu
jądro ogoniaste ciało modzelowate
Układ nerwowy
komora boczna
wzgórze torebka wewnętrzna
komora trzecia B1
skorupa
gałka blada przedmurze
ciało migdałowate
podwzgórze szlak wzrokowy
lejek (przysadki)
Ryc. B 1.1-8. Przekrój czołowy przez mózg na wysokości ciała migdałowatego (według Kahlego).
baty (gyrus dentatus), leżący nad ciałem modzelowa- impulsy są przewodzone przez nerw trójdziel-
tym zakręt obręczy (gyrus cinguli) i opuszki węchowe ny (V, n. trigeminus), twarzowy (VII, n. facialis)
(bulbus olfactorius). Od hipokampa odchodzi szlak i nerw smakowy (IX, n. glossopharyngeus; bardziej
włókien, tzw. sklepienie (fornix), biegnący łukowato poprawną nazwą jest nerw językowo-gardłowy),
do podwzgórza, a szczególnie do ciał suteczkowatych który dodatkowo odgrywa rolę w funkcjach we-
(corpora mamillaria). Do układu limbicznego zalicza getatywnych. Nerw błędny (X, n. vagus) unerwia
się ponadto jądra podkorowe, przede wszystkim ją- jako główny nerw układu przywspółczulnego klatkę
dro migdałowate (nucleus amygdalae), a także inne piersiową i trzewia jamy brzusznej. Impulsy rucho-
funkcjonalnie przyporządkowane struktury mózgu. we przewodzą nerwy III (n. oculomotorius = nerw
Układ limbiczny ma podstawowe znaczenie dla okoruchowy)), IV (n. trochlearis = nerw bloczkowy)
emocjonalnej oceny treści doznań i śladów pamięcio- i VI (n. abducens = nerw odwodzący), unerwiające
wych, a także dla afektywnej regulacji zachowania. zewnętrzne mięśnie oka, a także nerwy XI (n. ac-
cessorius = nerw dodatkowy) i XII (n. hypoglossus
Nerwy czaszkowe. Jako nerwy czaszkowe określa = nerw podjęzykowy).
się 12 par nerwów wychodzących z podstawy mózgu
oraz w okolicy pnia mózgu. Nerwy czaszkowe pełnią Komory mózgu i płyn mózgowo-rdzeniowy.
bardzo różnorodne funkcje: częściowo przenoszą one We wnętrzu mózgu znajdują się wolne przestrze-
informacje z narządów zmysłów, częściowo prze- nie nazywane komorami mózgowymi. Wyróżnia
wodzą impulsy czuciowe z narządów czuciowych się cztery komory: dwie komory boczne w przodo-
z okolic głowy do ośrodkowego układu nerwowego, mózgowiu, trzecią komorę położoną w pośrodko-
a część przewodzi impulsy ruchowe do mięśni głowy wej części międzymózgowia oraz komorę czwartą
i szyi. w tyłomózgowiu. Komory mózgowe są połączone
Do nerwów sensorycznych należą: nerwy węchowe i tworzą wspólny układ, który jest wypełniony pły-
(I, nervi olfactori), nerw wzrokowy (II, n. opticus) nem mózgowo-rdzeniowym (liquor cerebrospina-
i nerw słuchowo-równoważny (VIII, n. statoacusti- lis). Te wewnętrzne przestrzenie płynowe są po-
cus; bardziej poprawną nazwą jest nerw przedsion- łączone z zewnętrznymi przestrzeniami otaczają-
kowo-ślimakowy = n. vestibulo-cochlearis; przyp. cymi cały ośrodkowy układ nerwowy (zob. ryc. B
tłum.). Zarówno sensoryczne, jak i motoryczne 1.1-9).
MUTSCHLER-2009.indd 153 2010-01-07 22:11:26
154 Układ nerwowy
1.1.4. Budowa rdzenia kręgowego
komora boczna (1 i 2) splot naczyniówkowy
Liczący 40–45 cm długości, leżący w kanale kręgo-
wym rdzeń kręgowy dzieli się na część szyjną, pier-
siową, lędźwiową i krzyżową. Na przekroju poprzecz-
nym składa się z podobnej do motyla istoty szarej
oraz otaczającej ją istoty białej (zob. ryc. B 1.1-10).
Rdzeń kręgowy pełni funkcję przewodzenia i przełą-
czania pobudzeń.
Istota szara (substantia grisea) składa się tak jak
otwór w mózgu głównie z ciał komórek nerwowych, pod-
międzykomorowy czas gdy istota biała (substantia alba) zawiera wstę-
pujące i zstępujące drogi przewodzeniowe.
komora trzecia W obrębie istoty szarej – na jej przekroju poprzecz-
splot naczyniówkowy nym można obustronnie wyróżnić róg przedni, róg
wodociąg mózgu tylny oraz łączący je róg boczny. Z rogów przednich
(Sylwiusza) zbiornik wychodzą włókna ruchowe (aksony motoryczne), na-
komora czwarta móżdżkowo-rdzeniowy tomiast do rogów tylnych wchodzą włókna czuciowe.
Te ostatnie przenoszą docierające nimi pobudzenia
Ryc. B 1.1-9. Wewnętrzne (kolor pomarańczowy) i zewnętrzne albo do mózgu, albo do ruchowych komórek rogów
(kolor szarobrązowy) przestrzenie płynowe, tj. wypełnione pły-
przednich. Neurony rogów bocznych są elementami
nem mózgowo-rdzeniowym (według Kahlego).
wegetatywnego układu nerwowego.
W substancji białej można wyróżnić po każdej po-
łowie rdzenia sznur (powrózek) przedni, boczny i tyl-
Płyn mózgowo-rdzeniowy jest wytwarzany przez sploty ny. W ich obrębie przebiegają zstępujące i wstępujące
naczyniówkowe (plexus chorioidus), które leżą w komorach aksony zgrupowane zależnie od ich funkcji w drogi
mózgowych i swoją budową oraz wyglądem przypominają
kosmki łożyska – stąd pochodzi ich nazwa. Leżące w ko- nerwowe (tractus).
morach uwypuklające się do ich wnętrza sploty żylne skła- Nerwy ruchowe i czuciowe rdzenia kręgowego
dają się z wielokrotnie zwiniętych naczyń kapilarnych oraz (formalnie mowa tu jest raczej o korzonkach ner-
tkanki łącznej. Są one pokryte warstwą komórek nabłonko- wowych niż nerwach – przyp. tłum.), które wchodzą
wych typu sześciennego (kostkowego, brukowego – przyp. do kanału kręgowego lub z niego wychodzą przez
tłum.), pokrytych miejscowo rąbkiem szczoteczkowym.
otwory międzykręgowe, łączą się krótko przed jego
Opony mózgowe. Ośrodkowy układ nerwowy jest od swej opuszczeniem w nerwy rdzeniowe zaopatrujące ob-
zewnętrznej powierzchni otoczony trzema powłokami na- wód.
zywanymi oponami (meninges). Zewnętrzna, mocna i trwa-
ła opona twarda (dura mater) składa się z kolagenowej
tkanki łącznej z licznymi elastycznymi włóknami.
Obie delikatne i cienkie wewnętrzne powłoki, tzn.
do mózgu
opona pajęcza (arachnoidea) i opona miękka (pia mater) drogi
tworzą razem oponę miękko-pajęczą mózgu lub rdzenia róg tylny
czuciowe
(leptomeninx). Od opony twardej opona miękko-pajęcza
jest oddzielona małą szczeliną zwaną przestrzenią (jamą)
podtwardówkową (cavum subdurale).
Opona pajęcza składa się z łącznotkankowej błony po-
zbawionej naczyń krwionośnych i z podobnego do gąbki
szkieletu z delikatnej, włóknistej tkanki łącznej, która łączy
się ze ściśle przylegającą do mózgu oponą miękką. W ten
sposób mózg zawieszony jest niejako w przestrzeni podpa-
jęczynówkowej.
Opona miękka jest delikatną, bogato unaczynioną błoną
łącznotkankową, która leży na powierzchni ośrodkowego
układu nerwowego i wnika również w bruzdy. Pomiędzy
pasmami tkanki łącznej opony miękkiej i pajęczej znajdują droga
się liczne szczeliny tkankowe, które są wypełnione płynem róg przedni ruchowa
mózgowo-rdzeniowym. Cały system tych połączonych
ze sobą szczelin tkankowych nazywany jest przestrzenią Ryc. B 1.1-10. Schematyczny przekrój poprzeczny rdzenia krę-
podpajęczynówkową. gowego.
MUTSCHLER-2009.indd 154 2010-01-07 22:11:26
Układ nerwowy 155
1.1.5. Budowa obwodowego zaznaczyć, że obie części tworzą funkcjonalną całość
układu nerwowego i ścisły podział nie jest możliwy, ponieważ istnieje
ścisła integracja między neuronami somatycznymi
i autonomicznymi. Wiele wzajemnych powiązań ist-
Poza kanałem kręgowym nerwy rdzeniowe rozdzie-
Układ nerwowy
nieje np. pomiędzy zjawiskami psychicznymi i we-
lają się na trzy gałęzie (zob. ryc. B 1.1-11):
getatywnymi (rumienienie się, blednięcie, kołatanie
■ gałąź tylna (grzbietowa), która unerwia czuciowo serca, itp.).
skórę w okolicy kręgosłupa i ruchowo część mięśni
przykręgosłupowych, Układ czuciowy (sensoryczny). Ta część układu
nerwowego służy do przyjmowania, przekazywania
■ gałąź przednia (brzuszna) – odpowiedzialna za czu-
i przetwarzania informacji, które docierają do organi-
ciowe i ruchowe unerwienie pozostałej części tuło-
zmu w formie różnorodnych bodźców. Przekazywa- B1
wia i kończyn,
nie i przetwarzanie informacji zachodzi w ten sposób,
■ gałąź, która stanowi połączenie współczulnego że bodziec wyzwala powstanie potencjału receptoro-
układu nerwowego z układem somatycznym (for- wego, który ostatecznie prowadzi do wyzwolenia po-
malnie są to dwie gałęzie łączące – biała i szara tencjału czynnościowego.
– odchodzące od wspomnianej wcześniej gałęzi W obrębie synapsy dochodzi do uzależnionego
brzusznej – przyp. tłum.). od częstotliwości docierających potencjałów czyn-
nościowych uwalniania neuroprzekaźnika, który
Przednie gałęzie mają więc największy obszar uner- (przy odpowiedniej jego ilości) wyzwala potencjał
wienia. Wykazują one charakterystyczną tendencję czynnościowy w następnym neuronie. Za pomocą
do tworzenia splotów nerwowych, do których wcho- połączonych jeden za drugim neuronów – impulsy
dzą nerwy rdzeniowe wychodzące z kilku segmentów nerwowe docierają do obszarów mózgu odpowie-
rdzenia i z których wyodrębniają się odmiennie zor- dzialnych za przetwarzanie informacji. Swoiste drogi
ganizowane nerwy obwodowe. somatosensoryczne, tzn. otrzymujące informacje z re-
ceptorów skóry, mięśni i stawów, kończą się w zakrę-
cie zaśrodkowym (gyrus postcentralis) oraz wtórnie
w sąsiadujących obszarach korowych. W przeciwień-
korzeń tylny gałąź grzbietowa stwie do tego informacje z tzw. wyższych organów
zmysłowych docierają do specyficznych projekcyj-
korzeń przedni gałąź nych pól korowych.
brzuszna Poza swoistym układem istnieje też nieswoisty
układ czuciowy, którego zadaniem jest ogólna akty-
wacja kory mózgowej. Impulsy ze wszystkich wstę-
pień współczulny pujących dróg czuciowych przebiegają ponadto przez
gałąź łącząca kolaterale tworu siatkowatego (formatio reticularis).
Docierają one do wzgórza (thalamus) i stąd ulegają
rozlanemu rozprzestrzenieniu na całą korę mózgo-
wą. Ten nieswoisty strumień pobudzeń ma decydu-
Ryc. B 1.1-11. Korzenie i gałęzie pary nerwu rdzeniowego jące znaczenie dla utrzymania odpowiedniego stanu
(według Clary). Gałąź tylna (grzbietowa): ramus dorsalis; gałąź przytomności i jasności stanu świadomości. Jego
przednia (brzuszna): ramus ventralis; gałąź łącząca: ramus com- przerwanie, co może być wynikiem zastosowania
municans.
znieczulenia ogólnego (narkozy), prowadzi do utra-
ty świadomości. U osoby śpiącej można poprzez
drażnienie określonych struktur układu nieswoistego
wywołać reakcję budzenia (w ten sposób za pomocą
1.1.6. Funkcje somatycznego układu
bodźców świetlnych, dźwiękowych, zimna lub ciepła
nerwowego – można obudzić śpiącą osobę). Z tego powodu nie-
swoisty układ czuciowy nazywa się wstępującym ak-
Po tym wprowadzeniu poświęconym w przeważającej tywującym układem siatkowatym (ARAS – ascendig
mierze relacjom anatomicznym zostaną omówione reticular activating system).
funkcje somatycznego i autonomicznego układu ner- Ponadto nieswoisty układ siatkowaty pełni w cza-
wowego. Podział ten wydaje się sensowny i koniecz- sie czuwania ważne zadanie: pozwala na kierowanie
ny do zrozumienia jego znaczenia. Należy jednak uwagi na określone zjawiska zewnętrzne, tzn. odby-
MUTSCHLER-2009.indd 155 2010-01-07 22:11:27
156 Układ nerwowy
■ motoryką postawną i podporową, tzn. niezależną
róg tylny od woli aktywnością mięśniową, która prowadzi
włókno dośrodkowe typu Ia do stabilizowania pozycji ciała i utrzymania rów-
nowagi,
wrzecionko ■ zautomatyzowanymi sekwencjami ruchowymi zwią-
mięśniowe zanymi z określonym programem ruchowym de-
terminowanym genetycznie (oddychanie, mimika
twarzy) lub wyuczonym (chodzenie, bieganie, jazda
na rowerze i in.),
neuron ruchowy α
■ motoryką celu, tzn. ukierunkowanymi ruchami do-
róg przedni (α-motoneuron) wolnymi wynikającymi z wewnętrznej potrzeby.
płytka nerwowo- Dawniej uważano, że motoryka celu jest regulowana
-mięśniowa jedynie przez korę ruchową okolicy zakrętu przed-
środkowego (gyrus pracentralis) i wychodzącą z niej
drogą piramidową (ryc. B 1.1-13), podczas gdy inne
obszary związane z ruchem, w szczególności zwoje
podstawne z przynależnymi im drogami, odpowie-
Ryc. B 1.1-12. Budowa monosynaptycznego łuku odruchowe-
dzialne są za ruchy bezwolne (niezależne od świa-
go (według Thewsa, Mutschlera, Vaupela). domości). Z tego powodu dzielono ponadrdzeniowy
układ ruchowy na:
■ piramidowy,
wa się selekcja strumienia pobudzeń, przy czym in-
formacje, na których nie jest skupiona uwaga, ulegają ■ pozapiramidowy.
tłumieniu.
Według obecnego stanu wiedzy – funkcje wszyst-
Układ ruchowy (motoryki dowolnej). Układ moto- kich uczestniczących struktur są ze sobą związane
ryki dowolnej dzieli się na: do tego stopnia, że taki podział z punktu widzenia
■ układ ruchowy rdzeniowy,
■ układ ruchowy ponadrdzeniowy.
Za pomocą odruchów rdzeniowy układ ruchowy kora ruchowa
uczestniczy w kontroli napięcia mięśniowego. thalamus
Przez pojęcie odruchu rozumie się takie same (stereoty-
powe) reakcje organizmu na określony bodziec czuciowy. prążkowie
Reakcja taka zachodzi w obrębie tzw. łuku odruchowego. gałka blada
Jego najprostszą formą jest monosynaptyczny łuk odrucho-
wy. Składa się on z receptora rozciągowego wrzecionka pień
mięśniowego, wstępującej drogi prowadzącej do rdzenia mózgu
kręgowego (włókna czuciowe Ia) oraz synapsy znajdują-
cej się w rogu przednim z odśrodkową drogą podążającą droga droga
do mięśnia, na której końcu znajduje się efektor (mięsień) korowo-rdzeniowa korowo-rdzeniowa
(zob. ryc. B 1.1-12). przednia boczna
Jeżeli dojdzie do nagłego rozciągnięcia wrzecionek
mięśniowych (np. przez przegięcie stopy), to impulsy rdzeń szyjny
w nim wywołane wędrują dośrodkową (aferentną) drogą
do rdzenia kręgowego, gdzie zostają przełączone na drogi
ruchowe, co ostatecznie prowadzi do skurczu właściwych
tym wrzecionkom włókien mięśniowych.
Poza odruchami monosynaptycznymi istnieją też liczne rdzeń lędźwiowy
odruchy polisynaptyczne (np. skóry brzucha, odruchy ki-
chania, kasłania i opróżniania pęcherza moczowego).
Ponadrdzeniowy układ motoryczny jest odpowie-
dzialny za nadrzędną kontrolę ruchu. Kieruje on: Ryc. B 1.1-13. Schemat drogi piramidowej (według Schmidta).
MUTSCHLER-2009.indd 156 2010-01-07 22:11:27
Układ nerwowy 157
fizjologii nie wydaje się dalej uzasadniony. Mimo wych nasilają się wszystkie procesy służące organi-
to nadal używa się terminu układ pozapiramido- zmowi w spoczynku: zwiększa się aktywność gruczo-
wego ruchu, ponieważ różne leki, a szczególnie łów trawiennych i mięśniówki jelit, a zmniejsza się
neuroleptyki, a także zespół Parkinsona, wywołują wydolność układów krążenia i oddychania.
Układ nerwowy
zaburzenia niezależnych od woli aktywności mięśni
przez wpływ lub uszkodzenie/zanik neuronów do- Ośrodki autonomiczne. Jak to już zostało opisane,
paminergicznych w obrębie zwojów podstawy. istnieją w podwzgórzu nadrzędne ośrodki układu we-
getatywnego odpowiedzialne za regulację ciśnienia
krwi, oddychania i temperatury ciała. Ponadto stąd
1.1.7. Funkcje autonomicznego kontrolowana jest także gospodarka wodna i liczne
(wegetatywnego) procesy metaboliczne.
W rdzeniu przedłużonym znajdują się ważne ośrod- B1
układu nerwowego ki odruchowe. Za pomocą dróg aferentnych ośrodki
te otrzymują informacje z chemo- i baroreceptorów
Autonomiczny układ nerwowy służy utrzymaniu op-
oraz receptorów rozciągowych; ponadto określone
tymalnej wewnętrznej równowagi organizmu („sy-
parametry (np. ciśnienie cząsteczkowe gazów, war-
stem świata wewnętrznego”). Kieruje on funkcjami,
tość pH) są rejestrowane w tych ośrodkach. Nato-
które nie są zależne od woli i świadomości, takimi
miast odpowiedni wzorzec pobudzenia przekazywa-
jak:
ny przez drogi eferentne wpływa na funkcje narzą-
■ układ krążenia przez nasilenie lub osłabienie pracy dów wewnętrznych.
serca, a szczególnie przez zwężanie i rozszerzanie
naczyń krwionośnych, Układ nerwowy wegetatywny obwodowy. Od-
środkowa (eferentna) część obwodowego układu we-
■ oddychanie przez podwyższanie lub obniżanie czę-
getatywnego składa się w przypadku układu współ-
stotliwości oddechów oraz przez zwężanie lub roz-
czulnego i przywspółczulnego podobnie z dwu neu-
szerzanie mięśni oskrzeli,
ronów (zob. ryc. B 1.1-14): pierwszy neuron przewo-
■ perystaltyka żołądka i jelit, dzi pobudzenia z ośrodkowego układu nerwowego
do zwoju wegetatywnego, gdzie następuje przełącze-
■ napięcie wszystkich innych mięśni gładkich (np.
nie na drugi neuron podążający już do narządu wyko-
pęcherzyka żółciowego, moczowodów, pęcherza
nawczego. Na podstawie położenia względem zwoju
moczowego, macicy),
– pierwszy neuron określany jest jako przedzwojowy,
■ wydzielanie gruczołów potowych, ślinowych, gru- drugi jako zazwojowy.
czołów w żołądku i jelitach oraz pozostałych. Odmiennymi w przypadku układu współczulnego
i przywspółczulnego są:
Ponadto układ ten uczestniczy w regulacji metaboli-
■ lokalizacja początku neuronu przedzwojowego,
zmu komórkowego.
Według kryteriów morfologicznych i funkcjonal- ■ położenie zwojów,
nych w obrębie wegetatywnego układu nerwowego
■ rodzaj substancji neuroprzekaźnikowej w synap-
można wyróżnić dwie jego części, układ współczul-
sach neuronów zazwojowych.
ny i przywspółczulny, które w obrębie narządów
unerwionych zwykle podwójnie, tzn. współczulnie
i przywspółczulnie (ale nie zawsze!) – wywołują Ciała neuronów przedzwojowych włókien współczul-
przeciwstawne (antagonistyczne) działania (zob. tab. nych są zlokalizowane w rogach bocznych piersiowej
1.1-2). Autonomiczne unerwienie poszczególnych i górnej lędźwiowej części rdzenia, natomiast włókien
narządów zostało przedstawione na ryc. B 1.1-14. przywspólczulnych – w pniu mózgu i części krzyżo-
Pobudzenie układu współczulnego wywołu- wej rdzenia. Zwoje współczulne leżą w pobliżu ośrod-
je – w dużym uproszczeniu – reakcje ergotropowe, kowego układu nerwowego (przede wszystkim pod
a przywspółczulnego – reakcje trofotropowe. postacią pnia współczulnego biegnącego po obu stro-
W przypadku reakcji ergotropowej zwiększeniu nach kręgosłupa), a zwoje przywspółczulne w pobliżu
ulega zdolność do wysiłku i do dawania sobie rady docelowych narządów wykonawczych lub bezpośred-
z otoczeniem/środowiskiem: serce, układ krążenia nio w ich obrębie (tzw. zwoje śródścienne). Podczas
i oddychanie są aktywowane, glikogen ulega mobi- gdy we wszystkich wegetatywnych zwojach zarówno
lizacji, zmniejsza się natomiast aktywność układu współczulnych, jak i przywspółczulnych, przekazy-
pokarmowego. Odwrotnie przy reakcjach trofotropo- wanie pobudzenia następuje przy pomocy acetylo-
MUTSCHLER-2009.indd 157 2010-01-07 22:11:28
158 Układ nerwowy
oko, gruczoł łzowy
ślinianka przyuszna
ślinianka
podżuchwowa
i podjęzykowa
serce
płuca, oskrzela
naczynia brzuszne
żołądek
wątroba
trzustka
nerki
jelito cienkie,
jelito grube
pozostała część jelita
grubego, prostnica,
pęcherz moczowy,
narządy płciowe
układ współczulny: (włókna przedzwojowe) (włókna zazwojowe)
układ przywspółczulny: (włókna przedzwojowe) (włókna zazwojowe)
Ryc. B 1.1-14. Schemat współczulnego i przywspółczulnego układu nerwowego (według Waldeyera).
MUTSCHLER-2009.indd 158 2010-01-07 22:11:28
Układ nerwowy 159
Tabela B 1.1-2. Wpływ pobudzenia układu współczulnego i przywspółczulnego na narządy wegetatywne
Narząd Działanie po pobudzeniu układu
współczulnego przywspółczulnego
Układ nerwowy
Serce
częstotliwość skurczu wzrost obniżenie
siła skurczu wzrost obniżenie (jedynie przedsionki)
Naczynia krwionośne
wieńcowe rozszerzenie rozszerzenie
skóry zwężenie rozszerzenie
płuc zwężenie rozszerzenie
B1
mózgu lekkie zwężenie –
mięśni szkieletowych rozszerzenie –
trzewne zwężenie –
Płuca
mięśniówka oskrzeli rozkurcz skurcz
Gruczoły ślinowe wydzielanie gęstej śliny obfite wydzielanie rzadkiej śliny
Układ pokarmowy
perystaltyka osłabienie nasilenie
mięśnie zwieracze skurcz rozkurcz
Wątroba glikogenoliza –
Pęcherzyk żółciowy rozkurcz skurcz
Pęcherz moczowy
mięsień zwieracz skurcz rozkurcz
mięsień wypieracz rozkurcz skurcz
Macica
różnie - zależnie od fazy cyklu różnie - zależnie od fazy cyklu
Oczy
mięsień rozwieracz źrenicy skurcz –
mięsień zwieracz źrenicy – skurcz
Gruczoły łzowe – wydzielanie
choliny, to neuroprzekaźnikiem zazwojowym w ukła- w mięśniach tzw. wielokrotnej jednostki ruchowej
dzie współczulnym jest noradrenalina, a w układzie (multi-unit-type) – pobudzenie związane jest z uwol-
przywspółczulnym – acetylocholina. nieniem substancji przekaźnikowej z zakończenia
nerwu zazwojowego. Mięśnie typu single-unit wy-
Wpływ układu wegetatywnego na napięcie mięśni stępują przede wszystkim w ścianie narządów jami-
gładkich. Mięśnie gładkie są unerwione wyłącz- stych (np. w układzie pokarmowym, pęcherzu mo-
nie przez włókna układu wegetatywnego. W przy- czowym, przewodzie moczowym, macicy), a mięś-
padku mięśni gładkich tzw. typu pojedynczej jed- nie typu multi-unit – w tętnicach, tęczówce, skórze
nostki ruchowej (single-unit-typ), które zdolne są i nasieniowodach.
do automatycznego wywoływania pobudzenia, układ
współczulny i przywspółczulny modyfikuje jedy-
nie ich aktywność spontaniczną i napięcie. Z kolei
MUTSCHLER-2009.indd 159 2010-01-07 22:11:29
160 Układ nerwowy
1.1.8. Unerwienie jelit
(układ nerwowy trzewny) układ układ układ
przywspółczulny współczulny przywspółczulny
pnia mózgu piersiowo-lędźwiowy krzyżowy
Regulacja motoryki układu pokarmowego jest zapew-
niona także bez unerwienia współczulnego czy przy-
współczulnego, tzn. również przy przecięciu odpo-
wiednich włókien nerwowych. Oznacza to, że system przed-
wegetatywny pełni tu jedynie funkcję modyfikującą kręgosłupowo
(zob. powyżej). Kontrola i koordynacja narządu do-
celowego – układu pokarmowego – podlega zasad-
niczo tzw. układowi nerwowemu trzewnemu, którego
komórki nerwowe położone są głównie w splocie
podśluzówkowym (plexus submucosus, splot Meiss-
nera) – znajdującym się pod warstwą śluzówki, oraz mięśniówka
w splocie warstwy mięśniowej jelita (plexus myente- jelito cienkie
jelito grube
ricus, splot Auerbacha) – rozmieszczonym pomiędzy żołądek
warstwami mięśni podłużnych i okrężnych. Układ
nerwowy trzewny składa się z neuronów aferentnych, cholinergiczne, nieadrenergiczne,
których aksony wykazują właściwości receptorowe, pobudzające niecholinergiczne,
z interneuronów oraz eferentnych neuronów rucho- hamujące
adrenergiczne,
wych oraz wydzielniczych. Łącznie na układ nerwo- hamujące interneuron cholinergiczny,
wy trzewny składa się ok. 108 neuronów, tzn. z liczby serotoninergiczny,
podobnej do liczby neuronów w rdzeniu kręgowym. dośrodkowe peptydergiczny, pobudzający
Odpowiednim bodźcem dla wyzwolenia ruchu jelit
jest rozciąganie ścian żołądka lub jelit, które prowa-
dzi do pobudzenia neuronów aferentnych. Po przełą- Ryc. B 1.1-15. Schemat trzewnego układu nerwowego (zmodyf.
według Burnstocka).
czeniu na włókna aferentne wywołany zostaje skurcz
proksymalny i jednocześnie dochodzi do rozluźnie-
nia mięśni gładkich dystalnie od masy pokarmowej,
przez co jest ona transportowana w kierunku dood-
bytniczym. W zjawisku tym uczestniczą włókna ner- tej wynika, że jest to układ zamknięty, w który in-
wowe, które nie zawierają ani noradrenaliny, ani ace- gerują w sposób modulujący układy współczulny
tylocholiny jako neuroprzekaźnika i dlatego są zwane i przywspółczulny. Niektóre motoneurony (neurony
włóknami NANC (not adrenergic, not cholinergic). ruchowe) układu trzewnego, szczególnie w żołądku
Do licznych neuroprzekaźników tych neuronów nale- i jelicie końcowym, są jednocześnie zazwojowymi
żą natomiast m.in. serotonina i substancja P – wpły- neuronami przywspólczulnymi.
wające na skurcz mięśni oraz tlenek azotu NO (nitric Zazwojowe neurony współczulne hamują gene-
oxide), naczynioruchowy peptyd jelitowy VIP (vaso- ralnie przewodzenie pobudzenia w neuronach pobu-
active intestinal peptide) i trifosforan adenozyny ATP dzających. Mięśniówka gładka zwieraczy ulega nato-
– jako związki o działaniu rozkurczającym. miast pod ich wpływem ciągłemu skurczowi, wsku-
Na ryc. B 1.1-15 przedstawiono schematycznie tek czego dochodzi do funkcjonalnego oddzielenia
organizację układu nerwowego trzewnego. Z ryciny różnych części układu pokarmowego.
MUTSCHLER-2009.indd 160 2010-01-07 22:11:29
Układ nerwowy 161
1.2. Leki wpływające na sferę psychiczną
(leki psychotropowe)
Układ nerwowy
Mimo że schorzenia psychiczne należą do naj- wówczas, gdy na jej końcu dochodzi ponownie do pełnego
częstszych i równocześnie szczególnie znaczących wyzdrowienia; natomiast rzutem określa się przebieg, gdy
po nim utrzymują się pozostałości choroby.
obrazów chorobowych, ich podział nie zawsze był
i nie jest do końca jednoznaczny. Oprócz różnych
Do połowy ubiegłego stulecia medycyna była niemal-
międzynarodowych klasyfikacji nadal w poszczegól-
że bezradna wobec większości chorób psychicznych,
nych krajach stosowane bywają podziały wywodzące
zwłaszcza w przypadku psychoz. Dopiero wprowa-
się z lokalnych szkół psychiatrycznych. Liczni psy-
dzenie leków psychotropowych w latach pięćdzie- B1
chiatrzy posiłkują się Międzynarodową Klasyfikacją
siątych XX wieku pozwoliło osiągnąć zdecydowany
Chorób w jej 10 wersji (ICD-10; International Clas-
postęp w leczeniu pacjentów chorych psychicznie
sification of Diseases) – opracowana przez Światową
(m.in. zmniejszenie stosowania środków przymusu,
Organizację Zdrowia WHO lub – szczególnie w ame-
ułatwienie sprawowania opieki i umieszczania w za-
rykańskim kręgu językowym – Podręcznikiem Diag-
kładach leczniczych, skrócenie pobytu w klinice, uła-
nostyki i Statystyki w wersji IV (DSM-IV; Diagnostic
twienie procesu powrotu do społeczeństwa). Jednakże
and Statistical Manual) Amerykańskiego Towarzy-
przyczynowa farmakoterapia nadal nie jest do końca
stwa Psychiatrycznego (APA = American Psychiatric
możliwa. Znane są bowiem jedynie częściowe aspek-
Association). Ponieważ same definicje schorzeń psy-
ty przyczyn chorób psychicznych i mechanizmu
chicznych bywają bardzo zmienne, a niektóre wcześ-
działania leków psychotropowych. Ale coraz bardziej
niej powszechnie stosowane pojęcia, np. psychoz
pewne są dowody na to, że:
czy nerwic, wydają się obecnie polemiczne – zaleca
się stosowanie ogólnych określeń w rodzaju zaburzeń ■ u podstaw chorób psychicznych leżą zaburzenia
psychicznych. neuroprzekaźnictwa – zwłaszcza zaburzenia do-
tyczące aromatycznych monoamin, tj. dopaminy,
Przez pojęcie psychozy (rzadziej używane z powodu trud- noradrenaliny i serotoniny – a w ich następstwie
ności z jednoznacznym zdefiniowaniem, lecz nadal często zmiany w dystrybucji oraz gęstości receptorów,
stosowane w warunkach klinicznych i praktyki lekarskiej)
rozumie się choroby dotyczące umysłu i psychiki, które pro- ■ leki psychotropowe w wyniku interakcji z fizjo-
wadzą do strukturalnej zmiany całości przeżywania, w zna- logicznymi substancjami przekaźnikowymi lub
czącym stopniu zmieniają osobowość dotkniętych nimi ich receptorami wpływają na regulację układu
osób, występują często pod postacią faz i rzutów oraz które
można różnicować z innymi zaburzeniami psychicznymi nerwowego i dzięki temu przynajmniej częścio-
– częściowo na podstawie objawów chorobowych, często wo przywracają zaburzoną równowagę w zakresie
jednak tylko według ich przebiegu. O fazie można mówić neuroprzekaźnictwa.
Tabela B 1.2-1. Podział leków psychotropowych
Synonim Główne wskazania
neuroleptyki trankwilizatory duże schizofrenie, manie,
(leki neuroleptyczne) zespoły psychoorganiczne,
stany pobudzenia psychoruchowego
i stany lękowe,
alkoholowy zespół abstynencyjny
leki przeciwdepresyjne depresje
leki uspokajające, leki trankwilizujące, zaburzenia nerwicowe,
anksjolityki trankwilizatory małe, zaburzenia psychowegetatywne,
ataraktyki stany lękowe
substancje psychostymulujące leki tonizujące psychikę, narkolepsja,
środki pobudzające zespół deficytu uwagi i z hiperaktywnością
substancje psychomimetyczne środki psychodysleptyczne,
iluzjogeny, halucynogeny
MUTSCHLER-2009.indd 161 2010-01-07 22:11:29
162 Układ nerwowy
Różne grupy leków psychotropowych zostały zesta- (m.in. słowotok lub otamowanie mówienia, dzi-
wione w tab. B 1.2-1. waczne wypowiedzi, manieryzmy, neologizmy,
perseweracje – ciągłe powtarzanie),
■ w sferze afektywnej (m.in. zobojętnienie, nadwraż-
1.2.1. Schizofrenie liwość, depresyjność, drażliwość, utrata kontaktu,
ambiwalencja uczuciowa),
1.2.1.1. Podstawy psychopatologii
■ przeżywania własnej osoby (m.in. autyzm, deperso-
nalizacja, poczucie wyobcowania i ulegania wpły-
Schizofreniami określa się grupę zaburzeń psychicz-
wom, rozpad struktury osobowości).
nych, w których przebiegu ujawniają się wielowar-
stwowe zaburzenia struktury osobowości z charak-
Do warunkowanych nadczynnością dopaminergiczną
terystycznymi zmianami myślenia, odczuwania oraz
w obrębie układu mezolimbicznego objawów dodat-
relacji w stosunku do otoczenia. Prawdopodobień-
kowych/towarzyszących zalicza się:
stwo zachorowania na nie wynosi około 1% i war-
tość ta jest niezależna od rasy, kultury i warunków ■ omamy (halucynacje; słuchowe, wzrokowe, zapa-
zewnętrznych. Etiologia i patogeneza mają charakter chowe i smakowe),
wieloczynnikowy; jak to pokazują badania na bliźnię-
■ urojenia (prześladowcze, trucia, seksualne i in.),
tach – zwykle obecny jest udział czynnika genetycz-
nego. Niekorzystna stresowa sytuacja psychospołecz- ■ zaburzenia motoryki i napędu (tzw. objawy katato-
na sprzyja pierwszemu zachorowaniu oraz kolejnym niczne, takie jak bezruch i zatrzymanie mówienia,
nawrotom. Około 60% zachorowań na schizofrenię zahamowanie napędu, ale również niepokój/pobu-
rozpoczyna się między okresem dojrzewania a 30 ro- dzenie psychoruchowe, stereotypie ruchowe).
kiem życia.
W przypadku jednej trzeciej pacjentów – rokowanie
Na poziomie patofizjologicznym bierze się pod jest niekorzystne; w przypadku dalszych około 40%
uwagę – oprócz zmian w zakresie przekaźnictwa przypadków – w obrazie klinicznym z trudem roz-
GABA-ergicznego, glutaminergicznego, choliner- poznaje się później objawy psychotyczne, a u kolej-
gicznego i serotoninergicznego – w szczególności nych 20% pacjentów następuje (prawie) całkowite
zaburzenia układu dopaminergicznego. Niewystar- wyzdrowienie.
czającej aktywacji receptorów D2 w układzie mezo-
kortykalnym towarzyszy nadmierna aktywacja tych Wyróżnia się następujące postacie kliniczne schizofrenii:
samych receptorów w układzie mezolimbicznym. ■ (obecnie najczęstszą) postać paranoidalno-omamową
Stąd też duże dawki agonistów dopaminergicznych (formalnie Klasyfikacja ICD-10 wyróżnia postać parano-
w przypadku wcześniej istniejącej wrażliwości idalną – przyp. tłum.) ze szczytem zachorowań w czwar-
tej dekadzie życia, w której przebiegu na pierwszym pla-
mogą wyzwolić ujawnienie się objawów schizo- nie występują urojenia i omamy,
frenii, ale nie powodują wyzwolenia się samej cho-
roby. ■ schizofrenię prostą, w której obserwuje się niemal wy-
łącznie objawy osiowe, z trudnym do zidentyfikowania
początkiem, pełzającym przebiegiem i złym rokowa-
Wśród objawów klinicznych schizofrenii wyróżnia niem,
się:
■ hebefrenię, która ujawnia się w wieku młodzieńczym
■ objawy osiowe (objawy negatywne, podstawowe), i której towarzyszą niedorzeczne i błazeńskie zachowa-
nia, spłycenie afektu i odhamowanie,
■ objawy dodatkowe/towarzyszące (objawy pozytyw- ■ postać katatoniczną z dominacją objawów ruchowych,
ne),
■ schizofrenię rezydualną z długo utrzymującymi się obja-
wami negatywnymi.
które w przypadku każdego pacjenta mogą być nasi-
lone w różnym stopniu.
Zaburzenia schizoafektywne. Termin ten dotyczy
epizodycznie pojawiających się zaburzeń, w któ-
Do objawów osiowych – powstających w wyniku
rych przebiegu ujawniają się zarówno objawy psy-
niedoczynności dopaminergicznej układu mezokor-
chotyczne, jak i objawy afektywne (depresyjne i/lub
tykalnego – zalicza się zaburzenia.
maniakalne; zob. poniżej). Tym samym zaburzenia
■ myślenia (m.in. roztargnienie, rozpad, otamowanie te zajmują pośrednie miejsce między schizofreniami
myślenia, zaburzenia toku myślenia) i mówienia a zaburzeniami afektywnymi. W zakresie objawów
MUTSCHLER-2009.indd 162 2010-01-07 22:11:30
Układ nerwowy 163
paranoidalno-omamowych zbliżają się one do schi- wy, a opieka i obchodzenie się z chorym są znacznie
zofrenii, z kolei swym łagodnym przebiegiem bar- ułatwione. Ponadto neuroleptyki działają zapobie-
dziej przypominają zaburzenia afektywne. gawczo na nawroty choroby.
Oprócz działania neuroleptycznego w wąskim
Układ nerwowy
znaczeniu neuroleptyki charakteryzują się – różnie
nasilonym – działaniem uspokajającym i tłumiącym
1.2.1.2. Leki neuroleptyczne
objawy wegetatywne.
(neuroleptyki) Ze względu na strukturę chemiczną i jednocześnie
właściwości farmakologiczne rozróżnia się:
Neuroleptyki, zwane często lekami przeciwpsycho-
■ klasyczne (dawne) neuroleptyki (fenotiazyny, po-
tycznymi, są substancjami, które potrafią – szczegól-
nie u pacjentów z rozpoznaniem schizofrenii – ła-
chodne fenotiazyny, butyrofenony, difenylobutylo- B1
piperydyny),
godzić objawy psychiczne (psychotyczne), bez wy-
wierania istotnego wpływu na świadomość i funkcje ■ tzw. atypowe neuroleptyki (trójcykliczne atypowe
intelektualne. neuroleptyki, benzamidy, risperidon, zyprazydon,
Neuroleptyki tłumią stany pobudzenia psychoru- aripiprazol).
chowego i zmniejszają napięcia afektywne, lęk i po-
strzeganie złudzeniowe. Pomaga to pacjentowi na- Mechanizm działania. Mechanizm działania prze-
brać dystansu do choroby, tzn. jest on teraz w stanie ciwpsychotycznego neuroleptyków został poznany
sam rozpoznać swój stan jako chorobliwy. Nowsze – podobnie jak patogeneza schizofrenii – jedynie
(atypowe, zob. poniżej) neuroleptyki poprawiają po- częściowo. Bezsprzeczna jest jednak to, że neuro-
nadto objawy negatywne. leptyki ingerują w przekaźnictwo synaptyczne i tym
Dzięki leczeniu neuroleptykami stan chorobowy samym wpływają modulująco na współdziałanie róż-
szybko staje się zwykle dla pacjenta mniej dokuczli- nych neuronów.
A
haloperidol
przestrzeń
zewnątrz-
komórkowa
D2,D4 D2 D2 przestrzeń
Gi wewnątrz-
Gi Gi komórkowa
drogi/struktury mezolimbiczne drogi/struktury drogi/struktury
między istotą czarną a prążkowiem guzowo-lejkowe
objawy pozytywne ↓, działania niepożądane pod postacią prolaktyna ↑
nadmierne uspokojenie, apatia zaburzeń pozapiramidowych (mlekotok, brak miesiączki)
B
olanzapina
przestrzeń
zewnątrz-
komórkowa
D2,D4 5-HT2 D2 przestrzeń
Gq wewnątrz-
Gi Gi komórkowa
drogi/struktury mezolimbiczne kora przedczołowa drogi/struktury guzowo-lejkowe
objawy pozytywne ↓ objawy negatywne ↓, prolaktyna ↑
działania niepożądane pod postacią (mlekotok, brak miesiączki)
objawów pozapiramidowych ↓,
zwiększenie masy ciała
Ryc. B 1.2-1. Mechanizm działania i wynikające z niego efekty „klasycznych” oraz atypowych neuroleptyków na przykładzie halope-
ridolu i olanzapiny.
MUTSCHLER-2009.indd 163 2010-01-07 22:11:30
164 Układ nerwowy
Tabela B 1.2-2. Względne powinowactwo neuroleptyków do receptorów in vitro (według Bandelowa)
Neuroleptyk Receptor
D1 D2 D3 D4 5-HT2 M1 α1 H1
amisulprid 0 +++ +++ ? 0 0 0 0
aripiprazol + +++ +++ ++ ++ 0 + +
benperidol 0 +++ ++ ? 0 0 + 0
chloropromazyna + ++ +++ + +++ ++ ++ ++
chlorprotiksen ++ ++ + ? +++ + + +++
klopentiksol ++ +++ ++ ? +++ 0 +++ +++
klozapina ++ + ++ +++ ++ +++ + +++
flufenazyna + +++ +++ + ++ 0 + +
haloperidol + +++ ++ ++ 0 0 + 0
melperon 0 + + ++ +++ 0 + +
olanzapina ++ +++ + ++ +++ ++ ++ ++
perfenazyna + +++ +++ ? + 0 + ++
pimozid 0 +++ +++ + 0 0 0 0
pipamperon 0 + + ? ++ 0 + +
kwetiapina + ++ + 0 ++ 0 +++ +++
risperidon + ++ + + +++ 0 ++ +++
sulpiryd 0 ++ +++ ? 0 0 0 0
tiorydazyna + ++ ++ + ++ +++ ++ +
ziprazydon 0 ++ ++ ++ +++ + ++ +++
zotepina 0 ++ ++ ++ +++ + ++ +++
+++ wysokie powinowactwo, ++ średnie powinowactwo, + nieznaczne powinowactwo, 0 bardzo małe powinowactwo aż do jego braku
Podczas gdy praktycznie już niestosowana jako tycznego (moc neuroleptyczną) na związki o działa-
neuroleptyk rezerpina osłabia zdolność magazynowa- niu słabym, średnio silnym (średnim), silnym (dużej
nia monoamin, a tym samym prowadzi do zubożenia mocy) i bardzo silnym (bardzo dużej mocy). Za sub-
pęcherzyków presynaptycznych w neuroprzekaźnik, stancję o referencyjnym charakterze przyjmuje się
dla większości innych neuroleptyków podstawowym chloropromazynę, której moc działania neuroleptycz-
mechanizmem działania jest hamowanie funkcji re- nego określa się jako 1. W tab. B 1.2-3 zestawiono
ceptorów – szczególnie receptorów dopaminergicz- niektóre neuroleptyki pod względem ich działania
nych. W tab. B 1.2-2 zestawiono powinowactwo do re- neuroleptycznego. W przypadku dawnych (klasycz-
ceptorów różnych neuroleptyków, z których wynika nych) neuroleptyków, a w mniejszym zakresie (lub
profil ich działania i działań niepożądanych. W przy- wcale) atypowych neuroleptyków (zob. poniżej)
padku klasycznych neuroleptyków działanie przeciw- – wraz ze wzrostem siły działania neuroleptycznego
psychotyczne polega przede wszystkim na blokadzie nasilają się pozapiramidowe działania niepożądane
receptorów D2 (i D3), natomiast w przypadku tzw. (zob. poniżej), zwiększa się ich działanie uspokajają-
atypowych neuroleptyków dochodzi do tego bloko- ce, jak również niepożądane objawy ze strony układu
wanie innych receptorów, szczególnie receptorów wegetatywnego.
5-HT2 (zob. ryc. B 1.2-1 A i B).
Należy jednak wskazać, że indywidualna reakcja pacjen-
Siła działania neuroleptycznego. Z klinicznego ta na neuroleptyki jest bardzo różna. Ponadto niezależnie
od mocy neuroleptycznej – działanie przeciwpsychotycz-
punktu widzenia, a także w aspekcie zróżnicowane- ne wszystkich neuroleptyków jest zasadniczo podob-
go działania terapeutycznego, neuroleptyki można ne. Z drugiej jednak strony leki o słabej mocy działania
podzielić ze względu na ich siłę działania neurolep- – w związku z niepożądanymi działaniami wegetatyw-
MUTSCHLER-2009.indd 164 2010-01-07 22:11:30
Układ nerwowy 165
nymi – nie mogą być stosowane w wystarczająco dużych Tabela B 1.2-3. Podział neuroleptyków ze względu na siłę dzia-
dawkach, tak aby móc osiągnąć pełny zakres działania łania neuroleptycznego
przeciwpsychotycznego.
Układ nerwowy
Wskazania. Poza schizofreniami – będącymi głów- I. Neuroleptyki o słabej mocy działania
nym wskazaniem neuroleptyki mogą być stosowane amisulprid
w małych dawkach w stanach lękowych i wzmożo- chloropromazyna
nego napięcia wewnętrznego. W podanych wskaza- chlorprotiksen
niach – ze względu na niewielkie obciążenie substan- lewomepromazyna
cją aktywną – są zwykle dobrze tolerowane. Jednak melperon
z obawy przed wystąpieniem – aczkolwiek rzadkim pipamperon
– objawów pozapiramidowych częściej stosowa- prometazyna B1
ne są leki uspokajające lub leki przeciwdepresyjne. protypendyl
W dalszym ciągu neuroleptyki nadają się do terapii sulpiryd
np. stanów pobudzenia ujawniających się w przebie- tiorydazyna
gu majaczenia. II. Neuroleptyki o średniej mocy działania
Stosowanie neuroleptyków w premedykacji klopentiksol
do znieczulenia ogólnego, jak również w ramach klozapina
neuroleptanalgezji lub neuroleptanestezji zostało perazyna
omówione w rozdziale B 1.7.3.3. Ich zastosowanie ziprazydon
w przypadku wymiotów opisano w rozdziale B 1.11. zotepina
zuklopentiksol
Dawkowanie. Reakcja na neuroleptyki jest różna III. Neuroleptyki o dużej mocy działania
u poszczególnych pacjentów, dlatego dawkę należy
benperidol
dopasować indywidualnie. Również dawki podtrzy-
bromperidol
mujące powinno się ustalać indywidualnie. Z tego flupentiksol
względu dawki podane w tab. od B 1.2-4 do B 1.2-7 flufenazyna
należy traktować jako wartości orientacyjne. fluspirylen
haloperidol
Działania niepożądane. Wśród najważniejszych olanzapina
działań niepożądanych terapii za pomocą neuro- perfenazyna
leptyków należy wymienić przede wszystkim wy- pimozyd
stępujące w przypadku klasycznych neuroleptyków risperidon
zaburzenia pozapiramidowe, a w dalszej kolejności sertindol
zaburzenia ze strony układu wegetatywnego i hor-
monalne oraz reakcje alergiczne. Ponadto dość czę-
sto mogą ujawnić się objawy niepożądane natury
psychicznej.
W przypadku objawów pozapiramidowych wa-
runkowanych blokadą receptorów dopaminowych
w prążkowiu rozróżnia się: ni mimicznych. Dyskinezy wczesne występują pra-
wie wyłącznie w początkowej fazie po rozpoczęciu
■ wczesne dyskinezy,
leczenia neuroleptykami.
■ wywołany neuroleptykami zespół Parkinsona, Ryzyko wystąpienia i nasilenie objawów farma-
kologicznie wyzwolonego zespołu Parkinsona uza-
■ akatyzję,
leżnione jest od rodzaju zastosowanego preparatu (w
■ późne dyskinezy, przypadku klasycznych neuroleptyków czynnikiem
sprzyjającym jest moc ich działania neuroleptyczne-
■ złośliwy zespół neuroleptyczny.
go), od zastosowanej dawki oraz od indywidualnej
predyspozycji samego pacjenta. W opanowywaniu
Wczesne dyskinezy charakteryzują się m.in. takimi wczesnych dyskinez, a także objawów farmakolo-
objawami, jak: niesymetryczne wysuwanie języka, gicznie wyzwolonego zespołu Parkinsona – najle-
porażenie mięśni okoruchowych, wzmożone napięcie piej sprawdzają się preparaty przeciwcholinergiczne.
mięśni karku i szyi oraz hyperkinezy w obrębie mięś- Nie powinno się ich jednak stosować profilaktycznie
MUTSCHLER-2009.indd 165 2010-01-07 22:11:31
166 Układ nerwowy
razem z neuroleptykami, gdyż z jednej strony osła- a także wysoką gorączką. Terapia polega na poda-
biają one działanie neuroleptyków, a z drugiej strony waniu dantrolenu, agonistów dopaminergicznych
– wczesne dyskinezy występują jedynie u części pa- (np. bromokryptyny) lub amantadyny.
cjentów poddanych leczeniu.
Pojęciem akatyzji określa się subiektywnie mę- Działania niepożądane ze strony układu wege-
czące poczucie niepokoju, które związane jest z nie- tatywnego wynikają z zablokowania receptorów
możnością pozostania przez chwilę w spoczynku adrenergicznych i muskarynowych receptorów cho-
– głównie na siedząco. Zwyczajowo akatyzje obser- linergicznych – co ostatecznie powoduje zmniejsze-
wuje się później – już po ujawnieniu się objawów nie przekaźnictwa w obrębie układu współczulnego
zespołu Parkinsona. Ponieważ leki przeciwparkin- i/lub przywspółczulnego. W wyniku zahamowania
sonowe pomagają jedynie w nieznacznym stopniu receptorów α-adrenergicznych mogą ujawnić się:
na objawy akatyzji, konieczne jest dokonanie zmiany stany zamroczenia, zaburzenia regulacji ortosta-
stosowanego neuroleptyku lub też zmniejszenie jego tycznej, wyrównawcza tachykardia oraz zatkany
dawki. Przejściową poprawę można uzyskać, podając nos; efektami działania przeciwmuskarynowego są:
leki z grupy benzodiazepin. Można także spróbować suchość w ustach, zaburzenia akomodacji, zmniej-
podać pacjentowi propranolol. szenie pocenia się, zaparcie i zaburzenia oddawania
Późne dyskinezy charakteryzują się nieprawidło- moczu.
wymi i często stereotypowymi ruchami typowymi dla
zaburzeń hiperkinetycznych – będącymi przeciwień- Wśród zaburzeń hormonalnych będących skut-
stwem hipokinetycznego farmakologicznie wyzwolo- kiem blokady receptorów dopaminergicznych
nego zespołu Parkinsona. Wprawdzie zaburzenia tego w obrębie przysadki mózgowej można wymienić
typu mogą wystąpić we wszystkich grupach pacjen- hiperprolaktynemię, brak miesiączki, zahamowa-
tów, jednakże szczególnie predysponowani do wy- nie owulacji, ginekomastię, jak również osłabienie
stąpienia późnych dyskinez są pacjenci z cechami libido i potencji, a wśród psychicznych objawów
organicznego uszkodzenia mózgu, a także pacjenci niepożądanych – brak napędu, a przede wszystkim
w podeszłym wieku. Szczególne znaczenie w zakre- stany depresyjne.
sie zapobiegania wystąpieniu tych objawów niepo-
żądanych ma stosowanie neuroleptyków w możliwie Dalsze objawy niepożądane zostaną zaprezentowane
niewielkich, ale jeszcze skutecznych dawkach. Te- w trakcie omawiania poszczególnych grup leków.
rapia profilaktyczna za pomocą leków antycholiner-
gicznych stosowana przez długi czas sprzyja powsta- Przeciwwskazania. Neuroleptyki są przeciwwska-
waniu późnych dyskinez. zane w przypadku ostrych zatruć lekami o działaniu
hamującym ośrodkowy układ nerwowy oraz alkoho-
Jako przyczynę ujawnienia się późnych dyskinez przyj- lem.
muje się, że w wyniku blokady postsynaptycznych re-
ceptorów dopaminergicznych dochodzi do zwiększenia
się ilości tych receptorów (tzw. regulacja w górę recep- Interakcje. Neuroleptyki nasilają działanie innych
torów dopaminergicznych). W dalszej kolejności przez leków działających hamująco na ośrodkowy układ
hamowanie presynaptyczne nasileniu ulega uwalnianie nerwowy (np. środków stosowanych do znieczu-
dopaminy z pęcherzyków ją gromadzących. W wyniku lenia ogólnego, leków nasennych, silnie działają-
długotrwałego zażywania neuroleptyków zmniejsza się
również GABA-ergiczna neurotransmisja w obrębie ukła-
cych leków przeciwbólowych lub alkoholu), leków
du pozapiramidowego. antycholinergicznych oraz blokerów receptorów
α- i β-adrenergicznych.
W terapii późnych dyskinez – oprócz powolnego
przerywania terapii – brane może być pod uwagę Zatrucia neuroleptykami. Zatrucia neuroleptyka-
przestawienie na klozapinę oraz podanie tiaprydu. mi objawiają się – w zależności od rodzaju substan-
cji działającej – pod postacią senności, pobudzenia,
Szczególnie niebezpieczny – aczkolwiek rzadko obniżenia lub zwiększenia ciepłoty ciała, ciężkich
występujący – jest złośliwy zespół neuroleptyczny zaburzeń pozapiramidowych, hipotonii, tachykardii
(mimo leczenia ryzyko zejścia śmiertelnego wynosi lub bradykardii, jak również dość często pod postacią
około 20%!). Zaburzenie to charakteryzuje się cięż- napadów uogólnionych drgawek.
kimi zaburzeniami pozapiramidowymi (sztywność W terapii stosuje się antycholinergicznie działają-
mięśniowa – rigor; osłupienie – stupor), zaburze- ce leki przeciwparkinsonowe oraz diazepam – w celu
niami świadomości i zaburzeniami układu krążenia, opanowania drgawek.
MUTSCHLER-2009.indd 166 2010-01-07 22:11:31
Układ nerwowy 167
1.2.1.2.1. Klasyczne neuroleptyki Do tioksantenów zalicza się chlorprotiksen, flupen-
tiksol i zuklopentiksol.
1.2.1.2.1.1. Fenotiazyny
i pochodne fenotiazyny Chlorprotiksen i lewopromazyna – oprócz działania
Układ nerwowy
neuroleptycznego – charakteryzują się ponadto pew-
nym, chociaż niezbyt dużym, działaniem przeciwde-
Pierwsze wprowadzone do terapii neuroleptyki – fe-
presyjnym.
notiazyny oraz ich pochodne – azafenotiazyny i tiok-
santeny składają się z niemal płaskiego układu trzech
pierścieni (trójcykliczne neuroleptyki; zob. tab. B
1.2.1.2.1.2. Butyrofenony
1.2-4). W zależności od łańcucha bocznego wyróżnia
i difenylobutylopiperydyny
się następujące rodzaje fenotiazyn: B1
■ z otwartym łańcuchem bocznym (typu chloropro- Butyrofenony. Opracowane na bazie petydyny leki
mazyny), z grupy butyrofenonów są w większości bardzo sku-
tecznymi neuroleptykami, które pod względem swojej
■ z piperydynoalkilowym łańcuchem bocznym (typ
mocy działania odpowiadają silnie działającym feno-
pekazyny),
tiazynom typu perfenazyny. Szczególne znacznie w tej
■ z piperazynoalkilowym łańcuchem bocznym (typu grupie ma haloperidol (zob. tab. B 1.2-5), którego siła
perfenazyny). działania jest ok. 50 razy większa niż chloropromazy-
ny. Oprócz działania przeciwpsychotycznego haloperi-
Chloropromazyna, najstarszy ze znanych neuro- dol charakteryzuje się silnym działaniem przeciwwy-
leptyków, w dużym stopniu utraciła swoje pierwotne miotnym. Podobnie skuteczny jest także w przypadku
znaczenie. Blokuje rozliczne typy receptorów – stąd schorzeń neurologicznych przebiegających z hiperki-
też wykazuje bardzo szeroki zakres działania: cha- nezami (np. choroba tików, pląsawica). Dalszym wska-
rakteryzuje się ośrodkowym działaniem hamującym zaniem do stosowania haloperidolu są organicznie wa-
i przeciwpsychotycznym, ponadto działa przeciw- runkowane zaburzenia zachowania i stany niepokoju.
wymiotnie, miejscowo znieczulająco, blokuje zwoje Podany doustnie haloperidol jest szybko i całko-
układu wegetatywnego, działa antycholinergicznie, wicie resorbowany z przewodu pokarmowego. Mimo
przeciwadrenergicznie oraz przeciwhistaminowo. to – w związku z efektem tzw. pierwszego przejścia
Chloropromazyna zaburza w dalszej kolejności re- (first pass) – biodostępność leku wynosi jedynie oko-
gulację temperatury ciała poprzez wpływ na ośro- ło 60%. Procent wiązania leku z białkami osocza wy-
dek termoregulacji: przez schłodzenie ciała z ze- nosi 90%. Czas połowiczej eliminacji wynosi 12–35
wnątrz może dojść do hipotermii, z kolei wysoka godzin. W wyniku procesów oksydacji dochodzi
temperatura otoczenia może wywołać hipertermię do odłączenia grupy alkilowej w miejscu atomu azo-
organizmu. tu i powstania kwasu p-fluoro-benzoilo-propionowe-
Innymi związkami podobnymi do chloropromazy- go. Jedynie około 1% leku wydalane jest przez nerki
ny są: lewopromazyna i prometazyna. w niezmienionej postaci.
Lekiem pochodnym jest bromperidol.
Ze związków fenotiazynowych typu pekazyny do- Niektóre inne preparaty (melperon, pipamperon,
stępna w handlu jest jedynie tiorydazyna – neurolep- benperidol) zostały ujęte w tab. 1.2-5.
tyk o słabej mocy działania (i ona jednak – w związ-
ku z niekorzystnym działaniem na serce – została Difenylobutylopiperydyny. Zastępując w butyrofe-
w ostatnim okresie wycofana ze sprzedaży – przyp. nonach tlen w grupie karbonylowej atomem wodoru
tłum.). i resztą p-fluorofenylową, otrzymuje się neuroleptyki
typu difenylobutylopiperydyny, które w porównaniu
Perfenazyna i inne pochodne fenotiazyny (pera- z butyrofenonami wykazują dłuższy czas działania.
zyna, i flufenazyna), z piperazyną jako podstawową Należą do nich stosowany pozajelitowo fluspirylen
substancją czynną, wykazują z reguły większą siłę oraz skuteczny po podaniu doustnym pimozyd (zob.
działania neuroleptycznego niż chloropromazyna. tab. B 1.2-6). Czas działania wynosi w przypadku
pimozydu ok. 24 godzin, w przypadku fluspirylenu
Jedynym przedstawicielem pochodnych azafenotia- – 1 tydzień. Długi czas działania jest uwarunkowany
zyny jest protypendyl. w znacznej mierze powolną biotransformacją i krąże-
niem jelitowo-wątrobowym.
MUTSCHLER-2009.indd 167 2010-01-07 22:11:31
168 Układ nerwowy
Tabela B 1.2-4. Fenotiazyna i neuroleptyki wykazujące pokrewieństwo chemiczne z fenotiazynami
Wzór strukturalny Nazwa Preparaty Okres Średnia
międzynarodowa handlowe półtrwania dawka
(godz.) dzienna (mg)*
l. Fenotiazyny typu chloropromazyny
S
N R1
R2
R1 R2
CH3 prometazyna Atosil, 12 50-150
Promethazine-
H CH2 N -neuraxpharm,
CH3
Proneurin,
CH3 Prothazin, Diphergan
CH3 chloropromazyna Propaphenin ca. 30 75-150 (-500)
Cl N
CH2 CH3
CH3 CH3 lewomepromazyna Levium, 20 30-75 (-300)
Levomepromazin-
OCH3 N -neuraxpharm,
CH2 CH3
Neurocil, Tisercin
II. Fenotiazyny typu pekazyny
S
N R1
R2
R1 R2
tiorydazyna Melleril, 21-24 50-200 (-500)
Thioridazin-
SCH3 CH2 -neuraxpharm
N
CH3
III. Fenotiazyny typu perfenazyny
N R1
R2
R1 R2
perazyna Perazin-neuraxpharm, 8-16 50-150 (-600)
CH3 Taxilan, Perazin,
N
Pernazinum,
N Pernazyna
H CH2
N CH2OH perfenazyna Decentan, 10 8-24 (-64)
Perphenazin-
Cl N -neuraxpharm,
CH2
Trilafon
flufenazyna Dapotum, Fluphenazin- 33 3-6 (-24)
N CH2OH -neuraxpharm D,
N Lyogen, Lyrodin Depot,
CF3
CH2 Mirenil
* Dawki znajdujące się w nawiasach można stosować w trakcie terapii prowadzonej w warunkach stacjonarnych
MUTSCHLER-2009.indd 168 2010-01-07 22:11:31
Układ nerwowy 169
Tabela B 1.2-4. Fenotiazyna i neuroleptyki wykazujące pokrewieństwo chemiczne z fenotiazynami (kontynuacja)
Wzór strukturalny Nazwa Preparaty Okres Średnia
międzynarodowa handlowe półtrwania dawka
Układ nerwowy
(godz.) dzienna (mg)*
IV. Azafenotiazyny
S
N N R1
R2
R1 R2 B1
CH3 protypendyl Dominal 2,5 120-320 (-960)
N
H CH2 CH3
V. Tioksanteny
R1
R2
R1 R2
CH3 chlorprotiksen Chlorprothixen Holsten, 8-12 15-45 (-300)
Chlorprothixen-
Cl CH2 N -neuraxpharm,
CH3
Truxal, Chlorprotixen
CF3 OH flupentiksol Fluanxol, 22-36 1-2 (-6)
N flupendura,
CH2 N Flupenixol
-nauraxpharm
OH zuklopentiksol Ciatyl-Z, 20 10-40 (-150)
Cl N Clopixol,
CH2 N Clopixol,
Acuphase
* Dawki znajdujące się w nawiasach można stosować w trakcie terapii prowadzonej w warunkach stacjonarnych
1.2.1.2.2. Tak zwane neuroleptyki atypowe jątkiem amisulpirydu i prawdopodobnie ziprasidonu;
zob. poniżej) jest znaczące zwiększenie się masy ciała
Atypowe neuroleptyki charakteryzują się tym, u pacjenta.
że w porównaniu ze starszymi neuroleptykami trój- W tab. B 1.2-7 zestawione zostały neuroleptyki
pierścieniowymi, a także butyrofenonami i difenylo- atypowe.
butylopiperydynami, albo prawie wcale nie wywołu-
ją pozapiramidowych objawów niepożądanych, albo
w znacznie mniejszym stopniu, jak również mają 1.2.1.2.2.1. Atypowe neuroleptyki
korzystny wpływ na objawy negatywne. Stąd też po- trójcykliczne
siadają one odmienny i częściowo bardziej korzystny
profil działania – w porównaniu z klasycznymi neu- Substancją prekursorową dla grupy atypowych
roleptykami. Częstym i niejednokrotnie ograniczają- neuroleptyków jest klozapina – pochodna fenotia-
cym długotrwałe stosowanie leku działaniem niepo- zyny w szerokim znaczeniu tego słowa. Wyróżnia
żądanym licznych atypowych neuroleptyków (z wy- się ona tym, że często bywa skuteczna w przy-
MUTSCHLER-2009.indd 169 2010-01-07 22:11:32
170 Układ nerwowy
Tabela B 1.2-5. Butyrofenony
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
R1
O
R2
N
F
R1 R2
melperon Eunerpan, 5 50-200
H CH3 Melneurin,
Melperon-neuraxpharm
Melperon-ratiopharm
halopendol Haldol-Janssen, 12-35 5-10
Cl Haloperidol HEXAL,
OH Haloperidol-neuraxpharm,
Halopendol-ratiopharm,
Haloperidol
Br brompendol Impromen, Tesoprel 36 4-8
OH
O pipamperon Dipiperon, 4 120-360
Pipamperon-n
NH2 N -euraxpharm
NH benpendol Benpendol-neuraxpharm, 8 3-6
H O Glianimon
N
padkach, w których za pomocą innych neurolep- Dawkowanie należy ustalać indywidualnie (ty-
tyków nie można uzyskać zadowalającego efektu. powa dawka początkowa: 12,5–25 mg/dzień; dawki
Klozapina praktycznie nie wywołuje objawów podtrzymujące 200–450 mg/dzień).
pozapiramidowych. Znacząco mocniej oddziałuje Poza typowymi dla innych neuroleptyków przeciw-
na receptory D4 niż na D2, a ponadto silnie hamuje wskazaniami – klozapiny nie można stosować również
działanie receptora 5-HT2, jak również receptorów u pacjentów z zaburzeniami procesu hematopoezy, jak
muskarynowych, adrenergicznych α1 i histami- również – z powodu działania przeciwcholinergiczne-
nowych H1 (zob. tab. 1.2-2). Ponieważ w trakcie go – w atonii pęcherza moczowego i układu pokarmo-
zażywania klozapiny znacznie częściej niż w przy- wego. Przeciwwskazana jest również w ciężkich zabu-
padku innych neuroleptyków ujawnia się działanie rzeniach czynności wątroby i nerek. U pacjentów z ja-
niepożądane pod postacią agranulocytozy (> 1%), skrą z wąskim kątem przesączania w trakcie leczenia
jej stosowanie możliwe jest tylko przy wdrożeniu klozapiną konieczne jest przeprowadzanie regularnych
odpowiednich procedur zabezpieczających (m.in. kontroli ciśnienia śródgałkowego.
pisemnej informacji dla pacjenta, regularnej kon-
troli leukocytozy). Chemicznie spokrewnioną i podobną w zakresie pro-
Klozapina w wysokim odsetku ulega biotransfor- filu powinowactwa do określonych receptorów i kli-
macji za pomocą izoenzymu CYP1A2 cytochromu nicznej skuteczności do klozapiny jest olanzapina.
P-450 i jest wydalana głównie przez nerki; okres Jest dobrze wchłaniana w przewodzie pokarmowym,
półtrwania jest osobniczo zmienny i wynosi około lecz podlega – w wyniku bezpośredniej glukuroni-
6 godzin. zacji oraz utleniania w obrębie izoenzymu CYP1A2
MUTSCHLER-2009.indd 170 2010-01-07 22:11:32
Układ nerwowy 171
Tabela B 1.2-6. Difenylobutylopiperydyny
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
F
F
B1
R
O fluspirylen Fluspi, ca. 300 1,5-2
NH Fluspirylen beta, (co 7 dni)
Imap
N
N
O pimozyd Orap 55 1-2
NH
dziennie
N
oraz (w mniejszym zakresie) CYP2D6 – bardzo wy- działanie neuroleptyczne, ale ma również właściwości
raźnemu efektowi pierwszego przejścia rzędu 40%. przeciwdepresyjne. Nie wykazuje on działania uspo-
Okres półtrwania w osoczu wynosi około 30 godzin; kajającego, lecz wzmaga napęd i poprawia nastrój.
lek wydalany jest głównie przez nerki. W przypadku zażycia doustnego sulpiryd jest
W przypadku podania większych dawek może wchłaniany jedynie częściowo i długo (biodostęp-
dojść do ujawnienia się objawów zespołu Parkinsona ność wynosi około 30%) i lek praktycznie nie ulega
lub akatyzji – jednakże znacząco rzadziej przy terapii procesowi transformacji. Okres półtrwania w osoczu
haloperidolem. Także ryzyko ujawnienia się zmian wynosi 6–8 godzin. Jest wydalany w formie niezmie-
w obrazie krwi jest wyraźnie mniejsze niż w przy- nionej – głównie przez nerki.
padku klozapiny i nie różni się od innych atypowych W dawkowaniu 150–300 mg jest on stosowany
neuroleptyków. w przypadku chorób psychosomatycznych, zabu-
rzeń napędu i afektu, zaburzeń depresyjnych, a tak-
Innymi atypowymi neuroleptykami o podobnych że zawrotów głowy. W dużych dawkach (600–1200
właściwościach są kwetiapina i zotepina. Ostatni mg/dzień) sulpiryd zaleca się w przypadkach ostrych
z wymienionych leków – podobnie jak ziprasidon i przewlekłych schizofrenii.
(zob. poniżej) – wykazuje silne hamowanie wychwy- Różnorodne działanie ujawniające się w przy-
tu zwrotnego noradrenaliny. padku stosowania różnych dawek można wyjaśnić
w ten sposób, że sulpiryd w małych dawkach blokuje
przede wszystkim dopaminergiczne receptory presy-
1.2.1.2.2.2. Benzamidy naptyczne i w ten sposób zwiększa wyrzut dopaminy,
tymczasem w większych dawkach hamuje działanie
Sulpiryd – pochodna benzamidu o strukturze che- receptorów dopaminergicznych zarówno presynap-
micznej całkowicie różniącej się od dotychczas opi- tycznych, jak i postsynaptycznych.
sywanych neuroleptyków, jest wysoce selektywnym Jako działania niepożądane mogą wystąpić: nad-
antagonistą receptorów D2. Wykazuje on nie tylko mierne uspokojenie, zatrzymanie miesiączki i mle-
MUTSCHLER-2009.indd 171 2010-01-07 22:11:32
172 Układ nerwowy
Tabela B 1.2-7. Atypowe neuroleptyki
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
klozapina Clozapin HEXAL, 6 200-450
CH3 Clozapin-neuraxpharm,
N Elcrit, Leponex,
Klozapol
N
N
Cl
NH
olanzapina Zyprexa, 30 5-20
CH3
Zolafren,
N Olzapin,
Zalasta,
N Zolaxa
N
CH3
NH S
O kwetiapina Seraquel, 6 300-600
CH2OH Ketrel
N
N
N
zotepina Nipolept 15 75-300
N(CH3) 2
O
Cl
sulpiryd Dogmatil, 7 600-1200
Sulpirid-neuraxpharm,
O NH
N Sulpirid-ratiopharm,
Sulpirid Stada,
H3CO C2H5 Sulpiryd
SO 2NH2
amisulprid Amisulprid HEXAL, 12-17 400-800
O NH Amisulprid Lich,
N Amisulprid STADA,
H3CO Solian
C2H5
SO 2C2H5
NH2
MUTSCHLER-2009.indd 172 2010-01-07 22:11:33
Układ nerwowy 173
Tabela B 1.2-7. Atypowe neuroleptyki (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
H3C N risperidon Risperdal, Rispolept, 3 (-20) 4-6
Diasperid, Lioxam,
F N Mepharis, Rispen,
N Risperon, Rispofren,
O Risset, Speridan,
Torendo Q-Tab, Ziperid
O N
Cl ziprazidon Zeldox 4-5 80-160
B1
NH
O
N
N
S N
aripiprazol Abilify 75 15-30
O NH O
Cl N
Cl N
kotok, zaburzenia oddawania moczu i zaburzenia do receptorów D2 jest około dziesięciokrotnie słab-
widzenia, reakcje alergiczne oraz bezsenność. Przy sze. Ponadto powoduje on blokowanie receptorów
stosowaniu dużych dawek – podobnie jak w przy- α1 i H1; z kolei pozostaje nieskuteczny w przypadku
padku innych neuroleptyków, choć wyraźnie rzadziej receptorów muskarynowych.
– mogą występować dyskinezy oraz zaburzenia po- Po zażyciu doustnym risperidon jest szybko wchła-
zapiramidowe. niany. Biodostępność leku jest wyraźnie zależna
W przypadku pacjentów z pobudzeniem, padacz- od sprawności metabolizowania leku (polimorfizm
ką i epizodami maniakalnymi – stosowanie sulpirydu izoenzymu CYP2D6). Wynosi ona ok. 80% w przy-
jest przeciwwskazane. padku osób z powolnym metabolizmem leku i około
Zawierające aluminium leki-preparaty przeciw 65% u osób szybko metabolizujących. Jednym z me-
nadkwasocie oraz sukralfat zmniejszają wchłanianie tabolitów jest 9-hydroksy-risperidon, który wykazuje
sulpirydu. te same właściwości farmakodynamiczne co substan-
cja macierzysta (okres półtrwania w osoczu wynosi
Pochodnym lekiem jest amisulprid, którego działa- w przypadku risperidonu – 3 godziny, podczas gdy
nie zostało potwierdzone głównie w zakresie działa- 9-hydroksy-risperidonu – aż 24 godziny). Wydalanie
nia na objawy negatywne. Jak wspomniano powyżej następuje głównie przez nerki.
– w przeciwieństwie do innych atypowych neurolep- Typowe dawkowanie podano w tab. B 1.2-7.
tyków – nie prowadzi on do zwiększenia masy ciała. W przypadku zalecanego dawkowania ryzyko wy-
Jego biodostępność wynosi ok. 50%; czas półtrwa- stąpienia objawów pozapiramidowych jest względ-
nia w osoczu – ok. 17 godzin. Amisulprid wydalany nie niewielkie; w dużych dawkach zwiększa się ono
jest głównie przez nerki. jednak i jest porównywalne z tym u butyrofenonów.
Często obserwowane są: bezsenność, stany lękowe
oraz bóle głowy. Innymi działaniami niepożądanymi
są m.in. nadmierna senność, dolegliwości żołądko-
1.2.1.2.2.3. Risperidon, ziprazidon wo-jelitowe, dysfunkcje seksualne i zaburzenia regu-
lacji ortostatycznej.
Risperidon – pochodna benzisoksazolu – jest silnym W niewydolności wątroby i nerek, w chorobie
antagonistą receptorów 5-HT2A. Jego powinowactwo Parkinsona, guzach wytwarzających prolaktynę oraz
MUTSCHLER-2009.indd 173 2010-01-07 22:11:33
174 Układ nerwowy
chorobach serca i układu krążenia – risperidon nale- 1.2.1.2.3. Neuroleptyki o przedłużonym
ży stosować z wyjątkową ostrożnością. działaniu
Ziprazidon jest farmakologicznie spokrewnioną
Dzięki przemianie klasycznych leków neurolep-
z risperidonem pochodną benzisotiazolu. Poza blo-
tycznych w estry – za pomocą kwasów węglowych
kowaniem receptorów ziprazidon powoduje również
o dłuższych łańcuchach oraz przez specjalne galeno-
zahamowanie wychwytu zwrotnego serotoniny i nor-
we przygotowanie nietypowego neuroleptyku, jakim
adrenaliny ze szczeliny synaptycznej – dzięki czemu
jest risperidon – otrzymano neuroleptyki o długim
ma także działanie przeciwdepresyjne.
(przedłużonym) czasie działania wynoszącym 1–2(–
Biodostępność po doustnym podaniu wynosi 60%,
4) tygodnie. Należą do nich następujące preparaty:
wiązanie z białkami osocza sięga wartości ponad
dekanonian flupentiksolu, dekanonian flufenazyny,
99%, okres półtrwania wynosi 4–5 godzin. W związ-
fluspirylen, dekanonian haloperidolu, enantan perfe-
ku z tym ziprazidon musi być zażywany dwa razy
nazyny, dekanonian zuklopentiksolu.
dziennie.
Działania niepożądane i ograniczenia w zakresie
stosowania są podobne jak w przypadku risperidonu.
1.2.1.2.4. Zalecenia dotyczące kryteriów
stosowania neuroleptyków
1.2.1.2.2.4. Aripiprazol
Celem terapii schizofrenii jest poprawa w zakresie
objawów pozytywnych oraz negatywnych, wraz z po-
Aripiprazol odróżnia się od dotychczas opisanych
prawą rokowań. Główną rolę odgrywa tu stosowanie
atypowych neuroleptyków odmiennym działaniem
neuroleptyków – oprócz koniecznych aktywności
na receptor D2 – działa on bowiem zarówno jako
psychoterapeutycznych (m.in. aktywnego przekazy-
częściowy agonista, jak i antagonista tego recepto-
wania pacjentowi informacji o naturze choroby, po-
ra. Ponadto jest on silnym antagonistą receptorów
mocy w pokonywaniu sytuacji stresowych i dawaniu
5-HT2A. Ponadto nie wykazuje działania przeciw-
sobie rady z chorobą).
cholinergicznego, a jedynie słabe działanie antago-
Neuroleptyki o słabej mocy działania wskazane
nistyczne w obrębie receptorów H1 i α1. Opisany
są w przypadku pobudzenia psychoruchowego i agi-
profil działania wyjaśnia więc, dlaczego aripipra-
tacji lękowej.
zol u pacjentów ze schizofrenią działa w struktu-
Średnio mocno działające neuroleptyki stosowa-
rach mezolimbicznych na skutek zwiększonego
ne są m.in. w schizofreniach hebefrenicznych i schi-
napięcia dopaminergicznego jako antagonista re-
zofrenicznych stanach zejściowych, a także w ostrych
ceptorów D2, podczas gdy w strukturach mezo-
fazach maniakalnych.
kortykalnych z powodu zmniejszonej aktywności
Związki o działaniu silnym i bardzo silnym
dopaminergicznej – jako częściowy agonista, jak
zalecane są do zastosowania w ostrych zespołach
również dlaczego rzadko prowadzi do zwiększenia
psychotycznych, np. zaburzeniach paranoidalnych
masy ciała i jedynie okazjonalnie do zaburzeń or-
i urojeniowo-omamowych, a ponadto w przypadku
tostatycznych.
schizofrenii o przewlekłym przebiegu.
Aripiprazol ulega szybkiemu i dobremu wchła-
W terapii stanów ostrych może być niekiedy wska-
nianiu (biodostępność rzędu 85%), wiązanie z biał-
zane zastosowanie neuroleptyku o dużej mocy dzia-
kami osocza osiąga wartości powyżej 99%, a okres
łania łącznie z preparatem o silnym działaniu uspo-
półtrwania w osoczu wynosi 75%. Głównym meta-
kajającym.
bolitem jest jeszcze częściowo skuteczny dehydro-
W przypadku złego tolerowania klasycznych neu-
aripiprazol.
roleptyków, jak również w przypadku nasilonych
Dawkowanie wynosi 15–30 mg/dzień.
objawów negatywnych oraz przy nowym ustalaniu
W przypadku równoczesnego podania preparatów
leczenia – zaleca się stosowanie atypowych neuro-
hamujących izoenzymy CYP3A4 (np. ketokonazolu)
leptyków.
lub CYP2D6 (np. chinidyny lub fluoksetyny) należy
zmniejszyć dawkę aripiprazolu o połowę, z kolei przy
Neuroleptyki o przedłużonym działaniu w przy-
równoczesnym podawaniu induktorów izoenzymu
padku konieczności stosowania terapii długotermi-
CYP3A4 (np. karbamazepiny, rifampicyny, prepara-
nowej mają tę zaletę, że łatwiejszy jest nadzór nad
tów dziurawca) dawkę należy podwoić.
zażywaniem leku (iniekcja wykonana przez lekarza
bądź też zażycie w obecności lekarza lub personelu
MUTSCHLER-2009.indd 174 2010-01-07 22:11:34
Układ nerwowy 175
medycznego). Wiadomo bowiem, że liczni pacjenci w stanie obniżenia wszystkich aktywności: nic go nie
krótko po wypisaniu z kliniki całkowicie zaprzestają cieszy, ma poczucie beznadziejności, nie ma apetytu,
zażywania neuroleptyków lub też znacznie zmniej- ma problemy ze snem. Dodatkowo dochodzi często
szają dawkę zaordynowanych im leków – co osta- do zahamowania napędu, niejednokrotnie połączone-
Układ nerwowy
tecznie prowadzi do nawrotu choroby. Przy stoso- go z dręczącym poczuciem wewnętrznego niepokoju.
waniu neuroleptyków o przedłużonym działaniu Myślenie jest monotonne i krąży zwykle wokół włas-
możliwe jest zmniejszenie częstotliwości leczenia nego samopoczucia. Pacjent czuje się przy tym do ni-
stacjonarnego. Jednakże w przypadku nieumiejętne- czego niezdolny i bezwartościowy, a wobec bliskich
go przestawienia pacjenta na neuroleptyki o prze- – zdradza poczucie winy.
dłużonym działaniu – zwiększa się ryzyko przedaw- Nie tak rzadko depresja ujawnia się jednak nie tyle
kowania i związanych z nim nasilonych działań nie- w objawach psychicznych, ile w somatycznych, ta-
pożądanych. Ponadto należy uwzględnić, że w razie kich jak poczucie rozbicia, dolegliwości gastryczne, B1
konieczności przerwania dalszego stosowania neu- bóle serca. Taka postać depresji ukryta za objawami
roleptyku – jego działanie i działania niepożądane somatycznymi określana bywa mianem depresji ma-
ustępują powoli. skowanej.
Czas trwania faz wynosi zwykle 3–9 miesięcy; róż-
nice indywidualne są przy tym bardzo duże.
1.2.2. Zaburzenia afektywne Szczególnie znaczące i wymagające poważnego
potraktowania jest niebezpieczeństwo podjęcia próby
samobójczej przez pacjenta z depresją.
W przypadku zaburzeń afektywnych mamy do czy-
nienia z obrazem choroby, na który składają się zmia- Powszechny dawniej podział na różne postacie depresji (or-
ny emocjonalne, nastroju i napędu, mogące wyrażać ganiczne, objawowe, neurotyczne/nerwicowe, reaktywne,
się pod postacią przeciwstawnych sobie – depresji endogenne) został w znacznym stopniu zarzucony, ponie-
(melancholii) lub manii. Zaburzenia afektywne mają waż często nie jest możliwe jednoznaczne różnicowanie.
przebieg fazowy, a remisje mają zwykle charakter peł- W zależności natomiast od liczby i nasilenia objawów roz-
różnia się depresję lekką, średniego stopnia i ciężką. Dla
ny i nie pozostawiają po sobie żadnych utrwalonych różnicującej terapii farmakologicznej znaczenie mają na-
zaburzeń osobowości. W przypadku przebiegów jed- stępujące zespoły depresyjne:
nobiegunowych występują jedynie fazy depresyjne
■ depresja lękowa z pobudzeniem ruchowym, w której do-
(znacznie częściej) lub fazy maniakalne (obecne kla- minują stany niepokoju, lęk i lękliwość,
syfikacje właściwie wykluczają możliwość istnienia
jednobiegunowych manii; manie są uznawane zwy- ■ zespół depresyjny z zahamowaniem napędu i apatią, cha-
rakteryzujący się zmęczeniem, utratą energii i zdolności
czajowo za epizody choroby afektywnej dwubiegu- odczuwania,
nowej – przyp. tłum.); przebiegi dwubiegunowe doty-
czą sytuacji występowania epizodów zarówno depre- ■ depresja urojeniowa (psychotyczna) z urojeniami zubo-
żenia i winy oraz czasem z omamami i myśleniem typu
syjnych, jak i maniakalnych wyraźnie oddzielonych paranoidalnego,
od siebie czasowo (niekiedy jednak przejście między
fazami może być niemal natychmiastowe – tzw. od- ■ depresyjne osłupienie, przy którym pacjent nie jest już
zdolny do kontaktu ze swoim otoczeniem,
wrócenie fazy; zwyczajowo nie ma również równo-
wagi między ilością faz depresyjnych i maniakalnych ■ depresje sezonowe, które występują jedynie w miesią-
– przyp. tłum.). Zaburzenia afektywne zaliczane cach zimowych (między listopadem a marcem) i wynika-
ją ze zmniejszonego nasłonecznienia,
są do najczęściej występujących zaburzeń psychicz-
nych: zachorowalność wynosi powyżej 10%. Choruje ■ depresja starcza, która wyróżnia się występowaniem ob-
prawie dwukrotnie więcej kobiet niż mężczyzn. jawów lęku i paranoidalnych – oprócz zaburzeń wegeta-
tywnych i funkcji poznawczych,
Przez pojęcie depresji rozumie się obniżenie na-
stroju, które jest nieadekwatne do życiowych okolicz- ■ już wcześniej wymieniona depresja maskowana.
ności, a tym samym nieuzasadnione, i które ostatecz-
nie prowadzi do zaburzenia (zahamowania) całej sfe- Mania charakteryzuje się z kolei podwyższonym na-
ry afektywnej. To właśnie odróżnia depresję od smut- strojem, zwiększonym napędem i gonitwą myśli;
ku, który stanowi adekwatną reakcję psychiczną samopoczucie odnośnie do stanu somatycznego
na określone wydarzenie. (Smutek może być jednak jest podwyższone, a sami pacjenci mają tendencję
początkową fazą depresji wyzwolonych lub warun- do przeceniania swoich możliwości. Zdarzają się
kowanych sytuacyjnie bądź tzw. depresji reaktyw- również pacjenci z zachowaniem pretensjonalnym,
nych – przyp. tłum.). W depresji pacjent znajduje się drażliwi i agresywni.
MUTSCHLER-2009.indd 175 2010-01-07 22:11:34
176 Układ nerwowy
Gdy zaburzenie jest jedynie nieznacznie lub Kolejnym mechanizmem działania leków przeciw-
umiarkowanie nasilone – można mówić o zaburze- depresyjnych jest różnie nasilone blokowanie po-
niach typu hipomanii. szczególnych receptorów, zwłaszcza α-adrenergicz-
nych oraz histaminergicznych i dopaminergicznych
(zob. tab. 1.2-8). Mechanizm działania leku określa
1.2.2.1. Leki przeciwdepresyjne w sposób istotny jego profil klinicznego działania
(np. uspokajające działanie wynikające z hamowania
Przez pojęcie leków przeciwdepresyjnych rozumie receptorów H1, przeciwlękowe działanie ujawniające
się substancje czynne, które są w stanie spowodować się na skutek blokady receptorów 5-HT2). Niektóre
poprawę objawów depresji. W zależności od rodza- trójcykliczne leki przeciwdepresyjne (np. amitryp-
ju substancji czynnej wykazują one różnie nasilone tylina) – ale już nie inhibitory wychwytu zwrotnego
działanie w zakresie: – są w stanie również hamować otwieranie kanałów
sodowych.
■ ustępowania objawów depresyjnych i poprawy na-
stroju,
Inhibitory monoaminooksydazy – przez blokowanie
■ pobudzania lub hamowania aktywności psychoru- enzymu MAO – prowadzą do zwiększenia stężenia
chowej. monoamin w obrębie pęcherzyków kolbki presynap-
tycznej komórki nerwowej.
Mechanizm działania. Podobnie jak przy neuro-
leptykach – dokładny mechanizm działania leków Początkowo hamowanie wychwytu zwrotnego adre-
przeciwdepresyjnych nie jest znany. Mimo to istnieją naliny łączono z działaniem polegającym na zwięk-
liczne wyniki badań eksperymentalnych i klinicznych szeniu napędu, podczas gdy hamowanie wychwytu
wykazujących, że leki przeciwdepresyjne ingerują zwrotnego serotoniny miało być odpowiedzialne
w metabolizm neuroprzekaźników, jak również w in- za poprawę (podwyższenie) nastroju. Istnienie
terakcje między neuroprzekaźnikiem a receptorem. jednak takiej korelacji mimo zgodności profilów
Większość leków przeciwdepresyjnych hamuje wy- działania wielu leków przeciwdepresyjnych –
chwyt zwrotny (reuptake) noradrenaliny i/lub seroto- ale nie wszystkich! – z ich szczególnym wpływem
niny ze szczeliny synaptycznej do aksoplazmy (zob. na zwrotny wychwyt jest bardzo wątpliwe. Niektó-
ryc. B 1.2-2). Siła hamowania wychwytu zwrotnego re nowsze leki przeciwdepresyjne mimo zróżnico-
jest różna w przypadku poszczególnych leków (zob. wanego wpływu na zwrotny wychwyt noradrenali-
ryc. B.1.2-3). ny i serotoniny niemal nie różnią się od siebie pod
presynaptyczny neuron noradrenergiczny presynaptyczny neuron serotoninergiczny
MAO-A inhibitor MAO-A
MAO
metabolit NA 5-HT metabolit
NA transporter transporter 5-HT
NA 5-HT
α Gi NRI SSRI
2 Gi T 1D
5-H
wenlafaksyna,
imipramina
α1 β1 5-HT2A/2C
Gq Gs Gq
regulacja aktywności regulacja aktywności
neuronalnej neurony postsynaptyczne neuronalnej
Ryc. B 1.2-2. Mechanizm działania leków przeciwdepresyjnych. SSRI – selektywne inhibitory wychwytu zwrotnego serotoniny;
NRI – inhibitory wychwytu zwrotnego noradrenaliny; 5-HT – serotonina; NA – noradrenalina; MAO – monoaminooksydaza.
MUTSCHLER-2009.indd 176 2010-01-07 22:11:34
Układ nerwowy 177
względem klinicznego działania. Z kolei kilka in-
nych leków, np. mianseryna, praktycznie w ogóle
nie hamuje wychwytu zwrotnego neuroprzekaźni- paroksetyna
ków. Tym samym wpływem na zwrotny wychwyt fluwoksamina
Układ nerwowy
neuroprzekaźników można jedynie częściowo
klomipramina
tłumaczyć efekt przeciwdepresyjny. Twierdzenie
to zdaje się potwierdzać obserwacja, że zahamowa- fluoksetyna
nie wychwytu zwrotnego danego neuroprzekaźnika amitryptylina
ujawnia się w niedługim okresie od podania leku,
podczas gdy efekt przeciwdepresyjny ujawnia się imipramina
dopiero po okresie oczekiwania wynoszącym 2–3 trazodon
tygodnie. Dlatego obecnie przyjmuje się, że w wy- B1
1 10 100 1000 10000
niku wywołanego działaniem leków przeciwdepre-
względna siła działania
syjnych zmienionego stężenia monoamin w szczeli-
nie synaptycznej oraz blokady receptorów zmienia serotonina noradrenalina
się gęstość różnych receptorów dla poszczególnych
neuroprzekaźników (np. regulacja w dół recepto-
Ryc. B 1.2-3. Hamowanie różnymi lekami przeciwdepresyjnymi
rów β i 5-HT2). Zgodnie z tym działanie przeciw-
wychwytu zwrotnego noradrenaliny i serotoniny ze szczeliny
depresyjne należy raczej tłumaczyć regulacyjnym synaptycznej.
wpływem na ośrodkowe noradrenergiczne i seroto-
ninergiczne przekaźnictwo nerwowe.
właściwości polegające na blokowaniu receptorów
Na podstawie opisanych mechanizmów działania określonych neuroprzekaźników (z trójpierścienio-
rozróżnia się: wymi lekami przeciwdepresyjnymi jako najważ-
niejszymi przedstawicielami),
■ nieselektywne inhibitory zwrotnego wychwytu se-
rotoniny/noradrenaliny posiadające dodatkowo ■ nieselektywnych antagonistów receptorów α2,
Tabela B 1.2.-8. Względne powinowactwo leków przeciwdepresyjnych do receptorów in vitro (wybór; zmodyf. według Bandelowa)
Lek Receptor
przeciwdepresyjny
5-HT2A 5-HT2C 5-HT3 α1 α2 H1 D2
amitryptylina +++ ++ + +++ ++ + +
klomipramina + + + ++ + + +
dezipramina + + 0 + + + +
doksepina +++ ++ + ++ +++ + +
fluoksetyna 0 + 0 0 0 0 0
fluwoksamina 0 0 0 0 0 0 0
imipramina + + + + +++ ++ +
maprotylina + + 0 ++ + ++ +
mianseryna +++ +++ + +++ ++ +++ +
mirtazapina +++ +++ +++ + +++ + 0
nortryptylina +++ + ? + +++ ++ +
paroksetyna 0 0 0 0 0 0 0
sertralina 0 0 0 0 0 0 0
wenlafaksyna ? ? ? 0 0 0 ?
+++ wysokie powinowactwo, ++ średnie powinowactwo, + niewielkie powinowactwo, 0 bardzo małe powinowactwo aż do jego braku, ? nie opubliko-
wano danych
MUTSCHLER-2009.indd 177 2010-01-07 22:11:35
178 Układ nerwowy
■ selektywne inhibitory zwrotnego wychwytu seroto- tycholinergiczny – poprzez blokadę cholinergicznych
niny/noradrenaliny (SNRI – serotonin/noradrena- receptorów muskarynowych; ponadto hamują rów-
line reuptake inhibitors), nież receptory α1-adrenergiczne. Tym można tłuma-
czyć część ich działań niepożądanych.
■ selektywne inhibitory zwrotnego wychwytu seroto-
niny (SSRI – selective serotonine reuptake inhibi- Opipramol wykazuje duże powinowactwo do receptorów
tors), sigma i małe powinowactwo do receptorów D2, w ogóle
nie hamuje wychwytu zwrotnego serotoniny i noradrena-
■ (selektywne) inhibitory zwrotnego wychwytu nora- liny.
drenaliny (NRI – noradrenaline reuptake inhibi-
tors),
Początek działania. Poprawa nastroju ze zmniej-
■ inhibitory monoaminooksydazy, szeniem nasilenia objawów depresyjnych ujawnia
się wyraźnie dopiero po 2–3 tygodniach stosowania.
■ pozostałe leki przeciwdepresyjne.
Natomiast objawy antycholinergiczne i antyadrener-
giczne występują szybko i są szczególnie wyraźne
Wskazania. Poza zaburzeniami depresyjnymi – nie-
na początku leczenia.
które leki przeciwdepresyjne stosuje się w przypadku
uogólnionych zaburzeń lękowych, napadów paniki,
Farmakokinetyka. Trójcykliczne leki przeciwde-
w fobiach i zaburzeniach jedzenia. Trójcykliczne
presyjne wchłaniają się szybko i dobrze. Biodostęp-
leki przeciwdepresyjne – w związku z ich zdolnością
ność jest jednak często obniżona w wyniku dużego
blokowania kanałów sodowych – nadają się również
efektu pierwszego przejścia. Ze względu na właści-
do stosowania w terapii bólów neuropatycznych.
wości lipofilne objętość dystrybucji leków jest duża.
W biotransformację leków włączone są liczne pro-
Objawy uboczne, przeciwwskazania i interakcje zo-
cesy biochemiczne, np. N-demetylacja, N-oksydacja,
staną omówione dla poszczególnych grup lekowych.
hydroksylacja pierścieni i łańcuchów bocznych oraz
wiązanie z kwasem glukuronowym. Wydalanie na-
stępuje głównie z moczem.
1.2.2.1.1. Trójcykliczne
leki przeciwdepresyjne
Dawkowanie. Przeciętne dawki dzienne podano
Liczne leki przeciwdepresyjne – podobnie jak fenotiazyny w tab. B 1.2-9.
i tioksanteny – są trójpierścieniowymi difenyloaminowymi
i difenylometanowymi pochodnymi związków podstawo- Działania niepożądane. Z powodu działania anty-
wych. Różnica polega jednak na tym, że ich potrójny pier- cholinergicznego mogą wystąpić: suchość w ustach,
ścień jest wielokątowy. zatkany lub suchy nos, zaburzenia akomodacji, za-
parcia i zaburzenia oddawania moczu. Jeszcze waż-
Do trójcyklicznych (trójpierścieniowych) leków prze- niejsze są zaburzenia sercowo-naczyniowe, takie jak
ciwdepresyjnych należą (zob. tab. B 1.2-9): spadki ciśnienia krwi, tachykardia oraz warunkowane
blokadą kanałów sodowych – zaburzenia przewodze-
■ imipramina,
nia przedsionkowo-komorowego. W przypadku prze-
■ dezipramina, dawkowania może dojść do groźnych, nawet zagra-
żających życiu, zaburzeń rytmu serca (zob. poniżej).
■ trimipramina,
W przypadku pacjentów z istniejącymi już wcześniej
■ klomipramina, objawami uszkodzenia mięśnia sercowego stosowa-
nie trójcyklicznych leków przeciwdepresyjnych po-
■ opipramol,
winno być szczególnie ostrożne.
■ amitryptylina, Ponadto mogą występować zaburzenia dotyczące
samego ośrodkowego układu nerwowego, np. stany
■ amitryptylinoksyd (tlenek amitryptyliny),
pobudzenia i splątania, bezsenność lub też nadmierne
■ nortryptylina, zmęczenie, drżenie mięśniowe (tremor), drgawki pa-
daczkowe, mioklonie, zwiększenie się apetytu i wy-
■ doksepina.
nikające z niego zwiększenie masy ciała.
Poza działaniem na główne objawy depresyjne – trój- Innymi działaniami niepożądanymi są zaburzenia
cykliczne leki przeciwdepresyjne wywołują efekt an- funkcji wątroby (wzrost poziomu transaminaz, chole-
MUTSCHLER-2009.indd 178 2010-01-07 22:11:35
Układ nerwowy 179
Tabela B 1.2-9. Trójcykliczne leki przeciwdepresyjne
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
I. Pochodne dihydrodibenzazepiny
N
R
R B1
CH3 imipramina Imipramin-neuraxpharm, 6-18 50-150
Pryleugan,
N
CH2 CH3 Tofranil
CH3 dezipramina Petylyl 14-25 50-150
N
CH2 H
CH3 CH3 trimipramina Stangyl, 23 50-150
Trimineurin,
N Trimipramin-1A-Pharma,
CH2 CH3
Trimipramin-neuraxpharm
Ia. Substytuowane pochodne dihydrodibenzazepiny
N
R Cl
R
klomipramina Anafranil, Clomipramin-CT, ok. 30 50-150
CH3 Clomipramin-neuraxpharm,
N Clomipramin-ratiopharm,
CH2 CH3 Anafranil SR
II. Pochodne dibenzazepiny
N
R
R
OH opipramol Insidon, Opipramol-HEXAL, 6-9 150-300
N Opipramol-neuraxpharm,
N
Opipramol-ratiopharm, Pramolan,
CH2 Sympramol
III. Pochodne dihydrodibenzocykloheptadienu
R
R
amitryptylina Amineurin, 9-25 50-150
CH3
Amitriptylin beta,
N Amitriptylin-neuraxpharm,
CH CH3 Saroten, Amitryptylinum
MUTSCHLER-2009.indd 179 2010-01-07 22:11:35
180 Układ nerwowy
Tabela B 1.2-9. Trójcykliczne leki przeciwdepresyjne (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
III. Pochodne dihydrodibenzocykloheptadienu
R
R
CH3 amitryptylinoksyd Amioxid-neuraxpharm, 2 60-150
(tlenek amitryptyliny) Equilibrin
N
CH CH3
O
CH3 nortryptylina Nortrilen 15-40 30-150
N
CH H
IV. Pochodne dibenzoksepiny
O
R
R
doksepina Aponal, 8-25 20-150
CH3 Doxepin-neuraxpharm,
N
Doxepin-ratiopharm, Mareen,
CH2
CH3 Doxepin
staza), zaburzenia funkcji płciowych (opóźniony wy- i drgawkami. W ciężkich przypadkach może dojść
trysk, brak orgazmu), a także reakcje alergiczne. do zatrzymania akcji serca lub oddechu. Obraz
zatrucia odpowiada w znacznym stopniu zatru-
Przeciwwskazania. Przeciwwskazaniem do stoso- ciu atropiną. Jako antidotum podaje się dożylnie
wania trójcyklicznych leków przeciwdepresyjnych inhibitor cholinesterazy – salicylan fizostygminy
są stany majaczeniowe, zatrucia alkoholem i lekami w dawce 1–2 mg, który prowadzi do zwiększenia
nasennymi, jak również – z powodu komponenty w organizmie stężenia acetylocholiny – co powinno
przeciwcholinergicznej – jaskra i problemy z odda- być przeprowadzone w warunkach intensywnego
waniem moczu. nadzoru medycznego i pod kontrolą EKG (w razie
potrzeby kolejne dawki mogą być podawane w od-
Interakcje. Przy równoczesnym podawaniu trójcy- stępach 1 godziny). Dodatkowo w terapii tachykar-
klicznych leków przeciwdepresyjnych nasila się dzia- dii i zaburzeń rytmu serca można stosować inhibi-
łanie alkoholu, substancji sympatykomimetycznych tory receptorów β-adrenergicznych; z kolei drgawki
oraz antycholinergicznych; efekty leków sympatyko- mogą być opanowywane za pomocą benzodiazepin,
mimetycznych (adrenolitycznych) ulegają natomiast np. diazepamu.
osłabieniu.
Zatrucia. Zatrucia z użyciem trójcyklicznych le- 1.2.2.1.2. Czterocykliczne
ków przeciwdepresyjnych objawiają się groźnymi leki przeciwdepresyjne
zaburzeniami sercowo-naczyniowymi (duże spadki
ciśnienia krwi, tachykardia, zaburzenia rytmu ser- Do czterocyklicznych (czteropierścieniowych) leków
ca), a także hipertermią, stanami majaczeniowymi przeciwdepresyjnych należą (zob. tab. B 1.2-10):
MUTSCHLER-2009.indd 180 2010-01-07 22:11:36
Układ nerwowy 181
Tabela B 1.2-10. Czterocykliczne leki przeciwdepresyjne
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
mianseryna Mianserin, 10-40 30-90
Mianserin-neuraxpharm,
Mianserin-ratiopharm, Tolvin,
N H Deprexolet, Lerivon,
Miansemerck, Norserin
N
CH3
B1
mirtazapina Mirtazapin-neuraxpharm, 20-40 15-45
Mirtazapin-ratiopharm,
N Mirtazapin STADA, Remergil,
N H Esprital, Martastad,
Mirtazapine Teva,
N Mirzaten, Remeron
CH3
maprotylina Ludiomil, ok. 40 75-150
Maprolu,
Maprotilin-neuraxpharm,
Maprotilin-ratiopharm
CH3
NH
■ maprotylina, mianseryny – 10–40 godzin, a mirtazapiny – 20–40
godzin. W związku z ujawniającym się na początku
■ mianseryna,
stosowania efektem tłumiącym – czterocykliczne leki
■ mirtazapina. przeciwdepresyjne mogą być stosowane w depre-
sjach z lękiem i pobudzeniem. Ich pozostałe właści-
Profil działania maprotyliny podobny jest do pro- wości odpowiadają w szerokim zakresie lekom trój-
filu trójcyklicznych leków przeciwdepresyjnych, jed- cyklicznym, choć ich działanie antycholinergiczne
nakże lek hamuje wychwyt zwrotny głównie nora- jest wyraźnie mniej nasilone. Jednakże w przypadku
drenaliny. Z kolei mianseryna działa przede wszyst- stosowania mianseryny, lecz nie mirtazapiny, częściej
kim przez hamowanie presynaptycznych receptorów niż przy innych lekach przeciwdepresyjnych opisy-
α2-adrenergicznych i wynikające z tego nasilenie wy- wane było ryzyko wystąpienia agranulocytozy i ane-
rzutu noradrenaliny i serotoniny. Ponadto w słabym mii aplastycznej; z kolei w przypadku maprotyliny
stopniu hamuje wychwyt zwrotny serotoniny i działa istnieją wzmianki o podwyższonym ryzyku wyzwo-
jako bloker receptorów 5-HT2, 5-HT3 i H1. Mirta- lenia drgawek.
zapina – pirydylowa pochodna mianseryny – od-
różnia się od substancji macierzystej jeszcze silniej-
szym blokowaniem presynaptycznym receptorów α2 1.2.2.1.3. (Selektywne) inhibitory
i 5-HT3, brakiem hamowania wychwytu zwrotnego wychwytu zwrotnego serotoniny/
serotoniny i wyraźnie słabszym działaniem antagoni- noradrenaliny
stycznym w obrębie receptora H1. (SNRI = serotonin/noradrenalin
reuptake inhibitors)
Maprotylina ulega powolnej, ale całkowitej re-
sorpcji. Biodostępność mianseryny wynosi natomiast Do tej grypy leków przeciwdepresyjnych należą
jedynie 20–30%, a mirtazapiny 50%. Demetylowane wenlafaksyna oraz duloksetyna, które podobnie
metabolity wszystkich trzech substancji oraz tlenek jak leki trójcykliczne, hamują wychwyt zwrotny
N-maprotyliny są nadal czynne farmakologicznie. serotoniny i noradrenaliny, jednak nie posiadają
Podawane w literaturze okresy półtrwania w osoczu żadnego istotnego powinowactwa do receptorów
wynoszą w przypadku maprotyliny ok. 40 godzin, adrenergicznych, cholinergicznych czy histamino-
MUTSCHLER-2009.indd 181 2010-01-07 22:11:36
182 Układ nerwowy
1.2.2.1.4. Selektywne inhibitory wychwytu
zwrotnego serotoniny
N(CH3) 2 (SSRI = selective serotonin
OH reuptake inhibitors)
H3CO Lekami przeciwdepresyjnymi selektywnie hamują-
cymi wychwyt zwrotny serotoniny są (zob. tab. B
wenlafaksyna 1.2-11):
■ citalopram,
■ fluoksetyna,
■ fluwoksamina,
O NHCH3
■ paroksetyna,
S
■ sertralina.
duloksetyna Ich zakres działania jest bardzo podobny; wszystkie
pozbawione są nadmiernego działania uspokajające-
go, lecz działają raczej aktywizująco. Różnice far-
makokinetyczne polegają na tym, że poszczególne
wych. Ujawnienie się działania klinicznego – przy selektywne inhibitory zwrotnego wychwytu seroto-
zastosowaniu dużej dawki początkowej – następuje niny z różną siłą hamują poszczególne izoenzymy
nieco szybciej niż w przypadku trójcyklicznych le- cytochromu P-450. Najsłabsze działanie blokujące
ków przeciwdepresyjnych. wykazują citalopram oraz escitalopram.
Obie substancje czynne są w dużym odsetku re- Leki z grupy SSRI są szybko i dobrze wchłaniane
sorbowane, jednakże znacząca część leków ulega – ulegając silnej biotransformacji (w części do ak-
metabolizowaniu już w wyniku pierwszego przejścia tywnych metabolitów). Średnie okresy osoczowego
przez wątrobę (biodostępność wenlafaksyny wynosi półtrwania wynoszą w przypadku citalopramu – 33–
zaledwie 12%, duloksetyny 30–80%). Wenlafaksyna 37 godz., escitalopramu – 22–32 godz., fluoksetyny
ulega biotransformacji głównie w obrębie izoenzymu – 4 dni (a jej aktywnego metabolitu norfluoksetyny
CYP2D6; dulokesetyna – również w obrębie izoen- – 7 dni), fluwoksaminy – 15, paroksetyny 24 i ser-
zymu CYP2D6, a ponadto przez izoenzym CYP1A2 traliny – 26 godz. Wydalanie następuje głównie przez
– do metabolitów zachowujących częściową aktyw- nerki.
ność farmakologiczną. Okresy półtrwania w osoczu Ze względu na korzystne właściwości terapeutycz-
wynoszą dla wenlafaksyny 5, dla O-demetylowanej ne SSRI są często stosowane w terapii zaburzeń de-
wenlafaksyny – 11, a dla duloksetyny 11–16 godzin. presyjnych (fluoksetyna jest najczęściej stosowanym
Wydalanie leku następuje głównie przez nerki – pod na świecie lekiem przeciwdepresyjnym).
postacią metabolitów. Typowe dawkowanie podano w tab. B 1.2-11.
W przypadku wenlafaksyny pacjenci leczeni w wa- Wśród innych działań niepożądanych należy wy-
runkach ambulatoryjnych otrzymują dwa razu dzien- mienić: zaburzenia snu, zawroty głowy, drżenie mięś-
nie 37,5–75 mg postaci zwykłej leku lub raz dzien- niowe, nadmierne pocenie się, zmianę fazy w epizod
nie 75–100 mg postaci o przedłużonym działaniu; maniakalny (rzadko) oraz dość często ujawniające się
pacjenci leczeni stacjonarnie otrzymują początkowo zaburzenia żołądkowo-jelitowe (nudności, wymioty).
trzy razy dziennie 50 mg, przy czym maksymalna Objawy ze strony serca i układu krążenia są natomiast
dawka dzienna wynosi 375 mg. Duloksetyna jest po- – w porównaniu z trójcyklicznymi lekami przeciwde-
dawana dziennie w dawce 60 mg. presyjnymi – rzadkie. W przypadku paroksetyny ist-
Jako działania niepożądane mogą wystąpić m.in.: nieją pojedyncze doniesienia o ciężkich zaburzeniach
nudności, zawroty głowy, bezsenność, drżenie mięś- funkcji wątroby.
niowe, nadmierna nerwowość, zaparcia, zwiększone Lekki SSRI podwyższają stężenie osoczowe sto-
pocenie się i zaburzenia wytrysku. sowanych równocześnie trójcyklicznych leków prze-
Równoczesne zażywanie inhibitorów monoamino- ciwdepresyjnych, haloperidolu, fenytoiny, karbama-
oksydazy jest przeciwwskazane. zepiny, diazepamu lub soli litu.
MUTSCHLER-2009.indd 182 2010-01-07 22:11:36
Układ nerwowy 183
Tabela B 1.2-11. Selektywne inhibitory wychwytu zwrotnego serotoniny
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
F citalopram Cipramil, 33-37 20
Citalopram HEXAL,
Citalopram-ratiopharm,
Citalopram STADA, Aurex,
CH 3 Cilon, Citabax, Cital, Citalec,
N Citaratio, Citaxin, Oropram 22-32 10
O CH 3
NC escitalopram Cipralex, Lexapro B1
O NH fluoksetyna Fluctin, Fluoxetin beta, 96 20-60
CH 3 Fluoxetin-neuraxpharm,
Fluoxetin-ratiopharm, Andepin,
F3C Apo-Flox, Fluoksetyna, Bioxetin,
Deprexetin, Prozac, Salipax,
Seronil
O
N NH 2
OCH 3 fluwoksamina Fevarin, 15 100-300
Fluvo HEXAL,
F3C Fluvoxamin-neurax
F O O
O
paroksetyna Paroxat, Paroxetin beta, 24 20-40
Paroxetin-ratiopharm,
NH Seroxat, ParoMerck,
Paxeratio, Paxtin,
Cl Rexetin, Xetanor
Cl sertralina Gladem, Sertralin-CT, 26 50
CH 3 Sertralin-ratiopharm, Zoloft,
NH Asentar, Asertin, Luxeta,
Sertagen, SertaHEXAL, Sertiva,
Setaloft, Setaratio, Stimuloton,
Zotral
Równoczesne stosowanie selektywnych inhibito- nym inhibitorem wychwytu zwrotnego noradrenaliny
rów wychwytu zwrotnego serotoniny z inhibitorami jest pochodna morfoliny – reboksetyna – stosowana
monoaminooksydazy (zob. poniżej) jest przeciw- przede wszystkim u pacjentów z depresją z zahamo-
wskazane z powodu zagrożenia toksycznym wzro- waniem psychoruchowym.
stem stężenia serotoniny. Podana doustnie reboksetyna wchłania się szybko
i w ponad 90%. Wiązanie z białkami jest wysokie i wy-
nosi około 95%. Substancja czynna metabolizowana
1.2.2.1.5. (Selektywne) inhibitory wychwytu jest w obrębie izoenzymu CYP3A4 do nieaktywnych
zwrotnego noradrenaliny metabolitów (głównie w procesach O-dealkilowania
(NRI = noradrenalin reuptake i hydroksylacji). Okres półtrwania w osoczu wynosi
inhibitors) 12–14 godz. Wydala się głównie przez nerki.
Zwykłe dawkowanie wynosi dwa razy dziennie po
Także za pomocą substancji, które hamują wyłącznie 4 mg.
wychwyt zwrotny noradrenaliny można uzyskać efekt Działaniami niepożądanymi są bezsenność, za-
przeciwdepresyjny, który w dużym stopniu odpowia- wroty głowy, suchość w ustach, nadmierne pocenie
da równoczesnej blokadzie wychwytu zwrotnego się, tachykardia, problemy z oddawaniem moczu i za-
serotoniny i noradrenaliny. Tego rodzaju selektyw- burzenia potencji.
MUTSCHLER-2009.indd 183 2010-01-07 22:11:37
184 Układ nerwowy
Przeciwwskazane jest równoczesne stosowanie Wskazaniem do ich stosowania są depresje z zaha-
reboksetyny z lekami o wąskim zakresie działania te- mowaniem i fobie społeczne, a ponadto – oporność
rapeutycznego, które metabolizowane są za pomocą na leczenie trójcyklicznymi lekami przeciwdepresyj-
izoenzymów CYP3A4 lub CYP2D6 (np. leki prze- nymi.
ciwarytmiczne, trójcykliczne leki przeciwdepresyj- Średnie dawki dzienne tranylcyprominy wynoszą
ne) lub też z inhibitorami monoaminooksydazy. 10–20 mg, a moklobemidu 300–600 mg.
Wśród działań niepożądanych stosowania obu le-
ków należy wymienić zaburzenia snu, nudności i bóle
głowy. W trakcie stosowania tranylcyprominy mogą
dodatkowo ujawniać się pojedyncze przypadki wy-
stąpienia omamów, drgawek, zapaleń wątroby, zabu-
OC2H5 rzeń obrazu krwi oraz dolegliwości żołądkowo-jeli-
O towych. W przypadku leczenia moklobemidem dość
H często występuje suchość w ustach.
H
O W zagrożeniu próbą samobójczą, ostrych stanach
NH splątaniowych oraz u pacjentów z guzem chromo-
chłonnym oraz tyreotoksykozą – stosowanie MAO-I
reboksetyna
jest przeciwwskazane. Nie powinny być one stosowa-
ne również z petydyną lub klomipraminą.
Powyżej wspomniano o istnieniu interakcji mię-
dzy tranylcyprominą a środkami spożywczymi za-
wierającymi tyraminę. Równoczesne stosowanie tra-
nylcyprominy z amfetaminami lub innymi pośredni-
mi sympatykomimetykami również może prowadzić
1.2.2.1.6. Inhibitory monoaminooksydazy do ciężkich incydentów zdrowotnych (zaburzenia
Inhibitory monoaminooksydazy (oksydazy mono-
aminowej) hamują proces oksydatywnej degradacji
monoamin. W wyniku hamowania enzymu zwiększa
się synaptyczne stężenie dopaminy, noradrenaliny,
adrenaliny i serotoniny w mózgu.
Oprócz nieselektywnej tranylcyprominy, hamu- NH2
jącej nieodwracalnie zarówno monooksydazę ty-
pu A, jak i B, stosowany jest również selektywny
tranylcypromina
odwracalny inhibitor monoaminooksydazy A – mo-
klobemid. Zaletą tego drugiego leku – w porównaniu
z trancylprominą – jest to, że równoczesne spożycie
pożywienia bogatego w tyraminę (np. sera) nie pro-
wadzi do ciężkich interakcji (wzrostu ciśnienia, kryzy O O
hipotonicznej). Związane jest to z tym, że do degra- NH
N
dacji tyraminy wystarczająca jest jeszcze niezabloko-
wana monoaminooksydaza B, a ponadto moklobemid Cl
może być wyparty z wiązania z monaminooksyda- moklobemid
zą A pod wpływem wysokich stężeń tyraminy (ty-
ramina jest zwiększającą ciśnienie substancją sym-
patykomimetyczną). Stąd też – w przeciwieństwie
do terapii tranylcyprominą – stosowanie moklobemi-
du nie wymaga utrzymywania specjalnej diety.
Tranylcypromina i moklobemid są dobrze i szybko rytmu serca, kryzy hiper- lub hipotoniczne, hiperter-
wchłaniane; w dużym odsetku ulegają jednak bio- mia). W przypadku równoczesnego spożycia alko-
transformacji. Okres półtrwania w osoczu wynosi holu lub preparatów zawierających rezerpinę mogą
w przypadku tranylcyprominy 1,5–2,5 godz., w przy- czasem wystąpić reakcje nietolerancji. Równoczesne
padku moklobemidu – 1–3 godz. Wydalanie następu- użycie tranylcyprominy z preparatami zawierającymi
je głównie przez nerki. tryptofan grozi wystąpieniem objawów majaczenia.
MUTSCHLER-2009.indd 184 2010-01-07 22:11:37
Układ nerwowy 185
1.2.2.1.7. Pozostałe leki przeciwdepresyjne
1.2.2.1.7.1. Trazodon O
N N N
Układ nerwowy
Triazolopirymidynowa pochodna trazodon jest selektyw-
nym, lecz jedynie słabo działającym inhibitorem wychwytu N N Cl
zwrotnego serotoniny. Ponadto blokuje on receptory 5-HT2,
z czym można wiązać jego działanie uspokajające.
Trazodon po zażyciu doustnym jest szybko i całkowicie
wchłaniany, ulegając niemal całkowitej biotransformacji trazodon
w wątrobie – do częściowo aktywnych metabolitów. Okres
półtrwania w osoczu wynosi około 6 godz. Wydalanie na-
stępuje głównie przez nerki. B1
Wskazaniem są zespoły lękowo-depresyjne.
Średnia dawka dzienna wynosi 200–400 mg.
Wśród objawów niepożądanych opisano uczucie zmę- Typowe dawkowanie przy stosowaniu jako lek
czenia, bóle głowy i zawroty, zaburzenia ortostatyczne, przeciwdepresyjny wynosi co najmniej 600–900 mg
drgawki (rzadko), jak również szczególnie znaczące powi- ekstraktu dziennie.
kłanie, jakim jest priapizm (długotrwały bolesny wzwód).
Przeciwwskazaniami są ostre zatrucia alkoholem, leka-
Korzystne jest ogólne dobre tolerowanie wyciągu
mi przeciwbólowymi i lekami psychotropowymi, jak rów- przez pacjentów. W wyniku działania niepożądanego
nież zespół rakowiaka i zdekompensowana niewydolność może dojść do uczulenia na światło (zwłaszcza u pa-
serca. cjentów o jasnej karnacji); ponadto mogą się ujawnić
alergiczne reakcje skórne oraz dolegliwości żołądko-
wo-jelitowe.
1.2.2.1.7.2. Dziurawiec Klinicznie znaczące, częściowo mogące zagrażać
życiu interakcje polegają na tym, że w przypadku
Wyciąg z dziurawca należy do najczęściej stosowa- równoczesnego spożywania wyciągu z dziurawca
nych preparatów przeciwdepresyjnych w przypadku z lekami przeciwzakrzepowymi (np. fenprokumo-
lekkich (do średnio nasilonych) depresji. Ponadto sto- nem), innymi lekami przeciwdepresyjnymi (ami-
sowany bywa w samodzielnym leczeniu dolegliwości tryptyliną, nefazodonem, nortryptyliną, paroksety-
psychowegetatywnych, nerwowego niepokoju i lęku. ną, sertraliną), cyklosporyną, digoksyną, indinawi-
Mimo licznych badań nie udało się jednoznacznie rem i innymi inhibitorami proteaz, doustnymi środ-
wyjaśnić, które z substancji zawartych w wyciągu kami antykoncepcyjnymi – dochodzi do osłabienia
odpowiadają za efekt przeciwdepresyjny. Hypery- działania tych preparatów! Efekt ten wynika przede
cyna, według której początkowo standaryzowano wszystkim z indukcji izoenzymu CYP3A4 oraz gli-
wyciąg dziurawca, nie jest w sposób miarodajny od- koproteiny P.
powiedzialna za efekt przeciwdepresyjny. Natomiast
hyperforyna – w stężeniu możliwym do osiągnięcia
również w warunkach in vivo – wykazuje efekt niese-
lektywnego hamowania wychwytu zwrotnego seroto-
niny, noradrenaliny, dopaminy i GABA, jak również
– chociaż w mniejszym stopniu – kwasu glutamino-
wego. Bardzo prawdopodobne jest, że w wyciągu H3C CH3
znajdują się inne, dotychczas niezidentyfikowane
związki, które mogą być odpowiedzialne za efekt te-
rapeutyczny. O OH
Kwestią bardzo problematyczną jest to, że obok
H3C CH(CH3) 2
dobrze zbadanych i w odpowiedniej ilości dawkowa-
nych preparatów występują w handlu takie (i często O O
CH3 CH
3
sprzedawane są poza aptekami), które zawierają bar- CH3
dzo różne ilości wyciągu, a ich skuteczność nie zo- H3C H3C CH3
stała udokumentowana. Ponadto należy podkreślić,
hiperforyna
że potencjalnie zagrażające życiu schorzenie, jakim
jest depresja, nie powinno być leczone samodzielnie
przez pacjenta, a stosowanie wyciągu z dziurawca
w ciężkich depresjach jest przeciwwskazane.
MUTSCHLER-2009.indd 185 2010-01-07 22:11:38
186 Układ nerwowy
1.2.2.1.8. Zalecenia dotyczące kryteriów lub neuroleptyk o słabej mocy działania; z kolei w za-
stosowania leków burzeniach depresyjnych z objawami psychotycznymi
przeciwdepresyjnych konieczne jest dodatkowe włączenie neuroleptyku.
Ważnym zaleceniem jest stosowanie leków prze-
ciwdepresyjnych przez wystarczająco długi okres
Celem terapii przeciwdepresyjnej jest osiągnięcie
– aż do całkowitego ustąpienia objawów chorobo-
zadowalającego efektu terapeutycznego, tzn. opano-
wych (remisji). W zakresie terapeutycznej skutecz-
wanie lub przynajmniej zmniejszenia nasilenia symp-
ności nadal nic nie może prześcignąć klasycznych
tomatyki depresyjnej, poprawa umiejętności pacjenta
trójcyklicznych leków przeciwdepresyjnych, jednak
w pokonywaniu codziennych zadań oraz zapobiega-
nowe związki są zwykle lepiej tolerowane – stąd też
nie nawrotowi. Cel ten jest możliwy do osiągnięcia
są obecnie znacznie częściej stosowane. W przypad-
jedynie po zastosowaniu całej koncepcji terapeutycz-
ku oporności na leczenie zalecane jest sprawdzenie
nej, która musi obejmować – oprócz obowiązkowej
poziomu (stężenia) leku w osoczu; do rozważenia po-
farmakoterapii prowadzonej za pomocą leków prze-
zostaje zmiana rodzaju stosowanego leku.
ciwdepresyjnych oraz procedur psychoterapeutycz-
Terapia podtrzymująca stan remisji powinna być
nych – także metody fizykalne (np. terapię światłem,
prowadzona przez okres 6–12 miesięcy; z kolei pro-
elektrowstrząsy) oraz deprywację snu.
filaktyka nawrotu w przypadku powtarzających się
Właściwe zastosowanie leków przeciwdepresyj-
faz depresyjnych powinna być kontynuowana przez
nych wymaga wiedzy dotyczącej zakresu ich dzia-
co najmniej 5 lat.
łania. Wybór preparatu jest uzależniony od rodzaju
najważniejszych objawów stwierdzanych u pacjenta.
Na ich podstawie można wyróżnić dwa podstawowe
zespoły kliniczne z objawami lękowo-depresyjnymi 1.2.2.2. Leki stosowane
z pobudzeniem (agitacją) oraz objawami depresyjny- w profilaktyce choroby
mi z zahamowaniem. W tab. B 1.2-12 zestawiono leki afektywnej oraz w terapii manii
przeciwdepresyjne zalecane do stosowania w obu
głównych postaciach zaburzeń depresyjnych. W celu 1.2.2.2.1. Sole litu
opanowania lęku można włączyć – na możliwie krót-
ki okres – leki uspokajające z grupy benzodiazepiny Sole litu (np. octan, węglan litu), które oprócz karba-
mazepiny (zob. poniżej) zaliczane są do grupy tzw.
stabilizatorów nastroju (mood stabilizers), nadają się
do stosowania zarówno w profilaktyce psychoz afek-
Tabela B 1.2-12. Określone (celowane) zespoły depresyjne,
tywnych (choroby afektywnej dwubiegunowej), jak
w których stosuje się odpowiednie leki przeciwdepresyjne (wy-
bór; zmodyf. według Benkerta i Hippiusa)
neuroprzekażnik-
Leki przeciwdepresyjne dla -receptor
zespołów depresyjnych zespołów depresyjnych
z zahamowaniem z pobudzeniem PIP PIP2 PLC Gq
psychoruchowym psychoruchowym i lękiem PI
dezipramina amitryptylina DAG
inozytol
fluoksetyna amitryptylinoksyd
IP3 aktywacja
fluwoksamina doksepina kinazy
IP1 IP2 białkowej C
imipramina maprotylina
moklobemid mianseryna lit mobilizacja
wewnątrzkomórkowego
nortryptylina trazodon lit wapnia Ca2+
paroksetyna trimipramina
Ryc. B 1.2-4. (Częściowy) mechanizm działania jonów litu; PLC
tranylcypromina – fosfolipaza C, PI – fosfatydyloinozytol, PIP – monofosforan fo-
sfatydyloinozytolu, PIP2 – 4,5-difosforan fosfatydyloinozytolu,
wenlafaksyna IP3 – trifosforan inozytolu, IP2 – difosforan inozytolu, IP1 – mo-
nofosforan inozytolu, DAG – diacylglicerol.
MUTSCHLER-2009.indd 186 2010-01-07 22:11:38
Układ nerwowy 187
i zaburzeń schizoafektywnych, przy czym muszą być podawaniu leczniczym już po 2–3 dniach. Dawka
one stosowane przez dłuższy okres, a także w terapii podtrzymująca uzależniona jest od osiągniętego stę-
faz maniakalnych. Na ujawnienie się ich efektu tera- żenia w osoczu.
peutycznego trzeba czekać nieco ponad 1 tydzień.
Układ nerwowy
Działania niepożądane. Nawet w przypadku do-
Mechanizm działania. Podobnie jak w przypadku in- kładnego ustalenia poziomu litu w osoczu – zgodnie
nych leków psychotropowych – mechanizm działania z powyższymi zaleceniami – na początku leczenia
jonów litu znany jest jedynie częściowo. Udowodnio- mogą ujawnić się działania niepożądane pod posta-
no, że jony litu ingerują w metabolizm fosfatydylo- cią nudności, dolegliwości żołądkowo-jelitowych,
inozytolu (PI-turnover; zob. ryc. 1.2-4). Prowadzą osłabienia mięśniowego oraz drobnofalistego drżenia
w ten sposób do zablokowania fosfatazy 1-polifosfo- mięśniowego (tremor), które mogą być opanowane
rano-inozytolowej oraz fosfatazy monofosforano-ino- za pomocą nieselektywnych inhibitorów receptorów B1
zytolowej – chroniąc tym samym przed oderwaniem β-adrenergicznych. W przypadku długotrwałej terapii
się ostatniej reszty fosforanowej od cząsteczki inozy- często obserwuje się u pacjentów przyrost masy ciała,
tolu. Powoduje to niewystarczający dostęp do inozy- jak również powstanie wola (zob. mechanizm dzia-
tolu – potrzebnego do budowy fosfolipidu będącego łania), które może być dobrze opanowane za pomo-
składnikiem błony komórkowej – 4,5-difosforanu- cą hormonów tarczycy. W dalszej kolejności mogą
fosfatydyloinozytolu (PIP2). Tym samym działanie ujawnić się zmiany skórne, zaburzenia w EKG oraz
neuroprzekaźników – wykorzystujące metabolizm EEG.
fosfatydyloinozytolu – może ulec niekompetycyjne-
mu osłabieniu. Ponadto jony litu osłabiają induko- Przeciwwskazania. W przypadku niewydolności ne-
wane tyreotropiną i adiuretyną powstawanie cAMP rek, ciężkich chorób serca i układu krążenia, choroby
oraz wpływają na dobowy rytm aktywności różnych Addisona i zaburzeń gospodarki sodu – stosowanie
receptorów. soli litu jest przeciwwskazane. Także w czasie ciąży
nie powinny być one stosowane, gdyż zwiększają ry-
Farmakokinetyka. Doustnie podane jony litu są do- zyko powstania wad rozwojowych płodu.
brze wchłaniane. Przenikalność litu do wnętrza komó-
rek odpowiada przenikalności jonów sodu, natomiast Interakcje. Saluretyki podwyższają – w wyniku
jony litu wykazują mniejsze powinowactwo do pomp zmniejszonego wydalania – poziom litu w osoczu
jonowych – stąd też znacznie trudniej podlegają (ak- i tym samym zwiększają niebezpieczeństwo zatrucia
tywnemu) przemieszczaniu na zewnątrz komórek. litem. Również liczne niesteroidowe leki przeciwreu-
Dlatego gromadzą się w przestrzeniach wewnątrz- matyczne, np. diklofenak, ibuprofen lub indometacy-
komórkowych. Ich wydalanie następuje głównie na, ale już nie kwas acetylosalicylowy (!), a także inhi-
przez nerki. Współczynnik wchłaniania zwrotnego bitory acetylocholinoesterazy zmniejszają klirens litu.
w układzie kanalików nerkowych zależy od zawarto- Natomiast acetazolamid zwiększa nerkowe wydalanie
ści sodu w moczu. W przypadku wysokiego stężenia jonów litu i w ten sposób osłabia ich działanie.
jonów sodu wydalanie litu również jest większe; przy
niskim stężeniu sodu – wydalanie litu jest również Zatrucie litem. Zatrucie litem objawia się wymiota-
niskie. Średni okres półtrwania w osoczu wynosi 24 mi, utrzymującymi się biegunkami, zawrotami głowy,
godziny i podlega znacznym zmianom – w zależno- osłabieniem i grubofalistym drżeniem mięśniowym.
ści od ilości spożywanego sodu, funkcji nerek oraz W ciężkich przypadkach może dojść do wystąpienia
wieku pacjenta. śpiączki oraz drgawek. Terapia polega na zastosowa-
niu wymuszonej diurezy przy równoczesnej podaży
Dawkowanie. Wąski zakres działania terapeutyczne- soli kuchennej lub na hemodializie.
go wymaga indywidualnego dawkowania i dokładnej
kontroli stężenia litu w osoczu. W terapii profilak-
tycznej stężenie to winno wynosić 0,6–0,8 mmol/l, 1.2.2.2.2. Leki przeciwpadaczkowe
podczas terapii czynnych objawów maniakalnych
stężenie powinno osiągać poziom 1,0–1,2 mmol/l. Takie leki przeciwpadaczkowe, jak karbamazepi-
Profilaktykę litem rozpoczyna się od dawki 10–20 na, okskarbamazepina i kwas walproinowy nadają
mmol litu; terapia w stanach maniakalnych wymaga się, podobnie jak jony litu, do stosowania zarówno
stosowania dawki 30–40 mmol dziennie. Kontrolę w profilaktyce, jak i w terapii manii. Dokładny me-
stężenia litu w surowicy krwi należy przeprowadzić chanizm działania w tym wskazaniu nie jest jeszcze
po tygodniu stosowania profilaktyki litowej, a przy całkowicie wyjaśniony. Przypuszcza się, że działanie
MUTSCHLER-2009.indd 187 2010-01-07 22:11:38
188 Układ nerwowy
to związane jest z mechanizmem blokowania róż- pomieszczeniach, np. w windzie (klaustrofobia).
nych napięciowozależnych kanałów jonowych (prze- W przypadku fobii społecznych lęk przed oceną do-
de wszystkim – sodowych) i warunkowanym tym konywaną przez innych ludzi prowadzi do unikania
zmniejszeniem się neuronalnej częstotliwości wy- kontaktów społecznych.
ładowań, jak również działaniem antagonistycznym
w obrębie receptorów glutaminergicznych i adeno- Zespół lęku uogólnionego. W tym zaburze-
zynowych. Na ujawnienie się efektu terapeutycznego niu chodzi o uogólniony, niezwiązany z obiektem
należy czekać 2–4 dni. lub sytuacją, utrzymujący się lęk, który skądinąd
Dawkowanie w terapii manii wynosi w przypad- nie jest szczególnie nasilony – w różnym stopniu
ku karbamazepiny i okskarbamazepiny 800–1600 prowadząc do ograniczenia pacjenta w zakresie peł-
(–2000) mg/dzień, a kwasu walproinowego 759(–1500) nienia funkcji społecznych i zawodowych. Pacjenci
mg/dzień. Dla celów profilaktyki wystarczające są lękają się np. mogącego spotkać ich w przyszłości
już wyraźnie mniejsze dawki. nieszczęścia, są nerwowi i skarżą się na niepokój oraz
wegetatywną nadpobudliwość (np. tachykardię, nasi-
lone pocenie się).
1.2.2.2.3. Neuroleptyki
Zaburzenia typu paniki. Istotną cechą zaburzenia
U pacjentów z nasilonymi postaciami zaburzeń ma- typu paniki (epizodycznego napadowego lęku) są na-
niakalnych możliwe jest – szczególnie na początku wracające ciężkie napady lęku (ataki paniki), które
terapii – skojarzone zastosowanie litu i leków neuro- nie są ograniczone do jakiejś określonej sytuacji i dla-
leptycznych. Zaletą jest szybkie ujawnienie się dzia- tego są niemożliwe do przewidzenia. Do najbardziej
łania terapeutycznego oraz – jeżeli jest to konieczne istotnych objawów należą, tak jak w przypadku in-
– możliwość podania parenteralnego. Zalecane w tym nych zaburzeń lękowych: zawroty głowy, uczucie du-
wskazaniu są atypowe neuroleptyki, np. olanzapina. szenia się, bóle w klatce piersiowej, kołatanie serca
i uczucie wyobcowania.
1.2.3. Choroby neurotyczne Zaburzenia typu natręctw. W przypadku natręctw
(nerwice) i zaburzenia (zaburzeń obsesyjno-kompulsyjnych – przyp. tłum.)
dochodzi do powtarzających się (w wyniku ciągłego
z obciążenia powtarzania) bezsensownych czynności. W przypadku
przymusu mycia np. stale myte są ręce aż do uszko-
Przez (kwestionowane) pojęcie nerwic rozumiane
dzenia skóry, w przypadku przymusu liczenia liczone
są choroby z objawami dotyczącymi psychiki, a tak-
są bez przerwy najprzeróżniejsze przedmioty, w przy-
że często czynnościowymi objawami somatycznymi
padku przymusu porządku ciągle na nowo układane
– bez dającego się wykryć organicznego podłoża,
są rzeczy w szafach. Pacjent dostrzega wprawdzie
które powstają na skutek zaburzeń w rozwiązywaniu
bezsensowność swojego postępowania, nie jest jed-
konfliktów (wewnętrznych) lub błędnie sterowanych,
nak w stanie się przed tym bronić. Jeśli zaniecha wy-
nieadekwatnych procesów uczenia się.
konywania tych czynności, wpada w lęk nie do znie-
Według Freuda często chodzi o sytuacje konfliktowe wy- sienia.
wodzące się z wczesnego dzieciństwa, które zostają usu-
nięte ze świadomości. Powstawanie nerwicy jest jednak Neurastenia (zespół psychowegetatywny, dystonia
procesem bardzo złożonym i często nie da się go powiązać wegetatywna, nerwowość). Przez pojęcie to rozumie
z określonym wydarzeniem (traumą). Oprócz czynników się spadek wydolności (organizmu) z wegetatywnymi
konstytucjonalnych istotny udział ma wpływ otoczenia.
Jako czynniki wpływające na ujawnienie się nerwicy nale- i psychicznymi zaburzeniami powstałymi w wyniku dłu-
ży brać pod uwagę zarówno sytuacje obciążające (w rozu- gotrwałego nadmiernego przeciążenia. Pacjenci cierpią
mieniu stresu), jak i sytuacje odciążające (np. zakończenie na osłabienie koncentracji, brak ochoty do robienia cze-
egzaminów, początek urlopu). gokolwiek, drażliwość, bóle głowy, zaburzenia snu
oraz z powodu różnorodnych objawów wegetatywnych
Zaburzenia typu fobii. Są one szczególną posta- (np. dolegliwości żołądkowych, zaparcia, bólów ser-
cią zaburzeń lękowych, wywoływanych w znacznej ca). Jeżeli objawy dotyczą głównie jednego narządu,
mierze lub wyłącznie przez określone sytuacje, które np. serca, mówi się o nerwicy narządowej.
obiektywnie nie są niebezpieczne. Przykładem jest lęk Czynnościowe zaburzenia narządowe można rozu-
przed przebywaniem w tłumie oraz na otwartej prze- mieć jako somatyczny korelat stanu emocjonalnego
strzeni (agorafobia, lęk przestrzeni) lub w ciasnych napięcia.
MUTSCHLER-2009.indd 188 2010-01-07 22:11:39
Układ nerwowy 189
Zaburzenia konwersyjne (reakcje histeryczne). W przy- ■ wywołują stan równowagi (wyrównania),
padku tych zespołów dominują objawy somatyczne
o demonstracyjnym charakterze i celowym ukierunkowa- ■ w możliwie najmniejszym stopniu wpływają na
niu: problem (psychiczny) jest rozwiązywany na drodze zdolność myślenia i sprawność (umysłu).
somatycznej. Obok zjawisk ruchowych, np. (pozornych)
Układ nerwowy
porażeń, występują również zaburzenia czucia i funkcjo- Ponadto większość leków uspokajających cechuje się
nowania narządów zmysłów (np. zaburzenia czucia typu
parestezji, „ślepota”). Ponadto mogą występować padacz- następującym działaniem:
kopodobne napady (bez utraty przytomności, bez urazów) ■ nasennym,
oraz stany psychicznego pobudzenia.
■ przeciwdrgawkowym,
Zaburzenia hipochondryczne. Hipochondryczne posta-
wy objawiają się lękliwą obserwacją samego siebie i oba- ■ zmniejszającym napięcie mięśniowe.
wą przed chorobą, przy czym dotyczy to przede wszystkim B1
serca, przewodu pokarmowego, organów moczowo-płcio- Idealny lek uspokajający powinien się charaktery-
wych oraz mózgu. Niegroźne wegetatywne zaburzenia
czynnościowe są nadmiernie oceniane i w ten sposób ule- zować tym, że dla szerokiego zakresu dawkowania
gają nasileniu – w mechanizmie błędnego koła (circulus ujawniałby jedynie działanie rozluźniające i przeciw-
vitiosus). lękowe – bez wywoływania oszołomienia, senności,
zaburzeń chodu (ataksji) czy też zaburzeń mówienia.
Jadłowstręt psychiczny i bulimia. Są to choroby Niestety, taki idealny preparat nie istnieje.
zaczynające się w okresie dojrzewania lub w wie- Ze względu na często spotykane sytuacje streso-
ku młodzieńczym, które charakteryzują się przede we oraz powszechnie występujące, zwykle warun-
wszystkim odmową przyjmowania pokarmu lub wil- kowane psychicznie, zaburzenia neurowegetatywne
czym głodem, a następnie czynnie wywoływanymi wy- – leki uspokajające znalazły bardzo szerokie zasto-
miotami – powodującymi wychudzenie. Zaburzenia sowanie i nadal stanowią grupę bardzo często prze-
występują głównie u młodych dziewcząt i kobiet. Pa- pisywanych leków psychotropowych. Ich utrzymu-
cjentki nie mogą albo też nie chcą przyjąć roli dorosłej jąca się na wysokim poziomie sprzedaż wskazuje
kobiety. Wychudzenie zapobiega wykształceniu się wyraźnie na kryjące się w nich niebezpieczeństwo:
kobiecych kształtów ciała i tym samym obowiązko- ponieważ stosowane bywają bez wyraźnej potrzeby
wi bycia kobietą. (Niektóre opisy psychopatologicz- do radzenia sobie z problemami życia codziennego
ne zamieszczone w niniejszej książce są niezwykle – wiele osób nie potrafi sobie bez nich poradzić i za-
uproszczone, aby nie powiedzieć strywializowane, żywa te leki w sposób nawykowy. Prawidłowy roz-
i mogą u klinicysty wywoływać poczucie niedosytu wój osobowości człowieka wymaga jednak bycia
lub nawet nieprawidłowości. Bezcelowe byłoby jed- poddawanym rozmaitym napięciom psychicznym,
nak modyfikowanie oryginalnego tekstu książki pod przeżywania wzlotów i upadków, a także krytyczne-
kątem zwiększenia jego poprawności – tu: psychia- go stawiania czoła problemom świata zewnętrznego.
trycznej. Zainteresowani przedmiotem powinni jed- Dlatego osiągana za pomocą leków uspokajających
nak sięgnąć do podręczników psychiatrii i zapoznać „niewzruszoność” (ataraksja) często nie jest niczym
się z zamieszczonymi tam opisami zaburzeń klinicz- innym jak obojętnością. Tak jak leki uspokajające
nych – przyp. tłum.). są cennym narzędziem terapii w przypadku rzeczy-
wistego wskazania, tak również należy ostrzegać
przed ich bezkrytycznym stosowaniem.
1.2.3.1. Leki stosowane w terapii
zaburzeń lękowych Profil działania. Wymienione powyżej jakościo-
we cechy (sposoby działania) są bardzo podobne
1.2.3.1.1. Leki trankwilizujące (uspokajające)/ w odniesieniu do wszystkich leków uspokajających,
leki przeciwlękowe (anksjolityki) szczególnie w najważniejszej grupie – benzodiazepin
(zob. poniżej). Poszczególne substancje z grupy ben-
Leki uspokajające (trankwilizujące, trankwilizatory, zodiazepin różnią się między sobą głównie odmienną
uspokajające, przeciwlękowe, anksjolityki, atarak- mocą oraz farmakokinetyką – jednak jedynie nie-
tyki) są substancjami, które nie wykazując działania znacznie w zakresie profilu działania. Niejednokrot-
przeciwpsychotycznego: nie jedynie kwestią dawkowania jest to, jakie efekty
działania ujawniają się na pierwszym planie.
■ działają uspokajająco,
■ opanowują nadmiar lęku i (wewnętrznego) napię- Miejsce działania. Leki uspokajające – w zwykłym
cia, dawkowaniu – ingerują w funkcje układu limbiczne-
MUTSCHLER-2009.indd 189 2010-01-07 22:11:39
190 Układ nerwowy
go, a wtórnie – w obrębie układu siatkowatego. Leki po leki. W celu zapobieżenia temu efektowi rebound
zmniejszają ilość impulsów elektrycznych w obu (z odbicia) leki uspokajające muszą być odstawia-
wymienionych okolicach i osłabiają indukowane, ne stopniowo. Rzadko natomiast występuje fizyczne
szczególnie bodźcami psychicznymi, pobudzenia uzależnienie ze zwiększeniem dawki (tzw. high-dose-
neuronów wegetatywnych (tzw. rozprężenie psycho- -dependency).
wegetatywne). Podobnie jak inne substancje psychotropowe –
również leki uspokajające są nadużywane przez oso-
Wskazania. Leki uspokajające stosowane są w sta- by uzależnione.
nach niepokoju, lęku i napięcia, jak również w do-
legliwościach psychosomatycznych. Łącznie z lekami Przeciwwskazania. Leki uspokajające są przeciw-
przeciwdepresyjnymi leki uspokajające mogą być sto- wskazane w przypadku ataksji, miastenii, jak również
sowane w początkowej fazie leczenia depresji lękowej w ostrych zatruciach alkoholem, opioidami i lekami
z pobudzeniem psychoruchowym (agitacją). Nie po- nasennymi.
winno się stosować podobnych terapii skojarzonych
na stałe (w przewlekły sposób). Interakcje. Ośrodkowe działanie hamujące leków
Czynnościowe zaburzenia snu są kolejnym zakre- sedatywnych, nasennych, jak również neuroleptyków
sem zastosowania. jest nasilane przez równoczesne stosowanie leków
W związku z ich zdolnością zmniejszania napięcia uspokajających. Podobnie nasileniu ulega działanie
mięśniowego – leki uspokajające nadają się do długo- leków zwiotczających mięśnie.
terminowej terapii skurczów mięśniowych lub wzmo-
żonego napięcia mięśniowego. W przypadku pacjen-
tów z porażeniem poprzecznym ze spastycznymi nie- 1.2.3.1.1.1. Benzodiazepiny
dowładami leki uspokajające z grupy benzodiazepin
należą do leków podstawowych. Benzodiazepiny są zdecydowanie najważniejszą gru-
Stosowanie leków uspokajających w przypadku pą leków uspokajających. Ich działanie jest związane
padaczki zostało opisane w rozdziale B l.9, a ich stoso- z nienaruszonym pierścieniem złożonym z siedmiu
wanie przy znieczuleniu ogólnym – w rozdziale 1.7. atomów. W dalszej kolejności znaczenie ma struktura
laktamowa i podstawnik przyjmujący elektrony przy
Działania niepożądane. Ze względu na ośrodkowe układzie dwupierścieniowym. W tab. B 1.2-13 zesta-
działanie hamujące mogą ujawniać się – szczególnie wione zostały 1,4-benzodiazepiny.
w początkowym okresie terapii – następujące dzia-
łania niepożądane: uczucie zmęczenia, ataktyczne Większość związków chemicznych wywodzi się
zaburzenia chodu, oszołomienie, zawroty głowy oraz od diazepamu – będącego pochodną 1,4-benzodiaze-
osłabienie funkcji intelektualnych. W przypadku pa- piny – albo od jego metabolitów. Z wzorów struktu-
cjentów w podeszłym wieku mogą ujawniać się za- ralnych wyraźnie wynika, że w przypadku związków
burzenia koordynacji, a także paradoksalne reakcje analogowych wprowadzono często jedynie nieznacz-
pobudzenia i splątania, którym mogą towarzyszyć ne zmiany strukturalne. Np. oksazepam jest meta-
omamy. Z powodu działania polegającego na zmniej- bolitem diazepamu; z kolei w prazepamie grupa
szeniu napięcia mięśniowego leki uspokajające mogą N-metylowa diazepamu została zastąpiona grupą cy-
prowadzić – również u starszych osób – do poważne- klopropylometylenową. Występująca w bromazepa-
go zagrożenia spowodowanego upadkami (przyczy- mie zwykła reszta fenylowa jest zastąpiona bioizoste-
na złamań szyjki kości udowej). W przypadku długo- rycznym podstawnikiem pirydylowym. W niektórych
trwałego zażywania mogą nasilać apetyt i prowadzić nowszych substancjach, np. alprazolamie, wprowa-
do zwiększenia masy ciała, jak również wywoływać dzono dodatkowy pierścień.
utratę libido, zaburzenia owulacji i zaburzenia cyklu
miesiączkowego. Klobazam jest jedyną 1,5-benzodiazepiną, która za-
Najbardziej problematyczne jest jednak częste sadniczo wykazuje takie same właściwości działania
ujawnianie się przyzwyczajenia (uzależnienia psy- jak 1,4-benzodiazepiny.
chicznego) – bez zwiększenia dotychczas stosowanej Podobnie jak w przypadku innych leków psycho-
dawki leku (tzw. low-dose-dependency). Polega ono tropowych – działanie benzodiazepin ma charakter
na tym, że – szczególnie w przypadku długotrwałe- jedynie objawowy, a nie przyczynowy. Ich stosowa-
go zażywania – (nagłe) odstawienie preparatu po- nie musi mieć charakter celowy i podlegać okresowej
nownie wyzwala lub też nasila poczucie lęku i bez- krytycznej kontroli. Długotrwałe stosowanie zwięk-
senność, co powoduje, że pacjent ponownie sięga sza ryzyko uzależnienia psychicznego.
MUTSCHLER-2009.indd 190 2010-01-07 22:11:39
Układ nerwowy 191
Mechanizm działania. Wykazano, że mechanizm
A kompleks receptora GABAAA
działania benzodiazepin i substancji pochodnych - kanał chlorkowy
polega na nasileniu hamujących funkcji GABA-er- GABA BZD
przestrzeń
gicznych neuronów. Benzodiazepiny i substancje zewnątrz-
Układ nerwowy
analogowe ingerują w układ kwasu gamma-ami- komórkowa
nomasłowego GABA, a mianowicie w taki sposób,
przestrzeń
że nasilają hamującą funkcję neuronów GABA-er- wewnątrz-
gicznych. Udało się zidentyfikować istnienie specy- komórkowa
ficznych miejsc wiązania dla benzodiazepin (błęd-
nie określanych jako receptory benzodiazepinowe) B Cl-
w obrębie podjednostek α receptorów GABAA-er-
gicznych rozmieszczonych w całym ośrodkowym B1
układzie nerwowym, a występujących w większej
gęstości w czołowej i potylicznej korze mózgowej,
w hipokampie i móżdżku. Benzodiazepiny reagują
przy tym nieselektywnie z różnymi podjednostkami
Cl-
CH3
O
N
Cl N
O
D Cl-
klobazam
Cl-
α poszczególnych podtypów receptorów GABAA. Ryc. B 1.2-5. Mechanizm działania benzodiazepin (BZD).
Benzodiazepiny działają w miejscach wiązania jako A – bez połączenia GABA i BZD kanał chlorkowy pozostaje zamknięty;
agoniści (zob. ryc. B 1.2-5): w wyniku interakcji B – Połączenie z GABA prowadzi do otworzenia kanału i napływu jonów
benzodiazepin w ich miejscach wiązania zwiększa chlorkowych; C – w przypadku połączenia jedynie z BZD kanał pozosta-
się powinowactwo GABA do odpowiedniego miej- je zamknięty; D – równoczesne związanie z GABA i BZD prowadzi do
nasilonego napływu jonów chlorkowych.
sca wiązania – będącego częścią kanału chlorko-
wego (tzw. kompleks receptora GABAA z kanałem
chlorkowym). Dzięki tym allosterycznym wzajem-
nym oddziaływaniom benzodiazepiny zwiększają
siłę wiązania GABA, a tym samym nasilają również Kinetyka. W przypadku zażycia doustnego benzo-
siłę działania GABA. Dochodzi do tego w ten spo- diazepiny są szybko i dobrze wchłaniane. Substancja
sób, że zwiększa się prawdopodobieństwo otworze- prekursorowa chlordiazepoksyd zostaje w organizmie
nia się kanału chlorkowego, przez co zwiększona przekształcona – z odszczepieniem grupy metyloami-
ilość jonów chloru może napłynąć do wnętrza ko- nowej – w aktywną formę laktamową (demoksepam).
mórki. Prowadzi to do hiperpolaryzacji odpowied-
nich komórek z następczym obniżeniem się ich po- W przypadku diazepamu następuje szybka demety-
budliwości. lacja, a następnie wolniejszy proces hydroksylacji
do oksazepamu, który jest wydalany głównie z mo-
Działanie benzodiazepin może być zniesione za po- czem – pod postacią glukuronianu. Ponadto – choć
mocą odpowiednich antagonistów. w mniejszym zakresie – oksazepam może być utwo-
MUTSCHLER-2009.indd 191 2010-01-07 22:11:39
192 Układ nerwowy
Tabela B 1.2-13. Benzodiazepiny
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
CH3 diazepam Diazepam-ratiopharm, 20-50 5-15
O Diazepam STADA,
N Faustan, Valium,
Relanium, Relsed
Cl N
H
chlordiazepoksyd Librium, 10-48 10-50
Multum,
N CH3 Radepur,
N Elenium
Cl N
O
prazepam Demetrin, ok. 80 20-30
Mono Demetrin
O
N
Cl N
H oksazepam Adumbran, 5-12 10-40
O Oxa-CT,
N Oxazepam-ratiopharm,
OH Praxiten, Oxam,
Cl N Oxazepam
klorazepan dipotasowy Tranxilium, 2 5-20
H OH Cloranxen,
N O- Tranxene
COO-
2K
Cl N
H lorazepam Lorazepam-neuraxpharm, 12-24 1,5-3
O Lorazepam-ratiopharm,
N Tavor, Tolid, Lorafen
OH
Cl N
Cl
MUTSCHLER-2009.indd 192 2010-01-07 22:11:40
Układ nerwowy 193
Tabela B 1.2-13. Benzodiazepiny (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
Układ nerwowy
(godz.) dzienna (mg)
H klonazepam Antelepsin, 30-40 4-8
O
N Rivotril,
Clonazepamum
O2N N
Cl
B1
H bromazepam Bromazanil, 8-20 3-6
O Durazanil,
N Lexotanil,
Normoc,
N
Lexotan,
Br Sedam
H3C N alprazolam Alprazolam-ratiopharm, 10-25 0,75-1,5
Alprazolam Sandoz,
N
N Cassadan, Tafii, Afobam,
Alprazomerck, Alprox,
Neurol, Neurol SR, Xanax,
Cl N Xanax SR, Zomiren
rzony z diazepamu w wyniku pierwotnej hydroksyla- Klorazepan dipotasowy jest w soku żołądkowym
cji i następczej demetylacji (zob. ryc. B 1.2-6). w znacznym stopniu metabolizowany do demetylo-
W trakcie biotransformacji diazepamu powstają- diazepamu. Tym samym nie jest on również nową
ce metabolity – demetylodiazepam i temazepam oraz substancją.
oksazepam – są nadal czynne i mogą być stosowane Etapem determinującym prędkość biotransforma-
jako samodzielne leki. cji 1,4-benzodiazepin jest uzależniony od posiadanej
Po podaniu prazepamu nie stwierdza się go w nie- przez nie budowy strukturalnej proces N-dezalkilacji,
zmienionej postaci w organizmie. Głównym metaboli- C3-hydroksylacji lub sprzęgania z kwasem glukuro-
tem jest w tym przypadku także demetylodiazepam. nowym w obrębie C3.
H
H
N CH3 O
N N
Cl N Cl N
O O
chlordiazepoksyd demoksepam
MUTSCHLER-2009.indd 193 2010-01-07 22:11:40
194 Układ nerwowy
Benzodiazepiny są wydalane głównie pod posta-
CH3 cią hydroksylowaną lub jako glukuroniany – głównie
O przez nerki.
N
diazepam Dawkowanie. Średnie dzienne dawki zostały podane
Cl N
w tabeli B 1.2-13.
1.2.3.1.1.2. Hydroksyzyna
CH3 H
O O Hydroksyzyna należy do leków uspokajających z grupy
N N difenylometanu. Jest spokrewniona z różnymi lekami prze-
OH ciwhistaminowymi i przeciwskurczowymi – posiadając
Cl N Cl N tym samym właściwości przeciwhistaminowe i uspokajają-
ce, a ponadto przeciwdrgawkowe, przeciwwymiotne, anty-
cholinergiczne i miejscowo znieczulające. Jako anksjolityk
nie zdobyła ona takiego znaczenia jak inne leki uspokajają-
temazepam ce; stosowana bywa natomiast w świądzie i pokrzywce.
demetylodiazepam Działania niepożądane odpowiadają w znacznym stop-
(nordazepam) niu tym występującym u innych działających ośrodkowo
H leków przeciwhistaminowych.
O
N
OH
oksazepam
Cl N Cl
N
H N C2H4OH
O O
N
O O COOH hydroksyzyna
Cl N
HO OH
OH
glukuronian oksazepamu
1.2.3.1.1.3. Buspiron
Ryc. B 1.2-6. Biotransformacja diazepamu.
Buspiron zalecany jest w leczeniu ostrych i prze-
wlekłych stanów lękowych, jednakże ujawnienie
się efektu terapeutycznego jest wyraźnie wolniejsze
(1–3 tygodnie), a siła działania mniejsza niż w przy-
Silnie lipofilny demetylodiazepam charakteryzuje
padku benzodiazepin. Buspiron nie działa na układ
się długim okresem połowiczego rozpadu – wynoszą-
GABA-ergiczny, wykazuje natomiast częściowo ago-
cym około 100 godzin. W przypadku wszystkich ben-
nistyczne działanie na receptory 5-HT1A – powodując
zodiazepin, które metabolizowane są do demetylodia-
zahamowanie uwalniania serotoniny – oraz blokuje
zepamu, istnieje niebezpieczeństwo kumulacji. Okres
receptory D2. W przeciwieństwie do benzodiazepin
półtrwania postaci hydroksylowanych jest krótszy
nie wykazuje działania uspokajającego, zmniejszają-
i wynosi np. dla oksazepamu – 5–12 godz., dla lora-
cego napięcie mięśniowe czy przeciwdrgawkowego.
zepamu – 12–24 godz. Szczególne znaczenie ma to,
Nie prowadzi także do uzależnienia.
że wraz z wiekiem może znacząco wydłużać się okres
Po podaniu doustnym – buspiron jest dobrze
półtrwania niehydroksylowanych postaci (co jest skut-
wchłaniany, jednak podlega wysokiemu efektowi
kiem spowolnionego metabolizmu – przyp. tłum.).
pierwszego przejścia. W wątrobie jest metabolizowa-
Również zaburzenia funkcji wątroby wydłużają czas
ny oksydatywnie przez izoenzym CYP3A4. Ponad-
eliminacji leku.
to dochodzi do powstawania glukuronianów. Okres
MUTSCHLER-2009.indd 194 2010-01-07 22:11:40
Układ nerwowy 195
chwytu zwrotnego serotoniny, szczególnie z grupy
SSRI i SNRI, jak również trójcykliczne leki prze-
ciwdepresyjne, przede wszystkim klomipraminę
i imipraminę. W przypadku zaburzeń lęku uogól-
Układ nerwowy
N nionego należą one do środków pierwszego wyboru.
W napadach paniki i fobiach powinny być stosowane
O N N w okresie początkowym łącznie z benzodiazepinami,
N a po 2–3 tygodniach stosowane samodzielnie (mo-
N
noterapia) w terapii podtrzymującej i zapobieganiu
O nawrotowi. Ich zaletą w porównaniu z benzodiazepi-
nami jest brak potencjału uzależniającego.
buspiron B1
1.2.4. Leki psychostymulujące
(psychotonizujące,
psychoanaleptyki)
Leki psychostymulujące zwiększają psychiczną ak-
półtrwania w osoczu wynosi ok. 2,5 godz. Wydalanie
tywność. Usuwają uczucie zmęczenia i wewnętrznego
następuje przez nerki i z kałem.
napięcia, jak również zwiększają zdolność koncentra-
Dawkowanie wynosi na początku leczenia trzy
cji i wydolności psychicznej. Ze względu na ich dzia-
razy dziennie po 5 mg, a w razie potrzeby można
łanie polegające na zmniejszeniu zapotrzebowania
zwiększyć dzienną dawkę do 20–30 (–60) mg – po-
na sen powstaje niebezpieczeństwo – szczególnie
dawaną w kilku dawkach podzielonych.
przy stosowaniu dużych dawek – deficytu snu i wyni-
Wśród działań niepożądanych opisano m.in. dole-
kającego z tego całkowitego wyczerpania, gdy tylko
gliwości żołądkowo-jelitowe, bóle i zawroty głowy,
fizyczne rezerwy organizmu ulegną zużyciu. W przy-
nasilone pocenie się, bezsenność, uczucie osłabienia
padku znacznego przedawkowania stają się trucizną
i ginekomastię.
drgawkową.
Buspiron przeciwwskazany jest w przypadku cięż-
kich zaburzeń funkcji wątroby i nerek, w ostrym
napadzie jaskry z wąskim kątem przesączania oraz
w znanych z wywiadu napadach drgawkowych 1.2.4.1. Kofeina
i w miastenii.
Ze względu na niebezpieczeństwo wywołania kry- Spośród występujących w roślinach pochodnych
zy nadciśnieniowej buspiron nie powinien być stoso- ksantynowych: kofeiny, teofiliny i teobrominy – ko-
wany łącznie z inhibitorami MAO. feina ma najsilniejsze działanie polegające na ośrod-
kowej stymulacji. Mniej skuteczna jest teofilina, pod-
czas gdy teobromina w ogóle nie działa pobudzająco
1.2.3.1.2. Inhibitory wychwytu serotoniny, na układ ośrodkowy (zob. tab. B 1.2-14).
trójcykliczne leki przeciwdepresyjne Kofeina jest regularnie spożywana przez dużą
część populacji pod postacią napojów pobudzających
Oprócz leków uspokajających (przeciwlękowych) (kawy, herbaty, „coli”). Jest najczęściej spożywa-
w zaburzeniach lękowych stosuje się inhibitory wy- ną na świecie substancją o działaniu pobudzającym
Tabela B 1.2-14. Profil działania pochodnych ksantyny
Substancja Stymulacja OUN Zwiększenie częstości akcji Rozszerzenie Zwiększenie
serca i jego kurczliwości oskrzeli diurezy
kofeina +++ + + +
teofilina ++ +++ +++ +++
teobromina – ++ ++ ++
MUTSCHLER-2009.indd 195 2010-01-07 22:11:41
196 Układ nerwowy
tami wydalanymi w moczu są ksantyny dwu- i mo-
nometylowe, oraz tri-, di- i monometylowany kwas
moczowy.
O
CH3 Nawet w przypadku codziennej podaży kofeiny
H3C N
N nie ujawniają się żadne utrzymujące się organiczne
N
uszkodzenia. Osoby labilne wegetatywnie mogą jed-
O N
nak reagować już na niewielkie dawki kofeiny – bez-
CH3
sennością, wewnętrznym niepokojem, tachykardią
i ewentualnie biegunką. Z drugiej jednak strony ko-
kofeina
feina u osób w podeszłym wieku sprzyja np. zaśnięciu
– prawdopodobnie w wyniku poprawy krążenia móz-
gowego związanego ze zwiększeniem się wydolno-
O O
H CH3 ści serca. Mimo to dokładny mechanizm tego efektu
H3C N H N
N N nie jest jeszcze dobrze poznany. Duże dawki kofeiny
N N
wywołują niepokój, gonitwę myśli i drżenie mięśnio-
O N O N
we, jak również zaburzenia rytmu serca. Prawdziwe
CH3 CH3
zatrucia kofeiną są jednak rzadkie.
W badaniach eksperymentalnych na zwierzętach
teofilina teobromina – bardzo duże dawki kofeiny wykazywały działanie
teratogenne. W przypadku kobiet ciężarnych spoży-
wanie ponad 600 mg kofeiny dziennie grozi ryzykiem
zwiększenia się częstości poronień oraz przedwczes-
nych porodów.
ośrodkowo. W celach terapeutycznych kofeina stoso-
wana jest w preparatach łączonych z lekami przeciw-
bólowymi. 1.2.4.2. Leki stosowane
w terapii zaburzeń
Po spożyciu kawy działanie kofeiny ujawnia się stosunko-
wo szybko – osiągając swoje maksimum po 30 minutach
hiperkinetycznych i narkolepsji
i ustępuje w czasie 2–3 godzin. Przy spożywaniu herbaty
ujawnienie się działania jest opóźnione, a czas działania Podstawy patofizjologiczne. Typowy dla dzie-
wydłużony. ci z zespołem deficytu uwagi z hiperaktywnością
(ADHD – attention deficit and hyperactivity disor-
Kofeina w zwykłych dawkach 50–200 mg działa der) jest już we wczesnym okresie dziecięcym ujaw-
głównie na korę półkul mózgowych. U osób zmę- niający się brak lub osłabienie uwagi i koncentracji,
czonych pod wpływem blokady receptorów ade- a także niepokój ruchowy (nadaktywność ruchowa)
nozynowych ustępują objawy zmęczenia i zwiększa wraz z zaburzeniami zachowań społecznych (nieak-
się wydolność umysłowa. Poprawa sprawności psy- ceptowanie adekwatnych do wieku norm społecz-
chicznych po spożyciu kofeiny u osób wypoczętych nych) i pogorszenie się w zakresie osiągnięć szkol-
i w pełni przytomnych jest zwykle niewielka. nych. Zaburzeniem tym dotknięci są głównie chłopcy
W większych dawkach kofeina pobudza ośrodek (stosunek chłopców do dziewcząt wynosi 8:1; współ-
naczynioruchowy i oddechowy. Mimo to nie docho- czynnik całkowitej chorobowości osiąga około 4%).
dzi do wzrostu ciśnienia, gdyż – w wyniku działania
obwodowego – równoczesnemu rozszerzeniu ulegają Należąca do grupy zaburzeń snu tzw. hipersomnii
naczynia krwionośne w skórze, nerkach i wieńcowe. narkolepsja – charakteryzuje się napadami snu (nie-
Kofeina nasila ponadto procesy glikogenolizy i lipo- możliwy do opanowania przymus zaśnięcia/zasypia-
lizy. nia w ciągu dnia) z częściową lub całkowitą utratą
W przypadku naczynioruchowych bólów głowy ko- napięcia mięśni szkieletowych (katapleksja) oraz
rzystne działanie kofeiny związane jest rozkurczem omamami hipnagogicznymi (występującymi w trak-
mózgowych naczyń krwionośnych i zmniejszeniem cie zasypiania). Napad snu trwa zwykle 10–20 minut,
ciśnienia płynu mózgowo-rdzeniowego. a po nim którym następuje wybudzenie się wypoczę-
Po podaniu doustnym kofeina jest szybko i całko- tego pacjenta. Już na początku snu występują fazy
wicie wchłaniana, a wewnątrz organizmu ulega częś- REM. Podłoże genetyczne tego zaburzenia uważane
ciowej demetylacji i utlenieniu. Głównymi metaboli- jest za pewne.
MUTSCHLER-2009.indd 196 2010-01-07 22:11:41
Układ nerwowy 197
1.2.4.2.1. Amfetaminy amfetamin 10–30 godz. (zależnie od wartości pH
i substancje pokrewne moczu), dla metylofenidatu – 2–2,5 godz. Wydalanie
(„aminy budzące”) następuje głównie przez nerki.
Dawkowanie metylofenidatu należy indywidual-
Układ nerwowy
nie dopasować (powolne zwiększanie dawki). Śred-
Związki należące do tej grupy (zob. tab. B. 1.2-15):
nie dawki dzienne podano w tab. B 1.2-15.
amfetamina, metamfetamina (oba preparaty niedo-
Jako działania niepożądane występują zaburzenia
stępne w handlu) oraz metylofenidat – wywodzą się
ośrodkowego układu nerwowego (lęk, bóle głowy,
z katecholamin lub efedryny. W wyniku braku grup
drżenie mięśni, brak apetytu), suchość w jamie ust-
hydroksylowych wzrasta wyraźnie lipofilność, stąd
nej, tachykardia, wzrost ciśnienia krwi i reakcje aler-
też substancje te mogą łatwo przechodzić przez ba-
giczne.
rierę krew-mózg.
Przy nadciśnieniu od stopnia średnio ciężkiego B1
do ciężkiego, w nadczynności tarczycy, jadłowstręcie
Ich działanie opiera się przede wszystkim na wyrzu-
psychicznym, tachykardii, schizofreniach, zaburze-
cie katecholamin – a tym samym substancje te są po-
niach lękowych oraz guzie chromochłonnym – amfe-
średnio działającymi sympatykomimetykami. Szcze-
taminy są przeciwwskazane.
gólnie wyraźnie widoczne jest ośrodkowe działanie
Stężenie w osoczu trójpierścieniowych neurolep-
pobudzające, które w znacznym stopniu determinuje
tyków i leków przeciwdepresyjnych oraz fenytoiny
obraz kliniczny. Dodatkowo związki te ujawniają ob-
i primidonu – przy równoczesnym podaniu amfeta-
wodowe efekty sympatykomimetyczne.
min – ulega podwyższeniu w wyniku zahamowaniu
Jak w prawie żadnej innej grupie leków – zalety
biotransformacji.
i wady zależą od sposobu użycia i wskazań. W przy-
padku zespołu deficytu uwagi z nadpobudliwością
ruchową, jak również w narkolepsji – metylofenidat
jest cennym i nieprowadzącym do uzależnienia le-
kiem. Jednak amfetaminy są niejednokrotnie naduży-
1.2.4.2.2. Atomoksetyna
wane jako narkotyk lub środek dopingujący. W tych
przypadkach charakteryzują się dużym potencjałem Nową substancją czynną stosowaną w terapii zespołu
uzależniającym i są niebezpieczne. Dlatego amfe- ADHD jest atomoksetyna – środek hamujący presy-
taminy podlegają przepisom odpowiedniej ustawy naptyczny transporter noradrenaliny. W przeciwień-
o przeciwdziałaniu narkomanii. stwie do metylofenidatu, który działa bardzo szybko,
atomoksetyna ujawnia swoje działanie dopiero po
Amfetaminy są szybko i dobrze (w ponad 90%) tygodniu leczenia. Aż do ujawnienia się pełnej sku-
wchłaniane; okres półtrwania w osoczu wynosi dla teczności leku może minąć 4–6 tygodni.
Tabela B 1.2-15. Amfetaminy i substancje pokrewne
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
CH3 amfetamina – 10-30 5-10
NH2
CH3 metamfetamina – 10-15 3-9
HN
CH3
COOCH3 metylofenidat CONCERTA, 2-2,5 10-20 (-60)
H Equasym,
Medikinet,
Ritalin
HN
MUTSCHLER-2009.indd 197 2010-01-07 22:11:42
198 Układ nerwowy
część efektów związana jest z pobudzeniem ośrodkowych
receptorów adrenergicznych α1.
Modafinil jest szybko wchłaniany; okres półtrwania
w osoczu wynosi 10–13 godzin. Metabolity są nieskutecz-
CH3
ne. Wydalanie następuje głównie przez nerki.
Dawkowanie wynosi 200–400 mg rano.
O Działaniami niepożądanymi są typowe oznaki ośrod-
CH3 kowej stymulacji: bóle głowy, niepokój, bezsenność, lęk;
NH w dalszej kolejności opisywano hiperkinezy, nasilone po-
cenie się, tachykardię.
W trakcie leczenia inhibitorami receptorów adrenergicz-
atomoksetyna nych α1, a także w przypadku istniejących w wywiadzie
uzależnień stosowanie modafinilu jest przeciwwskazane.
Po podaniu doustnym atomoksetyna jest wchłania- O O
na szybko i niemal całkowicie (biodostępność wynosi S
NH2
– w zależności od efektu pierwszego przejścia – 65–
95%). Powstający w obrębie izoenzymu CYP2D6
główny metabolit 4-hydroksyatomoksetyna działa
podobnie silnie jak substancja macierzysta. Średni modafinil
okres półtrwania w osoczu wynosi ok. 3,5 godziny
u osób z szybkim metabolizmem i 21 godzin u wol-
no metabolizujących. Wydalanie następuje głównie
przez nerki pod postacią O-glukuronianu 4-hydrok-
syatomoksetyny.
Terapię u dzieci i osób młodocianych o wadze 1.2.4.2.4. Zalecenia dotyczące kryteriów
do 70 kg rozpoczyna się od dawki całkowitej 0,5 stosowania leków
mg/kg; młodzież o masie powyżej 70 kg rozpoczyna psychostymulujących
leczenie z dawką 40 mg i dawkę tę utrzymuje przez w zespole ADHD i narkolepsji
okres przynajmniej 7 dni. W zależności od skutecz-
ności i tolerowania leku można następnie powoli
W przypadku terapii dzieci hiperkinetycznych za po-
zwiększać dawkę leku. Zalecana podtrzymująca daw-
mocą leków psychostymulujących leczenie powin-
ka dzienna wynosi 1,2 mg/kg lub 80 mg.
no być zindywidualizowane i prowadzone zgodnie
Jako działania niepożądane opisano suchość w us-
ze ścisłymi zaleceniami, jak również zintegrowane
tach, nudności i wymioty, zaburzenia żołądkowo-jeli-
z całościowym planem leczenia. Rodzicom należy
towe, osłabienie apetytu, bezsenność, wahania nastro-
objaśnić wspomagający charakter leczenia; równo-
ju, zaburzenia oddawania moczu, zapalenie skóry.
cześnie należy zdjąć z rodziców obawy przed wytwo-
W przypadku jaskry z wąskim kątem przesączania
rzeniem się uzależnienia u dziecka. Tylko w ten spo-
atomoksetyna jest przeciwwskazana. Ponadto nie po-
sób zapewni się jego konsekwentne podawanie, które
winna być równocześnie stosowana z inhibitorami
jest wymagane. Podając ostatnią dawkę leku przed
MAO.
godziną 16, można znacząco zmniejszyć ryzyko wy-
Inhibitory izoenzymu CYP2D6 mogą podwyż-
stąpienia zaburzeń snu. Ze względu na anorektyczne
szać stężenie atomoksetyny w osoczu. W przypadku
działanie amin budzących wskazane jest okresowe
skojarzonego podania atomoksetyny z substancjami,
przeprowadzanie kontroli wagi ciała.
które wpływają na stężenie noradrenaliny (np. leków
Również w przypadku narkolepsji – stosowanie
przeciwdepresyjnych), możliwe jest ujawnienie się
leków psychostymulujących jest jedynie częścią ca-
efektów synergistycznych.
łego schematu terapii. Ważne jest ponadto włączenie
w proces terapii krewnych pacjenta, stosowanie się
do zasad prawidłowej higieny snu (regularne – tj.
1.2.4.2.3. Modafinil
o stałej porze – kładzenie się spać i wstawanie) oraz
Modafinil jest kolejną substancją o działaniu psychostymu- unikanie pracy zmianowej. Leki psychostymulujące
lującym, która jest stosowana w terapii narkolepsji. Do- nie są w stanie opanować objawu katapleksji. W tym
kładny mechanizm działania nie jest znany. Przynajmniej celu lepiej nadają się trójcykliczne leki przeciwde-
MUTSCHLER-2009.indd 198 2010-01-07 22:11:42
Układ nerwowy 199
presyjne, inhibitory wychwytu zwrotnego seroto- pojmowania i koncentracji, orientacji i sfery afektyw-
niny oraz działający tłumiąco na ośrodkowy układ nej, jak również zmiany w zakresie struktury osobo-
nerwowy oksybat sodu (HO–(CH2)3–COOH – kwas wości – prowadzące do całkowitego rozpadu osobo-
4-hydroksymasłowy, sól sodowa tego kwasu), za po- wości. Wraz z wiekiem znacząco zwiększa się czę-
Układ nerwowy
mocą którego możliwe jest zredukowanie epizodów stość występowania zaburzeń otępiennych. Objawy
katapleksji o 90%. otępienia stwierdza się u około 2% sześćdziesięcio-
pięciolatków i aż u 30% dziewięćdziesięciolatków.
Oksybat sodu występuje fizjologicznie w mózgach ssaków Z etiopatogenetycznego punktu widzenia wyróż-
i jest tam określany terminem gamma-hydroksy-maślanu nia się:
(GBH). Jego dokładne działanie nie jest znane. Potwier-
dzone jest, że działa on jako agonista receptorów GBH oraz ■ otępienie w chorobie Alzheimera (otępienie typu
GABAB oraz wpływa na transdukcję sygnału w obrębie
szeregu innych układów neuroprzekaźnikowych. W przy-
alzheimerowskiego; DAT), B1
padku podania leku przed zapadnięciem w sen nocny ■ otępienie naczyniowe (warunkowane zmianami na-
– zwiększa ona udział snu wolnofalowego ze stadiów 3 i 4 czyniowymi),
oraz zmniejsza częstotliwość występowania faz snu REM.
Zwykle bywa stosowany łącznie z substancjami stymulują- ■ mieszane postacie otępienia (obejmujące elementy
cymi. obu wcześniej wymienionych typów otępienia),
Po podaniu doustnym oksybat sodu jest szybko wchła-
niany i metabolizowany poprzez postać aldehydu do kwa- ■ otępienia w przebiegu innych schorzeń neurozwy-
su bursztynowego (biodostępność ok. 25%). Tylko ok. 5% rodnieniowych (np. w pląsawicy Huntingtona lub
ulega wydaleniu przez nerki w niezmienionej postaci.
Początkowa dawka, przy kładzeniu się do łóżka wynosi chorobie Parkinsona),
2,25 g, oraz ponownie 2,4–4 godz. później. W zależności ■ (wtórne) otępienia ujawniające się po lub w trakcie
od tolerancji i skuteczności można dawkę zwiększać lub
zmniejszać (maksymalna dawka dzienna – 9 g). różnych chorób zakaźnych/zapalnych (np. otępie-
Jako działania niepożądane mogą ujawnić się m.in.: nie w zakażeniu wirusem HIV) urazach głowy, gu-
zawroty i bóle głowy, nudności, senność, stany spląta- zach mózgu lub zatruciach (np. alkoholem).
nia, wymioty i (rzadko) depresja ośrodka oddechowego.
W przypadku nielegalnego stosowania jako narkotyku im- Choroba Alzheimera (morbus Alzheimer) jest naj-
prezowego może dojść do powstania uzależnienia i obja-
wów odstawiennych. częstszą przyczyną zaburzeń otępiennych. Więcej
U pacjentów z niedoborem dehydrogenazy sukcyny- niż 50% wszystkich zespołów otępiennych jest wa-
losemialdehydu – otrzymujących opiaty lub barbiturany runkowanych tą chorobą. Ostateczne ustalenie roz-
– użycie oksybatu sodu jest przeciwwskazane. poznania jest przy tym możliwe dopiero po zgonie
Alkohol, podobnie jak leki uspokajające, np. benzodia- pacjenta. W trakcie obdukcji ujawniają się nasilone
zepiny, nasilają działanie oksybatu sodu. Dlatego należy
unikać równoczesnego ich przyjmowania. zaniki tkanki nerwowej mózgu (głównie w obrębie
płatów czołowych i ciemieniowych). Dzięki badaniu
histologicznemu stwierdza się wewnątrzkomórko-
we wiązki włókienek (neurofibryle), które zawierają
bogate w reszty fosforanowe białka neurofibrylarne/
1.2.5. Leki stosowane w terapii neurowłókienkowe (białko tau); z kolei w przestrzeni
zaburzeń otępiennych pozakomórkowej stwierdza się tzw. płytki i włókien-
(leki przeciwotępienne) ka, które zbudowane są ze złogów specyficznego amy-
loidu – składającego się z toksycznie działającego
1.2.5.1. Podstawy patofizjologiczne białka β-amyloidu (Aβ). Białko to powstaje z białka
prekursorowego – białka prekursorowego dla amylo-
idu (APP).
Zespołami otępiennymi (demencjami) nazywamy
schorzenia, które występują zwykle dopiero w po-
deszłym wieku oraz które charakteryzują się jakoś- Rozkład APP w mniejszej wielkości polipeptydy następu-
je pod wpływem rozmaitych enzymów, tzw. sekretaz. Sek-
ciowym i ilościowym pogorszeniem się sprawności retaza α prowadzi do tworzenia rozpuszczalnego białka
funkcji umysłowych, a także pojawieniem się ogra- APP (sAPP), które – co ciekawe – odgrywa pozytywną rolę
niczeń w zakresie zachowań społecznych. Zaburzenia np. w zakresie funkcji troficznych i neuroprotekcyjnych.
otępienne w zaawansowanych stadiach uniemożliwia- Tymczasem sekretazy β i γ są odpowiedzialne za powstanie
ją pacjentowi samodzielne funkcjonowanie. Wśród białka β-amyloidu, które prowadzi do śmierci komórki. (Stąd
też inhibitory sekretaz β i γ stanowią punkt wyjścia do ba-
najważniejszych objawów otępienia należy wymienić dań nowych leków służących terapii choroby Alzheimera).
stopniowo ujawniające się i stale narastające zaburze- Mutacje w zakresie APP, jak również w obrębie tzw. genów
nia napędu, uczenia się, pamięci, myślenia, zdolności preseniliny sprzyjają tworzeniu się β-amyloidu.
MUTSCHLER-2009.indd 199 2010-01-07 22:11:42
200 Układ nerwowy
Uszkodzenia komórek nerwowych dotyczą głównie
neuronów cholinergicznych (szczególnie w obrębie ją-
dra podstawnego Meynerta, skąd wywodzi się około NH2
90% wszystkich cholinergicznych dróg prowadzących
do kory nowej półkul mózgowych), których brak wy-
raźnie warunkuje ujawnienie się zaburzeń uczenia się N
i pamięci. Odpowiednie objawy ujawniają się jednak takryna
dopiero wtedy, gdy dojdzie do uszkodzenia ok. 70%
neuronów dotkniętych procesem chorobowym.
U podłoża otępień naczyniowych leżą zaburzenia H3CO
ukrwienia mózgu powodujące tzw. udary. Najważ- O
N
niejszą postacią w tej grupie otępień (20% wszyst- H3CO
kich postaci) jest otępienie wielozawałowe polega-
jące na występowaniu licznych drobnych zawałów donepezil
(udarów) mózgowych – ujawniających się na podło-
żu miażdżycowo zmienionych naczyń mózgowych.
Postacie mieszane (choroba Alzheimera + otę-
pienie naczyniowe) stanowią 20% postaci zaburzeń OH
H
otępiennych.
O
Wyraźnie rzadziej występują inne wymienione po-
H3CO
stacie otępienia.
N
CH3
1.2.5.2. Substancje wpływające na galantamina
neurotransmisję cholinergiczną
Ponieważ – jak to zostało wyjaśnione – choroba Alzhe-
imera polega zasadniczo na ograniczeniu przekaźni-
ctwa nerwowego głównie w układzie cholinergicznym C2H5 CH3
– usiłuje się wykorzystać w celach terapeutycznych N O
H3C N(CH3) 2
podawanie prekursorów acetylocholiny (fosfatydy-
locholinę, dimetyloaminoetanol), agonistów choli- O
nergicznych (agonistów receptorów muskarynowych riwastygmina
M1) lub inhibitorów acetylocholinoesterazy. Możliwe
do zastosowania terapeutycznego okazały się substan-
cje z dwóch ostatnich grup.
Z wymienionej grupy leków do terapii otępienia typu
podwyższenie transaminaz w surowicy krwi). Z tego też
alzheimerowskiego o zaawansowaniu od łagodnego powodu takryna wkrótce po wprowadzeniu do stosowania
do umiarkowanego zostały dopuszczone następujące została wycofana z rynku.
ośrodkowo działające substancje:
■ donepezil, Donepezil, galantamina i riwastygmina są wyraź-
nie lepiej tolerowane i dlatego mogą być stosowane
■ galantamina,
w większych dawkach – co poprawia również ich sku-
■ riwastygmina. teczność. Oprócz hamującego działania na acety-
locholinoesterazę galantamina wpływa na presy-
Takryna, pierwszy z grupy inhibitorów acetylocholino- naptyczne receptory nikotynowe i zwiększa w ten
esterazy wprowadzony do terapii jako lek przeciwotę- sposób uwalnianie acetylocholiny; skądinąd opisana
pienny, poprawiał wprawdzie funkcje poznawcze (łącz- właściwość podawana jest w wątpliwość. Podobnie
nie z funkcjami pamięciowymi) oraz korzystnie wpły- jak w przypadku innych leków przeciwotępiennych,
wał na zaburzenia zachowania u pacjentów z chorobą
Alzheimera, jednak jego zalety były istotnie ograniczone skuteczność kliniczna jest ograniczona.
ujawniającymi się działaniami niepożądanymi – szcze- Inhibitory acetylocholinoesterazy są dobrze wchła-
gólnie działaniem hepatotoksycznym (częste przejściowe niane, jednak podlegają nasilonemu efektowi pierw-
MUTSCHLER-2009.indd 200 2010-01-07 22:11:43
Układ nerwowy 201
szego przejścia. Okres półtrwania w osoczu wynosi bardzo szybko oddzielić się od miejsca wiązania, aby
w przypadku donepezilu – około 70 godzin, galan- zapobiec zbyt długotrwałej blokadzie kanału, a tym
taminy – 6 godzin, a riwastygminy – ok. 1 godziny. samym przedłużającemu się wypadnięciu funkcji.
Wydalanie następuje głównie przez nerki.
Układ nerwowy
Dawkowanie donepezilu w trakcie pierwszych 4–6 Związkiem spełniającym powyższe założenia jest me-
tygodni wynosi 5 mg podawane wieczorem; później mantyna. Na podstawie licznych kontrolowanych ba-
dawkę zwiększa się do 10 mg. Galantaminę począt- dań, w których przebiegu uzyskano poprawę w zakre-
kowo podaje się w dawce 8 mg/dzień; później – w za- sie funkcji poznawczych, napędu, umiejętności spo-
leżności od tolerancji – zwiększa się ją do 16–24 mg/ łecznych i codziennych czynności, memantyna jest jak
dzień. Na początku leczenia riwastigminą podaje się dotąd jedynym zarejestrowanym preparatem do terapii
ją w dawce 1,5 mg dwa razy dziennie; po przynaj- umiarkowanych i ciężkich postaci otępienia.
mniej 14 dniach dawka może być zwiększona na dwa B1
razy dziennie po 3 mg (maksymalna dawka dzienna
wynosi 12 mg).
Wśród działań niepożądanych wymienia się brak
apetytu, zawroty głowy, bezsenność, koszmary senne,
nudności, wymioty, nadmierne pobudzenie, splątanie,
drgawki mięśniowe, jak również infekcje górnych NH2
dróg oddechowych i układu moczowo-płciowego.
Farmakodynamiczne interakcje występują przy
H3C
równoczesnym podaniu z cholinomimetykami i pole-
gają na zwiększeniu siły działania; z kolei przy rów- H3C
noczesnym stosowaniu substancji przeciwcholiner-
gicznych – obserwuje się osłabienie siły działania. memantyna
Z interakcjami farmakokinetycznymi u donepezilu
należy liczyć się w przypadku stosowania substra-
tów izoenzymu CYP3A4, np. chinidyny, makrolitów
i doustnie stosowanych azolowych leków przeciw-
grzybiczych. Wymienione leki podwyższają stężenie
donepezilu w osoczu.
Memantyna po doustnym podaniu ulega szybkie-
mu i całkowitemu wchłonięciu; wydalanie następuje
głównie przez nerki – w niezmienionej postaci.
1.2.5.3. Niekompetycyjni antagoniści Terapię rozpoczyna się od dawki 5 mg/dzień
NMDA przez jeden tydzień, później 10 mg/dzień, a od trze-
ciego tygodnia kontynuuje się leczenie za pomocą
dawki 15–20 mg/dzień. Dawka podtrzymująca wy-
W przypadku degeneracyjnych chorób mózgu za waż-
nosi 20–30 mg.
ny czynnik patogenetyczny uznaje się: nadmierne
Wśród działań niepożądanych stwierdza się za-
pobudzenie receptorów NMDA, czego skutkiem
wroty i bóle głowy, nudności, niepokój i nadmierną
jest przeładowanie komórek nerwowych jonami wap-
pobudliwość.
nia (excitotoxity = tzw. toksyczność z pobudzenia).
W przypadku ciężkich stanów splątania, padacz-
Dlatego też antagoniści receptorów NMDA, jeżeli
ki i ciężkich zaburzeń funkcji nerek – memantyna
tylko cechują się poniższymi właściwościami, wydają
jest przeciwwskazana.
się obiecującymi substancjami o działaniu neuropro-
Memantyna nasila działanie równocześnie przyj-
tekcyjnym. Aby nie wpływać (lub też w niewielkim
mowanych neuroleptyków, leków przeciwcholiner-
stopniu) na prawidłową neurotransmisję glutaminer-
gicznych, lewodopy, agonistów dopaminergicznych
giczną – nie powinny reagować z właściwym miej-
i amantadyny.
scem wiązania kwasu glutaminowego, lecz winny
wiązać się – w niekompetycyjny sposób – we wnę-
trzu sterowanego przez NMDA kanału jonowego
w miejscu wiązania magnezu (jest to możliwe jedy- 1.2.5.4. Leki nootropowe
nie w sytuacji, gdy doszło już do pobudzenia i sam
kanał jonowy został otwarty; tzw. use-dependency). Tym niejednoznacznie zdefiniowanym terminem –
Ponadto antagoniści NMDA powinni być w stanie leki nootropowe (neurotropowe) określa się substan-
MUTSCHLER-2009.indd 201 2010-01-07 22:11:43
202 Układ nerwowy
cje, które bez (lub też z bardzo niewielkim) ośrodko- 1.2.5.5. Ginkgo biloba
wego działania stymulującego wpływają na metabo-
lizm mózgu (np. przez poprawę właściwości transpor-
Suche wyciągi z liści drzewa Ginkgo biloba – miło-
towych błony komórkowej, zwiększenie utylizacji
rzębu dwuklapowego (drzewa świątynnego) należą
glukozy bądź aktywację metabolizmu nukleotydów,
do najczęściej stosowanych środków farmakologicz-
fosfolipidów i/lub białek). Skutkiem tego jest popra-
nych w leczeniu otępienia. Najważniejszymi substan-
wa sprawności różnych funkcji mózgowych – szcze-
cjami składowymi wyciągu są glikozydy flawonowe
gólnie pamięci, koncentracji, uwagi, osądu i orienta-
(m.in. izoramnetyna, olejek kamforowy, kwercetyna)
cji. Do leków nootropowych, które z chemicznego
oraz laktony terpenowe (ginkgolidy A, B i C, bilo-
punktu widzenia należą do bardzo wielu różnych klas
balid). Większość różnych dostępnych w handlu pre-
substancji, zalicza się (zob. tab. B 1.2-16):
paratów jest standaryzowana w odniesieniu do poje-
■ nicergolinę, dynczej (lub dwukrotnej albo też trzykrotnej) dawki,
zawierającej 8,8–10 mg glikozydów flawonowych
■ piracetam,
i 2–2,8 mg laktonów terpenowych.
■ pyritinol. Nowsze badania z zastosowaniem standaryzowa-
nych dawek wyciągu przyniosły wyniki nieodbie-
Mimo że zarówno w doświadczeniach na zwierzę- gające od rezultatów osiąganych za pomocą innych
tach, jak i w badaniach na ludziach stwierdzono wiele leków przeciwotępiennych. Mimo to nadal kliniczne
korzystnych efektów dotyczących przemiany materii znaczenie tych wyników jest dyskusyjne.
w mózgu lub określonych funkcji mózgu, skutecz-
ność tych leków przy zaburzeniach sprawności móz-
gu jest nadal kwestionowana. Powodem jest to, że
wprawdzie uzyskiwane efekty są często statystycznie 1.2.5.6. Akceptory (wolnych) rodników
znaczące, ale równocześnie uważane są za mało zna-
Jedną z możliwych przyczyn uszkodzenia komórek mózgu
czące klinicznie. jest tworzenie się wolnych rodników. Dlatego przyjmuje
Średnie dawki dzienne zostały podane w tab. B się, że działanie chroniące komórki nerwowe można osiąg-
1.2-16. nąć za pomocą substancji, które bądź zapobiegają powsta-
Tabela B 1.2-16. Leki nootropowe
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
N nicergolina Ergobel, Nicergolin-CT, 2,5 10-20
Nicergolin-neuraxpharm,
O Nicerium, Sermion,
Br
Adavin, Circulat
O
H3CO
N CH
3
H
N
H3C
CONH2 piracetam Nootrop, Normabrain, 5-6 2400-4800
Piracetam-neuraxpharm,
N O Piracetam-ratiopharm,
Lucetam, Memotropil,
Nootropil
N CH3 pyritinol Encephabol 2-8 600
CH2OH
HO S
S OH
CH2OH
H3C N
MUTSCHLER-2009.indd 202 2010-01-07 22:11:43
Układ nerwowy 203
waniu rodników, bądź też je unieszkodliwiają. Jednakże loamidu kwasu lizergowego, LSD) – wypróbowując
nie udało się jak dotychczas udowodnić istnienia klinicz- go na sobie. Hofmann wyizolował również z mek-
nych korzyści w razie stosowania takich akceptorów rodni-
sykańskiego „boskiego” grzyba teonanacatl (Psilocy-
ków (np. witaminy A, C i E).
be mexicana) psylocynę i psylocybinę – rozpoznając
Układ nerwowy
ich działanie halucynogenne.
Zaprezentowane w tab. B 1.2-17 pochodne indo-
1.2.5.7. Pozycja
lu wykazują podobne jakościowo działanie, różnią
leków przeciwotępiennych się jednak między sobą w zakresie siły działania.
Na przykład do wywołania stanu psychotycznego
Skuteczna terapia zespołów otępiennych – za po-
potrzeba jedynie lizergidu 0,5–1 μg/kg m.c. i aż
mocą powyżej opisanych leków przeciwotępien-
30–100 μg psylocybiny na kg masy ciała. Charak-
nych – jest obecnie możliwa jedynie w niewielkim
terystyczne jest zniesienie poczucia czasu, ponadto B1
lub ograniczonym zakresie. Przyczyną jest to, że
ujawniają się omamy słuchowe i wzrokowe. Nastrój
objawy otępienia ujawniają się zwykle wówczas,
może się zmieniać między euforycznym i dysfo-
gdy większość dotkniętych chorobą neuronów zo-
rycznym (horrortrip). Działanie utrzymuje się zwy-
stała już zniszczona/uszkodzona, a tym samym
kle przez 6–8 godzin. Za pomocą neuroleptyków
jest już (za) późno na wdrożenie efektywnej przyczy-
można szybko i całkowicie opanować objawy psy-
nowej terapii. Ponadto należy zauważyć, że nie tyl-
chotyczne.
ko układ cholinergiczny, lecz również liczne inne
układy neuroprzekaźnikowe dotknięte są procesem Meskalina. Substancja ta jest czynnym składnikiem róż-
chorobowym, a równoczesne modyfikowanie kilku nych, występujących w Meksyku kaktusów (z rodzaju
układów jest farmakologicznie mało prawdopodob- Anhalonium). Jej działanie odpowiada działaniu lizergidu.
ne. Mimo to możliwe jest za pomocą leków przeciw- Meskalina wymaga jednak około 10 000 razy większych
otępiennych zmniejszenie przez pewien czas (ogólnie dawek (5–10 mg/kg masy ciała).
Wśród wegetatywnych działań niepożądanych po zaży-
– w ciągu pół roku) nasilenia objawów chorobowych, ciu tej substancji spokrewnionej z noradrenaliną obserwu-
poprawienia codziennego funkcjonowania pacjenta je się: nudności, zawroty głowy, rozszerzenie źrenic oraz
oraz przesunięcie terminu przeniesienia go do domu zlewne poty.
pomocy. Do pochodnych meskaliny, które zaliczane bywają rów-
Szczególne znaczenie w przyszłej terapii zaburzeń nież do analogów amfetaminy, należą „narkotyki artystów”,
np. metylenodioksy-metamfetamina (ekstazy). Substancje
otępiennych będzie więc miała wczesna diagnosty- tego typu zaczynają odgrywać na scenie narkotykowej co-
ka tego rodzaju zaburzeń oraz wprowadzenie leków, raz większą rolę .
które byłyby w stanie spowolnić progresję choroby
podstawowej.
Cannabis indica. Z konopi indyjskich (Cannabis
sativa var. indica) pozyskuje się marihuanę oraz ha-
szysz. Marihuana składa się z wysuszonych kwiato-
1.2.6. Substancje stanów i końcówek łodyżek, z kolei haszysz jest wy-
psychozomimetyczne suszoną żywicą kwiatową. Skuteczność 1 mg mari-
(psychodysleptyczne, huany odpowiada około 200 mg haszyszu. Główną
halucynogenne) substancją działającą w nich zawartą jest ∆9-tetra-
hydrokannabinol.
Środki psychozomimetyczne wywołują u zdrowych
osób – w sposób ostry i przejściowy – stan podob- Jest interesujące, że ze zwierzęcej i ludzkiej tkanki
ny do ostrej psychozy paranoidalnej. Oprócz zabu- udało się sklonować dwa receptory kannabinoidowe
rzeń w zakresie relacji do otoczenia i poczucia jaźni (receptory CB), z którymi wiążą się tetrahydrokanna-
– występują doznania omamowe; rzeczywistości binol i jego analogi. Należą one do grupy receptorów
nie można odróżnić od spostrzeżeń złudzeniowych. sprzężonych z białkiem G i składają się z 472 (re-
Nadużywanie środków psychozomimetycznych, ceptor CB1) lub 360 aminokwasów (receptor CB2).
które na podstawie odpowiednich przepisów krajo- Te z kolei należą do grupy receptorów sprzężonych
wych zaliczane są zwykle do środków odurzających, z białkiem G i przez to białko o działaniu hamującym
jest znaczne. Nie mają one znaczenia leczniczego. działają na cyklazę adenylanową. Udało się zidenty-
fikować endogenne ligandy tych receptorów, do któ-
Pochodne indolu. W 1943 roku A. Hofmann stwier- rych należą etanolamid arachidonylowy (anandamid)
dził silne psychotropowe działanie lizergidu (diety- oraz glicerol arachidonylowy.
MUTSCHLER-2009.indd 203 2010-01-07 22:11:44
204 Układ nerwowy
Tabela B 1.2-17. Psychodysleptyki (z wyjątkiem haszyszu)
Wzór strukturalny Nazwa Występowanie
I. Pochodne indolu
N(CH3) 2 N,N-dimetylotryptamina (syntetyczna pochodna)
NH
OH N(CH3) 2
psylocyna Psilocybe mexicana
NH
PO3H2 psylocybina Psilocybe mexicana
O N(CH3) 2
NH
N(CH3) 2 bufotenina wydzielina gruczołów śluzowych ropuchy
HO
NH
O N(C2H5) 2 lizergid (półsyntetyczna pochodna)
N
CH3
H
HN
II. Pochodne fenyloetyloaminy
H3CO NH2 meskalina gatunki Anhalonium
H3CO
OCH3
O NH metylenodioksy-metamfetamina (syntetyczna pochodna)
CH3 (ekstazy)
O CH3
Marihuanę i haszysz zażywa się głównie poprzez jówek, nieżyt oskrzeli, dolegliwości astmatyczne,
ich palenie, rzadziej przez spożywanie pod postacią ataksja i drżenia mięśniowe (tremor). W przypadku
napojów. Ich działanie jest silnie warunkowane czyn- przewlekłego nadużywania istnieje niebezpieczeń-
nikami zewnętrznymi (wpływ grupy środowisko- stwo degradacji struktury osobowości. Największym
wej), strukturą osobowości zażywającego, sposobem zagrożeniem ze strony haszyszu jest jednak to, że
zażycia i, naturalnie, dawką. Po fazie poczucia pobu- może on służyć jako „narkotyk wejściowy”, który
dzenia lub odprężenia następuje zwykle stan rzekomo pomaga „przesiąść się” na silniejsze substancje.
zwiększonej zdolności postrzegania połączony z uro- Terapeutyczne stosowanie konopi lub tetrahydro-
jeniowymi wyobrażeniami. Duże dawki wywołują kannabinoli m.in. w leczeniu bólów jest dyskutowane
podobne efekty jak lizergid. w rozdz. B 1.5.3.
Objawami somatycznymi – obserwowanymi
u osób często palących haszysz – są: zapalenie spo-
MUTSCHLER-2009.indd 204 2010-01-07 22:11:44
Układ nerwowy 205
Jedynym długotrwale skutecznym środkiem w tera-
pii nadwagi jest – oprócz zwiększenia aktywności
HO (CH2) 4CH3 fizycznej – zmniejszenie spożycia kalorii, co jednak
H trudno zrealizować licznym pacjentom mimo posia-
Układ nerwowy
H3C danej wiedzy w zakresie konsekwencji wynikających
O z nieprzestrzegania przepisów dietetycznych. Farma-
H koterapia może być stosowana jedynie jako środek
H3C CH3
podtrzymujący i czasowo ograniczony. Można przy
tym wyróżnić dwie zasadniczo odmienne możliwości
Δ9-tetrahydrokannabinol terapeutyczne:
■ zmniejszenie poczucia głodu – prowadzące B1
do zmniejszenia ilości przyjmowanego pokarmu
lub
■ ograniczenie procesu trawienia z następczym
1.2.7. Dodatek: leki przeciw otyłości wchłanianiem składowych części pokarmowych.
Podstawy patofizjologiczne. Około 20–40% popu-
lacji w krajach zachodnich cierpi na spowodowaną 1.2.7.1. Leki zmniejszające łaknienie
zwiększoną podażą kalorii nadwagę, tj. ma wagę cia- (anorektyczne, odchudzające)
ła przekraczającą określone normy. Można to przed-
stawić jako przekroczenie prawidłowej masy w pro- Do zmniejszenia uczucia głodu, a tym samym i ape-
centach lub za pomocą wskaźnika masy ciała (zob. tytu można stosować leki zmniejszające łaknienie
tab. B 1.2-18). (anorektyczne). Te albo nasilają działanie anorek-
tycznych (zmniejszających apetyt) substancji endo-
Przyjmuje się, że prawidłową masę ciała (masa wg Broki) gennych (lub też same działają anorektycznie), albo
wylicza się za pomocą wzoru Broki: hamują działanie substancji oreksygennych (zwięk-
prawidłowa masa (kg) = długość ciała (cm) – 100. szających apetyt) (zob. tab. 1.2-19).
Wskaźnik masy ciała (BMI – body mass index) jest definio-
wany jako: Ośrodkowo działające sympatykomimetyki. Do
związków pierwszej z wymienionych grup nale-
BMI = masa ciała (kg) : wysokość2 (m2). żą przede wszystkim ośrodkowo działające bez-
pośrednie sympatykomimetyki. Pokładane w tych
Ponieważ nadwaga i otyłość są czynnikami sprzyjają-
substancjach nadzieje albo się nie spełniły, albo też
cymi powstawaniu zmian miażdżycowych w obrębie
ścian naczyń krwionośnych, nadciśnienia, cukrzy-
cy i zmian zwyrodnieniowych stawów – muszą być Tabela B 1.2-19. Substancje wpływające na apetyt
poważnie traktowane jako czynniki ryzyka powinny
być ograniczane.
Anoreksygenne Oreksygenne
(hamujące apetyt) (zwiększające apetyt)
noradrenalina neuropeptyd Y
Tabela B 1.2-18. Klasyfikacja masy ciała na podstawie typo-
serotonina endorfina β
wych parametrów
cholecystokinina (CCK) dynorfina
peptyd 1 podobny do glukagonu peptyd pokrewny Agouti
Kategoria Stopień Nadwaga BMI (glukagon-like peptide 1) (AgRP – Agouti-related peptide);
otyłości wg Broki (%) (kg/m2) antagonista (α-MSH)
hormon stymulujący melanocyty α hormon koncentrujący melaninę
prawidłowa masa ciała 0 0 20-24,9 (α-MSH – α-melanocyte- (MCG – melanin-concentrating
-stimulating hormone) hormone)
Nadwaga
peptyd pokrewny kokainie- agoniści receptora CB1
umiarkowana I 0-20 25-29,9 -amfetaminie (CART – cocaine-
-amphetamine-related peptide)
znaczna II 20-70 30-40 leptyna grelina
ekstremalna III > 70 > 40 peptyd YY3-36 (PY3-36)
MUTSCHLER-2009.indd 205 2010-01-07 22:11:44
206 Układ nerwowy
w bardzo niewielkim stopniu, gdyż ich skuteczność go izoenzymu – indukcji (np. rifampicyna, fenytoina,
ulega szybkiemu osłabieniu, istnieje znaczące ryzy- fenobarbital) lub hamowania (np. makrolidy, azolo-
ko uzależnienia i dodatkowo ujawniają się dalsze we leki przeciwgrzybicze).
poważne działania niepożądane, np. bezsenność,
nadmierna pobudliwość, zawroty głowy, tachykar- Rimonabant. Do drugiej grupy leków zmniejsza-
dia, zaburzenia rytmu serca lub nadciśnienie. Stąd jących łaknienie – związków hamujących działanie
też większość substancji należących do tej grupy oreksygenne – zalicza się rimonabant, będący an-
(m.in. fenfluramina lub fentermina) musiały zostać tagonistą anandamidu i glicerolu arachidonylowego
wycofane z rynku leków. w obrębie receptorów kannabinoidowych 1. Osta-
teczna ocena tej substancji nie jest obecnie możliwa.
Sibutramina. Do grupy preparatów zmniejszających Po podaniu doustnym ulega dobremu i szybkiemu
łaknienie zalicza się ponadto sibutraminę – wprowa- wchłanianiu – będąc wydalany później z kałem. Bio-
dzoną początkowo jako lek przeciwdepresyjny, bę- transformacja następuje głównie w CYP3A4 oraz za
dący inhibitorem noradrenaliny i serotoniny (SNRI). pomocą amidohydrolazy. Okres półtrwania w osoczu
Czy sibutramina – w związku z innym mechanizmem u osób otyłych oceniany jest na 16 dni (w przypad-
działania – ma rzeczywistą przewagę w porównaniu ku osób z wagą prawidłową czas ten jest wyraźnie
z bezpośrednimi sympatykomimetykami, pozostaje krótszy).
kwestią otwartą. Po rocznej terapii stwierdzano jednak Roczna terapia za pomocą pojedynczej dawki 20
zmniejszenie masy ciała prawie o 4 kg w porównaniu mg dziennie prowadziła do zmniejszenia się masy
z grupą placebo. ciała średnio o 8,8 kg w porównaniu z 2,9 kg w grupie
Biodostępność sibutraminy wynosi ok. 80%; okres placebo.
półtrwania substancji macierzystej w osoczu sięga
1 godziny, natomiast obu aktywnych metabolitów:
mono- i didemetylowanej sibutraminy – 14–16 go-
dzin. Wydalanie następuje głównie przez nerki.
Cl
O
N N
N NH
Cl
CH(CH3) 2 CH3
N(CH3) 2
Cl
sibutramina Cl
rimonabant
Dawkowanie wynosi 10 mg (maksymalnie 15 mg)
raz dziennie.
Jako działania niepożądane ujawniają się często
– podobnie jak w przypadku bezpośrednich sympa-
tykomimetyków – zaburzenia sercowo-naczyniowe
(wzrost ciśnienia, tachykardia, palpitacje), a po-
Najczęściej obserwowanymi działaniami niepo-
nadto – bezsenność, depresyjne obniżenie nastroju,
żądanymi były nudności, zawroty głowy, depresyjne
nudności, zaparcia lub reakcje skórne.
obniżenie nastroju, zaburzenia lękowe i biegunka.
Sibutramina jest przeciwwskazana w przypadku
W przypadku ciężkich depresji nie powinno się
choroby wieńcowej, niewydolności serca, zaburzeń
zażywać rimonabantu; w przypadku zaburzeń funkcji
rytmu serca, udaru mózgu, nadczynności tarczycy,
wątroby i nerek, a także w terapii skojarzonej z silny-
ciężkiej niewydolności wątroby i nerek, guza chro-
mi inhibitorami CYP3A4 lek powinien być zażywany
mochłonnego i przerostu tarczycy.
z dużą ostrożnością.
Ponieważ jest biotransformowana głównie przez
CYP3A4, może dochodzić do interakcji z substancja- Rimonabant – oprócz zmniejszania łaknienia – wykazuje
mi metabolizowanymi również w obrębie tego same- działanie antydiabetogenne.
MUTSCHLER-2009.indd 206 2010-01-07 22:11:45
Układ nerwowy 207
1.2.7.2. Orlistat niem kału, parcia stolca i jego nietrzymanie. Ponadto
orlistat może osłabiać wchłanianie witamin rozpusz-
czalnych w tłuszczach. Stąd też należy zwracać uwa-
Działanie orlistatu polega na całkowicie innej zasa-
gę na odpowiednią podaż witamin.
dzie niż w przypadku leków zmniejszających łaknie-
Układ nerwowy
W zespole złego wchłaniania i zastoju żółci, jak
nie: jest on inhibitorem lipaz żołądkowo-jelitowych.
również w okresie karmienia – orlistat jest przeciw-
Poprzez kowalentne wiązanie z resztą serynową
wskazany.
w aktywnym centrum enzymu (pierścień β-laktono-
W przypadku równoczesnego zażywania orlistatu
wy) zapobiega miejscowo, tj. w świetle żołądka i jelit,
i leków zmniejszających krzepliwość krwi – głównie
rozkładowi (tzw. inhibitor asymilacyjny) tłuszczów
z grupy dikumarolu – należy kontrolować wartości
(triglicerydów) do tłuszczów prostych (monoglicery-
INR.
dów) i kwasów tłuszczowych, które dopiero mogą być B1
wchłonięte przez ścianę jelit. Niewchłonięte tłuszcze
(ok. 30%) zostają wydalone z kałem, w wyniku czego
osiąga się zmniejszenie ilość kalorii dostarczanych
do organizmu. W badaniach klinicznych zmniejsze-
nie masy ciała w grupie otrzymującej lek było więk-
sze niż w grupie placebo, a ponadto ponowny przy- NH
OHC CH(CH3) 2
rost masy ciała po wcześniejszym jego zmniejszeniu O
w grupie otrzymującej orlistat był mniejszy. O O O
Po podaniu doustnym orlistat praktycznie nie ule- H3C(CH2) 5 (CH2) 10CH3
ga wchłanianiu i niemal całkowicie jest wydalany
z kałem (z tego ponad 80% w postaci niezmienio- orlistat
nej). W przypadku odstawienia leku – aktywność li-
paz błyskawicznie powraca, gdyż enzymy te ulegają
syntezie na nowo i są wydzielane do światła układu
pokarmowego.
Orlistat jest wskazany wraz z nieznacznie hipo-
kaloryczną dietą do stosowania u pacjentów bez
dodatkowych czynników ryzyka z BMI > 30 kg/m2 Naturalnie pojawia się pytanie, czy sensowne jest za pomo-
cą leku w rodzaju orlistatu prowadzić do częściowej niewy-
oraz u pacjentów z innymi czynnikami ryzyka z BMI
dolności trzustki, aby w ten sposób doprowadzić do reduk-
> 28 kg/m2. cji masy ciała. Istnieje tu ten sam problem co w przypadku
Dawkowanie wynosi 120 mg bezpośrednio przed, stosowania przez otyłe osoby z cukrzycą typu II doustnych
podczas lub do jednej godziny po spożyciu głównych środków przeciwcukrzycowych o działaniu insulinotropo-
posiłków. wym. Mimo że w zasadzie pożądany efekt terapeutyczny
mógłby być osiągnięty na drodze fizjologicznej dzięki
Wśród działań niepożądanych, które zwiększa-
odpowiedniej diecie – w praktyce efekt ten jest uzyskiwa-
ją się wraz z zawartością tłuszczów w pożywieniu ny niekiedy przy wykorzystaniu dodatkowych środków,
i zmniejszają się wraz z czasem leczenia – wymienia których celowość stosowania jest trudna lub niemożliwa
się cuchnące tłuste/oleiste stolce, wzdęcia z oddawa- do uzasadnienia.
MUTSCHLER-2009.indd 207 2010-01-07 22:11:45
208 Układ nerwowy
1.3. Substancje wpływające na sen
(środki nasenne, hipnotyczne)
1.3.1. Podstawy fizjologiczne stadia snu: na fazę zasypiania (stadium I), fazę lekkie-
go snu (stadium II) oraz dwie fazy snu głębokiego (sta-
Czuwanie i sen. Nawet przy wyłączeniu wpływu dia III i IV). Ten przebiegający pod postacią falistą sen
wszystkich czynników pochodzących z otoczenia jest przerywany szczególnymi fazami snu, w których
utrzymuje się endogennie sterowana cykliczność czyn- występują salwy szybkich ruchów oczu i dlatego nazy-
ności układu krążenia i przemiany materii, zmian tem- wane są one fazami REM (od rapid eye movements).
peratury ciała oraz aktywności mózgu, która warunko- Sen REM charakteryzuje się nasiloną aktywnością
wana jest endogennymi oscylatorami (biologiczny ze- elektryczną mózgu, podczas gdy pozostałe parametry
gar, wewnętrzny zegar). Poprzez zewnętrzne synchro- odpowiadają snowi głębokiemu (minimalne napięcie
nizatory czasowe (Zeitgeber – „dawca czasu”), takie mięśniowe, wysoki próg wybudzania). Dlatego też
jak cykliczność jasności, ciemności i aktywności (pra- stadia snu REM, które w ciągu nocy wydłużają się
ca dzienna – nocny spoczynek), endogenne (okołodo- od 5–10 do 20–30 minut, nazywane są również snem
bowe) rytmy dopasowują się do 24-godzinnego rytmu paradoksalnym. Fazy REM są okresami, w których
dobowego. Również rytm spania-czuwania jest takim występują marzenia senne. Jak to jest obecnie wiado-
pierwotnym okołodobowym procesem, który jest syn- me, dla dobrego samopoczucia niezbędny jest prawid-
chronizowany zewnętrznymi regulatorami czasu. Naj- łowy przebieg poszczególnych stadiów snu. Zwłasz-
ważniejszą strukturą sterującą (master clock) układu cza trwające przez dłuższy czas znaczące skrócenie lub
okołodobowego jest jądro nadskrzyżowaniowe (nucle- zniesienie snu stadium REM prowadzi do poważnych
us suprachiasmaticus; zob. poniżej). następstw.
Osoba śpiąca jest niewrażliwa na zewnętrzne bodź- Czas trwania snu wolnofalowego i snu fazy REM
ce, a ponadto wyłączona jest świadomość. Jednakże zmniejsza się wraz z wiekiem (zob. ryc. B 1.3-1).
między snem a znieczuleniem ogólnym (narkozą) Starsze osoby potrzebują mniej snu niż ludzie mło-
istnieją istotne różnice: sen jest niezbędnym do ży- dzi. Fakt ten ma znaczenie dla oceny zaburzeń snu.
cia aktywnym procesem, w trakcie którego w prawie Również zachowania związane ze snem zmieniają się
wszystkich narządach przebiegają procesy regenera- wraz z wiekiem (zob. ryc. B 1.3-2). Wraz z wiekiem
cyjne i odbudowy, natomiast w przypadku znieczule- dochodzi w trakcie snu do częstszych przebudzeń,
nia ogólnego ma się do czynienia z zahamowaniem zmniejszeniu ulega liczba faz snu głębokiego oraz
czynności ośrodkowego układu nerwowego. We śnie snu REM.
utrzymują się odruchy ochronne (np. odruch kaszlu),
w narkozie dochodzi do ich zniesienia. Osobę śpiącą Neuronowe podstawy stanów sen-czuwanie.
można w każdej chwili obudzić, osobę wprowadzoną Wprawdzie znane są liczne szczegóły dotyczące pro-
w narkozę – nie. cesów neurofizjologicznych leżących u podstawy
rytmu sen-czuwanie oraz przebiegu faz snu, jednak
Rodzaje snu i jego fazy. Na podstawie pomiarów współdziałanie poszczególnych części ośrodkowego
neurofizjologicznych, w szczególności badania elek- układu nerwowego w trakcie stanu czuwania oraz
troencefalograficznego, można wyróżnić dwa ro- snu nie jest jeszcze do końca w pełni zrozumiałe.
dzaje snu: Jest rzeczą pewną, że w przypadku snu nie docho-
dzi do ogólnego hamowania czynności ośrodkowego
■ sen wolnofalowy, „synchronizowany” (sen fazy
układu nerwowego, lecz zmienia się stosunek aktyw-
NREM/Non-REM),
ności określonych grup neuronalnych. Również zło-
■ sen paradoksalny lub sen fazy REM. żone procesy fizjologiczne nie są sterowane przez po-
jedynczy ośrodek snu, lecz raczej bierze w tym udział
Pełny cykl obejmujący fazę snu NREM i REM okre- wiele okolic mózgu.
śla się terminem podstawowego cyklu odpoczynku
i aktywności (BRAC – Basic-Rest-Activity-Cycle). Główną rolę w kontroli nad cyklicznością przebiegu stanów
sen-czuwanie odgrywa jądro nadskrzyżowaniowe w pod-
wzgórzu. Jest ono położone bezpośrednio nad skrzyżowa-
Sen wolnofalowy można – również na podstawie ba- niem nerwów wzrokowych (chiasma opticum; otrzymuje
dania elektroencefalograficznego – podzielić na różne przez kolaterale (bocznice) drogi wzrokowej informacje
MUTSCHLER-2009.indd 208 2010-01-07 22:11:45
Układ nerwowy 209
nowo- 2-3 10-14 18 30 45 60 90
rodek r.ż. r.ż. r.ż. r.ż. r.ż. r.ż. r.ż.
24 godz.
Układ nerwowy
stan czuwania
16
B1
8 stadia snu
od I do IV
faza REM
0
16 12 10 8 1/2 7 3/4 7 6 5 3/4
godz.
przeciętny czas trwania snu / 24 godz.
Ryc. B 1.3-1. Czas trwania snu wolnofalowego i paradoksalnego w zależności od wieku (według Leutnera).
o oświetleniu siatkówki, stąd też ulega wpływowi jasnoś- W zapadnięciu w sen NREM prawdopodobnie uczestni-
ci lub ciemności otoczenia. Ponadto istnieją połączenia czą neurony jądra pasma samotnego (nucleus tractus solita-
od tego jądra do szyszynki – uwalniającej melatoninę (zob. rii), które hamują wstępujący siatkowaty układ aktywujący
poniżej), która z kolei oddziałuje zwrotnie na jądro nad- (ARAS) i tym samym zmniejszają stopień stanu czuwa-
skrzyżowaniowe. Zlokalizowane tutaj neurony wykazują nia. Proces zasypiania zależy przy tym od poprzedzającej
periodyczną aktywność. Jest ona modulowana przez układ go aktywności fizycznej lub umysłowej, przyjęcia pokar-
wzrokowy i synchronizowana z zależnymi od pory dnia mu, temperatury mózgu i innych czynników. W celu wyjaś-
wahaniami jasności. nienia ośrodkowego efektu zmęczenia postulowano istnie-
Okołodobowy „dawca czasu”, jądro nadskrzyżowanio- nie „substancji snu”, która miałyby gromadzić się w stanie
we prawdopodobnie nie wywiera bezpośredniego wpływu czuwania pod wpływem wymienionych czynników. Pewne
na czynności fizjologiczne, lecz steruje aktywnością innych jest, że GABA i adenozyna uczestniczą we wprowadzaniu
struktur mózgowych, m.in. pola ostatniego (area postrema) do snu i jego podtrzymywaniu. Natomiast histamina przez
w podwzgórzu. Udział tej struktury wynika również z tego, pobudzenie receptorów H1 działa hamująco na sen.
że u zwierząt przez drażnienie prądem elektrycznym tej
okolicy można wywołać sen. Ponadto istnieją połączenia Melatonina, hormon wytwarzany w pinealocytach
z jądra nadskrzyżowaniowego do pnia mózgu, zwłaszcza szyszynki w wyniku metoksylowania i N-acetylowa-
do jąder szwu i do miejsca sinawego (locus coerulesus), jak
również poprzez boczne wzgórze do kory mózgowej. Do- nia serotoniny, jest biogenną aminą, której wydzie-
piero współdziałanie neuronów tych obszarów umożliwia lanie warunkowane jest rytmem dzień-noc: oświetle-
kontrolę okresowych zmian czynności, które są podłożem nie, przekazywane za pośrednictwem fotoreceptorów
stanu snu lub czuwania. siatkówki, wywołuje hamowanie współczulnych
Różne obserwacje i wyniki badań eksperymentalnych włókien z górnego zwoju szyjnego, w wyniku czego
dostarczyły ponadto wskazówek co do przekaźników
biorących udział w kontroli snu. Tak więc wychodzące zmniejsza się uwalnianie noradrenaliny, a następnie
z jąder szwu neurony uwalniają serotoninę. (Jeżeli zosta- melatoniny. W ciemności wzrasta natomiast ilość
nie zniesione uwalnianie serotoniny, to przejściowo wystę- syntetyzowanej i uwalnianej melatoniny. Melato-
puje bezsenność). Dla stadium snu REM znaczenie mają nina działa nasennie poprzez zintegrowane z błoną
prawdopodobnie cholinergiczne neurony w moście, które komórkową receptory melatoniny (ML) oraz procesy
poprzez drogi zstępujące hamują rdzeniowe motoneurony
i tym samym wywołują typowe dla snu REM zwiotczenie transdukcji powiązane z układem białka G i cyklazy
mięśni. Ponadto przyjmuje się, że zakończenie faz REM adenylanowej. W ten sposób melatonina bierze udział
jest wywoływane aktywnością neuronów noradrenergicz- w regulacji rytmu sen-czuwanie. Ostatnio melatonina
nych w miejscu sinawym. w postaci o opóźnionym działaniu została dopuszczo-
MUTSCHLER-2009.indd 209 2010-01-07 22:11:45
210 Układ nerwowy
1.3.2. Zaburzenia snu
NHCOCH3 Do zaburzeń snu, na które skarży się 15–25% doro-
H3CO słych osób, należą dyssomnie występujące pod nastę-
pującymi postaciami:
NH
■ bezsenność (insomnia; brak snu lub sen dający nie-
melatonina dostateczny odpoczynek),
■ nadmierna senność (hipersomnia; utrzymująca się
nieprawidłowo duża potrzeba snu),
■ zaburzenia rytmu snu i czuwania (brak synchroni-
na do krótkotrwałej terapii zaburzeń snu u pacjentów
zacji między indywidualnym rytmem sen-czuwa-
po 55 roku życia. Dawkowanie wynosi 2 mg zażywa-
nie a rytmem sen-czuwanie otoczenia)
ne 1–2 godziny przed położeniem się spać. Powinna
być stosowana przez okres trzech tygodni.
jak również istotnie rzadziej występujących para-
somnii, do których zalicza się sennowłóctwo (som-
nambulizm), lęk nocny (pavor nocturnus) oraz kosz-
dzieci mary senne.
wybudzenie
Z farmakoterapeutycznego punktu widzenia szcze-
REM
gólne znaczenie mają zaburzenia typu bezsenności.
1
stadia snu
W zależności od ich etiopatogenezy wyróżnia się:
2
■ bezsenność idiopatyczną, w której nie stwierdza się
3
żadnych czynników wyzwalających i która przy-
4 puszczalnie jest skłonnością uwarunkowaną gene-
tyczną predyspozycją,
1 2 3 4 5 6 7
■ bezsenność psychofizjologiczną – jako następ-
młodzi dorośli stwo reakcji stresowej na emocjonalne obciążenie
wybudzenie (np. obciążenie psychospołeczne),
REM ■ bezsenność sytuacyjną, która jest wywołana bodź-
1 cami pochodzącymi z otoczenia (np. zmiana po-
stadia snu
2 gody, hałas, praca zmianowa, nieregularny tryb
3 życia),
4 ■ bezsenność objawową – jako objaw towarzyszący
lub następstwo choroby somatycznej (organicznej)
1 2 3 4 5 6 7 lub psychicznej (m.in. depresji, zespołów lęko-
wych),
osoby w podeszłym wieku
■ bezsenność farmakogenną lub toksyczną – spowo-
wybudzenie dowaną stymulacją wywołaną przyjęciem substan-
REM cji ośrodkowo pobudzających (np. kofeiny, środ-
1 ków hamujących łaknienie, amin analeptycznych),
stadia snu
2 ■ zespół bezdechu sennego (epizodycznie występu-
3 jące fazy zatrzymania oddychania w trakcie snu,
4 które trwają ponad 10 sekund).
1 2 3 4 5 6 7 Z klinicznego punktu widzenia jest również ważne,
godziny snu czy w przypadku rozpoznawanej bezsenności chodzi
o zaburzenia zasypiania czy też podtrzymywania
Ryc. B 1.3-2. Zmiany przebiegu snu wraz z wiekiem (według (przebiegu) snu (tu z kolei wyróżnić można postacie
Anschütza). Stadia snu REM – kolor żółty. polegające na częstym śródnocnym lub przedwczes-
MUTSCHLER-2009.indd 210 2010-01-07 22:11:46
Układ nerwowy 211
nym porannym budzeniu się z niemożnością ponow- – fizjologiczny przebieg snu, przy czym najbardziej
nego zaśnięcia – przyp. tłum.). dotknięte bywają stadium IV snu wolnofalowego
oraz faza snu REM. Podczas gdy barbiturany prowa-
dziły do skrócenia głównie snu REM, benzodiazepi-
Układ nerwowy
1.3.3. Leki nasenne ny wydłużają fazę II i III snu wolnofalowego, z kolei
skróceniu ulega czas trwania fazy IV snu głębokiego
(zob. tab. 1.3-1).
Wiele należących do starszych generacji, aczkolwiek
W przypadku środków nasennych, które upo-
wcześniej powszechnie stosowanych leków nasen-
śledzają sen fazy REM – odstawienie po dłuższym
nych, np. barbituranów, zostało obecnie prawie całko-
stosowaniu prowadzi do wywołania nadmiaru fazy
wicie zastąpionych przez leki nasenne z grupy benzo-
REM (tzw. efekt REM-rebound), tj. ulega wydłuże-
diazepin (zob. poniżej) oraz podobnie działające sub-
niu całkowity czas trwania fazy snu REM. Istnieje B1
stancje. Ich decydującą zaletą jest to, że w przypadku
przy tym niebezpieczeństwo wystąpienia majacze-
ich samodzielnego zażycia (nadużycia) – w przeci-
nia (delirium) z odstawienia, które wzrasta wraz
wieństwie do dawnych substancji – nie jest możliwe
ze zwiększaniem się zjawiska REM-rebound. Leki
dokonanie próby samobójczej.
nasenne, które zmniejszają sen fazy REM, wydłuża-
ją najczęściej również okres latencji REM, tzn. czas
trwania snu od zaśnięcia do wystąpienia pierwszej
Wymagania stawiane idealnemu lekowi nasenne-
fazy REM.
mu. Od idealnego leku nasennego wymaga się, aby:
Ponieważ liczne preparaty nasenne ulegają bardzo
■ wywoływał stan podobny do snu fizjologicznego, długotrwałemu rozkładowi, po ich zażyciu następne-
tj. aby nie zmieniał fizjologicznego profilu snu, go dnia mogą ujawniać się nierzadko takie objawy,
jak poczucie zmęczenia i rozbicia (zjawisko kaca
■ w przypadku przedawkowania – nie wpływał
– hangover); istnieje też ryzyko kumulacji.
na inne funkcje ośrodkowego układu nerwowego
Ponadto w przypadku licznych leków nasennych
(oraz funkcje innych narządów),
po pewnym czasie ich stosowania (zwykle po 2–4 ty-
■ nie ulegał kumulacji, godniach) zmniejsza się ich skuteczność. Powoduje
to, że pacjent zwiększa dawkę zażywanego leku lub
■ nie wywoływał następnego dnia żadnych nieko-
też zażywa dodatkowo inny lek.
rzystnych działań późnych (tzw. hangover,
■ przy długotrwałym stosowaniu nie zmniejszała się Interakcje. Należy zwrócić szczególną uwagę
jego skuteczność. na możliwe interakcje leków nasennych z wielo-
ma innymi lekarstwami. Prowadzą one do nasile-
Porównanie pożądanych cech z opisanymi poniżej nia działania wszystkich leków cechujących się
rzeczywistymi właściwościami leków nasennych ośrodkowym działaniem hamującym, np. leków
wykazuje, że taki idealny środek nasenny obecnie przeciwhistaminowych lub ośrodkowo działających
nie istnieje (zob. tab. B 1.3-1). leków przeciwnadciśnieniowych, jak również alko-
holu. Ciężkie działania niepożądane mogą ujawnić
Właściwości leków nasennych. Wszystkie substan- się także przy równoczesnym stosowaniu leków
cje działające nasennie zaburzają – w różnym stopniu psychotropowych.
Tabela B 1.3-1. Działanie leków nasennych (zmodyf. według Bausta)
Działanie na fizjologiczny sen
Substancja stadia snu Latencja Efekt REM- Utrata siły
II III IV REM REM -rebound działania
barbiturany ↑ ∅ ↓ ↓↓ ↑ ↑↑ ++
wodzian chloralu ↑ ↓ ↑ ∅ ∅ ∅ +
benzodiazepiny ↑ ↑ ↓ (↓) ∅ ∅ (+)
zolpidem, zopiklon ↑ ↑↓* ↑↓* (↓) ∅ ∅ (+)
↑ zwiększenie ↓ zmniejszenie ∅ bez efektu + efekt obecny (+) słaby efekt * zależnie od dawki
MUTSCHLER-2009.indd 211 2010-01-07 22:11:46
212 Układ nerwowy
1.3.3.1. Benzodiazepiny i ich pochodne głym) ich odstawieniu – na skutek efektu rebound
(z odbicia) może dojść do ujawnienia się bezsenno-
ści, stanów lękowych, zawrotów głowy, nudności,
Do benzodiazepin lub ich pochodnych – obecnie naj-
splątania i innych objawów związanych z ośrodko-
ważniejszych i najczęściej stosowanych leków na-
wym układem nerwowym.
sennych – należą (zob. tab. B 1.3-2):
■ temazepam,
■ flurazepam, 1.3.3.2. Zaleplon, zolpidem, zopiklon
■ lormetazepam,
Zaleplon – pirazolopirymidyna, zolpidem – imidazo-
■ nitrazepam,
pirydyna, oraz zopiklon – cyklopirolon są związkami
■ flunitrazepam, o podobnym mechanizmie działania jak benzodiaze-
piny. Podobnie jak benzodiazepiny – choć bardziej
■ triazolam,
selektywnie – wiążą się w sposób antagonistycz-
■ brotizolam. ny jedynie z podjednostką α1 kompleksu receptor
GABAA – kanał chlorkowy. Różnice w profilu działa-
Profil działania. Leki stosowane jako środki na- nia – w porównaniu z benzodiazepinami – są niewiel-
senne pod względem swoich właściwości farmako- kie. Ze względu na znacząco mniejsze powinowactwo
logicznych odpowiadają innym lekom z tej grupy, do podjednostek α2 receptorów zlokalizowanych
np. diazepamowi. Oznacza to, że oprócz działania w obrębie rdzenia kręgowego – ich działanie prze-
nasennego cechują się one zasadniczo także innymi ciwdrgawkowe i polegające na zmniejszeniu napięcia
właściwościami benzodiazepin. Jak już to wcześniej mięśniowego jest mniej nasilone. Mimo to – szczegól-
zostało wspomniane, w typowych dawkach wywie- nie w przypadku osób w podeszłym wieku – istnieje
rają niewielki wpływ na sen fazy REM, natomiast ryzyko wystąpienia niepewności w chodzeniu i upad-
wydłużeniu ulegają stadia II i III snu wolnofalowego; ków. Potencjał uzależnienia się od wymienionych le-
skraca się natomiast stadium IV. ków jest – zgodnie z dotychczasowym doświadcze-
niem – niższy niż w przypadku benzodiazepin, mimo
Farmakokinetyka. Leki wymienione w tab. B 1.3.-2 to WHO uznaje ryzyko nadużywania i uzależnienia
leki ulegają szybkiemu i dobremu wchłanianiu. Dla od nich na podobnym poziomie jak benzodiazepin.
oceny preparatu pod kątem ewentualnego ujawnienia
się efektu hangover (działania późne/odległe) nale- Wszystkie trzy substancje – po podaniu doustnym
ży uwzględnić zarówno okres półtrwania w osoczu są dobrze wchłaniane i w wysokim odsetku ulegają
substancji macierzystej, jak i powstających z niej ak- biotransformacji. Biodostępność leży na poziomie
tywnych metabolitów. Okres półtrwania brotizolamu 70–80%. Okres półtrwania w osoczu wynosi dla za-
i jego metabolitów wynosi 3,5–7 godzin, lormetaze- leplonu 1 godzinę, dla zolpidemu – 1,5–2,5 godziny,
pamu i temazepamu 8–16 godzin, nitrazepamu i fluni- a dla zopiklonu 3,5–6 godzin (ze względu na krót-
trazepamu (obie substancje ulegają biotransformacji ki okres półtrwania zaleplon nadaje się jedynie jako
poprzez redukcję grupy nitrowej do grupy aminowej środek ułatwiający zaśnięcie). Wydalanie zaleplonu
z wytworzeniem nieczynnych metabolitów) 15–30 i zolpidemu następuje przez nerki i z żółcią, podczas
godzin, a flurazepamu łącznie z aktywnym metaboli- gdy zopiklon wydalany jest głównie przez nerki.
tem dealkiloflurazepamem 50–100 godzin. Skądinąd Dawkowanie zaleplonu u osób dorosłych w wieku
należy zauważyć, że okresy półtrwania leków w oso- powyżej 65 lat wynosi 5 mg, u dorosłych w wieku
czu są zwykle dłuższe niż okres skuteczności danego poniżej 65 lat – 10 mg, zolpidemu u dorosłych w wie-
leku. ku powyżej 65 lat – 5–10 mg, u dorosłych w wieku
poniżej 65 lat – 10–20 mg, natomiast zopiklonu u do-
Dawkowanie. Przeciętne dawki nasenne zostały po- rosłych w wieku powyżej 70 lat – 3,75–7,5 mg i u do-
dane w tab. B 1.3-2. rosłych w wieku poniżej 70 lat – 7,5–15 mg. Leki te
Działania niepożądane, przeciwwskazania, inter- zażywa się tuż przed położeniem się spać.
akcje. Te zostały już opisane w rozdziale B 1.2.3.1.1. Działania niepożądane i przeciwwskazania odpowia-
Mimo to należy ponownie zwrócić uwagę, że w przy- dają w większości tym stwierdzanym w przypadku
padku długotrwałego zażywania tych leków i po (na- benzodiazepin.
MUTSCHLER-2009.indd 212 2010-01-07 22:11:47
Układ nerwowy 213
Tabela B 1.3-2. Benzodiazepiny i ich pochodne stosowane jako środki nasenne
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
CH3 temazepam Planum, 8-16 20-40
O Pronervon T,
N
Remestan,
OH Temazepam-CT,
Cl N Signopam
B1
N(C2H5) 2 flurazepan Dolmadorm, 50-100 15-30
O Staurodorm Neu
N
Cl N
CH3 lormetazepam Ergocalm, 8-16 0,5-2
O Lormetazepam AL.,
N
Lormetazepam-
OH -ratiopharm,
Cl N Noctamid,
Noctofer,
Loretam
Cl
H nitrazepam Etan N, 15-30 2,5-10
O Mogadan,
N Nitrazepam AL,
Radedorm
O2N N
CH3 flunitrazepam Fluninoc, 15-30 0,5-2
O Flunitrazepam-
N
-neuraxpharm,
Flunitrazepam-
O2N N -ratiopharm,
Rohypnol,
Flunimerck
F
H3C N brotizolam Lendormin 4,5-7 0,125-0,25
N
S N
Br
N
Cl
MUTSCHLER-2009.indd 213 2010-01-07 22:11:47
214 Układ nerwowy
nie zaleca się bezkrytycznego zapisywania tych leków
CN jako środka do samodzielnej terapii.
N
N N
1.3.3.4. Inne syntetyczne leki nasenne
O Pochodne kwasu barbiturowego (barbiturany). Barbi-
N CH3
turany – jak to już wcześniej zauważono – utraciły swo-
je pierwotne znaczenie jako leki nasenne i uspokajające.
C2H5 Praktycznie nie powinny być już dłużej stosowane jako leki
nasenne i do uspokojenia. Natomiast metoheksital i tiopen-
zaleplon
tal są w dalszym ciągu znaczącymi środkami do znieczu-
lenia ogólnego (narkozy); z kolei fenobarbital pozostaje
również ważnym lekiem przeciwpadaczkowym.
Wodzian chloralu (chloral hydrat) – wodzian aldehydu
N
trójchlorooctowego jest jednym z najstarszych środków
CH3
N nasennych, który w związku z miejscowym działaniem
H3C drażniającym i gorzkim, ostrym smakiem – podaje się
N(CH3) 2
doustnie w kapsułkach lub doodbytniczo w oleistym roz-
O tworze. Z szybko wchłoniętego wodzianu aldehydowego
– w wyniku redukcji – powstaje w organizmie trójchloro-
zolpidem etanol – będący główną substancją działającą i nieczynny
trójchlorowany kwas octowy. Zaletą jest to, że wodzian
chloralu nie wpływa na sen fazy REM lub w każdym razie
O nieznacznie (zob. tab. B 1.3-1). Okres półtrwania w osoczu
N N trójchloroetanolu wynosi 4–12 godzin.
N Cl Oprócz działania nasennego – wodzian chloralu bywa
wykorzystywany w stanach pobudzenia i drgawkowych.
N O
O Przeciętna dawka dla dorosłych wynosi 0,5–1,5 g. Za-
kres dawek terapeutycznych jest mały (dawka śmiertelna:
N 6–10 g).
Jako możliwe działania niepożądane zostały opisane
N stany splątaniowe, nudności i reakcje alergiczne. Podobnie
CH3
jak to się ma z innymi związkami chlorowcopochodnymi
istnieje ryzyko uczulenia mięśnia sercowego na katecho-
zopiklon laminy.
W przypadku przewlekłego stosowania może ujawnić
się uzależnienie.
Wodzian chloralu jest przeciwwskazany w przypadku
ciężkich zaburzeń funkcji wątroby, w niewydolności nerek
(niebezpieczeństwo kumulacji!) i niewyrównanej niewy-
dolności serca.
1.3.3.3. Leki przeciwhistaminowe H1
OH
Przede wszystkim należące do starszej generacji leki
przeciwhistaminowe H1 wywołują na skutek blokady Cl3C OH
ośrodkowych receptorów H1 działanie uspokajające
i nasenne. Dlatego też wodzian chloralu
■ difenhydramina oraz
■ doksylamina
Talidomid był uważany za najbezpieczniejszy środek
zostały swego czasu wprowadzone do wolnej sprze- nasenny, ponieważ nawet przy dużym przedawkowaniu
daży (bez recepty) na rynku jako leki nasenne, zanim nie występowały żadne niebezpieczne objawy zatrucia. Tym
nie zostały zastąpione przez inne leki nasenne, które bardziej zaskakujące było alarmujące odkrycie, że substan-
cja ta jest odpowiedzialna za nagły skokowy wzrost liczby
już wymagały przepisywania na receptę. Również poważnych wad rozwojowych u płodów. Ponadto okazało
dla tych substancji obowiązują wymienione wyżej się, że przyjmowanie talidomidu przez dłuższy okres może
kryteria stosowania leków nasennych. Zdecydowanie doprowadzić do ciężkiego zapalenia wielonerwowego
MUTSCHLER-2009.indd 214 2010-01-07 22:11:47
Układ nerwowy 215
1.3.4. Kryteria stosowania leków
O nasennych
N O Leki nasenne powinny być stosowane jedynie wów-
Układ nerwowy
NH
czas, gdy nie można opanować przyczyny zaburzeń
O O
snu lub też kiedy inne środki, np. trening autogenny,
talidomid okazały się nieskuteczne. Osoby w podeszłym wie-
ku powinny być poinformowane o tym, że ich za-
potrzebowanie na sen w sposób fizjologiczny uległo
z częściowo nieodwracalnymi zmianami. Po wykryciu tych
zmniejszeniu i stąd konieczny czas snu uległ skróce-
działań niepożądanych wycofano z rynku wszystkie prepa- niu. W przypadku łagodnych zaburzeń snu powinno B1
raty nasenne zawierające talidomid. się spróbować rozpocząć leczenie od stosowania le-
ków pochodzenia roślinnego, szczególnie tych za-
wierających walerianę – choćby z powodu silnego
1.3.3.5. Leki nasenne efektu placebo. Jeżeli przepisanie leku nasennego
pochodzenia roślinnego jest rzeczywiście konieczne, to należy sprawdzić,
jakiego rodzaju zaburzenia snu występują u pacjen-
Jako leki nasenne pochodzenia roślinnego stosowa- ta: zaburzenia zasypiania czy też zaburzenia prze-
ne są wyciągi z korzenia kozłka lekarskiego, szyszek biegu/trwania snu. W celu ułatwienia zaśnięcia na-
i gruczołów wydzielniczych (otarte z wysuszonych leży stosować preparaty o szybko ujawniającym się
szyszek włoski gruczołowe) chmielu, liści melisy działaniu i krótkim czasie działania (leki ułatwiające
oraz kwiatostanów męczennicy. Najlepiej udoku- zaśnięcie), np. brotizolam, triazolam lub zaleplon.
mentowane jest działanie waleriany. Dla pozostałych Natomiast w przypadku zbyt płytkiego snu i przy
leków roślinnych brakuje wiarygodnych wyników przedwczesnym budzeniu się należy stosować leki
badań klinicznych. o dłuższym czasie działania (leki podtrzymujące sen),
np. lormetazepam, temazepam lub zopiklon. Długo
Kozłek lekarski. Jako lek wykorzystywany jest głów- działające leki nasenne, np. flurazepam lub nitraze-
nie korzeń kozłka lekarskiego (Valeriana officinalis). pam, są wskazane wówczas, gdy trzeba uzyskać rów-
Mimo licznych badań nie udało się stwierdzić, któ- nież efekt uspokajający w trakcie (następnego) dnia
ry ze związków składowych w pierwszej kolejności lub przynajmniej nie jest on niepożądany. W przy-
odpowiedzialny jest za ujawniające się działanie padku ordynowania benzodiazepin i ich pochod-
uspokajające lub nasenne. Do tych efektów przypusz- nych osobom w podeszłym wieku należy sprawdzić,
czalnie przyczyniają się kwas walerianowy i kwas czy trzeba indywidualnie dopasować (zmniejszyć)
acetoksywalerianowy, a także produkty rozkładu wa- dawkę – z powodu osłabionych procesów eliminacji.
lepotriatów (tzw. baldrinale) oraz olejek eteryczny. Ponadto starsze osoby należy poinformować o możli-
W badaniach na synaptosomach udało się za pomocą wości występowania upadków – z powodu działania
wyciągu z waleriany wykazać zwiększenie wyrzutu polegającego na zmniejszeniu napięcia mięśniowego.
i równocześnie zmniejszenie wchłaniania zwrotnego Zalecając leki nasenne, należy ponadto za każdym
GABA; z kolei kwas walerianowy wchodził w inter- razem zwracać uwagę na to, żeby były one w miarę
akcje z receptorem GABAA. U pacjentów ujawnia się możliwości stosowane w sposób czasowo ograniczo-
pewna poprawa snu (szybsze osiągnięcie głębokich ny. Jednak w przypadku ciężkich przewlekłych zabu-
faz snu oraz nieznaczne zwiększenie się czasu trwa- rzeń snu może być niekiedy konieczne długotrwałe
nia snu głębokiego). Toteż wyciągi z kozłka lekar- leczenie – także ze względu na niebezpieczeństwo
skiego nadają się do terapii łagodnych zaburzeń snu. wystąpienia wtórnych chorób, np. depresji. W końcu
Typowe dawkowanie przy zaburzeniach zasypia- nie można pominąć faktu, że leki nasenne są naduży-
nia wynosi 200–600 mg suchego wyciągu. wane tak jak żadna inna grupa leków.
MUTSCHLER-2009.indd 215 2010-01-07 22:11:48
216 Układ nerwowy
1.4. Analeptyki
Analeptyki pobudzają określone okolice ośrodkowego Należy liczyć się z następującymi działaniami niepożą-
układu nerwowego – głównie ośrodek oddechowy danymi: z zaburzeniami ośrodkowymi (np. bólami i zawro-
i naczynioruchowy (wazomotoryczny) – znajdujące się tami głowy, lękiem), zaburzeniami oddychania, objawami
w obrębie rdzenia przedłużonego (medulla oblongata). żołądkowo-jelitowymi i zaburzeniami rytmu serca.
W większych dawkach stają się truciznami drgawkowy- Przeciwwskazaniami są padaczka, nadciśnienie, choro-
mi. Obecnie prawie całkowicie utraciły swoje wcześniej- ba niedokrwienna serca, niewyrównana niewydolność ser-
sze znaczenie. ca, choroby układu oddechowego, udar mózgu.
W przypadku zatrucia opiatami preferuje się obecnie
antagonistów opioidowych, przy ośrodkowej zapaści krą-
żeniowej – substancje działające obwodowo (zob. sympa-
C2H5
tykomimetyki), a w przypadku depresji ośrodka oddecho-
wego – sztuczne oddychanie. N O
Doksapram jest jedynym dostępnym w handlu lekiem N
analeptycznym. Okres półtrwania w osoczu wynosi 5–15 O
minut. Stosowany jest w dawce 0,5–1,5 mg/kg m.c. poda-
wany powoli dożylnie lub 1–5 mg/min w ciągłym wlewie
w celu stymulacji oddychania u pacjentów z występującym doksapram
po znieczuleniu lub wyzwolonym lekami zahamowaniem
oddychania.
MUTSCHLER-2009.indd 216 2010-01-07 22:11:48
Układ nerwowy 217
1.5. Analgetyki (leki przeciwbólowe)
Za leki przeciwbólowe uznaje się substancje, które 1.5.1.1. Przyczyny bólu, rodzaje bólu
Układ nerwowy
zmniejszają lub też znoszą odczuwanie bólu, nie wy-
wołując przy tym działania narkotycznego. Na pod- Ból jest nieprzyjemnym doznaniem zmysłowym i po-
stawie mocy i mechanizmu działania oraz działań wstaje wówczas, gdy bodźce mechaniczne, termicz-
niepożądanych – rozróżnia się dwie główne grupy ne, chemiczne lub elektryczne przekroczą wartość
leków przeciwbólowych: graniczną (próg bólowy) – prowadząc (zwykle)
do uszkodzenia tkanek z uwolnieniem mediatorów
■ leki przeciwbólowe opioidowe (analgetyki opioi-
bólu (substancje algogenne; zob. poniżej) oraz na-
dowe, opioidy, opiaty, narkotyczne środki przeciw-
stępczego wstępującego (aferentnego) przewodzenia B1
bólowe = narkoanalgetyki, hipnoanalgetyki, silnie
impulsów bólowych. Wyzwalanie bodźca bólowego,
działające analgetyki) – głównie z ośrodkowym,
jego dalsze przewodzenie i przetwarzanie w obrębie
lecz również obwodowym działaniem,
ośrodkowego układu nerwowego określane jest mia-
■ nieopioidowe leki przeciwbólowe (słabe/małe anal- nem czucia (zmysłu) bólu (nocycepcji).
getyki) – charakteryzujące się działaniem zarówno Ze względu na etiologię i patofizjologię można
obwodowym, jak i ośrodkowym, a ponadto posia- wyróżnić trzy ważne rodzaje doznań bólowych:
dające właściwości przeciwgorączkowe i przeciw-
■ fizjologiczny ból z receptorów bólowych (fizjolo-
zapalne.
giczny ból nocyceptywny/nocyceptorowy),
Wcześniejszy podział na ośrodkowe, silnie dzia- ■ patologiczny ból z receptorów bólowych (patolo-
łające i obwodowe, słabo działające analgetyki giczny ból nocyceptywny/nocyceptorowy),
jest na podstawie nowszych danych przestarzały,
■ ból neuropatyczny.
gdyż opiody posiadają także działanie obwodowe,
a leki przeciwbólowe nieopioidowe wykazują dzia-
Fizjologiczny ból z receptorów bólowych powstaje
łania ośrodkowe.
jako sygnał ostrzegawczy w przypadku oddziaływania
bodźców mechanicznych (np. ucisku), chemicznych
(np. kwasu) lub termicznych (np. gorąca) na zdrową
1.5.1. Patofizjologia bólu tkankę. Reakcja bólowa jest wyzwalana przez po-
budzenie zakończeń czuciowych nocyceptorów (re-
Ból jest jednym z najczęstszych objawów miejsco- ceptorów bólu/receptorów bólowych). Prowadzi ona
wego uszkodzenia tkanek lub objawem choroby, do natychmiastowej odruchowej reakcji ruchowej
a zarazem najczęstszą przyczyną wizyty u lekarza. (np. cofnięcie ręki po dotknięciu przez nią gorącej
Wprawdzie ból, zwłaszcza ostry (zob. poniżej), płyty kuchennej; reakcja ucieczkowa), która zapobie-
spełnia zazwyczaj pożyteczną rolę ostrzegawczą ga uszkodzeniu tkanki (zob. ryc. B 1.5-1 A). Fizjo-
i ochronną oraz stanowi dla lekarza ważny ob- logiczny ból ma żywotne znaczenie, aby spostrzegać
jaw w ustalaniu rozpoznania, ale może być też wszelkiego rodzaju zranienia i aby móc zapobiegać
on, zwłaszcza jeśli przybierze postać przewlekłą, wynikającemu z niego dalszemu okaleczeniu.
niekorzystny i stać się przyczyną udręki. Dla od-
czuwających go chorych ból jest stale uciążliwy. Patofizjologiczny ból z receptorów bólowych (ból
Dlatego też konsekwentne opanowywanie bólu ma zapalny) powstaje w wyniku uszkodzenia tkanki lub
szczególne znaczenie. W przebiegu niektórych cho- też procesu zapalnego, i może wyrażać się pod po-
rób, np. w końcowej fazie choroby nowotworowej, stacią bólu spoczynkowego (resting pain), hiperal-
adekwatne zwalczanie bólu staje się nawet jedynym gezji i allodynii. Przez pojęcie hiperalgezji rozumie
możliwym postępowaniem lekarskim. się nasilone odczuwanie bólu na bodziec uszkadza-
Wrażliwe na bodźce bólowe są, oprócz całej po- jący, przy czym hiperalgezja ograniczona do miejsca
wierzchni skóry i większej części błon śluzowych, uszkodzenia tkanki nazywa się pierwotną, natomiast
również liczne tkanki i narządy wewnętrzne. Jednak- obejmująca otoczenie określana jest jako wtórna.
że istnieją także narządy pozbawione receptorów bó- Mechanizmy zarówno pierwotnej, jak i wtórnej hi-
lowych, np. mózg lub wątroba. peralgezji są złożone i polegają na procesach ob-
MUTSCHLER-2009.indd 217 2010-01-07 22:11:48
218 Układ nerwowy
obwód OUN skutek
A. Fizjologiczny ból nocyceptywny ból
np. ukłucie igłą krótko-
trwały
badanie elektrofizjologiczne
bodziec nie- bodziec (czas)
nocyceptywny nocyceptywny
B. Patofizjologiczny ból nocyceptywny ból
(ból w stanie zapalnym)
np. zapalenie utrzymujący się
badanie elektrofizjologiczne
(allodynia) (hiperalgezja)
bodziec bodziec (czas)
nienocyceptywny nocyceptywny
C. Ból neuropatyczny ból
uszkodzenie
nerwu obwodowego niepra-
lub OUN widłowy
mechanoreceptory
badanie elektrofizjologiczne (allodynia)
samoistne wyładowania (czas)
Ryc. B 1.5-1. A-C. Rodzaje bólu zgodnie z ich etiologią i patogenezą (według Cervera i in. oraz Schaible’a i in.). A – fizjologiczny
ból nocyceptywny. Potencjały czynnościowe występują dopiero po zadziałaniu bodźca nocyceptywnego. B – patofizjologiczny ból
nocyceptywny. Już bodźce niebędące bodźcami nocyceptywnymi wyzwalają potencjały czynnościowe (allodynia). Z kolei bodź-
ce nocyceptywne prowadzą do wyzwolenia większej ilości potencjałów czynnościowych (hiperalgezja). C – ból neuropatyczny.
Spontaniczne wyładowania po uszkodzeniu nerwów.
MUTSCHLER-2009.indd 218 2010-01-07 22:11:48
Układ nerwowy 219
wodowej i ośrodkowej sensybilizacji (zwiększania bodźce termiczne, chemiczne i mechaniczne ulegają
wrażliwości) w układzie przetwarzającym bodźce zamianie na sygnały elektryczne (potencjały czyn-
bólowe. W przypadku gdy ból może być wywoła- nościowe). Nocyceptory zmieniają przy tym swoje
ny przez bodźce, które zwykle nie wywołują wra- właściwości elektrofizjologiczne: albo bezpośrednio
Układ nerwowy
żeń bólowych – można mówić o allodynii (np. ból – np. w wyniku aktywowania kanałów jonowych,
przy dotknięciu skóry w oparzeniu słonecznym; zob. albo pośrednio – za pomocą wewnątrzkomórkowych
ryc. B 1.5-1 B). dróg transdukcji sygnału, co ostatecznie prowadzi
do zmiany progu pobudzenia (sensybilizacja) recep-
Bóle neuropatyczne powstają w sytuacji, kiedy doj- torów bólowych. Na ryc. B 1.5-2 pokazano w sche-
dzie do uszkodzenia nerwów w wyniku zmiażdżenia, matyczny sposób wolne nocyceptywne zakończenia
ucisku (np. wypadnięcia dysku międzykręgowego), czuciowe (nocyceptory) wraz z najważniejszymi dla
przecięcia (np. w wyniku amputacji), zapalenia ich pobudzenia i sensybilizacji mediatorami. B1
(np. w półpaścu) lub też zaburzeń metabolicznych
(np. w przebiegu cukrzycy). W obrębie błon uszko- Po ostrym uszkodzeniu tkanek ze zniszczonych ko-
dzonych nerwów oraz w ich pobliżu dochodzi do po- mórek zaczynają się uwalniać ATP i jony wodoru
wstania nowych białek receptorowych i kanałowych, (protony) oraz serotonina z płytek krwi. Wymienio-
z następowym generowaniem ekotopowych impul- ne mediatory mogą pobudzić odpowiednie receptory
sów nerwowych (spontaniczne potencjały czynnoś- bólowe w sposób bezpośredni (fizjologiczny ból no-
ciowe w zranionej okolicy uszkodzonego nerwu). cyceptywny):
W wyniku utrzymującej się aktywności uszkodzone-
■ ATP (trifosforan adenozyny), występujący w stę-
go nerwu – w ośrodkowym układzie nerwowym po-
żeniach milimolarnych, ulega natychmiastowemu
wstają zmiany neuroplastyczne. W tych warunkach
uwolnieniu ze wszystkich uszkodzonych komórek,
ośrodkowe neurony nocyceptywne mogą być pobu-
aktywuje receptory purynowe P2X3, które w wy-
dzone także przez niskoprogowe mechanoreceptory
niku napływu jonów wapniowych i sodowych ge-
(allodynia dotykowa) lub receptory zimna (allodynia
nerują w nocyceptorze potencjały czynnościowe
wywoływana zimnem). Bóle neuropatyczne mają
i tym samym wyzwalają natychmiastowe wrażenie
nieprawidłowy charakter i mogą być bardzo dotkli-
bólu (ryc. B 1.5-3).
we. Ponadto mogą im towarzyszyć ruchowe i czucio-
we objawy ubytkowe. Bóle powstające przy uszko- ■ Protony pobudzają kanały bramkowane jonami
dzeniu neuronów ośrodkowych są określane jako bóle wodorowymi H+ (ASIC – acid-sensing ion chan-
ośrodkowe. Szczególną postacią bólów ośrodkowych nel) oraz tzw. kanały związane z waniloidowym
są bóle fantomowe albo bóle deaferencyjne, do któ- receptorem przejściowego potencjału TRPV1
rych dochodzi w wyniku niedostatecznego/braku do- (TRP – transient receptor potential vanniloid),
pływu bodźców aferentnych (np. po amputacji) pro- w wyniku czego również dochodzi do napływu
wadzącego do nieprawidłowej pobudliwości i ak- jonów wapniowych i sodowych (ryc. B 1.5-3).
tywności neuronów tylnego rogu rdzenia kręgowego W przypadku obniżenia się wartości pH poniżej 6
(zob. ryc. B 1.5-1C). dochodzi do odczuwania bólu, które zwiększa się
w przypadku dalszego obniżania się stężenia H+.
Kanały TRP stanowią rodzinę białek, do których
należy receptor waniloidowy typ 1 (VR1) i stąd
1.5.1.2. Powstawanie bólu
określany jest łącznie jako kanał TRPV1. Receptor
i jego przetwarzanie pobudzany jest przede wszystkim przez protony,
uszkadzające gorąco i kapsaicynę (substancję za-
Ból wywołany jest przez uszkodzenie tkanki lub zabu- wartą w papryce i nadającą jej ostry smak), a zwią-
rzenia metabolizmu tkankowego. Przy tym dochodzi zany z nim kanał jest wykorzystywany głównie
do uwalniania się z uszkodzonych komórek substan- przez jony wapnia i sodu (napływ do wnętrza ko-
cji endogennych, tzw. mediatorów bólu, lub następu- mórki). Endogenne mediatory lipidowe, np. anan-
je tam ich syntetyzowanie. Substancje te pobudzają damid lub prostaglandyny, mogą również wpły-
receptory bólowe lub je uwrażliwiają. Przez pojęcie wać na te kanały. Uszkadzające zimno odbierane
receptorów bólu/bólowych (nocyceptorów) rozumie jest przez inne receptory z tej rodziny, np. kana-
się wolne zakończenia włókien czuciowych C i Aδ, ły TRPM8 (TRPM – kanał TRP melastatynowy;
które odbierają w tkankach bodźce bólowe. Nocy- TRPM8 – kanał TRPM aktywowany także mento-
ceptory wyposażone są w liczne kanały jonowe oraz lem – przyp. tłum.). Również transdukcja (uszka-
receptory dla mediatorów bólu, za pomocą których dzających) bodźców mechanicznych posiłkuje się
MUTSCHLER-2009.indd 219 2010-01-07 22:11:49
220 Układ nerwowy
uszkodzenie tkanki,
uraz,
zapalenie
naskórek
korzeń włosa skóra
właściwa
5-HT
powiększenie: zob. ryc. B 1.5-4 prostaglandyny bradykinina
tkanka
nacz ATP histamina
ynie
krw
ionośn substancja P
e H+ NGF
substancja P
CGRP
powiększenie: zob. ryc. B 1.5-6 ekspresja genów
zwój rdzeniowy bodziec receptor
NGF TrkA
do mózgu
bradykinina B1, B2
serotonina (5-HT) 5-HT3
ATP P2X3
H+ TRPV1, ASIC
prostaglandyna EP, IP
gorąco TRPV1
zimno TRPM8
rdzeń kręgowy
bodziec mechaniczny TRPA1
Ryc. B 1.5-2. Po uszkodzeniu tkanki lub urazie dochodzi do uwolnienia wielu mediatorów, które prowadzą do procesu zapalnego.
Mediatory te aktywują lub uwrażliwiają nocyceptory. Na dole po prawej stronie ryciny zestawione zostały najważniejsze mediato-
ry lub bodźce wraz z aktywowanymi przez nie receptorami lub kanałami (bliższe informacje w tekście). NGF (nerve growth factor)
– czynnik wzrostu nerwów; 5-HT – serotonina; ATP – trifosforan adenozyny; H+ – proton; CGRP (calcitonin gene-related peptide) – pep-
tyd związany z genem kalcytoniny; TrKA – receptor A kinazy tyrozynowej; TRP (transient receptor potential) – przejściowy potencjał
receptorowy; ASIC (acid-sensing ion channel) – kanały bramkowane jonami H+; EP – receptor prostaglandyny E; IP – receptor prosta-
glandyny I (prostacykliny) (według Juliusa i Basbauma).
częściowo receptorami (kanałami) z rodziny TRP nerwowego – neurony serotoninergiczne mające
(TRPA1; kanał TRP ankyrynowy – przyp. tłum.) swój początek w jądrach szwu działają hamująco
(zob. ryc. B 1.5-3). na odczuwanie bólu.
■ Serotonina, uwalniana z trombocytów, pobudza W przypadku ostrego bólu nie uczestniczą jeszcze
receptory 5-HT3, które również przyczyniają się żadne komórki immunokompetentne. Te zostają ak-
do napływu wapnia i sodu. Na obwodzie serotonina tywowane i dołączają się dopiero w ramach procesu
jest bardzo skutecznym przekaźnikiem wyzwalają- zapalnego. Bóle zapalne (patologiczny ból nocycep-
cym ból. Natomiast w obrębie ośrodkowego układu tywny) prowadzą spontanicznie do aktywacji i mi-
MUTSCHLER-2009.indd 220 2010-01-07 22:11:49
Układ nerwowy 221
Układ nerwowy
gorąco kwas (H+) bodziec zimno
mechaniczny
naskórek
B1
skóra korzeń
właściwa włosa
ATP
(uszkodzenie komórki)
tkanka
podskórna TRPM8
TRPV1 TRPA1
ASIC P2X3
+
Ca2+ Na+ Na+ Ca2+ Na
Ca2+ Na+ Ca2+
wolne czuciowe Ca2+ Na+
zakończenie nerwu:
nocyceptor
(w dużym powiększeniu)
Na+
Nav 1,8/1,9
potencjał czynnościowy
do rogu tylnego rdzenia kręgowego
Ryc. B 1.5-3. Wolne czuciowe, nocyceptywne zakończenie nerwu, które pobudzane jest za pomocą różnych bodźców (gorąco, kwas,
bodziec mechaniczny, zimno) (bliższe informacje w tekście, według Marchanta i in.). ATP – trifosforan adenozyny; ASIC (acid-sensing
ion channel) – kanały bramkowane jonami wodorowymi H+; TRP (transient receptor potential) – kanał receptora przejściowego po-
tencjału; P2X3 – receptor P2X3-ATP.
gracji z krwi określonych komórek (granulocytów ■ Cytokininy TNF-α (czynnik martwicy nowotwo-
obojętnochłonnych, makrofagów, komórek tucznych), rów – tumor necrosis factor) i IL-β (interleukina β)
które z kolei uwalniają lub syntetyzują mediatory za- prowadzą do bezpośredniego aktywowania nocy-
palne i bólowe – w ten sposób wyzwalając objawy ceptorów. Łączą się z odpowiednimi receptorami
zapalenia: obrzęk, zaczerwienienie, ból i przeczulicę (receptor TNF-α i receptor IL-1β) komórek zapal-
(zob. ryc. B 1.5-4). nych (np. makrofagów) i wywołują m.in. aktywa-
cję czynników transkrypcji (np. NKκB), które na-
Granulocyty obojętnochłonne (neutrofile) są pierw- stępnie w jądrze komórkowym rozpoczynają pro-
szymi komórkami zapalnymi, które wwędrowują ces trankrypcji genów zapalnych. Najważniejszym
z krwi do tkanki dotkniętej procesem zapalnym. Pro- z farmakologicznego punktu widzenia dla bólów
dukują głównie cytokininy (TNF-α, IL-β i in.), czyn- zapalnych produktem genowym jest cyklooksyge-
nik wzrostu nerwu (NGF – nerve growth factor) i leu- naza 2 (COX-2), której ekspresja ulega masywnej
kotrieny, które są głównymi mediatorami bólowymi indukcji. COX-2 katalizuje przemianę kwasu ara-
(zob. ryc. B 1.5-4). chidonowego do prostaglandyn. Skutkiem jest nad-
mierna produkcja prostaglandyn (głównie prosta-
MUTSCHLER-2009.indd 221 2010-01-07 22:11:49
222 Układ nerwowy
uwalnianie m
ediatoró
w
bradykinina stymulu
uwalnianie je
substancja P
mediatorów makrofag
komórka tuczna neutrofil
TNF-α PLA2
AA IL-1β
COX-2 ↑
T
Y
R
receptor
IL-1β cytokininowy NGF
NGF LTs
bradykinina
histamina NGF TNF-α
PGs
z.B. PGE2
TRPV1
TRPV1 TrkA EP2
B2 H1
Gq Gq Gs
cykleany-
ad owa
P
lan
Gq
T cAMP
NK1
za
P +
PKC Y Ca2+ Na
Ca2+ Na +
R
PKC
PKA ATP
PKC
PKC
Na+
P 1,9
substancja P 1,8/
Ca2+ ↑ Na v
Na+
nacz
ynie
krw
ionośn
e
wynaczynienie osocza potencjał czynnościowy
do rogu tylnego rdzenia kręgowego
Ryc. B 1.5-4. W bólach zapalnych (patofizjologiczny ból nocyceptywny) aktywowaniu ulegają granulocyty obojętnochłonne (neu-
trofile), makrofagi i komórki tuczne, które uwalniają określone mediatory (bliższe informacje w tekście). NGF (nerve growth factor)
– czynnik wzrostu nerwu; TNF-α (tumor necrosis factor) – czynnik martwicy nowotworów; IL-1β – interleukina 1β; LT – leukotrieny;
PLA2 – fosfolipaza A2; AA (arachidonic acid) – kwas arachidonowy; COX-2 – cyklooksygenaza 2; PG – prostaglandyny; EP2 – receptor
prostaglandyny E typu 2; cAMP – cykliczny AMP (monofosforan adenozyny); PKA – kinaza białkowa A; TRP (transient receptor potential)
– kanał receptora przejściowego potencjału; TrkA – receptor A kinazy tyrozynowej; PKC – kinaza białkowa C; NK1 – receptor neurokini-
ny 1; Naν 1,8/1,9 – izoformy napięciowozależnych kanałów sodowych; B2 – receptor bradykininy 2; H1 – receptor histaminy typu 1.
glandyny E2, PGE2). PGE2 pobudza znajdujące się A (PKA). Jej aktywacja wywołuje w nocycepto-
na błonie nocyceptora związane z białkiem G re- rze fosforylację swoistych napięciowozależnych
ceptory EP z następczą aktywacją cyklazy adenyla- kanałów sodowych Naν 1,8/1,9 (izoformy kana-
nowej – powodującą nadmierną produkcję cAMP. łów sodowych ulegające specyficznej ekspresji
Ponadto dochodzi do stymulacji kinazy białkowej jedynie w obrębie nocyceptorów) oraz kanałów
MUTSCHLER-2009.indd 222 2010-01-07 22:11:50
Układ nerwowy 223
TRPV1. W efekcie końcowym dochodzi z jednej W przypadku aktywowania lub sensybilizacji nocy-
strony do obniżenia progu pobudliwości błony no- ceptorów – w sposób opisany powyżej – ich same
cyceptorów i tym samym potencjały czynnościo- obwodowe zakończenia wytwarzają mediatory bó-
we są łatwiej wyzwalane (rekrutacja tzw. śpiących lowe. Najważniejszym syntetyzowanym przez no-
Układ nerwowy
nocyceptorów). Z drugiej strony obniża się rów- cyceptory mediatorem bólu jest neuropeptyd sub-
nież próg bólu wywołanego gorącem w kanałach stancja P. Z jednej strony neuropeptydy pobudzają
TRPV1 – co powoduje, że temperatury zwykle nocyceptory przez odpowiednie dla nich receptory,
nieodbierane jako bolesne, zaczynają wyzwalać przy czym przykładowo substancja P aktywuje recep-
doznania bólowe (zob. ryc. B 1.5-4). tor neurokininy 1 (NK-1). Z drugiej strony zmuszają
także komórki tuczne i makrofagi do dalszej synte-
■ Również czynnik wzrostu nerwu (NGF – nerve
zy i uwalniania mediatorów zapalnych i bólowych.
growth factor) nie aktywuje nocyceptorów pośred-
Ponadto substancja P wywołuje rozszerzenie naczyń B1
nio. Łaczy się z receptorem A kinazy tyrozynowej
przedwłosowatych oraz przesiękanie osocza z naczyń
(TrkA) na błonie nocyceptora i prowadzi do fosfo-
zawłosowatych (zob. ryc. B 1.5-4).
rylacji kanałów TRPV1 – wykorzystując do tego
drogę metaboliczną za pomocą kinazy białkowej
Impulsy nerwowe, pochodzące z nocyceptorów
aktywowanej mitogenem (MAP – mitogen activa-
skóry, mięśni szkieletowych i stawów, przewodzo-
ted protein) oraz kinazy białkowej C (PKC) (zob.
ne są włóknami bezrdzeniowymi C oraz włóknami
ryc. B 1.5-4). Stąd też NGF jest kluczowym me-
rdzeniowymi Aδ w kierunku do rdzenia kręgowego
diatorem uczestniczącym w powstaniu termicznej
(zob. ryc. 1.5-5). Przewodzenie impulsów bólowych
hiperalgezji (zob. powyżej).
przychodzących z okolicy trzewnej następuje w prze-
■ Leukotrieny (w tym zasadniczo leukotrien B4) ważającej mierze za pomocą włókien C.
prowadzą do sensybilizacji nocyceptorów praw-
dopodobnie również przez fosforylację kanałów Wstępujące (aferentne) włókna kończą swoją drogę
TRPV1. w rogach tylnych rdzenia kręgowego, gdzie uwalniają
m.in. neuropeptydy w rodzaju substancji P oraz po-
Również makrofagi i komórki tuczne stanowią ważne budzający aminokwas – kwas glutaminowy. Gluta-
źródło mediatorów bólowych. Makrofagi albo znaj- minian może po stronie postsynaptycznej aktywować
dują się już w tkance zmienionej procesem zapalnym, jonotropowe receptory NMDA (N-metylo-D-aspara-
albo wwędrowują do niej z krwi – będąc zwabiane ginianu), jonotropowe receptory non-NMDA (recep-
tam przez chemotaktyczne cytokininy i chemokini- tory AMPA i kainowe) oraz metabotropowe recepto-
ny. Produkują one głównie cytonikiny TNF-α i IL-β, ry glutaminergiczne. Receptor NMDA jest zwykle
NGF oraz prostaglandyny. Również one przyczynia- zamknięty w wyniku bloku magnezowego. Dopiero
ją się istotnie do powstania bólów zapalnych (zob. w wyniku częściowej depolaryzacji neuronu rdzenio-
powyżej). wego poprzez substancję P (w receptorze NK-1) oraz
Komórki tuczne wyzwalają w szczególności NGF poprzez glutaminian (w receptorze AMPA) może
(zob. powyżej) i histaminę, która również może pro- dojść do zniesienia bloku magnezowego receptorów
wadzić do uwrażliwienia (sensybilizacji) nocycepto- NMDA, w wyniku czego dochodzi do masywnego
ra (zob. ryc. 1.5-4). napływu jonów wapniowych i całkowitej depolary-
Kolejnym, bardzo ważnym mediatorem bólowym zacji.
jest bradykinina. Powstaje ona w osoczu w wyniku Z rogu tylnego rdzenia kręgowego informacja
proteolitycznego rozpadu kininogenu i w wyniku przekazywana jest albo bezpośrednio, albo przez
wynaczynienia (przesączania) przedostaje się do za- neuron wstawkowy (interneuron) na następny neu-
palnej tkanki. Poprzez swoje rozmieszczone na bło- ron, którego akson ulega skrzyżowaniu i podąża
nie nocyceptorów receptory (B1 i B2) aktywowana do góry pod postacią drogi rdzeniowo-wzgórzowej
jest kinaza białkowa C (PKC), która fosforyluje ka- (bocznej; tractus spino-thalamicus lateralis) (zob.
nały TRPV1 i w ten sposób doprowadza do obniżenia ryc. 1.5-5). Ostatnie przełączenie następuje w okolicy
progu odczuwania temperatury dla bodźca bólowego, jąder bocznych wzgórza, skąd seria impulsów trafia
jakim jest gorąco. Ponadto bradykinina może stymu- do sensorycznych pól projekcyjnych kory mózgowej,
lować w makrofagach fosfolipazę A2, która uwalnia tu – do zakrętu zaśrodkowego (gyrus postcentralis).
kwas arachidonowy z błony fosfolipidowej i w ten Wymieniona okolica kory mózgowej wraz ze wzgó-
sposób pobudza metabolizm prostaglandyn. W ten rzem odpowiedzialna jest za świadome odbieranie
sposób bradykinina stymuluje syntezę prostaglandyn wrażeń bólowych, w szczególności za lokalizację
de novo (zob. ryc. 1.5-4). i ocenę nasilenia odbieranych bodźców bólowych.
MUTSCHLER-2009.indd 223 2010-01-07 22:11:51
224 Układ nerwowy
zstępujący
kora układ hamujący
wzgórze
miejsce sinawe
przednia część mostu noradrenalina
PAG
enkefaliny
GABA
jądra szwu
serotonina
jądro siatkowate
przyolbrzymiokomórkowe
rdzeń przedłużony enkefalina
serotonina
sznur
przednio-boczny sznur
grzbietowo-boczny
H+, NGF, PGE2, SP,
rdzeń kręgowy 5-HT, BK, Hist i in.
glutaminian, SP, PG, skóra
NO, 5-HT, NA, ENK,
GABA, CGRP,
adenozyna i in.
Ryc. B 1.5-5. Wstępujący układ nocyceptywny i zstępujący układ antynocyceptywny. PAG (periaqueductal gray) – istota szara oko-
łowodociągowa; BK – bradykinina; Hist – histamina; SP – substancja P; NA – noradrenalina; NGF (nerve growth factor) – czynnik
wzrostu nerwu; NO – tlenek azotu; PG – prostaglandyny; 5-HT – serotonina; GABA – kwas gamma- aminomasłowy; CGRP (calcitonin
gene-related peptide) – peptyd związany z genem kalcytoniny; ENK – enkefalina (według Cousinsa i in.).
W reakcjach emocjonalnych, które są wyzwalane
przez ból, uczestniczy układ limbiczny. Wegetatyw- nie nocyceptorów prowadzi do ich uwrażliwienia (nad-
ne reakcje towarzyszące bólowi są sterowane przez wrażliwość/sensybilizacja obwodowa), a także zmian
podwzgórze. wrażliwości w obrębie rdzenia kręgowego (nadwraż-
liwość/sensybilizacja ośrodkowa). W nadwrażliwości
obwodowej obniża się próg pobudzenia w następstwie
nasilonego tworzenia lub uwalniania mediatorów bó-
1.5.1.3. „Pamięć bólu” lowych (zob. powyżej oraz ryc. B 1.5-4). W ramach
ośrodkowej sensybilizacji w rdzeniu kręgowym w wy-
Ból jest zjawiskiem fizjologicznym niezbędnym z ży- niku serii krótkotrwałych lecz intensywnych bodźców
ciowego punktu widzenia. W wyniku procesu chroni- bólowych dochodzi do aktywowania wewnątrzkomór-
fikacji traci on jednakże swój ostrzegawczy charakter kowych kinaz (np. PKA, PKC), które fosforylują ka-
i staje się bólem patologicznym. Powtarzane pobudza- nały jonowe i receptory (zob. ryc. 1.5-6). Szczególne
MUTSCHLER-2009.indd 224 2010-01-07 22:11:51
Układ nerwowy 225
znaczenie mają aktywowane glutaminianem receptory w obrębie interneuronów hamujących, tak więc przez
NMDA, które – po tym, jak ulegną procesowi fosfo- PGE2 zostaje uniemożliwione endogenne hamowanie
rylacji – przemieszczają się w okolicę synaptycznej (zob. ryc. B 1.5-7).
błony komórkowej i tam łatwiej ulegają pobudzeniu. Jeżeli nie zapobiegnie się opisanym procesom
Układ nerwowy
W wyniku przedłużonego prawdopodobieństwa otwo- ośrodkowej sensybilizacji lub też nie zostaną one
rzenia receptorów NMDA napływa wapń do wnętrza wystarczająco wcześnie przerwane, ból może ulec
neuronów, co prowadzi do zwiększenie pobudliwości chronifikacji („pamięć bólu”, „pamięć doznań bólo-
neuronów. Ta wczesna faza ośrodkowej sensybilizacji wych”) i źle poddawać się farmakoterapii.
prowadzi do tego, że odpowiednie neurony rogów tyl-
nych reagują na bodźce bólowe już w sposób nisko-
progowy.
W późnej fazie dochodzi do indukcji tzw. genów 1.5.1.4. Jakość doznań bólowych B1
natychmiastowej wczesnej odpowiedzi (IEG – im-
mediate early genes), jak c-fos i c-jun. Białka FOS Ze względu na miejsce powstania ból można podzie-
i JUN po translacji dimeryzują i ulegają jako czyn- lić również na somatyczny i narządowy (wisceralny).
niki transkrypcji (AP-1) wiązaniu w obrębie jądra O bólu somatycznym mówi się wówczas, gdy wra-
komórkowego neuronów z właściwymi sekwencjami żenia bólowe pochodzą ze skóry, mięśni, stawów,
odpowiednich genów, które kodują np. nowe białka kości lub tkanki łącznej. Jeśli bodziec zlokalizowany
receptorowe (np. receptor NMDA) i białka kanałowe jest w obrębie skóry, wyzwalane doznanie określane
(np. kanały sodowe). Te wędrują w kierunku błony jest jako ból powierzchniowy. Natomiast pochodzący
neuronów, gdzie zmieniają elektrofizjologiczne właś- z mięśni, stawów, kości i tkanki łącznej ból określany
ciwości błony i prowadzą tym samym do nadpobud- jest jako głęboki.
liwości neuronów. Innym ważnym czynnikiem tran- Ból powierzchniowy, taki jak powstający przy
skrypcji jest już wymieniony jądrowy czynnik kappa ukłuciu skóry szpilką, ma charakter bólu ostrego,
(NKκB), który wpływa m.in. na indukcję ekspresji jest dobrze zlokalizowany i szybko ustaje po zakoń-
COX-2 (zob. ryc. 1.5-7). czeniu działania bodźca. Znaczenie tzw. początko-
Ponadto w późnej fazie ośrodkowej sensybiliza- wego bólu polega na tym, że zazwyczaj zapoczątko-
cji uczestniczy interleukina IL-1β, która wprawdzie wuje on odruchową reakcję unikania (jak np. przy
w związku ze swoją wielkością nie może pokonać cofaniu nogi po nastąpieniu na ostry przedmiot)
bariery krew-mózg, lecz poprzez receptory IL-1β ko- i co za tym idzie – ochronę organizmu przed dal-
mórek śródbłonka błony krew-mózg prowadzi do in- szymi uszkodzeniami. Po tym bólu następuje często,
dukcji COX-2. Podobnie jak na obwodzie – w miej- zwłaszcza przy dużym nasileniu bodźca, w krótkim
scu zapalenia (zob. powyżej), tak samo na poziomie odstępie czasu ból wtórny o charakterze bólu tępego
rdzeniowym – COX-2 ulega indukcji przez cytokini- lub piekącego, który jest trudny do zlokalizowania
ny (przede wszystkim przez IL-1β). Napływ jonów i powoli ustępuje.
Ca2+ (zob. powyżej), w którym pośredniczy receptor Ból głęboki jest również odczuwany jako tępy,
NMDA, aktywuje fosfolipazę A2 – przygotowującą trudny do zlokalizowania i zazwyczaj promieniujący
kwas arachidonowy do kontrolowanej przez nowo do otoczenia. Najbardziej znanym przykładem bólu
wytworzony COX-2 syntezy prostaglandyn. Prosta- głębokiego jest ból głowy, który – w swych różnora-
glandyny (głównie PGE2) aktywują teraz na poziomie kich postaciach – stanowi najczęstszą postać bólu.
rdzenia receptory EP, które w dalszym ciągu wymu- Bólowi wtórnemu oraz głębokiemu często towa-
szają uwalnianie glutaminianu i substancji P w okoli- rzyszą objawy afektywne lub wegetatywne, takie jak
cy presynaptycznej, ale również w części postsynap- złe samopoczucie, nudności, nadmierne pocenie oraz
tycznej mogą poprzez cAMP aktywować PKA i w ten obniżenie ciśnienia tętniczego.
sposób fosforylować kanały jonowe i receptory (zob. Ból wisceralny lub trzewny przypomina swym
powyżej i ryc. B 1.5-7). Ponadto PGE2 stymuluje ko- charakterem ból tępy, a towarzyszącymi objawami
mórki mikrogleju do produkcji IL-1β, która z jednej wegetatywnymi – ból głęboki. Występuje on m.in.
strony ponownie wpływa na dalszą indukcję COX-2, przy rozciągnięciu organów umiejscowionych w ja-
a z drugiej strony zmienia elektrofizjologiczne właś- mie brzusznej, skurczach mięśni gładkich, upośle-
ciwości błony komórkowej neuronów. dzeniu ukrwienia i w stanach zapalnych.
Niedawno wykazano, że PGE2 może hamować Oprócz miejsca powstawania – ważnym kryterium
aktywność receptorów glicyny. Receptory glicynowe oceny bólu jest również czas jego utrzymywania się.
w obrębie rdzenia kręgowego znajdują się głównie Ból ostry – pełniący ewidentnie funkcję ostrzegaw-
MUTSCHLER-2009.indd 225 2010-01-07 22:11:51
226 Układ nerwowy
Wczesna faza ośrodkowej sensybilizacji
inter
neu
ron
ha m
ują
cy
glicyna
PGE2
cy
EP 2 a kla
Cl- Gs landeny-za
ow
receptor a
glicynowy P
BDNF TrkB cAMP
T ATP
Y
R PKA
PKC
NMDA
P
glutaminian do mózgu
AMPA Ca2+
P
zakończenie aferentnego Ca2+
włókna nocyceptywnego
neuron projekcyjny
Ca2+
substancja P PKC
Ca2+ Cav
Gq
1
NK
T
Y
R
receptor IL-1
Ryc. B 1.5-6. Wczesna faza ośrodkowej sensybilizacji/uwrażliwienia (bliższe informacje w tekście). EP – receptor prostaglandyny
E typu 2; PKA – kinaza białkowa A; PKC – kinaza białkowa C; NK1 – receptor neurokininy 1; cAMP – cykliczny AMP (monofosfo-
ran adenozyny); ATP – trifosforan adenozyny; TrkA – kinaza tyroksynowa – receptor A; BDFN (brain derived neurotrophic factor)
– neurotropowy czynnik pochodzenia mózgowego; NMDA – (receptor) N-metylo-D-asparaginian(u); AMPA – (receptor) kwas(u)
α-amino-3-hydroksy-5-metylo-4-isoksazolepropionowy(ego); Caν – napięciowozależny kanał wapniowy.
MUTSCHLER-2009.indd 226 2010-01-07 22:11:52
Układ nerwowy 227
Późna faza ośrodkowej sensybilizacji
Układ nerwowy
inter
neu
ron
ham
ują
c y
B1
glicyna PGE2
EP 2 cy
a kla
Cl- Gs landeny-za
ow
a
receptor
glicynowy np.
P
cAMP PGE2
ATP
BDNF
COX-2
TrkB
T
Y
R
PKA
NMDA np. COX-2 mRNA
P
Ca2+ ik transkrypcyjny
p50 p65
czynn p. NF-κB
z.B COX-2,
NK-1, TrKB,
FOS, JUN
zakończenie aferentnego
n
AMPA
Ca2+
P
włókna nocyceptywnego glutaminian
DNA
Ca2+
neuron projekcyjny
substancja P
ów
aktywaskrypcyjnych
Ca2+
cja czynnik
Cav jądro
komórkowe
tran
T
Y
R
receptor IL-1
IL-1β
Ryc. B 1.5-7. Późna faza ośrodkowej sensybilizacji/uwrażliwienia (bliższe informacje w tekście). Skróty jak na ryc. B 1.5-6.
MUTSCHLER-2009.indd 227 2010-01-07 22:11:53
228 Układ nerwowy
czą – ma ograniczony czas trwania i szybko zanika („Wprawdzie nadal boli, lecz ból nie jest już on tak
po ustaniu wyzwalającej go przyczyny. Najczęściej dokuczliwy”). Trójcykliczne leki przeciwdepresyjne
jest on dobrze zlokalizowany, a jego nasilenie zależy okazały się szczególne cenne jako koanalgetyki lub
od natężenia wywołującego bodźca. pomocnicze leki przeciwbólowe.
Ból przewlekły występuje albo pod postacią bó-
lów (długo)trwałych (np. bóle pleców, bóle nowotwo-
rowe), albo bólów ciągle nawracających (np. migre- 1.5.1.7. Endogenny
nowe bóle głowy, bóle w klatce piersiowej w dusz- układ hamowania bólu
nicy bolesnej). Na ogół ból uważa się za przewlekły,
jeśli dolegliwości utrzymują się dłużej niż trzy mie- Oprócz wstępującego układu przenoszenia bólu ist-
siące. Bóle przewlekłe mogą, w miarę upływu czasu, nieje również zstępujący układ hamujący ból (układ
przewyższyć wywołujące je uszkodzenie i stworzyć antynocyceptywny, układ przeciwbólowy), które-
samodzielny zespół chorobowy. Czynniki psycholo- go włókna nerwowe pochodzą z różnych poziomów
giczne i środowiskowe odgrywają przy tym istotną ośrodkowego układu nerwowego (zob. ryc. B 1.5-5).
rolę. Zadaniem tego układu antynocyceptywnego jest osła-
bianie synaptycznego przetwarzania impulsów bólo-
wych i tym samym zmniejszenie odczuwania bólu.
1.5.1.5. Reakcje na ból Stymulowanie receptorów opioidowych przez endo-
genne peptydy opioidowe (zob. poniżej) aktywuje ten
Bólowi niejednokrotnie towarzyszą reakcje wegeta- endogenny układ antynocyceptywny. Jak to uwidocz-
tywne. Zazwyczaj dochodzi do wyrzutu katecholamin niono na ryc. B 1.5-5 – ważne ośrodki tego układu
i tym samym do pobudzenia układu sympatycznego. znajdują się w substancji szarej okołowodociągowej
Czynność serca ulega przyspieszeniu, ciśnienie krwi (PAG – periaqueductal gray) oraz miejscu sina-
wzrasta, źrenice stają się szerokie. Bardzo nasilone wym (locus coeruleus). Tylko część włókien zmie-
reakcje wegetatywne występują w przebiegu bólów rza bezpośrednio do rdzenia kręgowego, inna część
trzewnych, np. w kolce żółciowej, której towarzyszą ulega przełączeniu w jądrach rdzeniowych. Z jądra
nudności, wymioty, obfite poty i spadek ciśnienia tęt- szwu wielkiego (nucleus raphe magnus) oraz jądra
niczego. szwu grzbietowego (nucleus raphe dorsalis) wycho-
Ponadto bodziec bólowy wyzwala również reak- dzą serotoninergiczne zstępujące drogi hamujące,
cje ruchowe. Do nich zaliczana jest wcześniej wy- a z miejsca sinawego noradrenergiczne zstępujące
mieniona reakcja ucieczki. Ból głęboki, a także ból drogi hamujące. W dalszej kolejności synaptyczne
trzewny, mogą w niektórych okolicznościach wywo- przekazywanie pobudzenia bodźców bólowych ulega
ływać zwiększenie napięcia mięśniowego. hamowaniu w obrębie istoty galaretowatej (substan-
Ostatecznie ból posiada składową emocjonalną tia gelatinosa) rdzenia kręgowego przez neuron za-
(afektywną), której nasilenie wykazuje dużą zmien- wierający metenkefalinę jako neuroprzekaźnik (zob.
ność zarówno osobniczą, jaki zależną od sytuacji poniżej).
(zob. Ocena nasilenia bólu). Dzięki układowi hamowania bólu można wy-
jaśnić, dlaczego bóle w warunkach stresowych
(np. po zranieniu w czasie wypadku drogowego)
1.5.1.6. Ocena nasilenia bólu pozostają początkowo niezauważone, lecz zaczyna-
ją być odczuwane dopiero po obniżeniu pobudzenia.
Bodźce bólowe o porównywalnym nasileniu są oce- Endogenne układy hamujące doznania bólowe mają
niane przez różne osoby bardzo odmiennie: podczas oczywiście za zadanie przejściowo zapobiegać do-
gdy jeden pacjent mówi o silnych (aż niemożliwych znaniom bólowym upośledzającym sprawność w sy-
do zniesienia) bólach, inny ocenia je tylko jako nie- tuacjach, w których niezbędna jest zdolność do na-
znaczne. Przypuszczalnie oprócz różnic w aktywno- tychmiastowego działania.
ści układów hamujących przekaźnictwo bodźców bó- Międzyosobnicze różnice w zakresie aktywności
lowych (zob. poniżej) odpowiedzialne są za to różne układu hamowania bólu są prawdopodobnie istotną
emocjonalno-afektywne sposoby przetwarzania bólu. przyczyną odmiennej wrażliwości na bodźce bólowe
Dlatego niektóre stany lękowe można opanowywać występujące u poszczególnych osób.
za pomocą leków psychotropowych. Leki te zazwy-
czaj same nie działają bezpośrednio przeciwbólo- Endogenne peptydy opioidowe. Endogenne pepty-
wo, zmieniają jednak przeżycia związane z bólem dy opioidowe (endogenne morfiny, endorfiny) są pro-
MUTSCHLER-2009.indd 228 2010-01-07 22:11:54
Układ nerwowy 229
dukowanymi wewnątrz ciała agonistami receptorów dów wiąże się z działaniem receptora opioidowego μ.
układu hamującego ból – receptorów opioidowych Pobudzenie receptorów κ ma prowadzić do analgezji,
(zob. poniżej). Do tych poli- i oligopeptydów należą: zwężenia źrenicy i sedacji, z kolei stymulacja recep-
torów δ ma wyzwalać także dysforię oraz omamy.
■ β-endorfina złożona z 31 aminokwasów,
Układ nerwowy
Wszystkie receptory opioidowe w przypadku sty-
■ dynorfina złożona z 17 lub 13 aminokwasów, mulacji hamują sprzężone z białkiem G (Gi/o) cyklazy
■ pentapeptydy metionino- i leucyno-enkefaliny, któ- adenylanowe. Powodują one:
re składają się z pięciu końcowych aminokwasów
■ presynaptycznie hamowanie uwalniania neuroprze-
endorfiny (met-enkefalina) lub dynorfiny (leu-en-
kaźników – poprzez zmniejszenie prawdopodo-
kefalina).
bieństwa otwarcia kanałów wapniowych,
Endorfiny powstają w mózgu, w przysadce oraz ■ postsynaptycznie hiperpolaryzację neuronów – B1
w nadnerczach z trzech białek prekursorowych: pro- głównie w wyniku zwiększenia prawdopodobień-
opiomelanokortyny (POMC), proenkefaliny i prody- stwa otwarcia kanałów potasowych.
norfiny.
Endogenne peptydy opioidowe i opioidy działają W ten sposób dochodzi do hamowania dalszego
na te same receptory – receptory opioidowe (zob. po- przewodzenia informacji nocyceptywnej (zob. ryc.
niżej). Związki te wykazują więc podobne właściwo- 1.5-8).
ści farmakodynamiczne. Różnią się jedynie właści- Przed kilkoma laty sklonowano kolejny receptor,
wościami farmakokinetycznymi. Na przykład enke- wykazujący duże podobieństwo strukturalne do zna-
faliny jako peptydy są bardzo szybko hydrolizowane nych receptorów opioidowych, z którym nie ulegają
przez proteazy i dlatego są skuteczne przeciwbólowo wiązaniu klasyczny agoniści opioidowi, tacy jak mor-
jedynie przy podaniu dokomorowym. fina. Receptor ten otrzymał najpierw nazwę ORL1
Mechanizm działania endorfin polega na tym, (opioid receptor-like – receptor podobny do opioi-
że hamują one uwalnianie mediatorów bólowych dowego), a obecnie określany jest jako receptor N/
(np. glutaminianu, substancji P), w wyniku czego OFQ. Nocyceptyna (syn. orfanina FQ; zob. powyżej)
zmniejszają ilość przewodzonych nocyceptywnych jest selektywnym agonistą receptora N/OFQ. W ba-
potencjałów czynnościowych. daniach eksperymentalnych na zwierzętach nocycep-
W 1995 r. sklonowano nowy endogenny peptyd tyna wykazuje działanie zarówno anty-, jak i prono-
opioidowy (nocyceptynę; syn. orfanina FQ), który cyceptywne.
w związku z długością łańcucha peptydowego wy-
noszącą 17 aminokwasów wykazuje względnie duże Receptory kannabinoidowe. Receptory kannabino-
podobieństwo sekwencyjne do dynorfiny A. idowe występują zarówno w ośrodkowym układzie
nerwowym (np. móżdżek, mózg, przysadka, rdzeń
Receptory opioidowe. Receptory opioidowe wystę- kręgowy), jak i na obwodzie (np. migdałki, śledziona,
pują z różną gęstością zarówno pre-, jak i postsynap- trzustka, szpik, macica, płuca). Jak to już opisano –
tycznie, w ośrodkowym układzie nerwowym i na ob- od niedawna znane są dwa receptory kannabinoidowe
wodzie. Szczególnie duża liczba receptorów opioi- (CB1 i CB2). Receptor CB1 występuje głównie, lecz
dowych została znaleziona w układzie limbicznym, nie wyłącznie – w ośrodkowym układzie nerwowym;
wzgórzu, podwzgórzu i prążkowiu, a także w tworze tymczasem receptor CB2 identyfikowany jest głów-
siatkowatym pnia mózgu oraz istocie galaretowatej nie na immunokompetentnych komórkach układu
rdzenia kręgowego. krwiotwórczego, w migdałkach i śledzionie. Recep-
Podobnie jak w przypadku receptorów dla innych tory kannabinoidowe hamują sprzężone z białkiem G
neuroprzekaźników również wśród receptorów opio- (Gi/o) cyklazy adenylanowe. Tym samym receptory
idowych rozróżniono typy, które określa się jako kannabinoidowe wywołują – podobnie jak receptory
receptory μ (mi), κ (kappa) i δ (delta). Stosowane opioidowe – presynaptycznie hamowanie uwalniania
są również inne oznaczenia: OP1 (dla δ), OP2 (dla neuroprzekaźników oraz postsynaptycznie hiperpola-
κ) oraz OP3 (dla μ). Wśród receptorów opioidowych ryzację odpowiednich komórek. Ponieważ receptory
znane są liczne podtypy (μ1, μ2, δ1, δ1, κ1, κ2, κ3,). kannabinoidowe występują w wielu okolicach mózgu
Receptor opioidowy μ pośredniczy głównie w prze- – wywoływane przez nie efekty mają charakter róż-
kazywaniu takich efektów, jak analgezja, depresja norodny. Kannabinoidy wywierają wpływ na pamięć,
oddechowa, zwężenie źrenicy, euforia, zaparcie i bra- emocje i motorykę, jak również na sferę odczuwania
dykardia. Również przeciwkaszlowe działanie opioi- bólu (nocycepcję).
MUTSCHLER-2009.indd 229 2010-01-07 22:11:54
230 Układ nerwowy
bez morfiny aferentne włókna z morfiną aferentne włókna
nerwowe typu C nerwowe typu C
Ca2+ morfina Ca2+
μ Cav μ Cav
Gi PKA↑ P Gi PKA ↑
ATP ATP ↑
cAMP↑ [Ca2+]↑ cAMP ↑
ade
[Ca2+]
cyk ylano
ad cyk
n
en la
laz wa
yla za
a
no
wa
glutaminian
+
Ca2+
+
Ca2+
Na K
NMDA NMDA
neuron neuron
rogu tylnego + μ rogu tylnego μ
Gi
Gi
K
↑
[Ca2+]↑ 2+
[Ca ]
hiper- K+
depolaryzacja polaryzacja
morfina
odczuwanie brak
odczuwania
bólu bólu
Ryc. B 1.5-8. Molekularny mechanizm działania morfiny (opioidów) na poziomie rdzenia kręgowego. Poprzez wiązanie z presynap-
tycznym receptorem opioidowym sprzężonym z białkiem G – morfina obniża prawdopodobieństwo otworzenia kanałów wapnio-
wych i tym samym hamuje uwalnianie neuroprzekaźnika (np. glutaminianu) z aferentnych włókien bólowych typu C. W obszarze
postsynaptycznym morfina prowadzi przez zwiększenie prawdopodobieństwa otwarcia jonów wapnia do hiperpolaryzacji neuro-
nów (prawa część rysunku). Dla lepszego wyjaśnienia lewa cześć rysunku prezentuje sytuację, w której brak jest morfiny. ATP – tri-
fosforan adenozyny; cAMP – cykliczny AMP (monofosforan adenozyny); PKA – kinaza białkowa A; Caν – napięciowozależny kanał
wapniowy; NMDA – (receptor) N-metylo-D-asparaginian(u).
1.5.1.8. Wpływ leków na odczuwanie bólu ■ obwodowa analgezja za pomocą leków przeciwbó-
lowych opioidowych;
W zależności od przyczyny bólów istnieją nastę-
■ zapobieganie powstawaniu impulsów w nocycep-
pujące możliwości farmakologicznego wpływania
torach przez stosowanie znieczulenia powierzch-
na ich odczuwanie (zob. ryc. 1.5-9):
niowego lub znieczulenia nasiękowego;
■ hamowanie bólu przez ingerencję w obrębie ośrod-
■ hamowanie przewodzenia impulsów w czuciowych
kowego układu nerwowego za pomocą leków
drogach nerwowych przez zastosowanie znieczule-
przeciwbólowych opioidowych, leków przeciw-
nia przewodzeniowego,
bólowych nieopioidowych, trójcyklicznych leków
przeciwdepresyjnych lub innych leków przeciwbó- ■ zapobieganie ośrodkowej sensybilizacji przez leki
lowych; przeciwbólowe opioidowe i leki przeciwbólowe
nieopioidowe,
■ zapobieganie sensybilizacji nocyceptorów przez
zahamowanie syntezy prostaglandyn za pomocą ■ wpływ na przeżywanie bólu przez stosowanie le-
kwaśnych leków przeciwbólowych nieopioido- ków przeciwbólowych opioidowych, neurolepty-
wych; ków i leków przeciwdepresyjnych.
MUTSCHLER-2009.indd 230 2010-01-07 22:11:54
Układ nerwowy 231
mózg ■ Trójcykliczne leki przeciwdepresyjne i niektóre
leki przeciwpadaczkowe stosowane są w terapii
bólów neuropatycznych.
■ Triyptany stosowane są w terapii migrenowych bó-
Układ nerwowy
lów głowy.
Profilaktyka bólu jest lepsza niż leczenie bólu, tzn.
terapia antycypacyjna winna być preferowana przed
opioidy, nieopioidowe leki
przeciwbólowe, reaktywną terapią bólu. Zgodnie z podaną zasadą
leki psychotropowe – w przypadku operacji leki przeciwbólowe powinny
być zaordynowane w wystarczających dawkach jesz-
rdzeń kręgowy cze przed wystąpieniem bólów. W przypadku prze-
B1
wlekłych bólów, szczególnie w bólach nowotworo-
wych, leki przeciwbólowe nie powinny być ordyno-
wane na życzenie, lecz według stałego planu terapii
środki znieczulające – w wystarczająco dużych dawkach i w regularnych
miejscowo stosowane odstępach czasu – pod postacią preparatów o przedłu-
przewodowo
żonym działaniu. Bóle przewlekłe wymagają ponadto
włókna nerwowe typu C
dodatkowego podawania leków pomocniczych.
uszkodzona
tkanka Zgodnie ze stopniowym schematem Światowej Orga-
BK nizacji Zdrowia (WHO), opracowanym na potrzeby
środki znieczulające terapii bólów nowotworowych, który można jednak
NGF miejscowo stosowane zastosować w innych stanach bólowych jako pewną
powierzchniowo, wytyczną, terapia bólu powinna być przeprowadzona
H+ nieopioidowe leki
przeciwbólowe, opioidy w następujący sposób (zob. ryc. B 1.5-10):
PG
Ryc. B 1.5-9. Schemat różnych farmakologicznych możliwości stopień 3
wpływania na odczuwanie bólu. BK – bradykinina, PG – prosta-
silne leki opioidowe
glandyna; NGF – czynnik wzrostu nerwu.
± nieopioidowe leki przeciwbólowe
± leki wspomagające
jeżeli bóle nadal się utrzymują
stopień 2
1.5.1.9. Kryteria stosowania leków
przeciwbólowych słabe leki opioidowe
± nieopioidowe leki przeciwbólowe
Podstawową przesłanką skutecznego zastosowania ± leki wspomagające
leków przeciwbólowych jest dokonanie rozeznania
w zakresie rodzaju bólu, czasu jego trwania i obja- jeżeli bóle nadal się utrzymują
wów towarzyszących (zob. ryc. B 1.5-1): czy mamy stopień 1
do czynienia z bólem ostrym czy przewlekłym, jaka
jest lokalizacja bólu oraz jakie wykazuje on nasilenie, nieopioidowe leki przeciwbólowe
jaka jest etiopatogeneza bólu i czy pacjent skarży się ± leki wspomagające
na zaburzenia czucia?
■ Opioidowe leki przeciwbólowe nadają się dosko- ból
nale do terapii bólów pourazowych, pooperacyj-
nych i nowotworowych. Ryc. B 1.5-10. Schemat stopniowej farmakoterapii bólów no-
wotworowych wg WHO. Leki wspomagające/pomocnicze (ko-
■ Nieopioidowe leki przeciwbólowe wskazane analgetyki) są preparatami niemającymi formalnie działania
są do stosowania głównie w przypadku patofizjolo- przeciwbólowego (np. leki przeciwdepresyjne, neuroleptyki,
gicznych bólów pochodzenia zapalnego. leki przeciwdrgawkowe i inne).
MUTSCHLER-2009.indd 231 2010-01-07 22:11:55
232 Układ nerwowy
■ w pierwszym (1.) stopniu zaleca się stosowanie ■ euforia lub dysforia: w zależności od (wyjściowe-
nieopioidowych leków przeciwbólowych samo- go) nastroju opioidy prowadzą do podwyższenia
dzielnie lub łącznie z innym wspomagającym/po- nastroju (euforii) lub drażliwości (dysforii);
mocniczym lekiem przeciwbólowym (koanalge-
■ depresja oddechowa: opioidy hamują działanie
tykiem);
ośrodka oddechowego;
■ na drugim (2.) poziomie powinno stosować się
■ działanie przeciwkaszlowe: opioidy blokują ośro-
słabo działający lek opioidowy (np. tramadol lub
dek kaszlu;
tilidin) – samodzielnie lub w połączeniu z nieopioi-
dowym lekiem przeciwbólowym i/lub preparatem ■ sztywność mięśni;
wspomagającym/pomocniczym;
■ wymioty: opioidy pobudzają początkowo ośrodek
■ na trzecim (3.) poziomie pacjent otrzymuje silnie wymiotny, później jednak prowadzą do jego hamo-
działający lek opioidowy samodzielnie lub w połą- wania – a tym samym ujawniają działanie przeciw-
czeniu z nieopioidowym lekiem przeciwbólowym wymiotne;
i/lub lekiem wspomagającym.
■ zwężenie źrenic: opioidy prowadzą do zwężenia
źrenic – poprzez pobudzenie przywspółczulnej
Terapia bólu pozostaje tym samym postacią leczenia
części jądra nerwu okoruchowego;
zindywidualizowanego. Rodzaj leku przeciwbólowe-
go, drogę podania, dawki i odstępy między podania- ■ działanie antydiuretyczne: opioidy zwiększają wy-
mi ustala się indywidualnie u określonego (często dzielanie hormonu antydiuretycznego.
cierpiącego na wiele innych chorób) pacjenta.
Efekty obwodowe:
■ analgezja: opioidy ujawniają działanie przeciwbó-
1.5.2. Opioidowe leki przeciwbólowe lowe również przez działanie na obwodowe recep-
tory opioidowe;
Opioidowe leki przeciwbólowe (opioidy, opiaty, ■ opóźnione opróżnianie się żołądka;
narkotyczne leki przeciwbólowe, hipnoanalgety-
■ zaparcie: opioidy osłabiają motorykę i zwiększają
ki, silnie działające leki przeciwbólowe) działają
napięcie mięśni gładkich przewodu pokarmowego;
przeciwbólowo w ten sposób, że łączą się w spo-
sób agonistyczny z jednym lub kilkoma typami ■ skurcz zwieraczy w obrębie dróg żółciowych;
receptorów opioidowych. Ze względu na wspólne
■ zwiększenie napięcia mięśniówki pęcherza moczo-
miejsce wychwytu opioidowych leków przeciwbó-
wego oraz zwieraczy pęcherza moczowego (zatrzy-
lowych w obrębie receptora opioidowego – profil
manie moczu);
działania wszystkich leków z tej grupy jest podob-
ny, ale nie identyczny. Ponadto różnią się między ■ zaburzenia ortostatyczne: opioidy obniżają na-
sobą w zakresie farmakokinetyki. Ich działanie pięcie mięśniówki naczyń krwionośnych (spadek
jest różnorodne, gdyż receptory opioidowe wystę- ciśnienia) z zagrożeniem reakcjami ortostatyczny-
pują w licznych organach i tkankach. mi;
■ uwalnianie histaminy: opioidy mogą – poprzez
Efekty ośrodkowe: wyrzut histaminy – prowadzić do zaczerwienie-
nia skóry, powstania pęcherzy i ujawnienia się
■ analgezja (działanie przeciwbólowe): opioidy ha-
świądu skóry, jak również – szczególnie u pacjen-
mują na poziomie rdzenia kręgowego impulsy bó-
tów z astmą oskrzelową – mogą wywołać skurcz
lowe, aktywują endogenny układ przeciwbólowy
oskrzeli.
oraz zmieniają odczuwanie bólu w obrębie układu
limbicznego (bóle nie są odczuwane już jako bar-
dzo nieprzyjemnie i zagrażające); W dalszej kolejności – w przypadku opioidów – ist-
nieje ryzyko rozwinięcia się psychicznego i fizycz-
■ sedacja (nadmierne uspokojenie): opioidy zmniej-
nego uzależnienia, jak również ujawnienia się zja-
szają uwagę i zdolność koncentracji, jednak nie wy-
wiska tolerancji. Jednak w przypadku zgodnego
wołują żadnej amnezji;
z zaleceniami oraz prawidłowego stosowania leków
■ anksjoliza (redukcja/zmniejszenie lęku): opioidy – ryzyko ujawnienia się uzależnienia jest stosunko-
wykazują działanie przeciwlękowe; wo niewielkie.
MUTSCHLER-2009.indd 232 2010-01-07 22:11:55
Układ nerwowy 233
1.5.2.1. Farmakologiczny Tabela B 1.5-1. Farmakologiczny podział opioidów należących
podział opioidowych leków do 3-stopnia drabiny analgetycznej WHO
przeciwbólowych
receptor receptor receptor
μ δ κ
Układ nerwowy
Opioidy różnią się między sobą w zakresie:
czyści agoniści
■ powinowactwa do receptorów, morfina +++ + +
hydromorfon +++ + +
■ siły działania, L-metadon +++
■ czasu działania. oksykodon +++ + +
petydyna ++ + +
W zależności od powinowactwa do różnych typów mieszani agoniści/ B1
antagoniści
receptorów opioidowych oraz ich wewnętrznej ak-
tywności (intrinsic activity) – opioidy dzieli się na- pentazocyna – + ++
stępująco (zob. tab. B 1.5-1): częściowi agoniści
■ czyści (pełni) agoniści (np. morfina), buprenorfina (+++) ––
■ mieszani agoniści i antagoniści (np. pentazocyna), czyści antagoniści
nalokson ––– – ––
■ częściowi agoniści (np. buprenorfina),
+ agonista; ilość znaków plus odpowiada względnemu
■ czyści antagoniści (np. nalokson). powinowactwu do danego receptora
( ) częściowy agonista
Klasyczne opioidowe leki przeciwbólowe (np. mor- antagonista; ilość znaków minus odpowiada względnemu
fina) działają jako czyści agoniści przede wszystkim powinowactwu do danego receptora
receptorów opioidowych μ. Poprzez zmiany w struk-
turze cząsteczkowej, w szczególności przez wpro-
wadzenia grupy allilowej lub innego nienasyconego
podstawnika w miejsce grupy metylowej przy atomie żądanych, np. depresji oddechowej. Jeżeli – głównie
azotu – uzyskano związki, które działają odmiennie po długim stosowaniu dużych dawek czystych agoni-
na poszczególne populacje receptorów opioidowych stów receptorów μ – dojdzie do zmiany na lek o częś-
– czysto (w pełni) agonistycznie lub czysto antago- ciowym agonistycznym i antagonistycznym działaniu
nistycznie albo też częściowo agonistycznie/ antago- w obrębie tych receptorów – mogą ujawnić się w okre-
nistycznie. Czyści antagoniści (aktywność wewnętrz- ślonych warunkach objawy zespołu odstawiennego.
na = 0), np. nalokson (Naloxonum hydrochloricum) Ostatecznie nadzieje na stworzenie silnie działa-
znoszą działanie opioidów, ponieważ wiążą się jących opioidów bez niebezpieczeństwa uzależnienia
wprawdzie mocniej z receptorami opioidowymi, lecz – jak dotąd – nie zostały spełnione.
nie wywołują żadnego działania. Substancje tego typu Stosowanie preparatów o opóźnionym/przedłu-
stosowane są m.in. w opanowywaniu zatruć opioida- żonym działaniu (typu retard) zmniejsza znaczą-
mi (zob. poniżej). co niebezpieczeństwo uzależnienia i nadużywania
Związki o mieszanym mechanizmie działania – za- leków opioidowych, ponieważ substancja czynna
równo z komponentą agonistyczną, jak i antagonistycz- z preparatów retard uwalnia się wolniej i ze wzglę-
ną, takie jak pentazocyna wykazująca działanie agoni- du na utrzymywanie się stężenia leku w przybliże-
styczne w stosunku do receptorów κ i równocześnie niu na podobnym poziomie zapobiega powstawaniu
antagonistyczne działanie w stosunku do receptorów μ pików stężenia w osoczu (z działaniem euforyzują-
– zostały stworzone z założeniem, że ich potencjał uza- cym!). Ponadto powolne zmniejszanie się poziomu
leżniający mógłby być niższy niż czystych agonistów substancji w osoczu zmniejsza ryzyko ujawnienia się
receptorów μ. Podobny cel przyświecał badaniom nad objawów odstawiennych.
rozwojem częściowych agonistów, np. buprenorfiny.
Podobnie jak morfina – buprenorfina działa przeciw- Wskazania. Leki opioidowe są środkiem z wyboru
bólowo poprzez receptory opioidowe μ, jednakże w terapii silnych bólów (np. śród- i pooperacyjnych,
z mniejszą aktywnością wewnętrzną, tzn. buprenorfina nowotworowych). Ze względu na różnorodność do-
nie jest w stanie osiągnąć maksymalnej skuteczności stępnych postaci galenowych – opioidy mogą być
przeciwbólowej morfiny (efekt pułapowy – ceiling). podawane drogą doustną, parenteralnie lub w po-
Efekt pułapowy dotyczy również objawów niepo- bliże rdzenia kręgowego (podpajęczynówkowo – in-
MUTSCHLER-2009.indd 233 2010-01-07 22:11:55
234 Układ nerwowy
tratekalnie, nadtwardówkowo – zewnątrzoponowo ■ depresja oddechu/oddechowa (tłumienie ośrodka
– epiduralnie). Wszystkie te możliwości podania leku oddechowego): objaw ten jest wyrażony znacznie
mają szczególne znaczenie wobec rozmaitych postaci słabiej u pacjentów z dolegliwościami bólowy-
bólów i ich intensywności. mi niż u pacjentów bez bólów, gdyż ból stymulu-
Z powodu równocześnie wywoływanego działa- je ośrodek oddechowy. Wynika z tego, iż celem
nia psychosedatywnego (trankwilizującego) opioi- terapii nie powinno być całkowite opanowanie
dy nadają się w szczególny sposób do stosowania objawów bólowych. Szczególna ostrożność naka-
w przebiegu zawału serca oraz ostrego obrzęku zana jest przy stosowaniu opioidów u pacjentów
płuc, ponieważ powodują przerwanie błędnego koła z obturacyjną chorobą płuc oraz rozedmą. Rów-
(circulus vitiosus) prowadzącego do duszności, lęku, nież niemowlęta i dzieci reagują w sposób szcze-
pogorszenia się ekonomiki pracy serca – związane- gólnie wrażliwy na opioidowe leki przeciwbólo-
go ze stymulacją układu współczulnego – i nasilenia we. W takim przypadku zalecane jest stosowanie
się obrzęku płuc. Ponadto opioidy mogą być stoso- częściowych agonistów, np. jak buprenorfiny, gdyż
wane w przypadku bólów występujących w choro- w przypadku hamowania oddechu wykazuje ona
bach układu ruchu (w zmianach zwyrodnieniowych efekt pułapowy (ceiling);
stawów, reumatoidalnym zapaleniu stawów). Dzieje
■ spadek ciśnienia krwi (hipotensja): na objaw ten
się tak szczególnie w przypadkach, kiedy to scho-
należy zwracać szczególną uwagę w przypadku hi-
rzenia towarzyszące lub też przeciwwskazania ogra-
powolemii lub u pacjentów otrzymujących równo-
niczają możliwość zastosowania niesteroidowych
cześnie leki obniżające ciśnienie;
(zob. poniżej) leków przeciwzapalnych. Opioidy
odgrywają również dużą rolę w terapii bólów neu- ■ zaparcia: spastyczne zaparcie jest jednym z kli-
ropatycznych. nicznie szczególnie ważnych objawów niepożąda-
Oprócz efektu przeciwbólowego w terapii moż- nych, który występuje w przypadku przewlekłej
na wykorzystać również inne działania opioidów. terapii opioidami (np. w terapii bólów nowotwo-
Opioidy – poza leczeniem przeciwbólowym – sto- rowych) i musi być prawie zawsze opanowywany
suje się w terapii substytucyjnej u osób uzależnio- za pomocą leków przeczyszczających (zob. tab.
nych od narkotyków (metadon, buprenorfina; heroina 1.5-3);
w modelowym projekcie), w biegunce (np. lopera-
■ działanie polegające na zatrzymaniu moczu.
mid) oraz jako leki przeciwkaszlowe.
Ze względu na to działanie niepożądane należy
okresowo kontrolować stan wypełnienia pęcherza
Dawkowanie. Dawkowanie opioidów jako leków
moczowego, gdyż może dojść do jego przepeł-
przeciwbólowych ma charakter indywidualny – za-
nienia, czego pacjent może nie wyczuwać m.in.
leżny od charakterystyki bólu, jego intensywności
w związku z efektem przeciwbólowym;
i czasu trwania. Ze względu na rozwój tolerancji
(zob. poniżej) konieczne jest z czasem zwiększenie ■ zwężenie źrenic (mioza);
dawki. W przypadku przewlekłego stosowania opio-
■ świąd;
idów – leczenie powinno być prowadzone według
określonego planu, a nie na żądanie (zob. powyżej), ■ (nadmierne) pocenie się;
a sam pacjent powinien prowadzić „kalendarzyk”
■ uzależnienie: psychiczne i fizyczne uzależnienie
bólu. Należy w tym miejscu podkreślić, że opioidy
w przypadku kontrolowanego i prawidłowego sto-
przedostają się przez łożysko, a także do mleka mat-
sowania nie stanowi zwykle poważnego problemu.
ki. Przeciętne, pojedyncze dawki ogólnie stosowa-
Prawdziwym problemem zastosowania opioidów
nych opioidów zostały podane w tab. B 1.5-2.
nie jest powstanie uzależnienia w trakcie terapii
przeciwbólowej, lecz ich wykorzystanie jako nar-
Działania niepożądane. Działania niepożądane są kotyków. Osoby stają się uzależnione z powodu
wynikiem pobudzenia receptorów opioidowych i moż- euforyzującego działania opioidów oraz ekstre-
na je wyprowadzić z powyżej omówionego profilu malnie nieprzyjemnych objawów abstynencyjnych
działania: po ich odstawieniu. Osoby uzależnione od opioi-
dów (morfiniści) – charakteryzujące się labilnym
■ nudności i wymioty (szczególnie na początku tera-
nastrojem i bladożółtym wyglądem – błyskawicznie
pii);
przyzwyczajają się do dużych dawek morfiny, które
■ sedacja (nadmierne uspokojenie; również na po- mogą sięgać 1 grama (!) morfiny dziennie (rozwój
czątku terapii); tolerancji). W zaawansowanym stadium występu-
MUTSCHLER-2009.indd 234 2010-01-07 22:11:56
Układ nerwowy 235
Tabela B 1.5-2. Opioidowe leki przeciwbólowe i przeciwkaszlowe
Wzór strukturalny Nazwa Preparaty handlowe Główne Średnia
międzynarodowa zastosowania dawka
Układ nerwowy
dzienna (mg)
l. Pochodne morfiny
HO morfina Morphin HEXAL, lek przeciwbólowy 10-60
Morphin-ratiopharm, parenteralnie,
M-STADA, MST, Doltard, 60-120
O H MST Continus, Sevredol, doustnie
CH3 Vendal retard, lub doodbytniczo
N
Morphini sulfas
HO
B1
H3CO kodeina Gelonida, Nelodon P, lek przeciwkaszlowy, 60-120
Paracetamol comp. STADA, lek przeciwbólowy (jako fosforan
talvosilen, Antidol, kodeiny)
Ascodan, Efferalgan
O H
CH3 codeine, Thiocodin,
N Codeinum phosphoricum,
Nurofen Plus
HO
O O diamorfina = heroina substytucja
(obecnie
CH3 w badaniach
O H klinicznych)
CH3
CH3 N
O O
II. Pochodne dihydromorfiny
H3CO dihydrokodeina DHC Mundipharma, lek przeciwkaszlowy, 20-60
Paracodin, Tiamon Mono lek przeciwbólowy, (jako winian
DHC Continus dihydrokodeiny)
O H
CH3
N
HO
HO hydromorfon Dilaudid, Jurnista, lek przeciwkaszlowy 2 (parenteralnie),
Palladon 8–32
(kapsułki typu retard)
O H
CH3
N
H3CO oksykodon OXYGESIC lek przeciwbólowy 20-40
O OH
CH3
N
H3CO hydrokodon Dicodid lek przeciwkaszlowy 10-15
O H
CH3
N
MUTSCHLER-2009.indd 235 2010-01-07 22:11:56
236 Układ nerwowy
Tabela B 1.5-2. Opioidowe leki przeciwbólowe i przeciwkaszlowe (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Główne Średnia
międzynarodowa zastosowania dawka
dzienna (mg)
III. Grupa petydyny
H3C O
N
R2
R1
R1 R2
petydyna Dolantin, lek przeciwbólowy 25-50
H OC2H5 Dolargan,
Dolcontral
IV. Grupa metadonu
R1
R2
R1 R2
lewometadon L-Polamidon lek przeciwbólowy 2,5-7,5
CH3
O
CH3
CH2 N
C2H5
CH3
piritramid Dipidolor lek przeciwbólowy 15-60
CH2 N
CONH2
N CN
V. Grupa fentanylu
VI. Mieszani/częściowi agoniści
HO
pentazocyna Pentazocinum, lek przeciwbólowy 30-90
Fortral
CH3
H3C N CH3
CH3
HO buprenorfina SUBUTEX, Temgesic, lek 0,3-0,9
Norspan (plaser), przeciwbólowy (parenteralnie)
Transtec PRO (plaster), 0,2-0,8
O Bunondol, Transtec (podjęzykowo)
0,8-1,6
N (przezskórnie)
H3CO
H3C C(CH3) 3
OH
MUTSCHLER-2009.indd 236 2010-01-07 22:11:56
Układ nerwowy 237
Tabela B 1.5-2. Opioidowe leki przeciwbólowe i przeciwkaszlowe (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Główne Średnia
międzynarodowa zastosowania dawka
dzienna (mg)
Układ nerwowy
tilidyna Tilidin AL. Comp., lek przeciwbólowy 100-300
(w skojarzeniu Tilidin HEXAL Comp.,
z naloksonem) Tilidin-ratiopharm plus,
COOC2H5 Valoron N
N(CH3) 2
tramadol Tramadolor, lek przeciwbólowy 100-300
Tramadol-ratiopharm, B1
OCH3
Tramal, Tramundin,
OH Noax Uno, Pratram,
N(CH3) 2
Poltram, Tramahexal,
Travictol, Zaldiar
ją bezsenność, wychudzenie, impotencja, drżenie nu, racematu metadonu, buprenorfiny, a ostatnio
mięśniowe, zaburzenia koordynacji oraz zaburze- nawet także heroiny (zob. poniżej) – zmniejszyć
nia psychiczne: pacjent degraduje się fizycznie związaną z nadużywaniem środków odurzających
i psychicznie. Jeżeli osobie uzależnionej odstawi kryminogenność oraz zwiększyć skuteczność tera-
się nagle opioid – w ciągu godzin ujawniają się ob- pii odwykowej polegającej na długotrwałym poda-
jawy abstynencyjne, takie jak niepokój, depresja, waniu stopniowo zmniejszanych dawek opioidu.
stany lękowe, poczucie marznięcia lub nadmierne Po przeprowadzonej detoksykacji osób uzależnio-
pocenie się i nasilone łzawienie. Po okresie 24–48 nych od opioidów w terapii odwykowej opartej
godzin zespół abstynencyjny, który może być wy- na oddziaływaniach psychoterapeutycznych sku-
jaśniony zarówno odhamowaniem neuronów nora- teczną farmakologiczną pomocą może być włącze-
drenergicznych w miejscu sinawym, jak i warunko- nie naltreksonu. Podobnie jak nalokson (zob. po-
wanym efektem z odbicia po odstawieniu opioidów niżej) jest on antagonistą receptorów opioidowych
zwiększeniem aktywności cyklazy adenylanowej, i zapobiega łączeniu się innych agonistów, takich
osiąga swój punkt maksymalnego nasilenia. Zespół jak heroina, z receptorem. Jego biodostępność przy
ten charakteryzuje się nudnościami, wymiotami, doustnym podaniu wynosi 10–40% i jest znamien-
biegunkami, nasileniem oddychania, zwiększenie nie większa od biodostępności naloksonu (ok. 2%),
częstotliwości pracy serca i skurczowego ciśnienia tak więc naltrekson może być stosowany doustnie.
krwi, zwiększeniem temperatury i odwodnieniem –
■ tolerancja: w trakcie długotrwałego stosowania
jako objawami wegetatywnej dysfunkcji. W dalszej
opioidów może wystąpić rozwój tolerancji, tzn.
kolejności występują drgawki mięśniowe i skurcze
jelitowe. Opisane objawy abstynencyjne mogą wy-
stąpić u osób uzależnionych również po podaniu
antagonistów opioidowych. W sumie trwa to około
tygodnia, zanim objawy abstynencyjne całkowi-
cie ustąpią. Nasilenie symptomatyki odstawiennej
udaje się złagodzić za pomocą α2-adrenomimety- HO
ku – klonidyny. Leczenie odwykowe jest możliwe
do przeprowadzenia wyłącznie w warunkach sta- O OH
cjonarnych. Oprócz odstawienia środka uzależnia- N
jącego niezbędne jest wdrożenie metod psychote-
O
rapeutycznych i postępowania resocjalizacyjnego.
W przypadku tzw. programów substytucyjnych, naltrekson
które od pewnego czasu przeprowadzane są w wie-
lu krajach, usiłuje się za pomocą pozostającego
pod państwową kontrolą stosowania lewometado-
MUTSCHLER-2009.indd 237 2010-01-07 22:11:57
238 Układ nerwowy
konieczne jest zwiększenie dawki, aby uzyskać Zgon następuje w wyniku porażenia ośrodka odde-
ten sam efekt przeciwbólowy. Rozwój tolerancji chowego (dawka śmiertelna morfiny u osoby dorosłej
dotyczy również działań niepożądanych: nudności, nieuzależnionej od leku wynosi < 0,1 g w przypadku
sedacji i depresji oddechowej – co powoduje, że podania pozajelitowego oraz 0,3–1,5 g przy podaniu
występują one głównie na początku terapii i z cza- doustnym; dla noworodka podanie już 2–3 kropli na-
sem zmniejsza się ich nasilenie. Tolerancja nie roz- lewki opiumowej może być śmiertelne!).
wija się jednak w stosunku do takich objawów, jak W przebiegu terapii zatrucia morfiną najważniej-
zaparcia czy zwężenie źrenic, które z tym samym szym elementem jest opanowanie braku tlenu (niedo-
nasileniem ujawniają się nawet po długotrwałej tlenienia). Oprócz sztucznego oddychania – ustaloną
ekspozycji na opioidy (zob. tab. B 1.5-3. Rozwój wartość terapeutyczną ma podanie antagonisty opio-
tolerancji wiąże się z mechanizmami adaptacyj- idowego – naloksonu. Dawkowanie wynosi począt-
nymi receptorów opioidowych, w których ważną kowo 0,4–2 mg dożylnie, domięśniowo lub podskór-
funkcję zdają się pełnić receptory NMDA. Toteż nie, następnie jeśli to niezbędne 0,4–2 mg co dwie
u niektórych pacjentów możliwe jest osłabienie do trzech minut. Z powodu zagrożenia wystąpieniem
tolerancji opioidowej za pomocą ketaminy – anta- niebezpiecznego dla życia zespołu ostrej abstynen-
gonisty NMDA. cji u osób uzależnionych należy stosować mniejsze
dawki przy jednocześnie krótszych odstępach między
Przeciwwskazania wynikają zasadniczo z zakresu kolejnymi podaniami.
działań niepożądanych. W stanach chorobowych,
w których należy unikać tłumienia ośrodka oddecho-
wego, jak też w ostrej porfirii wątrobowej – stoso-
wanie opioidów jest przeciwwskazane. Do względ-
nych przeciwwskazań należy niedoczynność tarczycy
– ze względu na nasilenie działania narkotycznego,
wrzodziejące zapalenie jelita grubego i zapalenie
trzustki. W przebiegu mocznicy zwiększa się wraż-
HO
liwość na opioidy.
Interakcje. Równoczesne spożycie środków o ośrod- O OH
CH2
kowym działaniu hamującym, a także alkoholu nasila N
określone działania niepożądane opioidów.
O
Ostre zatrucie opioidami. Ostre zatrucie opioidami nalokson
charakteryzuje się głęboką śpiączką z płytkim, a nie-
kiedy prawie niewyczuwalnym oddechem oraz mak-
symalnym zwężeniem źrenic (typowa triada objawów:
utrata przytomności, depresja oddechu i zwężenie
źrenic), jak również sinicą, zimną skórą i hipotermią.
1.5.2.2. Opium
Tabela B 1.5-3. Częste działania niepożądane terapii za pomocą
leków opioidowych Opium jest wysuszonym na powietrzu mlecznym sokiem
niedojrzałych torebek nasiennych (makówek) maku lekar-
skiego (Papaver somniferum). Zawiera ponad 20 alkaloi-
Początek W trakcie terapii dów, których proporcje mogą być bardzo zmienne. Głów-
terapii długoterminowej
nym alkaloidem jest morfina; z pozostałych ważniejszych
Nudności ↑↑ - alkaloidów towarzyszących należy wymienić: noskapinę
(dawniej nazywaną narkotyną), kodeinę, papawerynę, te-
Wymioty ↑ - bainę i narceinę.
Opium bywa jeszcze bardzo rzadko stosowane pod po-
Poczucie zmęczenia/ ↑ -
znużenia stacią nalewki opiumowej w celu uspokojenia motoryki
jelitowej w biegunkach. Ze względu na zawartość innych
Zaparcia ↑ ↑↑↑ konieczne leki alkaloidów, zwłaszcza papaweryny, podawanie opium pro-
przeczyszczające wadzi do zaparcia atonicznego (w przeciwieństwie do za-
parcia spastycznego – wywoływanego przez morfinę).
MUTSCHLER-2009.indd 238 2010-01-07 22:11:57
Układ nerwowy 239
1.5.2.3. Silnie działające leki opioidowe 3-glukuronian hydromorfonu nie ma działania prze-
stopnia 3. wg WHO ciwbólowego. Okres półtrwania wynosi w przybliże-
niu 3 godziny.
Układ nerwowy
Morfina jest od samego początku standardową sub- Lewometadon jest prawie 4-krotnie silniejszy od mor-
stancją opioidową. Może być podana na drodze po- finy i dłuższy jest także jego okres działania. W rów-
zajelitowej (parenteralnie, np. dożylnie, podskórnie, noważnych dawkach przeciwbólowych (ekwianalge-
nadtwardówkowo), doustnie pod postacią preparatów tycznych) jego objawy niepożądane wydają się nieco
retard oraz doodbytniczo. słabsze. Również objawy abstynencyjne rozwijają się
Wchłanianie z przewodu pokarmowego następuje wolniej i są mniej nasilone. Po zażyciu doustnym lek
względnie powoli. Ponadto morfina charakteryzuje jest dobrze wchłaniany, a jego biodostępność jest nie-
się silnym efektem pierwszego przejścia. Biodostęp- mal całkowita. Okres półtrwania jest indywidualnie B1
ność po podaniu doustnym preparatów niemających zmienny i wynosi między 20 a 60 godzin (w pojedyn-
przedłużonego działania wynosi ok. 25%, tymcza- czych przypadkach nawet do 100 godzin).
sem biodostępność związków typu retard wzrasta
do 40%). Jednakże morfina już w trakcie pierwsze- Oksykodon działa nieco silniej od morfiny. Biodo-
go pasażu przez wątrobę ulega w znaczącym stopniu stępność po podaniu doustnym uzależniona jest od po-
przekształceniu w 6-glukuronian morfiny, który wy- staci galenowej i jest większa o 40–80% od morfiny.
kazuje podobne działanie agonistyczne wobec recep- Czas połowiczej eliminacji wynosi 3–56 godzin.
torów μ (zob. ryc. B 1.5-11). Głównym metabolitem Ostatnio do stosowania dopuszczony został pre-
jest nieczynny 3-glukuronian morfiny. Wydalanie na- parat złożony typu retard zawierający oksykodon
stępuje głównie przez nerki pod postacią glukuronia- wraz z naloksonem (TARGIN; 10 mg oksykodonu
nów. Okres półtrwania wynosi 2–3 godziny. z 5 mg naloksonu lub 20 mg oksykodonu z 10 mg
Pojedyncza dawka przy podaniu dożylnym (po- naloksonu). Nalokson może połączyć się w sposób
woli) wynosi 5–10 mg, przy podaniu podskórnym antagonistyczny z receptorami opioidowymi znaj-
10–30 mg, a przy podaniu doustnym (postać retard) dującymi się po stronie wewnętrznej ściany jelit
30–60 mg. U chorych z niewydolnością nerek kumu- i w ten sposób częściowo zapobiega zaparciom in-
lacji ulega czynny 6-glukuronian morfiny. Dlatego dukowanym opioidami. Po wchłonięciu nalokson
też należy zmniejszyć dawkę morfiny lub lepiej za- w wyniku pierwszego przejścia przez wątrobę ulega
stosować inny lek opioidowy (zob. tab. 1.5-4). niemal całkowitemu rozkładowi i dlatego nie zno-
si przeciwbólowego działania oksykodonu. Ponie-
Diamorfina (heroina, diacetylomorfina) ulega szyb- waż jednak oksykodon osiąga receptory opioidowe
kiej hydrolizie przez 6-monoacetylomorfinę do morfi- od strony zewnętrznej jelit – koniecze jest przepro-
ny. Heroina i 6-monoacetylomorfina pokonują barierę wadzenie dalszych obserwacji, aby stwierdzić, jak
krew-mózg szybciej niż morfina, gdyż są względnie dalece klinicznie korzystna jest tego rodzaju kombi-
lipofilne. W następstwie szybkiej penetracji heroiny nacja obu leków.
do ośrodkowego układu nerwowego bardzo łatwo
powoduje ona uzależnienie. Ponieważ może być Petydyna działa około 5 razy słabiej od morfiny.
w prosty sposób i z dużą wydajnością wytworzona Jej biodostępność oceniana jest na poziomie 50%,
z morfiny, a ponadto jest od niej wyraźnie silniejsza a okres półtrwania – na 2–6 godzin. Ważnym meta-
– stosowana jest nielegalnie jako środek odurzający. bolitem jest norpetydyna, mająca właściwości drgaw-
W większości krajów wytwarzanie i rozprowadzanie korodne. Ponieważ norpetydyna – po wielokrotnym
heroiny jest prawnie zakazane (z wyjątkiem wykorzy- podaniu petydyny – ulega kumulowaniu w organi-
stania w specjalnych programach odwykowych, zob. zmie, petydyna nie nadaje się do stosowania w dłu-
powyżej). Natomiast w Anglii diamorfina jest nadal gotrwałej terapii. Zasada ta jest ważna szczególnie
stosowana jako lek przeciwbólowy. W ostatnim okre- w przypadku pacjentów z niewydolnością nerek.
sie podejmowane są badania modelowe nad wyko-
rzystaniem heroiny do substytucji u pacjentów z cięż- Piritramid wykazuje podobnie silne działanie jak
kimi postaciami uzależnienia od opioidów. morfina i stosowany jest często w Niemczech – z po-
wodu względnie dobrej sterowalności – w dożylnej
Hydromorfon jest 7,5 razy silniejszy niż morfina. terapii bólów pooperacyjnych („miareczkowanie
Ze względu na silny efekt pierwszego przejścia – bio- bólu”). Jego okres półtrwania wynosi 4–10 godzin.
dostępność po podaniu doustnym preparatu typu re- W handlu niedostępna jest doustna postać tego
tard wynosi w przybliżeniu 40%. Główny metabolit leku.
MUTSCHLER-2009.indd 239 2010-01-07 22:11:58
240 Układ nerwowy
HOOC
O
HO O
~60% HO OH
O H
CH3
N
HO
HO
O H 3-monoglukuronian morfiny (nieczynny przeciwbólowo)
CH3
N
HO
HO
morfina O H
CH3
~10% N
HOOC O O
HO OH
OH
6-monoglukuronian morfiny (agonista receptorów μ)
Ryc. B 1.5-11. Główne drogi metabolizmu morfiny.
Buprenorfina, endoetylenowa pochodna morfinianu, we niż morfina (duża moc). Jednakże charakteryzuje
jest częściowym agonistą receptorów opioidowych μ się niższą aktywnością wewnętrzną niż pełny agonista
i ma do nich wysokie powinowactwo. Stąd też wyka- jakim jest morfina, co powoduje, że nigdy nie osiąga
zuje prawie 40 razy silniejsze działanie przeciwbólo- maksymalnego efektu analgetycznego (skuteczności)
Tabela B 1.5-4. Właściwości farmakokinetyczne niektórych opioidów
Biodostępność po po- Okres półtrwania Problemy w przypadku Problemy w przypadku
daniu doustnym BA (%) (godz.) niewydolności wątroby niewydolności nerek
morfina 15-50 1,5-4,5 F ↑, Cl ↓, t1/2 ↑ kumulacja M-6-G
6-monoglukuronian morfiny – 1-2 – kumulacja M-6-G
petydyna 40-60 3-7 F ↑, Cl ↓, t1/2 ↑ kumulacja norpetydyny
kodeina ~50 ~3 morfina ↓ zob. morfina
dihydrokodeina 10-35 ~4 brak danych Cmax ↑, AUC ↑
oksykodon 40-80 2,5-4,5 Cl ↓, t1/2 ↑ metabolity ↑
tramadol 60-75 5-6 t1/2 ↑ t1/2 ↑ (?)
O-demetylotramadol – ~9 t1/2 ↑ –
tilidyna (pralek) ~6 3-5 (nortilidyna) nortilidyna ↓ AUC (nortilidyna) ←
→
metadon 40-99 20-60 t1/2 ↑, AUC ←
→ t1/2 ↑, AUC ↑
buprenorfina 50-55 s.l. 12-16 brak danych kinetyka ←
→
pentazocyna <20 2-5 (-10)* F ↑, Cl ↓, t1/2 ↑ kinetyka ←
→
hydromorfon 40 ~3 Cl ↓, t1/2 ↑ metabolity ↑
* – przy krążeniu wątrobowo-jelitowym; ↓ zmniejszone; ↑ zwiększone lub kumulacja; ← → niezmienione;
Cl – współczynnik oczyszczania (klirens); AUC – pole pod krzywą (stężenie/czas); Cmax – maksymalne stężenie w osoczu;
t1/2 – czas półtrwania; s.l. – podjęzykowo; M-6-G – 6-monoglukuronian morfinyl.
MUTSCHLER-2009.indd 240 2010-01-07 22:11:58
Układ nerwowy 241
jaki uzyskuje się w przypadku morfiny (efekt puła- B 1.7.2.6. Tabletki do ssania (lizaki) z fentanylem
powy – ceiling). Buprenorfina działa również agoni- stosowane są w terapii bólów przebijających.
stycznie z małym powinowactwem względem recep-
torów N/OFQ (zob. powyżej) oraz antagonistycznie Przezskórne systemy terapeutyczne. W przewle-
Układ nerwowy
na receptory opioidowe κ. Zasadniczo buprenorfina kłej terapii bólu (np. w chorobie nowotworowej)
ma podobny potencjał uzależniający jak inne opioidy dostępne są pochodzące od różnych producentów
i dlatego do niej również odnoszą się odpowiednie plastry zawierające fentanyl lub buprenorfinę – pod
przepisy dotyczące środków odurzających. postacią tzw. przezskórnych systemów terapeutycz-
Poza terapią bólu buprenorfina– podobnie jak meta- nych (TTS). Po założeniu pierwszego plastra upły-
don – stosowana jest w leczeniu substytucyjnym osób wa około 12–24 godzin, zanim osiągnięte zostanie
uzależnionych od narkotyków. Ze względu na duże odpowiednio wysokie stężenie leku w osoczu, a tym
powinowactwo do receptorów μ i silne wiązanie mię- samym – zanim wystąpi działanie przeciwbólowe. B1
dzy agonistą a receptorem – heroina jest wypierana Później stężenia fentanylu w osoczu utrzymują się
z receptorów lub też nie może związać się z recep- na względnie stałym poziomie przy zmienianiu pla-
torami μ, czego skutkiem jest niewywołanie (lub też stra co trzy dni. Zaparcia i sedacja występują w przy-
w niewielkim zakresie) objawów euforii. padku przezskórnego podawania fentanylu rzadziej
Ze względu na właściwości lipofilne buprenorfina niż przy doustnym stosowaniu morfiny. U chorych
jest dobrze wchłaniana, lecz w związku ze względnie z podwyższoną temperaturą ciała (np. w stanach go-
wysokim efektem pierwszego przejścia jej biodostęp- rączkowych, przy użyciu elektrycznych poduszek,
ność po podaniu doustnym jest niewielka (ok. 15%). po korzystaniu z sauny, solarium itp.) zwiększa się
Tymczasem biodostępność po podaniu podjęzyko- resorpcja substancji aktywnej przez skórę, w wyniku
wym (lingwetki) wynosi ok. 55%, a okres połowiczej czego częściej mogą być obserwowane typowe dla
eliminacji 12–16 godzin. opioidów objawy niepożądane. Zawierające fentanyl
Ponieważ buprenorfina dość powoli oddziela się plastry są dostępne w 5 różnych zakresach dawko-
(dysocjuje) od receptora, w przypadku przedawko- wania (szybkość uwalniania odpowiednio: 12, 25, 50,
wania lub też zatrucia, aby znieść depresję ośrodka 75 i 100 μg/godz.), natomiast plastry z buprenorfiną
oddechowego, konieczne są względnie duże dawki w 3 zakresach dawkowania (szybkość uwalniania:
czystych antagonistów opioidowych w rodzaju na- 35, 52,5 i 70 μg/godz.).
loksonu.
Pentazocyna jest czystym (pełnym) agonistą recep-
1.5.2.4. Słabo działające
torów opioidowych κ, mającym dodatkowe działanie
antagonistyczne względem receptorów opioidowych
leki opioidowe stopnia 2. wg WHO
μ, oraz wykazuje efekt pułapowy. Należy do leków
przeciwbólowych z grupy benzomorfanów, w któ- Kodeina stosowana jest jako lek przeciwbólowy nie-
rych pierścień C morfinanu jest jeszcze częściowo mal wyłącznie w terapii skojarzonej z nieopioidowy-
zachowany. Pentazocyna posiada jedną trzecią siły mi lekami przeciwbólowymi. Efekt przeciwbólowy
przeciwbólowego działania morfiny. W przeciwień- wiąże się z demetylacją cząsteczki kodeiny (za po-
stwie do tej ostatniej podwyższa ciśnienie tętnicze mocą CYP2D6) do morfiny (w 10%). W przypadku
i przyspiesza czynność serca. W związku z antagoni- osób z wolnym metabolizmem (slow metabolizers)
stycznym działaniem wobec receptorów μ – zmianie CYP2D6 (polimorfizm genetyczny) działanie prze-
terapii z pentazocyny na czystego agonistę (np. mor- ciwbólowe leku może być niewystarczające; z kolei
finę) towarzyszy początkowo zmniejszenie skutecz- u osób z bardzo szybkim metabolizmem (ultra rapid
ności czystego agonisty. Z kolei w przypadku zmiany metabolizers) może dojść do zatrucia morfiną. Bio-
z pełnego agonisty na pentazocynę mogą ujawnić się dostępność po zażyciu doustnym wynosi 40–60%,
objawy odstawienia (abstynencyjne). Stąd też pen- a jej okres półtrwania 2–3 godziny.
tazocyna nie jest żadnym zamiennym środkiem dla
osób uzależnionych. Ze względu na nasilony efekt Dihydrokodeina jest prawie trzykrotnie silniejsza
pierwszego przejścia jej biodostępność sięga jedy- w zakresie działania przeciwbólowego niż kodei-
nie ok. 20%. Jej okres półtrwania szacowany jest na na. W wątrobie ulega przemianie m.in. do dihydro-
2–4 godz. morfiny, która cechuje się znacznym potencjałem
uzależniającym. DHC charakteryzuje się nasilonym
Dożylne stosowanie fentanylu, alfentanylu, sufen- efektem pierwszego przejścia (biodostępność rzędu
tanylu i remifentanylu zostało omówione w rozdz. 20%), a jej okres półtrwania wynosi 3–5 godzin.
MUTSCHLER-2009.indd 241 2010-01-07 22:11:58
242 Układ nerwowy
Tilidyna jest prolekiem. Wykazuje bardzo słabe 1.5.3. Dronabinol
działanie agonistyczne. Właściwą substancją działa-
jącą jest nortilidyna – powstająca w wyniku oksyda- Dyskutowane jest podawanie dronabinolu, stereo-
tywnej demetylacji w obrębie atomu azotu. Tilidyna izomeru 9-tetrahydrokanabinolu (THC), jako obie-
stosowana jest w stałych proporcjach w preparatach cującego postępowania uzupełniającego w terapii
łączonych z naloksonem (50 mg tilidyny + 4 mg bólu. W Niemczech nadal nie ma co prawda w han-
naloksonu). U podstaw tego rodzaju terapii skoja- dlu gotowego preparatu dronabinolu, ale jest on
rzonej leży następująca przesłanka: tilidyna ulega dostępny w postaci substancji recepturowej (pod-
aktywacji w wyniku pierwszego przejścia przez wą- legającej przepisom o środkach odurzających!).
trobę, z kolei nalokson jest w wyniku silnego efektu Ponieważ prawodawca nie ustalił żadnych wskazań
pierwszego przejścia inaktywowany. W przypad- dla dronabinolu, lekarz może przepisać go w mak-
ku pozajelitowego podania preparatu (w przebiegu symalnej ilości 500 mg na 30 dni dla każdego sta-
uzależnienia) lub też spożycia nadmiernych dawek nu chorobowego, w którym zastosowanie uznane
leku drogą doustną – nalokson prowadzi do zanta- jest za celowe. Opisano korzystne efekty wspoma-
gonizowania działania (nor)tilidyny i tym samym gającego podawania dronabinolu przede wszystkim
zapobiega nadużywaniu. Stosowanie takich skoja- w terapii stwardnienia rozsianego oraz lekoopor-
rzonych preparatów u pacjentów z niewydolnością nego świądu. Terapeutyczna korzyść została po-
wątroby nie jest wskazane, gdyż powstaje wówczas twierdzona w badaniach klinicznych u pacjentów
zbyt mało skutecznej nortilidyny, a równocześnie z różnymi stanami bólowymi – choć nie na pozio-
nalokson przy pierwszym przejściu przez wątrobę mie znamiennym. Ponieważ posiada on niewielki
nie ulegnie pełnej dezaktywacji. Stosowanie prepa- zakres terapeutyczny i charakteryzuje się znaczny-
ratu jest natomiast możliwe u osób z niewydolnością mi objawami niepożądanymi (np. sedacja, stany za-
nerek (zob. tab. B 1.5-4). mroczenia – zob. poniżej), stosowany jest w prze-
Praktycznie nie są znane przypadki uzależnienia wlekłej terapii bólu jako środek co najwyżej wspo-
od tego typu terapii skojarzonej opartej na połączeniu magający.
tilidyny z naloksonem w postaci retard. Dronabinol wiąże się nieselektywnie z receptora-
mi kannabinoidowymi (CB1, CB2) oraz wpływa w ten
Tramadol dostępny jest na rynku pod postacią race- sposób na uwalnianie neuroprzekaźników i neuromo-
miczną i należy do najczęściej stosowanych na świe- dulatorów.
cie opioidów. Jest on słabym agonistą receptorów Po doustnym podaniu dostępność biologiczna
opioidowych μ [ok. 1/6000 w porównaniu z morfiną; dronabinolu jest bardzo słaba z powodu zjawiska
(+)-tramadol >> (–)-tramadol]. O-demetolotramadol pierwszego przejścia i wynosi 5–10%. W związku
(metabolit M1) posiada wprawdzie jeszcze większe z tym badane są możliwości podawania leku innymi
powinowactwo do receptorów opioidowych μ niż drogami. Dronabinol przechodzi przez barierę krew-
substancja macierzysta, lecz działanie przeciwbó- -mózg, przez łożysko oraz do mleka matki. Ponad-
lowe tramadolu wiąże się również z hamowaniem to rozmieszcza się w tkankach bogatych w tłuszcze
wychwytu zwrotnego monoamin (noradrenaliny, se- i ulega względnie powolnej redystrybucji do krwi
rotoniny). (+)-Tramadol hamuje głównie wychwyt i do innych tkanek. W wątrobie dronabinol metaboli-
zwrotny serotoniny, podczas gdy (–)-tramadol wpły- zowany jest w 95%. Główny metabolit – 11-hydrok-
wa na wychwyt noradrenaliny. Możliwa do osiągnię- sy-9-THC jest również związkiem czynnym. Okres
cia (maksymalna) siła działania jest wyraźnie słabsza półtrwania w osoczu wynosi 20–30 godz. Wydalanie
niż w przypadku morfiny, lecz z drugiej strony działa- metabolitów dronabinolu odbywa się w jednej trze-
nie depresyjne na ośrodek oddechowy a także wpływ ciej przez nerki, a mniej więcej w dwóch trzecich
uzależniający są również wyraźnie słabsze. Tramadol ze stolcem.
– szczególnie w dużych dawkach – dość często wy- Ponieważ osobnicza wrażliwość na dronabinol
wołuje nudności i/lub wymioty – prawdopodobnie jest bardzo zmienna, zaleca się stosowanie dawek
w wyniku zahamowania wychwytu zwrotnego sero- wzrastających od dwóch razy dziennie po 2,5 mg
toniny. lub 5 mg doustnie do pięciu razy dziennie po 10 mg
Tramadol w przypadku podania doustnego – jako dawki maksymalnej.
jest wchłaniany w 95%, jego doustna biodostępność Jako działania niepożądane obserwowano tachy-
szacowana jest na poziomie 70%, a okres półtrwania kardię i suchość w jamie ustnej – jako wynik działa-
wynosi ok. 5 godzin. Ulega procesowi N- lub O-de- nia na uwalnianie i biotransformację acetylocholiny.
metylacji, po czym jest sprzęgany z kwasem glukuro- Ponadto donoszono o istnieniu działań ośrodkowych,
nowym lub siarkowym. takich jak upośledzenie sprawności psychofizycznej
MUTSCHLER-2009.indd 242 2010-01-07 22:11:58
Układ nerwowy 243
i pamięci. Mimo że potencjał uzależniający drona- Ich stosowanie jest wskazane jedynie w przypadku
binolu jest względnie słaby – objawy abstynencyjne suchego kaszlu, gdyż na skutek wyłączenia odruchu
mogą wystąpić już po odstawieniu dawek leczni- kaszlowego nie grozi niebezpieczeństwo gromadze-
czych (lęk, niepokój, bezsenność, ślinotok, biegunka, nia się wydzieliny w oskrzelach.
Układ nerwowy
wzrost ciśnienia śródgałkowego). Możliwe jest też Leki przeciwkaszlowe będące pochodnymi opioi-
uzależnienie fizyczne. dów zestawione zostały w tab. B 1.5-2, niektóre inne
Nie opisano dotąd istotnych klinicznie interakcji przedstawiono w tab. B 1.5-5.
dronabinolu z innymi lekami. Jednakże w tym zakre-
sie konieczne są dalsze badania. Kodeina, której działanie przeciwbólowe zostało osła-
bione przez eteryfikację grupy hydroksylowej pier-
ścienia fenolowego morfiny, mimo to wykazuje jej (tj.
morfiny) pełne działanie przeciwkaszlowe. Sprawą B1
1.5.4. Dodatek: leki dyskusyjną jest, czy efekt przeciwkaszlowy – podob-
przeciwkaszlowe (antitussiva) nie jak działanie przeciwbólowe – nie jest wywoływa-
ny głównie przez metabolity morfiny (zob. powyżej).
Środki łagodzące bodziec kaszlowy (kaszel) zmniej-
szają częstość i nasilenie kaszlu – w wyniku tłumienia Dihydrokodeina należy w Niemczech do najczęściej
odruchu kaszlowego – co może być skutkiem hamo- przepisywanych leków przeciwkaszlowych. Podob-
wania tzw. ośrodka kaszlu i/lub blokowania „recep- nie jak kodeina, może być stosowana również jako
torów kaszlowych” w obrębie drzewa oskrzelowego. lek przeciwbólowy (zob. powyżej).
Tabela B 1.5-5. Leki przeciwkaszlowe o różnorodnej budowie chemicznej
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
CH3 CH3 klobutinol wycofany z rynku 1,5-3,5 120
N
CH3
HO CH3
Cl
pentoksyweryna Sedotussin 2-6 100-150
C2H5
O N
O C2H5
O
H3CO dekstrometorfan Hustenstiller-ratiopharm, 3-4 22-88
NeoTussan, Silomat BMP,
Wick Husten-Pastillen, Acodin,
Actifed, Dexatussin, Gripex,
H Grypostop, Robitussin, Tabcin,
CH3
N Tussal Antitussicum, Tusidex,
TussiDrill
O noskapina Capval 4-5 75-150
H
O N
CH3
OCH3 H
O
OCH3
O OCH3
MUTSCHLER-2009.indd 243 2010-01-07 22:11:59
244 Układ nerwowy
Hydrokodon powinien być stosowany w ciężkich Pewnymi wyjątkami są tu nefopam i flupirtyna,
przypadkach, gdyż może prowadzić do uzależnienia. których profil działania odbiega od innych nieopio-
Noskapina, alkaloid maku lekarskiego, cechuje się idowych leków przeciwbólowych.
działaniem przeciwkaszlowym porównywalnym z ko-
deiną, nie ma natomiast działania przeciwbólowego. Dawniej powszechnie używane określenie „słabo
Opisano ciężkie, lecz rzadko występujące działania działające leki przeciwbólowe” nie jest odpowiednie
niepożądane (np. zespół Stevensa-Johnsona). dla posiadanych przez tę grupę właściwości, ponie-
waż zwłaszcza w bólach wywołanych procesami za-
Dekstrometorfan jest prawoskrętnym enancjome- palnymi (zob. ryc. B 1.5-1 B) ich działanie przeciw-
rem metyloeteru leworfanolu. Związek oddziałuje bólowe jest często lepsze niż opioidów.
na receptory opioidowe dopiero w przypadku prze-
dawkowania. Z tego też powodu w niektórych kra- W celu lepszego zrozumienia działania nieopioido-
jach obserwowano nadużywanie dekstrometorfanu. wych leków przeciwbólowych poniżej przedstawiono
Ze względu na antagonistyczne działanie na receptor patofizjologiczne procesy leżące u podłoża gorączki
NMDA dekstrometorfan jest coraz częściej stosowa- i stanów zapalnych, a następnie omówiono ważniej-
ny w leczeniu bólów neuropatycznych. sze właściwości tych związków.
Klobutinol wykazuje wprawdzie pewne strukturalne
podobieństwo do metadonu, nie ma jednak działania
ani przeciwbólowego, ani depresyjnego na ośrodek 1.5.5.1. Patofizjologia gorączki i zapalenia
oddechowy. Ponieważ (tymczasowe) wyniki niedaw-
no przeprowadzonego badania klinicznego wykazały, Termoregulacja i gorączka. Zadaniem termoregu-
iż klobutinol prowadzi do wydłużenia odstępu QTc lacji jest utrzymanie ciepłoty organizmu (tj. ciepłoty
w zapisie EKG – niemiecki urząd leków wycofał lek wewnątrz tułowia i głowy) na stałym należnym po-
z rynku. ziomie, wynoszącym średnio 37°C – pomimo wahań
zachodzących w wytwarzaniu, jego przyjmowaniu
Pentoksyweryna jest zasadowym estrem, który i utracie. Struktury odpowiedzialne za przetwarza-
nie wykazuje zarówno niepożądanego działania na- nie informacji dotyczącej ciepłoty są umiejscowione
sennego, jak i działania depresyjnego na czynność w przedniej części podwzgórza. Bodźce napływające
oddechową. z termoreceptorów skóry i narządów wewnętrznych
podlegają tu integracji i – w przypadku odchyleń
od wartości należnej – są przetwarzane w sygnały
1.5.5. Nieopioidowe regulujące. W przypadku przegrzania (np. wskutek
wysiłku fizycznego) poprzez wzmożone pocenie
leki przeciwbólowe
i nasilenie przepływu krwi przez skórę – następuje
stopnia 1. wg WHO zwiększenie oddawania ciepła; natomiast przy zagro-
żeniu przechłodzeniem – oddawanie ciepła (przede
Leki przeciwbólowe należące do tej grupy – okre- wszystkim wskutek skurczu naczyń obwodowych)
ślane niekiedy jako małe lub błędnie jako obwodo- zostaje zmniejszone oraz następuje nasilenie wytwa-
we (obwodowo działające) analgetyki – mają mimo rzania ciepła.
różnorodnej budowy chemicznej bardzo podobny Przez pojęcie gorączki rozumie się termoregula-
profil działania: oprócz działania przeciwbólowego cję z przestawieniem wartości należnej temperatury
wykazują również składową przeciwgorączkową; na wyższy poziom. Gorączka jest objawem towa-
ponadto nieopioidowe leki przeciwbólowe będące rzyszącym niemal wszystkim zakażeniom. Części
pochodnymi kwasowymi, a także niebędące kwa- składowe patogennych mikroorganizmów, np. en-
sami inhibitory COX-2 wykazują działanie prze- dotoksyny bakterii gram-ujemnych, a także wirusy
ciwzapalne. Natomiast praktycznie nie występują mogą wywoływać gorączkę. Uczestniczące w tym
u nich wcale właściwości psychotropowe i uspoka- procesie związki określane są wspólnym mianem
jające (sedatywne) – typowe dla opioidowych leków egzogennych pirogenów. Molekularne mechanizmy
przeciwbólowych. Ze względu na swój profil dzia- leżące u podłoża odczynów gorączkowych są złożo-
łania – zakres stosowania nieopioidowych leków ne. Współistnieje tu wzajemne oddziaływanie układu
przeciwbólowych jest duży i leki z tej grupy należą immunologicznego, układu endokrynnego i ośrodko-
do najczęściej stosowanych preparatów farmakolo- wego układu nerwowego. Cytokiny, takie jak inter-
gicznych. leukina-1 (IL-1), IL-6 i czynnik martwicy nowotworu
MUTSCHLER-2009.indd 244 2010-01-07 22:11:59
Układ nerwowy 245
alfa (TNF-α – tumor necrosis factor), odgrywają tutaj ■ myszy pozbawione COX-2 („znokautowane” gene-
główną rolę. Podczas występowania odczynu gorącz- tycznie) po podaniu egzogennego pirogenu nie rea-
kowego ich stężenia zarówno w miejscu uszkodze- gują odczynem gorączkowym.
nia tkanki (głównie IL-1), jak i w układzie krążenia
Układ nerwowy
oraz w mózgu ulegają zwiększeniu. Przyjmują one Bezpośrednio po przestawieniu wartości należnej
na siebie rolę humoralnego przekaźnika pomiędzy na wyższy poziom prawidłowa temperatura cia-
tkankami obwodowymi i ośrodkowym układem ner- ła wynosząca 37°C odczuwana jest jako wychło-
wowym, w związku z czym są też nazywane krąże- dzenie. Powoduje ona skurcz skórnych naczyń
niowymi pirogenami. W mózgu cytokiny – poprzez krwionośnych, dreszcze oraz subiektywne uczu-
wpływ na swoiste receptory – indukują aktywność cie zimna. Po obniżeniu gorączki (powrót do pra-
cyklooksygenazy-2 (COX-2) i co za tym idzie – syn- widłowej wartości należnej) panująca temperatura
tezę prostaglandyn (głównie PGE2). PGE2 jest „koń- ciała jest natomiast odczuwana jako zbyt wysoka. B1
cowym przekaźnikiem” odczynu gorączkowego. Obfite pocenie się, rozszerzenie naczyń skórnych
Pobudzając receptory prostaglandynowe, zwiększa i subiektywne uczucie gorąca charakteryzują fazę
ona zależny od cAMP wzrost przemiany komórkowej odgorączkowania.
neuronów ośrodka termoregulacyjnego podwzgórza.
W następstwie dochodzi do podwyższenia wartości
Zapalenie. Na różnego rodzaju czynniki szkod-
należnej ciepłoty ciała.
liwe (noksy) – noksy chemiczne lub fizyczne, za-
każenia drobnoustrojami lub pasożytami – tkanka
Kaskada powstawania gorączki (produkcja cytokiny
w miejscu uszkodzenia reaguje odczynem zapalnym
– indukcja aktywności COX-2 w mózgu – synteza
(zapaleniem). Przebiegające przy tym procesy cha-
PGE2) została ostatecznie potwierdzona w doświad-
rakteryzuje ścisłe powiązanie reakcji naczyniowych
czeniach na zwierzętach:
i komórkowych oraz odczynów obronnych zarówno
■ antagoniści IL-1 i leki hamujące aktywność COX-2 specyficznych, jak i niespecyficznych antygenowo.
(wybiórcze i nieselektywne) działają przeciwgo- Jako tzw. objawy podstawowe (ostrej) reakcji zapal-
rączkowo, nej występują symptomy opisane już ponad 2000 lat
Tabela B 1.5-6. Mediatory procesu zapalnego, pochodzenie i działanie (według Thewsa, Mutschlera, Vaupela)
Mediator Pochodzenie Podstawowe działania
histamina komórki tuczne, rozszerzenie naczyń, zwiększenie
zasadochłonne granulocyty przepuszczalności naczyń
serotonina płytki krwi agregacja płytek, złożony wpływ na układ
naczyniowy, hiperalgezja
czynniki dopełniacza C3a, C5a produkty rozpadu układu dopełniacza uwalnianie histaminy z komórek tucznych,
chemotaksja, nasilenie przepuszczalności naczyń
bradykinina końcowy produkt rozpadu układu rozszerzenie naczyń, zwiększenie przepuszczalności
kalikreina-kinina naczyń, hiperalgezja
prostaglandyny E2, F2α granulocyty, makrofagi, rozszerzenie (PGE2) lub zwężenie (PGF2α)
komórki śródbłonka, neurony naczyń, sensybilizacja nocyceptorów, hiperalgezja
leukotrieny granulocyty, makrofagi, chemotaksja, zwiększenie przepuszczalności naczyń
komórki tuczne
czynnik aktywujący płytki (PAF) granulocyty, makrofagi, aktywacja granulocytów i płytek, chemotaksja,
komórki tuczne, płytki krwi zwiększenie przepuszczalności naczyń
tlenek azotu (NO) makrofagi, komórki śródbłonka, rozszerzenie naczyń, w pewnych warunkach
neurony działanie cytotoksyczne; hiperalgezja
wolne rodniki granulocyty, makrofagi uśmiercanie bakterii, niszczenie
macierzy tkankowej
MUTSCHLER-2009.indd 245 2010-01-07 22:11:59
246 Układ nerwowy
temu przez Aulusa Corneliusza Celsusa, a następnie czaniu uszkodzenia i przywrócenie stanu pierwot-
rozszerzone o piąty objaw podstawowy (functio la- nego. Mogą mieć one również wpływ szkodliwy,
esa) przez Galena: np. w przewlekłych zapaleniach reumatycznych.
Na ryc. B 1.5-12 przedstawiono schematycznie
■ zaczerwienienie (rubor),
jeszcze raz omawiane powyżej zjawiska występu-
■ obrzęk (tumor), jące w procesie zapalnym.
■ (miejscowe) podwyższenie ciepłoty (calor),
■ ból (dolor) oraz 1.5.5.2. Właściwości farmakologiczne
■ upośledzenie czynności (functio laesa). nieopioidowych
leków przeciwbólowych
Objawy te są następstwem, wywołanych przez
szkodliwy bodziec, zaburzeń ukrwienia w obrę- Klasyfikacja. Nieopioidowe leki przeciwbólowe dzie-
bie mikrokrążenia z ucieczką składników osocza li się na dwie grupy. Pierwsza obejmuje substancje,
do przestrzeni międzykomórkowej (wysięk) na sku- które oprócz działania przeciwbólowego i przeciwgo-
tek zwiększonej przepuszczalności naczyń włosowa- rączkowego mają również wyraźne działanie przeciw-
tych. Do tego dochodzi pobudzenie i uwrażliwienie zapalne. Leki należące do tej grupy leków – oprócz
nocyceptorów przez uwolnione mediatory zapalenia niektórych selektywnych inhibitorów COX-2 (koksy-
(zob. tab. B 1.5-6). bów; zob. poniżej) – należą do pochodnych kwaso-
Już na początku – proces zapalny może się co- wych. W związku z ich działaniem przeciwzapalnym
fać, jeśli dojdzie do usunięcia czynnika szkodli- określane bywają mianem niesteroidowych leków
wego (np. zniszczenia toksyn bakteryjnych) lub przeciwzapalnych (NLPZ; NSAID – non-steroidal
zakończenia jego szkodliwego działania (np. unie- anti-inflamatory drugs). Kwasowe NLPZ posiadają
możliwienia dalszego oddziaływania szkodliwego fragmenty cząsteczkowe o charakterystyce lipofilnej,
czynnika fizycznego). Częstokroć jednak do po- oraz hydrofilnej, ich wartość pKa znajduje się między
czątkowych zaburzeń ukrwienia i wysięku osocza 3 a 6, aż do ponad 99% wiążą się z białkami osocza.
dołącza się migracja elementów komórkowych krwi Z chemicznego punktu widzenia do niesteroidowych
(m.in. granulocytów i monocytów) do przestrze- leków przeciwzapalnych zalicza się: salicylany, po-
ni pozakomórkowej, jak też rozplem histiocytów chodne kwasu octowego i propionowego, oksykamy,
i fibroblastów. Procesy te służą pierwotnie zwal- fenamaty i in. (zob. poniżej).
czynnik szkodliwy
(noksa)
uszkodzenie
tkanki
zaburzenia uwolnienie mediatorów migracja leukocytów,
mikrokrążenia (zapalenia) proliferacja komórek
miejscowe zwiększenie przepuszczal- wysięk pobudzenie i sensybilizacja
poszerzenie naczyń ności naczyń nocyceptorów
zaczerwienienie podwyższenie ciepłoty obrzęk upośledzenie czynności ból
(rubor) (calor) (tumor) (functio laesa) (dolor)
Ryc. B 1.5-12. Patogeneza i objawy zapalenia (wdług Thewsa, Mutschlera, Vaupela).
MUTSCHLER-2009.indd 246 2010-01-07 22:11:59
Układ nerwowy 247
Sprawą wątpliwą jest, czy kwasowe NLPZ cechują obojętne lub nieznacznie zasadowe pH. Nie posiadają
się szczególnie dobrą skutecznością przeciwzapalną, one równocześnie właściwości lipofilnych i hydrofil-
ponieważ gromadzą się w tkance o kwasowym pH nych. Z białkami osocza wiążą się wyraźnie słabiej
i z toczącym się tam procesem zapalnym. Hipoteza niż leki z grupy NLPZ. Do leków z tej grupy zalicza
Układ nerwowy
opiera się na tym, że w kwasowym podścielisku ist- się paracetamol i pochodne pirazolowe – fenazon,
nieje porównywalnie większy odsetek niezdysocjo- propyfenazon, metamizol oraz fenylobutazon.
wanego i zdolnego do przeniknięcia przez błony leku
niż może dotrzeć do miejsca działania. Eksperymen-
talnie nie udało się jednak jak dotąd potwierdzić tej
1.5.5.2.1. Niesteroidowe leki przeciwzapalne
teorii. Wykazano jedynie, że ze względu na stosun-
(NLPZ; NSAID – non steroidal
kowo dużą zawartość zewnątrzkomórkowego białka
w zakwaszonej tkance (wysięk osocza!) oraz bardzo
anti-inflammatory drugs) B1
wysoką wartość wiązania NLPZ z białkami osocza
Mechanizm działania n.l.p.z. Dla zrozumienia
(> 98%) – dochodzi do nasycenia lekiem związanym
mechanizmu działania oraz działań niepożądanych
z białkiem. Pytanie, czy kwasowe otoczenie w tkan-
n.l.p.z. szczególne znaczenie mają pochodzące z lat
ce z procesem zapalnym zwiększa również w sposób
70. XX w. wyniki badań Vane’a i Ferreiry potwier-
klinicznie istotny zawartość niezwiązanej z białkami
dzające, że związki te oddziałują na biosyntezę
i tym samym farmakologicznie aktywnej frakcji kwa-
prostaglandyn. Niesteroidowe leki przeciwzapalne
sowych NLPZ – nie znalazło odpowiedzi w dotych-
w dawkach leczniczych hamują – jak to przedsta-
czasowych badaniach. Przeciwko podanej powyżej
wiono na ryc. B 1.5-13 – aktywność cyklooksyge-
hipotezie świadczy także obserwacja kliniczna, że
naz (syntaz prostaglandyn H, PGH-syntaz), czyli
niemające kwasowego charakteru inhibitory COX
enzymów kontrolujących przekształcanie kwasu
(np. celecoksyb, etorikoksyb; zob. poniżej) są do-
arachidonowego w cykliczne nadtlenki (prostaglan-
kładnie tak samo skuteczne przeciwzapalnie jak kwa-
dyna H2), które są prekursorami zarówno prosta-
sowe NLPZ.
glandyn, jak i tromboksanu A2 oraz prostacykliny.
Jak to już zostało opisane, prostaglandyny w sposób
Do drugiej grupy nieopioidowych analgetyków za-
istotny uczestniczą w powstawaniu bólu, gorączki,
licza się leki przeciwbólowe o działaniu przeciwgo-
a także reakcji zapalnych. Dlatego też związki blo-
rączkowym – niebędące pochodnymi kwasów, które
kujące syntezę prostaglandyn działają:
w dawkach terapeutycznych nie powodują hamo-
wania procesów zapalnych. Są to substancje mające ■ przeciwbólowo,
fosfolipidy
fosfolipidaza A2
kwas arachidonowy i inne
nienasycone kwasy tłuszczowe
cyklooksygenazy lipooksygenazy epoksygenazy wolne rodniki
NLPZ (COX-1, COX-2) (CYP450)
PGI2 PGE2 TxA2 PETE, epoksydy izoprostany
leukotrieny,
lipoksyny
Ryc. B 1.5-13. Możliwe drogi metabolizowania kwasu arachidonowego i hamowanie biosyntezy prostaglandyn przez NLPZ. PGE2
– prostaglandyna E2; PGI2 – prostacyklina; TxA2 – tromboksan A2, PETE – kwas hydro-peroksy-eikozatetraenowy.
MUTSCHLER-2009.indd 247 2010-01-07 22:12:00
248 Układ nerwowy
■ przeciwgorączkowo, COX-2 jest natomiast enzymem szybko indukowa-
nym (powoływanym do życia; gen dla COX-2 jest ge-
■ przeciwzapalnie.
nem natychmiastowej wczesnej odpowiedzi, immedia-
te early gen) przez rozmaite czynniki (np. cytokiny)
Ponieważ prostaglandyny są syntetyzowane pra-
i ujawnia się w znacznej ilości w przebiegu zapalenia,
wie we wszystkich komórkach lub tkankach, gdzie
reakcji bólowej i innych uszkodzeń tkanek. Hamowa-
uczestniczą w różnorodnych procesach fizjologicz-
nie COX-2 wyjaśnia działanie przeciwzapalne, prze-
nych (zob. tab. B 1.5-7) – łatwo zrozumieć, że dzia-
ciwbólowe i przeciwgorączkowe niesteroidowych
łania lecznicze i określone działania niepożądane
leków przeciwzapalnych, tymczasem liczne działania
niesteroidowych leków przeciwzapalnych są ze sobą
niepożądane (np. nadżerki lub owrzodzenia w ukła-
nierozłącznie związane.
dzie pokarmowym) wynikają głównie z hamowania
Opisany wpływ na syntezę prostaglandyn wyjaśnia także COX-1. Te obserwacje eksperymentalne doprowa-
przyczynę, dla której różnorodne metody galenowe, np. wy- dziły do stworzenia NLPZ selektywnych względem
tworzanie tabletek opornych na sok żołądkowy albo stoso- COX-2 (tzw. koksybów).
wanie czopków zamiast postaci doustnych – nie są w stanie
poprawić tzw. żołądkowej tolerancji tych leków (tj. rzad- Z wyjątkiem koksybów wszystkie dostępne w handlu
szego wywoływania działań niepożądanych ze strony prze-
wodu pokarmowego lub w mniejszym nasileniu). Wywo- postacie NLPZ w dawkach leczniczych hamują ak-
ływane przez te leki uszkodzenia błony śluzowej żołądka tywność obu izoform COX (tj. COX-1 i COX-2).
zależą bowiem przede wszystkim od ogólnoustrojowego Jednak okazało się, że również COX-2 może
hamowania syntezy prostaglandyn. występować w sposób konstytutywny w niektórych
narządach, takich jak rdzeń kręgowy, nerki, śródbło-
Przez długi czas zakładano, że istnieje tylko jedna, nek naczyń lub macica. Ponadto w znacznych iloś-
niezmienna postać cyklooksygenazy. W l990 roku zi- ciach może być on wytwarzany w trakcie licznych
dentyfikowano dwie, różniące się w swych funkcjach procesów adaptacyjnych (np. w miejscu gojenia się
izoformy (postacie izomeryczne) cyklooksygenazy rany lub wrzodu, w macicy w procesie zagnieżdża-
(COX-1 i COX-2). (U zwierząt znaleziona została nia się jaja płodowego, jak również w komórkach
także postać COX-3, która u człowieka prawdopo- śródbłonka naczyń; zob. ryc. B 1.5-14). Tym samym
dobnie nie występuje). COX-1 – będąca enzymem działań niepożądanych NLPZ (zob. poniżej) w ża-
konstytutywnym (stale obecnym) – uczestniczy w syn- den sposób nie można wiązać jedynie z hamowa-
tezie prostaglandyn, np. w żołądku, płytkach krwi lub niem COX-1.
w nerkach. Zahamowanie tego enzymu przez NLPZ Także w przebiegu proliferacji komórek nowo-
powoduje ujawnienie się działań niepożądanych tworowych (np. komórek raka jelita grubego) COX-2
w obrębie wymienionych organów. odgrywa istotną rolę, z czego zdają się wynikać nowe
Tabela B 1.5-7. Działania prostaglandyn i następstwa zahamowania ich syntezy
Działanie prostaglandyn Działanie inhibitorów Efekt kliniczny
syntezy prostaglandyn
sensybilizacja nocyceptorów osłabienie sensybilizacji nocyceptorów działanie przeciwbólowe
zmniejszenie wydzielania soku żołądkowego, zwiększenie wytwarzania soku żołądkowego uszkodzenie śluzówki, ewentualnie wrzód
cytoprotekcja
spowolnienie czynności motorycznej jelit przyspieszenie motoryki jelit biegunki
nasilenie nerkowego wydalania sodu zmniejszenie wydalania sodu, zatrzymanie wody obrzęki, podwyższenie ciśnienia krwi
nasilenie agregacji płytek krwi hamowanie agregacji płytek zapobieganie udarom, zwiększone
przez tromboksan A2 niebezpieczeństwo krwawienia
zwiększenie napięcia mięśnia macicy zmniejszenie nasilonego napięcia mięśnia macicy znoszenie bólu towarzyszącego
miesiączkowaniu
MUTSCHLER-2009.indd 248 2010-01-07 22:12:00
Układ nerwowy 249
bodziec fizjologiczny fizjologiczna adaptacja bodziec zapalny
Układ nerwowy
glukokortykosteroidy
COX-1 COX-2 COX-2
konstytutywny konstytutywny regulowany indukowany
B1
trombocyty
(TxA2)
nerki, żołądek PGs PGs
(PGE2)
śródbłonek naczyń
(PGI2)
rdzeń kręgowy, nerki, macica zapalenie
enzym utrzymujący homeostazę gojenie się ran ból
(„housekeeping enzyme”) śródbłonek naczyń i inne gorączka
Ryc. B 1.5-14. Ekspresja, regulacja i funkcja cyklooksygenazy 1 (COX-1) i cyklooksygenazy 2 (COX-2). PG – prostaglandyny; PGI2
– prostacyklina, TxA2 – tromboksan A2.
potencjalne wskazania do stosowania selektywnych ne do (farmakologicznego) zamknięcia przetrwałego
inhibitorów COX-2. (tj. drożnego jeszcze po urodzeniu) przewodu tętni-
czego Botalla (ductus arteriosus; krótkiego przewodu
Farmakokinetyka. Większość substancji z grupy łączącego tętnicę płucną z aortą), gdyż w utrzymaniu
typowych niesteroidowych leków przeciwzapalnych drożności tego przewodu wyraźnie uczestniczą właś-
ulega szybkiemu i dobremu wchłanianiu. Dla określo- nie prostaglandyny (PGE2, PGI2).
nych zastosowań terapeutycznych istotne znaczenie
mogą mieć ich bardzo różne czasy połowiczej elimi- Działania niepożądane. Mechanizm działania kla-
nacji, które zostały zebrane w tab. B 1.5-8. sycznych niesteroidowych leków przeciwzapalnych
warunkuje ujawnienie się wspólnych wszystkim le-
Wskazania. Niesteroidowe leki przeciwzapalne kom działań niepożądanych, takich jak:
wskazane są w następujących przypadkach:
■ zaburzenia żołądkowo-jelitowe (niestrawność i in.),
■ różne stany bólowe (np. bóle głowy i zębów), nadżerki układu pokarmowego aż do owrzodzeń,
krwawienia i perforacje (pęknięcia wrzodu),
■ bóle w przebiegu choroby zwyrodnieniowej sta-
wów, ■ reakcje skórne,
■ migrena, ■ zaburzenia czynności nerek z retencją jonów sodu
i wody z następczym tworzeniem się obrzęków
■ gorączka, a przede wszystkim
i wzrostem ciśnienia krwi,
■ patofizjologiczne (warunkowane procesem zapal-
■ zahamowanie agregacji płytek krwi,
nym) bóle nocyceptywne, włącznie z występujący-
mi w związku z chorobą reumatyczną. ■ objawy ze strony ośrodkowego układu nerwowego
np. zawroty i bóle głowy,
Również w terapii bólów związanych z przerzutami
■ zmniejszenie aktywności motorycznej macicy,
nowotworowymi do kości niesteroidowe leki prze-
ciwzapalne okazały się bardzo efektywne. Inhibitory ■ powikłania sercowo-naczyniowe (zawał serca, udar
syntezy prostaglandyn mogą być ponadto zastosowa- mózgu),
MUTSCHLER-2009.indd 249 2010-01-07 22:12:00
250 Układ nerwowy
Tabela B 1.5-8. Nieopioidowe leki przeciwbólowe/przeciwzapalne
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna
(mg)
l. Salicylany
kwas acetylosalicylowy Alka-Seltzer, Aspirin ~0,25 1500-3000
COOH ASS-ratiopharm,
O CH3 ASS STADA, Acard, Ascodan,
Asprocol, Calcipirina,
O Etopiryna, Polocard,
Polopiryna, Upsarin
COOH diflunisal Dolobid 9-13 1000
OH
F F
II. Pochodne kwasu octowego
indometacyna Indo-CT, Indometacin BC, 2-11 150-200
COOH Indometacin Sandoz,
H3CO Indomet-ratiopharm,
Elmetacin, Metindol
CH3
N
Cl
O
acemetacyna (prolek) Acemetacin-CT, ~5 90-180
Acemetacin Heumann,
O COOH
Acemetacin STADA,
Rantudil,
H3CO
CH3
N
Cl
diklofenak Diclac, 1-2 100-150
COOH Diclofenac-ratiopharm,
Diclofenac Sandoz,
NH Voltaren, Arthrotec, Dcialc,
Cl Cl Dicloratio, Diky, Majamil,
Naclofen, Olfen,
Rewodina, Voltanec
Cl lonazolak Argun ~6 600
COOH
N
N
MUTSCHLER-2009.indd 250 2010-01-07 22:12:01
Układ nerwowy 251
Tabela B 1.5-8. Nieopioidowe leki przeciwbólowe/przeciwzapalne (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półłtrwania dawka
(godz.) dzienna
Układ nerwowy
(mg)
III. Pochodne kwasu propionowego
CH3 ibuprofen Aktren, Dolgit, Dolormin, 1,5-2,5 1200-2400
IbuHEXAL, Acatar Zatoki,
COOH Ibalgin, Ibufen, Ibum, Ibupar,
Ibuprofen, Ibuprom, Metafen,
(H3C) 2HC
Modafen, Nurofen, B1
Rhinafen
deksibuprofen Deltaran, Dolomagon, 1,5-2,5 600-900
CH3 (S-ibuprofen) Seractil
COOH
(H3C) 2HC
CH3 flurbiprofen Dobendan Direkt, 3-4 35-43,75
Flurbiprofen,
COOH Strepsils Intensive
ketoprofen Alrheumun, Gabrilen, 1,5-2,5 100-200
O CH3 Ketoprofen CT, Spondylon,
Fastum, Profenid, Febrofen,
COOH Ketonal, Ketoprom, Ketores,
Profenid
O CH3
deksketoprofen Sympal 1,5-2,5 50-75
S-ketoprofen)
COOH
CH3 naproksen Aleve, 13-15 500-1000
(S-naproksen) Naproxen CT, Proxen,
COOH Apo-Napro, Naproxen,
Anapran Neo
H3CO
kwas tiaprofenowy Surgam 1,5-3 600
O CH3
S
COOH
IV. Oksykamy
O O piroksykam Piroxicam HEXAL, ~50 10-20
Piroxicam-ratiopharm, (20-120)
S CH3 Piroxicam STADA, Feldene,
N
Flamexin
NH N
OH O
MUTSCHLER-2009.indd 251 2010-01-07 22:12:01
252 Układ nerwowy
Tabela B 1.5-8. Nieopioidowe leki przeciwbólowe/przeciwzapalne (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna
(mg)
O O meloksykam Meloxicam HEXAL, ~20 7,5-15
S CH3 Meloxicam-ratiopharm,
N Meloxicam STADA, Mobec,
NH N Aglan, Aspicam, Lormed,
Melobax, Meloksam,
OH O S Movalis
CH3
O O lornoksykam Telos 3-5 12-16
S CH3
N
Cl
S NH N
OH O
V. Pochodne aniliny
paracetamol Paracetamol AL, 1,5-3 1000-4000
NH CH3
Paracetamol-ratiopharm,
Perfelgan, Acenol, Apap, Calpol,
O
HO Cefalgin, Codipar, Coldrex,
Efferalgan, Febrisan, Fervex,
Gripex, Kofepar, Panadol,
Paracetamol, Saridon, Tabcin,
Theraflu
VI. Pochodne 2,3-demetylo-1-fenylo-3-pyrazolino-5-onu
R CH3
O N CH3
N
R
fenazon Migräne-Kranit, 11-12 1000-4000
H Migränin Phenazon,
Antotalgin
(H3C) 2HC propyfenazon DEMEX, Cefalgin, 1-3 1000-4000
Saridon
H3C metamizol Analgin, Metamizol HEXAL, 2-4 (dla MAA*) 1000-4000
O N PyraHEXAL, Novalgin,
Novaminsulfon-ratiopharm,
O S
Pyralgin, Pyralginum,
OH Spasmalgon, Tolargin
* MAA – metyloaminoantypiryna (4-metyloamonofenazon)
■ wyzwolenie napadu astmy u szczególnie predyspono- rzenie się mających działanie skurczowe w obrębie
wanych pacjentów, głównie u cierpiących na astmę oskrzeli leukotrienów (reakcja rzekomoalergiczna).
oskrzelową, co związane jest z zahamowaniem cy-
klooksygenaz i następczą podażą substratu dla lipo- Działania niepożądane selektywnych inhibitorów
oksygenaz – czego ostatecznym skutkiem jest two- COX-2 (zob. poniżej) są zasadniczo podobne do tych
MUTSCHLER-2009.indd 252 2010-01-07 22:12:02
Układ nerwowy 253
występujących w przebiegu stosowania nieselektyw- niejsze działanie przeciwgorączkowe i przeciwza-
nych NLPZ, poza działaniami związanymi z brakiem palne, a w szczególności zahamowanie agregacji
hamowania COX-1 (owrzodzenia, krwawienia, per- płytek krwi (zahamowanie syntezy tromboksanu
foracje) i zahamowaniem agregacji płytek. A2). Kwas acetylosalicylowy jest w związku ze swy-
Układ nerwowy
mi właściwościami jednym z częściej stosowanych
Przeciwwskazania. NLPZ są przeciwwskazane leków przeciwbólowych/przeciwzapalnych i – obok
w chorobie wrzodowej żołądka i dwunastnicy, astmie klopidogrelu – najważniejszym inhibitorem agre-
i skazie krwotocznej. Nie powinny być również sto- gacji płytek. Inaktywuje on cyklooksygenazy przez
sowane w ostatnich tygodniach ciąży – z powodu za- nieodwracalną acetylację reszty serynowej (w przy-
grożenia związanego z przedwczesnym zamknięciem padku COX-1 – w pozycji 530, a w przypadku
przewodu tętniczego Botalla; w ciężkim uszkodzeniu COX-2 – w pozycji 516). Dojrzałe płytki krwi po-
wątroby i nerek mogą być stosowane z wielką ostroż- siadają wyłącznie ważną dla syntezy tromboksanu B1
nością. Nie powinny być podawane łącznie z pochod- – COX-1. Ponieważ są pozbawione jąder komórko-
nymi kumaryny (zob. poniżej). wych, co uniemożliwia regenerację uszkodzonego
enzymu, gwarantują utrzymywanie się działania an-
Interakcje. W przypadku równoczesnego stosowania tyagregacyjnego – mimo krótkiego okresu półtrwa-
niesteroidowych leków przeciwzapalnych z innymi nia ASA (ok. 15 min; zob. poniżej) – przez okres
lekami może dojść do następujących interakcji: kilku dni, zanim dojrzeje następna pula trombocy-
tów. W zapobieganiu zawałom serca wystarczają-
■ glukokortykosteroidy zwiększają niebezpieczeń-
ce są niewielkie dawki kwasu acetylosalicylowego
stwo powikłań żołądkowo-jelitowych, gdyż w trak-
(100 mg na dobę).
cie gojenia się owrzodzeń wyzwolonych przez
NLPZ dochodzi do wzmożonej indukcji COX-2,
Kinetyka. Po podaniu doustnym ASA ulega szybkie-
która pod wpływem glukokortykosteroidów ulega
mu wchłanianiu – w dużym odsetku. Już w trakcie pa-
zahamowaniu;
sażu przez śluzówkę przewodu pokarmowego docho-
■ zmniejsza się urykozuryczne działanie probeneci- dzi do częściowego oderwania się reszty acetylowej.
du i równocześnie spowolnieniu ulega wydalanie W ten sposób powstający kwas salicylowy również
inhibitorów syntezy prostaglandyn typu kwaso- posiada właściwości przeciwbólowe. W wątrobie
wego. – w wyniku dalszej hydrolizy estru – powstają glu-
kuroniany estrowe i eterowe, jak również glicynian
Ponadto NLPZ: kwasu salicylowego (kwas salicylurowy) (zob. ryc.
1.5-15). Jedynie niewielka część ASA ulega utle-
■ zmniejszają moczopędne działanie saluretyków,
nieniu do kwasu gentyzynowego. Okres półtrwania
■ nasilają działanie hipoglikemiczne doustnych le- w osoczu ASA wynosi 15 minut, natomiast okres pół-
ków przeciwcukrzycowych, trwania powstającego kwasu salicylowego po małych
dawkach ASA – 2–3 godziny. W przypadku stosowa-
■ spowalniają eliminację metotreksatu i tym samym
nia dużych dawek ASA dochodzi do wysycenia en-
zwiększają jego toksyczność,
zymów wątrobowych i jego powolniejszej eliminacji
■ zmniejszają wydalanie jonów litu, (przejście z kinetyki rzędu pierwszego na kinetykę
rzędu zerowego). Wydalanie metabolitów ASA nastę-
■ osłabiają działanie przeciwzakrzepowe pochod-
puje głównie przez nerki.
nych kumaryny,
■ zmniejszają efekt antyhipertensyjny leków prze- Dawkowanie. W stanach bólowych i gorączkowych
ciwnadciśnieniowych, szczególne z grupy inhibito- dawkowanie wynosi 1,5–3 g ASA dziennie, w cho-
rów ACE (konwertazy angiotensyny). robach reumatycznych konieczne są dawki dobowe
rzędu 4–6 g, które jednak w związku z toksycznością
żołądkową są źle znoszone. W profilaktyce zawałów
1.5.5.2.1.1. Kwas acetylosalicylowy serca podaje się dawkę 100 mg dziennie.
(ASA – acetylsalicyl acid)
Specyficzne działania niepożądane. Częstymi
Przez estryfikację fenolowej grupy hydroksylowej działaniami niepożądanymi są: zgaga, dolegliwości
kwasu salicylowego za pomocą kwasu octowego żołądkowe i mikrokrwawienia śluzówki żołądka.
uzyskuje się nie tylko lepszą miejscową tolerancję Działania te zależne są od dawki i ujawniają się przy
(zmniejszenie działania drażniącego), ale także sil- stosowaniu ASA częściej niż w przypadku innych
MUTSCHLER-2009.indd 253 2010-01-07 22:12:02
254 Układ nerwowy
ASA i ibuprofenu w profilaktyce zawału serca ibu-
COOH profen może się związać z katalitycznym centrum
O CH3 cyklooksygenazy-1 przed ASA. W zależności więc
od sposobu zażywania możliwe jest zniesienie kar-
O
dioprotekcyjnego działania ASA. Z tego powodu
kwas acetylosalicylowy
pacjenci, którzy zażywają ASA w profilaktyce za-
wału serca, powinni (przy odpowiednim wskazaniu)
otrzymywać nie więcej niż jedną dawkę ibuprofe-
nu w ciągu dnia i zażyć ją co najmniej 2 godziny
COOH po przyjęciu kwasu acetylosalicylowego. Równo-
OH czesne przyjmowanie paracetamolu, diklofenaku
lub celekoksybu nie wpływa natomiast na działanie
kwasu acetylosalicylowego polegające na hamowa-
kwas salicylowy niu agregacji trombocytów.
O Zatrucia. W ostrym zatruciu kwasem acetylosalicy-
COOH
O NH lowym obserwuje się początkowo hiperwentylację,
OH OH
OH zlewne poty i pobudzenie, później postępujące po-
HO rażenie czynności oddechowej, utratę przytomności,
podwyższenie ciepłoty (hipertermię) i odwodnienie.
kwas salicylurowy kwas gentyzynowy W wyniku hiperwentylacji dochodzi do nadmierne-
go wydychania ditlenku węgla, w następstwie czego
Ryc. B 1.5-15. Biotransformacja kwasu acetylosalicylowego. dochodzi do oddechowej zasadowicy, która zostaje
wyrównana przez zwiększone nerkowe wydalanie
wodorowęglanu. Z postępującym zatruciem docho-
NLPZ. U pacjentów z dną należy liczyć się z nasi- dzi jednak do kwasicy metabolicznej wskutek nasila-
loną retencją kwasu moczowego – czego przyczyną jącego się porażenia czynności oddechowej oraz roz-
jest konkurencja o nośnik kwasowy. przęgnięcia oksydatywnej fosforylacji z nasilonym
Ciężkie działania niepożądane (szum w uszach, wytwarzaniem CO2.
upośledzenie słuchu, zawroty głowy, nudności, wy- Leczenie zatrucia, oprócz metod zmierzających
mioty, silne krwawienia z przewodu pokarmowego, do zahamowania dalszego wchłaniania ASA, ma
owrzodzenia) występują głównie w przypadku stoso- na celu przywrócenie prawidłowej równowagi kwa-
wania wysokich dawek ASA przez dłuższy okres czas sowo-zasadowej i zwiększenie wydalania salicyla-
i w przypadku zmniejszenia stosowanej dawki leku nów. Uzyskuje się to przez wlew wodorowęglanu
mają charakter odwracalny. Ponadto należy zwrócić sodu, dzięki któremu jednocześnie zwiększa się re-
uwagę, że wysokie dawki ASA prowadzą również zerwa alkaliczna oraz nasila się nerkowe wydalanie
do obniżenia poziomu protrombiny. Nie można wy- salicylanów – przez podwyższenie pH moczu.
kluczyć, że stosowanie ASA u dzieci z infekcją wi- W przypadku zatruć zagrażających życiu wykonu-
rusową może prowadzić do wystąpienia bardzo rzad- je się hemodializę.
kiego zespołu Reye’a (uszkodzenie wątroby z encefa-
lopatią, śmiertelność > 50%), stąd też ASA nie powi-
nien być stosowany u dzieci. 1.5.5.2.1.2. Pochodne kwasu octowego
Rzadko obserwowane przy stosowaniu ASA
prawdziwe reakcje alergiczne należy wiązać z zanie- 1.5.5.2.1.2.1. Indometacyna i acemetacyna
czyszczeniami substancji czynnej, zwłaszcza silnie
alergogennym bezwodnikiem kwasu acetylosalicy- Lekiem prototypowym dla pochodnych kwasu octo-
lowego, i można ich w większości uniknąć stosując wego z przeważającym działaniem przeciwzapalnym
preparaty zawierające czysty kwas acetylosalicy- i przeciwreumatycznym jest indometacyna – bardzo
lowy. Zwrócono już uwagę na rzekomo-alergiczne silny inhibitor obu cyklooksygenaz ze słabą preferen-
reakcje występujące przy stosowaniu ASA (zob. cją w stosunku do COX-1 (zob. tab. 1.5-8). Indometa-
powyżej). cyna jest wchłaniana szybko i prawie w całości. Wią-
zanie z białkami osocza – podobnie jak w przypadku
Interakcje z innymi niesteroidowymi lekami innych NLPZ – jest wysokie (> 95%); w związku
przeciwzapalnymi. Przy jednoczesnym zażywaniu ze zróżnicowanym zwrotnym krążeniem jelitowo-
MUTSCHLER-2009.indd 254 2010-01-07 22:12:02
Układ nerwowy 255
wątrobowym jej okres półtrwania w osoczu wynosi wynoszą 12,5–25 mg; terapia przeciwreumatyczna
3–11 godzin (średni okres działania 4–6 godzin). Je- wymaga stosowania już większej dawki 50–100 mg/
dynie około 15% substancji ulega wydaleniu w nie- dzień leku, który musi być przepisany na receptę.
zmienionej postaci wraz z moczem; przeważająca Do terapii nieswoistych zapaleń zewnętrznie
Układ nerwowy
część ulega jednak eliminacji przez nerki i z żółcią umiejscowionych tkanek i miejscowych bólów oka
– pod postacią nieaktywnych metabolitów (procesy diklofenak został dopuszczony również w postaci
O-demetylacji, sprzęgania z kwasem glukuronowym kropli do oczu.
oraz N-dezacetylacji).
Dawka dobowa wynosi 50–150 (–200) mg.
Częstość występowania działań niepożądanych 1.5.5.2.1.2.3. Pochodne
sięga ponad 30%. Szczególnie dolegliwości żołąd- kwasu arylopropionowego
kowo-jelitowe są działaniami niepożądanymi wystę- B1
pującymi częściej niż w trakcie stosowania innych Pochodne kwasu 2-arylopropionowego posiada-
NLPZ. W dalszej kolejności obserwowane są rów- ją asymetryczny atom węgla. Enancjomery typu S
nież zaburzenia czuciowe, bóle głowy w okolicy czo- hamują działanie cyklooksygenaz 100–1000 razy
łowej, zaburzenia widzenia i zawroty. silniej niż analogiczne enancjomery R. Stąd też
Prolekiem dla indometacyny jest acemetacyna w niektórych krajach oprócz mieszanek racemicz-
– ester indometacyny z kwasem glikolowym. nych dostępne są czyste enancjomery S (S-ibupro-
fen, S-ketoprofen). Dotychczas nie udało się jed-
nak jednoznacznie wykazać, że pochodne kwasów
1.5.5.2.1.2.2. Diklofenak 2-arylopropionowych podawane w postaci S-enan-
cjomerów są lepiej znoszone niż podwójna daw-
W celu uzyskania środków lepiej znoszonych niż ka odpowiedniego racematu. Naproksen dostępny
indometacyna zsyntetyzowano wiele kolejnych aro- jest na rynku jedynie pod postacią enancjomeru S,
matycznych lub heteroaromatycznych pochodnych mimo to nie wywołuje mniej poważnych działań
kwasu octowego o właściwościach przeciwbólo- niepożądanych ze strony przewodu pokarmowego
wych, jednak tylko częściowo spełniły one oczeki- niż na przykład racemiczna postać ibuprofenu. Nie-
wania. które pochodne kwasów arylopropionowych ulegają
Jednym ze szczególnie często (w Niemczech ok. in vivo specyficznej, zależnej od charakteru związku
65% udziału w rynku) stosowanych niesteroidowych jednokierunkowej przemianie (inwersji) enancjo-
leków przecizapalnych jest diklofenak. Jest to bardzo meru R do enancjomeru S.
silny inhibitor cyklooksygenazy – wykazujący jedy-
nie nieznaczną preferencję do COX-2. Jego wchła- Ibuprofen jest najlepiej przebadanym związkiem
nianie z przewodu pokarmowego zmienia się w za- z grupy pochodnych kwasów 2-arylopropionowych.
leżności od galenowej formy leku. W związku z efek- Jest względnie słabym, nieselektywnym inhibitorem
tem pierwszego przejścia biodostępność po spożyciu cyklooksygenaz. Badania epidemiologiczne wykaza-
doustnym wynosi jedynie 30–80% (średnio 50%). ły, że ibuprofen – ze wszystkich tradycyjnych NLPZ
Diklofenak jest szybko metabolizowany (procesy – cechuje się najmniejszym ryzykiem wystąpienia
hydroksylacji i koniugacji); jego okres półtrwania ciężkich żołądkowo-jelitowych działań niepożą-
w osoczu wynosi 1,5 godziny. Wydalanie metabolitów danych. Doprowadziło to do sytuacji, że ibuprofen
następuje głównie przez nerki, jak również z żółcią. w małych dawkach zyskuje coraz większe znaczenie
Duże badania epidemiologiczne potwierdziły, że w terapii chorób reumatycznych jako lek przeciwbó-
diklofenak wywołuje mniej ciężkich żołądkowo-jeli- lowy typu OTC. Substancja cechuje się wysokiego
towych powikłań (zob. powyżej) niż np. indometa- stopnia wiązaniem z białkami osocza (> 98%) i ma
cyna. Jednakże diklofenak – częściej niż inne NLPZ krótki okres półtrwania (ok. 2 godzin). Metabolity
– prowadzi do zwiększenia się poziomu enzymów są nieaktywne, a ich wydalanie następuje głównie
wątrobowych. Pozajelitowe podanie diklofenaku przez nerki pod postacią glukuronianów. Szczególnie
może wywołać szok anafilaktyczny. szybki początek działania ma po podaniu doustnym
Aby zapobiec powstawaniu owrzodzeń przewodu sól D,L-lizynowa, która ze względu na dobrą rozpusz-
pokarmowego, do stosowania został wprowadzo- czalność w wodzie szybciej rozpuszcza się w obrębie
ny preparat złożony zawierający diklofenak ze stałą przewodu pokarmowego niż wolna postać kwaso-
dawką 0,2 mg mizoprostolu. wa i dlatego potrzebuje mniej czasu, aby osiągnąć
Mniejsze dawki które nie wymagają przepisywa- skuteczny poziom w osoczu. Ponieważ S-ibuprofen
nia na receptę (analgetyk OTC – over the counter) hamuje cyklooksygenazy (COX-1 i COX-2) około
MUTSCHLER-2009.indd 255 2010-01-07 22:12:03
256 Układ nerwowy
100 razy silniej niż R-enancjomer – na rynek wpro- Przeciętne dawki dobowe w leczeniu przeciwreu-
wadzono również czysty enancjomer (deksibupro- matycznym wynoszą 20 mg w przypadku piroksyka-
fen). Jednak S-ibuprofen stosowany w połowie dawki mu i tenoksykamu, 7,5-15 mg w przypadku meloksy-
preparatu racemicznego powoduje podobnie nasilone kamu i 12–16 mg dla lornoksykamu.
objawy niepożądane.
Pojedyncza dawka wynosi 200–400 mg w przeciw-
bólowych preparatach OTC lub 400–800 mg w lecze- 1.5.5.2.1.5. Niesteroidowe leki
niu przeciwreumatycznym. Odpowiadające im maksy- przeciwzapalne
malne dawki dobowe wynoszą 1200 albo 2400 mg. selektywne względem COX-2
Pozostałe leki tej grupy zestawiono w tab. B 1-31. (koksyby)
Inne związki będące pochodnymi kwasu 2-arylopro- Działanie przeciwzapalne, przeciwbólowe i przeciw-
pionowego zostały ujęte w tab. B 1.5-8. Jako lek nie- gorączkowe klasycznych NLPZ związane jest głów-
recepturowy dostępny jest naproksen w pojedynczej nie z hamowaniem COX-2, z kolei COX-1 odpowiada
dawce 200 mg (co odpowiada 220 mg naproksenu- za występowanie licznych objawów niepożądanych.
Na) i maksymalnej dawce dziennej 600 mg. Obserwacja ta była przesłanką do poszukiwań se-
lektywnych inhibitorów COX-2 jako nowych NLPZ
– charakteryzujących się mniejszym ryzykiem wy-
1.5.5.2.1.4. Oksykamy stąpienia działań niepożądanych. Tego rodzaju sub-
stancjami, które w dawkach terapeutycznych hamują
Do leków tej grupy należą piroksykam, meloksy- jedynie COX-2, są:
kam i lornoksykam (zob. tab. B 1.5-8). Wpraw-
■ celekoksyb,
dzie nie należą one do kwasów karboksylowych,
ale w związku z wykazywaną tautomerią ketonowo- ■ etorikoksyb,
-enolową są to jednak związki o charakterze kwaso-
■ lumirakoksyb,
wym. Piroksykam i lornoksykam są nieselektywny-
mi inhibitorami cyklooksygenaz. Meloksykam – po- ■ parekoksyb.
dobnie jak diklofenak – hamuje aktywność COX-2
nieco silniej niż aktywność COX-1. Tę niewielką Dane farmakokinetyczne koksybów zostały zesta-
preferencję w stosunku do COX-2 można częściowo wione w tab. B 1.5-9. Wskazania do ich stosowania
wykorzystać w przypadku stosowania dawek do 7,5 są zasadniczo identyczne jak u nieselektywnych
mg/dzień. Ponieważ jednak leczenie przeciwreuma- NLPZ, choć nie wszystkie koksyby zostały dopusz-
tyczne wymaga stosowania dawek dziennych rzędu czone w pełnym zakresie terapeutycznym, w jakim
15 mg meloksykamu i więcej – lek ten nie posiada stosowane są klasyczne NLPZ. W związku z bra-
jakichś szczególnych zalet w porównaniu z innymi kującym działaniem hamującym na COX-1 – se-
NLPZ. Okres połowiczej eliminacji z osocza wynosi lektywni inhibitory COX-2 wywołują mniej dzia-
w przypadku lornoksykamu 3–5 godzin, meloksyka- łań niepożądanych niż tradycyjne niesteroidowe
mu ok. 20 godzin, a piroksykamu – ok. 40 godzin. leki przeciwzapalne, co jednak wcale nie oznacza,
Tabela B 1.5-9. Dane farmakokinetyczne dotyczące selektywnych inhibitorów COX-2
Objętość dystrybucji (l) tmax (godz.) t1/2 (godz.) Uwagi
celekoksyb ~400 2-4 6-12 nadwrażliwość na sulfonamidy
etorikoksyb ~120 ~1-2 20-26 długi czas półtrwania
lumirakoksyb ~13 1-3 2-6 dopuszczenie zawieszone,
kwas, hepatotoksyczność
rofekoksyb ~90 2-4 12-18 wycofany z handlu!
waldekoksyb ~50-90 2-4 6-11 wycofany z handlu!
tmax – czas potrzebny do osiągnięcia maksymalnego stężenia w osoczu; t1/2 – okres połowiczej eliminacji
MUTSCHLER-2009.indd 256 2010-01-07 22:12:03
Układ nerwowy 257
że są zupełnie wolne od niepożądanych działań, ce jako pozytywna. Wszystkie koksyby powinny być
gdyż również COX-2 pełnią pewne fizjologiczne stosowane w możliwie najmniejszych dawkach, przez
funkcje. możliwie jak najkrótszy okres, gdyż ryzyko powikłań
Do najczęstszych działań niepożądanych zalicza sercowo-naczyniowych wzrasta prawdopodobnie
Układ nerwowy
się infekcje górnych dróg oddechowych, biegunki, wraz z dawką i czasem stosowania terapii. W czasie
niestrawność, uczucie ucisku w nadbrzuszu i bóle ciąży koksyby są przeciwwskazane.
głowy. Obwodowe obrzęki i zwyżki ciśnienia krwi
ujawniają się podobnie często, z kolei powikłania żo- Celekoksyb, pochodna sulfonamidów, posiada zmien-
łądkowo-jelitowe występują rzadziej niż w przypad- ną biodostępność po podaniu doustnym – osiagają-
ku tradycyjnych NLPZ. W dalszej kolejności mogą ca wartość 50–70%. Po 3 godzinach osiąga maksy-
ujawniać się epizody natury sercowo-naczyniowej malne stężenie w osoczu i ulega metabolizowaniu
(zawał serca, udar mózgu). głównie przez CYP2C9 do nieczynnych metaboli- B1
Rofekoksyb – w związku z jednoznacznie po- tów. Okres połowiczej eliminacji wynosi 6–12 go-
twierdzonym podwyższonym ryzykiem powikłań dzin. Ponieważ CYP2C9 podlega genetycznemu po-
sercowo-naczyniowych – został wycofany z rynku. limorfizmowi stężenie celekoksybu w osoczu u osób
Leżące u podstaw tych zaburzeń mechanizmy nie zo- wolno metabolizujących jest podwyższone. Ponadto
stały jednak w pełni wyjaśnione. Ostatnio opubliko- celekoksyb hamuje CYP2D6, w związku z czym na-
wane prace potwierdzają, że także klasyczne NLPZ leży zwracać uwagę na interakcje z substratami tego
powodują powikłania sercowo-naczyniowe. Wynika izoenzymu.
z tego, że w określonych okolicznościach wszystkie Na podstawie stwierdzenia, że COX-2 wywie-
niesteroidowe leki przeciwzapalne – zarówno selek- rają znaczący wpływ hamujący na wzrost różnych
tywne, jak i nieselektywne inhibitory COX – są ob- komórek nowotworowych i polipów jelita grubego
ciążone ryzykiem tego powikłania. W przeciwień- – w ostatnim czasie celekoksyb dopuszczono do te-
stwie do rofekoksybu – relacja korzyści do ryzyka rapii rodzinnej polipowatości gruczolakowatej jeli-
pozostałych dostępnych na rynku koksybów została ta grubego (FAP – familial adenomatous polyposis)
oceniona przez odpowiednie instytucje dopuszczają- – w celu zmniejszenia ilości polipów.
O O O O
S S
H2N H3C
N Cl
N
CF3
N N
H3C H3C
celekoksyb etokrikoksyb
O OH O
O O O
H3C S S
HN CH3 H2N CH3
NH H5C2 O
O O
F Cl N N
lumirakoksyb parekoksyb waldekoksyb
MUTSCHLER-2009.indd 257 2010-01-07 22:12:03
258 Układ nerwowy
Dawkowanie celekosybu w zmianach zwyrodnie- dobieństwo strukturalne z naproksenem (zob. tab. B
niowych stawów wynosi 1 × 200 mg, w reumatoidal- 1.5-8). Jako prolek nabumeton nie posiada miejsco-
nym zapaleniu stawów – 2 × 100–200 mg, a w przy- wego działania drażniącego. Natomiast wywoływa-
padku FAP – 2 × 400 mg. ne przez czynny metabolit, zależne od hamowania
Ponieważ celekoksyb jest pochodną sulfonamidu syntezy prostaglandyn, ogólnoustrojowe działania
– u predysponowanych osób może dojść do ujawnie- niepożądane nie odróżniają się od działań innych
nia się alergii sulfonamidowej. NLPZ. Wbrew wcześniejszym przypuszczeniom
6-MNA nie jest wybiórczym inhibitorem COX-2.
Etorikoksyb posiada niemal pełną biodostępność Okres połowiczej eliminacji z osocza aktywnego
po podaniu doustnym i osiąga po 1–2 godzinach metabolitu 6-MNA osiąga wartość 20–25 godzin.
maksymalne stężenie w osoczu. Substancja jest me- Dawka dzienna wynosi 2 × 500 mg.
tabolizowana głównie przez CYP3A4 – w większości
do nieczynnych metabolitów. Okres półtrwania wy- Aceklofenak jest estrem diklofenaku, a pomimo
nosi 20–26 godzin. Pacjenci z zespołami bólowymi to nie jest prolekiem. Jest on metabolizowany głów-
i dodatkowo nadciśnieniem nie powinni być leczeni nie do 4′-hydroksyaceklofenaku. Inne metabolity,
za pomocą etorikoksybu. takie jak diklofenak lub 4′-hydroksydiklofenak, sta-
Dzienne dawkowane w zmianach zwyrodnienio- nowią mniej niż 5% podanej dawki. Aceklofenak
wych stawów wynosi jednorazowo 60 mg, w przebie-
gu reumatoidalnego zapalenia stawów jednorazowo
90 mg, a w ostrym dnawym zapaleniu stawów jedno-
razowo 120 mg (maksymalnie przez 8 dni).
Parekoksyb jest dobrze rozpuszczalnym w wodzie,
O
podawanym parenteralnie prolekiem dla waldekok-
sybu, powstającego w wyniku enzymatycznego roz- CH3
kładu, o czasie półtrwania ok. 20 minut. Okres pół-
H3CO
trwania waldekoksybu wynosi ok. 6–11 godzin. Pare-
koksyb jest wskazany do krótkotrwałej terapii bólów nabumeton
pooperacyjnych.
Zalecana dawka wynosi 40 mg i.v., a maksymalna
dawka dzienna 80 mg.
Lumirakoksyb jest jako pochodna diklofenaku jedy- O O COOH
ną kwasową pochodną w grupie koksybów. W zakre-
sie skuteczności i działań niepożądanych – lumira-
koksyb jest porównywalny z pozostałymi dostępnymi
na rynku koksybami. Jego okres półtrwania – wyno- NH
szący 2–6 godzin – jest względnie krótki. Podobnie Cl Cl
jak diklofenak prowadzi do podwyższenia poziomu
enzymów wątrobowych (zob. powyżej). Na podsta-
wie doniesień o ciężkich reakcjach ze strony wątroby aceklofenak
lumirakoksyb został niedawno ponownie wycofany
z rynku.
Zalecana dawka dzienna wynosi w przypadku
zmian zwyrodnieniowych stawów 100 mg.
O CH3
N COOH
1.5.5.2.1.2.6. Inne niesteroidowe leki
przeciwzapalne
HO
Nabumeton nie jest co prawda kwasem, lecz keto- oksaceprol
nem, jednak w organizmie ulega przemianie do właś-
ciwego czynnego związku kwasu 6-metoksy-2-naf-
tylooctowego (6-MNA), który wykazuje ścisłe po-
MUTSCHLER-2009.indd 258 2010-01-07 22:12:03
Układ nerwowy 259
w dawkach leczniczych jest prawdopodobnie słabym rapeutycznych – jest nieselektywnym inhibitorem
inhibitorem cyklooksygenazy; zmniejszanie przez lek cyklooksygenaz. Względna selektywność narządo-
tworzenia prozapalnych prostaglandyn jest następ- wa hamowania COX jest wyjaśniona tym, że jedy-
stwem zahamowania syntezy interleukiny-l. Okres nie w ośrodkowym układzie nerwowym występuje
Układ nerwowy
połowiczej eliminacji z osocza wynosi 4 godziny. wystarczająca aktywność FAAH. Sam paracetamol
Maksymalne dawki dobowe w leczeniu zwyrod- w stężeniach terapeutycznych nie działa na cyklook-
nieniowych zmian kostno-stawowych lub reuma- sygenazy lub też to działanie jest słabe.
toidalnego zapalenia stawów wynoszą ok. 200 mg.
Czy aceklofenak jest lepiej znoszony niż inne NLPZ
pierwszej generacji – w tej kwestii istnieją odmienne 1.5.5.2.2.1. Pochodne aniliny
oceny.
Z pochodnych aniliny istotne znaczenie ma jedynie B1
Oksaceprol (N-acetylohydroksyprolina) in vitro nie wymieniony już paracetamol (p-hydroksyacetanilid).
wpływa na uwalnianie kwasu arachidonowego ani Wcześniej często stosowana fenacetyna, będąca jego
też na tworzenie jego metabolitów. Jako mechanizm eterem etylowym, nie jest obecnie używana ze wzglę-
przeciwzapalnego działania dyskutowane jest hamo- du na wywoływane przy nadużywaniu uszkodzenia
wanie adhezji granulocytów. Zgodnie z tym typowe nerek (zapalenie śródmiąższowe, martwica broda-
dla NLPZ działania niepożądane – ulcerogenność wek, niewydolność nerek).
i nefrotoksyczność – obserwowane są rzadko. Okres
połowiczej eliminacji z osocza oceniany jest na 2 go- Paracetamol charakteryzuje się dobrym działaniem
dziny. W dawkach od 3× 200 mg do 3 × 400 mg/dzień przeciwgorączkowym i dość słabym działaniem prze-
oksaceprol stosowany jest w chorobach degeneracyj- ciwbólowym. W przeciwieństwie do NLPZ (zob.
nych i zapalnych stawów. powyżej) działanie przeciwzapalne paracetamolu
Działaniami niepożądanymi tego związku są głów- jest jednak bardzo słabe. Wyjaśnieniem jest brak ha-
nie zaburzenia żołądkowo-jelitowe (bóle brzucha, mującego działania na aktywność cyklooksygenazy
nudności, utrata łaknienia, biegunki) i alergiczne od- w miejscu procesu zapalnego (zob. powyżej). Efekt
czyny skórne. przeciwbólowy zależy głównie od działania ośrodko-
wego (zob. powyżej).
Po podaniu doustnym paracetamol jest szybko
1.5.5.2.2. Leki przeciwgorączkowe i w całości wchłaniany z przewodu pokarmowego.
i przeciwbólowe Okres półtrwania w osoczu wynosi 2–3 godz. Wią-
niebędące kwasami zanie z białkami wynosi tylko ok. 30%. Eliminacja
następuje głównie poprzez biotransformację. Głów-
Mechanizm działania. Molekularny mechanizm nymi metabolitami są glukuronian i siarczan. Tylko
działania leków przeciwbólowych o działaniu prze- w niewielkim odsetku (ok. 3%) paracetamol jest wy-
ciwgorączkowym i niemających budowy kwasowej dalany z moczem.
nie został ostatecznie wyjaśniony. Szybko przeni- Pojedyncza dawka wynosi u dorosłych (w mono-
kają one barierę krew-mózg i na poziomie rdzenia preparatach) 500–1000 mg. U dzieci dawka pojedyn-
kręgowego oraz nadrzędnych ośrodków nerwowych cza nie powinna przekraczać 10 mg/kg m.c., a dawka
hamują wyzwalaną bodźcami nocyceptywnymi dobowa – 50 mg/kg m.c.
syntezę prostaglandyn. Na obwodzie i w miejscu W zazwyczaj stosowanych dawkach paracetamol
zapalenia nie dochodzi do istotnego zahamowania jest na ogół dobrze tolerowany.
syntezy prostaglandyn. Stąd też nie wykazują one W zatruciach paracetamolem na pierwszym planie
działania przeciwzapalnego, jak również nie wy- znajduje się działanie hepatotoksyczne. Przy stosowa-
zwalają typowych dla klasycznych NLPZ działań niu dawek terapeutycznych paracetamol zostaje wy-
niepożądanych, takich jak toksyczność w obrębie dalony głównie pod postacią glukuronianów i siarcza-
układu żołądkowo-jelitowego lub hamowanie agre- nów (zob. powyżej). Gdy jednak w wyniku przyjęcia
gacji płytek krwi. większych dawek lub też w przebiegu niewydolności
Paracetamol w organizmie ulega deacetylizacji wątroby wyczerpie się zdolność wątroby do koniu-
do 4-aminofenolu, który – katalizowany przez ami- gowania z kwasem glukuronowym lub siarkowym
dohydrolazę kwasów tłuszczowych (FAAH) – rea- – wówczas w wyniku mikrosomalnej oksydacji do-
guje w ośrodkowym układzie nerwowym z kwasem chodzi do powstawania toksycznych metabolitów
arachidonowym z wytworzeniem arachidonoilofe- paracetamolu, z których najważniejszym jest N-ace-
nolaminy (AM-404). AM-404 – w stężeniach te- tylo-p-benzochinonoimina. Związek ten u pacjentów
MUTSCHLER-2009.indd 259 2010-01-07 22:12:04
260 Układ nerwowy
ze zdrową wątrobą może być przejęty przez grupy fenazonu. Ten ostatni jest w następstwie acetylacji
SH glutationu z wytworzeniem nietrujących koniu- (w zależności od genotypu NAT 2) częściowo prze-
gatów. Po wyczerpaniu zasobów glutationu – w wy- kształcany w 4-acetyloaminofenazon (zob. ryc. B
niku niewydolności wątroby lub też na skutek zbyt 1.5-16). Okres półtrwania 4-metyloaminofenazonu
dużej dawki paracetamolu – metabolity chinonoimi- wynosi 2–5 godz. Wydalanie metabolitów następuje
ny wiążą się z białkami hepatocytów. W wyniku tego głównie przez nerki. Obserwowane czasami czerwo-
dochodzi do ujawnienia się reakcji cytotoksycznych ne zabarwienie moczu wynika z tworzenia metabolitu
(martwicy komórek wątroby). Środki prowadzące – kwasu rubazonowego.
do indukcji enzymatycznej – stymulujące syntezę cy- Pojedyncza dawka wynosi przy podaniu doustnym
tochromu P-450 – zmniejszają próg stężenia toksycz- 0,5–1 g, a przy podaniach parenteralnych 1–2,5 g.
nego paracetamolu. Najcięższym działaniem niepożądanym metami-
Dawki powyżej 10 g (co odpowiada 3-krotnej zolu, tak zresztą jak i innych pochodnych pirazolu,
maksymalnej dawce dziennej) prowadzą u osoby jest agranulocytoza. Dane dotyczące częstości wy-
dorosłej do ciężkiej, a w przypadku niewdrożenia stępowania agranulocytozy przy stosowaniu meta-
odpowiedniej terapii – nawet śmiertelnej, martwicy mizolu bardzo się różnią, mimo że lek ten znajduje
komórek wątroby. U pacjentów z wcześniej uszko- się w obrocie od wielu już lat. Wydaje się również,
dzoną wątrobą (np. w wyniku spożywania alkoholu) że występuje znaczne zróżnicowanie geograficzne.
toksyczne mogą okazać się już dawki rzędu 6 g. W przypadku zachodniej Europy (i przypuszczalnie
W leczeniu zatruć paracetamolem potwierdzono Polski) ryzyko wystąpienia może wynosić 0,01%.
skuteczność dostarczycieli (donorów) grup SH. Obok W krajach skandynawskich donoszono o istotnie
metioniny stosuje się też cysteaminę i acetylocyste- wyższym ryzyku ujawniania się działań niepożąda-
inę. nych. W przypadku dłuższego podawania metami-
Czy przy długotrwałym stosowaniu paracetamol zolu bezwzględnie konieczna jest kontrola obrazu
wykazuje istotnie słabsze działanie nefrotoksyczne krwi.
od fenacetyny – nie zostało dotychczas jednoznacz- Innymi działaniami niepożądanymi są zmiany
nie potwierdzone. na skórze i błonach śluzowych. Ostre zatrucie może
prowadzić do drgawek.
1.5.5.2.3. Pochodne pirazolu Fenazon i propyfenazon posiadają podobną sku-
(fenazony i pochodne teczność i wykazują działania niepożądane podobne
pirazolidyno-3,5-dionu) do metamizolu.
Do związków tej grupy należą oprócz prototypowych Fenylobutazon – pochodna pirazolidyno-3,5-dionu
fenazonów, w tym samego fenazonu, jego izopropy- – jest w postaci soli sodowej dobrze rozpuszczalny.
lowe pochodne – propyfenazon i metamizol (zob. tab. Może on być podawany pod postacią iniekcji, a także
B 1.5-8), a także pochodna pirazolidyno-3,5-dionu doustnie. Po podaniu doustnym lek jest niemal cał-
– fenylobutazon. kowicie wchłaniany. Wiązanie z białkami jest bardzo
wysokie (> 98%). Okres półtrwania wynosi ok. 70
Metamizol – w związku z posiadaną resztą kwasu godz. Głównymi metabolitami obecnymi w osoczu
metanosulfonowego – jest jako sól dobrze rozpusz- są również dobrze działające przeciwzapalnie oksy-
czalny w wodzie. Podany dożylnie działa skutecznie fenbutazon i γ-hydroksyfenylobutazon. Wydalanie
nawet w silnych bólach, a w następstwie dodatkowego metabolitów następuje głównie przez nerki.
działania spazmolitycznego jest też skuteczny w kol- Z powodu częstych i niekiedy poważnych dzia-
kach. Trzeba jednak wyraźnie podkreślić, że przy łań niepożądanych (zob. poniżej), w pewnych oko-
dożylnym podaniu 50-proc. roztworów, zwłaszcza licznościach prowadzących do zejścia śmiertelnego,
w szybkich wstrzyknięciach, może wystąpić wstrząs. wskazania do stosowania fenylobutazonu są obecnie
Niezbędne jest więc ścisłe przestrzeganie wskazań w znacznym stopniu ograniczone. Można go jesz-
i powolne wstrzykiwanie (najlepiej w postaci krótko- cze nadal stosować w ostrych napadach dny oraz
trwałego wlewu). w ostrych rzutach choroby Bechterewa (zesztywnia-
Po podaniu doustnym związek jest szybko rozkła- jące zapalenie stawów kregosłupa).
dany hydrolitycznie, a powstający 4-metylofenazon Dawkowanie wynosi w ostrym napadzie dny
– będący substancją czynną – jest całkowicie wchła- 400–800 mg/dzień przez 3 dni, a w chorobie Bechte-
niany. Przez odalkilowanie azotu grupy aminowej rewa 200–400 mg/dzień, przy czym okres stosowania
w pozycji 4 ulega on biotransformacji do 4-amino- nie może przekroczyć tygodnia.
MUTSCHLER-2009.indd 260 2010-01-07 22:12:04
Układ nerwowy 261
CH3 CH3 H O H
N CH3 H N CH3 H N CH3 N CH3
HO3S H3C
Układ nerwowy
O N O N O N O N
N CH3 N CH3 N CH3 N CH3
metamizol 4-metyloamino- 4-amino- 4-acetyloamino-
fenazon fenazon fenazon
B1
Ryc. B 1.5-16. Biotransformacja metamizolu.
Działania niepożądane występują znacznie częś- Działaniami niepożądanymi są czasem ujawniają-
ciej niż w przypadku metamizolu: należy się z nimi ce się zaburzenia ośrodkowe (upoczucie zmęczenia,
liczyć u co trzeciego pacjenta, a w 10% przypadków zawroty głowy) oraz zaburzenia żołądkowo-jelitowe
są one tak silne, że zmuszają do odstawienia leku. (nudności, zaparcia, ale również biegunka). Rzadziej
Do ważniejszych interakcji należy wypieranie le- występują: suchość w ustach, nadmierne pocenie się,
ków przeciwzakrzepowych typu dikumarolu, jak też reakcje skórne, zaburzenia widzenia oraz podwyższe-
doustnych leków przeciwcukrzycowych z połączeń nie poziomu enzymów wątrobowych.
białkowych związane ze zwiększeniem niebezpie- Flupirtyna jest przeciwwskazana u pacjentów z en-
czeństwa odpowiednio krwawień lub występowa- cefalopatią wątrobową lub zastojem żółci, jak rów-
niem stanów hipoglikemicznych. Działanie leków nież w miastenii.
przeciwzakrzepowych jest też zwiększone przez bez- Działanie etanolu, leków uspokajających, leków
pośrednie hamujące krzepliwość działanie fenylobu- zwiotczających mięśnie i leków przeciwzakrzepo-
tazonu. wych ulega nasileniu przy równoczesnym stosowa-
niu flupirtyny.
1.5.5.2.4. Flupirtyna
H3C(CH2) 3 O
Flupirtyna odróżnia się od wcześniej opisanych
nieopioidowych leków przeciwbólowych tym, N
O
że nie prowadzi do hamowania syntezy prostaglan- N
dyn oraz – przy zwykłym dawkowaniu – nie ma
działania przeciwgorączkowego i przeciwzapalne-
go. Jest pochodną triaminopirydyny, która oprócz
umiarkowanie silnych właściwości przeciwbólo- fenylobutazon
wych wykazuje też ośrodkowe działanie miorelak-
sacyjne. Mechanizm działania został jedynie częś-
ciowo poznany. Flupirtyna ma w sposób selektywny
zwiększać prawdopodobieństwo otwarcia neuronal-
nych kanałów potasowych. W wyniku zwiększenia
F
przepuszczalności potasu neurony ulegają hiperpo-
laryzacji i w ten sposób są słabiej pobudzane przez NH N NH2
bodźce nocyceptywne. O
Po spożyciu doustnym flupirtyna ulega w 90% NH OC2H5
wchłonięciu i jest wydalana głównie przez nerki.
Pojedyncza dawka wynosi przy podaniu doust- flupirtyna
nym 100 mg; maksymalna dawka dzienna – 600 mg.
U pacjentów z niewydolnością nerek nie powinno się
przekraczać maksymalnej dawki dziennej 300 mg.
MUTSCHLER-2009.indd 261 2010-01-07 22:12:04
262 Układ nerwowy
bólowy w połączeniu z kofeiną, środkami nasennymi,
1.5.5.2.5. Zikonotyna
lekami rozkurczowymi i/lub innymi substancjami.
Według niektórych autorów nadużywanie leków prze-
Zikonotyna jest syntetycznym, złożonym z 25 L-ami- ciwbólowych jest związane właśnie ze stosowaniem
nokwasów analogiem ω-konopeptydu – trucizny śli- tego rodzaju preparatów złożonych (m.in. zawierają-
maka morskiego Conus magnus. Substancja blokuje cych substancje o działaniu psychotropowym). Nad-
na poziomie rdzenia kręgowego neuronalne presy- używanie leków przeciwbólowych nie jest bezpiecz-
naptyczne kanały wapniowe typu N, a tym samym ne, gdyż m.in. może prowadzić do uszkodzenia nerek
hamuje uwalnianie mediatorów bólowych. Ziko- (tzw. nefropatia analgetyczna).
notyna działa przeciwbólowo – hamując rdzeniową Ostatnio badanie przeprowadzone u pacjentów
transmisję nocyceptywną. Nie jest nieopioidowym z migrenowymi bólami głowy wykazało, że leczenie
lekiem przeciwbólowym, nie należy do grupy leków skojarzone za pomocą kwasu acetylosalicylowego,
przeciwbólowych stopnia 1. wg WHO, nie ma także paracetamolu i kofeiny było bardziej skuteczne niż
działania przeciwgorączkowego czy też przeciwza- każda z substancji stosowana z osobna lub połączenie
palnego. Zikonotyna została dopuszczona w terapii bez kofeiny. Oceny skuteczności określonych kombi-
silnych, przewlekłych bólów – do podawania podpa- nacji lekowych są często kontrowersyjne i jak dotąd
jęczynówkowo (intratekalnie). niemożliwe jest dokonanie ostatecznej oceny terapii
Zikonotyna nie jest metabolizowana w płynie skojarzonej.
mózgowo-rdzeniowym; dopiero po trafieniu do ukła-
du krążenia ulega proteolitycznemu rozkładowi przez
peptydazy. Po podaniu podpajęczynówkowym – 1.5.6. Terapia bólów
w zależności od cyrkulacji płynu – ulega eliminacji neuropatycznych
z ośrodkowego układu nerwowego ze średnim okre-
sem półtrwania 4–5 godzin. Okres połowiczej elimi-
Bóle neuropatyczne (np. neuralgia po przebyciu
nacji z osocza wynosi 1,3 godziny.
półpaśca, neuropatia cukrzycowa, neuropatia po-
Zikonotynę podaje się we wlewie za pomocą cew-
urazowa) różnią się w zakresie etiologii i patoge-
nika dokanałowego w dawce początkowej 2,4 μg/
nezy od bólów zapalnych. Stąd też niesteroidowe
dzień. Minimalny odstęp czasowy między kolejny-
leki przeciwzapalne, jak również paracetamol i me-
mi zwiększającymi się dawkami wynosi 24 godziny
tamizol nie są skuteczne w tym wskazaniu lub też
(zalecany jest odstęp 48 godzin). Maksymalna dawka
ich skuteczność jest niewielka. W terapii bólów neu-
dzienna wynosi 21,6 μg. Zikonotyna powinna być po-
ropatycznych stosowane są zasadniczo niektóre leki
dawana na drodze podpajęczynówkowej tylko przez
przeciwdepresyjne, leki przeciwdrgawkowe/przeci-
doświadczonego lekarza.
padaczkowe i opioidy – stosowane albo w monote-
Jako bardzo częste działania niepożądane (> 10%)
rapii, albo w terapii skojarzonej za pomocą 2 lub 3
opisano: bóle i zawroty głowy, nudności, oczopląs,
substancji z wymienionych grup lekowych.
splątanie, zaburzenia chodu, zaburzenia pamięci,
Najlepiej przebadane zostało zastosowanie w te-
zamazane widzenie, astenię i senność. Ponadto ob-
rapii bólów neuropatycznych – trójcyklicznych leków
serwowano sercowe, nerkowe i płucne działania nie-
przeciwdepresyjnych typu amitryptyliny. Oprócz ha-
pożądane. Ponieważ zikonotyna została dopuszczona
mującego działania na wychwyt zwrotny serotoniny
do stosowania niedawno, brak jest ostatecznych da-
i noradrenaliny cechują się one działaniem polegają-
nych dotyczących jej tolerowania. Na razie nie obser-
cym na blokowaniu napięciowozależnych kanałów
wowano depresyjnego działania na oddychanie.
sodowych, co ma określony wpływ na ostateczną
Badania nad możliwymi interakcjami nie zostały
skuteczność terapii bólów neuropatycznych. Trójcy-
jak dotąd przeprowadzone. Mimo to nie należy stoso-
kliczne leki przeciwdepresyjne są skuteczne w róż-
wać zikonotyny jednocześnie z podawaną intratekal-
nych postaciach bólów neuropatycznych, przy czym
nie chemioterapią.
stosuje się dawki mniejsze niż te stosowane w te-
rapii przeciwdepresyjnej (zob. tab. B 1.5.10). Tym
samym redukcja bólów nie może być tłumaczona
1.5.5.2.6. Złożone leki przeciwbólowe działaniem przeciwdepresyjnym. Jednak rozliczne
działania niepożądane leków z tej grupy często wy-
Duża liczba preparatów przeciwbólowych znajdują- kluczają je z możliwości zastosowania. Inhibitory
cych się w handlu to preparaty złożone – zawierające wychwytu zwrotnego serotoniny i noradrenaliny
jednocześnie kilka analgetyków lub też lek przeciw- – duloksetyna i wenlafaksyna mają podobną sku-
MUTSCHLER-2009.indd 262 2010-01-07 22:12:04
Układ nerwowy 263
Tabela B 1.5-10. Leki stosowane w terapii bólów neuropatycz- oraz oksykodonu – leku opioidowego stopnia 3. wg
nych z zaleceniami w zakresie dawkowania u osób dorosłych WHO. Takie działania niepożądane, jak zaparcia mu-
(według Barona) szą być początkowo leczone w sposób objawowy.
Tabela B 1.5-10 zawiera przegląd substancji sto-
Układ nerwowy
Substancja Dawka Dawka sowanych w terapii neuropatii. Szczegóły farmako-
aktywna początkowa skuteczna dynamiczne i farmakokinetyczne, a w szczególności
(mg) (mg)
działania niepożądane tych substancji zostały opisa-
Leki przeciwdepresyjne ne w odpowiednich rozdziałach: B 1.2.2.1 (leki prze-
nortryptylina 10-25 50-75 ciwdepresyjne), B 1.9.2 (leki przeciwdrgawkowe)
i B 1.5.2 (opioidy).
dezypramina 10-25 50-75
B1
duloksetyna 30-60 60
wenlafaksyna 37,5 75-255 (ret./ER) 1.5.7. Terapia migreny
Leki przeciwpadaczkowe
gabapentyna 300 1200-2400 Przez pojęcie migreny rozumie się przewlekłe i na-
wracające, ujawniające się w sposób napadowy, czę-
pregabalina 75 150 sto (ale nie zawsze) występujące jednostronnie (he-
karbamazepina 100-200 600-1200 (ret.) mikrania) bóle głowy – mające zmienną częstość
ujawniania się, zmienne nasilenie i czas trwania.
lamotrygina 25 100-200
Schorzenie to dotyczny 16–24% kobiet i 6–8% męż-
Opioidy czyzn.
tramadol ret. 100-200 miareczkowanie
Na poziomie klinicznym migrena ujawnia się pod
morfina ret. 10-30 miareczkowanie
rozmaitymi postaciami. W przypadku najbardziej roz-
oksykodon ret. 5-20 miareczkowanie powszechnionej postaci – migreny bez aury (wcześ-
niejsza nazwa: migrena prosta) – ból głowy (zwykle
jednostronny) narasta powoli, utrzymuje się średnio
od 18 do 72 godzin, a towarzyszą mu nudności i wy-
mioty, jak również światłowstręt i nadwrażliwość
teczność. Selektywne inhibitory wychwytu zwrot- na dźwięki. Wykonywanie codziennych czynności
nego serotoniny typu fluoksetyny czy citalopramu nasila ból; zdolność do pracy zostaje ograniczenona.
nie są natomiast skuteczne lub też ich skuteczność W przypadku rzadszej postaci migreny z aurą
jest niewielka. (wcześniejsza nazwa: migrena klasyczna) – niedłu-
Stosowane w stanach bólów neuropatycznych leki go przed wystąpieniem bólów głowy – ujawniają się
przeciwdrgawkowe można podzielić na modulujące przejściowe, trwające 5–20 (rzadziej do 60) minut
kanały wapniowe (gabapentyna, pregabalina) oraz neurologiczne objawy ubytkowe: zaburzenia czucia
modulujące kanały sodowe (np. karbamazepina, la- (parestezje) i objawy oczne, np. pod postacią migota-
motrygina). Gabapentyna i pregabalina mogą być nia z upośledzeniem pola widzenia (zjawiska świet-
zastosowane w przypadku bardzo wielu bólów neu- lne w rodzaju migotania i błysków – przy równo-
ropatycznych. W wyniku interakcji w obrębie pod- czesnym osłabieniu ostrości widzenia). Czasem wy-
jednostki α2δ presynaptycznych napięciowozależ- stępują również zaburzenia mówienia lub porażenia.
nych kanałów wapniowych – wymienione preparaty Pozostałe objawy podobne są do tych występujących
hamują uwalnianie pronocyceptywnych neuroprze- w migrenie bez aury.
kaźników. Karbamazepina stosowana jest głównie Przyczyna migreny jest wieloczynnikowa. Po-
w neuralgii nerwu trójdzielnego. Na uwagę zasługu- twierdzony został udział komponenty genetycznej
je nieliniowa farmakokinetyka tej substancji. – dotyczącej wielu genów. W 60–70% przypadków
Opioidy z opóźnionym/przedłużonym czasem dzia- stwierdza się istnienie rodzinnego obciążenia tym
łania również są trwałym elementem terapii bólów schorzeniem. Określoną rolę odgrywają jednak rów-
neuropatycznych. Pozytywne badania kliniczne doty- nież czynniki środowiskowe.
czące skuteczności w neuralgii po półpaścu oraz neu- Patogeneza nie została w pełni poznana; na ryc. B
ralgii cukrzycowej istnieją szczególnie w przypadku 1.5-17 i B 1.5-18 zestawione zostały obecnie przyj-
tramadolu – leku opioidowego stopnia 2. wg WHO mowane poglądy na patogenezę migreny.
MUTSCHLER-2009.indd 263 2010-01-07 22:12:05
264 Układ nerwowy
aktywacja neuronów pnia
genetyczna mózgu (np. w jądrach szwu, bodźce
predyspozycja miejscu sinawym, istocie szarej endogenne i egzogenne
przywodociągowej)
szerząca się depresja osłabienie uwalnianie peptydu
neuronalna endogennej aktywności związanego z genem stymulacja
(spreading depression) antynocyceptywnej kalcytoniny, substancji pola najdalszego
P, VIP, NO
uwolnienie mediatorów
rozszerzenie zapalnych, nasilone poszerzenie niesteroi-
triptany naczyń dowe leki
naczyń krwionośnych
przeciw-
zapalne
wzmożona aktywność zapalenie
triptany neuronów nerwu neurogenne
trójdzielnego
aura ból głowy nudności, wymioty
Ryc. B 1.5-17. Poglądy na patogenezę napadu migreny (częściowo hipotetyczne) i punktu uchwytu substancji przeciwmigreno-
wych.
Czynnikami wyzwalającymi są m.in. stres psychicz- peptydu jelitowego (VIP – vasoactive intestinal
ny, spożywanie alkoholu, produkty spożywcze za- peptide), substancji P (SP), peptydu związanego
wierające tyraminę (np. czekolada, twardy ser) oraz z genem kalcytoniny (CGRP – calcitonin gene-re-
wahania hormonalne (szczególnie na początku okre- lated peptide) oraz tlenku azotu (NO) – wraz z wy-
su miesiączkowania). Wymienione czynniki prowa- nikającym stąd pobudzeniem aferentnych włókien
dzą prawdopodobnie do pobudzenia neuronów pnia C, a także
mózgu w jądrach szwu, miejsca sinawego (locus
■ wyzwalanego przez uwolnione mediatory odczynu
coeruleus) i istoty szarej okolicy przywodociągowej
zapalnego (zob. powyżej) neurogennego zapalenia
– co wyzwala reakcje patofizjologiczne. Stymulacja
okołonaczyniowego.
pola najdalszego (area postrema) prowadzi do nudno-
ści i wymiotów. Aura jest prawdopodobnie wyzwala-
Mechanizmy, które prowadzą do aktywacji układu
na zjawiskiem tzw. szerzącej się depresji neuronalnej
trójdzielno-naczyniowego, są tylko częściowo wyjaś-
(spreading depression). Jest to krótkotrwałe pobudze-
nione. Udowodniono, że najważniejszą rolę w patoge-
nie, rozprzestrzeniające się pod postacią fali od oko-
nezie migreny odgrywają receptory serotoninergiczne
lic potylicznych w kierunku ciemieniowym i skro-
typu 5-HT1B oraz 5-HT1D. W mięśniach gładkich we-
niowym, po którym następuje dłużej utrzymujące się
wnątrzczaszkowych naczyń krwionośnych rozmiesz-
zahamowanie korowych neuronów z niedokrwieniem
czone są m.in. receptory 5-HT1B. W przypadku pobu-
okolic dotkniętych omawianym zjawiskiem.
dzenia tych receptorów (zob. poniżej) – rozszerzone
w trakcie napadu migreny naczynia ulegają skurczo-
Właściwy migrenowy ból głowy jest następstwem:
wi. Receptory 5-HT1D mają być zlokalizowane głów-
■ podwyższonej aktywności neuronów nerwu trój- nie w presynaptycznych zakończeniach włókien typu
dzielnego w następstwie rozszerzenia naczyń spo- C i tam regulują wydzielanie mediatorów bólowych
wodowanego uwolnieniem naczynioruchowego i zapalnych.
MUTSCHLER-2009.indd 264 2010-01-07 22:12:05
Układ nerwowy 265
2. uwalnianie neuropeptydów
wewnątrzczaszkowe,
zewnątrzmózgowe
naczynia krwionośne
Układ nerwowy
ból głowy kora SP
fotofobie,
fonofobie
VIP
wzgórze 1. rozszerzenie światła naczyń B1
aktywacja autonomiczna: zwój nerwu
nudności, wymioty trójdzielnego
3. przekazywanie bodźców bólowych
jądro
ogoniaste
Ryc. B 1.5-18. Poglądy na powstawanie migrenowego bólu głowy (zmodyf. według Moskowitza oraz Hargreavesa). SP – substancja
P, VIP – naczynioruchowy peptyd jelitowy.
W rozpoznaniu różnicowym należy odróżnić od mi- 1.5.7.1. Terapia ostrego ataku migreny
greny inne zespoły bólów głowy, takie jak ból klaste-
rowy (ściśle jednostronne napady trudnych do znie- W trakcie ostrego napadu migreny pacjent powinien być
sienia bólów głowy, o charakterze świdrowania i pie- chroniony przed działaniem zewnętrznych bodźców
czenia i czasie trwania od 30 minut do 3–4 godzin), (zaciemnione i wyciszone pomieszczenie). W przy-
napięciowy ból głowy (tępy obustronny ból głowy padku lekkich i krótkotrwałych napadów migreny wy-
o umiarkowanym nasileniu, który często ustępuje starczająco skuteczne okazało się leczenie skojarzone
po lekach przeciwdepresyjnych typu amitryptyliny), za pomocą zwiększającego perystaltykę leku przeciw-
bóle głowy wywoływane przez leki (tępo ściskający, wymiotnego oraz nieopioidowego leku przeciwbó-
rozlany długotrwały ból, który nasila się pod wpły- lowego (zob. ryc. B 1.5-19). Dość skuteczne jest do-
wem obciążenia psychicznego). żylne podanie 1000 mg kwasu acetylosalicylowego.
Biorąc pod uwagę objawy kliniczne oraz poglądy Jako leki przeciwwymiotne o działaniu polegającym
na patogenezę – farmakologiczna terapia migreny na przyspieszeniu perystaltyki nadają się metoklo-
usiłuje wpływać na (opanowywać): pramid i domperidon. Tłumią one nie tylko nudności,
odruch wymiotny i wymioty, lecz również poprawia-
■ nudności i wymioty,
ją wchłanianie podawanych później – w prawidłowy
■ ból i stan zapalny, sposób – leków przeciwbólowych.
W przypadku średniociężkich i ciężkich oraz dłu-
■ zaburzenia naczyniowe.
gotrwałych napadów szybko i dobrze działającymi
lekami okazały się triptany, tj. agoniści receptorów
Wyróżnia się przy tym dwa rodzaje terapii:
5-HT1B/1D (zob. ryc. B 1.5-19). Prowadzą one do:
■ terapię ostrego napadu migreny,
■ skurczu rozszerzonych w wyniku napadu migre-
■ profilaktykę napadów migreny w okresie między- ny naczyń krwionośnych (obwodowy komponent
napadowym. działania),
MUTSCHLER-2009.indd 265 2010-01-07 22:12:05
266 Układ nerwowy
stopień 1
lek przeciwwymiotny lek przeciwbólowy O
CH3
(np. MCP lub + (np. ASA, paracetamol N
HN S
domperidon) lub ibuprofen)
H3C O CH3
NH
w przypadku braku efektywności sumatriptan
O
CH3
N S N
jedynie niewielkie nudności silne nudności
brak wcześniejszych wcześniejsze wymioty O CH3
NH
wymiotów
almotriptan
stopień 2 CH3
N
triptany triptany
O
triptan doustnie sumatriptan
HN S
6 mg podskórnie
H3C O
lub NH
sumatriptan
10 lub 20 mg donosowo naratriptan
lub
zolmitriptan donosowo 5 mg H3C
lub N
sumatriptan O
25 mg doodbytniczo
S
O
NH
Ryc. B 1.5-19. Terapia napadu migreny: MCP – metoklopramid,
ASA – kwas acetylosalicylowy (zmodyf. według Göbela). eletriptan
CH3
HN N
O
CH3
■ zahamowania uwalniania neuropeptydów z akty- O NH
wowanych zakończeń nerwu trójdzielnego,
zolmitriptan
■ zablokowania transmisji nocyceptywnej (bólowej)
w obrębie nerwu trójdzielnego w kierunku do jądra CH3
ogoniastego (nucleus caudatus; ośrodkowy kom- N N
ponent działania). N
N CH3
NH
Lekiem prototypowym dla triptanów jest sumatriptan.
W celu poprawienia niektórych właściwości farmako- rizatriptan
logicznych sumatriptanu – a także jego niewielkiej
doustnej biodostępności, krótkiego okresu półtrwania HN CH3
O
oraz względnie niskiej lipofilności i wynikającą z tego
faktu małej przenikalności do ośrodkowego układu H2N
nerwowego – zostali stworzeni nowi agoniści recep-
NH
torów 5-HT1B/1D. W tab. 1.5-11 dokonano porównania
ważnych właściwości farmakodynamicznych i farma- frowatriptan
kokinetycznych sumatriptanu oraz innych preparatów
takich jak: zolmitriptan, naratriptan, rizatriptan,
eletriptan, almotriptan i frowatriptan.
MUTSCHLER-2009.indd 266 2010-01-07 22:12:06
Układ nerwowy 267
Duże badania kliniczne przeprowadzone w kontro- Wśród działań niepożądanych triptanów obserwowa-
li do placebo potwierdziły skuteczność wszystkich no: przejściowe mrowienia, uczucie zimna, bóle, uczu-
triptanów w zakresie ostrych napadów migreny. Je- cie ciężaru, ucisku i ciasnoty w obrębie różnych okolic
żeli konieczne jest szybkie ujawnienie się działania, ciała (szczególnie w klatce piersiowej i szyi), a w dal-
Układ nerwowy
należy zaaplikować podskórnie sumatriptan (6 mg). szej kolejności – uderzenia gorąca, stany zamrocze-
Również przy podaniu donosowym (sumatriptan nia lub zawroty głowy, a także uczucie zmęczenia,
lub zolmitriptan) zapewnione jest szybsze działanie senność i zaburzenia widzenia. Ponadto może dojść
niż w przypadku podania doustnego. Jako skuteczne do krótkotrwałych skoków ciśnienia. W przypadku
u pacjentów z wczesnymi wymiotami sprawdziły się osób z określonymi predyspozycjami istnieje niebez-
czopki (z sumatriptanem). Lipofilne triptany (zob. pieczeństwo skurczu naczyń wieńcowych serca.
tab. B 1.5-11) działają szybciej i są zwykle bardziej Stąd też tryptany są przeciwwskazane u pacjen-
skuteczne niż związki mniej lipofilne, jednakże wy- tów ze skurczami naczyń wieńcowych, objawową B1
wołują również więcej ośrodkowych działań niepo- chorobą niedokrwienną serca, po przebytym zawale
żądanych. serca lub z nasilonym nadciśnieniem tętniczym oraz
Tabela B 1.5-11. Porównanie agonistów receptorów 5-HTIB/ID (triptanów)
sumatriptan zolmitriptan naritriptan rizatriptan
(Imigran, Cinie, (AscoTop, Zomig®) (Naramig®) (Maxalt®)
Sumigra)
dawka jednorazowa 50-100 mg doustnie 2,5 mg doustnie, 2,5 mg doustnie 5-10 mg doustnie
25 mg doodbytniczo 5 mg donosowo
20 mg donosowo
6 mg podskórnie
lipofilność (+) + ++ +
biologiczna dostępność po podaniu 14 40 60-70 40-45
doustnym (%)
tmax (godz.) 1,5-2,5 2-3 2-3 1-1,5
t1/2 (godz.) 2 ~3 6 2-3
skuteczność (%) 50-70 po podaniu doustnym 60-80 doustnie 60-68 62-71
80 po podaniu podskórnym
nawrót bólów (%) 30-40 po podaniu doustnym 30 doustnie 27 30-35
50 po podaniu podskórnym
eletriptan almotriptan frowatriptan
(Relpax®) (Almogran®) (Allegro®)
dawka pojedyncza 20-40 mg doustnie 12,5 mg doustnie 2,5 mg doustnie
lipofilność +++ +++ +
biologiczna dostępność ~50 ~70 20-30
po podaniu doustnym (%)
tmax (godz.)) 1-1,5 2,2-2,5 2-4
t1/2 (godz.)) 4-5 3-4 ~25
skuteczność (%) 64 (dla dawki 40 mg) 57-70 37-46
71 (dla dawki 80 mg)
nawrót bólów (%) 21 (dla dawki 80 mg) ~25 20
MUTSCHLER-2009.indd 267 2010-01-07 22:12:07
268 Układ nerwowy
w przebiegu choroby Raynauda i zarostową chorobą 1.5.7.2. Profilaktyka migreny
naczyń krwionośnych. Także w ciąży, w okresie kar-
mienia i u dzieci w wieku poniżej 12 lat – triptany
Profilaktyka migreny (leczenie w okresie międzyna-
nie powinny być stosowane.
padowym) powinna być przeprowadzona, jeżeli pa-
Triptanów nie należy podawać równocześnie z le-
cjent ma dwa do trzech napadów w miesiącu, czas
kami wpływającymi na metabolizm serotoniny (alka-
trwania i natężenie pojedynczych napadów stanowi
loidami sporyszu, inhibitorami MAO, inhibitorami
szczególne obciążenie i/lub napady ostre nie są w wy-
wychwytu zwrotnego serotoniny).
starczający sposób kontrolowane farmakologicznie.
Celem profilaktyki migreny jest zmniejszenie licz-
Alternatywą dla triptanów w leczeniu ciężkich na-
by i nasilenia jej napadów. Ocena skuteczności róż-
padów migreny może być stosowanie pochodnych
nych środków terapeutycznych jest utrudniona przez
ergotaminy (soli ergotaminy lub dihydroergotami-
to, że efekt działania placebo osiąga wartość rzędu
ny) – po wcześniejszym podaniu 20 mg metoklopra-
30–40%.
midu. Pochodne ergotaminy są podobnie jak triptany
Niefarmakologicznymi metodami profilaktyczny-
agonistami receptorów 5-HT1B/1D, ponadto wpływa-
mi jest unikanie wieczorem nadmiernego spożycia
ją na wiele innych receptorów (zob. ryc. B 1.5-20)
pokarmów i płynów, ograniczenie konsumpcji alko-
– niemających żadnego znaczenia w terapii migreny.
holu i uregulowanie snu.
Wyjaśnia to występowanie określonych dodatko-
wych działań niepożądanych, których nie wykazują
triptany. Do farmakologicznej terapii migreny stosuje się na-
Dawkowanie, np. winianu ergotaminy, wynosi 1–2 stępujące leki:
mg doustnie lub doodbytniczo.
■ leki β-adrenolityczne, przede wszystkim proprano-
W żadnym wypadku nie należy przekraczać mak-
lol i metoprolol,
symalnej dawki 4 mg winianu ergotaminy w jednym
napadzie lub 6 mg w ciągu tygodnia. ■ antagonistów wapnia, w szczególności flunaryzyna,
Nadużywanie ergotaminy prowadzi do przewle-
■ leki przeciwdepresyjne, np. amitryptylinę,
kłych bólów głowy, dolegliwości mięśniowych
i skurczów tętnic, w wyniku których może dojść m.in. ■ leki przeciwpadaczkowe – kwas walproinowy i to-
do ujawnienia się zgorzeli. piramat,
Ergotaminy Triptany
Receptory (np. dihydroergotamina) (np. sumatriptan)
nudności/wymioty,
5-HT1A ++++ + zaburzenia nastroju
5-HT1B +++ ++ działanie przeciwmigrenowe
5-HT1D ++++ +++
5-HT2A ++ – działania niepożądane
5-HT2C ++ – na naczynia,
adrenergiczne osłabienie,
α1 +++ –
α2 +++ – stany zamroczenia
β + –
dopaminergiczne
zaburzenia żołądkowo-jelitowe,
D1 + – nudności/wymioty
++++ wartości Ki (nM) = 0,1-1 ++ wartości Ki (nM) = 10-100 – wartości Ki (nM) > 1000
+++ wartości Ki (nM) = 1-10 + wartości Ki (nM) = 100-1000
Ryc. B 1.5-20. Różnice między pochodnymi ergotaminy i triptanami w zakresie selektywności wiązania z receptorami.
MUTSCHLER-2009.indd 268 2010-01-07 22:12:07
Układ nerwowy 269
■ niesteroidowy lek przeciwzapalny – naproksen. Również inny lek przeciwpadaczkowy – kwas wal-
proinowy (dawka dzienna 500–600 mg) okazał się
Ponadto przeprowadzono wiele prób terapii z in- efektywny w kontrolowanych badaniach. W Niem-
nymi lekami (były to np. cykladenalat, preparaty czech (i w Polsce) nie otrzymał on jeszcze zezwo-
Układ nerwowy
magnezu, lamotrygina, kandesartan, lizynopril, klo- lenia do stosowania we wskazaniu, jakim jest profi-
nidyna, toksyna botulinowa i wiele innych), których laktyka migreny. Należy zwracać szczególną uwagę
skuteczności w kontroli do placebo nie udało się na zaburzenia funkcji wątroby i układu krzepnięcia.
jednak potwierdzić. Ze względu na właściwości teratogenne – kwas wal-
proinowy może być stosowany u kobiet prowadzą-
Jedynie część leków β-adrenolitycznych (blokerów cych skuteczną antykoncepcję.
receptorów β-adrenergicznych) okazała się sku-
teczna w profilaktyce migreny. Najlepiej zbadany Po okresie terapii wynoszącym 6–9 miesięcy (stoso- B1
jest propranolol; innym dopuszczonym do stoso- wanie β-blokerów możliwe jest również przez dłuższy
wana w profilaktyce migreny β-blokerem jest meto- okres) leczenie profilaktyczne powinno być stopniowo
prolol. Dokładny mechanizm działania nie jest zna- odstawiane, a dalszy przebieg powinien być kontrolo-
ny; działanie na obwodowe receptory β-adrener- wany przez następne dwa, trzy miesiące. Gdy napady
giczne nie wydaje się odgrywać decydującej roli. migreny ponownie zwiększą swoją częstość i nasilenie
Propranolol i metoprolol są wskazane szczególnie – znów należy wdrożyć metody profilaktyczne.
u takich pacjentów, którzy oprócz migreny – cho-
rują jeszcze na nadciśnienie, chorobę niedokrwien- W przypadku migreny związanej z cyklem miesiącz-
ną serca lub jego przewlekłą niewydolność. Z kolei kowym – można ponadto spróbować przeprowadzić
pacjenci z astmą oskrzelową, depresją lub cukrzycą profilaktykę migreny za pomocą naproksenu (daw-
– nie powinni otrzymywać β-blokerów. Zwyczajo- ki: dwa razy 250–500 mg dziennie), zwracając uwagę
wa dawka dzienna propranololu wynosi 40–240 mg, na przeciwwskazania. Terapia powinna zaczynać się
metoprololu – 50–200 mg. kilka dni przed miesiączkowaniem i kończyć się kil-
ka dni po jego zakończeniu.
Zastosowanie antagonistów wapnia w profilaktyce
migreny jest częściowo kwestionowane. Za skutecz-
ną uważana jest flunaryzyna – antagonista wapnia 1.5.8. Schorzenia z kręgu choroby
z klasy blokerów przeładowania wapnia (calcium reumatycznej i ich terapia
overload blocker). Pacjenci z padaczką, chorobą
Parkinsona i/lub depresją nie powinni być jednak le-
1.5.8.1. Podstawy patofizjologiczne
czeni flunaryzyną. Dawka dzienna flunaryzyny wy-
nosi 5–10 mg. Antagonista wapniowy – nifedypina
Zbiorcze pojęcie schorzeń z kręgu choroby reuma-
okazała się nieskuteczna.
tycznej obejmuje schorzenia przebiegające w obrębie
stawów i otaczających je tkanek miękkich, przy czym
W szczególnych przypadkach (np. w przypadku mi-
często są to schorzenia ogólnoustrojowe. Rozróżnia się
greny z napięciowymi bólami głowy lub występującej
następujące postacie chorobowe (zob. tab. B1.5-12):
u pacjentów z depresją) w profilaktyce migreny mogą
być zastosowane również trójcykliczne leki prze- ■ zapalne,
ciwdepresyjne. Najwięcej korzystnych doświadczeń
■ zwyrodnieniowe,
dotyczy amitryptyliny (dawka dzienna 50–150 mg).
Jaskra z wąskim kątem przesądzania oraz gruczolak ■ pozastawowe.
prostaty są przeciwwskazaniami.
Obrazy kliniczne są niezwykle zróżnicowane
Niedawno do profilaktyki migreny dopuszczony zo- i zwłaszcza w postaciach zapalnych rozgraniczenia
stał lek przeciwpadaczkowy – topiramat. Czynnikami w okresie początkowym mogą być bardzo trudne.
ograniczającymi terapię może być utrata masy ciała Występują również tzw. przenikające się (nakłada-
oraz zaburzenia funkcji poznawczych. Z drugiej jed- jące się) zespoły. Dla podejmowanych metod lecz-
nak strony – topiramat może być wykorzystywany niczych decydujące znaczenie mają etiopatogeneza,
szczególnie u pacjentów z nadwagą. objawy kliniczne, diagnostyka immunologiczna oraz
Dawka dzienna topiramatu wynosi 25–100 mg. przebieg choroby.
MUTSCHLER-2009.indd 269 2010-01-07 22:12:07
270 Układ nerwowy
Tabela B 1.5-12. Schorzenia z kręgu choroby reumatycznej układzie nerwowym. Występuje ona szczególnie
często u dzieci i młodych osób. U podłoża choro-
I. Zapalne choroby reumatyczne by leży reakcja autoimmunologiczna w następstwie
uwrażliwienia (sensybilizacji) na określone antyge-
(ostra) gorączka reumatyczna ny paciorkowcowe. Na przykład wykazano immu-
(paciorkowcowa)
nologiczną reakcję krzyżową pomiędzy białkiem
reumatoidalne zapalenie stawów (przewlekłe zapalenie wielostawowe) paciorkowcowym i antygenem tkankowym mięśnia
z jego postaciami specjalnymi sercowego. Ponieważ jednak tylko niewielka część
chorych, którzy przeszli zakażenie paciorkowcowe,
łuszczycowe zapalenie stawów
zapada na gorączkę reumatyczną, w jej uwidocznie-
zesztywniające zapalenie stawów kręgosłupa niu się muszą uczestniczyć również inne czynniki.
(choroba Bechterewa) Zalicza, się do nich uwarunkowaną genetycznie
kolagenozy szczególną gotowość układu immunologicznego
toczeń rumieniowaty układowy do pobudzenia, niedożywienie, przemęczenie, wa-
zespół Sjögrena (pierwotny) runki klimatyczne.
postępująca twardzina układowa Jako ważne objawy kliniczne, które mogą wy-
zapalenie wielomięśniowe i skórno-mięśniowe
stępować obok siebie, ale też pojedynczo, trzeba
zapalenia naczyń wymienić: ostre zapalenie wielostawowe, zapalenie
polimialgia reumatyczna mięśnia sercowego, objawy skórne (pod postacią
guzkowe zapalenie tętnic różnorodnych rumieni) i pląsawicę (chorea minor).
ziarniniak Wegenera
Najczęstsze jest ostre zapalenie wielostawowe, ata-
infekcyjne zapalenia stawów kujące przede wszystkim duże stawy i zwykle ustę-
pujące bez następstw. Dużo bardziej niebezpiecz-
II. Zwyrodnieniowe choroby reumatyczne
niejsze jest przebiegające z zaburzeniami rytmu ser-
zwyrodnieniowa choroba stawów ca, dusznością, zmieniającymi się szmerami serca
zwyrodnienia jedno-, skąpo- i wielostawowe i innymi objawami zapalenie mięśnia sercowego,
mnogie zwyrodnienia stawów palców ponieważ w tym przypadku dochodzi do trwałych
zmiany degeneracyjne kręgosłupa szkód, przede wszystkim – wad zastawki mitralnej.
Pląsawicę Sydenhama, inaczej taniec świętego Wita,
zapalenia chrząstek stawowych z mimowolnymi bezcelowymi ruchami (zwłaszcza
osteochondrozy
zapalenie chrząstek kręgosłupa (spondyloza)
ramion i twarzy), zmniejszonym napięciem mięś-
choroba zwyrodnieniowa stawów kręgosłupa (spondyloartroza) niowym i zmiennością nastrojów, stwierdza się nie-
mal wyłącznie u dzieci i młodzieży.
III. Pozastawowe postacie choroby reumatycznej Przebieg gorączki reumatycznej jest zmienny,
(reumatyzm pozastawowy)
w 95% przypadków objawy ustępują w ciągu 3–6
reumatyczne zapalenie mięśni miesięcy. Niebezpieczeństwo nawrotu jest tym więk-
zapalenie kaletki maziowej
sze, im młodszy jest chory. Z każdym kolejnym na-
zapalenie ścięgna i pochewki ścięgna
zapalenie podskórnej tkanki tłuszczowej wrotem wzrasta zagrożenie uszkodzenia serca.
1.5.8.1.2. Reumatoidalne zapalenie stawów
1.5.8.1.1. Gorączka reumatyczna Reumatoidalne zapalenie stawów (RZS; wcześniej
określane jako: przewlekłe zapalenie wielostawo-
Ostra gorączka reumatyczna (ostre reumatyczne we) jest szczególnym znaczącym ogólnoustrojowym
zapalenie stawów; zob. poniżej) – z powodu terapii (systemowym) przewlekłym schorzeniem zapalnym
antybiotykowej rzadko występuje w krajach uprze- tkanki łącznej. Chorobą tą dotknięty jest 1% populacji
mysłowionych, lecz nadal jest dość częsta w krajach ludzkiej. Właściwa przyczyna choroby nie jest znana;
Trzeciego Świata. Jest schorzeniem wtórnym, które dyskutowane jest współdziałanie czynników predys-
ujawnia się 2–3 (a nawet do 5) tygodni po zakaże- pozycji genetycznej oraz czynnika zakaźnego. Auto-
niu β-hemolitycznymi paciorkowcami grupy A i któ- immunologiczne podłoże patogenetyczne wydaje się
re prowadzi do zmian zapalnych przede wszystkim udowodnione, mimo to przebiegi poszczególnych
w dużych stawach, sercu, skórze i ośrodkowym reakcji nadal są hipotetyczne.
MUTSCHLER-2009.indd 270 2010-01-07 22:12:08
Układ nerwowy 271
genetyczna predyspozycja czynniki środowiskowe ostry czynnik wyzwalający nieznane czynniki
Układ nerwowy
reakcja immunologiczna przeciwko własnej tkance stawowej
komórka kompleks
limfocyt T limfocyt B plazmatyczna autoprzeciwciało
przeciw własnej antygen-przeciwciało
limfokiny immunoglobulinie
endogenna
B1
immunoglobulina IgG
limfokiny
monokiny
aktywacja dopełniacza
makrofag
chemotaksja zniszczenie
komórki
monokiny granulocyt
mediatory zapalne, prostaglandyny, wolne rodniki tlenowe, enzymy niszczące chrząstkę stawową
w wyniku przewlekłego zapalenia:
staw prawidłowy zniszczenie chrząstki stawowej
i sąsiadującej z nią kości,
zwężenie szpary stawowej
zapalenie błony maziowej
Ryc. B 1.5-21. Poglądy na patogenezę reumatoidalnego zapalenia stawów. Monokiny (IL-1, IL-6, IL-8, TNF-α) uczestniczą w ujaw-
nieniu ostrej fazy procesu zapalnego i oddziałują m.in. z limfocytami T, podczas gdy limfokiny (IL-2, IL-4, IL-5, IL-9, IL-10, IFN-γ) są
odpowiedzialne raczej za przewlekły przebieg zapalenia.
Współczesne poglądy na patogenezę reumatoi- tycznych. Istnienie tych ostatnich daje się jednak wy-
dalnego zapalenia stawów zostały w uproszczony kazać jedynie u pewnej części chorych. IgG i czyn-
sposób przedstawione na ryc. B 1.5-21. Nieznany niki reumatyczne tworzą z dopełniaczem kompleksy
czynnik szkodliwy (być może bakteryjny, wiruso- immunologiczne, które ulegają fagocytozie poprzez
wy, być może zaburzenia przepuszczalności naczyń granulocyty i komórki torebki maziowej. Dochodzi
lub mechaniczne uszkodzenia komórek) uruchamia przy tym do uwolnienia cytokin, które podtrzymują
proces zapalny w torebce stawowej. W czasie tej po- proces zapalny. W reumatoidalnym zapaleniu sta-
czątkowej reakcji dochodzi do uwolnienia antygenu, wów uwalniane w obrębie stawu cytokiny mogą być
który aktywuje komórkowy system obronny). Proli- ze względu na funkcję patofizjologiczną podzielone
ferujące limfocyty B – po przekształceniu w komórki na 3 grupy:
plazmatyczne – wytwarzają przede wszystkim immu-
noglobulinę G (IgG), która ze swej strony indukuje ■ cytokiny prozapalne (IL-1, IL-2, IL-6, IL-8, IL-15,
wytwarzanie autoprzeciwciał, tzw. czynników reuma- IL-18, TNF-α i in.) regulują napływ i aktywują ko-
MUTSCHLER-2009.indd 271 2010-01-07 22:12:08
272 Układ nerwowy
mórki wykonawcze (efektorowe) procesu zapalne- stawów procesem chorobowym mogą zostać objęte
go, jak również zwiększają proliferację chondrocy- również inne narządy. Nierzadko można stwierdzić
tów i fibroblastów, np. patologiczne zmiany w obrębie mięśni (zaniki,
tworzenie się guzków), choroby oczu i obwodowe
■ cytokiny przeciwzapalne (IL-4, IL-10, IL-13,
neuropatie. W przypadkach o ciężkim przebiegu
TGF-β i in.) wybiórczo hamują działanie cytokin
– ze względu na uogólnione zmiany narządowe –
prozapalnych,
może dochodzić do zapalenia naczyń na tle autoim-
■ białka antycytokinowe (np. rozpuszczalny receptor munologicznym.
TNF-α lub receptor IL-1) mogą kontrolować pro-
ces zapalny poprzez wychwytywanie prozapalnych Do specjalnych postaci reumatoidalnego zapalenia
cytokin. stawów należą m.in.:
■ młodzieńcze reumatoidalne zapalenie stawów,
Ponieważ u chorych z reumatoidalnym zapaleniem
stawów można wykazać istnienie wszystkich trzech ■ zespól Felty’ego z powiększeniem śledziony i leu-
typów cytokin, wydaje się, że przewagę ilościową kopenią,
mają jednak cytokiny prozapalne.
■ (wtórny) zespół Sjögrena (zob. poniżej).
Pod wpływem procesu zapalnego błona mazio-
wa zaczyna przerastać i podobnie jak powierzchnia
chrząstki stawowej, zostaje pokryta włóknami fibryny.
Tworzy się kosmatopodobna tkanka ziarninowa okre- 1.5.8.1.3. Ujemne serologicznie zapalenia
ślana mianem łuszczki (pannus). Enzymy lizosomalne stawów i kręgosłupa (zapalenie
niszczą warstwę chrząstki, tak że w późnych stadiach stawów związane z HLA-B27)
pozbawione chrząstki elementy stawu zespalają się
między sobą. Dalszymi następstwami są ogniskowe Określane tym pojęciem obrazy chorobowe charak-
zniszczenia kości oraz obkurczanie się torebki stawo- teryzują się ścisłym związkiem z ludzkim antyge-
wej wskutek tworzenia się blizn. nem limfocytów (HLA) B27 i odpowiednią predys-
Przebiegająca pod postacią rzutów choroba roz- pozycją genetyczną. Czynniki reumatyczne zwykle
poczyna się najczęściej skrycie okresem zwiastuno- nie są obecne, stąd też określane są one jako serolo-
wym (prodromalnym), który charakteryzuje się utratą gicznie ujemne. Etiologia i patogeneza są w znacznym
łaknienia oraz masy ciała, uczuciem znużenia, ogól- stopniu nieznane. Jako przykłady tych chorób poniżej
nym osłabieniem i czasem gorączką. Odpowiednio zostało opisane łuszczycowe zapalenie stawów i ze-
do dalszego przebiegu klinicznego można za Stei- sztywniające zapalenie stawów kręgosłupa.
nbrockerem przyjąć podział na cztery stadia choro-
by: w okresie I występują zazwyczaj symetryczne Łuszczycowe zapalenie stawów (polyarthritis pso-
obrzmienia małych i średnich stawów z bolesnym riatica). Postępujące niszczące zapalenie wielosta-
ograniczeniem ich ruchomości i poranną sztywnoś- wowe towarzyszące łuszczycy występuje niekiedy
cią. Szczególnie często zmianami objęte są stawy równolegle do zmian skórnych, a niekiedy je wyprze-
śródręczno-palcowe i środkowe palców. W okresie dza lub też pojawia się po nich.
II chorobą dotknięte zostają zazwyczaj duże stawy; Zasadniczo zajęte mogą być dowolne stawy, za-
chorzy skarżą się oprócz bolesności w czasie ruchów zwyczaj jednak znajduje się zmiany w obrębie
również na bóle spoczynkowe. Czas trwania sztyw- wszystkich stawów jednego ciągu (np. palca ręki lub
ności porannej wydłuża się (3–4 godz.). Ruchomość stopy). W przypadku dłuższego przebiegu istnieje,
stawów jest ograniczona tylko w niewielkim stopniu. podobnie jak w reumatoidalnym zapaleniu stawów,
W okresie III stwierdza się obrzęki stawów znaczne- niebezpieczeństwo rozległych deformacji stawów.
go stopnia, deformacje z odchyleniami od osi stawu Podskórne guzki reumatyczne nie występują.
oraz podwichnięcia. Sztywność poranna może się
na tym etapie utrzymywać do pięciu godzin, a ogra- Zesztywniające zapalenie kręgosłupa. Określany
niczenia ruchomości stawów są znaczne. W okresie często jako zesztywniające zapalenie stawów kręgo-
IV jako dalszy objaw występuje włókniste lub kostne słupa (spondyloarthritis ankylopoetica) lub choroba
zesztywnienie stawów; dochodzi do (nasilonego) in- Bechterewa przewlekły stan zapalny tkanki łącznej
walidztwa, a sam pacjent wymaga opieki i pomocy. obejmuje przede wszystkim stawy biodrowo-krzyżo-
Reumatoidalne zapalenie stawów jest ogólno- we, drobne stawy oraz aparat więzadłowy kręgosłu-
ustrojową chorobą tkanki łącznej, dlatego oprócz pa. Poza objawami ze strony kręgosłupa – u prawie
MUTSCHLER-2009.indd 272 2010-01-07 22:12:08
Układ nerwowy 273
jednej trzeciej pacjentów zajęte są też stawy obwo- Przyczyną postępującej twardziny są podskórne, naczy-
dowe i inne narządy. Chorobą dotknięci są przede niowe i podśluzówkowe stwardnienia włóknikowe, tj. silne
zwiększenie i zagęszczenie warstwy odkładanych włókien
wszystkim młodzi mężczyźni. Etiologia jest niezna-
kolagenu. Kobiety są dotknięte chorobą dwukrotnie częś-
na; występują jednak czynniki dziedziczne. Skrycie ciej niż mężczyźni. W osoczu dochodzi do zwiększenia po-
Układ nerwowy
rozpoczynająca się choroba ujawnia się we wczes- ziomów makroglobuliny α2 i immunoglobuliny A, ponadto
nym stadium bólami w obrębie kręgosłupa lędźwio- udaje się wykazać obecność czynników przeciwjądrowych,
wego, bolesnością uciskową i przy oklepywaniu a w pojedynczych przypadkach – przeciwciał przeciwery-
trocytarnych.
stawów biodrowo-krzyżowych w następstwie stanu
Na początku choroby objawy często są zbliżone do ob-
zapalnego tego stawu oraz ograniczeniem rucho- jawów choroby Raynauda. Później na plan pierwszy wy-
mości kręgosłupa. Proces chorobowy postępuje naj- suwają się stwardnienie i sztywność skóry oraz podskór-
częściej od dołu ku górze, jednak w każdym stadium nej tkanki łącznej, które rozwijają się przede wszystkim
może ulec zatrzymaniu (przyjąć formę stacjonarną). na rękach i twarzy. Przy dalszym rozszerzaniu się procesu B1
chorobowego dochodzi do przykurczów, często z dziwacz-
W ciężkich przypadkach dochodzi do calkowitego
nymi nieprawidłowościami ustawienia palców lub dłoni,
skostnienia stawów i zesztywnienia kręgosłupa oraz utraty mimiki (twarz maskowata), zmniejszenia szpary
stawów bliskich tułowia. ustnej. Zajęcie narządów wewnętrznych nie zawsze kore-
luje ze zmianami skórnymi. Szczególnie często dotknięty
zmianami jest przewód pokarmowy. Zmiany twardzinowe
prowadzą do sztywności ściany przewodu pokarmowego,
1.5.8.1.4. Kolagenozy
przede wszystkim w obrębie dolnego odcinka przełyku,
żołądka i górnego odcinka jelita cienkiego z utrudnionym
Kolagenozy to grupa ogólnoustrojowych chorób au- połykaniem, spowolnieniem pasażu, złym wchłanianiem.
toimmunologicznych, w których obok zmian naczy- Rzadziej w procesie chorobowym uczestniczy tkanka łącz-
niowych (naczyniopatii) stwierdza się włóknikowate na płuc, serca, śledziony i nerek. Główną przyczyną zgo-
nów jest niewydolność serca, płuc lub nerek.
zwyrodnienia w tkankach. Należą do nich:
■ toczeń rumieniowaty układowy, W rzadko występującym zapaleniu wielomięśniowym
(polymiosistis) w krótkim czasie rozwija się obustronne,
■ pierwotny zespół Sjögrena, symetryczne osłabienie siły mięśniowej, obejmujące głów-
nie mięśnie pasa barkowego i biodrowego. Jeśli pojawią
■ postępująca twardzinę, się zmiany skórne w postaci płaskich, obrzękłych rumieni
barwy czerwonego wina oraz dodatkowo przebarwienia
■ zapalenie wielomięśniowe i skórnomięśniowe. lub odbarwienia, można rozpoznać zapalenie mięśniowo-
skórne (dermatomyositis). Szczególną uwagę zwraca to,
W przypadku układowego tocznia rumieniowatego (sy- że u stosunkowo dużego odsetka chorych stwierdza się no-
stemic lupus erythematosus – SLE) mamy do czynienia wotwory złośliwe.
ze schorzeniem o zmiennym obrazie klinicznym. Oprócz
skóry oraz stawów proces chorobowy obejmuje również
inne narządy. Choroba występuje najczęściej u kobiet
w wieku rozrodczym. 1.5.8.1.5. Zapalenia naczyń
Dla przyjęcia rozpoznania tocznia rumieniowatego waż-
ne jest potwierdzenie istnienia specyficznych autoprzeciw- Ta grupa chorób charakteryzuje się zapalnymi procesami
ciał. W immunodiagnostyce specjalne znaczenie ma róż- w ścianach naczyń i spowodowanymi przez nie zróżnico-
nicowanie tych przeciwciał przeciwjądrowych ze względu wanymi objawami, zależnymi od poszczególnych obrazów
na ich skojarzenie z objawami narządowymi. Przeciwciała chorobowych. Należą do nich: polimialgia (polymyalgia
skierowane przeciw dwuniciowemu DNA są patognomo- arteriica), guzkowe zapalenie tętnic (panarteriitis nodosa)
niczne dla SLE. i ziarniniak Wegenera (granuloma Wegeneri).
Choroba rozpoczyna się zazwyczaj wysoką gorącz-
ką, zapaleniem wielostawowym i rumieniem na twarzy Często występująca polymyalgia arteriitica obejmuje obra-
w kształcie motyla. Jednak zmiany na skórze, podobnie jak zy chorobowe określane jako zapalenie tętnic czaszkowych
zmiany w stawach, nie muszą występować. Dalszy prze- (arteriitis cranialis) i polimialgię reumatyczną. W przypad-
bieg charakteryzuje się zazwyczaj spontanicznymi remi- ku zapalenia tętnic czaszkowych procesem chorobowym
sjami i zaostrzeniami, spotyka się jednak również postacie objęte są przeważnie duże i średnie tętnice (skroniowa,
błyskawicznie przebiegające, prowadzące do zgonu. doczaszkowe), a sam proces przebiega pod postacią tzw.
zapalenia olbrzymiokomórkowego; z kolei w przypadku
Pierwotny zespół Sjögrena charakteryzuje się tzw. suchym polimialgii reumatycznej nie są znane jednoznaczne ce-
zespołem (sicca syndrom): suchość błon śluzowych oczu, chy anatomopatologiczne. W prawie jednej trzeciej przy-
jamy ustnej, a także innych błon śluzowych jest objawem padków stwierdza się jednoczesne występowanie obu tych
typowym. Jeśli zaburzenia te występują wraz z objawami jednostek. Jako objawy zapalenia tętnic czaszkowych wy-
innych chorób reumatycznych, np. z objawami reumatoi- stępują bóle głowy, zaburzenia widzenia (niebezpieczeń-
dalnego zapalenia stawów, mówi się o wtórnym zespole stwo zaniewidzenia!), zaburzenia przepływów mózgowych
Sjögrena. i porażenie mięśni ruchowych oka. W polimialgii wystę-
MUTSCHLER-2009.indd 273 2010-01-07 22:12:09
274 Układ nerwowy
pują obustronne, ostre bóle mięśni, barków i ramion, jak tym ulegać kalafiorowatopodobnym deformacjom.
również mięśni pośladków i ud. Odczyn opadania krwinek Występowaniu zmian sprzyjają nadmierne obcią-
czerwonych (OB) jest bardzo podwyższony. Chorobie czę-
żenie powierzchni stawowych, ostre lub przewlekłe
sto towarzyszą stany depresyjne.
czynniki urazowe, procesy zapalne stawów, choro-
Guzkowe zapalenie tętnic (panarteriitis nodosa, periarte- by metaboliczne, a także unieruchomienie. Zależnie
riitis nodosa) jest uogólnionym odczynem zapalnym śred- od lokalizacji mówi się o zwyrodnieniu stawów bio-
nich i małych tętnic z niedokrwienną martwicą, tworzeniem drowych (coxarthrosis), kolanowych (gonarthrosis),
tętniaków, zakrzepów i nacieków, które od błony środko-
barkowych (omarthrosis).
wej rozszerzają się się zarówno na błonę wewnętrzną, jak
i na przydankę. Mężczyźni chorują blisko trzykrotnie częś- Najczęściej spotykanym zwyrodnieniem dużych
ciej od kobiet. U połowy chorych stwierdza się przewlekłe stawów jest coxarthrosis. Odróżnia się przy tym pier-
wirusowe zapalenie wątroby typu B, a zapalenie naczyń wotne zwyrodnienie stawów biodrowych, występu-
jest w tym przypadku następstwem odkładania się kom- jące najczęściej u chorych otyłych, od zwyrodnienia
pleksów immunologicznych złożonych z białka wirusowego
wtórnego, którego najczęstszą przyczyną jest wro-
i endogennych przeciwciał w ścianie naczyń. Inne przypad-
ki wywołane są odczynem immunologicznym skierowanym dzony niedorozwój i nadwichnięcie stawu biodro-
przeciw lekom. wego. Zwyrodnienie stawu kolanowego najczęściej
Choroba niejednokrotnie rozpoczyna się skrycie. Wy- występuje obustronnie i zazwyczaj dotyczy kobiet
stępujące objawy są zmienne zależnie od tego, który na- z nadwagą.
rząd jest przede wszystkim objęty procesem chorobowym.
Obok wyżej wymienionych dużych stawów zmia-
Szczególnie często oprócz stanów podgorączkowych wy-
stępuje utrata masy ciała, jak też uczucie znużenia, bóle sta- nom mogą też ulegać małe i średnie stawy. Wśród
wowe i mięśniowe, zapalenia nerwowe, a przede wszystkim wielostawowych zwyrodnień małych stawów dotknię-
bóle w nadbrzuszu. W zaawansowanym stadium choroby te procesem zwyrodnieniowym są przede wszystkim
może dochodzić do zapalenia osierdzia, niewydolności ser- stawy palców (artroza Heberdena), wobec których
ca, zawałów serca, niewydolności nerek, krwawienia z jelit,
potwierdzony został autosomalny dominujący sposób
zapalenia trzustki, zawałów śledziony i nerek. Przy braku
leczenia – rokowanie jest złe. Śmierć następuje zazwyczaj dziedziczenia.
wskutek niewydolności serca lub nerek. Obraz kliniczny chorób zwyrodnieniowych cechu-
ją początkowa sztywność i ból przy dłuższym obcią-
W przypadku ziarniniaka Wegenera dochodzi do martwi- żeniu, jak też rozpoczynaniu ruchu, w późniejszym
czego, ziarninującego zapalenia małych naczyń tętniczych
okresie charakteryzuje się silnymi – często zależnymi
i żylnych – typowo dotykającego górnych i dolnych dróg
oddechowych oraz kłębuszków nerkowych (glomerulonep- od warunków pogodowych – bólami występującymi
hritis). Na chorobę zapadają głównie mężczyźni. Klinicznie również w spoczynku oraz nocą, a także upośledze-
stwierdza się różnorodne objawy laryngologiczne (katar, niem ruchomości stawów.
krwawienie z nosa, zapalenie uszu); zajęcie płuc charakte-
ryzuje się bólami w klatce piersiowej, kaszlem, odkrztusza- Zmiany zwyrodnieniowe w obrębie kręgosłupa. Zmia-
niem krwistej wydzieliny. W okresie uogólnienia choroby, ny zwyrodnieniowe kręgosłupa są zlokalizowane przede
w którym zajęte są również inne organy, występują m.in. wszystkim w półścisłych połączeniach (trzon kręgu – krą-
wychudzenie, gorączka, bóle i zapalenie stawowe. żek międzykręgowy – trzon kręgu) lub też stawach łuków
kręgowych. Są one spowodowane nadmiernym zużyciem
w następstwie przeciążenia, choć istotne znaczenie mają
również czynniki wrodzone.
1.5.8.1.6. Pozostałe choroby należące
do kręgu chorób reumatycznych Zmiany w obrębie krążków międzykręgowych określa się
jako chondrozy (zwyrodnienia chrząstki). Rozpoczynają
się one zubożeniem w płyn jądra galaretowatego (jądra
Choroba zwyrodnieniowa stawów (osteoarthrosis, miażdżystego, nucleus pulposus), wskutek czego zmniej-
arthrosis deformans). Tą nazwą obejmuje się zwyrod- sza się jego sprężystość i panujące w nim wewnętrzne ciś-
nieniowe choroby stawów obwodowych. U podłoża nienie. Otaczający je pierścień włóknisty (anulus fibrosus)
ulega rozluźnieniu, może dochodzić do jego naderwania
ich leżą objawy zużycia chrząstki stawowej, np. znie- lub nawet oderwania jego włókien. W następstwie istnie-
kształcenia włókniste lub dziurowate ubytki chrząstki. je niebezpieczeństwo przemieszczenia, uwypuklenia lub
Zawartość w chrząstce siarczanu 4-chondroityny się zaklinowania się jądra miażdżystego – zarówno w obrębie
zmniejsza, zwiększa się natomiast zawartość siarcza- otworów międzykręgowych, jak i w kierunku kanału krę-
nu keratyny. Obok zmian degeneracyjnych chrząstki gowego (wypadnięcie jądra miażdżystego).
Jeśli zwyrodnieniu ulega pokrywająca trzony kręgów
stwierdza się jednoczesne reaktywne zmiany przero- chrząstka, co powoduje czynnościowe usztywnienie pół-
stowe pod postacią skostniałych obwałowań brzeż- ścisłych połączeń i – w następstwie tego procesu – wtórne
nych („dziobów”) i stwardnienia kosmków błony zniszczenie krążków międzykręgowych, mówi się o os-
maziowej. Główka kości w obrębie stawu może przy teochondrozie. Zwyrodnienie stawów z odpowiadający-
MUTSCHLER-2009.indd 274 2010-01-07 22:12:09
Układ nerwowy 275
mi zmianami degeneracyjnymi trzonów kręgów w postaci po potwierdzeniu rozpoznania, tak aby już w fazie
tworzenia osteofitów w kształcie wyrostków i obwałowań początkowej agresywnie zapobiegać postępowi cho-
na brzegach trzonów określa się mianem spondyloz, a pro-
roby.
cesy przebudowy kości i stawów międzykręgowych nazy-
wane są spondyloartrozami. Następujące grupy leków stosowane są w terapii cho-
Układ nerwowy
Objawami klinicznymi zmian degeneracyjnych są bóle roby reumatycznej:
uciskowe, opukowe i przy naciąganiu uszkodzonego obsza-
ru kręgosłupa. Zwiększone napięcie mięśniowe powoduje ■ niesteroidowe leki przeciwzapalne (NLPZ),
nadwichnięcie i przymusowe ustawienia w nieprawidło-
■ glukokortykosteroidy (steroidowe leki przeciwza-
wym położeniu. Przy gwałtownych, niekontrolowanych
ruchach może dochodzić do unieruchomienia „stawów” palne),
kręgosłupa w niekorzystnych pozycjach, a poprzez ucisk
■ tzw. leki podstawowe lub induktory remisji (rów-
na korzonki nerwowe – do silnych stanów bólowych (kręcz
szyi, lumbalgia). Ucisk wypadniętego jądra miażdżystego nież – leki przeciwreumatyczne modyfikujące B1
na nerwy rdzeniowe wywołuje bóle neuralgiczne (neu- przebieg choroby; DMARD – disease modifying
ralgie), parestezje i wypadnięcie odruchów rdzeniowych. antirheumatic drugs),
W związku z budową anatomiczną i nasilonym obciąże-
niem szczególnie często objęte zmianami są: dolny odcinek ■ antybiotyki (w szczególności penicyliny),
kręgosłupa szyjnego oraz lędźwiowego (zespół szyjny i lę-
dźwiowy). Najczęstszym zespołem lędźwiowym jest lum- ■ tzw. związki hamujące degenerację chrząstki,
balgia (postrzał), czyli lumbago, często z towarzyszącą
■ substancje poprawiające ukrwienie do stosowania
neuralgią nerwu kulszowego (ischiasem).
miejscowego.
Zakaźne (infekcyjne) zapalenia stawów. Te postacie za-
paleń stawów powstają w przebiegu zakaźnych (infekcyj-
nych) procesów chorobowych, przy czym z treści pobranej
przy nakłuciu zajętych stawów często udaje się wyhodo- 1.5.8.2.1. Niesteroidowe leki przeciwzapalne
wać drobnoustrój, który wywołał zakażenie. Typowymi (NLPZ )
przykładami są: rzeżączkowe, gruźlicze i kiłowe zapalenia
stawów. Substancje należące do tej grupy leków zostały
już omówione w rozdz. B 1.5.5.2.1. Są one wskazane
we wszystkich zapalnych chorobach reumatycznych
Pozastawowe postacie chorób reumatycznych.
do leczenia objawowego (tłumienie reakcji zapal-
Przez to pojęcie rozumie się bolesne zmiany o cha-
nych, łagodzenie dolegliwości bólowych). Nieste-
rakterze przewlekłych zapaleń lub zwyrodnień wy-
roidowe leki przeciwzapalne nie wpływają jednak
stępujące w tkance łącznej około- lub pozastawowej,
na przebieg choroby. W przypadku utrzymujących
ścięgnach, pochewkach ścięgnistych lub torebkach
się bólów (np. w chorobie Bechterewa, w której sil-
stawowych, okostnej i mięśniach. Należą tu m.in.:
ne bóle ujawniają się często w godzinach nocnych)
bolesne stwardnienie mięśni (myogelosis), zapale-
wskazane jest stosowanie preparatów o długim
nie kaletek stawowych (bursitis), zapalenie ścięgien
okresie działania lub też preparatów typu retard;
i pochewek ścięgnistych (lendinitis, tendovaginitis)
z kolei przejściowe stany bólowe (np. w przypadku
i zapalenie podskórnej tkanki tłuszczowej (pannicu-
porannej bolesnej sztywności stawów) wymagają
litis).
stosowania raczej leków o krótkim okresie półtrwa-
nia. Działania niepożądane NLPZ ze strony układu
pokarmowego występujące w trakcie długotrwałej
1.5.8.2. Leczenie farmakologiczne chorób terapii często powodują jej przerwanie. Dlatego
reumatycznych pacjenci z podwyższonym ryzykiem w zakresie
tego rodzaju działań niepożądanych powinni otrzy-
Podczas gdy jeszcze 15 lat wcześniej wychodzono mywać preparaty złożone – zawierające klasyczne
z założenia, że człowiek nie umiera na reumatyzm, NLPZ wraz z inhibitorem pompy protonowej lub
obecnie wydaje się udowodnione, że reumatoidal- selektywny inhibitor COX-2. Należy zwracać uwa-
ne zapalenie stawów prowadzi jednak do skrócenia gę na podwyższone ryzyko powikłań sercowo-na-
oczekiwanej długości życia. W wyniku zastosowania czyniowych w trakcie terapii koksybami lub za po-
tzw. leków podstawowych (zob. poniżej), które sto- mocą NLPZ.
sowano wcześniej z pewnymi oporami, rokowanie Ponieważ zwyrodnieniowym chorobom reumatycz-
co do długości życia w przebiegu reumatoidalnego nym towarzyszy zwykle proces zapalny – leki z grupy
zapalenia stawów jest prawie niezmienione. Obec- NLPZ są często bardziej skuteczne niż leki o działa-
nie terapię podstawową wprowadza się bezpośrednio niu głównie przeciwbólowym (np. paracetamol).
MUTSCHLER-2009.indd 275 2010-01-07 22:12:09
276 Układ nerwowy
1.5.8.2.2. Glukokortykosteroidy Około 25% wszystkich pacjentów z chorobami
reumatycznymi otrzymuje glukokortykosteroidy.
Ta grupa hormonów kory nadnerczy została szczegó- Stosowane są one miejscowo pod postacią iniekcji
łowo omówiona w rozdziale B 2.7.3. dostawowych, doustnie w leczeniu uderzeniowym
lub pozajelitowo pod postacią leczenia pulsacyjne-
Około 1940 r. amerykański reumatolog Philip Hench do- go. Coraz częściej stosowane są – w małych dawkach
konał zaskakującej obserwacji, że chorzy z reumatoidal- (znacznie mniejszych od dawki progowej Cushinga)
nym zapaleniem stawów wkraczają w stan pewnej remisji,
kiedy chorują na żółtaczkę. To skłoniło go do postawienia – w terapii długoterminowej (zob. tab. B 1.5-13).
hipotezy, że zaobserwowana poprawa wyzwalana jest przez W przypadku przestawiania pacjenta na długotrwa-
tworzoną substancję endogenną. Dlatego też w 1948 r. pod- łe leczenie doustne dawka powinna być zmniejszana
jął on u chorej z ciężkimi reumatycznymi zmianami stawo- stopniowo przez okres kilku tygodni.
wymi próbę leczenia za pomocą odkrytego przed kilkoma Działania niepożądane terapii glukokortykoste-
laty „związku E” (kortyzolu). Sukces był zdumiewający
i przeszedł do historii medycyny jako „cud kortyzonowy”. roidami zostały przedstawione w rozdziale B 2.7.3.
Już jednak w swej pierwszej publikacji Hench wskazywał W zakresie reumatologii najważniejszym objawem
na możliwości i ograniczenia leczenia kortynolem: niepożądanym jest osteoporoza. Jej nasilenie wiąże
się z wielkością stosowanych dawek leków, długoś-
■ zależna od dawki poprawa kliniczna w ciągu kilku dni. cią okresu leczenia, skumulowaną dawką całkowitą
■ szybki nawrót dolegliwości po odstawieniu leczenia, oraz płcią pacjenta (kobiety są częściej dotknięte
chorobą). Dlatego też powinna być prowadzona te-
■ znaczne działania niepożądane przy długotrwałym sto- rapia towarzysząca – polegająca na podawaniu 1000
sowaniu. mg soli wapniowej oraz 400–800 IE witaminy D
dziennie.
Prace Philipa Hencha zostały w 1950 r. uhonorowane Na-
grodą Nobla z dziedziny medycyny. Glukokortykosteroidy nie wykazują bezpo-
średniego działania wrzodotwórczego. Ponieważ
jednak zwiększają o 3–4 razy ryzyko powstania
W dużych dawkach glukokortykosteroidy są wska-
wrzodów występujących przy stosowaniu NLPZ
zane w ostrych rzutach choroby reumatycznej, a tak-
– nie jest wskazane stosowanie leków z obu grup
że w przypadku złośliwych postaci choroby (np. przy
w terapii skojarzonej.
objęciu procesem chorobowym naczyń krwionoś-
nych). Są one w stanie ingerować w procesy regu-
lacyjne niektórych genów prozapalnych i w ten spo-
1.5.8.2.3. Tzw. leki podstawowe
sób hamują syntezę czynników zapalnych, jak rów-
(induktory remisji, leki
nież zapobiegają indukcji cyklooksygenazy COX-2
modyfikujące przebieg choroby;
– co określane jest mianem molekularnego mecha-
DMARD – disease modifying
nizmu działania. Stosowanie dużych dawek powinno
antirheumatic drugs)
być – jeżeli jest to tylko możliwe – ograniczone cza-
sowo, a dawka leku powinna być możliwie szybko
zmniejszona poniżej tzw. progu Cushinga (≤ 7,5 mg W przypadku przewlekłych zapalnych postaci cho-
odpowiednika prednizolonu). rób reumatycznych, szczególnie reumatoidalne-
Tabela B 1.5-13. Metody stosowania glukokortykosteroidów w reumatoidalnym zapaleniu stawów (według Krügera)
a
Sposób podawania Zakres dawki Przykładowe wskazania
długotrwałe leczenie doustne 7,5 mg i mniej chorzy w podeszłym wieku w przypadku
„małymi dawkami” („low-dose”) przeciwwskazań do stosowania NLPZ
doustne leczenie uderzeniowe (krótkotrwałe) dawka początkowa 20–60 mg ostry rzut
parenteralne leczenie pulsacyjne wlew 250–1000 mg – w ciągu bardzo ciężki ostry rzut,
trzech kolejnych dni powikłania zagrażające życiu
miejscowe wstrzyknięcia dostawowe 2–40 mg silny stan zapalny
(zależnie od wielkości stawu) pojedynczych stawów
a
Podane dawki są dawkami dobowymi w przeliczeniu na prednizolon
MUTSCHLER-2009.indd 276 2010-01-07 22:12:09
Układ nerwowy 277
Tabela B 1.5-14. Leki podstawowe i immunosupresyjne stosowane w leczeniu reumatoidalnego zapalenia stawów (uszeregowane
zgodnie z siłą działania)
Lek Siła działania Początek działania
Układ nerwowy
hydroksychlorochina, chlorochina słabo działające 3-6 miesięcy
związki złota doustnie słabo działające 3-6 miesięcy
sulfasalazyna średnio silnie działająca 2-3 miesiące
metotreksat silnie działający 1-2 miesiące
związki złota parenteralnie silnie działające 3-4 miesiące B1
D-penicylamina silnie działająca 3-4 miesiące
azatiopryna silnie działająca 2-3 miesiące
cyklosporyna silnie działająca 1-3 miesiące
cyklofosfamid bardzo silnie działający 1-2 miesiące
go zapalenia stawów, czynione są starania, aby za 1.5.8.2.3.1. Leki działające
pomocą tzw. leków podstawowych spowolnić pro- immunosupresyjnie,
ces chorobowy powodujący uszkodzenie stawów, immunomodulująco
a w idealnym przypadku doprowadzić nawet do re- i immunobiologicznie stosowane
misji. Leki podstawowe nie znajdują natomiast za- w terapii podstawowej
stosowania w chorobach reumatycznych typu zwy-
rodnieniowego. Ponieważ te substancje nie mają
Leki immunosupresyjne i immunomodulujące zo-
działania przeciwbólowego, a zwykle też przeciw-
staną dokładnie omówione w rozdziałach B 13.3
zapalnego – muszą być stosowane łącznie z NLPZ.
i 13.4. Z wyjątkiem leflunomidu i leków immunobio-
Wybór poszczególnych substancji lub leków zło-
logicznych – substancji tych nie stosowano pierwot-
żonych warunkowany jest rozmaitymi czynnikami
nie w leczeniu chorób reumatycznych; zostały one
chorobowymi, takimi jak nasilenie i przebieg choro-
wprowadzone do terapii innych schorzeń. Do terapii
by; pod uwagę należy wziąć również możliwe prze-
reumatoidalnego zapalenia stawów zostały wprowa-
ciwwskazania dotyczące poszczególnych leków.
dzone na podstawie założeń patofizjologicznych po-
Ważnym czynnikiem wymagającym uwzględnienia
twierdzających, iż schorzenia z kręgu choroby reu-
jest ponadto latencja wystąpienia działania (okres
matycznej są zaburzeniami autoimmunologicznymi.
utajenia między początkiem zażywania leku a ujaw-
Lekami z tej grupy są:
nieniem się efektów jego działania), gdyż wszystkie
leki podstawowe (z wyjątkiem leków biologicznych ■ metotreksat,
– zob. poniżej) rozpoczynają działanie nie od razu,
■ leflunomid,
lecz dopiero po wielu tygodniach i miesiącach sto-
sowania (zob. tab B 1.5-14). ■ cyklosporyna,
Określenie „leki podstawowe” nie powinno jed-
■ leki immunobiologiczne (tzw. biologicals, antago-
nak prowadzić do bezkrytycznego stosowania tych
niści receptorów TNF-α i in.),
substancji, gdyż przynajmniej część z nich wykazuje
znaczące działania niepożądane. Dla zastosowania ■ azatiopryna.
tych leków konieczną przesłanką jest potwierdzo-
ne rozpoznanie oraz udowodniony postęp choroby. Metotreksat. Metotreksat – antagonista kwasu folio-
Ich stosowanie winno być zastrzeżone dla doświad- wego uważany jest obecnie za lek pierwszego wyboru
czonych reumatologów. wśród leków podstawowych. Również w prowadze-
MUTSCHLER-2009.indd 277 2010-01-07 22:12:09
278 Układ nerwowy
7,5–15 mg (w pojedynczej dawce), a w przypadku
długotrwałego stosowania – 25 mg; podane dawki
CF3
są znacząco mniejsze od stosowanych w terapii prze-
O ciwnowotworowej.
NH
Ważnymi działaniami niepożądanymi są nudności,
N
wymioty, zapalenie skóry, zapalenie śluzówki jamy
O CH3 ustnej, zwiększenie poziomu transaminaz, zapalenie
płuc i zwiększona zapadalność na schorzenia zapal-
leflunomid
ne. Ponadto metotreksat jest teratogenny.
W celu zmniejszenia wątrobowych i żołądko-
wo-jelitowych działań niepożądanych należy dzień
po podaniu metotreksatu zaaplikować pacjentowi
CF3 kwas foliowy w dawce 5 mg.
O
N C
NH Leflunomid. Stworzony jako lek do terapii podsta-
HO CH3 wowej reumatoidalnego zapalenia stawów leflunomid
– będący pochodną izoksazolu – nie jest strukturalnie
aktywny metabolit podobny do innych immunomodulatorów. W wyniku
otwarcia pierścienia izoksalowego powstaje aktyw-
ny metabolit blokujący syntezę pirymidyny de novo
– w procesie odwracalnego zahamowania aktywno-
ści enzymu dehydrogenazy kwasu dihydroorotowego
niu skojarzonej terapii podstawowej – metotreksat (DHODH). Aktywowane limfocyty T są silniej – niż
jest substancją najczęściej stosowaną. Jego zaletą inne komórki – dotknięte zahamowaniem syntezy pi-
jest z jednej strony szybsze działanie – w porówna- rymidyny, gdyż przed rozpoczęciem procesu ich pro-
niu z innymi lekami podstawowymi (zob. tab. 1.5- liferacji poziom pirymidynowych nukleotydów musi
14; z wyraźnym działaniem należy liczyć się po 1–2 ulec ok. 8-krotnemu podwyższeniu. To podwyższone
miesiącach stosowania; pełne działanie ujawnia się zapotrzebowanie na pirymidynę nie może być pokry-
po 3–5 miesiącach), a z drugiej strony – prosty sche- te przy wykorzystaniu tzw. drogi ratunkowej (salvage
mat dawkowania. pathway) – przez tworzenie nukleotydów z 5-fosfo-
Metotreksat zmniejsza proliferację limfocytów rybozylo-1-difosforanu oraz ponownego wykorzysta-
oraz hamuje tworzenie czynników reumatycznych. nia odpowiednich zasad (recykling pirymidyn), lecz
Ponadto obniża syntezę cytokin oraz hamuje prze- wymaga nowej (de novo) syntezy pirymidyn.
chodzenie wielojądrzastych leukocytów z krwi Ze względu na długi okres półtrwania aktywnych
do tkanek. metabolitów (ok. 15 dni) – przez pierwsze trzy dni
Metotreksat wiązany jest w 50–60% z białkami stosuje się lek w dawce początkowej 100 mg/dzień.
osocza. Wolna część leku ulega przemieszczeniu Jako dawka podtrzymująca zalecanych jest 10–20
do wnętrza komórek – przy wykorzystaniu układu mg/dzień.
transportera kwasu foliowego; w komórce ulega Działaniami niepożądanymi są głównie biegun-
procesowi poliglutaminoilacji (wiązanie z resztami ki, nudności, łysienie, alergie oraz zwyżki ciśnienia
kwasu glutaminowego). Powstające poliglutaminia- krwi. Dlatego też ciśnienie krwi musi być skontro-
ny ulegają gromadzeniu wewnątrz komórek. Stąd też lowane przed rozpoczęciem terapii, a następnie mie-
działania niepożądane utrzymują się nawet po odsta- rzone w regularnych odstępach czasu w trakcie lecze-
wieniu leku. Eliminacja leku odbywa się w nerkach nia leflunomidem. Pojawiają się rzadkie doniesienia
w wyniku przesączania kłębuszkowego i czynnego o ciężkich uszkodzeniach wątroby, w związku z czym
wydalania. Związki kwasowe należące do grupy nie- leflunomid nie powinien być stosowany u pacjentów
steroidowych leków przeciwzapalnych zmniejszają z niewydolnością wątroby. Bardzo rzadko obserwo-
w zależności od dawki klirens nerkowy metotreksatu, wano zespół Stevensa-Johnsona. Aktywny metabo-
który również jest kwasem, a tym samym zwiększają lit leflunomidu jest inhibitorem CYP2C9. A zatem
jego toksyczność. również substancje metabolizowane przez CYP2C(
Metotreksat może być stosowany zarówno doust- (np. frenprokumon) – w przypadku równoczesnego
nie, jak i pod postacią iniekcji domięśniowej. Tygo- podania leflunomidu – będą rozkładane dużo wolniej.
dniowa dawka metotreksatu w przypadku jego stoso- W badaniach eksperymentalnych na zwierzętach
wania jako leku podstawowego wynosi początkowo – leflunomid wykazuje działanie teratogenne, wyko-
MUTSCHLER-2009.indd 278 2010-01-07 22:12:10
Układ nerwowy 279
rzystanie go w terapii u kobiet wymaga stosowania do zakażeń, nadciśnienie tętnicze, nadmiernie owło-
skutecznej antykoncepcji. sienie, przerost dziąseł, drżenia mięśniowe (tremor),
Łączne stosowanie leflunomidu z metotreksatem zaburzenia czucia (parestezje) i nudności/wymioty.
jest możliwe – wówczas terapia wpływa nie tylko Ponadto dyskutuje się nad problemem, w jakim stop-
Układ nerwowy
na syntezę pirymidyn, lecz także puryn. niu cyklosporyna może zwiększyć ryzyko powstania
nowotworu.
Cyklosporyna. Lek ten był początkowo przezna-
czony jako środek immunosupresyjny w medycynie Leki immunobiologiczne (biologicals). Stwierdzane
transplantacyjnej, jednak już od dobrych kilku lat u pacjentów z reumatoidalnym zapaleniem stawów
jest dopuszczony do stosowania w terapii reumato- podwyższone stężenie TNF-α (tumor necrosis factor
idalnego zapalenia stawów. W wyniku hamowania – czynnik martwicy guza), który może prowadzić
czynnika transkrypcyjnego NF-AT cyklosporyna do zapalenia błony maziowej i zniszczenia stawu, B1
blokuje głównie syntezę interleukiny-2 i w ten spo- było przesłanką do podjęcia starań w kierunku opra-
sób działa przeciwzapalne. Cyklosporynę stosuje się cowania strategii terapeutycznych działających bez-
głównie u pacjentów z ciężkim przebiegiem reuma- pośrednio przeciwko tej cytokinie. Ponieważ TNF-α
toidalnego zapalenia stawów, którzy w niewystar- reguluje także produkcję i wydzielanie IL-1 i IL-6
czającym stopniu reagują na leczenie metotreksatem – w wyniku blokady TNF-α można zahamować za-
– w terapii skojarzonej z tym lekiem w dawkach równo tworzenie IL-1 i IL-6, jak i migrację leukocy-
dziennych 2,5–3 mg/kg m.c. tów oraz ekspresję cząsteczek adhezyjnych (zob. ryc.
Wśród działań niepożądanych należy brać pod B 1.5-22).
uwagę głównie zaburzenia funkcji nerek (w dużych
dawkach cyklosporyna działa nefrotoksycznie), za- TNF-α wykazuje swoje działanie poprzez dwa różne
burzenia funkcji wątroby, zwiększoną skłonność receptory TNF (p55 o masie 55 kDa i p75 o masie
TNF-α
(z makrofagów)
hemokiny śródbłonkowe cytokiny:
cząsteczki adhezyjne: IFN-γ
(z makrofagów E-selektyna IL-1
i komórek śródbłonka) ICAM-1 IL-6
VCAM-1 TNF-α
(z makrofagów, komórek T,
(z komórek śródbłonka) fibroblastów)
wwędrowywanie aktywacja resorpcja białka ostrej
leukocytów kości fazy
(osteoblasty), (z hepatocytów)
proliferacja/
rozplem
przewlekły proces zapalny,
zapalenie błony maziowej (synovitis)
zniszczenie chrząstki stawowej i kości
Ryc. B 1.5-22. Kluczowe znaczenie prozapalnej cytokiny TNF-α w patogenezie reumatoidalnego zapalenia stawów.
MUTSCHLER-2009.indd 279 2010-01-07 22:12:10
280 Układ nerwowy
75 kDa), które znajdują się na powierzchni niemal świąd) oraz lekkie do średnio ciężkich zakażenia dróg
wszystkich komórek, jak również w formie rozpusz- oddechowych (katar, zapalenie gardła, kaszel). U oko-
czonej w osoczu. Rozpuszczalne receptory TNF speł- ło 10% pacjentów dochodzi do powstania przeciwciał
nieją funkcję naturalnych regulatorów aktywności IgG lub IgM skierowanych przeciw dwuniciowemu
TNF – poprzez wiązanie nadmiaru TNF-α. Ta natu- DNA (zob. przeciwciała przeciwjądrowe w toczniu
ralna regulacja ulega jednak zaburzeniu w przypad- rumieniowatyn układowym). Jak dotychczas żaden
ku pacjentów z reumatoidalnym zapaleniem stawów. z pacjentów nie miał jednak klinicznych objawów
Wynikiem tego rodzaju rozważań było zsyntetyzo- tocznia. Ponieważ ujawniły się ciężkie powikłania
wanie na drodze biologicznej różnych „antagonistów zakaźne (np. gruźlica) – przed zastosowaniem bloke-
TNF-α”, i wykorzystanie ich w terapii reumatoidal- rów czynnika TNF-α należy wykonać zarówno próbę
nego zapalenia stawów. Są to: tuberkulinową, jak i zdjęcie przeglądowe klatki pier-
siowej. Obserwowano pojedyncze przypadki pancy-
■ etanercept,
topenii i niedokrwistości aplastycznej.
■ infliksymab, Etanercept jest przeciwwskazany u pacjentów
z posocznicą oraz jawnymi zakażeniami, przewlekłą
■ adalimumab.
niewydolnością serca, chorobami demielinizacyjny-
mi i nowo stwierdzonymi chłoniakami.
Ponadto w terapii reumatoidalnego zapalenia stawów
dostępne są dwa inne leki immunobiologiczne: Infliksymab. Infliksymab jest stworzonym na dro-
dze rekombinacji, chimerycznym (mysz/człowiek)
■ anakinra (antagonista receptora IL-1),
przeciwciałem monoklonalnym (masa cząsteczko-
■ rytuksymab (przeciwciało przeciw antygenowi po- wa ok. 149 kDa), które jest skierowane przeciw
wierzchniowemu CD20). TNF-α. Stała część przeciwciała składa się z ludzkiej
immunoglobuliny IgG1, a obszar zmienny wywodzi
W przypadku wszystkich leków immunobiologicz- się z przeciwciała mysiego. Infliksymab hamuje ak-
nych panuje zasada, że z przyczyn finansowych po- tywność biologiczną TNF-α na drodze swoistego
winny być one stosowane dopiero po 6 miesiącach wiązania zarówno wolnego, jak i związanego z błoną
nieskutecznej lub też mało skutecznej terapii innymi komórkową TNF-α. Ponadto dochodzi do niszczenia
lekami podstawowymi. komórek TNF-α – co warunkowane jest działaniem
dopełniacza.
Etanercept. Etanercept jest dimerycznym białkiem Wskazania lecznicze są podobne do wskazań
fuzyjnym, które składa się z zewnątrzkomórkowej, etanerceptu. Pewnym problemem ujawniającym się
wiążącej ligand części ludzkiego receptora p75-TNF
oraz obszaru Fc ludzkiej immunoglobuliny IgG1 (zob.
ryc. B 1.5-23). Został stworzony na drodze rekombina-
cji – złożony jest z 934 aminokwasów i posiada masę
cząsteczkową równą 150 kDa. Jest rozpuszczalnym re- zewnątrzkomórkowa
ceptorem TNF, który w sposób swoisty wiąże TNF-α domena receptora p75
i w ten sposób zapobiega wiązaniu tej cytokiny z re-
ceptorami znajdującymi się na powierzchni komórek.
SS
SS
Etanercept jest wskazany u pacjentów z reumato- SS
idalnym zapaleniem stawów o nasilenia objawów S S
od umiarkowanego do znacznego, u których leki S S region Fc
podstawowe okazały się niewystarczające. Może być immunoglobuliny
stosowany w monoterapii lub w skojarzeniu z meto- IgG1
S S
treksatem. Prowadzi do szybko ujawniającej się, wy-
S S
raźnej poprawy w zakresie objawów klinicznych.
Okres półtrwania to 100–150 godzin.
Średnia dawka wynosi 25 mg (podskórnie) – poda-
wana dwa razy w tygodniu. etanercept
Częstymi działaniami niepożądanymi (ok. 40%)
są reakcje w miejscu wstrzyknięcia (ból, obrzęk, Ryc. B 1.5-23. Schematyczna struktura etanerceptu.
MUTSCHLER-2009.indd 280 2010-01-07 22:12:10
Układ nerwowy 281
w przypadku wielokrotnego stosowania infliksyma- IL-1. IL-1 jest ważną cytokiną o działaniu prozapal-
bu jest powstawanie przeciwciał przeciw temu leko- nym, której stężenie zwiększa się u pacjentów z reu-
wi. Większe dawki infliksymabu lub też dodatkowa matoidalnym zapaleniem stawów w czynnych fazach
immunosupresja za pomocą metotreksatu zapobie- choroby – zarówno w osoczu, jak i w płynach mazio-
Układ nerwowy
gają jednak niemal całkowicie tworzeniu się prze- wych. Ponadto u pacjentów tych dochodzi do zmniej-
ciwciał. szenia stężenia endogennych antagonistów recepto-
Czas półtrwania wynosi 9–10 dni. rów IL-1.
Obecnie infliksymab stosuje się jedynie w terapii Okres połowiczej eliminacji wynosi 4–6 godzin.
skojarzonej z metotreksatem w dawkach 3–10 mg/kg Anakinra wskazana jest w terapii reumatoidalnego
m.c. podawanych we wlewie w punkcie czasowym 0, zapalenia stawów – w skojarzeniu z metotreksatem.
a następnie 2 i 6 tygodni później; w przypadku terapii W sumie działanie anakinry wydaje się słabsze niż
podtrzymującej lek podawany jest co 8 tygodni. blokerów TNF-α. B1
Jako działania niepożądane opisano miejscowe Dawkowanie wynosi 100 mg podawanych pod-
i uogólnione reakcje poinfuzyjne (świąd, zaczerwie- skórnie raz dziennie.
nienie twarzy, pokrzywka, gorączka, uczucie zmę- Najczęściej obserwowanym działaniem niepożą-
czenia, bóle w klatce piersiowej, nudności), a tak- danym (ok. 70% przypadków) jest łagodna do umiar-
że lekkie do średnio ciężkich zakażenia górnych dróg kowanej reakcja w miejscu iniekcji (zaczerwienienie,
oddechowych. Innymi działaniami niepożądanymi zapalenie, ból). Bardzo często ujawniają się bóle gło-
są: nadciśnienie tętnicze, sztywność mięśni (rigor), wy. Obserwowane były również zmiany obrazu krwi
wysypki skórne i wypryski. U niewielkiego odsetka (neutropenie) i zakażenia – jako możliwe skutki dzia-
pacjentów, którzy dodatkowo otrzymywali leki im- łania immunosupresyjnego.
munosupresyjne, ujawniają się chłoniaki. Ponieważ Jak dotąd nie opisano interakcji z NLPZ, gluko-
– podobnie jak przy etanercepcie – istnieje niebezpie- kortykosteroidami lub innymi lekami podstawo-
czeństwo powikłań zakaźnych (np. gruźlica), przed wymi. Należy jednak ostrzec przed równoczesnym
włączeniem infliksymabu konieczne jest przeprowa- stosowaniem etanerceptu i anakinry – w związku
dzenie podobnych badań dodatkowych (zob. powy- ze zwiększonym ryzykiem wystąpienia neutropenii
żej). Ponadto opisano pojedyncze przypadki pancyto- oraz ciężkich infekcji; skądinąd terapia skojarzona
penii i niedokrwistości aplastycznej. wymienionymi substancjami nie przynosiła żadnej
Przeciwwskazania dotyczące stosowania infliksy- dodatkowej korzyści terapeutycznej w porównaniu
mabu odpowiadają tym dotyczącym etanerceptu. z monoterapią etanerceptem.
Adalimumab jest całkowicie ludzkim monoklonal- Rytuksymab jest monoklonalnym chimerycznym
nym przeciwciałem skierowanym przeciw TNF-α, ludzko-mysim przeciwciałem skierowanym prze-
które w przeciwieństwie do infliksymabu nie wyka- ciwko antygenowi powierzchniowemu CD20, które
zuje działania indukującego powstawanie przeciw- wytwarzane było początkowo dla terapii chłonia-
ciał. Lek został dopuszczony do stosowania w terapii ków nieziarniczych dodatnich antygenowo w za-
skojarzonej z metotreksatem lub też w monoterapii kresie CD20. Ostatnio rytuksymab – w skojarzeniu
reumatoidalnego zapalenia stawów – u pacjentów, z metotreksatem (10–25 mg raz w tygdniu) – został
którzy nie zareagowali pozytywnie na leczenie inny- dopuszczony do stosowania w terapii dorosłych
mi lekami podstawowymi, włącznie ze stosowanym pacjentów z ciężką postacią reumatoidalnego zapa-
samodzielnie metotreksatem. lenia stawów, u których nie udało się osiągnąć od-
Okres półtrwania został oszacowany na 14 dni. powiedniej poprawy w wyniku zastosowania innych
Dawkowanie wynosi 40 mg podawanych podskór- leków podstawowych, włącznie z jedną lub kilkoma
nie co 2 tygodnie. terapiami za pomocą inhibitorów TNF-α, bądź te
Działania niepożądane i przeciwwskazania odpo- wcześniejsze terapie nie były przez pacjenta tole-
wiadają tym dotyczącym infliksymabu. rowane. Rytuksymab eliminuje selektywnie komór-
ki (limfocyty) B, na których powierzchni obecny
Anakinra – stworzony na drodze rekombinacji anta- jest antygen CD20. Ponieważ komórki te odgrywają
gonista receptora ludzkiej interleukiny IL-1 – jest główną rolę w procesie autoimmunologicznym leżą-
po etanercepcie, infliksymabie i adalimumabie cym u podstaw reumatoidalnego zapalenia stawów
– czwartą znajdująca się w handlu „bilogiczną sub- (zob.: ryc. B 1.5-21), rytuksymab hamuje zarówno
stancją hamującą cytokiny”. W sposób kompetycyjny produkcję czynników reumatycznych, jak i prezen-
hamuje wiązanie interleukiny-1 (IL-1) z receptorem tację antygenu, a tym samym aktywację dopełniacza
MUTSCHLER-2009.indd 281 2010-01-07 22:12:11
282 Układ nerwowy
w przewlekłym zapalnym procesie immunologicz- (ICAM-1, VCAM-1) niezbędnych do przylgnięcia
nym RZS. (adhezji) leukocytów do śródbłonka ścian naczy-
Okres półtrwania jest bardzo zmienny i zdaje się niowych. Przylgnięcie do śródbłonka naczyniowego
zależeć m.in. od dawki. Dla typowych dawek stoso- jest z kolei podstawowym warunkiem migracji leuko-
wanych w terapii reumatoidalnego zapalenia stawów cytów z wnętrza naczynia do otaczającej tkanki. Je-
(zob. poniżej) – wynosi przeciętnie 20 dni. śli opuszczające krwiobieg leukocyty utkną w błonie
Cykl leczenia w przypadku reumatoidalnego zapa- maziowej, nasila się tu uwalnianie z nich mediatorów
lenia stawów składa się z dwóch dożylnych iniekcji procesu zapalnego. Zgodnie z przedstawionymi po-
po 1000 mg rytuksymabu podanych w odstępie 2 ty- glądami pod wpływem leczenia złotem obserwuje się
godni. W badaniach klinicznych nad dopuszczeniem obniżenie poziomów interleukiny-1.
leku większość pacjentów otrzymała w ciągu 6–12 Działanie ujawnia się zwykle dopiero po 3–4 mie-
miesięcy kolejny cykl leczenia. siącach; po ustąpieniu objawów terapia powinna być
Najczęstszymi działaniami niepożądanymi były kontynuowana przynajmniej przez pół roku do roku.
ostre reakcje poinfuzyjne, takie jak wzrost ciśnienia W związku z nasilonymi działaniami niepożąda-
krwi, wysypka skórna, świąd i dreszcze. Częstość nymi obecnie praktycznie nie ustawia się już żadnego
i nasilenie tych reakcji można było zmniejszyć do- pacjenta de novo na preparaty złota.
żylnym podaniem 100 mg metyloprednizolonu (30 Terapię prowadzi się na drodze podaży parente-
minut przed infuzją rytuksymabu). Ponadto występo- ralnej za pomocą zwiększającej się dawki: w pierw-
wały zakażenia dróg moczowych, górnych i dolnych szym tygodniu podaje się 20 mg, w drugim – 30
dróg oddechowych, skurcze oskrzeli, dyspepsja oraz mg, a w trzecim tygodniu – 40 mg, później 100 mg
rzadko obrzęk naczyniowo-nerwowy. co tydzień; lek podaje się głęboko domięśniowo (w
mięsień pośladkowy). Podawanie złota utrzymuje się
Azatiopryna jest prolekiem i ulega prawie całkowi- do uzyskania istotnej poprawy – dawka całkowita
tej biotransformacji do 6-merkaptopuryny. Działanie nie powinna przekroczyć 1600 mg aurotiojabłczanu
jest porównywalne z metotreksatem. Ze względu sodu. Po uzyskaniu działania dawka podtrzymująca
na bardziej niekorzystną relację między korzyścią wynosi 100 mg miesięcznie lub – alternatywnie – 50
a ryzykiem azatiopryna utraciła swoje znaczenie mg co dwa tygodnie.
i obecnie w reumatologii jest lekiem o charakterze Preparaty złota podawane doustnie działają znacz-
rezerwowym. nie słabiej od preparatów parenteralnych. Dawkowa-
Dawka dzienna wynosi 1–3 mg/kg m.c. nie auranofiny wynosi 6(–9) mg/dzień.
U 30% leczonych preparatami złota należy się
liczyć z możliwością ujawnienia się działań niepo-
1.5.8.2.3.2. Związki złota żądanych. Dotyczą one głównie krwi (leukopenia
i trombocytopenia), skóry (rumień, zapalenia skóry)
Związki złota należą do najbardziej skutecznych le- i śluzówki (zapalenie śluzówki jamy ustnej, zapale-
ków podstawowych (zob. tab. B 1.5-14). U niemałej nie dziąseł), wątroby (martwica komórek wątroby,
części chorych można za ich pomocą przez pewien zastoinowe zapalenie wątroby), oraz nerek (krwio-
czas uzyskać zatrzymanie – zwykle postępującego mocz, białkomocz). W przypadku wystąpienia tego
– procesu chorobowego. rodzaju objawów terapia musi być niezwłocznie
Do stosowania wprowadzone zostały jednowar- przerwana. Ciężkie komplikacje związane z zatru-
tościowe związki złota: aurotiojabłczan sodowy ciem złotem leczy się za pomocą wersenianu so-
– podawany pozajelitowo oraz auranofina. dowo-wapniowego, dimerkaprolu lub D-penicyla-
Mechanizm działania jest złożony. Jednowartoś- miny.
ciowe złoto posiada wysokie powinowactwo do grup
tiolowych (np. w cysteinie). Niektóre prozapalne
czynniki transkrypcji (np. AP-1, NF-κB) posiadają 1.5.8.2.3.3. Salazosulfapirydyna
wewnątrz swej dodatnio oddziałującej domeny wią- (sulfosalazyna)
żącej DNA reszty cysteinowe – łączące się z solami
złota (I). Związanie z jednowartościowym złotem Stosowana często u pacjentów z wrzodziejącym za-
zapobiega łączeniu się AP-1 i NF-κB z odpowied- paleniem jelita grubego i z chorobą Crohna oraz ma-
nimi sekwencjami DNA. Zgodnie z tym mecha- jąca udowodnioną skuteczność w tych jednostkach
nizmem można wykazać zahamowanie tworzenia chorobowych salazosulfapirydyna (sulfasalazyna)
śródbłonkowych (endotelialnych) białek adhezyjnych jest obecnie ponownie zalecana do leczenia reuma-
MUTSCHLER-2009.indd 282 2010-01-07 22:12:11
Układ nerwowy 283
toidalnego zapalenia stawów jako lek podstawowy, dających 100 g chlorochiny). Podane wartości odpo-
po tym jak już w przeszłości była stosowana w tym wiadają dawce dziennej 250 mg fosforanu chloro-
wskazaniu, a następnie została z niego wycofana. chiny (równe 155 mg chlorochiny) stosowanej przez
Mechanizm działania nie jest jasny. Zostało opi- okres 2 lat.
Układ nerwowy
sane, że sulfasalazyna hamuje aktywację czynnika Jako działania niepożądane ujawniają się wy-
transkrypcyjnego NF-κB. sypki skórne, dolegliwości żołądkowo-jelitowe,
Działanie ujawnia się już po 8–12 tygodniach. siwienie włosów, neuropatie i zaburzenia widzenia
Jest lekiem o średnio silnym działaniu i posiada pod postacią odwracalnego porażenia akomodacji
względnie dobrą relację między korzyściami a ryzy- i zmętnienia rogówki, jak również zwykle nieod-
kiem – dlatego może być stosowana zarówno w mo- wracalnych, ale skądinąd rzadko występujących re-
noterapii, jak i w terapii skojarzonej z innymi lekami tinopatii – powstających w wyniku odkładania się
podstawowymi. substancji czynnej w siatkówce. Stąd też konieczne B1
Dawkuje się w sposób stopniowo narastający jest przeprowadzanie w trakcie terapii chlorochiną
do utrzymywanej przez dłuższy czas dawki 2 g/dobę. regularnych kontroli okulistycznych w odstępach
Możliwe jest zwiększenie dawki do 3 g. 4–6 miesięcy.
Działania niepożądane są zależne od dawki: osoby Hydroksychlorochina jest preparatem analogo-
z wolnym przebiegiem procesu acetylacji są bardziej wym (dawka początkowa 400–600 mg/dzień; dawka
dotknięte ryzykiem wystąpienia objawów niepożą- podtrzymująca 200–400 mg/dzień), która jest nieco
danych, z których najczęstszymi są: nudności, wy- lepiej tolerowana niż chlorochina.
mioty, bóle i zawroty głowy, , oligospermia, objawy
alergiczne i immunologiczne, a także zaburzenia he-
matologiczne. W pojedynczych przypadkach może 1.5.8.2.3.5. D-penicylamina
wystąpić również agranulocytoza.
D-penicylamina – powstający w wyniku rozpadu pe-
nicylin, a obecnie syntetycznie uzyskiwany D-amino-
1.5.8.2.3.4. Hydroksychlorochina kwas – służyła początkowo jako środek chelatujący
i chlorochina (wiążący) w zatruciach metalami ciężkimi, jak rów-
nież w przewlekłym spichrzeniu miedzi (chorobie
Chlorochina i hydroksychlorochina wskazane są do Wilsona. Dopiero później zostało odkryte, że sub-
stosowania wyłącznie w reumatoidalnym zapaleniu stancja ta jest także skuteczna w reumatoidalnym za-
stawów o powolnym postępie choroby. Okres utaje- paleniu stawów.
nia między rozpoczęciem zażywania a ujawnieniem Podobnie jak inne merkaptany, D-penicylamina
się działania terapeutycznego jest względnie długi, potrafi rozkładać cząsteczki makroglobulin, a więc
a skuteczność – wyraźnie słabsza niż metotreksatu również i czynniki reumatyczne – co następuje w wy-
(zob. tab. 1.5-14). Stąd też obie substancje rzadko sto- niku rozszczepiania wewnątrzcząsteczkowych most-
sowane są w monoterapii; ich znaczenie w zakresie ków dwusiarczkowych. Wykazuje ona też działanie
terapii skojarzonej bywa natomiast coraz większe. supresyjne na mezenchymę, to znaczy upośledza
Dyskutowanym mechanizmem działania jest m.in. tworzenie tkanki łącznej wskutek blokowania po-
stabilizowanie błon lizosomalnych oraz hamowanie łączeń tworzących włókna białek prekursorowych,
aktywności enzymów lizosomalnych. Jednym z ta- jak również syntezy hydroksyprołiny. Na poziomie
kich enzymów jest kwaśna sfingomielinaza, która molekularnym D-penicylamina może tworzyć dime-
odgrywa rolę w kaskadzie przekazywania (trans- ry z resztami cysteinowymi domen wiążących AP-1-
dukcji) sygnału między receptorami TNF-α a akty-
wacją NF-κB.
Dawki chlorochiny podawane są w przeliczeniu
na masę ciała – co pozwala na zminimalizowanie ry-
zyka specyficznych działań niepożądanych (retinopa- SH COOH
tia; zob. poniżej) pojawiających się w trakcie terapii H3C
długoterminowej. Dzienna dawka nie powinna prze- CH3 NH2
kroczyć 4 mg fosforanu chlorochiny (co odpowiada D-penicylamina
2,5 mg czystej zasady) na kg masy ciała. Ponadto
skumulowana dawka u osoby dorosłej nie powinna
przekroczyć 160 g fosforanu chlorochiny (odpowia-
MUTSCHLER-2009.indd 283 2010-01-07 22:12:11
284 Układ nerwowy
DNA – i w ten sposób hamuje aktywność tego „poza- się przypadki reakcji anafilaktycznych i innych reak-
palnego” czynnika transkrypcyjnego. Jej skuteczność cji alergicznych, część tych preparatów została wy-
jest podobnie silna jak związków złota stosowanych cofana z rynku.
na drodze pozajelitowej. W związku z niekiedy ujaw- Sól sodowa kwasu hialuronowego pozyskiwana
niającymi się ciężkimi działaniami niepożądanymi z grzebienia kogutów oraz półsiarczan glukozaminy
(zob. poniżej) D-penicylamina stosowana jest zasad- zostały jednak dopuszczone do stosowania w terapii
niczo jedynie w przypadku bardzo wyraźnych wska- zmian zwyrodnieniowych stawów. Ich skuteczność
zań – jako preparat rezerwowy. (zob. powyżej) nie została jednak w pełni udowod-
Dawkowanie powinno być stopniowo zwiększane niona.
(dawka w pełni działająca – 600 mg/dzień).
Najważniejszymi działaniami niepożądanymi są:
zapalenie skóry, zapalenie śluzówki jamy ust- 1.5.8.2.6. Leki przeciwreumatyczne
nej, nudności, biegunka, zastój żółci, białkomocz, do stosowania miejscowego
krwiomocz, eozynofilia, cytopenie, polineuropatie,
zaburzenia smaku, gorączka oraz zaburzenia auto- Niesteroidowe leki przeciwzapalne – oprócz stoso-
immunologiczne (np. miastenia, toczeń rumienio- wania ogólnego – można stosować również miej-
waty uogólniony). scowo, np. pod postacią maści, żelów lub aerozoli;
D-penicylamina jest przeciwwskazana w przypad- przesłanką stosowania innych dróg aplikowania le-
ku chorych z wcześniej istniejącym uszkodzeniem ków jest zmniejszenie działań niepożądanych – wy-
nerek i zaburzeniami w obrębie układu krwiotwór- stępujących przy ogólnym podawaniu. Wskazaniami
czego, a także w czasie ciąży. są zapalenia ścięgien i pochewek ścięgnistych, pro-
cesy zapalne i zmiany zwyrodnieniowe stawów, a
także tępe urazy mechaniczne. Wnikanie substancji
1.5.8.2.3.4. Antybiotyki działającej do głębszych warstw tkanek ma jednak
zmienny charakter. Stąd też u znaczącej części pa-
W przebiegu (ostrej) gorączki reumatycznej – cjentów niesteroidowe leki przeciwzapalne osiąga-
w związku z etiologią paciorkowcową – wskazane ją w tkankach niewielkie stężenia. Stężenia w oso-
jest zastosowanie penicylin o wąskim zakresie dzia- czu znajdują się na znacznie niższym poziomie niż
łania lub w przypadku istnienia nietolerancji na pe- w przypadku stosowania tych samych dawek poda-
nicylinę – antybiotyków makrolidowych. Początko- wanych doustnie (< 5%). Stąd też nie trzeba obawiać
wa dawka dzienna dla dorosłych wynosi 1 Mega (= 1 się działań niepożądanych (zob. powyżej) występu-
milion IE). Po uzyskaniu poprawy dawka leku może jących przy ogólnym podawaniu niesteroidowych
zostać zmniejszona. leków przeciwzapalnych; czasem jednak występują
Leczenie za pomocą penicylin trzeba prowadzić objawy miejscowe (alergiczne reakcje skórne, świąd,
całymi latami – najlepiej korzystając z preparatów zaczerwienienie skóry). Pomimo ich bardzo częstego
typu depot. stosowania skuteczność lecznicza tych preparatów
Również w przypadku infekcyjnych zapaleń sta- jest oceniana krytycznie.
wów wskazane jest stosowanie antybiotyków.
Preparaty znajdujące się na rynku: diklofenak, hy-
droksyetylosalicylan, indometacyna, ibuprofen, keto-
1.5.8.2.5. Substancje tzw. hamujące profen i piroksykam.
proces degeneracji chrząstki
stawowej (chondroprotectiva) Innymi środkami stosowanymi miejscowo w leczeniu prze-
ciwreumatycznym są substancje wywołujące przekrwienie
W przypadku schorzeń stawowych zwykle dochodzi (olejki eteryczne, estry kwasu nikotynowego lub salicylo-
do przedwczesnego zużycia i zwyrodnienia chrząstki wego). W związku ze swym działaniem rozszerzającym
naczynia wywołują one zaczerwienienie (przekrwienie)
stawowej. Dlatego w celu zatrzymania tego procesu odpowiedniego obszaru skóry z towarzyszącym uczuciem
albo nawet uzyskania regeneracji chrząstki – podej- ciepła (tzw. rubefacientia). W jakim stopniu preparaty
mowane są próby polegające na wykorzystaniu pre- z tego rodzaju działaniem – oprócz efektu psychologiczne-
paratów zawierających wyciągi kostne, estry kwasu go – rzeczywiście wykazują skuteczność antyreumatyczną,
siarkowego z mukopolisacharydami lub siarczan trudno ocenić, ponieważ niemożliwe jest przeprowadzenie
kontrolowanych badań z użyciem placebo (zaczerwienie-
D-glukozaminy. Ponieważ skuteczność tego rodzaju nie skóry w razie zastosowania preparatu ze związkiem
preparatów nie została jednoznacznie potwierdzona, czynnym i brak takiego efektu po podaniu preparatu z pla-
a ponadto w trakcie ich stosowania mogą ujawniać cebo).
MUTSCHLER-2009.indd 284 2010-01-07 22:12:11
Układ nerwowy 285
1.5.8.2.7. Zróżnicowane stosowanie leków stosowane są w tym wskazaniu dość rzadko. W zapa-
przeciwreumatycznych w zapalnych leniu stawów występującym w łuszczycy – szczegól-
chorobach reumatycznych nie w przypadku ciężkiego przebiegu – koniecznym
jest czasami zastosowanie leków podstawowych.
Układ nerwowy
Jeżeli w uogólnionym toczniu rumieniowatym (lu-
W przebiegu ostrej gorączki reumatycznej – oprócz
pus erythematodes systemicus) nie doszło do zajęcia
leczenia antybiotykami – stosuje się dodatkowo nie-
ośrodkowego układu nerwowego, nerek i serca, nie-
steroidowe leki przeciwreumatyczne, a w przypad-
jednokrotnie wystarczy stosowanie niesteroidowych
ku zajęcia procesem chorobowym serca konieczne
leków przeciwzapalnych – łącznie z hydroksychlo-
jest włączenie glukokortykosteroidów.
rochiną. W razie niewystarczającego efektu wskaza-
Infekcyjne zapalenia stawów leczone są za po-
ne jest dodatkowe włączenie glukokortykosteroidu.
mocą odpowiednich środków przeciwbakteryjnych
W przypadku zmian toczniowych w nerkach oraz na- B1
– po wcześniejszym zidentyfikowaniu czynnika za-
silonych zmian zapalnych naczyń, konieczne jest do-
kaźnego, a silne dolegliwości bólowe są dodatkowo
datkowe wprowadzenie cyklofosfamidu.
opanowywane dzięki lekom przeciwbólowym.
Zespół Sjögrena wymaga zastosowania sztuczne-
W reumatoidalnym zapaleniu stawów stosuje się
go płynu łzowego oraz sztucznej śliny. W przypadku
obecnie – odmiennie od wcześniejszych zaleceń
silnych reakcji zapalnych wskazane jest przejściowe
– już we wczesnej fazie choroby lek podstawowy
podawanie glukokortykosteroidu wraz z niesteroido-
(najczęściej metotreksat), oprócz niesteroidowego
wym lekiem przeciwzapalnym.
leku przeciwzapalnego oraz stosowanego niekiedy
W początkowej fazie postępującej twardziny –
początkowo glukokortykosteroidu. W przypadku bra-
głównie w przypadku współwystępowania objawów
ku poprawy po zastosowaniu leków podstawowych
choroby Raynauda – zalecane jest stosowanie leków
konieczne jest przeprowadzenie leczenia skojarzone-
rozszerzających naczynia.
go (zob. ryc. B 1.5-24).
Zapalenie wielomięśniowe i zapalenie skórno-
W seronegatywnych zapaleniach stawów kręgo-
-mięśniowe powinny być w pierwszej kolejności le-
słupa najważniejszym środkiem terapii są niestero-
czone za pomocą glukokortykosteroidów. Jeżeli jest
idowe leki przeciwzapalne. Glukokortykosteroidy
to konieczne – można dodatkowo włączyć lek immu-
nosupresyjny.
W reumatycznych bólach wielomięśniowych glu-
kokortykosteroidy są lekami pierwszego wyboru.
MTX (lub LEF, SSZ, złoto) + steroidy + NLPZ W schorzeniu tym glukokortykosteroidy działają
szybko i efektywnie.
tak remisja po 3 nie Również do terapii guzkowego zapalenia tętnic
miesiącach? najlepiej nadają się glukokortykosteroidy, jednak dla
uzyskania remisji niejednokrotnie konieczne jest za-
zmniejszyć dawkę zwiększyć dawkę MTX stosowanie cyklofosfamidu.
steroidów/powoli lub
W uogólnionym ziarniniaku Wegenera leczenie
terapia skojarzona:
MTX + LEF, opiera się na zastosowaniu preparatów immunosu-
MTX + SSZ + HCQ, presyjnych.
tak MTX + CsA,
LEF + SSZ
remisja po 3 nie 1.5.8.2.8. Terapia niefarmakologiczna
miesiącach?
zmniejszyć dawkę steroidów/ zmiana leku lub leczenie Oprócz leczenia farmakologicznego w terapii chorób
powoli, zmniejszyć dawkę skojarzone z lekami reumatycznych duże znaczenie mają metody leczenia
leków podstawowych immunobiologicznymi fizykalnego (fizjoterapia, terapia pracą, naświetlania,
gimnastyka).
Ryc. B 1.5-24. Obecnie zalecany przez Niemieckie Towarzystwo
Reumatologii algorytm leczenia reumatoidalnego zapalenia
stawów. Sole złota i leki przeciwmalaryczne jako leki pierw- 1.5.9. Terapia dny moczanowej
szego wyboru powinny być stosowane jedynie w wyjątkowym
przypadku. MTX – metotreksat; LEF – leflunomid; SSZ – sulfa-
salazyna; HCQ – hydroksychlorochina; CsA – cyklosporyna A; Dna moczanowa (arthritis urica) jest objawem cho-
NLPZ – niesteroidowe leki przeciwzapalne. robowym hiperurykemii, tj. podwyższenia poziomu
MUTSCHLER-2009.indd 285 2010-01-07 22:12:11
286 Układ nerwowy
stężenia kwasu moczowego w osoczu powyżej war- Ostry napad dny, tj. ostre dnawe zapalenie stawów,
tości 6,4 mg/dl. występuje nagle i dość często w nocy. Najczęściej do-
tknięty jest paluch u stopy, rzadziej reakcje zapalne
Dna moczanowa pierwotna (w wąskim znaczeniu ujawniają się w stawach palców oraz w obrębie nad-
tego słowa) jest przewlekle przebiegającym, dzie- garstka. Skóra nad bolesnym stawem jest zaczerwie-
dzicznym zaburzeniem przemiany puryn z podwyż- niona, ponadto obserwuje się ciastowate obrzmienie.
szeniem ogólnej zawartości („puli”) kwasu moczo- Mogą ujawniać się objawy ogólne – gorączka, tachy-
wego w organizmie, jak również z wytrącaniem i gro- kardia, bóle głowy i wymioty.
madzeniem się moczanów w bogatych w kolagen Do ostrego napadu dny dochodzi wówczas, gdy
i mukopolisacharydy tkankach mezenchymalnych, w tkankach o niewielkim metabolizmie wytrącają się
a tym samym prowadzącym do nawracających zapa- kryształy moczanu sodowego, które następnie są fago-
leń stawów – ostatecznie o przewlekłym przebiegu cytowane przez leukocyty (zob. ryc. 1.5-25). Tworzą
z trwałymi deformacjami w obrębie stawów. się fagolizosomy, których błona pęka pod wpływem
Stwierdza się przy tym dwie możliwe przyczyny: kryształów. Dochodzi do uwolnienia enzymów lizo-
somalnych, które doprowadzają do autolizy (samo-
■ nasilone tworzenie kwasu moczowego w procesach
uszkodzenia) komórek biorących udział w opisywa-
przemiany materii
nych procesach, a w otaczających tkankach dochodzi
lub częściej do uszkodzeń i reakcji zapalnych. Warunkowane tym
obniżenie się wartości pH prowadzi ponownie do wy-
■ zaburzenie nerkowego wydalania kwasu moczowego.
trącania się moczanów sodu i nasila proces chorobo-
wy – na drodze mechanizmu błędnego koła (circulus
W przypadku zwiększonego wytwarzania kwasu mo-
vitiosus). Bez wdrożenia leczenia objawy ustępują
czowego albo upośledzeniu ulega resynteza nukleo-
dopiero po kilku dniach.
tydów purynowych z zasad purynowych, wskutek
czego hipoksantyna i guanina są w większym stop-
Bezobjawowy okres między napadami może utrzymy-
niu metabolizowane do kwasu moczowego, albo też
wać się przez tygodnie lub lata.
zniesieniu ulega mechanizm ujemnego sprzężenia
zwrotnego syntezy puryn, przez co zasady purynowe
W przypadku przewlekłej fazy dny – nasilenie napa-
są wytwarzane w dużych ilościach.
dów jest mniejsze, jednak nie udaje się uzyskać peł-
Zaburzenie wydalania polega na zmniejszeniu se-
nego ustąpienia objawów. Zwykle stwierdza się złogi
krecji kwasu moczowego w kanalikach nerkowych.
moczanowe (tzw. guzki dnawe = guzki artretyczne;
Oprócz komponenty dziedzicznej istotne znacze-
tophi) w małżowinie usznej, w obrębie dłoni i stóp.
nie mają również czynniki egzogenne, które nie mają
co prawda znaczenia przyczynowego, ale prowadzą Wtórna dna występuje jako powikłanie chorób,
do nasielania się objawów. Jako czynniki zewnętrz- w których przebiegu dochodzi do zmniejszenia wy-
ne pod uwagę muszą być brane: pozbawiony ruchu dalania kwasu moczowego lub też nasilonych prze-
wyniszczający tryb życia, nadużywanie alkoholu mian, w tym – rozkładu nukleoproteidów. Wśród tych
oraz błędy żywieniowe z otyłością. Chorobą dotknię- schorzeń można wymienić m.in. niewydolność nerek,
ci się głównie mężczyźni w średnim i zaawansowanym białaczki szpikowe i policytemię. Wtórna dna może
wieku. Należy podkreślić, że dna nie jest wyłącznie być następstwem radio- lub chemioterapii i związa-
schorzeniem stawów, lecz poważną chorobą ogól- nego z tymi terapiami nasilonego rozpadu komórek.
noustrojową. Stąd też u dużego odsetka pacjentów
z dną stwierdza się współistnienie innych schorzeń:
nadciśnienia, zaburzeń metabolizmu węglowodanów, 1.5.9.1. Terapia ostrego napadu dny
hiperlipoproteinemii, przedwczesnej miażdżycy,
stłuszczenia wątroby i uszkodzenia kanalików nerko- W terapii ostrego napadu dny stosuje się następujące
wych (nerka dnawa). preparaty:
■ niesteroidowe leki przeciwzapalne,
Kliniczne rozróżnia się w przebiegu dny:
■ czasem glukokortykosteroidy,
■ ostry napad dny,
■ kolchicynę.
■ okres między napadami – wolny od objawów (faza
międzynapadowa),
Leki przeciwzapalne. Największe doświadczenie
■ dnę przewlekłą (przewlekła faza choroby). w leczeniu ostrego napadu dny zebrano dzięki indome-
MUTSCHLER-2009.indd 286 2010-01-07 22:12:12
Układ nerwowy 287
wykrystalizowywanie się moczanu sodowego synowiocyty
(komórki błony
maziowej)
Układ nerwowy
kolchicyna
mechaniczne drażnienie komórki,
uwalnianie czynników chemotaktycznych moczan sodowy LTB4
leukocyt
PMN
migracja leukocytów
PG PG
B1
IL-1
fagocytoza, uwalnianie enzymów lizosomalnych,
tworzenie aktywnych związków tlenu enzymy
(rodników), leukotrienów, prostaglandyn i cytokin
ból IL-1 makrofag
NLPZ
obniżenie pH zapalenie obrzęk
zaczerwienie
Ryc. B 1.5-25. Błędne koło (circulus vitiosus) w patogenezie ostrego napadu dny moczanowej z możliwością jego przerwania przy
pomocy kolchicyny i niesteroidowych leków przeciwzapalnych (NLPZ). PMN – leukocyt z polimorficznym jądrem (wielojądrzasty);
PG – prostaglandyny; IL-1 – interleukina-1; LTB4 – leukotrien B4.
tacynie (początkowo 300 mg/dzień w trzech dawkach gocytarnej leukocytów i przerwaniu w ten sposób
podzielonych, następnie 100–150 mg/dz. przez 2–4 przerywa łańcucha reakcji prowadzących do ostrego
dni). Inne niesteroidowe leki przeciwzapalne – jeżeli napadu dny (zob. ryc. B 1.5-25). Prawdopodobnie
tylko podawane są w odpowiednich dawkach – mają kolchicyna oddziałuje również – podobnie jak przy
podobnie dobrą skuteczność. Ponieważ terapia ma hamowaniu mitozy – na mikrowłókienka komórki
charakter krótkotrwały – ciężkie działania niepożąda- i hamuje kurczliwość tubuliny – białka spokrewnio-
ne ujawniają się jedynie w pojedynczych przypadkach. nego z aktyną.
Skuteczne są również leki wybiórczo hamujące COX- Po doustnym podaniu kolchicyna jest dobrze
2 (np. etorikoksyb jeden raz dziennie w dawce 120 mg wchłaniana. Ponieważ ulega aktywnemu wydalaniu
stosowany przez maksymalnie 8 dni). do żółci oraz przez śluzówkę przewodu pokarmowe-
W przypadku istnienia przeciwwskazań dotyczą-
cych zastosowania niesteroidowych leków przeciw-
zapalnych można zastosować glukokortykosteroidy,
np. prednizolon w dawce dziennej 20–40 mg. H3CO
NH
Kolchicyna stosowana jest głównie w przypadkach H3CO CH3
z nie do końca potwierdzonym rozpoznaniem. Zwy- H3CO O
kle – z powodu działań niepożądanych – jest uważa- O
na za środek drugiego wyboru. Jako środek hamują- OCH3
cy mitozę łagodzi dolegliwości ostrego napadu dny
kolchicyna
– jednak nie obniżając poziomu kwasu moczowego
w osoczu i nie mając działania przeciwbólowego.
Jej działanie polega na zmniejszeniu aktywności fa-
MUTSCHLER-2009.indd 287 2010-01-07 22:12:12
288 Układ nerwowy
go do jego światła – podlega krążeniu jelitowo-wą-
trobowemu lub też jelitowo-jelitowemu. Kolchicyna
wiąże się ponadto bardzo silnie z białkami osocza
OH
i ulega powolnemu wydalaniu. Dlatego też istnieje
niebezpieczeństwo kumulacji leku. N N
Dawkowanie wynosi: początkowo 1–1,5 mg,
N NH
następnie co jedną, dwie godziny po 0,5–1 mg, aż
do ustąpienia bólów (maksymalna dawka dobowa allopurinol
8 mg).
Kolchicyna jest bardzo toksyczną substancją; daw-
ka śmiertelna dla dorosłego człowieka wynosi około
20 mg! Nawet w przypadku stosowania dawek tera-
peutycznych niekiedy nie można uniknąć biegunek –
będących objawami ostrego zapalenia żołądka i jelit. nol (okres półtrwania w osoczu ok. 40 minut) jest hy-
Zatrucie kolchicyną przypomina zatrucie arsenem: droksylowany pod wpływem oksydazy ksantynowej
charakterystyczne jest uczucie palenia, drapania do głównego metabolitu – oksypurinolu (czasem
w jamie ustnej z zaburzeniami przełykania; do tego błędnie określanego nazwą alloksantyny). Ten długo
dołączają kolki, wodniste biegunki, duszności, ta- działający metabolit (okres półtrwania ok. 14 godzin)
chykardia i wstrząs. Śmierć następuje zwykle po 2–3 jest główną substancją odpowiadającą za działanie
dniach w wyniku porażenia ośrodka oddechowego leku. W wyniku zahamowania oksydazy ksantynowej
lub niewydolności krążeniowej. Terapia ma charakter dochodzi do zwiększonego wydalania hipoksantyny
objawowy. i ksantyny z moczem, a poziom kwasu moczowego
we krwi i w moczu się zmniejsza. Dodatkową skła-
dową działania allopurinolu okazuje się również ha-
mowanie syntezy puryn de novo.
1.5.9.2. Terapia okresu bezobjawowego Przeciętna dawka wynosi 300 mg/dz.
dny oraz dny przewlekłej Allopurinol jest uważany za lek z wyboru w tera-
pii przewlekłej hiperurykemii.
Celem terapii jest obniżenie poziomu kwasu moczo- Jako działania niepożądane mogą ujawniać się
wego w surowicy poniżej wartości 5,5 mg/dl. zaburzenia żołądkowo-jelitowe oraz alergiczne reak-
W przypadku nieznacznie podwyższonego po- cje skórne (rzadko). Na początku terapii allopurino-
ziomu kwasu moczowego wystarczające są metody lem istnieje niebezpieczeństwo ostrego napadu dny,
związane z dietą. Polegają one na zmniejszeniu po- co wiąże się z mobilizacją złogów moczanowych
daży pokarmów bogatych w puryny (np. wątroba, i dodatkowym podniesieniem poziomu kwasu mo-
nerki, serce), ograniczeniu konsumpcji alkoholu czowego w osoczu.
oraz ograniczeniu ilości dostarczanych kalorii przy W czasie ciąży oraz w okresie karmienia allopuri-
nadwadze. nol nie powinien być stosowany.
Ważną interakcją jest hamowanie procesu elimi-
Często jednak nie da się w przebiegu dny uniknąć nowana azatiopryny i merkaptopuryny przez allo-
wdrożenia terapii długoterminowej. Terapia ta polega purinol (zwiększone ryzyko zmian w obrazie krwi
na zastosowaniu: – uszkodzenie szpiku). Obie wymienione pochodne
puryny – podobnie jak hipoksantyna i ksantyna – ule-
■ leków hamujących syntezę kwasu moczowego
gają biotransformacji pod wpływem oksydazy ksan-
(uricostatica) lub
tynowej, stąd też zablokowanie enzymu spowalnia
■ leków zwiększających wydalanie kwasu moczowe- ich metaboliczną eliminację.
go (uricosurica). Ponadto allopurinol nasila działanie pośrednio
działających leków przeciwzakrzepowych.
Leki hamujące syntezę kwasu moczowego (ury-
kostatyki). Jedynym stosowanym obecnie lekiem Leki zwiększające wydalanie kwasu moczowego
z tej grupy jest allopurinol. W małych dawkach (urykozuryki). W celu zwiększenia wydalania kwa-
jest kompetytywnym, a w dużych dawkach dodatko- su moczowego stosuje się:
wo niekompetycyjnym inhibitorem oksydazy ksanty-
■ probenecid,
nowej, która utlenia hipoksantynę poprzez ksantynę
do kwasu moczowego (zob. ryc. B.1.5-26). Allopuri- ■ benzbromaron.
MUTSCHLER-2009.indd 288 2010-01-07 22:12:12
Układ nerwowy 289
OH OH OH
oksydaza oksydaza
N ksantynowa N ksantynowa N
N N N
OH
Układ nerwowy
N NH HO N NH HO N NH
hipoksantyna ksantyna kwas moczowy
allopurinol, allopurinol,
oksypurinol oksypurinol
Ryc. B 1.5-26. Proces utleniania hipoksantyny do kwasu moczowego. B1
Probenecid oraz znacznie częściej stosowany benz- samego układu transportującego związki kwaso-
bromaron zwiększają wydalanie kwasu moczowe- we. Dlatego urykozuryczne działanie probenecidu
go z moczem – w wyniku zahamowania zwrotnego i benzbromaronu ulega osłabieniu pod wpływem
wchłaniania kanalikowego. Prowadzą do zmniejsze- salicylanów i saluretyków. Probenecid spowalnia
nia stężenia kwasu moczowego we krwi, nie docho- m.in. wydalanie penicylin i niesteroidowych leków
dzi do powstawania nowych guzków dnawych, a na- przeciwzapalnych o budowie kwasowej, z kolei
wet dochodzi do zanikania starych guzków. benzbromaron przyspiesza wydalanie oksypurinolu
Probenecid ulega niemal całkowitemu wchłania- (zob. powyżej).
niu; benzbromaron wchłaniany jest w 50%. Zarów-
no substancje macierzyste, jak i metabolity powstałe
w wyniku oksydatywnej biotransformacji – w przy-
padku benzbromanu m.in. tworzony wskutek utle-
nienia bocznego łańcucha etylowego drugorzędowy COOH
C3H7
alkohol i powstający z niego keton – dokładają się N
do efektu urykozurycznego. Okres półtrwania w oso- H7C3 S
czu wynosi w przypadku probenecidu – 4–6 godzin, O O
a metabolitów benzbromaronu – 12–35 godzin. probenecid
Dawki są stopniowo zwiększane. Probenecid
jest w pierwszym tygodniu stosowany w dawkach do-
bowych 500 mg, od drugiego tygodnia podaje się 1000
mg/dzień; benzbromaron podawany jest początkowo
w dawce 50 mg/dzień, później 100 mg na dobę. Br
O
Równocześnie aby zapobiec wytrącaniu kryszta-
OH
łów moczanu sodu w nerkach – podaje się wodoro-
węglan sodu lub cytrynian potasu, jak również zwięk- Br
sza się podaż płynów. W celu opanowania ostrych O C2H5
napadów dny, które mogą ujawnić się na początku
terapii urykozurycznej – w wyniku równoczesnego benzbromaron
hamowania kłębuszkowego wydalania i zwrotnego
wchłaniania kwasu moczowego – możliwe jest przej-
ściowe włączenie leków z grupy niesteroidowych le-
ków przeciwzapalnych. Leki złożone zawierające benzbromaron i allopu-
Jako działania niepożądane obserwowane są doleg- rinol. W związku z różnymi mechanizmami działania
liwości żołądkowo-jelitowe, oraz (rzadko) alergiczne możliwe jest łączne stosowanie benzbromaronu z al-
reakcje skórne. lopurinolem, jednak – jak to już zostało powiedziane
Interakcje ujawniają się w przypadku stosowania – lek urykozuryczny osłabia działanie allopurinolu
leków urykozurycznych wraz z innymi kwasowy- z powodu szybszego wydalania oksypurinolu. Tego
mi związkami wydalanymi w nerkach – w związ- rodzaju terapia skojarzona nie ma więc jakichś szcze-
ku z wykorzystywaniem w procesie wydalania tego gólnych zalet.
MUTSCHLER-2009.indd 289 2010-01-07 22:12:13
290 Układ nerwowy
1.6. Środki znieczulające miejscowo
Środki znieczulające miejscowo (do znieczulenia łaniem farmakologicznym środków znieczulających
miejscowego) znoszą w sposób odwracalny i miej- miejscowo była poddawana intensywnym badaniom.
scowo ograniczony pobudliwość wrażliwych na ból Według Löfgrena – odkrywcy środków znieczula-
(odbierających i przenoszących ból) zakończeń ner- jących miejscowo będących pochodną anilidyny
wowych oraz zdolność przewodzenia czuciowych – budowę najważniejszych środków do znieczulenia
włókien nerwowych. Skutkiem tego jest przejściowe miejscowego można wyprowadzić z przedstawionej
wyłączenie czucia bólu – jednak bez wpływu na stan na ryc. B 1.6-2 podstawowej struktury chemicznej.
świadomości. Gdyby ująć schemat Löfgrena w sposób jeszcze bar-
dziej ogólny:
Działanie środków znieczulających miejscowo na czucio-
we zakończenia włókien nerwowych jest niespecyficzne; reszta lipofilna – łańcuch łączący – reszta hydrofilna,
skądinąd poszczególne pobudliwe struktury są na nie róż-
nie wrażliwe. To, że np. nie dochodzi do zaburzeń czynności wówczas możliwe byłoby przyporządkowanie znieczu-
ruchowych w przypadku stosowania zwyczajowych dawek lającym miejscowo środkom pozbawionym atomu N –
środków znieczulających miejscowo – wiąże się głównie np. środka znieczulającego powierzchniowo polidoka-
z tym, że motoryczne włókna nerwowe posiadają większą
średnicę niż włókna czuciowe odpowiedzialne za przewo- nolu, C12H25–O–(CH2–CH2–O)8–9–CH2–CH2–OH).
dzenie bólu. Ponieważ skuteczność środków znieczulają-
cych miejscowo zmniejsza się wraz ze zwiększaniem śred- Reszta hydrofilna jest prawie zawsze dwu- lub trójrzędową
nicy włókna, blokowaniu ulegają najpierw cienkie przewo- grupą aminową. Łańcuch łączący, który może być bardzo
dzące ból włókna C i włókna Aδ oraz przedzwojowe włók- różnorodny, zawiera z reguły ugrupowanie atomów o cha-
na B. Dopiero większe dawki wpływają na osłabienie prze- rakterze polarnym (grupy estrowe lub amidowe). Reszta
wodzenia doznań ucisku i dotyku (włókna Aβ), a w końcu lipofilna ma zwykle budowę aromatyczną.
wpływają również ma funkcje ruchowe (włókna Aα) (zob.
ryc. B 1.6-1). W czuciowych zakończeniach nerwowych Mimo że schemat ten okazał się bardzo pożyteczny
odbierających ból (nocyceptywnych) dominującą ekspre- w powstawaniu nowych środków znieczulających
sją cechują się izoformy kanałów sodowych Naν 1,8/1,9, miejscowo o zastosowaniu klinicznym, nie powi-
które jak dotąd nie zostały zidentyfikowane w żadnej innej nien prowadzić do mylnego twierdzenia, że miejsco-
tkance. Jeżeli tylko udałoby się kiedykolwiek zsyntetyzo- we działanie znieczulające jest związane ze swoistą
wać selektywne inhibitory tych kanałów – otworzyłyby
się możliwości dla ubogiej w działania niepożądane terapii konfiguracją chemiczną. Liczne niepasujące do tego
bólu przeprowadzanej za pomocą środków znieczulających schematu substancje, np. alkaloid johimbiny, efekt
miejscowo. znieczulający miejscowo ujawniają jako działanie
niepożądane. Z drugiej strony istnieją także sub-
Zależność między budową strukturalną a działa- stancje, które można zaszeregować do powyższego
niem. Zależność miedzy budową chemiczną a dzia- schematu, a które pomimo to nie wykazują działania
Typ włókna A B C
(α β γ δ)
średnica (μm) 15 10 5 2 2 <1
mielinizacja tak tak nie
wrażliwość na środki znieczulające miejscowo + (Aδ +++) +++ ++++
Ryc. B 1.6-1. Charakterystyka włókien nerwowych.
MUTSCHLER-2009.indd 290 2010-01-07 22:12:13
Układ nerwowy 291
ną, przy czym duże znaczenie mają sensory napięcia
reszta aromatyczna łańcuch grupa aminowa (napięciowozależny kanał jonowy). Badania biofi-
(lipofilna) pośredni (reszta hydrofilna) zyczne i z zakresu biologii molekularnej wskazują
na istnienie przynajmniej jednego miejsca wiązania
Układ nerwowy
środków do znieczulenia miejscowego – w obrębie
O CH3
O H3CO N szóstego przezbłonowego segmentu domeny IV pod-
jednostki α kanału sodowego. To miejsce wiązania
O
może być osiągnięte przez środki znieczulające miej-
kokaina scowo jedynie od strony międzykomórkowej. Środek
do znieczulenia miejscowego za pomocą swoich reszt
hydrofilnych i lipofilnych wchodzi we wzajemne od-
O C2H5
działywanie z aminokwasami – fenyloalaniną i tyro- B1
N
O C2H5 zyną szóstego przezbłonowego segmentu (domeny
IV) i w wyniku tego zapobiega otworzeniu się kanału
H2N prokaina
sodowego. Jeżeli zablokowaniu ulegnie wystarczają-
ca ilość kanałów sodowych, nie jest możliwe osiąg-
nięcie progu pobudzenia i pobudzenie nie może być
CH3 dalej przewodzone. Może ono być przewodzone tyl-
NH C2H5 ko wtedy, gdy w wyniku nagłego zwiększenia prze-
N
O C2H5
puszczalności dla jonów sodu dochodzi do załamania
CH3 potencjału spoczynkowego.
lidokaina
Najważniejsze obecnie stosowane środki do znieczule-
nia miejscowego zawierają alifatyczną lub alicykliczną
Ryc. B 1.6-2. Podstawowa budowa środków znieczulających trzeciorzędową (lub drugorzędową) grupę aminową.
miejscowo (według Löfgrena). W wyniku tego w roztworze wodnym panuje równo-
waga pomiędzy protonowaną hydrofilną i nieprotono-
waną lipofilną postacią leku. Duże znaczenie dla moż-
liwości penetracji środka znieczulającego miejscowo
znieczulającego miejscowo. Szczególne znaczenie ma stan równowagi, który zależny jest od wartości pKa
mają bowiem właściwości fizykochemiczne środków substancji oraz od wartości pH środowiska. Wartości
znieczulających miejscowo, które są decydujące dla pKa leku wynoszą między 7,6 a 9. Zgodnie z równa-
zdolności ich wiązania z białkami lub lipidami błon niem Hendersona-Hasselbalcha przy pH 7,4 – zależnie
komórkowych. od pKa – jedynie 3–20% leku pozostaje w nieprotono-
wanej, rozpuszczalnej w lipidach postaci, która może
Mechanizm działania. Środki znieczulające miejscowo dyfundować przez błonę aksonu.
blokują napięciowozależne kanały sodowe i w ten sposób Tkanka z procesem zapalnym wykazuje niższą
uniemożliwiają istotny dla procesu depolaryzacji szybki
napływ jonów Na+ do komórki – tym samym hamują prze- wartość pH niż tkanka normalna, gdyż przez niedo-
wodzenie impulsów nerwowych (zob. ryc. B 1.6-3). Środki bór tlenu w wyniku tworzenia się obrzęku wydłuże-
znieczulające miejscowo, w stężeniach większych niż lecz- niu ulega droga dyfuzji – czego skutkiem jest nara-
nicze, mogą także blokować inne kanały jonowe, np. ka- stanie procesu beztlenowej glikolizy i zwiększenie
nały potasowe. Samo działanie na kanały jonowe wykazu- powstawania kwasu mlekowego (miejscowa kwasica
je postać protonowana leku. Aby móc zablokować kanał
sodowy, środki znieczulające miejscowo muszą najpierw mleczanowa). Środki do znieczulenia miejscowe-
dyfundować przez błonę komórkową w postaci nieproto- go są mniej skuteczne w obrębie tkanki z toczącym
nowanej (rozpuszczalnej w lipidach) do aksonu. Dlatego się procesem zapalnym (np. w przebiegu zapalnych
szczególną rolę odgrywają fizykochemiczne właściwości bólów zębów), gdyż równowaga między protonowa-
środków znieczulających miejscowo. nymi i nieprotonowanymi postaciami ulega przesu-
nięciu w kierunku formy protonowanej, co prowadzi
Kanały sodowe są białkami o budowie heterotrime- do zmniejszenia zdolności penetracji.
rycznej, które składają się z podjednostki α, β1 i β2
(zob. ryc. B 1.6-3). W podjednostce α można wydzie- Sposoby stosowania. Ze względu na sposób poda-
lić 4 homologiczne domeny (I-IV). Otworzenie biał- nia środka do znieczulenia miejscowego rozróżnia się
ka kanałowego wiąże się z jego zmianą konformacyj- znieczulenie:
MUTSCHLER-2009.indd 291 2010-01-07 22:12:13
292 Układ nerwowy
otwarty kanał Na+ zamknięty kanał Na+ zablokowany kanał Na+
(nieczynny) B + H+ BH+
Na+ Na+ przestrzeń
zewnątrz-
komórkowa
przestrzeń
wewnątrz-
komórkowa
Na+
B + H+ brak potencjału
szybka depolaryzacja BH+ czynnościowego
(potencjał czynnościowy)
podjednostka β1 podjednostka α podjednostka β2
ψ I II III IV ψ przestrzeń
H3N+ ψ ψ
ψ
ψ
ψψ
H3N zewnątrz-
komórkowa
+ + +
1 2 3 4 5 6 1 2 3 4 5 6 1 2 3 4 5 6 1 2 3 4 5 6
+ + +
przestrzeń
P BH+
-
OOC P COO- COO- wewnątrz-
komórkowa
H3N+ P
P
P P
ψ – miejsce glikozylacji; P miejsce fosforylacji
Ryc. B 1.6-3. Schematyczny molekularny mechanizm działania środków znieczulających miejscowo. B – środek znieczulający miej-
scowo (według Isoma i in. oraz Ragsdale’a i in.). Bliższe informacje w tekście.
■ powierzchniowe, Ponieważ w wyniku przypadkowego wstrzyk-
nięcia środka znieczulającego miejscowo do żyły
■ nasiękowe,
mogą wystąpić ciężkie powikłania (zob. poniżej),
■ przewodowe, stosowanie miejscowego znieczulenia dożylnego
jest dyskusyjne. Przed iniekcją środka do znieczu-
■ dożylne miejscowe w obrębie kończyn.
lenia miejscowego należy założyć mankiet aparatu
do mierzenia ciśnienia krwi, aby zatrzymać dopływ
W przypadku znieczulenia powierzchniowego śro- i odpływ krwi. Następnie wstrzyknięty środek znie-
dek znieczulający miejscowo jest podawany na ślu- czulający miejscowo przenika z żył do otaczają-
zówki lub powierzchnię rany i z tego miejsca dyfun- cych tkanek i w ciągu 10–15 minut wywołuje tam
duje do czuciowych narządów końcowych (recep- znieczulenie. Zatrzymanie krążenia powinno być
torów czuciowych) oraz do końcowych rozgałęzień utrzymane przez co najmniej 20–30 minut – tak
nerwowych. aby zapobiec rozprzestrzenieniu się większych ilo-
W znieczuleniu nasiękowym – środek znieczula- ści środka znieczulającego, który jeszcze nie uległ
jący miejscowo jest wstrzykiwany do tkanki lub też wchłonięciu, do sąsiadujących tkanek. Po przerwa-
tkanka nasiąka tym środkiem. W ten sposób zabloko- niu niedokrwienia miejscowe działanie znieczulają-
waniu ulegają nie tylko czuciowe narządy końcowe, ce ustępuje po kilku minutach.
lecz również mniejsze pnie nerwowe.
W znieczuleniu przewodowym nastrzykuje się Dodatkowe leki zwężające naczynia. W przeci-
w sposób celowy określone nerwy i w tym miejscu wieństwie do kokainy – pierwszego terapeutycznie
przerywa się przewodzenie pobudzenia. Szczegól- zastosowanego środka znieczulającego miejscowo
nymi formami znieczulenia przewodowego są znie- – większość syntetycznych anestetyków wykazuje
czulenie dordzeniowe, nadoponowe i przykręgosłu- działanie rozszerzające naczynia. Stąd też – z na-
powe. stępujących przyczyn – stosowane są często z sub-
MUTSCHLER-2009.indd 292 2010-01-07 22:12:14
Układ nerwowy 293
stancjami zwężającymi naczynia: zwężenie naczyń ■ reakcji alergicznych.
spowalnia rozprzestrzenianie się (odtransportowy-
wanie z miejsce podania) środka do znieczulenia Zbyt wysokie stężenie we krwi, będące skutkiem po-
miejscowego, zwiększa w ten sposób czas działania myłkowej iniekcji dożylnej, zbyt szybkiego wchła-
Układ nerwowy
i zmniejsza jego ogólną toksyczność. Prowadzi także niania lub podania zbyt dużej dawki środka do znie-
do zmniejszenia ukrwienia w obrębie pola operacyj- czulenia miejscowego, prowadzi do zaburzeń ze stro-
nego – co powoduje, że zabieg chirurgiczny staje się ny ośrodkowego układu nerwowego i serca.
prostszy i bardziej bezpieczny. Ośrodkowe objawy zatrucia – w okresie począt-
Wymienione zalety nie dotyczą jednak silnie ukrwio- kowym wiążą się z blokowaniem kanałów sodowych
nych okolic ciała, np. okolic głowy i szyi, jak również w neuronach hamujących (w wyniku czego powsta-
okolicy moczowo-płciowej oraz odbytu, gdzie środek ją objawy pobudzenia), a w okresie późniejszym
do znieczulenia miejscowego i lek zwężający naczy- – w przypadku cięższych zatruć – wynikają z po- B1
nia są błyskawicznie wchłaniane – czego skutkiem rażenia rozległych okolic ośrodkowego układu ner-
jest zwiększenie ich ogólnej toksyczności. wowego. W łagodnych przypadkach polegają one
Również w trakcie operacji w obrębie dystalnych na niepokoju, przymusie mówienia (gadatliwości,
części ciała (palce u rąk i stóp, nos, broda) – z po- słowotoku), wymiotach, drżeniach (tremor), stanach
wodu ryzyka uszkodzeń niedokrwiennych (zgorzel!) lękowych i majaczeniu (lub splątaniu), a w ciężkich
nie wolno stosować dodatkowych leków zwężają- przypadkach – na klonicznych drgawkach i porażeniu
cych naczynia. oddechu.
Jako leki zwężające naczynia stosuje się leki α-sym- Również w sercu leki do znieczulenia miejscowe-
patykomimetyczne (α-adrenomimetyczne) – przede go hamują przewodzenie pobudzenia. Na pierwszym
wszystkim adrenalinę i noradrenalinę, jak również – planie ujawniają się ujemne działania inotropowe,
chociaż rzadziej – analogi diuretyny hormonu tylnego dromotropowe i batmotropowe. W wyniku tego może
płata przysadki mózgowej, które już prawie nie wy- dojść do bradykardii, ewentualnie do bloku przed-
kazują działania antydiuretycznego, np. felypresynę. sionkowo-komorowego z efektem końcowym zatrzy-
Jako działania niepożądane stosowania dodatków mania akcji serca i drgawek z niedotlenienia. (Zasto-
α-sympatykomimetycznych obserwowano: uczucie sowanie lidokainy jako leku przeciwarytmicznego
lęku, niepokój, bóle głowy, wzrost ciśnienia krwi jest opisane w rozdziale B 4.3.3.2.2.1).
i zaburzenia rytmu serca. Także przy stosowaniu ana- Najważniejszym elementem postępowania te-
logów adiuretyny może ujawniać się niekorzystny rapeutycznego jest podanie tlenu do oddychania
wpływ na układ krążenia oraz reakcje nadwrażliwo- – w celu zapobieżenia hipo- lub anoksji, a w przy-
ści. Zaletą stosowania analogów adiuretyny jest jed- padku zatrzymania serca – zewnętrzny masaż serca
nak brak występowania interakcji z substancjami, wraz z prowadzeniem sztucznego oddychania. Jeżeli
które nasilają działanie katecholamin (np. trójpier- masaż serca nie okaże się efektywny w ciągu 2 mi-
ścieniowe leki przeciwdepresyjne). nut, należy podać 0,5–1 mg adrenaliny dożylnie
lub dotchawiczo. W przypadku wystąpienia drga-
Wytyczne dotyczące dawkowania. Należy do- wek sprawdzoną metodą jest powtarzane podawanie
kładnie przestrzegać stosowania przyjętych dawek chlorku suksametonium, a gdy drgawki nie są wywo-
maksymalnych (np. 300 mg lidokainy bez dodatku łane zatrzymaniem akcji serca i wynikającym z tego
i 500 mg z dodatkiem leku α-sympatykoomime- niedotlenieniem – wskazane jest dożylne podanie
tycznego). Podobna uwaga dotyczy również dodat- 10–20 mg diazepamu.
kowych leków zwężających naczynia. Maksymalna W przypadku zatrucia adrenaliną występują in-
jednorazowa dawka noradrenaliny lub adrenaliny tensywna bladość, zimny pot, tachykardia i znaczne
wynosi 0,25 mg. podwyższenie się ciśnienia krwi, a w rzadkich przy-
padkach – arytmia i migotanie komór; podczas gdy
Niebezpieczne powikłania i ich leczenie. Dotyczą one w przypadku przedawkowania noradrenaliny docho-
przede wszystkim narządów, w których środki do znie- dzi raczej do zwolnienia pracy serca (bradykardii).
czulenia miejscowego mogą zahamować przewodzenie Terapia uzależniona jest od występujących objawów:
pobudzenia. Ciężkie, a w określonych warunkach za- w przypadku tachykardii przeprowadza się ostrożną
grażające życiu powikłania mogą wystąpić w wyniku: dożylną iniekcję β-blokera, w znacznym zwiększeniu
ciśnienia krwi – podaje się leki o działaniu polega-
■ zbyt wysokiego stężenia we krwi środka znieczula- jącym na obwodowym rozszerzeniu naczyń, z kolei
jącego miejscowo lub leku sympatykoomimetycz- migotanie komór wymaga przeprowadzenia defibry-
nego o działaniu zwężającym naczynia, lacji.
MUTSCHLER-2009.indd 293 2010-01-07 22:12:14
294 Układ nerwowy
Reakcje alergiczne występują głównie w przypad- 1.6.1. Środki znieczulające miejscowo
ku leków do znieczulenia miejscowego typu estrowe- typu estrowego
go (zob. poniżej). Stąd też ich znaczenie wyraźnie się
zmniejszyło. Alergiczne reakcje mogą mieć charakter
łagodny (np. osutka pokrzywkowa) lub ciężki (skurcz Kokaina, alkaloid estrowy z liści krzewu kokai-
oskrzeli, wstrząs anafilaktyczny). W ich terapii wy- nowego, jest najstarszym środkiem stosowanym
korzystuje się leki przeciwhistaminowe typu H1 i glu- do znieczulenia miejscowego. W związku z działa-
kokortykosteroidy, a w przypadku wstrząsu anafilak- niem uzależniającym oraz niestabilnością podczas
tycznego podaje się dodatkowo adrenalinę (0,5–1 mg sterylizacji – jest uważana za przestarzałą. Ponie-
dożylnie). waż jednak służyła jako substancja modelowa pod-
Tabela B 1.6-1. Środki znieczulające miejscowo
Wzór strukturalny Nazwa Preparaty handlowe Preferowany
międzynarodowa rodzaj znieczulenia
I. Środki znieczulające miejscowo typu estrowego
benzokaina (etoform) Anaesthesin, Labocane, powierzchniowe
O
Orajel, Sapovent,
Septolete
OC2H5
H2N
C2H5
prokaina Pasconeural-Injectopas, nasiękowe i przewodowe
O Procain Deltaselect, Procain
N JENAPHARM, Prokain-loges,
O C2H5 Injectio Polocaini
hydrochlorici
H2N
O CH3 tetrakaina Opthocain-N Augentropfen, powierzchniowe
Ruskorex
N
O CH3
H3C(CH2) 3
NH
II. Środki znieczulające miejscowo typu amidów kwasowych
CH3 lidokaina Dynexan Mundgel, Lidoject, nasiękowe i przewodowe
NH C2H5 Xylocain, Xyloneural,
N Butapirazol, Calgel, Depo-
O -Medrol z lidokainą, Dicloratio,
C2H5 Emla, Lignocain,
CH3
Lignocainum hydrochloricum
CH3 CH3 prilokaina EMLA, Xylonest nasiękowe i przewodowe
NH C3H7
N
O H
CH3
mepiwakaina Meaverin, nasiękowe i przewodowe
Mecain,
NH MepiHEXAL,
N
Scandicain
O CH3
CH3
bupiwakaina Bucain, Bupivacain JENAPHARM, długotrwałe
CH3 Carbostesin, Dolanaest,
NH Bupivacaine, Bupivacainum
N hydrochloricum, Marcaine
O (CH2) 3CH3
CH3
MUTSCHLER-2009.indd 294 2010-01-07 22:12:14
Układ nerwowy 295
Tabela B 1.6-1. Środki znieczulające miejscowo (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Preferowany
międzynarodowa rodzaj
znieczulenia
Układ nerwowy
II. Środki znieczulające miejscowo typu amidów kwasowych
S-ropiwakaina Naropin nasiękowe i przewodowe
CH3
NH
N
O C3H7
CH3
CH3 artykaina Ultracain nasiękowe i przewodowe B1
O
S NH
NH C3H7
COOCH3 CH3
czas badań nad nowymi syntetycznymi środkami ku stosowania na dużych powierzchniach ran istnieje
znieczulającymi miejscowo, nadal w tym zakresie niebezpieczeństwo powstawania methemoglobiny,
pozostaje w kręgu zainteresowań. Jest ona poza tym a ponadto dosyć często obserwowane są objawy aler-
jedynym środkiem znieczulającym miejscowo, któ- giczne.
ry zwęża naczynia poprzez blokadę wychwytu neu-
ronalnego noradrenaliny ze szczeliny synaptycznej Prokaina (zob. tab. B 1.6-1) – dzięki wprowadzeniu
aksonu. grupy dietyloaminowej do bocznego łańcucha ben-
zokainy – jest jako chlorowodorek dobrze rozpusz-
Kokaina została wprowadzona w 1884 r. do okulisty- czalna w wodzie. W organizmie ulega – za pomocą
ki przez wiedeńskiego okulistę Kollera. Rok później esteraz – szybkiej hydrolizie do dietyloaminoetano-
Halsted i Hall zastosowali kokainę do znieczulenia lu, który wykazuje działanie rozszerzające naczynia
przewodowego. Z powodu szybko rozwijającego się krwionośne, oraz do kwasu p-aminobenzoesowego.
uzależnienia od kokainy, którego ofiarami stali się W handlu dostępne są 0,5-proc. roztwory do znieczu-
także Halsted i Hall, jeszcze przed ostatecznym wy- lenia nasiękowego oraz 1–2-proc. roztwory do znie-
jaśnieniem jej budowy (Willstätter, 1898), rozpoczę- czulenia przewodowego. Najwyższa dawka jednora-
to na dużą skalę zakrojone badania, mające na celu zowa wynosi 0,6 g s.c.
otrzymanie substancji o takim samym działaniu znie-
czulającym miejscowo, ale bez działania uzależniają- Tetrakaina jest około 10 razy bardziej skutecz-
cego i o mniejszej toksyczności. Rzeczywistym prze- na od prokainy, ale jest także dziesięć razy bardziej
łomem było otrzymanie przez Einhorna i Uhlfeldera od niej toksyczna. Stosowana jest jako lek znieczu-
w 1905 r. prokainy. Z powodu łatwej syntezy, dobrej lający powierzchniowo w okolicach oka. Działanie
rozpuszczalności i tolerancji tkankowej oraz znaczą- utrzymuje się przez wiele godzin. Tetrakaina wchła-
co mniejszej w stosunku do kokainy toksyczności, nia się bardzo szybko z uszkodzonej błony śluzowej
a także braku niebezpieczeństwa uzależnienia, proka- i dlatego przedawkowanie grozi zatruciem.
ina pozostaje standardową substancją typu estrowego
jako środek znieczulający nasiękowo. W 1944 r. Löf-
gren otrzymał lidokainę, pierwszy środek znieczula- 1.6.1. Środki znieczulające miejscowo
jący miejscowo typu amidu kwasowego. typu amidów kwasowych
Benzokaina (zob. tab. B 1.6-1) służy – w związku Środki znieczulające miejscowo typu amidów kwa-
z nieznaczną rozpuszczalnością w wodzie – jako lek sowych stanowią – w związku ze względnie długim
znieczulający wyłącznie powierzchniowo (5–20%, czasem działania oraz dobrą tolerancją w przypadku
np. w tabletkach do ssania, czopkach stosowanych dostosowania się do zasad dawkowania – najczęściej
w leczeniu hemoroidów, maściach lub pudrach). Za- stosowaną grupę preparatów do znieczulenia miej-
letą jest długo utrzymujące się działanie. W przypad- scowego (zob. tab. B 1.6-1).
MUTSCHLER-2009.indd 295 2010-01-07 22:12:15
296 Układ nerwowy
Lidokaina jest szybko i jednocześnie długotrwale ze stosowania preparatów sympatykomimetycznych
działającym środkiem znieczulającym miejscowo, (adrenomimetycznych). Poza tym lidokaina spraw-
którego działanie jest 4 razy silniejsze, a toksyczność dziła się również jako lek antyarytmiczny.
tylko 2 razy większa od prokainy. W przeciwieństwie Preparatami analogicznymi są prilokaina, mepi-
do środków znieczulających miejscowo typu estro- wakaina, bupiwakaina, które stosuje się jako długo
wego – lidokaina nie jest pierwotnie rozkładana za działające środki znieczulające, np. w przewlekłych
pomocą esteraz, lecz przede wszystkim ulega oksy- stanach bólowych, i artykaina. Mepiwakaina i bupi-
datywnej biotransformacji (m.in. dealkilacji w miej- wakaina dostępne są w sprzedaży pod postacią race-
scu atomu azotu). Stosuje się ją do znieczulenia matów. Ponieważ jednak R-enancjomery cechują się
powierzchniowego, nasiękowego i przewodowego większą toksycznością niż S-enancjomery – w przy-
– pod postacią roztworów 0,2–2-proc. Należy pod- padku ropiwakainy do terapii wprowadzono czysty
kreślić, że stosując lidokainę, można zrezygnować preparat S-ropiwakainy.
1.7. Środki do znieczulenia ogólnego
(anestetyki)
Podtlenek azotu został zsyntetyzowany po raz pierwszy ■ (zwykle także) napięcie mięśniowe.
w 1776 r. Mniej więcej 20 lat później Priestley i Davy
opisali jego właściwości przeciwbólowe i znieczulające.
W 1844 r. dentysta H. Wells w Hartford (Connecticut) Substancje, za pomocą których wywołuje się an-
na przedstawieniu wędrownego teatru, podczas którego estezję, określa się mianem środków do znieczulenia
największą atrakcją było wdychanie przez ochotników ogólnego (ogólnie znieczulającymi, anestetykami).
gazu rozweselającego (podtlenku azotu), dokonał odkrycia,
że jedna z osób biorących udział w tych próbach doznała W związku z możliwym sposobem podania wyróżnia
zranienia podudzia, lecz wcale nie wykazywała reakcji bó-
lowej. Następnego dnia w czasie przedstawienia Wells, in- się:
tuicyjnie zdając sobie sprawę z doniosłości tej obserwacji, ■ wziewne środki do znieczulenia ogólnego (aneste-
dokonał pod działaniem gazu rozweselającego usunięcia
zęba mądrości pacjentowi, który nie odczuł w ogóle bólu. tyki wziewne), które podawane są z powietrzem
Pięć tygodni później, po tym jak przekonał się u licznych do dróg oddechowych,
pacjentów o działaniu tego gazu, wystąpił ze swym odkry-
ciem publicznie: chciał wykonać bezbolesną ekstrakcję ■ iniekcyjne środki do znieczulenia ogólnego (aneste-
zęba w Szpitalu Ogólnym w Bostonie. Próba nie udała się, tyki iniekcyjne), które podawane są do światła na-
a Wells został wygwizdany. W1848 r. załamany popełnił czyń żylnych.
samobójstwo. Jednak rozwoju badań nad znieczuleniem
ogólnym nie udało się już zatrzymać. W tym samym miej- Przez pojęcie sterowania (sterowalności) znieczu-
scu, w którym Wellsowi się nie powiodło, już w paździer-
niku 1846 r. udało się Williamowi Mortonowi, dawnemu leniem ogólnym rozumie się możliwość dowolnego
współpracownikowi Wellsa, wykonać pierwsze kliniczne (w każdej chwili) zwiększania lub zmniejszania głę-
znieczulenie ogólne eterem. W 1847 r. w Edynburgu J. bokości anestezji. Terminem rozpiętości (szerokości)
Simpson wprowadził chloroform jako środek do anestezji. anestezyjnej leku opisuje się zakres swobody daw-
Kilka lat później we wszystkich salach operacyjnych na ca- kowania między osiągnięciem pożądanej głębokości
łym świecie operowano tylko w znieczuleniu ogólnym.
anestezji a początkiem stadium asfiksji (zob. poni-
żej). Środek do znieczulenia ogólnego jest więc tym
W przypadku znieczulenia ogólnego (anestezji = bardziej bezpieczny, im większa jest jego rozpiętość
dawniej narkozy) – w wyniku porażenia części anestezyjna. Nie należy jednak zapominać, że rozpię-
ośrodkowego układu nerwowego – odwracalnemu tość anestezyjna prawie wszystkich środków do znie-
wyłączeniu ulegają: czulenia ogólnego jest niewielka. Już 2–4-krotne
zwiększenie zwyczajowej dawki może prowadzić
■ odczuwanie bólu,
do zatrzymania czynności serca i oddychania.
■ świadomość (w rozumieniu szerokim – tj. przytom-
ność),
Właściwości idealnego anestetyku. Idealny środek
■ odruchy obronne, do znieczulenia ogólnego powinien spełniać następu-
MUTSCHLER-2009.indd 296 2010-01-07 22:12:15
Układ nerwowy 297
napięcie
stadium odychanie odruchy mięśniowe
szerokość źrenicy
mięśnie gładkie
Układ nerwowy
przeponowe
świadomość
szkieletowe
ruchy gałek
połykanie
piersiowe
spojówki
wymioty
rogówka
sekrecja
powieki
mięśnie
ocznych
stopnie
światło
brzuch
kaszel
I. analgezja 1.
2.
3. B1
II. pobudzenie ++++
1. ++++
+++
2. ++
+
III. tolerancja 3.
4.
IV. asfiksja
Ryc. B 1.7-1. Schemat poszczególnych stadiów znieczulenia ogólnego w przypadku zastosowania eteru (według Freya, Hügina,
Mayrhofera i in.) (w przypadku odruchów zastosowano pewnien skrót myślowy: i tak w przypadku „rogówki” chodzi o odruch rogów-
kowy, z kolei w przypadku „światła” autorzy mieli zapewne na myśli odruch źreniczy – przyp. tłum.).
jące wymagania (powinien mieć następujące właści- Żaden z obecnie dostępnych środków do znieczulenia
wości lub wykazywać następujące działania): ogólnego nie spełnia w całości wymienionych powy-
żej wymogów. Stąd też zwykle stosuje się równocześ-
■ dobre działanie przeciwbólowe (analgetyczne)
nie kilka anestetyków (zob. poniżej).
i znieczulające,
■ możliwie niewielki wpływ na układ oddechowy Stadia znieczulenia ogólnego. Jeżeli anestezja jest
i krążenia, przeprowadzana za pomocą jednego (wziewnego) an-
estetyku – jak to było wcześniej powszechnie stoso-
■ brak działania drażniącego w obrębie skóry i ślu-
wane – wraz ze zwiększającym się stężeniem środka
zówek,
do znieczulenia ogólnego w organizmie można wy-
■ brak biotransformacji z powstaniem szkodliwych różnić istnienie różnych stadiów anestezji (zob. ryc. B
metabolitów, 1.7-1), które miały praktyczne znaczenie dla prowadze-
nia znieczulenia ogólnego. W przypadku obecnie po-
■ niewielka toksyczność i tym samym duża rozpię-
wszechnie stosowanej anestezji złożonej (zob. poniżej)
tość (okno) terapeutyczna,
opisane stadia znieczulenia właściwie już nie występują.
■ szybkie występowanie działania (początek) i jego
ustępowanie (koniec działania) – a tym samym ła- W stadium analgezji (stadium I) – w stopniu pierwszym
twe sterowanie przebiegiem anestezji, dochodzi do osłabienia czucia bólu, w stopniu drugim
czucie bólu jest niemalże zniesione, a świadomość ulega
■ łatwe stosowanie przez anestezjologa, jednocześnie przytłumieniu. W stopniu trzecim występuje
niepamięć z brakiem czucia bólu.
■ korzystne właściwości fizyczne i chemiczne (trwa-
łość przechowywania, niepalność, niewybucho- W stadium pobudzenia (stadium II) odruchy są wzmożo-
wość), ne – w wyniku zahamowania wyższych ośrodków moto-
rycznych, oddech jest nieregularny, mogą wystąpić kaszel
■ nieszkodliwość dla środowiska w przypadku uwol- i wymioty. Często dochodzi do rozszerzenia się źrenic,
nienia się do atmosfery. a podwyższa się ciśnienie krwi i częstość pracy serca.
MUTSCHLER-2009.indd 297 2010-01-07 22:12:15
298 Układ nerwowy
znieczulenie ogólne
premedykacja pooperacyjna
zwiotczenie
terapia bólu
sedacja analgezja
mięśni
anksjoliza
anestezja
czas
Ryc. B 1.7-2. Całościowa koncepcja znieczulenia ogólnego.
Ponieważ stadium pobudzenia jest szczególnie niepożą- skojarzeniu) kilku i po części specyficznie działają-
dane, powinno trwać możliwie krótko. cych substancji móc uzyskać każdy z pożądanych
W stadium tolerancji (stadium III) wyłączone są – oprócz efektów osobno – niezależnie od innych. Zamiast
półkul mózgowych – również śródmózgowie i rdzeń krę- „pionowego” sposobu oceny dotyczącego osiąga-
gowy. Napięcie mięśni poprzecznie prążkowanych jest ob- nia poszczególnych stadiów znieczulenia ogólnego
niżone, odruchy są osłabione lub zniesione, ale funkcje au- jedynie za pomocą wzrastającego stężenia środka
tonomiczne rdzenia przedłużonego są w pełni zachowane: do anestezji – pojawiła się całościowa koncepcja
oddech jest regularny, krążenie stabilne. W tym stadium
przeprowadzane są zabiegi operacyjne. anestezjologiczna (zob. ryc. B 1.7-2), przy czym
W zależności od szerokości źrenic, ruchów gałek ocz- udaje się zmniejszyć dawki poszczególnych aneste-
nych, napięcia mięśni i oddychania – stadium tolerancji tyków, a tym samym również ich działania niepożą-
może być jeszcze podzielone na cztery stopnie (zob. ryc. dane. Wariantami znieczulenia ogólnego złożonego
B 1.7-1). są tzw. anestezja zrównoważona, neuroleptanalgezja
W stadium asfiksji (stadium porażenia, stadium IV) ule- i (całkowita) anestezja dożylna [(T)IVA]. Analgezja
gają porażeniu także ośrodki wegetatywne rdzenia przedłu- za pomocą opioidów jest we wszystkich postaciach
żonego: krążenie załamuje się, oddychanie się zatrzymuje. złożonej anestezji ich ważną częścią składową (zob.
Bez sztucznego oddychania i odpowiedniego postępowania poniżej).
objawowego – w ciągu kilku minut dochodzi do śmierci
pacjenta.
W trakcie budzenia się stadia anestezji przebiegają w od- Premedykacja. W przypadku znieczulenia ogólnego,
wrotnej kolejności. które przeprowadza się zgodnie z podanymi zasada-
mi, gotowość do reakcji neurowegetatywnych na stres
Znieczulenie ogólne złożone (anestezja kombi- związany z operacją i anestezją może być raczej
nowana/skojarzona). W okresie wcześniejszym podwyższona niż zmniejszona. Wynikające z tego
próbowano uzyskać wszystkie działania cząstko- zagrożenia muszą zostać opanowane za pomocą pre-
we znieczulenia ogólnego (analgezję, niepamięć medykacji, która jest znaczącym postępem w dzie-
– amnezję, sen, brak odruchów mięśniowych – are- dzinie anestezji. Dzięki premedykacji, przy pomocy
fleksję, rozluźnienie mięśni, hamowanie układu której pacjent jest przygotowywany do znieczulenia
autonomicznego) za pomocą tylko jednego środka ogólnego i która równocześnie pozwala zaoszczędzić
wziewnego (np. eteru lub chloroformu). Wymagane na samym środku do znieczulenia ogólnego, możliwe
w tym celu wysokie stężenia anestetyku wziewnego jest opanowanie (zmniejszenie):
miały jednak znaczący wpływ na metabolizm, funk-
■ nasilenia lęku i pobudzenia psychicznego – za po-
cje nerek i wątroby oraz układ sercowo-naczyniowy.
mocą leków przeciwlękowych (anksjolitycznych)
Stąd też obecnie przeprowadza się niemal wyłącznie
i neuroleptyków,
znieczulenia ogólne typu złożonego. W przypadku
nowoczesnej techniki anestezjologicznej dąży się ■ czucia bólu – dzięki lekom przeciwbólowym (anal-
do tego, aby za pomocą kombinacji (połączeniu, getycznych),
MUTSCHLER-2009.indd 298 2010-01-07 22:12:16
Układ nerwowy 299
■ odruchu wymiotnego i niebezpieczeństwa wstrząsu Już około 1990 r. Meyer i Overton przypuszczali, że an-
– stosując leki przeciwhistaminowe, estetyczne działanie leków do znieczulenia ogólnego po-
lega na wzajemnym oddziaływaniu z lipidami błonowy-
■ napięcia układu autonomicznego – lekami para- mi. Według współczesnych założeń efekt ten może być
sympatykolitycznymi (cholinolitycznymi) i sympa- wywołany blokadą kanałów jonowych (tzw. stabilizacja
Układ nerwowy
błony) w następstwie gromadzenia się anestetyku w war-
tykolitycznymi (adrenolitycznymi).
stwie lipidowej, w wyniku czego dochodzi do rozszerze-
nia się objętości błony. Należy jednak uwzględnić fakt,
że działanie ogólnie znieczulające może być następstwem
Dawniej podanie atropiny lub innego leku choli-
również wzajemnego oddziaływania z białkami błonowy-
nolitycznego, np. skopolaminy – w celu uniknięcia mi, np. przez wiązanie anestetyku z okolicami hydrofo-
niebezpiecznego, niekiedy prowadzącego nawet bowymi białka, co powoduje zmniejszenie możliwości
do śmierci odruchu cholinergicznego (zatrzymanie zmian konformacyjnych.
akcji serca), jak również w celu zmniejszenia wy- Kolejna interesująca hipoteza – pochodząca od Paulinga B1
i Millera – przyjmuje wzajemne oddziaływanie anestetyku
twarzania śliny i śluzu, było uważane za niezbędne.
i związanej z błoną wody. Cząsteczki wody dzięki asocjacji
Obecnie leków parasympatykolitycznych (cholinoli- dipolowej są w stanie tworzyć struktury „lodopodobne”.
tycznych) nie uważa się za niezbędną część składową W warunkach eksperymentalnych (np. pod wysokim ciś-
premedykacji – z następujących powodów: zwykła nieniem) cząsteczki hydrofobowe, np. anestetyki w postaci
dawka 0,5 mg atropiny i.m. podana 45 min przed roz- gazowej, mogą zostać uwięzione w takich strukturach wody
i tworzyć tzw. klatraty (związki okluzyjne). Następstwem
poczęciem znieczulenia ogólnego jest na ogół niewy-
tworzenia klatratów byłaby stabilizacja błony neuronalnej.
starczająca, aby zapobiec zwolnieniu czynności serca Przeciwko temu założeniu przemawia jednak fakt, że w wa-
warunkowanemu pobudzeniem nerwu błędnego. (Je- runkach znieczulenia ogólnego klatraty nie mają stabilnego
śli podczas operacji dojdzie do zagrażającego zwol- charakteru.
nienia czynności serca, zawsze możliwe jest natych-
miastowe uzyskanie jej przyspieszenia przez dożylne W sumie można stwierdzić, że działanie anestetyczne
podanie atropiny). Ponadto współczesne wziew- wziewnych środków do znieczulenia ogólnego opiera
ne środki do znieczulenia ogólnego (zob. poniżej) się na złożonym oddziaływaniu z lipidami, białkami
w znacznie mniejszym stopniu niż eter pobudzają i wodą – w obrębie błony komórkowej. Nowe wy-
wydzielanie śliny i śluzu. niki badań z zakresu biologii molekularnej sugerują,
Jeśli mimo to mają być zapobiegawczo podane że bardziej prawdopodobne jako wyjaśnienie dzia-
leki cholinolityczne, powinny być one podane dożyl- łania ogólnie znieczulającego jest ich bezpośrednia
nie krótko przed rozpoczęciem znieczulenia ogólne- interakcja z białkami niż odkładanie się w podwójnej
go. W ten sposób zaoszczędza się także pacjentowi błonie lipidowej. Mało specyficzny uchwyt działania
nieprzyjemnej suchości w jamie ustnej przed rozpo- wziewnych anestetyków warunkuje ich małą rozpię-
częciem anestezji. tość (szerokość) terapeutyczną oraz ich niekorzystny
wpływ na funkcje układu sercowo-naczyniowego
oraz metabolizm.
Ryzyko znieczulenia ogólnego. Dzięki wprowadze-
W przeciwieństwie do anestetyków wziewnych –
niu znieczulenia ogólnego złożonego i premedykacji
anestetyki dożylne wywołują tylko pojedyncze efekty
ryzyko związane z anestezją wyraźnie się zmniejszyło.
składowe z całościowej koncepcji znieczulenia ogól-
Mimo niewielkiej rozpiętości terapeutycznej więk-
nego; stąd też zawsze konieczne jest stosowanie te-
szości anestetyków – przy założeniu że są prawidło-
rapii skojarzonej obejmującej różne leki. To bardziej
wo podane – należy liczyć się z ryzykiem wystąpie-
selektywne działanie anestetyków dożylnych można
nia jednego warunkowanego anestezją zejścia śmier-
tłumaczyć tym, że prawie wszystkie leki z tej grupy
telnego na 105–106 zabiegów.
ujawniają swoje działanie poprzez specyficzne recep-
tory (zob. poniżej).
Mechanizm działania. Mechanizm działania środ-
ków do znieczulenia ogólnego nie jest do końca zna-
ny. Brak jest jak dotychczas teorii, która by w satys- Złośliwa hipertermia. W bardzo rzadkich przypad-
fakcjonujący sposób wyjaśniały, dlaczego chemicznie kach może dojść w trakcie znieczulenia ogólnego
całkowicie różniące się substancje – gazy szlachetne, do znacznego podniesienia się temperatury ciała.
gaz rozweselający, węglowodory, eter, barbiturany, Tego rodzaju powikłanie wiąże się z nasilonym uwal-
steroidy i in. – wywołują znieczulenie ogólne. Więk- nianiem jonów wapnia z siateczki sarkoplazmatycz-
szość teorii opiera się na wywoływanych przez an- nej pod wpływem wziewnego środka do znieczulenia
estetyki zmianach właściwości fizykochemicznych ogólnego (np. halotanu) lub leków zwiotczających
błon neuronalnych. mięśnie (np. suksametonium) u pacjentów z gene-
MUTSCHLER-2009.indd 299 2010-01-07 22:12:16
300 Układ nerwowy
tycznie warunkowanym zaburzeniem sprzężenia elek- C A/ C I
tro-mechanicznego. W okresie wcześniejszym hiper- N2O
termia miała niemal zawsze przebieg śmiertelny, jed- 1
desfluran
nak obecnie może być skutecznie opanowana dzięki
sewofluran
parenteralnemu podaniu dantrolenu.
0,8
izofluran
0,6
1.7.1. Anestetyki wziewne halotan
0,4
Zaletą znieczulenia ogólnego wziewnego jest łatwość
w jego sterowaniu. Sterowność (sterowalność) an-
estezji jest tym lepsza, im szybciej dana substancja 0,2
do znieczulenia ogólnego jest wchłaniana i wyda-
lana, tj. im krótszy jest czas potrzebny od wprowa-
dzenia do anestezji (od rozpoczęcia podawania leku) 0
5 10 15 20 25 30
do osiągnięcia określonej głębokości znieczulenia,
podanie (min)
oraz odwrotnie – od tego stadium anestezji do wybu-
dzenia się pacjenta. Anestetyki wziewne szczególnie Ryc. B 1.7-3. Stosunek stężenia pęcherzykowego (CA) aneste-
nadają się do podtrzymywania znieczulenia ogólne- tyku wziewnego do jego stężenia w powietrzu wdychanym
go, a z kolei szybko wchłaniane środki dożylne (zob. (CI). Narastanie następuje najszybciej w przypadku związków
poniżej) – odwrotnie – do wprowadzenia do aneste- źle rozpuszczalnych, a najwolniej – związków dobrze rozpusz-
zji. Szybkość wchłaniania i wydalania wziewnych czalnych.
anestetyków zależą głównie od gradientu między
stężeniem w powietrzu oddechowym i we krwi, jak
estetyku w powietrzu pęcherzykowym, a w ten spo-
również od rozpuszczalności anestetyku we krwi.
sób na głębokość znieczulenia.
Im mniejszy jest wskaźnik podziału krew/gaz, tym
szybciej następuje znieczulenie ogólne i tym szybciej
Anestetyki gazowe (np. gaz rozweselający) słabo
również ono ustępuje (zob. ryc. B 1.7-3). Wskaźniki
rozpuszczają się we krwi. Ze względu na koniecz-
podziału (dystrybucji) wziewnych leków do znieczu-
ne do osiągnięcia odpowiedniej głębokości anestezji
lenia ogólnego na granicy krew/gaz zostały zestawio-
wysokie stężenie w powietrzu oddechowym – szyb-
ne w tab. B 1.7-1.
ko ulegają wchłanianiu, jak też są szybko wydalane
Przy rozważaniu zagadnienia sterowania znieczu-
po zaprzestaniu podawania. Już po kilku minutach
leniem należy ponadto zwrócić uwagę na to, że głę-
osiągana jest wymagana głębokość znieczulenia
bokość anestezji, wywołanej określonym anestety-
ogólnego i równie szybko pacjent wybudza się po za-
kiem, jest uzależniona od jego stężenia w ośrodko-
kończeniu podawania anestetyku.
wym układzie nerwowym, a to z kolei zależy od:
■ stężenia anestetyku we wdychanym powietrzu, Tabela B 1.7-1. Wskaźniki podziału (dystrybucji) wziewnych an-
a w następstwie – w powietrzu pęcherzykowym, estetyków na granicy krew/gaz i olej/gaz w temperaturze 37°C.
MAC – minimalne stężenie pęcherzykowe przy 1 atmosferze
■ częstości i głębokości oddechu, u pacjentów w średnim wieku
■ przepuszczalności błon pęcherzykowo-kapilar-
nych, krew/gaz olej/gaz MAC (% obj.)
■ przepływu krwi w płucach i mózgu, halotan 2,4 224 0,8
■ rozpuszczalności anestetyku we krwi, enfluran 1,8 97 1,7
■ współczynnika podziału anestetyku pomiędzy krew izofluran 1,4 91 1,2
i tkankę mózgową.
sewofluran 0,7 47 1,7
desfluran 0,4 19 6,0
Przez zmiany wentylacji i/lub zmiany w ilości an-
estetyku dostarczanego we wdychanym powietrzu podtlenek azotu 0,5 1,4 105
anestezjolog może szybko wpływać na stężenie an-
MUTSCHLER-2009.indd 300 2010-01-07 22:12:16
Układ nerwowy 301
W przypadku stosowanych pod postacią par płyn- pin lub analgetyków opioidowych obniża wartości
nych anestetyków (np. eteru), które są zwykle MAC o 50 lub 65%. U osób z przewlekłą choro-
lepiej rozpuszczalne we krwi, znieczulenie ujaw- bą alkoholową zapotrzebowanie na wziewne środki
nia się zwykle przy zastosowaniu dużych stężeń do znieczulenia jest natomiast podwyższone.
Układ nerwowy
(tzw. zalania), tzn. gradient celowo ustawia się po-
czątkowo wysoko, tak aby skrócić czas potrzebny
do osiągnięcia żądanej głębokości anestezji. Następ- 1.7.1.1. Podtlenek azotu (N2O,
nie zmniejsza się stężenie anestetyku w powietrzu gaz rozweselający)
oddechowym – tak aby osiągnąć wartość potrzebną
do podtrzymywania znieczulenia. Faza wychodze-
Podtlenek azotu – bezbarwny i mało reaktywny gaz,
nia ze znieczulenia wywołanego parującymi aneste-
tykami – w przeciwieństwie do fazy wprowadza-
o słabym słodkawym zapachu i smaku, występu- B1
jący w handlu pod postacią skroploną, jest jednym
nia w stan znieczulenia – nie może być skrócona,
z najczęściej stosowanych oraz najmniej toksycznych
gdyż nie istnieją możliwości zwiększenia gradientu.
środków do znieczulenia ogólnego. Wykazuje silne
W przypadku niekiedy długo trwającej fazy wy-
działanie analgetyczne, ale stosunkowo słabe działa-
chodzenia ze znieczulenia ujawnia się odwracalne
nie narkotyczne i zupełnie pozbawiony jest działania
stadium pobudzenia (szczególnie w przypadku mo-
zwiotczającego mięśnie. Nawet dla wartości 75 pro-
noterapii). Ten stan jest dla pacjenta zarówno su-
cent objętościowych (% obj. = vol. %) gazu rozwese-
biektywnie nieprzyjemny (kaszel, wymioty, pobu-
lającego we wdychanym powietrzu nie jest możliwe
dzenie), jak i obiektywnie niekorzystny – z powodu
osiągnięcie głębokiego znieczulenia ogólnego. 70%
zwiększonego zagrożenia wystąpienia powikłań po-
obj. gazu rozweselającego we wdychanym powietrzu
operacyjnych.
stanowi jednak górną granicę, gdyż równocześnie
musi być podawane przynajmniej 30% obj. tlenu,
Minimalne pęcherzykowe stężenie anestetyków
aby zapobiec jego niedoborowi. Dlatego też pod-
wziewnych (MAC – minimal alveolar concentra-
tlenek azotu stosuje się łącznie z innymi środkami
tion). Jak już wspomniano, głębokość znieczulenia
do znieczulenia ogólnego (np. z izofluranem), a po-
ogólnego wywołanego anestetykiem wziewnym
nieważ nie ma on działania zwiotczającego mięśnie
zależy od jego ciśnienia parcjalnego w mózgu.
– częstokroć kojarzy się go również z lekami miore-
Nie może ono jednak być zmierzone bezpośrednio,
laksacyjnymi.
natomiast jest możliwe jego określenie pośrednio
badaniem warunków równowagi w stanie znieczule- Przy zastosowaniu sztucznego oddychania z nadciśnieniem
nia ogólnego, gdyż w tym przypadku ciśnienie par- (wartość MAC rzędu 105%) możliwe jest uzyskanie peł-
cjalne w mózgu jest identyczne jak w pęcherzykach. nego znieczulenia ogólnego (stadium III) także za pomocą
Stężenie w pęcherzykach odzwierciedla tym sa- mieszanki podtlenku azotu z tlenem; tego rodzaju metoda
mym stężenie w mózgu i dlatego może służyć także nie przyjęła się jednak do powszechnego zastosowania.
do porównania siły działania różnych anestetyków
wziewnych. Porównuje się przy tym tzw. minimal- Szybkość wchłaniania i wydalania gazu rozweselają-
ne stężenie pęcherzykowe różnych substancji. Przez cego jest bardzo duża, stąd też znieczulenie ogólne
pojęcie to należy rozumieć stężenie pęcherzykowe za pomocą tego anestetyku jest łatwe do sterowania.
anestetyku wziewnego (wyrażone w procentach Prawie nie wywiera on wpływu na ciśnienie krwi
wartości jednej atmosfery), dla którego 50% wszyst- i oddychanie, zupełnie nie wpływa na funkcje wą-
kich pacjentów nie reaguje już odruchami obronny- troby, nerek i jelit. Jednak w przypadku długo prze-
mi na nacięcie skóry. W tabeli B 1.7-1 zestawione prowadzanego znieczulenia, jakie zwykle jest ko-
zostały wartości MAC najważniejszych wziewnych nieczne w przypadku operacji chirurgicznych, lub też
anestetyków dla pacjentów w średnim wieku, u któ- przy powtarzanym stosowaniu – może niekorzystnie
rych nie było stosowane żadne inne leczenie dodat- wpłynąć na produkcję czerwonych i białych krwinek,
kowe. Różne wziewne środki stosowane do znie- aż do wywołania niedokrwistości megaloblastycznej.
czulenia ogólnego posiadają różną moc działania Aby zapobiec hipoksji dyfuzyjnej pod koniec znie-
anestetycznego, która koreluje z rozpuszczalnością czulenia podtlenkiem azotu – należy zwiększyć ilość
danej substancji w tłuszczach (wyrażoną pod posta- podawanego tlenu w trakcie wychodzenia ze znie-
cią współczynnika podziału olej/gaz). Wyższe war- czulenia ogólnego, gdyż w pierwszych minutach
tości MAC stwierdza się u noworodków – wyraźnie po przerwaniu podawania gazu dochodzi do znacz-
niższe niż u starszych pacjentów w wieku powyżej nego zwiększenia się ilości (uwalniania się) N2O
70 roku życia. Równoczesne podanie benzodiaze- w pęcherzykach płucnych – co jest wynikiem jego
MUTSCHLER-2009.indd 301 2010-01-07 22:12:17
302 Układ nerwowy
złej rozpuszczalności we krwi, a co prowadzi do roz- ogólnych z zastosowaniem enfluranu opisywano wy-
cieńczenia stężenia wdychanego tlenu. stępowanie toniczno-klonicznych skurczów mięśni
ze zmianami w EEG, tak więc nie powinien być on
stosowany u pacjentów predysponowanych (pacjent-
1.7.1.2. Eter dietylowy ki z zatruciem ciążowym, chorzy z padaczką).
(C2H2–O–C2H5, eter)
Dalsze badania rozwojowe nad enfluranem i izoflu-
Pomimo licznych wad – nietrwałości, niebezpieczeństwa ranem doprowadziły – w wyniku zastąpienia atomów
wybuchu, powolnego wchłaniania i wydalania, wymiotów chloru atomami fluoru – do stworzenia związków
po zakończeniu znieczulenia – eter był dawniej stosowany o mniejszej rozpuszczalności w wodzie: sewofluranu
bardzo często jako środek do anestezji, gdyż charakteryzu-
je się dużą rozpiętością znieczulenia i dobrym działaniem i desfluranu, które są podobnie łatwe w sterowaniu
zwiotczającym mięśnie, a ponadto znieczulenie za pomocą jak gaz rozweselający (zob. ryc. B 1.7-3). Zapewnia
eteru może być prowadzone bez dużych nakładów apara- to krótki czas wychodzenia ze znieczulenia ogólnego
turowych (eter zakrapia się na maskę pokrytą gazą). Poza pod koniec anestezji, dzięki czemu obie substancje
ośrodkowym działaniem zwiotczającym mięśnie eter wywo- – szczególnie desfluran – zyskują stale na znaczeniu
łuje także efekt kuraropodobny. Już w stadium III,l mięśnie
poprzecznie prążkowane są w znacznym stopniu zwiotczo- w zakresie chirurgii ambulatoryjnej. Ich działania
ne. Obecnie jednak – przynajmniej w krajach uprzemy- na układ oddechowy, sercowo-naczyniowy i mięśnio-
słowionych – eter nie jest już stosowany do znieczulenia wy przypominają właściwości izofluranu. Podobnie
ogólnego. jak izofluran – również sewofluran i desfluran – nasi-
Eter dietylowy jest silnie działającym środkiem do znie- lają działanie niedepolaryzujących leków zwiotczają-
czulenia ogólnego: wystarczy 3–4% objętościowych w po-
wietrzu oddechowym, aby uzyskać stadium tolerancji. Nie- cych mięśnie, tak że dawki tych ostatnich mogą być
mal nie wpływa na krążenie i oddychanie aż do stadium zmniejszone.
III,2. Naczynia skórne ulegają rozszerzeniu już w stadium I. Desfluran jest metabolizowany zaledwie w 0,02%
Stąd też możliwe jest wychłodzenie pacjenta w czasie trwa- – i nie jest ani nefro- ani hepatotoksyczny. Sewoflu-
nia znieczulenia ogólnego. Częstość pracy serca zwiększa ran w 5% ulega rozkładowi do fluorku i heksaflu-
się wskutek podwyższenia napięcia układu współczulnego.
Mogą wystąpić skurcze dodatkowe nadkomorowe, jednak oro-izopropanolu. Współczynnik biotransformacji
eter – w przeciwieństwie do halogenowanych węglowo- wątrobowej sewofluranu jest tym samym dwukrot-
dorów – nie zwiększa wrażliwości układu przewodzącego nie większy niż enfluranu i 250-krotnie większy
serca na katecholaminy. niż desfluranu. Sewofluran jest wśród nowoczes-
nych pochodnych eteru jedynym związkiem, który
nie ulega metabolizmowi do toksycznego kwasu tri-
1.7.1.3. Eter halogenowany fluorooctowego. Wadą desfluranu jest to, że silniej
niż sewofluran drażni drogi oddechowe, a z powodu
Izofluran i enfluran są niepalnymi, bezbarwnymi, niskiej temperatury wrzenia (23°C) trzeba stosować
o zapachu zbliżonym do eteru, cieczami o wysokim specjalny parownik.
ciśnieniu parowania, które względnie szybko ulegają
wchłanianiu i wydalaniu. Podobnie do halotanu (zob.
poniżej) wpływają depresyjnie na oddech (enfluran >
izofluran > halotan); ponadto mają też działanie uczu-
F F F F Cl
lające na katecholaminy (halotan > enfluran) oraz
F F
ujemne działanie inotropowe (halotan > izofluran > F O F O
enfluran). Izofluran i enfluran obniżają obwodowy F
Cl F
opór naczyniowy.
Współczynnik (stopień) biotransformacji izoflura- enfluran izofluran
nu jest niewielki – wynosi on jedynie około 0,2%;
z kolei w przypadku enfluranu współczynnik ten
F
jest względnie wysoki (ok. 3%). Jako metabolity F F
F F
enfluranu zostały zidentyfikowane: kwas difluoro-
F F
metoksy-difluorooctowy i fluorek, które są nefro- F O F O
F F
toksyczne. Nawet jeżeli stężenie fluorku po zastoso- F F
waniu enfluranu jest tak niskie, że nie osiąga progu sewofluran desfluran
neurotoksyczności. W Polsce jest on nadal używa-
ny, mimo że współczynnik metabolizmu izofluranu
jest o 15 razy mniejszy. Ponadto w trakcie znieczuleń
MUTSCHLER-2009.indd 302 2010-01-07 22:12:17
Układ nerwowy 303
1.7.1.4. Halogenowane węglowodory ■ kwas 4-hydroksymasłowy,
■ opioidy (fentanyl, alfentanyl, sufentanyl, remifen-
Jedynym jeszcze, lecz obecnie w Niemczech już niesto-
sowanym (zob. poniżej), halogenowanym węglowodorem tanyl),
jest halotan (F3C–CHClBr) – wrząca w temperaturze 50°C,
Układ nerwowy
■ dożylnie postacie benzodiazepin.
bezbarwna ciecz o przyjemnym zapachu. Pod wpływem
światła rozpada się do lotnych kwasów (konieczność prze-
chowywania w ciemnych naczyniach). Halotan działa słabo W przypadku wszystkich dożylnie podawanych an-
analgetycznie i miorelaksacyjnie. Zaletą jest to, że halotan: estetyków – cechą wspólną jest natychmiastowe dzia-
łanie oraz niewielka możliwość sterowania (mała ste-
■ z powietrzem lub tlenem nie tworzy wybuchowych mie-
rowalność) znieczuleniem. Ich zaletą jest psychiczne
szanek,
oszczędzanie pacjenta, gdyż w przypadku większości
■ działa silnie anestetycznie (konieczne dla podtrzymania substancji traci on natychmiast przytomność i oszczę- B1
stadium tolerancji stężenia w powietrzu oddechowym
wynoszą jedynie 0,5% objętościowych), dzone są mu zakładanie maski (w przypadku aneste-
zji wziewnej) oraz okres wybudzania się. Wykazują
■ nie powoduje podrażnienia śluzówek,
one jednak równocześnie wadę – podwyższone ryzyko
■ obniża napięcie mięśniówki oskrzeli (możliwe stosowa- anestezji: lek raz podany za pomocą iniekcji pozosta-
nie u pacjentów z astmą oskrzelową). je później poza jakąkolwiek kontrolą anestezjologa.
Wymienionym zaletom przeciwstawia się jednak wiele Przebieg anestezji związany jest już tylko z procesa-
wad, z powodu których halotan jest coraz rzadziej stoso- mi farmakokinetycznymi (dystrybucja, metabolizm,
wany: wydalanie). Wyjątek stanowią benzodiazepiny oraz
■ wąska rozpiętość anestezji, opioidy.
W przeciwieństwie do anestetyków wziewnych
■ działanie depresyjne (tłumiące) na oddychanie,
– siła działania anestetyków dożylnych nie może być
■ uczulenie (uwrażliwienie) mięśnia sercowego na kate- zmniejszona (zakończona) poprzez wpływ na wen-
cholaminy z wiążącym się z tym ryzykiem wystąpienia
tylację, lecz wiąże się głównie ze specyficznymi dla
zaburzeń rytmu serca (nawet do migotania komór),
tych substancji czynnikami, takimi jak redystrybucja
■ zależny od stężenia spadek ciśnienia tętniczego krwi i ob- w tkankach. Na przykład krótki czas działania tiobar-
jętości wyrzutowej serca w wyniku bradykardii i zmniej-
szenia siły skurczu serca, bituranów związany jest głównie z ich redystrybucją
z (silnie ukrwionego) ośrodkowego układu nerwo-
■ uszkodzenie wątroby w przypadku zastosowania wyso-
wego do (słabiej ukrwionych) mięśni (zob. poniżej).
kich stężeń halotanu lub też powtarzanych znieczuleń
ogólnych („halotan-hepatitis”) w wyniku reakcji tok- W przypadku wielokrotnej podaży leku na drodze
sycznych i/lub alergicznych (wskaźnik biotransforma- dożylnej istnieje niebezpieczeństwo kumulacji,
cji do 20%, główny metabolit – kwas trifluorooctowy; w związku z tym, że lipofilne anestetyki są metaboli-
w dalszej kolejności tworzenie reaktywnych produktów zowane względnie wolno.
pośrednich, które wiążą się kowalencyjnie z białkami or-
ganizmu)
■ praktyczny brak działania analgetycznego i jedynie słabe
działanie zwiotczające mięśnie.
1.7.2.1. Barbiturany N-metylowane
i tiobarbiturany
1.7.2. Anestetyki dożylne Z dużej grupy barbituranów do znieczulenia ogólne-
go stosowane są jedynie:
Anestetyki dożylne (infuzyjne, iniekcyjne) służą ■ metoheksital – N-metylowany barbituran,
głównie do wprowadzenia do anestezji. Do uzyski-
■ tiopental – tiobarbituran.
wania znieczulenia ogólnego na drodze podania do-
żylnego służą następujące substancje:
W związku ze słabą trwałością roztworów wodnych
■ barbiturany N-metylowane, występują one w handlu jako sole sodowe pod po-
stacią ampułek z suchą substancją i są podawane do-
■ tiobarbiturany,
żylnie jako 1- lub 2,5proc. silnie alkaliczny roztwór
■ etomidat, (wartość pH ok. 11).
■ propofol,
Korzystne jest to, że działanie narkotyczne obu sub-
■ ketamina, stancji rozpoczyna się bardzo szybko, pod koniec
MUTSCHLER-2009.indd 303 2010-01-07 22:12:17
304 Układ nerwowy
się częstość pracy serca. Zwykle zmniejsza się ciś-
nienie tętnicze krwi, a efekt ten występuje zwłaszcza
O NH O
u pacjentów z nadciśnieniem. W wyniku rozszerze-
H2C
N nia się żył dochodzi do wytworzenia się „puli żylnej”
H5C2 C C CH3
(venous pooling). Ponadto barbiturany mogą prowa-
CH3 O dzić do uwalniania histaminy z tkankowych komórek
tucznych, co może objawić się skurczem oskrzeli.
metoheksital
Zahamowanie odruchów obronnych nie jest szcze-
gólnie nasilone, również zwiotczenie mięśni jest nie-
O NH S wielkie.
H5C2 Podane do krwiobiegu metoheksital i tiopental
N
H7C3 H w znacznym stopniu wiążą się z białkami osocza.
CH3 O Leki ulegają dystrybucji zasadniczo w obrębie naj-
bardziej ukrwionych narządów, tj. w narządach,
tiopental w których udział objętości wyrzutowej serca jest naj-
większy (zob. ryc. B 1.7-5). W drugiej kolejności na-
stępuje szybka redystrybucja z ośrodkowego układu
nerwowego głównie do mięśni, które stanowią nie-
znieczulenia rzadko dochodzi do stanów pobudzenia mal połowę masy ciała. Ustalenie równowagi między
lub wymiotów, po zastosowaniu typowych dawek osoczem a mięśniami następuje w ciągu 15–30 mi-
pacjent wybudza się również szybko, a powikłania nut od iniekcji. To właśnie tego rodzaju redystrybu-
zdarzają się rzadko. cja, a nie jak wcześniej przyjmowano redystrybucja
do tkanki tłuszczowej, określa czas trwania działania
Barbiturany lub tiobarbiturany prowadzą w wyniku znieczulającego obu leków. W przypadku metoheksi-
działania na benzodiazepinowo-barbituranowy recep- talu dodatkowym czynnikiem jest biotransformacja.
tor kanału chlorkowego kompleksu GABA – jednakże Niewielkie ukrwienie tkanki tłuszczowej – mimo
w obrębie innego miejsca wiązania niż benzodiaze- dobrej rozpuszczalności metoheksitalu i tiopentalu
piny – do wzmożonego napływu (wnikania) jonów w tłuszczach – powoduje, że równowaga dystrybucji
chlorkowych, tym samym do hiperpolaryzacji ko- między osoczem a tkanką tłuszczową jest osiągana
mórek nerwowych. W większych dawkach prowadzą dopiero po 1,5–2,5 godz.
ponadto do nieselektywnego tłumienia procesów prze-
biegających w ośrodkowym układzie nerwowym. Metoheksital ulega w znaczącej części biotransfor-
Pacjenci zasypiają zwykle jeszcze w trakcie prze- macji w wątrobie do nieczynnych metabolitów (okres
prowadzania iniekcji, a utrata przytomności jest wywo- półtrwania 2 godziny). Z tiopentalu (okres półtrwania
łana hamowaniem wstępującego układu siatkowatego.
Pewną wadą jest to, że ani barbiturany, ani tiobarbi-
turany nie mają działania analgetycznego. W zakresie %
dawek subnarkotycznych zwiększają nawet czucie 100
bólu (hiperalgezja). W związku z niebezpieczeństwem krew
wystąpienia reakcji wegetatywnych na bodźce bólo- 80 mięśnie i skóra
OUN,
dobrze ukrwione tkanka
we – zabiegi chirurgiczne w anestezji barbituranowej tłuszczowa
60 narządy
lub tiobarbituranowej mogą być wykonywane jedynie
w przypadku jednoczesnego podania odpowiednich
dawek leków o silnym działaniu przeciwbólowym 40
(opioidów z grupy fentanylu; zob. poniżej).
Wpływ tłumiący na oddychanie zależny jest od 20
dawki; w przypadku przedawkowania może dojść
do całkowitego porażenia ośrodka oddechowego. 0
1/16 1/8 1/4 1/2 1 2 4 8 16 32 64 128 256
Dlatego zawsze musi istnieć możliwość wykonania czas (min)
i. v.
sztucznego oddychania. W obrębie serca barbitura-
ny i tiobarbiturany wykazują ujemne działanie ino- Ryc. B 1.7-5. Dystrybucja tiopentalu (po podaniu pod posta-
tropowe: zmniejsza się objętość wyrzutowa serca, cią bolusa) w różnych tkankach w zależności od czasu (według
w związku z czym na drodze odruchowej zwiększa Price’a i in.).
MUTSCHLER-2009.indd 304 2010-01-07 22:12:17
Układ nerwowy 305
5–6 godzin) powstaje w wyniku częściowej wymiany
siarki na tlen względnie długo działający pentobar-
bital.
N
Z właściwości kinetycznych metoheksitalu i tio-
Układ nerwowy
pentalu można wyciągnąć następujące wnioski H5C2OOC N
do praktycznego zastosowania w anestezjologii:
CH3
■ Powtórna iniekcja tiopentalu może w określonych
okolicznościach prowadzić do niebezpiecznej ku-
mulacji leku. etomidat
■ Nie należy zwiększać dawki u osób otyłych, gdyż
ośrodkowy kompartment nie jest u nich większy B1
niż w przypadku osób z prawidłową masą ciała.
przez R(+)-enancjomer występuje bardzo szybko
■ Stany wstrząsu są względnym przeciwwskazaniem,
i utrzymuje się dość krótko. Po szybkiej iniekcji do-
gdyż we wstrząsie redystrybucja do wówczas źle
żylnej lek wykazuje działanie ok. 15 razy silniejsze
ukrwionych mięśni przebiega bardzo wolno.
od tiopentalu. Również zakres działania terapeu-
■ W przypadku oznak zagrażającej śpiączki wątrobo- tycznego jest większy. Podobnie jak barbiturany (a
wej nie powinno się podawać metoheksitalu. w przeciwieństwie do ketaminy) – etomidat nie ma
działania analgetycznego. Prawie nie wywiera wpły-
Metoheksital i tiopental wskazane są do stosowa- wu na siłę skurczu serca oraz na ciśnienie krwi.
nia we wprowadzeniu do znieczulenia ogólnego, Etomidat ulega – podobnie jak większość innych
a wspólnie z substancjami o działaniu analgetycznym anestetyków dożylnych – szybkiej dystrybucji, a wią-
– w krótkotrwałych zabiegach chirurgicznych. zanie z białkami osocza wynosi 75%. Głównym me-
Dawkowanie metoheksitalu i tiopentalu zależy tabolitem jest wolny, niemający już działania aneste-
ściśle od wyników działania! (średnie dawkowanie tycznego kwas karbonylowy, który w 75% wydalany
soli sodowej metoheksitalu wynosi 1–2 mg/kg m.c, jest przez nerki, a w 15% z kałem.
a soli sodowej tiopentalu 3–5 mg/kg m.c.). W związku z brakiem działania analgetycznego
Jako działania niepożądane użycia metoheksitalu etomidat nie nadaje się do stosowania w monoterapii.
obserwowano głównie spadki ciśnienia, palpitacje Stosowany jest on do wprowadzania do znieczulenia
serca, ból i uszkodzenie nerwów w okolicy iniekcji, ogólnego – szczególnie u pacjentów z grup podwyż-
napady drgawkowe, nadmierne ślinienie się, niepokój szonego ryzyka w zakresie serca i układu krążenia.
i bóle głowy. Tiopental wywołuje natomiast nieprzy- Dawkowanie wynosi 0,15–0,3 mg/kg m.c.
jemne marzenia senne, jak również euforyczne pod- W przypadku niewystarczającej premedykacji
wyższenie nastroju. W pojedynczych przypadkach mogą wystąpić spontaniczne, niekontrolowane ruchy
obserwowano ciężkie reakcje alergiczne. mięśni. W dalszej kolejności obserwowane są doleg-
Dobitnie należy wskazać na to, że przypadkowa liwości ze strony żył oraz niekiedy ich zapalenie.
iniekcja do naczynia tętniczego prowadzi do cięż- Ponadto w wyniku hamowania 11β-hydroksylazy
kich zaburzeń ukrwienia i uszkodzenia tkanek, które etomidat już w zwyczajowych dawkach terapeutycz-
w określonych okolicznościach może spowodować nych prowadzi do obniżenia poziomu kortyzolu i al-
nawet utratę kończyny. dosteronu w osoczu.
W ciężkich zaburzeniach funkcji nerek i wątroby
(zob. powyżej), stanach śpiączki, w ostrych porfiriach
wątrobowych, ciężkiej hipowolemii i we wstrząsie,
jak również w stanie astmatycznym – stosowanie me- 1.7.2.3. Propofol
toheksitalu i tiopentalu jest przeciwwskazane.
Propofol jest źle rozpuszczalną w wodzie podsta-
wioną w pozycji 2 i 6 pochodną fenolu. Po podaniu
dożylnym prowadzi szybko do utraty przytomności,
1.7.2.2. Etomidat która przy zwykłym dawkowaniu (2–2,5 mg/kg m.c.)
utrzymuje się 5–8 minut. Podobnie jak inne anestety-
Etomidat jest estrem kwasu imidazolo-5-karbonylo- ki dożylne, również propofol nie wykazuje działania
wego. Działanie anestetyczne wywoływane jedynie analgetycznego.
MUTSCHLER-2009.indd 305 2010-01-07 22:12:18
306 Układ nerwowy
Wartości skurczowego i rozkurczowego ciśnienia matu i S-enancjomeru – prowadzi do stanu, który
krwi ulegają obniżeniu – odpowiednio o 10–20 i 5– określany jest pojęciem „anestezji rozszczepionej”
15 mm Hg; zmiany w zakresie częstości pracy serca (dissociative/dissociated anesthesia; równoczesne
są zwykle mniej nasilone. U większości pacjentów stany pobudzenia i hamowania w obrębie ośrodko-
występuje przejściowy bezdech, który w określonych wego układu nerwowego – przyp. tłum.). Ketamina
warunkach wymaga zastosowania kontrolowanego blokuje sterowane ligandami receptory NMDA (zob.
oddychania. poniżej) – poprzez działanie na miejsce wiązania fen-
cyklidyny (PCP). W wyniku tego dochodzi głównie
do przerwania dróg kojarzeniowych oraz rozłączenia
z korą mózgową i wzgórzem wzrokowym, podczas
gdy wpływ na układ limbiczny (rąbkowy) jest mniej-
OH szy. Pacjent wydaje się bardziej nieobecny duchowo
(H3C) 2HC CH(CH3) 2 niż śpiący.
Ketamina ma silne działanie analgetyczne. W cią-
gu 30–60 sekund po podaniu dożylnym dochodzi
propofol do całkowitej analgezji, która utrzymuje się jeszcze
po wyjściu ze znieczulenia ogólnego, jak również
do niepamięci (amnezji). Utrzymują się odruchy gar-
dłowe, a napięcie mięśniowe zmniejsza się jedynie
nieznacznie. Oczy pozostają często szeroko otwar-
te. Wyjątkowy wśród anestetyków jest aktywujący
Molekularny mechanizm działania propofolu na- wpływ ketaminy na układ krążenia, który może być
dal nie jest jasny. Dyskutowane są liczne mechani- korzystny (w przypadku zbyt niskiej objętości wy-
zmy: niespecyficzne działanie na błony lipidowe, rzutowej), lub niekorzystny (np. w przypadku osób
interakcja z białkami kanałów sodowych oraz nasi- z nadciśnieniem). Ciśnienie krwi i częstotliwość pra-
lenie działania hamującego GABA w ośrodkowym cy serca zwiększają się – głównie na samym początku
układzie nerwowym. – w związku z pobudzeniem układu współczulnego.
Dystrybucja propofolu przebiega w dwóch fa- W przypadku stosowania zwykłych dawek ketaminy
zach o różnych okresach półtrwania: okres półtrwa- jej wpływ na oddychanie jest niewielki.
nia pierwszej fazy wynosi kilka minut, drugiej – od Ketamina ulega szybkiej dystrybucji w organi-
pół godziny do godziny. Wiązanie z białkami osocza zmie; wiązanie z białkami osocza wynosi mniej niż
jest wysokie i wynosi 98%. Propofol ulega w wątro- 50%. Również w jej przypadku za zmniejszenie się
bie procesowi częściowej hydroksylacji w pozycji 4, (ustąpienie) działania odpowiedzialne są głównie
a wydalany jest głównie pod postacią siarczanów i procesy redystrybucji. Wydalanie następuje głównie
glukuronianów. przez nerki pod postacią metabolitów (produktów
Propofol stosowany jest do wprowadzania do an- hydroksylacji nadal jeszcze aktywnego metabolitu
estezji (dawkowanie: 1,5–2,5 mg/kg m.c.); ponadto – norketaminy oraz ich koniugatów).
jako składnik hipnotyczny, łącznie z opioidami z gru- Ketamina wskazana jest do wprowadzania do an-
py fentanylu nadaje się do wywołania tzw. całkowitej estezji w przypadku krótkotrwałych i bolesnych za-
anestezji dożylnej (zob. poniżej; dawkowanie: 0,1– biegów (np. w przypadku poparzeń), jak również
0,2 mg/kg m.c. – podawane we wlewie trwającym w medycynie ratunkowej i katastrof w przypadku
minutę). wystąpienia urazów masowych.
Jako działania niepożądane opisano m.in. spad-
ki ciśnienia, depresję oddechową, bradykardię aż
do zatrzymania akcji serca oraz reakcje alergiczne
na składniki pomocnicze preparatu (np. olej sojowy). CH3 CH3
HN HN
1.7.2.4. Ketamina Cl Cl
O O
ketamina esketamina
Ketamina różni się istotnie w zakresie niektórych
właściwości od innych dożylnych anestetyków. Znaj-
dująca się w handlu substancja – pod postacią race-
MUTSCHLER-2009.indd 306 2010-01-07 22:12:18
Układ nerwowy 307
Dawkowanie wynosi 1–3 mg/kg m.c. i.v. lub 4–8 idami. Stosowane w anestezji dawki wynoszą 60–90
mg/kg m.c. i.m. W przypadku konieczności dalsze- mg/kg m.c.
go prowadzenia anestezji można dostrzyknąć pół Ujawniającymi się działaniami niepożądanymi są
dawki. mioklonie, które mogą być opanowane przez podanie
Układ nerwowy
Jako działania niepożądane opisywane są nieprzy- barbituratów, U pacjentów z silnie ograniczoną funk-
jemne marzenia senne lub omamy występujące w fa- cją nerek może wystąpić hiponatremia i metaboliczna
zie wybudzania się; ich nasilenie może być znacząco zasadowica.
zmniejszone przez równoczesne podanie benzodiaze-
pin (np. midazolamu). Interesujące, że efekty oma-
mowe nie występują lub występują jedynie w bardzo 1.7.2.6. Opioidy
niewielkim nasileniu u dzieci i pacjentów w pode-
szłym wieku. Innymi działaniami niepożądanymi B1
Opioidy – od czasu ich wprowadzenia do anestezji
wartymi wymienienia są nudności, wymioty, zawroty
– znalazły szerokie zastosowanie. Podczas gdy star-
i bóle głowy.
sze substancje, np. morfina lub petydyna, obciążone
Ketamina jest przeciwwskazana u pacjentów
były poważnymi działaniami niepożądanymi, takimi
z dusznicą bolesną, podwyższonym ciśnieniem śród-
jak uwalnianie histaminy lub tłumienie układu serco-
gałkowym lub nadciśnieniem tętniczym, a podobnie
wo-naczyniowego, w przypadku nowszych substan-
także w przypadku zabiegów w obrębie górnych dróg
cji z grupy fentanylu (fentanyl, alfentanyl, sufentanyl,
oddechowych bez równoczesnego podania środków
remifentanyl) efekty te są wyraźnie słabsze. Opioidy
zwiotczających mięśnie, jak również w położnictwie
tego typu są we współczesnej anestezjologii stoso-
– w stanach zagrożenia rzucawką i w rzucawce po-
wane niemal w każdym znieczuleniu ogólnym. Po-
rodowej.
dobnie jak innym anestetykiem również „idealnemu”
S-(+)-ketamina blokuje receptory NMDA 3–4 razy
opioidowi stawia się znaczne wymagania:
silniej niż odpowiedni R-(+)-enancjomer. Doprowa-
dziło to do wprowadzenia na rynek preparatu S-(+)- ■ dobre sterowanie z szybkim występowaniem dzia-
ketaminy, która w porównaniu z racematem pozwala łania i jego ustępowaniem,
na zmniejszenie dawkowania o połowę, a ponadto
■ brak przebiegu działania o kształcie histerezy (zob.
powoduje znacząco mniej reakcji obudzeniowych
poniżej),
(przy wybudzaniu się ze znieczulenia).
■ brak kumulacji w przypadku ciągłej infuzji (we
wlewie) lub powtarzanych wstrzyknięć pod posta-
1.7.2.5. Kwas 4-hydroksymasłowy cią bolusów,
■ (szybki) metabolizm do nieczynnych metabolitów.
Poza terapią narkolepsji kwas 4-hydroksymasłowy
(kwas 4-hydroksybutanowy = kwas gamma-hydrok- Nowoczesne opioidy z grypy fentanylu zbliżają się
symasłowy = GHB) stosowany jest jako anestetyk do tych wymagań. Za pomocą alfentanylu i remi-
dożylny przy porodzie przez cesarskie cięcie, w chi- fentanylu maksymalne działanie uzyskuje się już po
rurgii powypadkowej, w przypadku długotrwałych 1–1,5 minuty po podaniu pod postacią bolusa, pod-
operacji, u chorych z niewydolnością wątroby, przed czas gdy okres latencji w przypadku fentanylu wy-
balonikowaniem serca, a także w chirurgii dziecię- nosi ok. 4 minut, a sufentanylu – ok. 3 minut. Al-
cej. Od kilku lat stosowany jest również w sposób fentanyl i remifentanyl wykazują w związku z tym
nielegalny jako „nokautujące krople” i „płynne jedynie niewielką histerezę (latencja między maksy-
ekstazy” (liquid ecstasy; mimo że substancja ani malnym stężeniem w osoczu a maksymalną efektem
chemicznie, ani farmakologicznie nie jest spo- działania) i stąd spełniają wymóg szybkiego rozpo-
krewniona z ekstazy). Kwas 4-hydroksymasłowy częcia działania.
podlega odpowiednim przepisom dotyczącym środ-
ków odurzających, a preparat handlowy Somsanit Farmakokinetyczne różnice pomiędzy poszczególny-
jest lekiem recepturowym (sama substancja jest jed- mi opioidami związane są głównie z ich właściwoś-
ną z wykorzystywanych w tzw. tabletkach gwałtu ciami fizykochemicznymi (zob. tab. B 1.7-2).
– przyp. tłum.).
Ponieważ kwas 4-hydroksymasłowy nie ma Ustępowanie działania jest przede wszystkim zależne
działania analgetycznego – w trakcie znieczulenia od czasu trwania podawania leku oraz podanej daw-
ogólnego musi być stosowany w skojarzeniu z opio- ki całkowitej. W celu jednoznacznej charakterystyki
MUTSCHLER-2009.indd 307 2010-01-07 22:12:18
308 Układ nerwowy
ustąpienia działania substancji wprowadzono pojęcie
okresu półtrwania zależnego od sytuacji. Określa on
H5C2 O
czas, który jest niezbędny do uzyskania zmniejszenia
o 50% stężenia opioidów w miejscu działania (OUN)
N po zakończeniu ich nieprzerwanej infuzji. Na ryc. B
N
1.7-6 pokazano, że okres półtrwania zależny od sytu-
acji dla fentanylu, alfentanylu i sufentanylu jest zwią-
zany z czasem trwania infuzji. Przykładowo: jeśli fen-
fentanyl tanyl będzie podawany w nieprzerwanej infuzji przez
2 godz., to muszą upłynąć 2 godz. od końca infuzji, do-
H5C2 O
póki stężenie fentanylu w mózgu ulegnie zmniejszeniu
OCH3 do połowy. Tylko dla remifentanylu okres półtrwania
N zależny od sytuacji (3–4 min) jest niezależny od czasu
O
N C2H5 trwania infuzji. Dlatego wśród wszystkich anestety-
N N ków dożylnych remifentanyl wykazuje jak dotąd naj-
N N lepszą sterowność śródoperacyjną. Istotną przyczyną
alfentanyl tego jest niezależny od funkcji wątroby i nerek meta-
bolizm do nieczynnych metabolitów – co przebiega
z udziałem nieswoistych esteraz. Z klinicznego punktu
H5C2 O widzenia jest jednak niekorzystne, że wraz z zakończe-
OCH3
N
niem infuzji remifentanylu niemal błyskawicznie mija
także jego działanie analgetyczne.
N S
Fentanyl, alfentanyl, sufentanyl i remifentanyl
są agonistami receptorów μ-opioidowych. Działania
sufentanyl pożądane (analgezja, sedacja) i niepożądane (depre-
sja oddechowa, bradykardia, sztywność klatki pier-
siowej, nudności, wymioty) są w związku z tym ja-
H5C2 O kościowo takie same jak dla morfiny. Tak jak w przy-
COOCH3 padku innych opioidów – ich działanie może być
N
zniesione przez antagonistę – nalokson. Sufentanyl
N OCH3 jest obecnie najsilniejszym stosowanym w praktyce
klinicznej analgetykiem opioidowym. Jest 7–10 razy
O
silniejszy od fentanylu lub remifentanylu, podczas
gdy siła działania alfentanylu wynosi jedynie od jed-
remifentanyl
nej trzeciej do jednej czwartej działania fentanylu.
Dla porównania: fentanyl jest ok. 100 razy bardziej
skuteczny niż morfina!
Tabela B 1.7-2. Porównanie danych fizykochemicznych i farmakokinetycznych dotyczących opioidów z grupy fentanylu
alfentanyl fentanyl sufentanyl remifentanyl
wartość pka 6,5 8,4 8,0 7,1
wskaźnik dystrybucji ~130 ~815 ~1750 ~18
kwas oktanowy/woda
wiązanie z białkami (%) 92 84 93 70
czas do wystąpienia działania (min) 1-1,5 4-5 2-3 1-1,5
objętość dystrybucji (l/kg) 0,5-1 4,0 2,9 0,2-0,4
klirens (ml/min/kg m.c.) 3-8 13 13 30-40
MUTSCHLER-2009.indd 308 2010-01-07 22:12:18
Układ nerwowy 309
100 H3C N
fentanyl
stężenia skutecznego do 50%
N
czas (min) do zmniejszenia
Układ nerwowy
75
alfentanyl Cl N
50
F
sufentanyl
25
midazolam
remifentanyl B1
0 100 200 300 400 500 600 N O
czas trwania infuzji (min)
N
OC2H5
Ryc. B 1.7-6. Zależny od sytuacji okres półtrwania opioidów
w zależności od czasu trwania infuzji. F N
CH3
O
1.7.2.7. Benzodiazepiny flumazenil
Benzodiazepiny – jak to już zostało opisane w roz-
dziale B 1.2.3.1.1.1 – nie są środkami do znieczulenia
ogólnego w wąskim tego słowa znaczeniu, a mimo
to są wykorzystywane w anestezjologii poza pre- nych od benzodiazepin mogą ujawnić się objawy
medykacją, również do wprowadzenia do anestezji abstynencyjne.
– podane dożylnie w dużych dawkach, oraz łącznie
z silnie działającymi analgetykami – do ataranalgezji
(zob. poniżej). 1.7.3. Szczególne postacie anestezji
W anestezjologii zastosowanie znalazł krótko
działający midazolam – w związku z łatwiejszą ste- 1.7.3.1. Anestezja zrównoważona
rowalnością w trakcie anestezji w porównaniu z dia-
zepamem. Jego okres półtrwania w osoczu wynosi Anestezja zrównoważona obejmuje procedurę znie-
1,5–2,5 godziny; głównym metabolitem jest farma- czulenia ogólnego polegającą na skojarzonym po-
kologicznie czynny hydroksy-midazolam, który wy- dawaniu opioidu z grupy fentanylu z anestetykiem
dalany jest przez nerki jako glukuronid. wziewnym (desfluranem), anestykiem dożylnym
o działaniu hipnotycznym (midazolamem) i ewentu-
Dawkowanie w przypadku wprowadzenia do aneste- alnie niedepolaryzującym środkiem zwiotczającym
zji wynosi 0,15–0,2 mg/kg m.c. mięśnie. Niekiedy dodaje się jeszcze inny silny an-
Zaletą jest to, że za pomocą antagonisty ben- estetyk wziewny (np. izofluran). Objawy ze strony
zodiazepin – flumazenilu – można w specyficzny serca i układu krążenia są w przypadku stosowania
sposób znieść działanie benzodiazepin na receptor. anestezji zrównoważonej zwykle bardzo niewielkie.
Flumazenil wypiera kompetycyjnie benzodiazepiny
z ich miejsca wiązania w kompleksie GABA-benzo-
diazepina-barbituran-kanał chlorkowy. W ten sposób 1.7.3.2. Całkowita anestezja dożylna
możliwe jest celowe sterowanie stanem czuwania
pacjenta oraz opanowywanie zaburzeń oddychania W przypadku całkowitej anestezji dożylnej (TIVA
wywoływanych ewentualnie przez benzodiazepiny. – total intravenous anesthesia) rezygnuje się w pełni
Flumazenil może być ponadto stosowany przy prze- ze stosowania lotnych anestetyków. Przesłanką że pro-
dawkowaniu benzodiazepin. wadzenia tego rodzaju anestezji jest to, iż stosowane
Przeciętna dawka flumazenilu wynosi 0,3–0,6 mg. substancje łatwo poddają się sterowaniu – szczególnie
Jako działania niepożądane flumazenilu wystę- korzystne wydaje się skojarzone stosowanie propofo-
pują nudności i wymioty, a po szybkim podaniu lu i remifentanylu. Początkowo podaje się we wlewie
– uczucie lęku i kołatanie serca. U osób uzależnio- dożylnym 0,3–0,5 μg/kg/min remifentanylu i 5–6 mg/
MUTSCHLER-2009.indd 309 2010-01-07 22:12:19
310 Układ nerwowy
kg/min propofolu. Po intubacji można zmniejszyć neuroleptanalgezji rozumie się postępowanie polega-
dawkę remifentanylu do 0,1 μg/kg/min, a propofolu jące na podaniu iniekcyjnym leku neuroleptycznego
do 2–4 mg/kg/min. Do przecinania skóry podwyższa równocześnie z opioidem – co prowadzi do stanu,
się dawkę remifentanylu do 0,25 μg/min. Sterowanie w którym pacjenci są uspokojeni i pozbawieni lęku,
śródoperacyjne jest indywidualne. Aby uniknąć do- jak również dalece obojętni w stosunku do otoczenia
znań bólowych odczuwanych przez pacjenta w czasie i przebiegających wydarzeń. Nie zawsze dochodzi
operacji, nie powinno się obniżać dawkowania propo- do zapadnięcia pacjenta w sen.
folu poniżej 2 mg/kg/min. W trakcie szczególnie bo- W neuroleptanalgezji mogą być przeprowadzane
lesnych zabiegów operacyjnych możliwe jest w razie małe zabiegi (np. endoskopie), lecz już nie duże za-
potrzeby natychmiastowe zwiększenie dawki podawa- biegi chirurgiczne – w związku z niewystarczającym
nego we wlewie remifentanylu. zwiotczeniem mięśni i stłumieniem odruchów.
W przypadku stosowania tej techniki anestezji do- W zmodyfikowanej postaci neuroleptanalgezji,
żylnej (IVA – intravenous anesthesia) oprócz dobrze tzw. ataranalgezji, zamiast leków neuroleptycznych
sterowalnego opioidu (np. remifentanylu) i aneste- stosuje się benzodiazepiny (zob. powyżej) w dużych
tyku dożylnego (np. propofolu) stosuje się również dawkach razem z opioidami.
anestetyk wziewny (np. gaz rozweselający). Dawko- W przypadku neuroleptanestezji – oprócz neuro-
wanie anestetyków dożylnych jest w związku z tym leptyku i opiodu – stosuje się dodatkowo gaz rozwe-
odpowiednio mniejsze. selający oraz ewentualnie środek zwiotczający mięś-
nie. Gaz rozweselający nasila działanie leku analge-
tycznego, a ponadto powoduje utratę przytomności.
1.7.3.3. Neuroleptanalgezja Jako lek neuroleptyczny wykorzystywany był
i neuroleptanestezja przede wszystkim droperidol, który w Niemczech
został wycofany z rynku prawdopodobnie w związku
Neuroleptanalgezja została niemal całkowicie wypar- z niewielkim obrotem (w Polsce droperidol również
ta przez inne techniki anestezjologiczne. Przez pojęcie nie jest już dostępny – przyp. tłum.).
MUTSCHLER-2009.indd 310 2010-01-07 22:12:19
Układ nerwowy 311
1.8. Środki zwiotczające mięśnie szkieletowe
(miorelaksacyjne)
Układ nerwowy
1.8.1. Podstawy anatomiczne Miofibryle zbudowane są z ułożonych równolegle
i fizjologiczne grubych i cienkich włókienek (filamentów). Prąż-
ki I tworzą wyłącznie filamenty cienkie, o średnicy
Budowa mikroskopowa włókien mięśni szkie- od 5 nm. W prążkach A występują zarówno grube,
letowych. Mięsień szkieletowy (zob. ryc. B 1.8-1) jak i cienkie filamenty (zob. ryc. B 1.8-2). Filamen-
składa się z włókien mięśniowych długości 5–12 cm ty grube zawierają cząsteczki miozyny – posiadające
i grubości 10–100 μm. Ich kurczliwym elementem cienką część ogonową i dwie części głowowe (głów- B1
są miofibryle, które przy obserwacji z boku, w wy- ki miozyny), natomiast filamenty cienkie zawierają
niku istnienia jasnych i ciemnych fragmentów (prąż- kuliste, łączące się w łańcuchy – cząsteczki aktyny,
ków), przyjmują obraz poprzecznego prążkowania jak również „białka regulacyjne” – troponinę i tropo-
(zob. ryc. B 1.8-2). Ciemne odcinki tych włókienek miozynę, które leżą w rowkach pomiędzy łańcuchami
są w świetle spolaryzowanym anizotropowe (po- aktynowymi.
dwójnie załamujące światło). Tworzą one prążki A, Dodatkowo, oprócz wymienionych struktur, w sar-
w których środku można ponadto rozpoznać cien- koplazmie włókien mięśniowych znajduje się układ
kie błony pośrodkowe (prążki M), a także obszary kanalików (układ sarkotubularny) składający się
o mniejszej gęstości (strefy H). Jasne, izotropowe z dwóch części: układu poprzecznego (układ T) i po-
odcinki włókienek tworzą prążki I, podzielone do- dłużnego (siateczka sarkoplazmatyczna). Jak wynika
datkowo przez linie Z (krążki Z). z nazw, przebieg poprzecznych kanalików jest pro-
Obszar włókna mięśniowego leżący pomiędzy stopadły, a podłużnych – równoległy do osi włókien.
dwiema liniami Z, tzn. najmniejsza jednostka mor- Układ T komunikuje się z przestrzenią pozakomór-
fologiczna i czynnościowa, określany jest jako sar- kową, natomiast układ podłużny nie ma z nią łącz-
komer. ności.
Przekazywanie pobudzenia w motorycznej płytce
końcowej. Przekazywanie pobudzenia między soma-
tomotorycznymi włóknami nerwowymi a komórkami
mięśni poprzecznie prążkowanych (przekaźnictwo
mięsień nerwowo-mięśniowe) odbywa się w obrębie moto-
rycznej płytki końcowej. Ta z kolei – jako jednostka
funkcjonalna – składa się z presynaptycznej części
nerwowej oraz postsynaptycznej części mięśniowej.
Na końcu włókno nerwowe traci osłonkę mielinową
włókno i dzieli się na liczne rozgałęzienia, które są otoczone błoną
presynaptyczną. W aksoplazmie znajdują się liczne pęche-
rzyki zawierające acetylocholinę. Zakończenie nerwowe
miofibryle zagłębia się wieloma małymi wypukleniami we włókno
mięśniowe, które w tym miejscu jest bogate w mitochon-
siateczka dria i jądra komórkowe – oznaki intensywnej przemiany
sarkoplazmatyczna materii. Przez liczne pofałdowania powierzchnia błony
włókna mięśniowego ulega znacznemu powiększeniu,
a tym samym powiększa się wielkość miejsca kontaktu.
układ T
(element kanalików
poprzecznych) Kiedy bodziec nerwowy dociera do motorycznej
płytki końcowej – dochodzi do błyskawicznego
cienki aktywator uwalniania acetylocholiny z pęcherzyków. W proce-
filament gruby (jony
(aktyna) wapnia) sie tym niezbędne są jony wapnia. Uwolniona ace-
filament
(miozyna) tylocholina dyfunduje szybko przez mierzącą około
500 Å szczelinę synaptyczną w kierunku błony post-
Ryc. B 1.8-1. Półschematyczna budowa mięśnia szkieletowego. synaptycznej, gdzie napotyka odpowiednie receptory
MUTSCHLER-2009.indd 311 2010-01-07 22:12:19
312 Układ nerwowy
A prążkowanie poprzeczne
prążki I prążki A prążki I
H I A I H
Z strefa H Z
A
aktyna
miozyna
Z M Z
B
filamenty miozynowe (grube)
B filamenty aktynowe (cienkie)
miozyna
aktyna
Ryc. B 1.8-3. Teleskopowopodobne przesunięcie względem
obrazy poprzeczne filamentów siebie mających stałą długość filamentów aktyny i miozyny pod-
C w różnych odcinkach sarkomeru czas biernego naciągania (A) i aktywnego skurczu (C); B – włó-
kienka w stanie spoczynkowym (zwiotczenia) (według Thewsa,
Mutschlera, Vaupela).
Sprzężenie elektromechaniczne i skurcz mięśnia.
Pobudzenia przekazywane przez motoryczną płytkę
Ryc. B 1.8-2. Schemat przyporządkowania prążkowania po- końcową są przenoszone przez układ T na układ po-
przecznego do ultrastruktury miofibryli: A – prążkowanie po- dłużny i uwalniają tam zmagazynowane jony wapnia.
przeczne, B – filamenty miozyny (grube linie) i aktyny (cienkie W wyniku podwyższenia stężenia wapnia dochodzi
linie), C – przekrój filamentu w różnych odcinkach sarkomeru do skurczu mięśnia (sprzężenie elektromechaniczne).
(według Thewsa, Mutschlera, Vaupela). Mianowicie włókienka (filamenty) miozynowy i ak-
tyny przesuwają się wzajemnie między sobą (mecha-
nizm poślizgowy włókienek; zob. ryc. B 1.8-3).
i oddziałuje z nimi – receptorami nikotynowymi (re- Przebiega to w następujący sposób (zob. ryc. B
ceptorami cholinergicznymi typu N). W ten sposób 1.8-4):
dochodzi do – już wcześniej opisanego – zwiększenia
■ Najpierw jony wapnia są wiązane z troponiną, któ-
przepuszczalności błony, a w następstwie tego do de-
ra przez to zmienia swą konformację.
polaryzacji błony płytki końcowej, do tzw. potencja-
łu płytki końcowej. W zdrowym mięśniu potencjały ■ W następstwie tego włókna tropomiozyny przecho-
płytki końcowej są zawsze nadprogowe, tzn. każdy dzą głębiej do rynienki pomiędzy włóknami aktyno-
presynaptyczny potencjał czynnościowy uwalnia wymi.
w mięśniu pewne pobudzenie, które przez błonę
■ W ten sposób staje się możliwe wiązanie części
włókna mięśniowego rozprzestrzenia się do okolicy
głowowych miozyny z filamentami aktynowymi
poza obszarem płytki końcowej.
(tzw. tworzenie mostków poprzecznych).
W ciągu 2 milisekund uwolniona acetylocholina
ulega pod wpływem acetylocholinoesterazy proceso- ■ Pod wpływem rozkładu ATP, który znajduje się
wi hydrolizy, po czym zostaje odtworzony potencjał w częściach głowowych miozyny, przez zmianę
spoczynkowy. o 45° nachylenia mostka poprzecznego następuje
MUTSCHLER-2009.indd 312 2010-01-07 22:12:20
Układ nerwowy 313
■ zapobieganie uwalnianiu acetylocholiny za pomo-
filament aktyny cą toksyny botulinowej, środków do znieczulenia
krążek Z miejscowego – stosowanych w wyższych stęże-
niach lub jonów magnezowych (kompetycyjny an-
Układ nerwowy
rozszcze- tagonizm z jonami wapnia),
pienie
ATP Mg2+ 2. w obszarze postsynaptycznym przez:
Ca2+ filament miozyny
■ blokadę receptorów acetylocholinergicznych zloka-
lizowanych na błonie postsynaptycznej przez środ-
ki działające kompetycyjnie hamująco względem
acetylocholiny (substancje stabilizujące), B1
■ długotrwałą depolaryzację płytek końcowych przez
środki zwiotczające mięśnie o działaniu depolary-
zacyjnym lub
■ zahamowanie sprzężenia elektromechanicznego
np. przez dantrolen.
Praktyczne znaczenie dla zwiotczania mięśni w prze-
Pi 15 nm +ATP biegu znieczulenia ogólnego mają jedynie:
ADP ■ środki zwiotczające mięśnie o działaniu stabilizu-
jącym, tj. zapobiegające procesowi depolaryzacji
(typu kurary),
Ryc. B 1.8-4. Schemat mechanizmu molekularnego skurczu ■ środki zwiotczające mięśnie o działaniu depolary-
mięśnia oraz faz cyklu mostkowania poprzecznego (według zującym (typu suksametonium).
Thewsa, Mutschlera, Vaupela).
Obwodowo działające środki zwiotczające mięśnie
są nadal stosowane w przypadkach, kiedy konieczne
jest uzyskanie rozluźnienia mięśni poprzecznie prąż-
przesunięcie filamentów aktynowych względem kowanych. Zdarza się to głównie przed większymi
miozynowych. zabiegami operacyjnymi (chirurgia jamy brzusznej
i klatki piersiowej). Dzięki środkom zwiotczającym
■ Ponowne wiązanie ATP znosi z kolei powstawanie
mięśnie można stosować mniejsze stężenia środków
mostków poprzecznych, jednakże pozwala – rów-
anestetycznych, a przy równoczesnym zastosowaniu
nież w wyniku rozkładu ATP – na ich ponowne two-
sztucznego oddychania – można zmniejszyć ryzyko
rzenie. Filamenty aktynowe ulegają następnie wciąg-
całego zabiegu znieczulenia ogólnego. Również dla
nięciu dalej w obręb filamentów miozynowych.
przeprowadzenia intubacji – niezbędne jest zastoso-
wanie środków zwiotczających mięśnie.
Przebieg skurczu jest zakończony, gdy uwolnio-
Poza znieczuleniem ogólnym – środki zwiotcza-
ne z sarkoplazmy jony wapnia zostaną z powrotem
jące mięśnie wskazane są w zatruciach (np. strych-
wchłonięte do układu podłużnego.
niną) lub w przebiegu chorób zakaźnych, które pro-
wadzą do skurczów mięśni szkieletowych (np. tężec,
wścieklizna). Stosuje się je również w psychiatrii
1.8.2. Środki zwiotczające mięśnie w ramach zabiegów elektrowstrząsowych – w celu
działające obwodowo zapobiegania pęknięciom mięśni i złamaniom ko-
ści, do których może niekiedy dochodzić w wyniku
Przekaźnictwo nerwowo-mięśniowe może być mo- nagle wywołanego stanu drgawkowego. Dalszym
dyfikowane (zob. ryc. B 1.8-5) wskazaniem, głównie w przypadku dantrolenu (zob.
poniżej), są stany spastyczne mięśni poprzecznie
1. w obszarze presynaptycznym przez: prążkowanych.
■ zahamowanie wychwytu zwrotnego choliny ze szcze-
liny synaptycznej przez hemicholinę,
MUTSCHLER-2009.indd 313 2010-01-07 22:12:21
314 Układ nerwowy
neuron mięśniowy
(motoneuron)
CoA
ace transfe
ACh
tylo raz
cho a
toksyna
lino
botulinowa
-
acetylo
Ca2+ CoA + cholina hemicholina
Ca2+
cholina + kwas octowy
ACh
Na+ Na+ acetylocholino- Na+
esteraza
postsynaptyczna
błona płytki końcowej
brak depolaryzacja depolaryzacja
depolaryzacji „potencjał płytki końcowej”
repolaryzacja repolaryzacja
stabilizujące środki depolaryzacja depolaryzujące środki
A zwiotczające mięśnie B zwiotczające mięśnie
skurcz mięśnia
Ryc. B 1.8-5. Mechanizm działania stabilizujących (niedepolaryzujących; A) i depolaryzujących (B) środków zwiotczających mięśnie.
Bliższe informacje w tekście.
1.8.2.1. Stabilizujące (niedepolaryzujące) mięśnie karku, tułowia i kończyn, a dopiero na sa-
środki zwiotczające mięśnie mym końcu mięśnie oddechowe. Jednak przy każdej
iniekcji omawianych środków – zwykle związki bis-
Stabilizujące środki zwiotczające mięśnie (zob. tab. czwartorzędowe są nieskuteczne po podaniu doust-
1.8-1) posiadają wprawdzie powinowactwo do re- nym – należy być przygotowanym na wystąpienie
ceptorów acetylocholinergicznych znajdujących się porażenia oddychania. Zwiotczenie mięśni może
w obrębie motorycznej płytki końcowej, nie posiada- być przeprowadzone jedynie wówczas, gdy istnieje
ją jednak aktywności wewnętrznej (intrinsic activity) możliwość natychmiastowego podjęcia sztucznego
acetylocholiny: w sposób kompetycyjny wypierają oddychania. Przed podaniem środka zwiotczają-
neuroprzekaźnik z miejsca wiązania w receptorze, cego należy bezwzględnie wyłączyć świadomość
zapobiegając w ten sposób depolaryzacji, a tym sa- pacjenta za pomocą środków do znieczulenia ogól-
mym również skurczowi mięśnia (zob. ryc. B 1.8-5). nego. Dla pacjenta krańcowo nieprzyjemna byłaby
Różne grupy mięśni charakteryzują się różną sytuacja narażenia go, przy pełnej świadomości,
wrażliwością na działanie środków z tej grupy le- na rozpoczynające się porażenie oddechu – bez rów-
ków. Najpierw na działanie środka zwiotczającego noczesnej możliwości zareagowania w jakikolwiek
reagują mięśnie oczu, języka i palców, następnie sposób.
MUTSCHLER-2009.indd 314 2010-01-07 22:12:21
Układ nerwowy 315
Tabela B 1.8-1. Środki zwiotczające mięśnie działające obwodowo
Wzór strukturalny Nazwa Preparaty handlowe
międzynarodowa
Układ nerwowy
l. Środki stabilizujące (niedepolaryzujące)
chlorek tubokuraryny
OH
OCH3
H3C CH3 O
N+
2 Cl –
+
B1
O NH
CH3
H3CO OH
chlorek alkuronium Alloferin
+ CH2
N
N
HOH2C H H
2 Cl –
H H CH2OH
N
N
H2C +
bromek pankuronium Pancuronium Deltaselect,
O Pancuronium Inresa,
Pancuronium Organon,
CH3 O CH3 Pancuronium-ratiopharm,
H3C
Curamed, Pancuronium,
CH3 CH H N+ Pavulon
+N 3 2 Br –
H H
O
H
O CH3
O bromek wekuronium Norcuron,
Vecuronium Inresa
CH3 O CH3
H3C
CH3 H N+
N Br –
H H
O
H
O CH3
O bromek rokuronium Esmeron
CH3 O CH3
O
CH3 H N+ Br –
N
H H
HO
H H2C
MUTSCHLER-2009.indd 315 2010-01-07 22:12:22
316 Układ nerwowy
Tabela B 1.8-1. Środki zwiotczające mięśnie działające obwodowo (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe
międzynarodowa
l. Środki stabilizujące (niedepolaryzujące)
benzenosulfonian atrakurium Atracurium DeltaSelect,
Atracurium HEXAL,
H3CO Tracrium
+N CH3 O O O
H3CO
–
O O
+N CH3 2 O S O
H3CO
H3CO
OCH3
H3CO
H3CO
OCH3
benzenosulfonian Nimbex
H3CO cis-atrakurium
+ N CH3 O O O
H3CO
–
O O
+N CH3 2 O S O
H3CO
H3CO
OCH3
H3CO
H3CO
OCH3
chlorek miwakurium Mivacron
H3CO
O
+ N CH3 O
H3CO O
H3CO O + CH3
H3CO N
H3CO 2 Cl –
OCH3 H3CO
H3CO
H3CO
OCH3
lI. Środki depolaryzujące
chlorek suksametonium Lysthenon,
O H3C CH3 Pantolax DeltaSelect,
H3C + O N+ Lysthenon, Chlorsuccillin
N O CH3 2 Cl –
H3C CH3 O
MUTSCHLER-2009.indd 316 2010-01-07 22:12:22
Układ nerwowy 317
Za pomocą środków hamujących działanie (blo- Wydalanie następuje w 80% w niezmienionej po-
kerów) cholinoesterazy, np. neostygminy, które pod- staci przez nerki.
wyższają stężenie acetylocholiny, można uzyskać Dawka początkowa wynosi 0,15 mg/kg m.c. i.v.,
efekty antagonistyczne do środków zwiotczających dawki powtórne – 0,025 mg/kg m.c. i.v.
Układ nerwowy
mięśnie. W osłabieniu siły mięśniowej (miastenii), ogólnym
Różne leki przeciwinfekcyjne (aminoglikozydy, wyniszczeniu, ciężkim uszkodzeniu miąższu wątroby
amfoterycyna B, linkozamidy), jak również chinidy- oraz niewydolności nerek – stosowanie chlorku alku-
na, ajmalina i pętlowe leki moczopędne nasilają blo- ronium jest przeciwwskazane.
kadę nerwowo-mięśniową wywołaną stabilizującymi
środkami zwiotczającymi mięśnie. Bromek pankuronium jest związkiem androsta-
Idealny, niedepolaryzujący środek zwiotczają- nowym zawierającym dwa czwartorzędowe atomy
cy mięśnie powinien się charakteryzować krótkim azotu. Ten około 5-krotnie silniej niż chlorek tubo- B1
czasem zadziałania (czas od zakończenia iniekcji kuraryny działający środek stabilizujący zwiotcza-
do wystąpienia maksymalnego efektu zwiotczające- jący mięśnie cechuje się szybkim czasem zadzia-
go), krótkim okresem działania i dużą jego siłą. Do- łania (krótkim okresem od podania do wystąpienia
tychczas żadna substancja nie odpowiada powyższym efektu) i średnio długim okresem działania. Środek
wymaganiom (zob. poniżej). sprawdził się głównie u pacjentów z grupy ryzyka
oraz znajdujących się we wstrząsie, a także w kar-
Chlorek tubokuraryny. Substancją prototypową dla sta- diochirurgii.
bilizujących środków zwiotczających mięśnie jest chlorek Uwalnianie histaminy w trakcie jego stosowania
tubokuraryny.
Terminem kurary określa się różne trucizny do strzał jest bardzo rzadkie.
stosowane przez Indian południowoamerykańskich. W za- Dawka początkowa wynosi 0,5 mg/kg m.c. i.v.,
leżności od sposobu przechowywania trucizny rozróżnia dawka powtórna – 0,03 mg/kg m.c. i.v.
się:
Bromek wekuronium jest monoczwartorzędową
■ tubokurarę otrzymywaną z rośliny rodzaju Chondroden-
dron i przechowywaną w rurkach bambusowych (bam- pochodną pankuronium. W przypadku porównywal-
boo tubes), nie silnej dawki – jego czas działania jest 2–3 razy
krótszy niż bromku pankuronium. Prawie nie ulega
■ kurarę kalebassową uzyskiwaną z roślin rodzaju Strych- kumulacji. Ponadto substancja ta nie wykazuje dzia-
nos i przechowywaną w wydrążonych tykwach łania blokującego zwoje nerwowe.
■ kurarę garnkową (przechowywaną w garnkach glinia- Wydalanie następuje w 50% z żółcią – stąd też śro-
nych), która nie ma znaczenia leczniczego. dek ten nadaje się do stosowana u pacjentów z niewy-
dolnością nerek.
Powyższe rodzaje kurary stanowią złożone mieszanki al- Uwalnianie histaminy obserwowane jest rzadko.
kaloidów. Dawka początkowa wynosi 0,08–0,1 mg/kg m.c.
Chlorek tubokuraryny (d-tubokuraryna) należy do alka-
loidów bis-benzylo-izochinolinowych. Za jej działanie od- i.v., dawka powtórna – 0,02–0,05 mg/kg m.c. i.v.
powiada czwartorzędowa grupa amoniowa i protonowany
trzeciorzędowy azot położony w odległości ok. 10 Å. Bromek rokuronium różni się od bromku wekuro-
W związku z występowaniem działań niepożądanych nium w zakresie 3 pozycji szkieletu steroidowego.
związanych z uwalnianiem histaminy (m.in. skurcz oskrze- Moc działania zwiotczającego mięśnie jest ok. 6 razy
li, obniżenie ciśnienia krwi) chlorek tubokuraryny został
wyparty przez poniżej opisane substancje. słabsza, a czas ujawnienia się działania znacznie krót-
szy niż w przypadku wekuronium.
Wydalanie rokuronium następuje w dużym stop-
Chlorek alkuronium jest allilową pochodną toksyfe- niu z żółcią; tylko ok. 20% substancji wydalane
ryny – alkaloidu z kurary kalebassowej. W porówna- jest w moczu.
niu z chlorkiem tybokuraryny uwalnianie histaminy Podobnie jak wekuronium – również rokuronium
jest znacząco słabsze: nawet przy 50-krotnym prze- praktycznie nie prowadzi do uwalniania histaminy.
dawkowaniu nie obserwowano zwiększenia ilości Dawka początkowa wynosi ok. 0,6 mg/kg m.c. i.v.,
wydzieliny w oskrzelach lub ujawnienia się skurczu dawka powtórna – ok. 0,15 mg/kg m.c. i.v.
oskrzeli. Działanie zwiotczające mięśnie jest silniej-
sze, lecz krótsze niż w przypadku chlorku tubokurary- Benzenosulfonian atrakurium, pochodna bisben-
ny. Mimo to przy powtarzanu podawania leku istnieje zylo-izochinolinowa, jest mieszaniną 10 stereoizo-
niebezpieczeństwo jego kumulacji. merów i podobnie jak wekuronium należy do krótko
MUTSCHLER-2009.indd 317 2010-01-07 22:12:23
318 Układ nerwowy
działających stabilizujących substancji zwiotczają- 1.8.2.2. Depolaryzujące środki
cych mięśnie. Dodatkowo ujawnia pewną farmako- zwiotczające mięśnie
kinetyczną osobliwość polegającą na tym, że podle-
ga w organizmie zależnemu od wartości pH nieen-
Depolaryzujące środki zwiotczające mięśnie – podob-
zymatycznemu przekształceniu Hofmanna (rozpad
nie jak acetylocholina – prowadzą do depolaryzacji
alifatycznych czwartorzędowych związków amo-
motorycznej płytki końcowej, uniemożliwiają jednak
nowych w trzeciorzędową aminę laudanozynę oraz
natychmiastową repolaryzację, gdyż znacznie wolniej
ester kwasu akrylowego) do substancji niemających
ulegają rozkładowi. Skutkiem tego jest zwiotczenie
działania zwiotczającego mięśnie. Za bardzo rzadko
mięśnia. Środki hamujące działanie cholinoesterazy
występujące przypadki drgawek odpowiedzialna jest,
nie działają jednak jako odtrutka, a raczej nasilają
jak wykazano w doświadczeniach na zwierzętach,
działanie!
właśnie laudanozyna. Dodatkowo występuje również
enzymatyczna hydroliza estrów. Podobnie jak tubo-
Chlorek suksametonium. Obecnie jedynym tera-
kuraryna, również atrakurium – choć nieco rzadziej
peutycznie stosowanym środkiem zwiotczającym
– prowadzi do uwalniania histaminy.
mięśnie o wyłącznie depolaryzującym działaniu
Dawkowanie wynosi początkowo 0,3–0,5 mg/kg
jest chlorek suksametonium (zob. tab. B 1.8-1), któ-
m.c. i.v., dawka powtórna – 0,1–0,2 mg/kg m.c. i.v.
ry formalnie może być uważany za podwójną (po-
łączoną) cząsteczkę acetylocholiny. Charakteryzuje
Benzenosulfonian cisatrakurium jest jednym z 10
się szybko występującym działaniem i krótkim cza-
stereoizomerów benzosulfonianu atrakurium o dzia-
sem działania – z tego też powodu środek ten dobrze
łaniu 3 razy silniejszym od niego. Podobnie jak atra-
nadaje się do stosowania przed intubacją pacjenta.
kurium, podlega eliminacji hofmannowskiej oraz
W organizmie ulega rozkładowi – przez jeszcze słabo
enzymatycznemu rozkładowi za pomocą esteraz.
działający zwiotczająco monocholinowy ester kwasu
Maksymalne stężenia laudanozyny sięgają poziomu
bursztynowego – do endogennych substancji: kwasu
1/5–1/6 stężenia występującego po podaniu atraku-
bursztynowego i choliny. Konieczna do przeprowa-
rium.
dzenia intubacji dawka, która ma być podana drogą
Benzenosulfonian cisatrakurium prowadzi znaczą-
dożylną w czasie 1 minuty, wynosi ok. l mg/kg m.c.
co rzadziej do uwalniania histaminy niż zdarza się to
Jako łagodne działania niepożądane występują
w przypadku benzenosulfonianu atrakurium.
– często do następnego dnia po iniekcji – bóle mięś-
Dawkowanie wynosi początkowo 0,1–0,2 mg/kg
ni, które polegają na bolesnych skurczach włókien-
m.c. i.v., dawka powtórna – 0,02–0,03 mg/kg m.c. i.v.
kowych na początku rozluźnienia mięśnia (depola-
ryzacja!) i którym można zapobiec przez podawanie
Chlorek miwakurium należy, podobnie jak ben- niewielkich i jeszcze nieprowadzących do blokady
zenosulfonian atrakurium, do grupy bisbenzylo- nerwowo-mięśniowej dawek stabilizujących środ-
izochinolinowej. Jest mieszaniną trzech izomerów ków zwiotczających mięśnie. U pacjentów z atypową
(trans-trans, cis-trans i cis-cis), przy czym działanie cholinoesterazą dochodzi do drastycznego wydłuże-
zwiotczające mięśnie wiąże się niemal wyłącznie nia czasu trwania blokady nerwowo-mięśniowej.
z izomerami trans-trans i cis-trans. Chlorek mi- Również u pacjentów z ciężkim uszkodzeniem
wakurium ulega hydrolitycznemu rozkładowi za wątroby i ogólnym wyniszczeniem wydłuża się czas
pomocą osoczowej cholinoesterazy. Metabolity działania chlorku suksametonium – w wyniku obni-
nie mają działania zwiotczającego mięśnie. U pa- żonego poziomu esterazy.
cjentów z heterozygotycznie atypową cholinoeste-
razą czas działania miwakurium wydłuża się o 30–
50%; w przypadku pacjentów z homozygotycznie 1.8.2.3. Dantrolen
atypową cholinoesterazą możne być on znacznie
bardziej wydłużony. Uwalnianie histaminy jest za- Obecnie dantrolen jest jedynym znajdującym się
leżne od dawki. w sprzedaży środkiem zwiotczającym mięśnie, któ-
Zalecany zakres dawkowania wynosi: początkowe ry obniża napięcie mięśniowe w wyniku częściowej
wstrzyknięcie jako bolus 0,07–0,25 mg/kg m.c., daw- blokady uwalniania jonów wapniowych z tzw. układu
ki powtórne – 0,1 mg/kg m.c. i.v. wydłużają blokadę podłużnego mięśnia.
nerwowo-mięśniową każdorazowo o 15 minut w cza- W przypadku podania doustnego jest wchłaniany
sie znieczulenia ogólnego. powoli i jedynie do wartości 20–30%. Głównym me-
MUTSCHLER-2009.indd 318 2010-01-07 22:12:23
Układ nerwowy 319
stosowania dużych dawek (ponad 200 mg na dzień)
istnieje niebezpieczeństwo wystąpienia omamów
i częstszego ujawniania się napadów u dzieci z móz-
O gowym porażeniem dziecięcym. W dalszej kolejności
Układ nerwowy
N opisane zostały również działania teratogenne.
O N NH
Ze względu na możliwe uczulenie na światło
O2N O pacjenci powinni unikać zwiększonej ekspozycji
na światło słoneczne.
dantrolen W przypadku schorzeń wątroby, upośledzonej funk-
cji płuc oraz ciężkiego uszkodzenia mięśnia sercowego
– stosowanie dantrolenu jest przeciwwskazane.
B1
1.8.2.4. Toksyna botulinowa
tabolitem jest 5-hydroksydantrolen. Okres półtrwania
w osoczu wynosi 8–10 godzin. Wydalanie następuje Toksyna botulinowa typu A hamuje zależne od jonów
głównie przez nerki pod postacią metabolitów. wapniowych Ca2+ uwalnianie acetylocholiny i tym
Dantrolen wskazany jest u pacjentów, u których samym prowadzi do nieodwracalnego zahamowania
utrzymuje się wzmożone napięcie mięśni szkieleto- przekaźnictwa nerwowo-mięśniowego. Jej działanie
wych (spastyczność) – będące wynikiem uszkodzeń polega na rozkładzie synaptobrewiny – białka uczest-
mózgu i rdzenia kręgowego (np. porażenia po uszko- niczącego w procesie uwalniania neuroprzekaźnika.
dzeniu poprzecznym rdzenia, mózgowe porażenie Dopiero po ponownym odtworzeniu zakończenia
dziecięce, stwardnienie rozsiane). Ponadto stosowa- nerwowego powraca prawidłowe przekaźnictwo
ny jest w przebiegu złośliwej hipertermii. bodźców.
Dawkowanie w stanach spastycznych powinno Miejscowe podanie może być zastosowane w le-
wzrastać powoli (od 2 razy dziennie po 25 mg) i osią- czeniu kurczu powiek (blepharospasmus), w terapii
gać po tygodniu maksymalnie 200 mg dziennie. Trze- występującego jednocześnie z kurczem powiek dysto-
ba się liczyć z tym, że wyraźne działanie lecznicze nicznego kurczu mięśni okołooczodołowych, jak rów-
wystąpi dopiero po tygodniu lub dwóch tygodniach. nież w przebiegu innych stanów spastycznych mięśni
Jako działania niepożądane ujawnić się mogą poprzecznie prążkowanych, np. kręczu szyi (karku).
– głównie na początku terapii – zmęczenie, zawroty Nowym wskazaniem jest nadmierne pocenie się.
głowy i uczucie osłabienia. W niektórych przypadkach Od kilku lat toksyna botulinowa stosowana jest co-
obserwowano działania hepatotoksyczne (o przebie- raz częściej jako zastrzyk przeciwzmarszczkowy –
gu podobnym do zapalenia wątroby). W przypadku do wygładzania fałd w okolicy twarzy. Po podaniu
Tabela B 1.8-2. Środki zwiotczające mięśnie działające ośrodkowo (poza benzodiazepinami)
Wzór strukturalny Nazwa Preparaty handlowe Średnia dawka
międzynarodowa dzienna (mg)
baklofen Baclofen AWD, 5-10
H2N COOH Baclofen dura, (powoli)
Baclofen-ratiopharm,
Baclofen, Lioresal
Cl
Cl tyzanidyna Sirdalud 2-4
NH NH
N
N
N S
MUTSCHLER-2009.indd 319 2010-01-07 22:12:23
320 Układ nerwowy
podskórnym dochodzi do porażenia drobnych seg-
mentów mięśni, w wyniku czego skóra leżąca powy-
żej zostaje wygładzona. Jednakże gra mimiczna prze- CH3
O
nosi się wówczas na sąsiednie (nieporażone) mięśnie, N
tak więc często po iniekcjach toksyny botulinowej
mogą powstawać nowe zmarszczki. Cl N
W leczeniu kurczu powiek dawka początkowa wy-
nosi 1,25–2,5 jednostek na jedno oko, maksymalna
dawka całkowita wynosi 100 jednostek. Leczenie
powinno być prowadzone przez specjalnie wyszko- tetrazepam
lonego lekarza.
Jako działania niepożądane mogą ujawnić się
m.in. opadanie powiek, obrzęk powiek i podwójne
widzenie.
leki przeciwlękowe i zostały już omówione w roz-
dziale B 1.2.3.1.1.1.
1.8.3. Środki zwiotczające mięśnie Pochodną benzodiazepiny stosowaną wyłącznie
działające ośrodkowo jako ośrodkowo działający lek zwiotczający mięśnie
jest tetrazepam. W miejscu reszty fenylowej posia-
Patologiczne podwyższenie napięcia mięśni szkie- da on resztę cykloheksylową. Jego dawka poczatko-
letowych (spastyczność, sztywność) jest wywołane wa wynosi 50 mg, a średnia dawka dzienna wynosi
wypadnięciem czynności neuronów hamujących 50–200 mg.
(np. w zatruciu strychniną) lub stałym pobudzeniem Mechanizm działania został poznany jedynie częś-
α-motoneuronów. To ostatnie może być wynikiem ciowo. Baklofen stymuluje receptory GABAB (ago-
uszkodzenia określonych okolic lub szlaków w móz- nista GABAB). Jak już opisano – benzodiazepiny
gu, a także uszkodzeń obwodowych, np. w procesach nasilają działanie GABA przez allosteryczne oddzia-
zapalnych w przebiegu schorzeń reumatycznych. ływanie na receptory GABAA (kanał chlorkowy).
Ośrodkowo działające środki zwiotczające mięś-
Bardzo zróżnicowane pod względem chemicznym nie są wskazane w przypadku bolesnego wzmożone-
substancje (zob. tab. B 1.8-2) z tej grupy leków pro- go napięcia mięśni szkieletowych, które mogą być
wadzą do zmniejszenia napięcia mięśni szkieletowych wywołane np. przez uszkodzenie krążka między-
przez działanie na ośrodkowe synapsy – a mianowicie kręgowego lub inne schorzenia układu ruchowego.
w wyniku hamowania odruchów wielosynaptycznych. Ponadto są one skuteczne w porażeniach spastycz-
Nie mają one natomiast większego wpływu na prze- nych, np. w chorobie Little’a czy stwardnieniu roz-
kaźnictwo nerwowo-mięśniowe w obrębie motorycz- sianym.
nej płytki końcowej. Uwagę kierowców należy zwrócić na sedatywną
Tabela B 1.8-2 zawiera stosowane preparaty, z wy- komponentę oraz wynikające z niej osłabienie szyb-
jątkiem używanych również do tego celu benzodiaze- kości reakcji – ujawniające się po zażyciu ośrodkowo
pin. Te ostatnie są przede wszystkim stosowane jako działających środków zwiotczających mięśnie.
MUTSCHLER-2009.indd 320 2010-01-07 22:12:23
Układ nerwowy 321
1.9. Padaczki i leki przeciwpadaczkowe
(przeciwdrgawkowe)
Układ nerwowy
Ogólne pojęcie padaczki obejmuje różne napadowo Pomiary przeprowadzane za pomocą mikroelek-
występujące, przewlekle nawracające schorzenia, trod wykazały, że w trakcie czynności napadowej
które polegają na podwyższonej pobudliwości (nad- ujawniają się charakterystyczne zmiany wewnątrz-
pobudliwości) neuronów ośrodkowych i wynikające- i zewnątrzkomórkowego stężenia jonów Na+, K+,
go z tego obniżenia progu drgawkowego – charak- Ca2+ i Cl–. Wewnętrzna pobudliwość komórki deter-
teryzując się nieprawidłowymi reakcjami ruchowymi minowana jest hamującymi kanałami K+ i Cl–, jak
(drgawki toniczne, kloniczne lub toniczno-kloniczne, również pobudzającymi kanałami Na+ i Ca2+. Zmia- B1
skurcze, stereotypie) i/lub zaburzeniami świadomości ny w zakresie przewodności i prawdopodobieństwa
albo utratą świadomości, jak również niekiedy nasi- otworzenia się tych kanałów, jak również zmiany
lonymi reakcjami wegetatywnymi. w zakresie zewnątrzkomórkowego środowiska jo-
Padaczki należą do najczęstszych przewlekłych nów mogą prowadzić do zwiększenia pobudliwości
schorzeń ośrodkowego układu nerwowego. Częstość komórki z ujawnieniem się w następstwie czynności
występowania padaczek na świecie wynosi 0,5–1% napadowej.
i na wartość tę nie mają istotnego wpływu czynniki Za pomocą badania elektroencefalograficznego
etniczne czy geograficzne. Mimo że padaczka może (EEG) możliwe jest bezpośrednie potwierdzenie roz-
wystąpić w każdym wieku – około 50% zachorowań poznania padaczki. Neuronalne potencjały czynnoś-
ujawnia się jeszcze przed 10 rokiem życia, a przed 20 ciowe, które powstają w trakcie czynności napadowej
rokiem pojawiają się prawie dwie trzecie wszystkich pojedynczych neuronów i całych ich grup, ulegają za-
przypadków. rejestrowaniu jako potencjały sumacyjne i są prezen-
towane pod postacią typowych dla padaczki zmian
zapisu EEG.
Zarejestrowany czas w trakcie wyładowania pa-
1.9.1. Patofizjologiczne
daczkowego określa się terminem fazy napadu (ic-
i kliniczne podstawy padaczek tal); okres bezpośrednio po tym – to faza ponapa-
dowa (postictal), a późniejsza wolna od wyładowań
Padaczki są w sposób napadowy ujawniającymi się – to faza międzynapadowa (interictal).
zaburzeniami mózgu, które warunkowane są niepra-
widłowo wysoką miejscową (ogniskową) lub uogól- Z etiologicznego punktu widzenia wyróżnia się nastę-
nioną synchronizacją czynności bioelektrycznej du- pujące postacie padaczek: objawowe (padaczki bę-
żej liczby neuronów. Mechanizmy hipersynchroniza- dące wyrazem choroby podstawowej, która może być
cji wyładowań padaczkowych polegają – oprócz bio- rozróżniona i zidentyfikowana pod względem mor-
elektrycznych właściwości neuronów – na swoistych fologiczno-histologicznym), kryptogenne (prawdo-
właściwościach i stanach funkcjonalnych dotkniętych podobnie objawowe padaczki, w przypadku których
schorzeniem obszarów mózgu, w których występują nie udało się – jak dotąd – zidentyfikować choroby
zaburzenia struktury sieci neuronalnej. Do czynników podstawowej) oraz idiopatyczne (padaczki związane
sprzyjających synchronizacji zalicza się określony z przypuszczalną lub potwierdzoną genetyczną pre-
wzór rozkładu kanałów jonowych w poszczególnych dyspozycją). Dzięki poprawie diagnostyki związanej
okolicach błony neuronów, podwyższony stopień z technikami neuroobrazowymi – przede wszystkim
sprzężenia neuronów z komórkami glejowymi za po- magnetycznym rezonansem jądrowym MRI – dawne
mocą połączeń szczelinowych typu gap junction (ne- padaczki krypotegenne stają się coraz częściej posta-
xus), jak również sprzęganie aktywności komórkowej ciami objawowymi.
za pomocą pól elektrycznych – przede wszystkim ko- Częstymi przyczynami padaczek objawowych
mórek piramidowych hipokampa i kory. Powstanie są korowe zaburzenia rozwojowe, guzy, zapalenia
czynności napadowej można powiązać tym samym mózgu, urazy czaszkowo-mózgowe, zmiany naczyń
z brakiem równowagi między neuronalnymi zjawiska- mózgowych, choroby metaboliczne, uszkodzenia
mi pobudzenia i hamowania. Za nieprawidłowe właś- wczesnodziecięce (w szczególności urazy okołoporo-
ciwości neuronów padaczkowych są odpowiedzialne dowe), choroby immunologiczne, rzadziej zapalenia
różne zaburzenia funkcjonalne błony komórkowej. naczyń krwionośnych oraz zatrucia.
MUTSCHLER-2009.indd 321 2010-01-07 22:12:24
322 Układ nerwowy
Coraz częściej diagnozowane są przyczyny genetyczne pa- W zakresie podziału padaczek są uwzględniane na-
daczek idiopatycznych. Należą do nich szczególnie mutacje stępujące kryteria: padaczkowe wyładowania neuro-
kanałów jonowych, przy czym zaburzeniem mogą być do-
nów występują od początku równocześnie, w rozleg-
tknięte rozmaite podtypu kanałów K+, Na+ i Ca2+. Przykła-
dowo mutacja dotyczącą nikotynowego receptora acetylo- łych obszarach obu półkul mózgowych i powodują
cholinergicznego prowadzi do dziedziczonej autosomalnie napad uogólniony. Jeśli jednak dominujące synchro-
dominująco padaczki czołowej z napadami występującymi niczne wyładowania grup neuronów są ograniczone
w nocy. Z kolei w łagodnej rodzinnej padaczce u niemow- do jednego początkowego miejsca, gdyż aktywacja
ląt stwierdza się mutację genów (KCNQ2 i KCNQ3) dla
hamujących neuronów GABA-ergicznych zmniej-
napięciowozależnych kanałów K+. W wyniku opóźnienia
wypływu K+ w trakcie fazy repolaryzacji wydłużeniu ule- sza ich rozprzestrzenianie się, występują napady
ga czas trwania potencjału czynnościowego, co prowa- ogniskowe, zwane częściej i bardziej poprawnie
dzi do zwiększenia napływu Ca2+ i tym samym ułatwia częściowymi. W przypadku zachowanej u pacjenta
powstawanie potencjałów padaczkowych. W przypadku świadomości napady określa się jako proste, przy
padaczki z napadami uogólnionymi i drgawkami gorącz-
zaburzonej świadomości – jako złożone. Z poje-
kowymi zidentyfikowano mutację dotyczącą podjednostek
neuronalnych kanałów Na+, które wywołują spowolnienie dynczego ogniska w niektórych przypadkach może
inaktywacji wyzwalającego potencjał czynnościowy napły- jednak dojść do synchronicznej aktywacji całej kory
wu Na+. Zidentyfikowane zostały także mutacje dotyczą- mózgowej obu półkul, przy czym fala pobudzenia
ce receptora GABAA oraz napięciowozależnych kanałów może się rozszerzyć przez jądra podstawne i wzgó-
chlorkowych, które towarzyszą młodzieńczej padaczce
rze lub przez układ limbiczny. Mówi się wówczas
z napadami mioklonicznymi lub określonym postaciom pa-
daczek z napadami uogólnionymi. o napadach wtórnie uogólnionych. Na podstawie
Mimo tej nowej wiedzy – zrozumienie procesu epilep- powyższych kryteriów podziału oraz specyfiki
togenezy pozostaje nadal bardzo niepełne. Nie jest jasne, ujawniających się objawów dokonano rozróżnienia
dlaczego warunkowane genetycznymi mutacjami trwałe na poszczególne postacie napadów (zob. poniżej).
zmiany w połączeniach neuronalnych i pobudliwości ujaw-
Śmiertelność pacjentów z napadami padaczkowymi
niają się jedynie okresowo (napadowo). Ponadto nie wia-
domo – dlaczego liczne zespoły padaczkowe charaktery- uogólnionymi jest 2–3 razy większa niż w populacji
zują się zależną od wieku datą pierwszego ujawnienia się, osób zdrowych.
a niektóre mogą ulec samoistnej remisji. Podane właści- Postać napadu (zob. ryc. B 1.9-1) w sposób istot-
wości pozwalają przypuszczać, że warunkowane wiekiem ny wpływa podczas terapii na wybór określonego
zmiany w obrębie ośrodkowego układu nerwowego mają
leku przeciwpadaczkowego; z kolei sama etiologia
duże znaczenie dla ujawniającego się obrazu klinicznego
schorzenia. padaczki ma szczególne znaczenie przy rokowaniu.
Napady częściowe (ogniskowe). W zależności
Wyzwolenie napadu padaczkowego może nastąpić od miejsca lokalizacji napadów częściowych możliwe
za pomocą różnych czynników zewnętrznych, które jest dokonanie podziału na padaczkę skroniową, ciemie-
podwyższają ośrodkową pobudliwość. W ekstremal- niową, czołową i potyliczną. Najczęściej diagnozowa-
nych stanach patologicznych (np. anoksja, znaczna ny jest zespół padaczki skroniowej (= padaczki płata
hipoglikemia, mocznica) napady drgawkowe wy- skroniowego; napady psychoruchowe), który zaliczany
jest do napadów częściowych złożonych. Charaktery-
stępują prawdopodobnie u wszystkich ludzi. Tego styczne są tu następujące objawy: dochodzi do zawężenia
rodzaju napady określane bywają terminem przygod- świadomości pacjenta – sprawia on wrażenie zamroczone-
nych. Indywidualna gotowość drgawkowa jest jednak go (napady zamroczeniowe), często poprzedzane są aurą
bardzo zróżnicowana i warunkowana jest genetyczną z ujawniającym się w okolicy nadbrzusza uczuciem cie-
predyspozycją. U 3% dzieci spotyka się w pierw- pła lub mrowienia, lękiem, objawami wegetatywnymi,
osobliwymi doznaniami smakowymi lub zapachowymi.
szych 5 latach życia napady padaczkowe w prze- U małych dzieci aura objawia się niejednokrotnie pod
biegu infekcji gorączkowych, szczególnie podczas postacią zachowań lękowych-symbiotycznych. Możliwe
wzrostu temperatury (drgawki gorączkowe). Napady są ponadto reakcje polegające na otamowaniu mówienia,
mogą być również wywołane – niezależnie od wieku automatyzmach okolicy ust i twarzy (ruchy żucia, mla-
– bodźcami sensorycznymi, np. szybkimi zmianami skania, przełykania lub inne ruchy języka), dystonicznych
zmianach ułożenia kończyny po stronie przeciwnej oraz
natężenia światła (mrugające światło) i widoku wzo- powoli zwężającej się świadomości. Ponapadowy okres
rów w paski, przez za długi lub za krótki sen, odsta- powrotu do stanu orientacji jest długi. Szczególną cechą
wienie alkoholu, niedotlenienie, leki (np. chlorochi- charakterystyczną napadów psychoruchowych jest ponad-
na, enfluran, sole litu, teofilina) i in. Napady takie to stałość w zakresie rodzaju i kolejności ujawniania się
zwane są odruchowymi. W przypadku terapii napa- poszczególnych objawów.
W przypadku zaliczanej do napadów prostych padacz-
dów przygodnych i odruchowych na pierwszym pla- ki ciemieniowej (= padaczki płata ciemieniowego) ujaw-
nie pozostaje opanowanie czynnika wyzwalającego, niają się po stronie przeciwnej zaburzenia czucia i/lub
a nie samo wydarzenie polegające na drgawkach. nieprawidłowe reakcje ruchowe. Gdy ognisko zlokalizo-
MUTSCHLER-2009.indd 322 2010-01-07 22:12:24
Układ nerwowy 323
z objawami ruchowymi
(napad jacksonowski, napad zwrotny)
Układ nerwowy
napady częściowe proste z objawami zmysłowymi
(bez utraty przytomności) lub somatosensorycznymi
napady napady częściowe złożone
ogniskowe (napady psychomotoryczne z objawami autonomicznymi
(częściowe) z utratą świadomości)
napady ogniskowe B1
wtórnie uogólnione
padaczka typu absence
dziecięcego
(piknolepsja)
absence
napady absence
napady miokloniczne wieku młodzieńczego
napady petit mal (zrywania miokloniczne, i dorosłych
impulsywne petit mal)
napady miokloniczno-
-astatyczne
napady napady typu
uogólnione salaam
napady
kloniczne
napady grand mal
(napady toniczno- napady
-kloniczne) toniczne
Ryc. B 1.9-1. Klasyfikacja postaci napadów padaczkowych i padaczek (w uproszczeniu).
wane jest w okolicy górnej krawędzi płaszcza – objawy Małe napady uogólnione (napady uogólnione nieświa-
ujawniają się w okolicy nogi, gdy w części pośrodkowej domości). Napady nieświadomości (absence, wyłączenia;
– ręki/ramienia, a w okolicy podstawno-bocznej – twarzy. dziecięca i młodzieńcza padaczka nieświadomości) charak-
Gdy objawy przemieszczają się (zwykle w obrębie kończy- teryzują się nagłym początkiem zawężenia świadomości
ny od jej części dystalnych do proksymalnych) – mówi się bez objawów aury oraz następczą amnezją. Wyłączenia
wówczas o napadach Jacksona. są zwykle znacznie krótsze niż w napadach częściowych
W przypadku padaczki czołowej (= padaczki płata czo- złożonych. Świadomość zanika na okres od kilku sekund
łowego) również może dochodzić do napadów jacksonow- do pół minuty – pacjent patrzy przed siebie i nie reaguje
skich. Mogą także wystąpić ruchy obrotowe i podnoszenia, na zwracanie się do niego. Nie tak rzadko obserwuje się
które na ogół połączone są ze zwrotem ciała w stronę prze- także drżenia powiek. Dzieci często upuszczają trzymane
ciwną do ogniska padaczkowego – stąd też zwane są na- w rękach przedmioty, a po zakończeniu napadu kontynuują
padami zwrotnymi (adwersyjnymi). Zwrotne ruchy gałek wcześniej wykonywaną czynność, jak gdyby nic się nie zda-
ocznych, głowy lub kończyn mogą występować razem rzyło. W przypadku wystąpienia wielu napadów nieświa-
z drgawkami toniczno-klonicznymi. domości jeden po drugim (zwykle w przebiegu padaczki
Padaczka potyliczna (= padaczki płata potylicznego) dziecięcej) mówi się o piknolepsji (piknoleptycznych napa-
– stanowiąca 5–10% wszystkich objawowych padaczek dach nieświadomości, absence petit mal). Czasem wystę-
częściowych – to najrzadsza postać padaczki. Dotyczy ona pują automatyzmy z ruchami tonicznymi, atonicznymi lub
płatów potylicznych, gdzie znajduje się także kora wzroko- klonicznymi. Możliwe jest również wystąpienie napadów
wa. Napadom wywodzącym się z tej okolicy zwykle towa- grand mal (zob. poniżej).
rzyszą omamy wzrokowe pod postacią utrzymujących się Młodociane napady miokloniczne (impulsive petit mal),
bądź migających plam lub prostych figur geometrycznych, które manifestują się często rano krótko po wstaniu z łóżka
ślepota i rzadziej toniczne lub kloniczne ruchy gałek ocz- lub w przypadku zmęczenia (deprywacja snu jest najczęst-
nych. szą przyczyną nawrotu tej postaci padaczki), objawiają się
MUTSCHLER-2009.indd 323 2010-01-07 22:12:24
324 Układ nerwowy
zwykle pod postacią symetrycznych skurczów rąk i ramion wany jako utrzymujący się, uogólniony napad drgawkowy
jakby w trakcie porażeń prądem (zrywania miokloniczne). trwający przez więcej niż 5 minut bądź też jako wystąpie-
W sposób typowy dochodzi do nierównego wyrzucenia nie dwóch lub więcej napadów w odstępie czasowym krót-
rąk nad głowę z rozcapierzeniem palców. Nogi mogą ulec szym od jednej godziny (zwykle co 5–15 minut) bez uzy-
podgięciu, co powoduje upadek. Pierwotnie świadomość skania restytucji w tym czasie, tzn. pacjent między kolejny-
jest zachowana, jednak w przypadku serii mioklonii lub mi napadami nie powraca w pełni do świadomości. Wraz
w stanie napadów mioklonicznych pacjent jest zamroczony ze zwiększającą się ilością napadów i czasem trwania stanu
i może – w trakcie wystąpienia napadu grand mal – rów- padaczkowego pogłębia się stan nieprzytomności pacjenta,
nież utracić świadomość. Inne napady padaczkowe mogą ujawniać się pod po-
stacią ogniskowego/częściowego stanu padaczkowego.
Padaczka napadów grand mal (duży uogólniony napad, Wprawdzie nie jest on tak zagrażający życiu, może jednak
epilepsia major). W przypadku dużego napadu wyróżnia prowadzić do utrwalonych deficytów funkcji w okolicach
się kilka jego faz. Zaczyna się on często objawami prodro- mózgu dotkniętych schorzeniem.
malnymi o różnym czasie trwania, do których należą: bóle
głowy, złe samopoczucie, osłabienie, niepokój, depresja.
Fakultatywnie bezpośrednio przed właściwym napadem wy-
stępuje – podobnie jak w napadach częściowych – tzw. aura 1.9.2. Leki przeciwpadaczkowe
z omami (halucynoidami) wzrokowymi i słuchowymi, a tak- (przeciwdrgawkowe)
że ogniskowe objawy ruchowe i czuciowe, np. skurcze, mro-
wienie. Po aurze, będącej markerem rozpoczynającego się
wyładowania padaczkowego, następuje toniczna faza napadu Leki przeciwpadaczkowe służą do objawowej terapii
– ponownie z nieobowiązkowym krzykiem początkowym. różnych postaci padaczki. Działają hamująco na po-
W tym momencie pacjent traci przytomność, upada budliwość neuronów i/lub tłumiąco na przestrzenne
na ziemię i w określonych warunkach może doznać wów-
rozszerzanie się napadu. Mogą przy tym wiązać się
czas poważnych obrażeń. W tej fazie istnieje również nie-
bezpieczeństwo dokonania bolesnych przygryzień języka. z kanałami jonowymi lub związanymi z nimi recep-
Od kilku sekund do minut później napad przechodzi w fazę torami dla neuroprzekaźników – w ten sposób zmie-
kloniczną z uogólnionymi drgawkami mięśni. W związku niając przepuszczalność błony komórkowej. Poprzez
ze współudziałem mięśni języka może powstawać ze śli- wpływ na wchłanianie zwrotne neuroprzekaźników
ny piana wydostająca się z ust; może dochodzić również
lub ich metabolizm – mogą również zmieniać stężenie
do oddania moczu, rzadziej także kału. Po napadzie pacjent
zapada zwykle w głęboki – krócej lub dłużej trwający – sen neuroprzekaźników w przestrzeni zewnątrzkomórko-
końcowy, któremu towarzyszą głęboki oddech, bladość wej. Z terapeutycznego punktu widzenia szczególnie
i zwężenie źrenic. Ze snu tego pacjent budzi się powoli i za- znaczący jest wpływ na napięciowozależne kanały
mroczony; skarży się na bóle głowy i bóle mięśniowe. Na+ oraz kanały należące do układu GABA-ergicz-
Jeśli 90% napadów występuje w pierwszych dwóch go-
nego. Wiele leków przeciwpadaczkowych ingeruje
dzinach po przebudzeniu, mówi się padaczce z napadami
grand mal podczas budzenia. Jeżeli 90% napadów ujaw- równocześnie w kilka miejsc układu neurotransmisyj-
nia się w trakcie snu – to ma się do czynienia z padaczką nego.
grand mal w czasie snu. Jeśli napady występują w ciągu
dnia i podczas snu, mówi się o padaczce z rozproszonymi Mechanizmy działania różnych leków przeciw-
napadami grand mal.
padaczkowych (zob. ryc. B 1.9-2). Takie leki prze-
Specyficzne padaczki dziecięce. Padaczka z napadami ciwpadaczkowe, jak: karbamazepina, okskarbazepi-
zgięciowymi salaam (napady skłonów, napady zgięciowe, na, kwas walproinowy, fenytoina i lamotrygina od-
skurcze niemowlęce, propulsive petit mal, zespół Westa), działują głównie na napięciowozależne kanały Na+;
występująca głównie w wieku niemowlęcym, charakte- w przypadku walproinianiów i topiramatu – efekt
ryzuje się gwałtownymi („błyskawicznymi”) zrywaniami
ten jest tylko częścią ich działania. Poprzez oddzia-
z szybkim, rzadziej powolnym ruchem tułowia do przodu
i skrzyżowaniem rąk (pozdrowienie arabskie salaam). ływanie na inaktywację kanałów Na+ dokonuje się
wpływ nie tylko na amplitudę i czas trwania pojedyn-
Zespół Lennoxa-Gastauta charakteryzuje się występowa- czego potencjału czynnościowego, lecz powoduje
niem rozmaitych postaci napadów. Najbardziej charakte- to zmniejszenie zdolności neuronów do generowania
rystycznymi dla obrazu choroby trzema postaciami napa-
salw wysokoczęstotliwych potencjałów czynnoś-
dów są: atypowe napady nieświadomości, (nocne) napady
toniczne i upadków. Ponadto u pacjentów mogą ujawniać ciowych. Stąd też działanie leków przeciwpadacz-
się trzy dalsze postacie napadów: miokloniczne, grand mal kowych jest lepiej widoczne w przypadku wysokiej
i rzadziej napady ogniskowe. Występuje zazwyczaj między częstotliwości wyładowań niż przy niskiej częstotli-
l a 7 rokiem życia. Nierzadko poprzedza go padaczka z na- wości (tzw. blokada zależna od użycia kanałów Na+;
padami zgięciowymi salaam.
use-dependent block).
Status epilepticus. Stan padaczkowy napadów grand mal W przypadku powstawania napadów nieświadomo-
jest ciężkim stanem wymagającym interwencji lekarskiej, ści (absence) ważną rolę odgrywają kanały Ca2+ typu
któremu towarzyszy wysoka śmiertelność. Jest on zdefinio- T. Tym właśnie można wyjaśnić działanie etosuksy-
MUTSCHLER-2009.indd 324 2010-01-07 22:12:24
Układ nerwowy 325
kanał Ca2+
Ca2+ typu L
etosuksymid,
soma gabapentyna mesuksymid
Układ nerwowy
Na+
karbamazepina, okskarbazepina,
fenytoina, lamotrygina,
walproinian, zonisamid kanał Ca2+
typu T
akson Ca2+
B1
glutaminian
receptor
AMPA
Na+, Ca2+ pobudzenie
Ca2+
Mg2+
Na+, Ca2+
kanał Ca2+ felbamat
typu N
receptor
NMDA
neuron presynaptyczny neuron postsynaptyczny
kanał Ca2+
typu N
Ca2+
walproinian,
wigabatryna Cl- zahamowanie
receptor
transaminaza GABA GABAA
GAT
GABA
GABA fenobarbital,
primidon,
klonazepam,
tiagabina, diazepam,
wigabatryna lorazepam
receptor
GABAA
Ryc. B 1.9-2. Schematyczny mechanizm działania leków przeciwpadaczkowych. Przedstawione zostały miejsca uchwytu różnych
substancji czynnych w obrębie glutaminergicznego lub GABA-ergicznego neuronu presynaptycznego oraz w obrębie dwóch neu-
ronów postsynaptycznych, które mogą należeć do układu glutaminergicznego lub GABA-ergicznego. Leki przeciwpadaczkowe blo-
kują napięciowozależne kanały Na+, kanały Ca2+ lub receptory NMDA, stymulują sterowane GABA kanały chlorkowe, hamują rozkład
GABA lub jego wchłaniane zwrotne do zakończeń aksonalnych. Bliższe informacje w tekście.
midu i mesuksymidu, gdyż wymienione leki prowadzą Benzodiazepiny, np. klonazepam, diazepam lub
do osłabienia prądów Ca2+ typu T w neuronach wzgó- lorazepam, oraz barbiturany, np. fenobarbital, są al-
rzowo-korowych. W zależności od potencjału błono- losterycznymi agonistami receptorów GABAA i wy-
wego neuronu – w związku z ich ingerencją w kanały wołują za pomocą wspomnianych receptorów hiper-
wapniowe – zapobiegają one powstawaniu niskoprogo- polaryzację błony komórkowej.
wych iglic Ca2+ i tym samym przeciwdziałają powsta- Felbamat blokuje miejsce wiązania glicyny w ob-
waniu wyłączeń (napadów nieświadomości). rębie receptora NMDA (zob. poniżej). Ponadto działa
MUTSCHLER-2009.indd 325 2010-01-07 22:12:25
326 Układ nerwowy
hamująco na napięciowozależne kanały Na+ i Ca2+ ■ kwas walproinowy,
oraz działa podobnie jak barbiturany na receptor
■ fenytoina,
GABA.
■ lamotrygina,
Kolejny mechanizm przeciwdrgawkowego działania
■ zonisamid.
wymienionych preparatów polega na ich wpływie
na kinetykę neuroprzekaźników lub neuromodula-
Karbamazepina, okskarbazepina. Karbamazepi-
torów. Na przykład wigabatryna działa hamująco
na jest dobrze wchłaniana (biodostępność wynosi
na enzym GABA-transaminazę i w ten sposób opóź-
70–80%). Wiązanie z białkami osocza sięga około
nia rozkład GABA w neuronach i komórkach glejo-
75%, a okres półtrwania w osoczu wynosi 12–24
wych. Poprzez hamowanie dopęcherzykowego trans-
godziny. Ze względu na zjawisko indukcji metaboli-
portu GABA wigabatryna zwiększa ponadto stężenie
zmu izoenzymu CYP3A4 karbamazepina – w wyniku
GABA w strukturach tkankowych. Z kolei tiagabi-
wielokrotnego zażywania – ulega wyraźnie szybszej
na blokuje w sposób selektywny wychwyt GABA
eliminacji. Aktywna farmakologicznie jest również
do neuronów i komórek glejowych.
karbamazepina 10,11-epoksydowana – metabolit
leku podstawowego.
Farmakokinetyka. Leki przeciwpadaczkowe ulegają
Karbamazepina jest lekiem pierwszego wyboru
zazwyczaj dobremu wchłanianiu. W długotrwałej te-
w przypadku prostych i złożonych napadów częścio-
rapii tymi lekami, szczególnie w przypadku podejrze-
wych. Wtórnie uogólnione napady mogą być opano-
nia braku współpracy pacjenta z lekarzem (non-com-
wywane bardziej skutecznie niż napady pierwotnie
pliance), przyjmowania zbyt dużych lub zbyt małych
uogólnione. Karbamazepina nie nadaje się natomiast
dawek, a także w przypadku ujawnienia się działań
do terapii napadów typu absence. Oprócz padacz-
niepożądanych, bardzo celowe jest kontrolowanie
ki – karbamazepinę stosuje się w neuralgii nerwu
ich stężenia w surowicy. Okresy półtrwania w suro-
trójdzielnego, w przypadku bólów neuropatycz-
wicy mogą się indywidualnie znacznie wahać.
nych, w profilaktyce nawrotów choroby afektywnej
dwubiegunowej oraz w celu zapobiegania napadom
Dawkowanie. Dawkowanie (zob. tab. 1.9-1) powin-
w trakcie alkoholowego zespołu odstawiennego.
no być dostosowane do obrazu klinicznego. W przy-
Do działań niepożądanych w trakcie terapii kar-
padkach utrzymujących się napadów powinno się
bamazepiną – ujawniających się szczególnie w okre-
zwiększać dawkę aż do granicy ujawnienia się dzia-
sie początkowym – zalicza się uczucie zmęczenia,
łań niepożądanych.
brak apetytu, odruch wymiotny, mdłości, bóle głowy,
zawroty głowy, zaburzenia widzenia i inne. Reakcje
Leki przeciwpadaczkowe a ciąża. Ryzyko wystą-
alergiczne (nadwrażliwości) lub objawy leukopenii,
pienia uszkodzeń teratogennych jest większe u dzie-
które mogą prowadzić do konieczności przerwania
ci (nieleczonych) kobiet chorych na padaczkę niż
terapii – są rzadkie.
u dzieci kobiet zdrowych. Ponieważ z drugiej stro-
W przypadku ciężkich zaburzeń funkcji wątro-
ny same leki przeciwpadaczkowe posiadają pewien
by, jak również przy istnieniu bloku przewodni-
potencjał działania teratogennego, nie jest zaskocze-
ctwa przedsionkowo-komorowego – karbamazepina
niem, że częstość powstawania wad rozwojowych
jest przeciwwskazana.
u dzieci – leczonych i nieleczonych – matek chorych
Jako induktor enzymu CYP3A4 – karbamazepina
na padaczkę nie różni się znamiennie, ale jest wyraź-
osłabia działanie innych substancji poddawanych ok-
nie większa niż w całej populacji. Ciąża nie jest więc
sydatywnej biotransformacji, takich jak klonazepam,
wskazaniem do przerwania terapii lekami przeciwpa-
etosuksymid, tiagabina, topiramat, hormonalne środ-
daczkowymi.
ki antykoncepcyjne, glukokortykosteroidy, haloperi-
dol lub teofilina.
1.9.2.1. Leki przeciwpadaczkowe głównie
blokujące napięciowozależne Okskarbazepina jest 10-ketonową pochodną
kanały sodowe karbamazepiny. W wątrobie ulega błyskawicznej
redukcji do właściwego związku działającego –
(jedno)hydroksylowanej pochodnej. Mimo że mecha-
Do tej grupy leków przeciwpadaczkowych (zob. tab.
nizm działania odpowiada temu znanemu u karbama-
1.9-1):
zepiny, jest lepiej od niej tolerowana i sukcesywnie
■ karbamazepina i okskarbazepina, zastępuje starszy preparat.
MUTSCHLER-2009.indd 326 2010-01-07 22:12:25
Układ nerwowy 327
Tabela B 1.9-1. Leki przeciwpadaczkowe
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
Leki przeciwpadaczkowe głównie blokujące napięciowozależne kanały sodowe
karbamazepina Carbamazepin HEXAL, 16-24 800-1200
Carbamazepin-ratiopharm,
Tegretal, Timonil, Amizepin,
N
Finlepsin, Neurotop,
O NH2 Tegretol
okskarbazepina Timox, 8-11 600-1200
B1
O
Trileptal,
Apydan
O NH2
COOH
kwas walproinowy Convulex, Ergenyl, Orfiril, 7-15 1200-2000
Valproat-neuraxpharm,
Absenor, Depakine, Dipromal,
H3C CH3 Orfiril, ValproLek,
Valpro-ratiopharm Chrono
O fenytoina Phenhydan, 10-60 300-400
NH Phenytoin AWD,
HN O Zentropil,
Phenytoinum
Cl lamotrygina Elmendos, Lamictal, 24-35 100-200
Cl Danoptin, Epiral, Lamepil S,
Lamexim, Lamilept, Lamitrin,
N Lamotrigine-S-ratiopharm,
N Lomox, Plexxo, Symla,
Triginet
H2N N NH2
zonisamid Zonegran 60-65 300-500
SO 2NH2
N
O
Leki przeciwpadaczkowe nasilające głównie działanie GABA
O
fenobarbital Luminal, 49-96 100-400
Luminaletten,
NH
Luminalum
H5C2
O NH O
O
primidon Mylepsinum, 5-15 750-1500
Primidon Holsten,
NH
Resimatil,
H5C2 Mizodin
O NH
COOH wigabatryna Sabril 5-8 2000-3000
H2C
NH2
MUTSCHLER-2009.indd 327 2010-01-07 22:12:25
328 Układ nerwowy
Tabela B 1.9-1. Leki przeciwpadaczkowe (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
tiagabina Gabitril 7-9 15-30
S CH3
S N
COOH
CH3
Inne leki przeciwpadaczkowe
H2N COOH gabapentyna Gabapentin HEXAL, 5-7 1200-2400
Gabapentin-ratiopharm,
Gabapentin STADA, Neurontin,
Gabapentin TEVA
NH2 pregabalina LYRICA 5-7 150-600
(H3C) 2HC COOH
OSO 2NH2
topiramat TOPAMAX, 18-24 100-500
Epiramat,
O O Topiramat-ratiopharm
CH3
H
O CH3
O
H
O H
H3C
CH3
O NH2 felbamat Taloxa 15-23 2000-4000
O NH2
O mesuksymid Petinutin 1,5-3 600-1200
N
CH3
H3C
O
O etosuksymid Petnidan, 48-60 1200-1500
H5C2 Suxilep
N
H
H3C
O
H5C2 CONH2 lewiracetam Keppra 6-8 1000-3000
N O
SO 2NH2 sultiam Ospolot 6-10 600-1200
O O
S
N
MUTSCHLER-2009.indd 328 2010-01-07 22:12:26
Układ nerwowy 329
Po podaniu doustnym okskarbazepina jest całko- Indukujące enzymy leki przeciwpadaczkowe: fe-
wicie wchłaniana. Okres półtrwania w osoczu sub- nobarbital, fenytoina, primidon lub karbamazepina,
stancji macierzystej wynosi 1–2,5 godziny, a hydro- podwyższają wydalanie kwasu walproinowego i tym
ksylowanej pochodnej – 8–11 godzin. samym osłabiają jego działanie. Kwas walproinowy
Układ nerwowy
Wydalanie następuje głównie przez nerki. zwiększa stężenie fenobarbitalu, co może wyrazić się
Wskazania, działania niepożądane i przeciwwska- pod postacią silnego uspokojenia (nadmiernej sedacji;
zania są podobne jak w przypadku karbamazepiny, szczególnie u dzieci). W przypadku prowadzonej tera-
natomiast potencjał możliwych interakcji okskarba- pii za pomocą fenytoiny dodatkowe użycie kwasu wal-
zepiny jest mniejszy niż u karbamazepiny. proinowego może zwiększyć ilość wolnej fenytoiny
Okskarbazepina i jej hydroksylowana pochodna (farmakologicznie aktywnej, niezwiązanej z albumi-
hamują CYP2C19 (stąd zwiększenie stężenia feno- nami frakcji), jednak bez podwyższania osoczowego
barbitalu i fenytoiny ) oraz indukują izoenzymy cy- stężenia całkowitej fenytoiny. W trakcie terapii skoja- B1
tochromowe CYP3A4 i CYP3A5, w wyniku czego rzonej za pomocą kwasu walproinowego z karbama-
zmniejsza się poziom felodypiny i doustnych środ- zepiną zostały opisane objawy, które można wytłuma-
ków antykoncepcyjnych. czyć nasileniem działania toksycznego karbamazepiny
przez kwas walproinowy. Kwas walproinowy wypiera
Kwas walproinowy. Kwas walproinowy działa diazepam z jego miejsc wiązania z albuminami oraz
nie tylko blokująco na kanały sodowe, lecz zwiększa hamuje jego metabolizm. W dalszej kolejności kwas
również stężenie GABA w szczelinie synaptycznej walproinowy blokuje również metabolizm lamotrygi-
(m.in. przez zahamowanie enzymów eliminujących ny, której dawkowanie musi być odpowiednio dopa-
GABA – np. transaminazy GABA). Jest całkowicie sowane. Równoczesne przyjmowanie leków zawiera-
wchłaniany. Okres półtrwania w osoczu wynosi 7–15 jących kwas walproinowy wraz z lekami przeciwza-
godzin; z kolei wiązanie z białkami osocza sięga 90%. krzepowymi lub kwasem acetylosalicylowym może
Około 20% leku wydalane jest pod postacią glukuro- powodować podwyższenie skłonności do krwawienia.
nianu. Ponadto kwas walproinowy ulega procesom Ponadto kwas acetylosalicylowy zmniejsza wiązanie
oksydatywnej transformacji. kwasu walproinowego z białkami osocza.
Wskazaniem do zastosowania kwasu walproino-
wego są przede wszystkim padaczki z napadami Fenytoina. Fenytoina cechuje się silnym działa-
pierwotnie uogólnionymi (grand mal, typu absen- niem przeciwdrgawkowym, ale w przeciwieństwie
ce), jak również – napady częściowe. Ponadto kwas do chemicznie spokrewnionych z nią barbituranów –
walproinowy może być skutecznie stosowany w za- jej działanie uspokajające jest bardzo słabe; niekiedy
burzeniach afektywnych dwubiegunowych, w napa- może działać nawet pobudzająco. Jej biotransforma-
dach paniki oraz w profilaktyce napadów migreny. cja ma charakterystykę wysycania się, tj. wraz z ros-
Jako działania niepożądane obserwowano: zabu- nącą dawką leku zwiększa się jego okres półtrwania
rzenia ośrodkowe (nadmierną senność, stany spląta- w osoczu.
nia, niepokój, omamy), dolegliwości żołądkowo-jeli- Fenytoina zalecana jest przede wszystkim w napa-
towe, (odwracalne) wypadanie włosów, zwiększenia dach grand mal występujących w czasie snu i niezależ-
masy ciała i zaburzenia krzepliwości krwi – jako nych od pory dnia, we wtórnie uogólnionych napadach
skutek trombocytopenii i zmniejszenia stężenia fi- grand mal, a w dalszej kolejności – w padaczce z napa-
brynogenu. Szczególnie ważne są zaburzenia funkcji dami Jacksona i w napadach psychomotorycznych.
wątroby (przede wszystkim u małych dzieci), które Dość częstym działaniem niepożądanym jest prze-
wprawdzie ujawniają się rzadko, lecz mogą przebie- rost śluzówki jamy ustnej (hiperplazja dziąseł) – ma-
gać z zejściem śmiertelnym. jący znaczenie kosmetyczne, lecz niejednokrotnie
Wobec powyższego zalecane są kontrole ilości przeszkadzający samemu pacjentowi. U 10% pacjen-
płytek krwi i innych parametrów krzepliwości krwi, a tów ujawnia się nadmierne owłosienie. Innymi dzia-
także funkcji wątroby. łaniami niepożądanymi są skórne reakcje alergiczne,
W przypadku schorzeń wątroby w wywiadzie, takie jak osteoporoza i osteomalacja. W przypadku
ciężkich (aktualnych) chorób wątroby i trzustki, za- przedawkowania może dość do ataksji chodu, zawro-
burzeń krzepliwości krwi oraz porfirii – kwas walpro- tów głowy, oczopląsu i zamazanej mowy, a także sta-
inowy jest przeciwwskazany. Szczególna ostrożność nów pobudzenia.
jest wskazana w sytuacji równoczesnego stosowa- W przypadku leukopenii oraz bloku przedsionko-
nia kwasu walproinowego z lekami, które wpływają wo-komorowego stopnia II i III – fenytoina jest prze-
na funkcję płytek krwi lub jej krzepnięcie. ciwwskazana.
MUTSCHLER-2009.indd 329 2010-01-07 22:12:26
330 Układ nerwowy
Doustne leki zmniejszające krzepliwość krwi, cy- ■ primidon,
metydyna, chloramfenikol, pochodne kumaryny, izo-
■ benzodiazepiny (klobazam, klonazepam, diazepam,
niazyd, sultiam, kwas walproinowy i inne – zwiększa-
lorazepam),
ją poziom fenytoiny w osoczu poprzez zahamowanie
izoenzymów cytochromowych i spowolnienie roz- ■ wigabatrynę,
kładu. Fentoina zwiększa toksyczność metotreksatu.
■ tiagabinę.
Lamotrygina. Lamotrygina posiada podobny mecha-
Fenobarbital. Należący do grupy barbituranów feno-
nizm działania do karbamazepiny. Charakteryzuje się
barbital nasila hamujące działanie GABA – na skutek
bardzo wysoką biodostępnością. Jej głównym meta-
allosterycznego oddziaływania w obrębie receptora
bolitem jest N-glukuronian, który ulega wydalaniu
GABAA (zob. ryc. B 1.9-2). W przypadku podania
głównie z moczem. Okres półtrwania w osoczu wy-
doustnego – lek jest niemal całkowicie wchłaniany
nosi 24–35 godzin.
(biodostępność rzędu 80–100%). Wiązanie z białkami
Lamotrygina stosowana jest jako monoterapia lub
osocza sięga 20–45%, a okres półtrwania w osoczu
terapia dodana (add-on) w przypadku napadów częś-
– 40–120 godzin. Głównym metabolitem jest nieak-
ciowych bez lub z ich wtórnym uogólnianiem się,
tywna p-hydroksylowana pochodna. Wydalanie na-
w napadach wtórnie uogólniających się oraz w ze-
stępuje głównie przez nerki.
spole Lennoxa-Gastauta.
Głównym wskazaniem są napady typu grand mal.
Jako działania niepożądane mogą ujawnić się: za-
Napady częściowe proste reagują lepiej na terapię
wroty i bóle głowy, nadmierna senność, dolegliwo-
fenobarbitalem niż napady częściowe złożone. Feno-
ści żołądkowo-jelitowe, a także wysypki skórne oraz
barbital stosuje się ponadto w opornym na inne lecze-
(rzadko) inne ciężkie reakcje alergiczne.
nie stanie padaczkowym (zob. poniżej).
U dzieci poniżej 2 roku życia lamotrygina jest prze-
Głównym działaniem niepożądanym jest nadmier-
ciwwskazana. Ostrożność zalecana jest u pacjentów
ne działanie uspokajające (sedacja). Ponadto istnieje
z niewydolnością wątroby i nerek.
ryzyko ujawnienia się móżdżkowej ataksji, a tak-
Karbamazepina, fenobarbital, fenytoina i primidon
że (choć rzadko) ciężkich powikłań skórnych (zespół
przyspieszają rozkład lamotryginy; z kolei kwas wal-
Stevensa-Johnsona).
proinowy hamuje jej biotransformację.
W przypadku ostrych zatruć środkami o ośrodko-
wym działaniu hamującym, w ciężkich zaburzeniach
Zonisamid. Mechanizm działania zonisamidu nie zo-
funkcji wątroby, mięśnia sercowego i nerek, w porfi-
stał w pełni wyjaśniony. Wydaje się on działać przede
rii, w stanie szoku oraz w stanie astmatycznym stoso-
wszystkim na napięciowozależne kanały Na+ i Ca2+
wanie fenobarbitalu jest przeciwwskazane.
– w ten sposób przerywając zsynchronizowane wyła-
Jako silny induktor enzymów cytochromowych
dowania neuronalne. Dodatkowo wywiera modulują-
– fenobarbital w związku z przyspieszeniem proce-
cy wpływ na neuronalne procesy hamowania związa-
su biotransformacji – zmniejsza siłę działania m.in.
ne z układem GABA-ergicznym.
karbamazepiny, lamotryginy, fenytoiny, tiagabiny
Zonisamid po zażyciu jest niemal całkowicie
i kwasu walproinowego, a w dalszej kolejności – do-
wchłaniany. Maksymalne stężenie w osoczu osiągane
ustnych leków zmniejszających krzepliwość krwi,
jest w ciągu 2–5 godzin po zażyciu. Okres półtrwania
doksycykliny, werapamilu, teofiliny oraz leków ste-
w osoczu wynosi ok. 65 godzin.
roidowych.
Zonisamid jest wskazany w terapii dodanej napadów
częściowych bez i z ich wtórnym uogólnianiem się.
Jako bardzo częste działania niepożądane mogą Primidon. Dezoksybarbituran – primidon jest w or-
ujawnić się: jadłowstręt, pobudzenie, drażliwość, de- ganizmie transformowany do dwóch głównych meta-
presja, ataksja, zawroty głowy, osłabienie pamięci, bolitów – fenyloetylomalonamidu oraz fenobarbitalu,
nadmierna senność i podwójne widzenie. choć pewne działanie farmakologiczne wykazuje tak-
że substancja macierzysta.
Wskazania primidonu są podobne do fenobarbi-
1.9.2.2. Leki przeciwpadaczkowe talu, jednakże skuteczność primidonu w napadach
nasilające głównie działanie GABA psychomotorycznych oraz mioklonicznych zdaje się
wyższa niż fenobarbitalu.
Do tej grupy leków przeciwpadaczkowych (zob. tab.
Na początku terapii mogą ujawniać się istotne
B 1.9-1) zalicza się:
działania niepożądane w rodzaju zawrotów głowy,
■ fenobarbital, nudności i stanów przypominających upojenie al-
MUTSCHLER-2009.indd 330 2010-01-07 22:12:26
Układ nerwowy 331
koholowe. Dlatego należy rozpocząć terapię od nie- łaniami niepożądanymi wigabatryna ma znaczenie
wielkich dawek primidonu. W pozostałym zakresie terapeutyczne właściwie jedynie w przypadku napa-
– działania niepożądane, przeciwwskazania i inter- dów zgięciowych salaam.
akcje primidonu nie odbiegają od tych spotykanych Szczególnie ostrożne stosowanie i ścisła kontrola
Układ nerwowy
w terapii fenobarbitalem. przy zażywaniu leku zalecane są w przypadku upo-
śledzenia funkcji nerek, znanym z wywiadu przeby-
Benzodiazepiny. Z całej grupy benzodiazepin jako tym epizodzie psychotycznym, hiperkinezach i zabu-
leki przeciwpadaczkowe stosuje się trzy długo dzia- rzeniach funkcji hormonalnych (endokrynopatiach).
łające substancje: klobazam, klonazepam, diazepam
i krótko działający lorazepam. Przede wszystkim Tiagabina. Tiagabina hamuje selektywnie wychwyt
służą one do ostrego przerwania zdarzenia padacz- zwrotny GABA ze szczeliny synaptycznej i w ten spo-
kowego (np. stanu padaczkowego), opanowania kry- sób nasila działanie GABA (zob. ryc. B 1.9-2). B1
zysowej sytuacji napadowej (np. niewyrównana pa- W przypadku podania doustnego jest dobrze resor-
daczka) lub przejściowego zastąpienia innych leków bowana (biodostępność > 90%). Wiązanie z białkami
przeciwpadaczkowych (np. w razie wystąpienia reak- osocza sięga 95%, a okres półtrwania w osoczu wyno-
cji alergicznej). Jedynie w przypadkach wyjątkowych si 7–9 godzin. Intensywna biotransformacja związku
benzodiazepiny stosowane bywają w terapii długo- przebiega w obrębie izoenzymu CYP3A oraz podle-
terminowej. Wadą jest, że ich siła działania znacząco ga procesowi sprzęgania z kwasem glukuronowym
się zmniejsza w przypadku przewlekle prowadzonej z wytworzeniem nieaktywnych metabolitów.
terapii. Tiagabina jest dopuszczona do terapii dodanej
Klonazepam może prowadzić u niemowląt i ma- u pacjentów z napadami częściowymi oraz wtórnie
łych dzieci do zwiększenia wydzielania śliny oraz uogólnionymi napadami toniczno-klonicznymi.
nadmiernej produkcji śluzu w obrębie drzewa oskrze- Jako działania niepożądane mogą wystąpić: za-
lowego. Stąd też ważna jest obserwacja małego pa- wroty głowy, nadmierna senność i astenia, w dalszej
cjenta pod tym kątem (niebezpieczeństwo utraty kolejności – labilność emocjonalna, drżenia mięśnio-
drożności dróg oddechowych). we (tremor), biegunki i drobne wybroczyny podskór-
ne (ekchymozy).
Wigabatryna. Wigabatryna jest pochodną GABA, Tiagabina jest przeciwwskazana w przypadku
która przez kowalencyjne wiązanie w obrębie centrum ciężkich zaburzeń funkcji wątroby.
aktywnego nieodwracalnie hamuje GABA-trans- Leki przeciwpadaczkowe, które prowadzą do in-
aminazę – enzym niezbędny w procesie rozkładu dukcji enzymatycznej, np. karbamazepina lub fenyto-
cząsteczki GABA (zob. ryc. B 1.9-2), a tym samym ina, przyspieszają biotransformację tiagabiny.
zwiększa stężenie GABA w obrębie szczeliny synap-
tycznej. Farmakologicznie skuteczniejszą częścią
składową znajdującego się w handlu racematu sub-
stancji jest jego forma enancjomerowa S-(+). 1.9.2.3. Leki przeciwpadaczkowe
W przypadku doustnego spożycia wigabatryna o innym mechanizmie działania
ulega szybkiemu wchłonięciu; biodostępność enan-
cjomeru S wynosi 50%, a okres półtrwania w osoczu Gabapentyna. Gabapentyna jest wprawdzie struk-
– 6–8 godzin. Substancja nie ulega wiązaniu z biał- turalnie podobna do cząsteczki neuroprzekaźnika
kami osocza. Wydalanie następuje w niezmienionej GABA, lecz nie ma ani pośrednich, ani bezpośred-
postaci – głównie przez nerki. nich właściwości GABA. Jako mechanizm działania
Wigabatryna została dopuszczona do stosowania leku dyskutowane jest hamowanie neuroprzekaźni-
jako monoterapia w napadach typu zgięciowego (sa- ctwa glutaminergicznego, jak również blokowanie
laam), a ponadto jako terapia dodana (add-on) w na- ośrodkowych kanałów wapniowych typu L (zob. ryc.
padach częściowych prostych i złożonych u doro- B 1.9-2).
słych, w napadach częściowych oraz w zespole Len- Z przewodu pokarmowego gabapentyna jest wchła-
noxa-Gastauta. niana za pomocą transportera aminokwasowego
Jako działania niepożądane obserwowano: uczu- (biodostępność wynosi 30–60%). Nie ulega biotrans-
cie zmęczenia i senności, rzadziej bóle głowy, zawro- formacji i nie wiąże się z białkami osocza. Okres po-
ty głowy, stany pobudzenia, zachowania agresywne łowiczej eliminacji wynosi około 5–7 godzin. Wyda-
i dolegliwości żołądkowo-jelitowe; ponadto dosyć lanie następuje głównie przez nerki.
często ujawniają się długo utrzymujące się zawęże- Gabapentyna została dopuszczona do monoterapii
nia pola widzenia. W związku z wymienionymi dzia- oraz do terapii dodanej prostych i złożonych napa-
MUTSCHLER-2009.indd 331 2010-01-07 22:12:27
332 Układ nerwowy
dów częściowych bez i z wtórnym ich uogólnianiem ctwa glutaminergicznego oraz nasileniu przekaźni-
się. Ze względu na brak wystarczającej skuteczności ctwa GABA-ergicznego, a także na słabej blokadzie
nie jest jednak lekiem przeciwpadaczkowym pierw- karboanhydrazy (tzw. działanie plejotropowe).
szego wyboru, lecz środkiem rezerwowym. (Poza Po podaniu doustnym topiramat ulega szybkiemu
terapią padaczki gabapentyna może być ponadto sto- i dobremu wchłanianiu (biodostępność rzędu 80%).
sowana w leczeniu bólów neuropatycznych). Okres półtrwania w osoczu wynosi 18–24 godz. Wy-
Działaniami niepożądanymi, jakie mogą ujaw- dalanie następuje głównie przez nerki.
nić się w trakcie stosowania gabapentyny, są: zabu- Topiramat jest stosowany w monoterapii oraz
rzenia ośrodkowe (np. uczucie zmęczenia, zawroty w terapii dodanej padaczek z napadami częściowymi
głowy, ataksja), w dalszej kolejności – nudności oraz uogólnionymi. W zakresie współczynnika po-
i wymioty, zwiększenie masy ciała, obrzęki, a tak- prawy podawane są różne dane. Efekty terapii długo-
że zaburzenia zachowania (np. drażliwość) – wy- terminowej nie zostały jeszcze wystarczająco dobrze
stępujące szczególnie często u dzieci z licznymi zbadane.
postaciami inwalidztwa. Wśród działań niepożądanych wymienia się zabu-
Stosowanie gabapentyny jest przeciwwskazane rzenia ośrodkowe (uczucie zmęczenia, zawroty gło-
w ostrym zapaleniu trzustki. wy, ataksję, zaburzenia myślenia ze spowolnieniem
Równoczesne zażywanie środków przeciw nad- toku), w dalszej kolejności – zmniejszenie masy ciała
kwasocie zawierających magnez lub aluminium i nasilenie tworzenia kamieni nerkowych (podwyż-
zmniejsza wchłanianie gabapentyny. szone ryzyko w przypadku skojarzonego spożywania
z tiamterenem). Stąd też w trakcie leczenia należy
Pregabalina. Środek ten jest również pochodną zwracać uwagę na przyjmowanie wystarczających
GABA. Podobnie jak gabapentyna, łączy się z dodat- ilości płynów.
kową podjednostką napięciowozależnych kanałów Karbamazepina i fenytoina przyspieszają rozkład
Ca2+ w ośrodkowym układzie nerwowym. topiramatu. Z drugiej strony topiramat zwiększa po-
Biodostępność po doustnym zażyciu wynosi oko- ziom fenytoiny w osoczu.
ło 90%. Maksymalne wartości stężenia w osoczu
osiągane są w ciągu 1 godziny po spożyciu. Stąd też Felbamat. Felbamat ingeruje prawdopodobnie
pregabalina ulega wydaleniu w niezmienionej postaci w miejsce wiązania glicyny w obrębie receptora
– głównie przez nerki. Średni okres połowiczej elimi- NMDA (zob. ryc. B 1.9-2) oraz dodatkowo nasila
nacji wynosi ok. 6 godzin. przekaźnictwo GABA-ergiczne.
Pregabalina jest stosowana w terapii dodanej Wchłanianie następuje szybko i niemal w całości.
w przypadku napadów częściowych bez i z wtórnym W wątrobie felbamat ulega procesowi częściowej
ich uogólnianiem się. Ponadto wskazana jest – jak hydrolizy, hydroksylacji oraz sprzęgania do częścio-
to już wcześniej opisano – w terapii obwodowych wo nieskutecznych metabolitów. Okres półtrwania
bólów neuropatycznych oraz zespołu lęku uogólnio- w osoczu wynosi 15–23 godz. Wydalanie następuje
nego. głównie przez nerki.
Częstymi działaniami niepożądanymi są: wzmo- Felbamat został jak dotychczas dopuszczony do le-
żony apetyt, zwiększenie masy ciała, euforia, spląta- czenia w ramach terapii dodanej u pacjentów z opor-
nie, drażliwość, zaburzenia chodu, uczucie upojenia, nym na leczenie zespołem Lennoxa-Gastauta – jeżeli
zawroty głowy, zamroczenie, senność, wymioty, su- brak jest innych możliwości terapeutycznych.
chość w ustach, zaparcia, wzdęcia, rozbicie, obwodo- Zagrażającymi życiu powikłaniami są anemia
we obrzęki, osłabienie libido i zaburzenia erekcji. aplastyczna (ryzyko wystąpienia 1:4000) oraz tok-
Ponieważ pregabalina nie jest u człowieka pra- syczne uszkodzenie wątroby (ryzyko wystąpienia
wie w ogóle metabolizowana – w badaniach in vivo 1:7000). Stąd też konieczne jest przeprowadzanie
nie obserwowano żadnych klinicznie istotnych in- co dwutygodniowych kontroli obrazu krwi oraz
terakcji między pregabaliną a fenytoiną, karbama- enzymów wątrobowych (transaminaz). Czasem
zepiną, kwasem walproinowym, lamotryginą, ga- ujawniają się takie objawy, jak zmęczenie, ataksja,
bapentyną, lorazepamem, oksykodonem czy etano- bóle i zawroty głowy, zaburzenia snu, oraz brak
lem. apetytu.
Stosowanie felbamatu jest przeciwwskazane u pa-
Topiramat. Przeciwpadaczkowe działanie topiramatu cjentów ze znanymi z wywiadu schorzeniami krwi
polega zarówno na blokowaniu napięciowozależnych i wątroby, u pacjentów z niewydolnością nerek, jak
kanałów sodowych, jak i na hamowaniu przekaźni- również u osób w wieku powyżej 65 lat.
MUTSCHLER-2009.indd 332 2010-01-07 22:12:27
Układ nerwowy 333
Felbamat obniża stężenie karbamazepiny w oso- Nie zostały opisane żadne interakcje in vivo mię-
czu, podwyższa z kolei poziom epoksydu karbamaze- dzy lewetiracetamem a innymi substancjami w obrę-
piny, fenytoiny i kwasu walproinowego. Odwrotnie bie cytochromu P-450.
karbamazepina i fenytoina obniżają stężenie felba- Sultiam. Sultiam jest lekiem przeciwpadaczkowym bez
uspokajająco-nasennego działania. Jako substancji hamu-
Układ nerwowy
matu w osoczu.
jącej aktywność karboanhydrazy jej działanie polega naj-
prawdopodobniej na efekcie metabolicznym wynikającym
Imidy kwasu bursztynowego (suksymidy). Oba z przesunięcia równowagi pH w kierunku kwasowym.
preparaty z grupy suksymidów – etosuksymid i me- Po podaniu doustnym sultiam wchłania się szybko i cał-
suksymid ujawniają działanie przeciwpadaczkowe kowicie – głównie w górnych odcinkach jelita cienkiego.
poprzez hamowanie napięciowozależnych kana- Maksymalne stężenia w osoczu występują po 1–5 godzi-
nach. Wydalanie w 80–90% następuję wraz z moczem; dal-
łów wapniowych typu T (szczególnie neuronów sze 10–20% – w wyniku sekrecji żółciowej – z kałem.
wzgórzowo-korowych). Tym samym ich mecha- Sultiam stosowany jest głównie w tzw. padaczce rolan-
B1
nizm działania różni się od innych leków przeciw- dycznej – w posiadającej dużą zdolność do spontaniczne-
padaczkowych. Podczas gdy mesuksymid ulega go ustępowania postaci padaczki występującej w wieku
biotransformacji do odpowiedzialnej za działanie wczesnodziecięcym i szkolnym, w zwykłych napadach
częściowych, a w terapii skojarzonej z innymi lekami
kliniczne N-demetylowanej pochodnej, z etosuk- przeciwpadaczkowymi – również w napadach typu grand
symidu powstają metabolity niemające działania mal.
biologicznego. Jako działania niepożądane obserwowano: zaburzenia
Etosuksymid (w mniejszym zakresie mesuksymid) czuciowe (parestezje), bóle głowy, nadmierną senność, jak
jest skuteczny w przypadku padaczki z napadami typu również rzadziej ujawniające się napadowopodobne zabu-
rzenia oddychania (hyperpnoe) i dolegliwości żołądkowo-
absence i wraz z kwasem walproinowym uznawany jelitowe.
jest w tym wskazaniu za lek z (pierwszego) wyboru. Sultiam jest przeciwwskazany w przypadku znanej ostrej
Jest natomiast nieskuteczny w napadach typu grand porfirii, u chorych z nadczynnością tarczycy oraz nadciś-
mal; w niektórych przypadkach może nawet służyć nieniem tętniczym.
do wywołania tego rodzaju napadów. W przypadku skojarzonego stosowania sultiamu z feny-
toiną spowolnieniu ulega rozkład fenytoiny w wątrobie,
Jako działania niepożądane mogą wystąpić: za- a tym samym dochodzi do podwyższenia poziomu feny-
wroty głowy, nudności, wymioty, dolegliwości ga- toiny w osoczu.
stryczne oraz skórne reakcje alergiczne. Przedawko-
wanie prowadzi głównie do objawów senności, choć
również do drażliwości, wahań nastroju i stanów po-
1.9.3. Zalecenia dotyczące
budzenia.
Kwas walproinowy podwyższa, a karbamazepina farmakoterapii padaczki
obniża stężenie suksymidów w osoczu.
Terapia farmakologiczna padaczki rozpoczyna się
Lewetiracetam. Mechanizm działania lewetira- zwykle po wystąpieniu u pacjenta dopiero drugiego
cetamu jest nieznany. Wchłanianie leku po doustnym lub nawet trzeciego napadu padaczkowego – o ile
podaniu jest szybkie. Lewetiracetam ulega częścio- istnieją przesłanki pozwalające przyjąć istnienie
wemu metabolizmowi na drodze hydrolizy i jest wy- przewlekłej choroby padaczkowej. Terapia lekowa
dalany głównie przez nerki. może być rozważona również wtedy, gdy istnieje
Lek jest wskazany do stosowania w ramach te- podejrzenie padaczki idiopatycznej (np. przy istnie-
rapii dodanej napadów częściowych z i bez wtórne- jącej predyspozycji genetycznej, gdy napad wystąpił
go uogólniania. Na skutek zwykle dobrej tolerancji w okresie pierwszych dwóch godzin po przebudze-
i istniejącej przynajmniej na początku dobrej sku- niu) lub też pierwszy napad zdarzył się w związku
teczności – stanowi pewną alternatywę dla innych ze zidentyfikowanym prawdopodobnym ogniskiem
leków przeciwpadaczkowych. Wadą lewetiracetamu padaczkowym związanym z uszkodzeniem mózgu
jest jednakże potencjalna zdolność prowokowania (m.in. stan po urazie czaszkowo-mózgowym lub
napadów grand mal oraz utrata skuteczności w ciągu zapaleniu mózgu). Leczenie nie wydaje się pilnie
tygodni lub miesięcy stosowania. wskazane, gdy napady występują rzadko, tj. jeden,
Najczęstszymi działaniami niepożądanymi są dwa razy w roku.
nadmierna senność, osłabienie (astenia), przymrocze-
nie, ataksja i drżenia mięśniowe. Dość często ujaw- Cele terapii. W przypadku prawie połowy wszyst-
niają się również psychiczne zaburzenia w rodzaju kich pacjentów możliwe jest za pomocą obecnie
depresji, agresji, wściekłości, lęku czy psychoz. dostępnych leków przeciwpadaczkowych całkowite
MUTSCHLER-2009.indd 333 2010-01-07 22:12:27
334 Układ nerwowy
opanowanie napadów. Jeżeli nie jest to możliwe, na- mi w warunkach podwójnie ślepej próby – równie
leży się przynajmniej starać, aby częstość napadów skuteczne jak klasyczne substancje aktywne, a po-
była jak najmniejsza. nadto charakteryzują się lepszą tolerancją.
Drugim celem terapii jest unikanie istotnych Przy braku odpowiedniej reakcji (poprawy)
działań niepożądanych prowadzonej farmakotera- na pierwszy z leków zastosowany w monoterapii na-
pii. Uwaga ta dotyczy przede wszystkim zaburzeń leży spróbować terapii drugim preparatem lub zasto-
funkcji poznawczych, jak również zwiększenia się sować terapię skojarzoną za pomocą dwóch substan-
masy ciała, hirsutyzmu lub rozrostu śluzówki dziąseł. cji aktywnych.
Ważne jest także zapobieganie wystąpieniu warunko-
wanych indukcją enzymów wątrobowych następstw Monoterapia za pomocą drugiego leku przeciwpadaczko-
hormonalnych (np. osteoporozie, zaburzeniom sek- wego ma szansę powodzenia przede wszystkim wówczas,
gdy pierwszy z leków – z powodu wystąpienia działań nie-
sualnym). Każdy lek przeciwpadaczkowy powinien pożądanych – nie mógł być zastosowany w maksymalnej
być stosowany w dawce – tuż przed osiągnięciem dawce. W przypadku objawowych padaczek z napadami
granicy ujawniania się działań niepożądanych; z dru- częściowymi – powstałymi w związku z uszkodzeniami
giej jednak strony ostatnia faza zwiększania dawki tkanki mózgowej – zwykle od samego początku konieczne
leku ze średniej na indywidualnie najwyższą możli- jest stosowanie terapii skojarzonej.
wą koresponduje jedynie z niewielkim zwiększeniem
się skuteczności leku. Dopasowanie dawki powinno Przy braku skuteczności terapii drugim lekiem lub
być przeprowadzone na podstawie oceny skuteczno- terapii skojarzonej – konieczne jest przeprowadze-
ści u danego pacjenta oraz klinicznej tolerancji leku, nie ponownej analizy rozpoznania, która ma na celu
a nie na podstawie jego stężenia w osoczu. Wydające wykluczenie dodatkowo lub wyłącznie występują-
się podwyższone z laboratoryjnego punktu widzenia cych napadów psychogennych. Najpóźniej do tego
poziomy leku w osoczu – przy dobrej tolerancji leku momentu należy przeanalizować problem zasadności
i kontroli napadów – nie uzasadniają zmniejszenia poddania pacjenta zabiegowi operacyjnemu. Wska-
dawki leku. zaniem do przeprowadzenia diagnostyki w zakresie
możliwości przeprowadzenia chirurgicznego leczenia
Terapia padaczek z napadami częściowymi. Kla- padaczki jest zasadniczo każda oporna na leczenie
syczne leki przeciwpadaczkowe: karbamazepina, fe- padaczka z napadami częściowymi.
nobarbital, fenytoina, primidon i kwas walproinowy
są dopuszczone do monoterapii padaczek z napada-
Terapia padaczek idiopatycznych z napadami
mi częściowymi bez i z wtórnym uogólnianiem się.
ogólnionymi. Lekiem pierwszego wyboru w tera-
Za nowe leki o podobnym profilu działania są uważa-
pii napadów grand mal pozostaje kwas walproino-
ne obecnie: gabapentyna, lamotrygina, okskarbaze-
wy, z kolei napadów nieświadomości typu absence
pina i topiramat. Wszystkie dotychczasowe badania
– etosuksymid. Uwzględniając indywidualny profil
pozwalają stwierdzić, że w przypadku tych różnych
działań niepożądanych – w przypadku niektórych
substancji aktywnych praktycznie nie ma większych
zespołów padaczkowych – można rozważyć użycie
różnic w zakresie – na ogół dobrej – skuteczności.
w początkowym okresie lamotryginy lub topirama-
W związku z bardzo licznymi obserwacjami i niski-
tu. Jako preparaty rezerwowe w przypadku oporno-
mi kosztami terapii – karbamazepina nadal uznawana
ści na leczenie dostępne fenobarbital i benzodiaze-
jest za lek pierwszego wyboru w wyżej wymienionych
piny oraz w przyszłości prawdopodobnie także – le-
wskazaniach. Z drugiej jednak strony uwzględniając
wetiracetam.
towarzyszące jej stosowaniu, niekiedy istotne dzia-
W przypadku niepowodzenia terapii pierwszego
łania niepożądane (m.in. alergiczne wysypki 5–8%
wyboru należy wziąć pod uwagę następujące rozwią-
leczonych) oraz skutki długoterminowe (np. osteopo-
zania:
roza wywołana nasilonym, warunkowanym indukcją
enzymów metabolizmem egzogennie dostarczanych,
– w przypadku utrzymywania się napadów typu ab-
jak również endogennych hormonów), jak również
sence: kwas walproinowy oraz
względnie niekorzystną farmakokinetykę – dogma-
tyczne uznawane karbamazepiny za lek pierwszego ■ etosuksymid/mesuksymid,
wyboru nie jest już tak sensowne. Wymienione nowe
■ lamotrygina lub
leki przeciwpadaczkowe są – zgodnie z kontrolowa-
nymi badaniami porównawczymi, przeprowadzony- ■ klobazam,
MUTSCHLER-2009.indd 334 2010-01-07 22:12:27
Układ nerwowy 335
– w przypadku utrzymywania się uogólnionych napa- 1.9.3.1. Terapia stanu padaczkowego
dów toniczno-klonicznych: (status epilepticus)
■ kwas walproinowy oraz lamotrygina,
Zagrażający życiu stan padaczkowy (status epilepti-
Układ nerwowy
■ kwas walproinowy oraz fenobarbital/primidon, cus) napadów grand mal wymaga wdrożenia natych-
miastowego leczenia – polegającego na dożylnym
■ monoterapia lamotryginą bądź terapia łączona la-
podaniu klonazepamu, diazepamu lub fenytoiny – bę-
motryginą z topiramatem, lewetiracetamen lub klo-
dących środkami pierwszego wyboru. Jeżeli w przy-
bazamem.
padku zastosowania koniecznych, ale dużych dawek
dojdzie do depresji układu oddechowego – pacjent
– w przypadku utrzymywania się napadów mioklo-
musi być poddany procedurze sztucznego oddycha-
nicznych: kwas walproinowy oraz
nia. Jeżeli nie uda się uzyskać wystarczającego efek- B1
■ fenobarbital/primidon, tu terapeutycznego, wskazane jest dodatkowe poda-
nie fenobarbitalu – również drogą wlewu dożylnego
■ klobazam,
(w razie potrzeby za pomocą iniekcji domięśniowej).
■ topiramat lub Również w przypadku stanu padaczkowego napa-
dów typu absence – klonazepam jest środkiem pierw-
■ lewetiracetam.
szego wyboru; alternatywnie może być podany (i.v.)
W przypadku niepowodzenia terapii drugiego rzutu kwas walproinowy.
należy rozważyć inne klinicznie i racjonalnie sen- W stanie padaczkowym napadów częściowych pro-
sowne zastosowanie kombinacji dwóch lub większej stych oraz częściowych złożonych postępuje się podob-
liczby wyżej wymienionych środków. nie jak w stanie padaczkowym napadów grand mal.
1.10. Zespół Parkinsona i leki przeciwparkinsonow-
1.10.1. Podstawy patofizjologiczne Z wyjątkiem postaci schorzenia, które są wyzwo-
lone działaniem niektórych leków (zob. poniżej),
Zespół Parkinsona jest po chorobie Alzheimera drugą u podłoża choroby leży postępujące zwyrodnie-
co do częstości zachorowań chorobą neurodegene- nie neuronów dopaminergicznych, przede wszyst-
racyjną z rosnącym współczynnikiem zachorowań. kim w obrębie istoty czarnej (substantia nigra).
Zaburzeniu ulegają przede wszystkim wykonywane Ta w związku z dużą zawartością żelaza i melaniny
sekwencje ruchowe – zarówno dowolne, jak i mi- ciemno zabarwiona okolica śródmózgowia (mesen-
mowolne. W większości chorują osoby w podeszłym cephalon) jest częścią składową jąder podstawy,
wieku; częstość zachorowań wśród mężczyzn i ko- które istotnie uczestniczą w specyficznej selekcji
biet jest podobna. Ryzyko zachorowania zwiększa się oraz przetwarzaniu motorycznych i niemotorycz-
wraz z wiekiem (zapadalność u osób powyżej 60 roku nych wzorców zachowań, a także blokują aktualnie
życia wynosi 1%, u ponad 70-latków – 2%, u ponad niewymagane wzorce aktywacji. Jako filtr – jądra
80-latków – 3%). Jednak chorobą Parkinsona są do- podstawy są przy tym powiązane złożonymi pętla-
tknięte nie tylko osoby starsze: 40% pacjentów zapada mi regulującymi, które wychodzą z kory mózgowej,
na chorobę przed 60 rokiem życia, a niektórzy nawet przechodzą przez jądra podstawne i wzgórze, podą-
przed 40 rokiem życia – jak to wykazuje lepsze me- żając z powrotem do płatów czołowych mózgu (zob.
tody diagnostyczne (za pomocą TK, MRI). W Niem- ryc. 1.10-1).
czech choruje 250 000 osób, przy czym każdego roku
diagnozuje się około 12 000 nowych przypadków Z wyjątkiem kory wzrokowej i słuchowej niemal wszyst-
zachorowania (w Polsce choruje 80 000–120 000, kie informacje (sensoryczne) docierają z kory mózgowej
przy czym każdego roku diagnozuje się około 8000 do prążkowia (striatum) – jako do stacji wejściowej do ją-
der podstawy – pod postacią dróg korowo-prążkowiowych
nowych przypadków). Ze względu na wydłużający się wykorzystujących pobudzającą neurotransmisję glutami-
czas życia ludzi – przewiduje się podwojenie liczby nergiczną. Przez stację wejściową jąder podstawy – część
chorych na całym świecie do 2040 r. siatkowatą istoty czarnej (substantia nigra pars reticularis)
MUTSCHLER-2009.indd 335 2010-01-07 22:12:27
336 Układ nerwowy
osoba zdrowa pacjent z chorbą Parkinsona
kora
mózgowa
THA
PUT
D1 D1
D2 D2
GPe
STN
GPi
śródmózgowie
SN SN
prawidłowa funkcja jąder podstawnych zaburzona funkcja jąder podstawnych w chorobie Parkinsona
drogi drogi drogi
dopaminergiczne GABA-ergiczne glutaminergiczne
Ryc. B 1.10-1. Pętle regulujące w obrębie jąder podstawnych, które uczestniczą w patomechanizmie choroby Parkinsona. SN – isto-
ta czarna (substantia nigra), PUT – skorupa (putamen), GPe – gałka blada zewnętrzna (globus pallidus externus), GPi – gałka blada
wewnętrzna (globus pallidus internus), STN – jądro podwzgórzowe (nucleus subthalamicus), THA – wzgórze (thalamus). Grubość linii
strzałek odpowiada stopniowi aktywacji neuronów.
oraz gałkę bladą wewnętrzną (globus pallidus internus) Modulowanie aktywności jąder podstawy umożliwia tym
– przepracowana w obrębie tych jąder informacja (koń- samym zarówno zmiany in plus, jak i in minus w zakre-
cowa) powraca hamującymi drogami GABA-ergicznymi sie aktywności ruchowej oraz realizowania odpowiednich
do wzgórza (thalamus), a stamtąd pobudzającymi gluta- wzorców ruchowych. Stąd też jądra podstawy mają duże
minergicznymi drogami do kory. Wymienione hamujące znaczenie dla części wykonawczej mózgu, który w szcze-
neurony GABA-ergiczne wywołują ostatecznie przez stłu- gólności odgrywa istotną rolę przy planowaniu i na począt-
mienie funkcji hamujących – pobudzenie wzgórza i kory. ku wykonywania każdej czynności ruchowej.
Podobny efekt netto zachodzi przy udziale dróg wychodzą-
cych z prążkowia i podążających przez część siatkowatą Ważne połączenie modulacyjne w obrębie jąder pod-
istoty czarnej do wzgórza. stawy stanowi dopaminergiczna droga rozpoczy-
Oprócz tego istnieje jeszcze pośrednie połączenie z prąż- nająca się komórkami nerwowymi w części zwar-
kowia do gałki bladej wewnętrznej i do części siatkowatej tej istoty czarnej (substantia nigra pars compacta)
istoty czarnej. W tym wypadku drogi wychodzące z prąż- i rzutująca w kierunku do prążkowia. Zaburzenie
kowia hamują w następstwie gałkę bladą zewnętrzną, przez pracy tej drogi wynikające z degeneracji tworzących
co dochodzi również do pośredniego pobudzenia jądra ją neuronów wywołuje zespół Parkinsona. Skutkiem
podwzgórzowego (nucleus subthalamicus). Ponieważ jądro
to drogą glutaminergiczną oddziałuje pobudzająco na gałkę wystąpienia niedoboru dopaminy są zaburzenia ak-
bladą wewnętrzną – ostatecznie dochodzi do zahamowania tywności wyładowań neuronów prążkowia. To z ko-
wzgórza i kory. lei powoduje przesunięcie równowagi na korzyść in-
MUTSCHLER-2009.indd 336 2010-01-07 22:12:27
Układ nerwowy 337
nych układów neuroprzekaźnikowych: cholinergicz- Różnorodne postacie zespołu Parkinsona można po-
nego, GABA-ergicznego i glutaminergicznego (zob. dzielić na trzy zasadnicze grupy:
ryc. B 1.10-1). Ostatecznie nasileniu ulega hamująca
■ rodzinny (dziedziczny) zespół Parkinsona (PARK 1
aktywność glutaminergiczna neuronów rzutujących
do PARK 8, PARK 10, 11) z zapadalnością rzędu
Układ nerwowy
w kierunku wzgórza. W wyniku tego zmniejsza się
5%,
pobudzająca aktywność glutaminergiczna dróg rzutu-
jących ze wzgórza do kory z typowymi dla choroby ■ idiopatyczny zespół Parkinsona (ok. 75% wszyst-
Parkinsona zaburzeniami ruchowymi. kich przypadków choroby),
Odwrotnie – wypadnięcie jądra podwzgórzowego lub gał- ■ objawowy (wtórny) zespół Parkinsona (ok. 15%
ki bladej zewnętrznej powoduje wzmożoną aktywność wszystkich przypadków) – powstały w wyniku m.in.
neuronów części siatkowatej istoty czarnej i wyzwala tym zatruć (np. tlenkiem węgla), wyzwolony niektórymi B1
samym zaburzenie z objawami wzmożonej aktywności ru- lekami (np. neuroleptykami lub solami litu) albo
chowej, np. pląsawicę Huntingtona. na skutek miażdżycy naczyń, zapalenia mózgu bądź
guzów.
W okresie początkowym utrata neuronów łączących
istotę czarną z prążkowiem może być jeszcze funkcjo- W przeciwieństwie do innych postaci – polekowy zespół
nalnie wyrównana przez mózg, jednak ginące komór- Parkinsona ustępuje zwykle po odstawieniu leku. Objawy
parkinsonowe obserwowane niekiedy u osób uzależnio-
ki nerwowe nie mogą być zastąpione przez kolejne. nych od narkotyków, które wiążą się z zanieczyszczeniem
To wyrównanie czynnościowe przestaje być możliwe, narkotyków pochodną tetrahydropirydyny (1-metylo-4-
gdy więcej niż połowa neuronów dopaminergicznych fenylo-1,2,3,6-tetrahydropirydyny = MFTP), są natomiast
ulegnie zanikowi. Kontrolne piętra układu ruchowe- nieodwracalne.
go we wzgórzu, jądrach podstawy i korze mózgowej
przestają funkcjonować jako skoordynowana całość Typowy dla pacjentów z chorobą Parkinsona jest chód
i ujawniają się typowe objawy choroby Parkinsona: małymi krokami, tułów nachylony do przodu, brak
współruchów ramion przy chodzeniu oraz brak mimi-
1. zaburzenia ruchowe z objawami pozytywnymi ki twarzy. Wszystkie ruchy rozpoczynane są z pew-
nym opóźnieniem i mogą być zatrzymane z pewną
■ wzmożenie napięcia (rigor),
trudnością.
■ drżenie spoczynkowe kończyn (tremor),
Post mortem znajduje się w licznych przypadkach złogi
■ posturalna niestabilność (niepewność przy staniu), białek w dopaminowych neuronach istoty czarnej, które
określane są terminem ciałek Levy’ego. Bardzo prawdopo-
■ zaburzenia mowy; dobne jest, że te błędnie pofałdowane białka uczestniczą
przyczynowo w chorobie Parkinsona.
oraz z objawami negatywnymi
■ hipo- i akinezja;
1.10.2. Leki stosowane
2. zaburzenia wegetatywne w chorobie Parkinsona
■ wzmożony ślinotok i łzawienie,
W zależności od określonych zmian patofizjolo-
■ podwyższone wydzielanie łoju (twarz jak posmaro- gicznych – terapia farmakologiczna zespołu Parkin-
wana maścią – nalana), sona jest możliwa dzięki (zob. ryc. B 1.10-2, tab. B
1.10-1):
■ zaburzona termoregulacja oraz wydzielanie potu,
■ lewodopie (zwykle w terapii skojarzonej z obwodo-
■ obniżone ciśnienie krwi,
wo działającymi inhibitorami dekarboksylazy),
■ czynnościowe zaburzenia pęcherza;
■ hamowaniu procesu metylacji dopaminy i lewodo-
py za pomocą inhibitorów COMT,
3. zaburzenia psychiczne
■ zapobieganiu rozpadowi dopaminy za pomocą in-
■ depresyjne obniżenia nastroju,
hibitorów monoaminooksydazy typu B,
■ wydłużenie procesów myślowych,
■ stymulowaniu ośrodkowych receptorów dopami-
■ w późnych okresach – ewentualnie spowolnienie nergicznych za pomocą bezpośrednich agonistów
toku myślenia (bradyfrenia) oraz otępienie. dopaminergicznych,
MUTSCHLER-2009.indd 337 2010-01-07 22:12:28
338 Układ nerwowy
komórka glejowa
prążkowie
COMT
neuron łączący DOPAC entakapon,
istotę czarną z prążkowiem HVS
COMT tolkapon
K+
MT
istota
czarna
Ca2+
↑
cAMP
selegilina MAO B
hamowanie
AC
Gi
D2
dopamina GABA-ergiczny neuron
w prążkowiu, cholinergiczny
interneuron
DDC
benserazyd,
karbidopa lewodopa
entakapon,
Gs
DAT
D1
tolkapon GABA-ergiczny
neuron w prążkowiu
AC
DDC pobudzenie
dopamina lewodopa lewodopa
COMT cAMP↑
3-OMD
COMT tkanki obwodowe mózgowe
naczynie
krwionośne
MT
nerka
lewodopa
wątroba
Ryc. B 1.10-2. Mechanizmy działania leków stosowanych w chorobie Parkinsona (przeciwparkinsonowych). Przez receptory D2 do-
pamina hamuje neurony GABA-ergiczne i interneurony cholinergiczne, a przez receptory D1 – aktywuje neurony GABA-ergiczne.
W komórkach glejowych dopamina ulega rozkładowi za pomocą enzymu katecholo-O-metylo-transferazy (COMT) do metoksytyra-
miny (MT), a za pomocą monoaminooksydazy typu B (MAO-B) i COMT – do octanu dihydroksyfenylowego (DOPAC) i kwasu homowa-
nilinowego (HVS). W wyniku blokady dekarboksylazy dopaminowej (DDC) – benserazyd i karbidopa hamują w tkankach obwodowych
(zob. także: ryc. B 1.10-3) przemianę lewodopy do dopaminy. Entakapon i tolkapon – w wyniku zahamowania COMT – zapobiegają
tworzeniu się zarówno MT, jak i 3-O-metylodopy (3-OMD). AC – cyklaza adenylanowa, DAT – transporter dopaminy.
■ blokowaniu receptorów NMDA w prążkowiu za po- przeciwcholinergicznych (antagonistów choliner-
mocą amantadyny, gicznych receptorów typu M).
■ hamowaniu muskarynowych receptorów choliner-
gicznych za pomocą ośrodkowo działających leków
MUTSCHLER-2009.indd 338 2010-01-07 22:12:28
Układ nerwowy 339
1.10.2.1. Lewodopa (L-DOPA), Mimo tych ograniczeń – oceniając ogólnie – wpro-
preparaty złożone z lewodopy wadzenie terapii lewodopą wyraźnie poprawiło ja-
i inhibitora dekarboksylazy kość życia i zwiększyło oczekiwaną długość życia
u pacjentów z chorobą Parkinsona.
Układ nerwowy
Najprostsze leczenie za pomocą podaży neuroprze- Po podaniu doustnym lewodopa ulega aktywnemu
kaźnika – dopaminy, która nie jest w wystarcza- wchłanianiu w obrębie jelita cienkiego. Maksymalne
jącej ilości syntetyzowana, nie jest możliwe, gdyż stężenie w osoczu – zależne jest od galenicznej posta-
nie może ona pokonać bariery krew-mózg. Tymcza- ci leku; w przypadku preparatów zwykłych osiągane
sem dla lewodopy – aminokwasu prekursorowego dla jest po 0,5–1 godzinie od podania, dla preparatów
dopaminy – istnieje aktywny mechanizm transportu o opóźnionym działaniu (retard) – po 1,5–2,5 godz.
(zob. ryc. B 1.10-3), dzięki któremu jest ona w stanie W przypadku wchłaniania do krwi i do ośrodkowego
przedostać się przez tę barierę do mózgu, gdzie może układu nerwowego lewodopa konkuruje z obojętny- B1
być wchłonięta przez jeszcze nieuszkodzone dopami- mi aminokwasami o aktywny mechanizm transportu
nergiczne zakończenia nerwowe dróg łączących isto- znajdujący się w ścianie jelita oraz w obrębie barie-
tę czarną z prążkowiem, jak również przez inne struk- ry krew-mózg. Dlatego też posiłki bogate w białko
tury mózgowia. W wyniku działania dekarboksylazy mogą prowadzić do niskich poziomów lewodopy
dopaminowej (DDC) od lewodopy ulega odłączeniu w osoczu oraz zmniejszenia się jej dostępności móz-
grupa karboksylowa i powstaje właściwa substan- gowej. Przyjmowanie leku powinno być więc czaso-
cja działająca – dopamina. Ponieważ przy podaniu wo przesunięte względem pory spożywania posiłków
jedynie lewodopy ponad 90% zaaplikowanej daw- (tj. 0,5–1 godz. przed lub 1,5–2 godz. po). W innym
ki nie dociera do ośrodkowego układu nerwowego, przypadku należy wprowadzić dietę ubogą w białko.
lecz ulega procesowi dekarboksylacji już na obwo- Okres półtrwania w osoczu wynosi 1,5–2,5 godz. Wy-
dzie, zapobiega się temu procesowi biotransformacji dalanie następuje prawie wyłącznie pod postacią me-
poza ośrodkowym układem nerwowym za pomocą tabolitów: dopaminy, kwasu homowanilinowego oraz
benserazydu lub karbidopy, które nie są w stanie kwasu dihydroksyfenylooctowego.
przeniknąć przez barierę krew-mózg. Równoczesne Zwykła doustna dawka dzienna wynosi początko-
podawanie lewodopy z inhibitorem dekarboksylazy wo 50 mg lewodopy plus 12,5 mg benserazydu lub
(lewodopa + benserazyd lub lewodopa + karbidopa) karbidopy. Dawka może być następnie – w zależno-
pozwala nie tylko na zmniejszenie koniecznej dawki ści od reakcji pacjenta i występujących działań nie-
lewodopy do jednej piątej, lecz także zmniejszają się pożądanych – zwiększana co kolejne 3 dni o 50 mg
znacznie obwodowe działania niepożądane lewodopy lewodopy oraz 12,5 mg inhibitora dekarboksylazy, aż
(zob. ryc. B 1.10-3). do maksymalnej dawki 3–4 razy dziennie 100–200
mg lewodopy. Maksymalna dawka dzienna nie po-
Jednak w wyniku wieloletniego leczenia zmienia się winna przekroczyć zwyczajowych 800 mg lewodopy
działanie lewodopy. O ile w pierwszych latach terapii i 200 mg środka blokującego dekarboksylazę.
zwyczajowe trzy dzienne dawki lewodopy są wystar- Jako działania niepożądane zależne od dawki oraz
czające do uzyskania utrzymującej się i dalece rów- od czasu trwania leczenia ujawniają się: zaburzenia
nomiernej ruchliwości, o tyle po 3–5 latach u więk- ruchowe (dys- i hiperkinezy), zaburzenia wegetatyw-
szości pacjentów zaczynają ujawniać się fluktuacje, ne (dolegliwości żołądkowo-jelitowe) oraz zaburze-
tj. wahania siły działania. Charakterystyczne są nagłe nia sercowo-naczyniowe (m.in. tachyarytmie i zabu-
zmiany od okresu dobrej ruchliwości do akinezy (tzw. rzenia ortostatyczne), a także zaburzenia psychiczne
zjawisko on-off). Ponadto oprócz opisanych powyżej (bezsenność, niepokój, pobudzenie, omamy).
fluktuacji u pacjentów znacząco zwiększa się ryzyko Wśród przeciwwskazań do stosowania lewodopy
ujawnienia się dyskinez z objawami ruchowymi typu należy wymienić zdekompensowane choroby endo-
pląsawiczego – w przypadku poprawy w zakresie ob- krynologiczne, nerek, wątroby i kardiologiczne, jak
jawów zespołu Parkinsona. Przyczyny tego rodzaju również schizofrenię.
wahań skuteczności leku upatruje się w postępującej Neuroleptyki i rezerpina osłabiają działanie lewo-
utracie neuronów dopaminergicznych, która nie może dopy. Leki przeciwnadciśnieniowe mogą wywoływać
być wyrównana dostarczaną lewodopą, oraz związa- większą ilość zaburzeń ortostatycznych. Podanie wi-
ną z tym zmniejszoną możliwością magazynowania taminy B6 prowadzi, na skutek zwiększenia aktyw-
w zakończeniach neuronów egzogennie dostarczonej ności dekarboksylazy, do częściowej utraty skutecz-
dopaminy, jak również w zmieniającej się czułości ności lewodopy. Działanie noradrenaliny i adrenaliny
i kinetyce receptorów. pod wpływem lewodopy ulega nasileniu.
MUTSCHLER-2009.indd 339 2010-01-07 22:12:29
340 Układ nerwowy
terapia za pomocą lewodopy
terapia za pomocą lewodopy + inhibitora dekarboksylazy
bariera bariera
krew-mózg krew-mózg
dekarbo- dekarbo- benserazyd
ksylaza ksylaza
karbidoba
działania niepożądane: działania niepożądane:
żołądkowo-jelitowe: ~50 % żołądkowo-jelitowe ~10 %
sercowo-naczyniowe: ~20 % sercowo-naczyniowe: ~ 5 %
lewodopa dopamina inne aminy biogenne
Ryc. B 1.10-3. Różnice w metabolizmie lewodopy do dopaminy bez i w obecności obwodowo działającego inhibitora dekarboksy-
lazy oraz wpływ na dawkowanie i działania niepożądane (według Birkmayera i Kappa).
1.10.2.2. Inhibitory COMT niemal w całości biotransformacji – szczególnie pro-
cesowi sprzęgania z kwasem glukuronowym. Wyda-
lanie entakaponu następuje głównie z kałem, z kolei
Do tej grupy leków – inhibitorów katecholo-O-me-
tolkaponu przez nerki (60%) i z kałem (40%)
tylo-transferazy (zob. ryc. B 1.10-3) należą enta-
kapon i tolkapon. Poprzez zablokowanie enzymu Preparat złożony zawierający lewodopę z karbidopą i en-
zapobiega się procesowi metylowania lewodopy takaponem (STALEVO) pozwala na zachowanie prostego
i dopaminy – w obrębie grupy M-hydroksylowej reżimu terapeutycznego u pacjentów z fluktuacjami typu
– do nieposiadających aktywności farmakologicz- end-of-dose (zob. poniżej).
nej metabolitów, co ostatecznie powoduje nasilenie
działania lewodopy. Podanie jedynie inhibitorów Jako działania niepożądane zażywania obu substan-
COMT – odpowiadając mechanizmowi działania cji, które spowodowane są zwykle podwyższonym
– jest nieskuteczne. poziomem dopaminy w osoczu i tym samym mogą
Po podaniu doustnym entakapon i tolkapon być opanowane przez zmniejszenie dawki lewodopy,
są szybko wchłaniane. Biodostępność sięga poziomu mogą wystąpić dyskinezy, nudności i bóle w obrębie
35% lub 65%, a okres półtrwania w osoczu wynosi jamy brzusznej. Ponadto opisywano zaburzenia mo-
około 22,5 lub 1,5–3 godz. Obie substancje ulegają toryki układu pokarmowego i suchość w ustach. Po-
MUTSCHLER-2009.indd 340 2010-01-07 22:12:29
Układ nerwowy 341
nieważ w przypadku tolkaponu dochodziło dodatko- bie wrzodowej. Oba inhibitory MAO-B nie powinny
wo do ciężkiego, niekiedy kończącego się śmiertelnie być stosowane łącznie z innymi inhibitorami MAO,
uszkodzenia wątroby – może być on stosowany jedy- inhibitorami wychwytu serotoniny, antagonistami
nie pod ścisłą kontrolą funkcji wątrobowych, głównie serotoniny lub opioidowymi (narkotycznymi) lekami
Układ nerwowy
wówczas, gdy inne leki przeciwparkinsonowe okaza- przeciwbólowymi.
ły się nieskuteczne.
W niewydolności wątroby, guzie chromochłon-
nym i złośliwym zespole neuroleptycznym – inhibi- 1.10.2.4. Agoniści receptorów
tory COMT są przeciwwskazane. Ponadto nie należy dopaminergicznych
ich stosować łącznie z nieselektywnymi inhibitorami
MAO lub z lekami, które są metabolizowane za po-
mocą COMT (np. noradrenalina lub adrenalina).
Główne działanie agonistów dopaminergicznych B1
u pacjentów z chorobą Parkinsona polega na stymu-
lowaniu receptorów D2 (zob. ryc. B 1.10-3). Roz-
maite substancje różnią się między sobą zarówno
1.10.2.3. Inhibitory MAO-B w zakresie właściwości farmakokinetycznych, jak
i w zakresie powinowactwa i aktywności wewnętrz-
Inna możliwość podwyższenia stężenia dopaminy nej w obrębie poszczególnych podtypów receptorów
w obrębie ośrodkowych receptorów dopaminergicz- dopaminergicznych oraz receptorów należących
nych polega na tym, aby zablokować rozkład dopa- do innych układów neuroprzekaźnikowych. Podczas
miny przez zahamowanie monoaminooksydazy typu gdy wcześniej stosowane one były głównie w bar-
B (MAO-B). Substancjami hamującymi ten enzym dziej zaawansowanych stadiach choroby Parkinsona
(inhibitory MAO-B; zob. ryc. B 1.10-3) są selegili- – łącznie z lewodopą (plus inhibitor dekarboksyla-
na i rasagilina. Blokada enzymu przez selegilinę ma zy), obecnie coraz częściej zaczyna się je stosować
początkowo charakter kompetycyjny, później jednak samodzielnie (w monoterapii) – zamiast lewodopy
polega na kowalencyjnym i nieodwracalnym wiąza- – już we wczesnych fazach choroby. Aby zapobiec
niu z enzymem; w przypadku rasagiliny ma charak- ujawnieniu się ciężkich działań niepożądanych zale-
ter pierwotnie nieodwracalny. W wyniku hamowania ca się powolne, stopniowe zwiększanie dawki leku,
MAO-B zmniejsza się również tworzenie reaktyw- czego skutkiem jest osiągnięcie pełnego działania
nych form tlenu. leku dopiero po kilku tygodniach stosowania. Czę-
Po podaży doustnej obie substancje ulegają szyb- stość takich działań niepożądanych, jak nudności
kiemu wchłanianiu. Mimo krótkiego okresu pół- i wymioty – powstających na skutek stymulacji re-
trwania w osoczu (ok. 1 godziny) czas działania ceptorów dopaminergicznych głównie w okolicy
leków wynosi 1–3 dni – co jest związane z nieod- pola ostatniego (area postrema) – może ulec istotne-
wracalną blokadą MAO-B. Podczas gdy selegili- mu zmniejszeniu na skutek podania obwodowo dzia-
na ulega biotransformacji do (–)-metamfetaminy łającego antagonisty dopaminergicznego, np. dom-
i (–)-amfetaminy, w trakcie biotransformacji rasa- peridonu.
giliny w obrębie CYP1A2 (N-dezalkilacja, hydrok-
sylacja) nie dochodzi do powstania amfetamin, lecz
aminoindanów. 1.10.2.4.1. Pochodne ergoliny
Za pomocą inhibitorów MAO-B można zapobiec
jedynie częściowo fluktuacjom warunkowanym le- Bromokryptyna jest wprawdzie w samodzielnym
wodopą. stosowaniu mniej skuteczna niż lewodopa, lecz nasi-
Jako działania niepożądane selegiliny mogą ujaw- la działanie lewodopy i w przypadku wczesnego roz-
nić się: suchość w ustach, zawroty głowy, zaburzenia poczęcia terapii skojarzonej zmniejsza ryzyko wy-
snu, arytmie nadkomorowe, blok przedsionkowo-ko- stąpienia fluktuacji. Mimo, że po doustnym podaniu
morowy i spadki ciśnienia, a w przypadku prepara- wchłanianie bromokryptyny jest dobre, ze względu
tów łączonych z lewodopą może dość do nasilenia na nasilony efekt pierwszego przejścia biodostępność
wymienionych działań niepożądanych. Po podaniu sięga zaledwie 20–30%. Wydalanie następuje głów-
rasagiliny opisane zostały m.in. bóle głowy, objawy nie z żółcią.
grypopodobne, złe samopoczucie, bóle karku i depre- Działania niepożądane zależne od dawki są po-
syjne obniżenie nastroju. dobne do tych u lewodopy. Dyskinezy ujawniają się
Selegilina jest przeciwwskazana w przypadku rzadziej, natomiast częściej występują zaburzenia
ograniczonej funkcji wątroby i nerek oraz w choro- psychiczne – szczególnie omamy, a także dolegliwo-
MUTSCHLER-2009.indd 341 2010-01-07 22:12:29
342 Układ nerwowy
ści ortostatyczne. W przypadku stosowania dużych czynności związanych z potencjalnym ryzykiem zra-
dawek mogą ujawnić się przejściowe zaburzenia nienia.
ukrwienia palców rąk i stóp. Opisywano również Leki wpływające na proces kanalikowego wy-
zwłóknienia płuc, pozaopłucnowe i zastawek serca. dzielania – mogą hamować wydalanie pramipeksolu.
W bardzo rzadkich przypadkach ujawniać się mogą Zjawisko to potwierdzono w przypadku cymetydyny
zaburzenia widzenia. i dotyczy ono przypuszczalnie również innych leków,
Preparatami pochodnymi o podobnym profilu które podlegają aktywnemu kanalikowemu wydziela-
działania oraz działaniach niepożądanych (zob. tab. niu, np. amantadyny, digoksyny, diltiazemu, trimeto-
B 1.10-1) są dihydroergokryptyna, kabergolina, li- primu i werapamilu.
suryd i pergolid. Podczas gdy dihydroergokryptyna,
kabergolina i lisuryd stymulują głównie receptory D2, Apomorfina została dopuszczona do stosowania u pacjen-
pergolid jest agonistą receptorów D1 i D2. tów z chorobą Parkinsona w przypadku ograniczających
funkcjonowanie powikłań ze strony układu ruchowego, któ-
re utrzymują się mimo indywidualnego ustawienia za po-
mocą lewodopy, inhibitorów dekarboksylazy i agonistów
1.10.2.4.2. Pozostali agoniści receptorów dopaminy. Apomorfina podawana jest podskórnie; z tkanki
dopaminergicznych podskórnej ulega szybkiemu i całkowitemu wchłonięciu,
co koreluje z szybkim ujawnieniem się efektów działania
(po 4–12 minutach). Krótki okres klinicznego działania
Agonista receptorów D2 (i D3) – ropinirol ma (ok. 1 godziny) tłumaczony jest szybkim wydalaniem leku.
we wczesnych fazach choroby podobną skuteczność Około 10% całkowitej dawki apomorfiny ulega metaboli-
jak lewodopa, a w bardziej zaawansowanej fazie cho- zowaniu poprzez jej sprzęganie z kwasem glukuronowym
roby wykazuje nieco mniejszą skuteczność. W przy- oraz siarkowym. Ponieważ apomorfina może w fazach
padku skojarzonego stosowania – skraca czas trwania on wywoływać ciężkie dyskinezy – jest przeciwwskaza-
na u pacjentów reagujących dyskinezami lub dystoniami
faz pogorszeń (off). już na podanie lewodopy.
Biodostępność ropinirolu wynosi około 50%,
szczytowe stężenie w osoczu jest osiągane średnio
po 1,5 godziny od zażycia. Metabolizm leku przez
utlenianie odbywa się głównie w obrębie izoenzymu 1.10.2.5. Amantadyna
CYP1A2. Metabolity ropinirolu wydalane są głównie
w moczu. Amantadyna – substancja pierwotnie wytworzona
U pacjentów otrzymujących ropinirol konieczne jako preparat stosowany w profilaktyce grypy – oka-
jest dopasowanie jego dawki – w sytuacji przyjmo- zała się skutecznym lekiem przeciwparkinsonowym.
wania równocześnie innych leków o działaniu hamu- Hamuje ona w sposób niekompetycyjny receptory
jącym na CYP1A2, np. ciprofloksacyny, enoksacyny NMDA, w wyniku czego zmniejsza się nierównowa-
lub fluwoksaminy. ga między hamowaniem dopaminergicznym a pobu-
dzeniem glutaminergicznym.
Pramipeksol działa na receptory D3 nawet silniej niż Amantadyna jest szybko i całkowicie wchłaniana.
na receptory D2. Lek okazał się efektywny zarówno Okres półtrwania w osoczu wynosi 10–15 godzin.
w monoterapii we wczesnej fazie choroby Parkinso- Wydalanie następuje głównie przez nerki – w 90%
na, jak i w terapii skojarzonej z lewodopą – w póź- w postaci niezmienionej.
niejszym okresie choroby. W monoterapii amantadyna nadaje się do stoso-
Po zażyciu doustnym – całkowita biodostępność wania w lekkich przypadkach, w których głównym
pramipeksolu sięga 90%. Maksymalne wartości objawem jest hipokineza; zwykle jednak stosowana
w osoczu osiągane są średnio po 2 godzinach. Prami- jest w terapii skojarzonej z lewodopą i innymi lekami
peksol jest wydalany przez nerki w niemal niezmie- przeciwparkinsonowymi. Amantadyna – pod posta-
nionej postaci. Okres połowiczej eliminacji wynosi cią kroplówki dożylnej – uważana jest za szczegól-
8–12 godz. W przypadku zmniejszonej wydolności nie skuteczną w opanowaniu kryzy akinetycznej (zob.
nerek nakazane jest ostrożne stosowanie leku. poniżej), gdyż w określonych okolicznościach może
Działanie niepożądane ropinirolu i i pramipekso- ratować pacjentowi życie.
lu są podobne do tych występujących w przypadku Działania niepożądane – w porównaniu z lewodo-
bromokryptyny, choć reakcje ortostatyczne i zwłók- pą – są mniejsze i utrudniają zwykle tylko początko-
nienia występują rzadziej. W związku z możliwoś- wy okres terapii. Może wówczas dojść do ujawnienia
cią wystąpienia ataków snu – zaleca się zaprzestanie się wewnętrznego niepokoju, dolegliwości żołądko-
prowadzenia samochodu, jak również wykonywania wo-jelitowych i czasem do stanów splątania.
MUTSCHLER-2009.indd 342 2010-01-07 22:12:30
Układ nerwowy 343
Tabela B 1.10-1. Leki przeciwparkinsonowe
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
Lewodopa i preparaty złożone z lewodopy i inhibitora dekarboksylazy
lewodopa Dopaflex 1 500-4000
HO COOH
NH2
HO
benserazyd lewodopa + benserazyd: 1,5 (lewodopa) 100/25-
OH NH2 Levopar, benserazyd: 800/200 B1
Madopar, brak danych
HO NH PK-Levo,
NH CH2OH
O
Restex
HO
karbidopa lewodopa + karbidopa: 100/25-
HO COOH Isicom, Levodopa comp. C STADA, 1,5 (lewodopa) 700/175
Levodop-neuraxpharm, Nacom, 2-3 (karbidopa)
H3C HN NH2 Nakom, Poldomet,
HO Sinimet CR
Inhibitory COMT
O
entakapon Comtess, 0,5 200-2000
HO C2H5 Comtan
N
CN C2H5
HO
NO2
O tolkapon Tasmar 2-3 300-600
HO
HO CH3
NO2
Inhibitory MAO-B
CH3 selegilina Antiparkin, Jutagilin, Movergan, 1,5 5-10
CH
N C Selegilin-ratiopharm, Api-Selin,
Jumex, Segan, Selerin,
CH3 Selgin, Selgres
H rasagilina AZILECT 0,6-2 1
CH
N C
Pochodne ergoliny
bromokryptyna Bromocriptin beta, 38 2,5-30
OH Bromocriptin HEXAL,
O N Bromocriptin-ratiopharm,
(H3C) 2HC
H Pravidel
O NH N
O
O
CH(CH3) 2
N
CH3
H
HN
Br
MUTSCHLER-2009.indd 343 2010-01-07 22:12:30
344 Układ nerwowy
Tabela B 1.10-1. Leki przeciwparkinsonowe (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Pochodne ergoliny (kontynuacja)
OH
α-dihydroergokryptyna Almirid, 10-15 10-60
(H3C) 2HC O Cripar
N
H
O NH N
O
O
CH(CH3) 2
H
N
CH3
H
HN
O NH kabergolina Cabaseril, 65 0,5-4
C2H5
Cabergolin STADA,
O N N(CH3) 2 Dostinex
H
N
CH2
H
HN
O lisuryd Dopergin 1-3 0,2-1,5
HN N(C2H5) 2
N
CH3
H
HN
SCH3 pergolid Parkotil, 7-16 0,05-3
Pergolid AL,
Pergolid HEXAL,
H Pergolid-ratiopharm
N
C3H7
H
HN
Pozostali agoniści receptorów dopaminergicznych
ropinirol Adartel, 3,5-10 0,75-9
Requip
C3H7
HN N
C3H7
O
N pramipeksol Sifrol 8-12 0,26-3,3
NH2
H7C3 S
NH
OH
apomorfina Apo-go 0,5 10-100
HO
N
H
CH3
MUTSCHLER-2009.indd 344 2010-01-07 22:12:30
Układ nerwowy 345
Tabela B 1.10-1. Leki przeciwparkinsonowe (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
Antagoniści receptora NMDA
NH2 amantadyna Amantadin AL., 10-30 100-900
Amantadin Hexal,
Amantadin-neuraxpharm,
PK-Merz, Amantix,
Viregyt K
C(CH3) 3 budypina Parkinsan 30 30-60 B1
N
Środki przeciwko dyskinezom
O O C2H5 tiapryd Tiaprid AbZ, 3 300-600
O
Tiaprid-CT,
S N Tiapridex,
H3C NH C2H5
Tiaprid-neuraxpharm
OCH3
W przypadku niewydolności nerek i ciężkich oraz objawy o charakterze egzogennej psychozy
stanów obniżonego ciśnienia krwi – amantadyna – nie należy stosować leków antycholinergicznych,
jest przeciwwskazana. gdyż mogą one nasilać wymienione objawy.
1.10.2.6. Ośrodkowo działające 1.10.2.7. Budypina
substancje antycholinergiczne
Budypina zajmuje pośrednie miejsce między aman-
Mające właściwości lipofilne trzeciorzędowe leki tadyną a ośrodkowo działającymi lekami przeciw-
(aminy) przeciwcholinergiczne (antagoniści muska- cholinergicznymi: z jednej strony ma działanie an-
rynowych receptorów cholinergicznych; zob. tak- tagonistyczne wobec receptorów NMDA, a z dru-
że: B 1.14.3.3) stosowane są do opanowania drże- giej strony jest również – choć słabym – antagoni-
nia mięśniowego (tremor), a w mniejszym zakresie stą muskarynowych receptorów cholinergicznych.
w leczeniu wzmożonego napięcia mięśniowego (ri- Nasila działanie lewodopy i zmniejsza drżenie
gor) i akinezy. Ponadto leki przeciwcholinergiczne mięśniowe.
są skuteczne w przypadkach wzmożonego pocenia Po doustnym podaniu budypina ulega wchłania-
się i nadmiernego wydzielania śliny. niu w 80%, jej biodostępność leży na poziomie 50%,
W terapii choroby Parkinsona dopuszczone zosta- a okres jej półtrwania w osoczu wynosi około 30 go-
ły następujące leki (zob. tab. B 1.10-2): biperiden, dzin. Powstająca z budypiny p-hydroksylowana po-
bornapryn, metyksen, procyklidyna i triheksyfe- chodna jest jeszcze biologicznie aktywna. Wydalanie
nidyl. następuje głównie przez nerki.
Leczenie należy rozpoczynać z powolnym zwięk- Dawkowanie jest ustalane indywidualnie. Lecze-
szaniem dawki leku, aż do osiągnięcia dawki, dla któ- nie powinno być rozpoczynane powoli od dawki
rej ujawnią się efekty terapeutyczne lub też działania 3 razy dziennie 10 mg. Jeżeli jest to konieczne, daw-
niepożądane – uniemożliwiające dalsze zwiększanie ka dzienna może być zwiększona dopiero po tygo-
dawki. U pacjentów z chorobą Parkinsona, u których dniu do 3 razy po 20 mg lub 2 razy po 30 mg.
ujawniają się wyraźne zaburzenia psychoorganicz- Działania niepożądane: nudności, suchość w us-
ne – szczególnie zaburzenia funkcji poznawczych tach, zaburzenia akomodacji i pamięci, a także oma-
MUTSCHLER-2009.indd 345 2010-01-07 22:12:31
346 Układ nerwowy
Tabela B 1.10-2. Leki przeciwparkinsonowe o działaniu antycholinergicznym
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
OH biperiden Akineton, 18 6-12
N Biperiden-neuraxpharm
OH procyklidyna Osnervan 12 10-20
N
OH triheksyfenidyl Artane, 6-10 6-16
Parkopan
N
bornapryn Sormodren 5 6-12
O N(C2H5) 2
S metyksen Tremarit 14 20-30
CH3
N
my wynikają głównie z antycholinergicznego działa- Lek – po podaniu doustnym – jest szybko i dobrze wchła-
nia leku. niany; czas półtrwania w osoczu wynosi 3–4 godziny.
Dawka dzienna tiaprydu powinna wynosić 300–600 mg.
W stanach splątania, miastenii, zdekompensowa- Efekt terapeutyczny ujawnia się dopiero po 4–6 tygodniach
nej niewydolności serca, hipokaliemii i hipomagne- leczenia.
zemii, jak również w przypadku wydłużenia odcinka Opisanymi działaniami niepożądaymi mogą być nie-
QT – budypina jest przeciwwskazana. znaczne spadki ciśnienia oraz (rzadko występujące) zatrzy-
Interakcje farmakokinetyczne opisano w stosunku manie miesiączkowania oraz mlekotok.
Tiapryd nasila działanie innych leków o działaniu tłumią-
do substancji rozkładanych za pomocą izoenzymu cym na ośrodkowy układ nerwowy. Należą do nich pochod-
CYP2D6, np. trój- i czterocyklicznych leków prze- ne morfiny, barbiturany, benzodiazepiny, leki przeciwlęko-
ciwdepresyjnych, neuroleptyków, leków przeciw- we, większość leków antyhistaminowych H1, ośrodkowo
arytmicznych, metoprololu oraz antybiotyków ma- działające leki przeciwnadciśnieniowe oraz klonidyna.
krolidowych.
1.10.3. Zalecenia dotyczące
1.10.2.8. Tiapryd farmakoterapii
Tiapryd cechuje się działaniem antagonistycznym wobec zespołu Parkinsona
receptora D2 i stosowany jest w przypadku późnych dyski-
nez wyzwolonych stosowaniem neuroleptyków, jak rów- Terapię choroby Parkinsona należy rozpoczynać od-
nież w przypadku ruchów pląsawiczych. powiednio wcześnie – adekwatnie do wieku – i pro-
MUTSCHLER-2009.indd 346 2010-01-07 22:12:31
Układ nerwowy 347
wadzić ją możliwie skutecznie. W zależności od wie- Terapia u pacjentów w wieku do 70 lat bez istotnej
ku, czasu trwania choroby i sytuacji społecznej – współchorobowości. Standardową terapią tej grupy
istotne znaczenie mogą mieć następujące cele tera- wiekowej jest monoterapia za pomocą agonisty dopami-
ny. Także skuteczność wcześniejszej terapii skojarzonej
peutyczne: za pomocą agonisty dopaminy z lewodopą – pozwalającej
Układ nerwowy
■ terapia objawów ruchowych, autonomicznych, po- na równoczesne zmniejszenie dawki lewodopy i poprawę
w zakresie fluktuacji związanych z lewodopą – udało się
znawczych i związanych z komunikacją, jak rów- potwierdzić przynajmniej dla niektórych preparatów.
nież natury psychicznej; Praktyczne przesłanki, np. możliwość przyjmowania
tylko jednej dawki leku przez młode, zawodowo czyn-
■ utrzymanie samodzielności w zakresie aktywności ne osoby lub wybór krótko działającego agonisty mający
codziennego życia, funkcjonowania rodzinnego na celu lepszą sterowalność terapii u osób w podeszłym
i społecznego oraz zawodowego; wieku – mogą być decydujące dla wyboru rodzaju terapii
(zob. tab. B 1.10-1). B1
■ zapobieganie konieczności sprawowania opieki W przypadku pacjentów, którzy mają być po raz pierw-
pielęgnacyjnej lub zmniejszenie zakresu tej opieki; szy ustawiani na terapię za pomocą pochodnej ergoliny, ko-
nieczne jest przeprowadzenie badania układu sercowo-na-
■ utrzymanie lub odzyskanie odpowiedniej jakości czyniowego, włącznie z przeprowadzeniem badania echo
życia; serca. Jego celem jest wykluczenie wcześniej istniejących
schorzeń zastawek serca.
■ unikanie dopaminergicznych działań niepożąda- W przypadku niewystarczającej skuteczności lub nieto-
nych (w jak największym stopniu). lerancji monoterapii agonistą dopaminy – zanim osiągnięta
zostanie wystarczająca dawka – możliwe jest wprowadze-
nie terapii skojarzonej (agonista dopaminy oraz lewodopa).
Terapia dopaminergiczna powinna być wprowadzo- Celem w tym przypadku jest objawowa terapia za pomocą
na wówczas, gdy na skutek zaburzeń motorycznych małej, ale skutecznej dawki lewodopy.
dojdzie do istotnego ograniczenia w zakresie zawo- Jeżeli konieczne jest uzyskanie szczególnie szybkiego
efektu terapeutycznego (np. przy zagrożeniu utratą pracy),
dowym lub też w istotnych codziennych aktywnoś- leczenie może zacząć się od zastosowania preparatu lewo-
ciach, do istotnego obniżenia jakości życia bądź ogra- dopy (zob. poniżej). Po maksymalnie 4–6 tygodniach nale-
niczenia w obszarze społecznym. ży włączyć dodatkowo agonistę dopaminy i tak dalece, jak
Strategia terapii powinna być ustalona indywi- to jest tylko możliwe, zmniejszyć dawkę lewodopy.
dualnie – przy uwzględnieniu wieku i współchoro- W przypadku mniej nasilonych objawów – rozpoczęcie
terapii dopaminergicznej może być często poprzedzone
bowości. Za rozpoczynaniem leczenia u młodych próbą monoterapii amantadyną lub selegiliną.
osób za pomocą agonistów receptorów dopamino-
wych przemawiają mniejsza częstość i mniejsze na- Terapia u pacjentów w wieku powyżej 70 lat lub u pa-
silenie ruchowych powikłań późnych, m.in. akinez cjentów ze współchorobowością z każdej grupy wie-
typu end-of-dose (występujących przed zażyciem kowej. Leczeniem standardowym w tych przypadkach
następnej dawki leku), fluktuacji on-off, zastyga- jest początkowo monoterapia za pomocą lewodopy (oraz
inhibitora dekarboksylazy). Może być ona później – w mia-
nia (freezing), dyskinez szczytu dawki (peak-dose), rę potrzeby – zastąpiona terapią skojarzoną.
dyskinez fazy off, w porównaniu z występującymi Tak jak u młodszych osób, w przypadku niewielkiej
w przebiegu terapii lewodopą. Z drugiej jednak stro- ilości objawów – istnieje możliwość rozpoczęcia terapii
ny agoniści dopaminergiczni mają bardziej nieko- amantadyną lub selegiliną zamiast lewodopą. U pacjen-
rzystny profil działań niepożądanych niż L-DOPA, tów z licznymi chorobami towarzyszącymi należy zwrócić
uwagę na ryzyko wywołania działań niepożądanych przez
szczególnie u pacjentów w podeszłym wieku cier- każdy z wymienionych leków.
piących na wiele innych chorób, a ponadto są mniej
skuteczni niż lewodopa. Terapia w przypadku występowania fluktuacji i dyski-
Zaburzenia snu, objawy bólowe i zaburzenia nez. Zmiany skuteczności działania oraz dyskinezy wyma-
depresyjne wyraźnie ograniczają jakość życia pa- gają zastosowania specjalnych środków terapeutycznych.
cjentów z chorobą Parkinsona; aktualne badania Należą do nich – w zależności od obecności określonych
objawów – m.in. stosowanie terapii skojarzonej, tj. prowa-
potwierdzają z kolei, że w okresie pierwszych czte- dzonej za pomocą wielu leków przeciwparkinsonowych,
rech lat chorowania dyskinezy nie stanowią dla nich dopasowanie liczby dawek podzielonych i wielkości dawki
dużego obciążenia, w okresie późniejszym nabiera- dziennej, stosowanie agonistów dopaminy na noc i parente-
ją one jednak sporego znaczenia. U pacjentów star- ralne podawanie lewodopy.
szych lub z zaburzeniami poznawczymi niezbędne
staje się stosowanie lewodopy bez równoczesnego Farmakoterapia drżenia mięśniowego (tremor). Jeżeli
mimo stosowania powyżej opisanej farmakoterapii w dal-
podawania inhibitorów dekarboksylazy, inhibitorów szym ciągu utrzymuje się wymagające leczenia drżenie
COMT w monoterapii oraz leków antycholinergicz- spoczynkowe, można wprowadzić lek antycholinergiczny
nych. lub budypinę – mieszanego antagonistę receptora NMDA
MUTSCHLER-2009.indd 347 2010-01-07 22:12:31
348 Układ nerwowy
i cholinergicznego receptora M, przy uwzględnieniu opisa- biegunka lub operacja. Tego rodzaju powikłanie może być
nych ograniczeń. także spowodowane nagłym przerwaniem zażywania le-
ków na chorobę Parkinsona. Kryza akinetyczna musi być
Terapia kryzy akinetycznej. Rzadka, ujawniająca się leczona w warunkach oddziału intensywnej terapii. Po-
zwykle w późnych stadiach choroby, zagrażająca życiu nieważ osoby dotknięte zaburzeniem nie mogą połykać
kryza akinetyczna charakteryzuje się nagle występującą, – muszą otrzymywać albo szybko rozpuszczalną lewodopę
całkowitą niemożliwością wykonywania ruchów przez pa- przez sondę żołądkową, amantadynę za pomocą wlewu lub
cjenta z chorobą Parkinsona. Czynnikiem wyzwalającym apomorfinę podaną podskórnie.
jest zwykle inna choroba, np. ciężka infekcja gorączkowa,
1.11. Leki przeciwwymiotne (antyemety-
1.11.1. Podstawy patofizjologiczne docierają do ośrodka wymiotnego w rdzeniu prze-
dłużonym (medulla oblongata), przez pobudzenie
chemoreceptorów pola najdalszego (area postre-
Nudności i wymioty są częstymi i zwykle niecharak- ma) położonego w rdzeniu przedłużonym lub przez
terystycznymi objawami towarzyszącymi w choro- pobudzenie narządu przedsionkowego (zob. ryc. B
bach, w których mogą być brane pod uwagę liczne 1.11-1). Jest to nieswoisty objaw, w którego powsta-
przyczyny (zob. poniżej). Ważnym diagnostycznym niu biorą udział receptory D2, 5-HT3 i H1, a także
elementem jest więc analiza przebiegu (nagłe, jed- M-cholinergiczne. Wymioty rozpoczynają się zwy-
norazowe lub nawracające) oraz dokładny wywiad kle głębokim wdechem; następnie odpowiednie
zarówno w zakresie obecnego schorzenia głównego, mięśnie zamykają wejście do tchawicy i do jamy
jak i wcześniej występujących schorzeń. nosowo-gardłowej – co zapobiega aspiracji masy
Wymioty są wywoływane przez bodźce aferen- pokarmowej. Światło przełyku poszerza się, a mięś-
tne z górnych części przewodu pokarmowego, które nie wpustu żołądka ulegają zwiotczeniu. Ostatecz-
traumatyczne wspomnienia,
ośrodkowy wyższe ośrodki lęki, przerażenie,
układ nerwowy antycypacja
ośrodek wymiotny jądro pasma samotnego
móżdżek (rdzeń) (nucleus tractus solitarii)
(5-HT3, D2, M, H1)
chemoreceptory
okolicy wyzwalającej
obwód (5-HT3, D2, M1, NK1)
(pole ostatnie)
ucho wewnętrzne żołądek, wagalne i współ-
(ruch) jelito cienkie czulne drogi
(5-HT3) wstępujące
drogi wstępujące nerwu
gardło językowo-gardłowego
(ból, zapach, cytostatyki, opiaty, i nerwu trójdzielnego
widok) substancje
parasympatykomimetyczne,
glikozydy nasercowe, substancje miejscowo
L-DOPA, bromokryptyna, drażniące gardło
apomorfina, emetyna i in. (cytostatyki, naświetlanie, (odruch
wirusy) wymiotny)
Ryc. B 1.11-1. Mechanizm powstawania wymiotów (zmodyf. według Goodmana i Glimana).
MUTSCHLER-2009.indd 348 2010-01-07 22:12:31
Układ nerwowy 349
nie występują skurcze żołądka, przepony i mięśni staminowych H1 lub leków antycholinergicznych.
brzucha, w wyniku czego zawartość żołądka zostaje Nudności i wymioty towarzyszące atakowi migreny
wyrzucona przez usta. opanowuje się za pomocą leków prokinetycznych
Jako przyczyny wymiotów można wymienić cho- (przyspieszających pasaż treści jelitowej poprzez
Układ nerwowy
roby żołądka, podrażnienie pęcherzyka żółciowego, zwiększenie częstotliwości lub siły skurczów jelita
przewlekłe zapalenie trzustki, mocznicę, śpiączkę cienkiego – przyp. tłum.).
wątrobową, podwyższenie ciśnienia wewnątrzczasz- W przypadku wymiotów towarzyszących chemio-
kowego (powstające np. w wyniku przebytego urazu terapii złośliwych guzów za pomocą cytostatyków
czaszkowo-mózgowego, krwawienia śródmózgo- szczególnie skuteczni okazują się antagoniści 5-HT3
wego, zapalenia opon lub w guzach mózgu), atak – podawani łącznie z deksametazonem.
migreny, reakcje psychowegetatywne na bodźce W przypadku terapii wymiotów pewnym proble-
wzrokowe, węchowe lub słuchowe, depresję, stany mem jest niekiedy dostarczenie odpowiedniej dawki B1
lękowe, jadłowstręt (anorexia nervosa), żarłoczność leku podawanego zwyczajowo drogą doustną. W ła-
(bulimia), leki o działaniu wymiotnym (cytostatyki, godnych przypadkach możliwe jest wtedy podanie
glikozydy naparstnicy, opoidy, apomorfina, i in.), leku przeciwwymiotnego (np. difenhydraminy nale-
spożycie nadmiernej ilości alkoholu, toksyny bak- żącej do leków przeciwhistaminowych H1) doodbyt-
teryjne oraz ogólne infekcje. W dalszej kolejności niczo. W ciężkich przypadkach może być konieczne
wymioty są także głównym objawem chorób loko- parenteralne podane leku wraz z dodatkowym wyrów-
mocyjnych (tzw. kinetoz), które mogą występować naniem niedoborów płynowych i elektrolitowych.
podczas wszelkiego rodzaju podróżowania. Wymioty Główne wskazania do poniżej opisanych substan-
występują często także we wczesnym okresie ciąży cji zostały zebrane w tabeli B 1.11-1. W przypadku
jako wymioty poranne (vomitus matutinus) lub niepo- leczenia wymiotów ciężarnych nie można zapominać
wściągliwe/niepohamowane wymioty ciężarnych (hy- o potencjalnym toksycznym dla płodu działaniu leku
peremesis gravidarum). W przypadku małych dzieci przeciwwymiotnego. Jeśli to możliwe należy zanie-
również ataki kaszlu mogą wyzwolić wymioty. chać podawania leków przeciwwymiotnych w pierw-
Po podaniu leków cytostatycznych i w następstwie szych trzech miesiącach ciąży.
napromieniowania w leczeniu chorób nowotworo-
wych – nudności i wymioty ujawniają się albo na- Leki przeciwhistaminowe H1. Difenhydramina lub
tychmiast, albo z kilkudniowym opóźnieniem. jej sól z 8-chloroteofiliną – dimenhydrynat – nadają
Zapowiedzią wymiotów jest wystąpienie potów, się głównie do stosowania w profilaktyce oraz w tera-
rozszerzenie źrenic, bladość, nadmierne ślinienie się, pii objawów choroby lokomocyjnej lub morskiej (ki-
nudności i odruch wymiotny. netoz). Wymienione substancje w przypadku wymio-
O ile pojedynczy epizod wymiotów właściwie tów ciężarnych powinny być w okresie pierwszych
nie ma żadnego znaczenia dla zdrowia, o tyle dłu- 16 tygodni ciąży stosowane wyjątkowo ostrożnie
gotrwale utrzymujące się wymioty mogą prowadzić – choć nie potwierdziły się podejrzenia co do ich te-
do zaburzeń gospodarki wodnej i elektrolitowej z al- ratogennego działania.
kalozą hipochloremiczną, skąpomoczem, odwodnie-
niem, podwyższeniem temperatury ciała, a nawet Skopolamina działa zapobiegawczo przeciwko ki-
śpiączką. Możliwymi do zaobserwowania objawami netozom pod postacią przezskórnego systemu tera-
towarzyszącymi są m.in. utrata masy ciała i wychu- peutycznego. Za pożądany efekt profilaktyczny od-
dzenie, bóle brzucha, biegunka, zawroty głowy, jak powiada jej ośrodkowe działanie antymuskarynowe
również zaburzenia widzenia i słuchu. w obrębie ośrodka wymiotnego.
Neuroleptyki. Perfenazyna, prometazyna, i halope-
ridiol mają szczególnie silne działanie przeciwwy-
1.11.2. Leki o działaniu
miotne. Efekt ten wiąże się głównie z blokowaniem
przeciwwymiotnym receptorów dopaminergicznych w okolicy pola naj-
dalszego (area postrema), częściowo także z hamo-
Leki przeciwwymiotne służą do powstrzymania waniem receptorów H1 i muskarynowych receptorów
odruchu wymiotnego i samych wymiotów. Wybór cholinergicznych (np. perfenazyna). Ponieważ leki
terapii wymiotów warunkowany jest – jeżeli tylko z grupy neuroleptyków charakteryzują się jednak
jest to możliwe do ustalenia – powyżej wymieniony- licznymi działaniami niepożądanymi – ich stosowa-
mi przyczynami. W przypadku zaburzeń równowagi nie w czasie ciąży powinno być ograniczone w wy-
zaleca się szczególnie stosowanie leków przeciwhi- jątkowych wskazaniach, np. w niepowściągliwych
MUTSCHLER-2009.indd 349 2010-01-07 22:12:32
350 Układ nerwowy
wymiotach ciężarnych z zaburzeniami gospodarki nosetron wyróżnia się długim osoczowym okresem
elektrolitowej. półtrwania wynoszącym 40 godzin.
Innym stosowanym przeciwwymiotnie środkiem Wydalanie następuje głównie przez nerki w posta-
z grupy neuroleptyków jest sulpiryd, który zalecany ci metabolitów.
jest w chorobie Ménière’a (napadowe zawroty gło- Jako działania niepożądane mogą ujawnić się bóle
wy z nudnościami, wymiotami, zaburzeniami słuchu głowy, zaparcia, objawy grypopodobne; do rzadkości
typu ucha wewnętrznego, szumami usznymi) oraz należą ciężkie reakcje nadwrażliwości oraz zaburze-
w innych stanach z przedsionkowymi zawrotami gło- nia rytmu serca.
wy. Pacjenci z zaburzeniami motoryki jelit nie powin-
ni otrzymywać antagonistów 5-HT3.
Prokinetyki. Metoklopramid, alizapryd i domperidon Interakcje występują przy równoczesnym podaniu
działają przeciwwymiotnie – podobnie jak neurolep- środków indukujących enzymy (fenytoina), prowa-
tyki – przez blokowanie receptorów dopaminergicz- dząc do osłabienia siły działania setronów.
nych w polu najdalszym. Poza działaniem przeciw-
wymiotnym metoklopramid i domperidon stosowane Antagoniści receptora neurokininy 1 (NK1). Apre-
są w przypadku zaburzeń opróżniania żołądka. pitant – pierwszy przedstawiciel tej klasy leków – ha-
muje pobudzenie receptora NK1 powstające w wyni-
Antagoniści 5-HT3 (setrony). W profilaktyce i tera- ku działania substancji P i zmniejsza nasilenie wy-
pii nudności i wymiotów (wymiotów wczesnych) wy- miotów wczesnych, a przede wszystkim wymiotów
wołanych lekami cytostatycznymi i napromienianiem, późnych w przebiegu chemioterapii, której towarzy-
jak również w przypadku wymiotów pooperacyjnych szą nasilone nudności. Lek stosuje się często w tera-
za leki pierwszego wyboru uznawani są antagoniści pii skojarzonej z antagonistami receptora 5-HT3 oraz
receptora 5-HT3, np. dolasetron, granisetron, on- deksametazonem.
dansetron i tropisetron. W celu zapobiegania ostrym Po podaniu doustnym jest dobrze wchłaniany.
wymiotom w przebiegu działającej silnie wymiotnie Jego biodostępność po podaniu doustnym wynosi
chemioterapii są one stosowane często łącznie z de- ok. 65%. Maksymalne stężenie w osoczu występuje
ksametazonem. Setrony doprowadziły do znaczącej po 4 godzinach. Aprepitant wiąże się silnie z białkami
zmiany w zakresie profilaktyki i terapii wymiotów in- – średnio do 97%.
dukowanych cytostatykami – powodując, że obecnie Jest prawie całkowicie metabolizowany w obrębie
same cytostatyki, takie jak cis-platyna, nie prowadzą izoenzymu CYP3A4. Metabolity są wydalane z mo-
do nasilonych wymiotów. czem oraz wraz z żółcił i kałem. Ostateczny okres
Po podaniu doustnym setrony wchłaniają się do- półtrwania wynosi 9–13 godz. Aprepitant indukuje
brze i szybko – będąc w dużym odsetku metabolizowa- ekspresję CYP3A4 oraz równocześnie miernie hamu-
ne. Dolasetron w wyniku procesu redukcji jest szyb- je działanie tego enzymu. Prowadzi to do możliwych
ko i w całości metabolizowany do farmakologicznie interakcji z lekami, które blokują CYP3A4, oraz tymi,
czynnej pochodnej. Okres półtrwania w osoczu wy- które ulegają rozkładowi w obrębie CYP3A4.
nosi dla ondansetronu 4–5 godzin, dla granisetronu
i tropisetronu oraz aktywnych metabolitów dolase-
tronu – 8–9 godz. W porównaniu z innymi – palo-
CF3
H3C
H3CO NH CF3
N O O
HN
N N
O N
N F
CH2 O
NH
HN
alizapryd aprepitant
MUTSCHLER-2009.indd 350 2010-01-07 22:12:32
Układ nerwowy 351
Tabela B 1.11-1. Leki przeciwwymiotne
Nazwa Preparaty handlowe Okres Średnia Główne wskazania
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
Leki przeciwhistaminowe H1
difenhydramina Emesan 4 50-150 profilaktyka i terapia nudności
i wymiotów, szczególnie
dimenhydrynat Vomex A, 4 50-300 w zapobieganiu kinetoz
Reisetabletten-ratiopharm,
Superpep, Vomacur
Antagoniści receptora muskarynowego
B1
skopolamina Scopoderm TTS 4 1,5 profilaktyka kinetoz
Neuroleptyki (wybór)
haloperidol Haldol-Janssen, 10-30 1-3 nudności i wymioty,
Haloper-CT w sytuacji gdy niemożliwe
jest zastosowanie leczenia
perfenazyna Decentan, 8-12 12-24 alternatywnego
Perphenazin-nauraxpharm
prometazyna Atosil, 10-40 25-100
Promethazin-neuraxpharm,
Proneurin,
Prothazin
sulpiryd Aminol, 7-10 150-300 zawroty głowy warunkowane
Dogmatil, obwodowo lub pobudzeniem
Sulpiryd HEXAL, przedsionka, np. choroba Ménière’a
Vertigo-Meresa
Prokinetyki
metoklopramid Cerucal, 2,5-4,5 25-40 nudności, wymioty;
MCP Sandoz, w dużych dawkach – w wymiotach
MCP STADA, wywołanych cytostatykami
Paspertin
alizapryd Vegentan 3 150-300 wymioty i nudności związane
z terapią cytostatykami,
naświetlaniami, wymioty
przed- i pooperacyjne
domperidon Domidon, 7-9 30-80 nudności, wymioty, uczucie pełności
DOMPERIDON HEXAL, joraz dolegliwości w nadbrzuszu
Domperidon AL, u pacjentów z czynnościową
Motilium dyspepsją
Antagoniści 5-HT3
dolasetron Anemet 7-9 100-200 zapobieganie i terapia nudności
i wymiotów w trakcie chemioterapii
granisetron Granisetron beta, 9 2 cytostatykami, naświetlania
Granisetron HEXAL, i po operacjach
Granisetron STADA,
Kevatril
ondansetron Axisetron, 3 8-16
Cellondan,
Ondansetron-ratiopharm,
Zofran
palonosetron Aloxi 40 0,25
tropisetron Navoban 8 5
MUTSCHLER-2009.indd 351 2010-01-07 22:12:32
352 Układ nerwowy
Tabela B 1.11-1. Leki przeciwwymiotne (kontynuacja)
Nazwa Preparaty handlowe Okres Średnia Główne wskazania
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Antagoniści receptora neurokininy 1 (NK1)
aprepitant Emend 9-13 80-125 zapobieganie ostrym i późnym
wymiotom w trakcie chemioterapii,
część całościowego schematu
terapeutycznego – obejmującego
również glukokortykosteroid
oraz antagonistę 5-HT3
Glikokortykoidy
deksametazon Fortecortin 5 10-24 wymioty pooperacyjne
i indukowane cytostatykami
Częstymi działaniami niepożądanymi są czkawka, Celem terapii objawowej skutków wymiotów
uczucie zmęczenia, zaparcia i bóle głowy. jest wyrównanie niedoborów płynowych, elektro-
Aprepitantu nie wolno stosować równocześnie litowych, opanowanie kwasicy lub zasadowicy.
z pimozydem, terfenadyną, astemizolem lub cisapry- W przypadku miernie nasilonych wymiotów sen-
dem. Hamowanie CYP3A4 za pomocą aprepitantu sowne jest stosowanie częstego doustnego dopaja-
może prowadzić do zwiększenia stężenia osoczowe- nia lub stałego nawadniania sondą nosowo-żołądko-
go wymienionych leków, czego konsekwencją może wą (u małych dzieci) za pomocą roztworów gluko-
być zagrażające życiu wydłużenie odcinka QT. zy i elektrolitów. W silnych wymiotach konieczne
jest nawadnianie za pomocą ciągłej dożylnej kro-
plówki roztworami glukozy i elektrolitów. Dawka
płynów infuzyjnych wyliczana jest z wartości zapo-
1.11.3. Zgodna z zaleceniami trzebowania podstawowego wynoszącego 1800 ml/
terapia wymiotów kg/dzień (u niemowląt: 120–140 ml/kg/dzień; u ma-
łego dziecka: 80–100 ml/kg/dzień) oraz szacowa-
Główną rolę odgrywa terapia choroby podstawowej nej utraty płynów (np. średnio ciężkie odwodnienie
wyzwalającej wymioty. Zakres różnych terapii przy- rzędu 5–10% odpowiada zapotrzebowaniu 50–100
czynowych obejmuje podanie antybiotyku (w przy- ml/kg). Początkowo należy podawać roztwór izoto-
padku zakażeń), zabiegi operacyjne (w przypadku niczny. W zakresie podaży elektrolitów należy się
spastycznego przerostowego zwężenia odźwiernika, kierować rodzajem odwodnienia (hipotoniczne, izo-
niedrożności jelit, zapalenia wyrostka robaczkowego toniczne, hipertoniczne).
i in.), postępowanie dietetyczne (w przypadku nieto-
lerancji białka zawartego w krowim mleku, choro- Leki przeciwwymiotne – tak jak to zostało powyżej
bach metabolicznych), eliminacja trucizny lub po- opisane – są stosowane w sposób dodany lub alterna-
danie antidotum (w przypadku zatruć egzogennych), tywny do tłumienia odruchu wymiotnego, jak rów-
jak również psychoterapię (w przypadku wymiotów nież w profilaktyce i terapii wymiotów.
psychogennych, jadłowstrętue psychicznego).
MUTSCHLER-2009.indd 352 2010-01-07 22:12:33
Układ nerwowy 353
1.12. Substancje wpływające na zwoje nerwowe
(układu autonomicznego)
Układ nerwowy
Pojęciem zwojów definiuje się leżące poza ośrodko- wym wzroście ciśnienia krwi jego długo utrzymują-
wym układem nerwowym skupienia komórek nerwo- cy się spadek; w przewodzie pokarmowym dochodzi
wych. Często termin ten jest ograniczony do zwojów do obniżenia napięcia lub atonii. Nikotyna wykazuje
układu autonomicznego. ponadto działanie ośrodkowe: po średnich dawkach
obserwuje się drobnofaliste drżenia, jak również
Leki działające w sposób stymulujący lub hamujący pobudzenie oddechu; duże dawki, np. w zatruciach,
na zwoje – poza nikotyną – utraciły swoje znacze- mogą wywołać drgawki i porażenie oddechu. Nikoty- B1
nie, gdyż nie jest możliwe (pożądane) swoiste od- na jest jedną z najważniejszych substancji prowadzą-
działywanie tylko na zwoje współczulne lub tylko cych do uzależnienia.
na przywspółczulne. W przypadku wszystkich zwo-
jów przenoszenie pobudzenia odbywa się na drodze
przewodnictwa cholinergicznego, tj. za pomocą ace-
tylocholiny.
Nikotyna, główny alkaloid tytoniu, w małych daw- N
kach prowadząc, podobnie jak acetylocholina, do de- CH3
N
polaryzacji błony postsynaptycznej – działa pobu-
dzająco na zwoje; z kolei w dużych dawkach blokuje nikotyna
zwoje na drodze desensybilizacji receptorów. Właś-
ciwości te wyjaśniają jej działania obwodowe: małe
dawki nikotyny powodują – na skutek stymulacji
zwojów – wzrost ciśnienia krwi, nasilenie wydzie-
lania kwasu żołądkowego, jak również zwiększenie Ostre i przewlekłe zatrucia nikotyną, a także postę-
napięcia w przewodzie pokarmowym. Po dużych powanie pomagające w odzwyczajeniu się od palenia
natomiast dawkach nikotyny występuje po początko- zostały omówione w rozdziale C 3.8.1.6.
1.13. Substancje wpływające na układ współczulny
Układ współczulny (sympatyczny, adrenergiczny), tj. wołane w ten sposób otwarcie napięciowozależ-
wyzwalająca reakcje ergotropowe część autonomicz- nych kanałów wapniowych Ca2+ typu N powoduje
nego (wegetatywnego) układu nerwowego, można napływ jonów wapniowych do wnętrza komórek
podzielić na układy: współczulno-nerwowy i współ- pozazwojowych. Skutkiem tego jest łączenie się
czulno-nadnerczowy, które umożliwiają rozmaite pęcherzyków gromadzących noradrenalinę z błoną
interakcje organizmu ze środowiskiem. Na ryc. B aksoplazmatyczną i wyrzut tego neuroprzekaźni-
1.13-1 przedstawiona została schematyczna budowę ka do szczeliny synaptycznej. W warunkach stresu
układu współczulnego. i nagłego zagrożenia – ponownie pod wpływem
acetylocholiny jako neuroprzekaźnika – dochodzi
Z licznych jąder w pniu mózgu szczególnie włók- do uwolnienia z rdzenia nadnerczy do krwiobiegu
na układu współczulnego, wywodzące się z miejsca adrenaliny, a w mniejszym zakresie również nora-
sinawego (locus coeruleus), kierują się do zwo- drenaliny, które wraz z krwią docierają do docelo-
jów współczulnych lub też do rdzenia nadnerczy. wych narządów.
Impuls nerwowy w zwojach współczulnych prze-
noszony jest za pomocą acetylocholiny na neuron Synteza, magazynowanie i uwalnianie dopaminy,
(po)zazwojowy, którego pobudzenie we współczul- noradrenaliny i adrenaliny. Katecholaminy: dopa-
nych tzw. żylakowatych rozszerzeniach prowadzi mina, noradrenalina i adrenalina są syntetyzowa-
do depolaryzacji błony aksoplazmatycznej. Wy- ne w organizmie w następujący sposób (zob. ryc. B
MUTSCHLER-2009.indd 353 2010-01-07 22:12:33
354 Układ nerwowy
przez dopa-dekarboksylazę do dopaminy. Dopamina
ośrodkowy układ nerwowy dociera dzięki aktywnemu transportowi do (magazy-
nujących) pęcherzyków synaptycznych, gdzie przez
β-hydroksylazę dopaminową w obrębie łańcucha
bocznego ulega hydroksylacji do noradrenaliny. Dal-
sze przekształcenie do adrenaliny – poza mózgiem
– jest niemożliwe w zakończeniach współczulnych,
neuron gdyż brak jest w nich N-metylotransferazy prze-
przedzwojowy
kształcającej noradrenalinę w adrenalinę.
Natomiast w komórkach chromafinowych rdzenia
acetylocholina nadnerczy, w których obecna jest N-metylotransfera-
za, dochodzi na drodze metylacji azotu noradrenaliny
rdzeń do powstawania adrenaliny.
zwój nadnerczy Etapem określającym szybkość reakcji w łańcu-
współczulny
chu syntezy jest aktywność hydroksylazy tyrozynowej.
Gdy zwiększa się uwalnianie noradrenaliny, wzrasta
także aktywność tego enzymu, natomiast jeśli zmniej-
sza się uwalnianie noradrenaliny, aktywność enzymu
wydzielanie adrenaliny również się zmniejsza.
neuron i noradrenaliny do krwiobiegu
(po)zazwojowy Neuronalne magazynowanie noradrenaliny nastę-
puje za pomocą nośników aminowych i pompy pro-
noradrenalina tonowej, która zużywając ATP, utrzymuje wysokie
wewnątrzpęcherzykowe stężenie protonów i w ten
sposób prowadzi do protonowania noradrenaliny
w pęcherzykach presynaptycznych.
mięśnie gładkie
Gdy dochodzi do depolaryzacji błony aksopla-
zmatycznej, wówczas noradrenalina najczęściej
Ryc. B 1.13-1. Schemat układu sympatycznego (współczulnego). z przekaźnikiem towarzyszącym (ATP, neuropeptyd
Y) – jak to już opisano – zostaje na drodze egzocy-
tozy uwolniona do szczeliny synaptycznej. Jej część
powoduje pobudzenie receptorów postsynaptycznych
1.13.-2): tyrozyna ulega wchłonięciu do aksoplazmy, w narządach wykonawczych i dzięki temu ujawnia
tam ulega hydroksylacji pierścień aromatyczny przez się właściwy efekt; inna część natomiast pobudza
hydroksylazę tyrozynową do dihydroksyfenyloala- presynaptyczne receptory adrenergiczne i w ten spo-
niny (dopa), która z kolei jest dekarboksylowana sób wpływa na uwalnianie neuroprzekaźnika na zasa-
hydroksylaza dekarboksylaza
NH2 tyrozyny HO NH2 dopy HO NH2
COOH COOH
HO HO HO
tyrozyna dopa dopamina
β-hydroksylaza
dopaminy
OH N-metylo- OH
HO NH
CH3
-transferaza HO NH2
HO HO
adrenalina noradrenalina
Ryc. B 1.13-2. Biosynteza dopaminy, noradrenaliny i adrenaliny.
MUTSCHLER-2009.indd 354 2010-01-07 22:12:34
Układ nerwowy 355
dzie sprzężenia zwrotnego (feed-back) (zob. poniżej). Badania molekularne wykazały ponadto, że wśród re-
Największa część noradrenaliny jest zwrotnie wchła- ceptorów α1 i α2, istnieją jeszcze dalsze podtypy.
niana – jeszcze przed osiągnięciem receptorów. W tab. B 1.13-1 zebrano rozmaite działania wyni-
kające z pobudzenia różnych współczulnych recepto-
Układ nerwowy
Wychwyt zwrotny i rozkład noradrenaliny. Bardzo rów α i β. Transdukcja sygnału w wyniku pobudzenia
szybka inaktywacja uwolnionej substancji przekaźni- tych receptorów również jest różna i dlatego ich me-
kowej następuje – jak to zostało opisane – głównie chanizm działania zostanie poniżej oddzielnie opisa-
(do 90%) przez wychwyt zwrotny do aksoplazmy, ny (zob. ryc. B 1.13.4).
a w dalszej kolejności poprzez metylację grupy OH Pobudzenie receptorów β powoduje – dzięki po-
znajdującej się w pozycji meta (m) pierścienia feno- średnictwu stymulującego białka G – aktywowanie
lowego (katecholowego) za pomocą enzymu COMT cyklazy adenylanowej i tym samym wzmożoną syn-
(katecholo-O-metylo-transferazy = metylotranferazy tezę cAMP. Cykliczny AMP pobudza z kolei zależną B1
katecholowej; czasem nazywanej również: katecho- od cAMP kinazę białkową, która prowadzi do fosfo-
lamino-O-metylo-transferazą), a także w wyniku ok- rylacji napięciowozależnych kanałów wapniowych.
sydatywnej dezaminacji przez monoaminooksydazę W sercu dochodzi – w wyniku pobudzenia receptorów
(MAO; zob. ryc. B 1.13-3). β1 – do nasilonego napływu jonów Ca2+ do komórek.
Ponadto zwiększa się wchłanianie Ca2+ do siateczki
Wychwyt zwrotny przekaźnika do aksoplazmy ma zna- sarkoplazmatycznej, a tym samym stopień wypeł-
czenie nie tylko dla szybkiego przerywania jego dzia- nienia zbiorników wapniowych. W obrębie mięśni
łania, lecz również zapobiega nadmiernemu opróż- gładkich dochodzi natomiast, poprzez receptory β2,
nieniu się zbiorników z neuroprzekaźnikiem. w wyniku fosforylacji małego wiążącego się z GTP
Rozkład za pomocą COMT do już farmakologicz- białka Rho, do reorganizacji cytoszkieletu komórko-
nie nieczynnego metabolitu – normetanefryny – prze- wego, a tym samym zmniejsza się stężenie wolnych
biega pozaneuronalnie w obszarze szczeliny synap- wewnątrzkomórkowych jonów wapnia – co prowadzi
tycznej, a w dalszej kolejności w wątrobie. Norme- do rozkurczu.
tanefryna ulega potem metabolizowaniu – poprzez Pobudzenie receptorów α1 prowadzi z kolei
aldehyd – na obwodzie do kwasu wanilinomigdało- do zwiększenia wewnątrzkomórkowego stężenia
wego, a w ośrodkowym układzie nerwowym do gli- Ca2+. W tym przypadku w wyniku aktywacji fosfo-
kolu 3-metoksy-4-hydroksy-fenyloetylowego. lipazy C – w czym pośredniczy białko G – nasileniu
Oksydatywna dezaminacja kontrolowana przez ulega synteza trifosforanu inozytolu (IP3) oraz dia-
MAO przebiega z kolei w mitochondriach zakończeń cyloglicerolu (DAG) oraz dochodzi do uwolnienia
komórek nerwowych, dodatkowo również w komór- jonów Ca2+ z siateczki sarkoplazmatycznej.
kach sąsiadujących z okolicą synaps oraz w wątro- Natomiast pobudzenie receptorów α2 – przy współ-
bie. Ponieważ substancje muszą najpierw przedostać działaniu hamującego biała G – prowadzi do zahamo-
się przez błonę mitochondriów – proces dezaminacji wania cyklazy adenylanowej, jak również do zablo-
przebiega wolniej niż O-metylacja. kowania kanałów Ca2+ i aktywowania kanałów K+.
Jedynie bardzo niewielkie ilości noradrenaliny
trafiającej do szczeliny synaptycznej docierają do na- Presynaptyczne receptory adrenergiczne. W ob-
czyń krwionośnych – stąd też jej działanie jest ogra- rębie synaps oprócz receptorów postsynaptycznych,
niczone miejscowo. Metabolity trafiają natomiast istnieją również receptory presynaptyczne, których
do krwiobiegu i są wydalane głównie przez nerki. pobudzenie wpływa na uwalnianie neuroprzekaź-
Tym samym ilość wydalanych z moczem katechola- nika. W nerwach współczulnych pobudzenie presy-
min i ich metabolitów pozwala na ocenę aktywności naptycznych receptorów α2-adrenergicznych hamuje
układu współczulnego, jak również daje pewne wska- uwalnianie noradrenaliny. Mechanizm ten służy deli-
zówki diagnostyczne co do możliwych schorzeń, któ- katnej regulacji przekazywania impulsów współczul-
rym towarzyszy zwiększenie lub też zmniejszenie nych i zapobieganiu nadmiernej stymulacji.
aktywności układu współczulnego.
Punkty uchwytu i klasyfikacja leków wpływają-
Receptory adrenergiczne (adrenoreceptory). cych na układ współczulny. Odpowiednio do mor-
W obrębie synaps współczulnych występują dwie fologicznej budowy i przekazywania pobudzenia
główne grupy receptorów adrenergicznych – recep- w układzie współczulnym – wpływ leków na współ-
tory α i β, przy czym wśród tych receptorów można czulny układ nerwowy jest możliwy w różnych miej-
wyróżnić jeszcze receptory α1 i α2 oraz β1, β2 i β3. scach.
MUTSCHLER-2009.indd 355 2010-01-07 22:12:34
356 Układ nerwowy
OH OH
HO NH2 H3CO NH2
COMT
HO HO
noradrenalina normetanefryna
MAO MAO
OH OH
aldehyd 3-metoksy-
HO O aldehyd 3,4-dihydroksy- H3CO O -4-hydroksy-
-fenyloglikolowy -fenyloglikolowy
H H
HO HO
a
ukc
ja acj
oks
yd
red
red oks
yda
u
kcj
cja
a
OH OH OH OH
HO OH HO O H3CO O H3CO OH
COMT
OH OH
HO HO HO HO
glikol 3,4-dihydroksy- kwas 3,4-dihydroksy- kwas wanilino- glikol 3-metoksy-4-hydroksy-
-fenyloetylenowy -migdałowy (DOMA) -migdałowy (VMA) -fenyloetylenowy (MOPEG)
(DOPEG)
COMT
Ryc. B 1.13-3. Rozkład noradrenaliny.
Agoniści receptorów adrenergicznych (bezpośred- ■ antagoniści receptora α-adrenergicznego (blokery
nie sympatykomimetyki/adrenomimetyki = bezpo- receptora α-adrenergicznego, blokery receptora α,
średnio działające leki sympatykomimetycznie/ad- α-blokery; leki α-adrenolityczne/α-sympatykoli-
renomimetycznie) pobudzają, podobnie jak noradre- tyczne) blokują receptory α-adrenergiczne,
nalina i adrenalina, receptory adrenergiczne (adreno-
■ antagoniści receptora β-adrenergicznego (blokery
receptory). W zależności od tego, na jakie receptory
receptora β-adrenergicznego, blokery receptora β,
substancje te wpływają, rozróżnia się:
β-blokery; leki β-adrenolityczne/β-sympatykoli-
■ agonistów α-adrenoreceptorów (agonistów recep- tyczne) blokują receptory β-adrenergiczne.
tora α-adrenergicznego; α-agoniści, α-sympatyko-
mimetyki, α-adrenomimetyki), Pojęciem antysympatykotoników (bądź sympatolity-
ków rzadko stosowane w polskiej farmakologii) obej-
■ agonistów β-adrenoreceptorów (agonistów recep-
muje różne leki lub grupy leków, z różnymi punktami
tora β-adrenergicznego; β-agoniści, β-sympatyko-
uchwytu, które wywierają wpływ hamujący na układ
mimetyki, β-adrenomimetyki),
współczulny. Leki te zmniejszają aktywność układu
współczulnego przez:
Pośrednio działające leki sympatykomimetyczne (ad-
renomimetyczne) uwalniają noradrenalinę ze zbior- ■ pobudzenie ośrodkowych i – w mniejszym stop-
ników i/lub hamują kompetycyjnie wychwyt zwrotny niu – obwodowych receptorów α-adrenergicznych
noradrenaliny ze szczeliny synaptycznej do aksopla- (klonidyna, metyldopa) lub
zmy.
■ blokadę aktywnego transportu z aksoplazmy do ma-
gazynów (rezerpina).
Antagoniści receptora adrenergicznego (leki adreno-
lityczne/sympatykolityczne, blokery adrenorecepto-
rów) blokują receptory adrenergiczne:
MUTSCHLER-2009.indd 356 2010-01-07 22:12:34
Układ nerwowy 357
Ca2+
ER
Układ nerwowy
ATP zmiana kanał Ca2+
rozkurcz RhoA organizacji
cytoszkieletu typu L
Gs RyR
cAMP PKA SR
β2 Ca2+
agonista β2 PKA
noradrenalina
skurcz Ca2+ B1
cAMP
α2 Gs
Gi
β1
agonista β1
Ca2+ drzewo oskrzelowe
serce
noradrenalina, adrenalina
zakończenie
współczulnego
aksonu
zakończenie współczulnego
aksonu
naczynia wieńcowe
Ca2+
tętnice,
tętniczki
noradrenalina, adrenalina
α2
β2
Gi
Gs
Ca2+
cAMP
noradrenalina ER rozkurcz
ATP
α1 PKA
Gq IP3 zmiana
organizacji RhoA
antagonista α1 Ca 2+ skurcz cytoszkieletu
IP3R
CaM
ER
MLCK
Ryc. B 1.13-4. Przykłady fizjologicznych działań, w których pośredniczą receptory adrenergiczne α i β. Noradrenalina i adrenalina
z kory nadnerczy oraz noradrenalina z zakończeń neuronalnych nerwów współczulnych prowadzą przez stymulację receptorów β2
do rozkurczu mięśniówki oskrzeli i naczyń wieńcowych. Stymulacja receptorów β1 mięśnia sercowego zwiększa siłę skurczu serca.
Tętnice i tętniczki narządów trzewnych ulegają skurczowi na skutek stymulacji receptorów α1 za pomocą noradrenaliny i adrenaliny.
Stymulacja receptorów α2 w zakończeniach aksonów neuronów współczulnych hamuje kanały Ca2+ typu N i prowadzi do samozaha-
mowania wydzielania noradrenaliny. IP3R – receptor trifosforanu inozytolu; MLCK – kinaza lekkich łańcuchów miozyny; PKA – kinaza
białkowa A; RhoA – małocząsteczkowe białko G Rho A; ER – siateczka (retikulum) endoplazmatyczna; SR – siateczka sarkoplazma-
tyczna; CaM – kalmodulina; RyR – receptor rianodynowy.
MUTSCHLER-2009.indd 357 2010-01-07 22:12:34
358 Układ nerwowy
Tabela B 1.13-1. Wpływ pobudzenia układu współczulnego na Tabela B 1.13-1. (Kontynuacja)
różne narządy (zmodyf. według Goodmana i Gilmana)
Narząd lub układ Działanie Główny receptor
Narząd lub układ Działanie Główny receptor układu adrenergiczny
układu adrenergiczny współczulnego uczestniczący
współczulnego uczestniczący w określonym
w określonym efekcie
efekcie
Nerki i drogi moczowe
Oczy
wydzielanie reniny zwiększenie β1
mięsień rozwieracz źrenicy rozszerzenie α1
(m. dilatator pupillae) mięśnie ściany pęcherza zwiotczenie β2
mięsień zwieracz źrenicy Ø
(m. sphincter pupillae)
zwieracz wewnętrzny skurcz α1
mięsień rzęskowy Ø
gruczoł łzowy Ø Narządy płciowe
Serce macica skurcze α1
węzeł zatokowy częstotliwość serca ↑ β1 zwiotczenie β2
mięśnie przedsionków kurczliwość ↑ β1 Metabolizm
węzeł przedsionkowo- szybkość β1 wątroba glikogenoliza ↑ β2
-komorowy przewodzenia ↑ glukoneogeneza ↑ β2
mięsień komór kurczliwość ↑ β1 komórki tłuszczowe lipoliza ↑ β2
Naczynia
skóra, błona śluzowa skóra, błona śluzowa α1 mięśnie szkieletowe glikogenoliza ↑ β2
mięśnie szkieletowe skurcz naczyń α1
rozkurcz naczyń β2 Ø brak działania
jama brzuszna skurcz naczyń α1
naczynia wieńcowe skurcz naczyń α1
rozkurcz naczyń β2
mózg skurcz naczyń α1
1.13.1. Noradrenalina i adrenalina
narządy płciowe ejakulacja α1
(nasieniowód)
Farmakologiczne działania noradrenaliny i adrenali-
nerki skurcz naczyń α1 ny są podobne, ale nie identyczne. Różnice wynika-
żyły skurcz naczyń α1 ją stąd, że siła działania obu substancji w przypadku
obu podtypów receptorów adrenergicznych jest różna
Przewód pokarmowy
(zob. tab. B 1.13-2).
gruczoły ślinowe małe wydzielanie α1
śliny śluzowej
Noradrenalina (norepinefryna) na skutek uogólnio-
gruczoły trawienne aktywacja amylazy β1 nego skurczu naczyń krwionośnych – z wyjątkiem
drogi żółciowe zwiotczenie β2 naczyń wieńcowych serca – podwyższa skurczowe
i rozkurczowe ciśnienie krwi. Podczas gdy w przy-
pasaż/napięcie spowolnienie/spadek α2, β2
padku izolowanego serca noradrenalina prowadzi
zwieracze skurcz α1 do zwiększenia częstości pracy (skurczów) oraz mi-
Trzustka
nutowej objętości wyrzutowej, w warunkach in vivo
ujawnia się efekt odwrotny – bradykardia. Ten po-
wydzielanie endokrynne sekrecja insuliny ↓ α2
czątkowo zaskakujący efekt można wytłumaczyć
Układ oskrzelowy w następujący sposób: wzrost ciśnienia krwi prowa-
mięśniówka zwiotczenie β2 dzi do odruchowego pobudzenia presoreceptorów
(baroreceptorów), które – za pośrednictwem nerwu
gruczoły ? β2
błędnego – wywołują w sercu zwrotną reakcję przy-
Skóra współczulną. W przypadku wyłączenia tej regulacji
gruczoły potowe wydzielanie cholinergiczne zwrotnej – przez podanie preparatu parasympatyko-
litycznego, np. atropiny – również in vivo uzyskuje
MUTSCHLER-2009.indd 358 2010-01-07 22:12:36
Układ nerwowy 359
Tabela B 1.13-2. Względna siła działania adrenaliny (A) i nora- gładką przewodu pokarmowego i oskrzeli adrena-
drenaliny (NA) na poszczególne receptory adrenergiczne lina działa rozkurczowo: perystaltyka jelit zwalnia,
wchłanianie tlenu w płucach poprawia się. Adrenali-
Podtyp receptora Siła działania na nie jest w stanie przedostać się przez barierę krew-
Układ nerwowy
mózg. Stąd też obserwowane po podaniu adrenaliny
α1 A ≈ NA efekty ośrodkowe (np. stany lękowe) mają charakter
α2 A ≥ NA czysto odruchowy.
W zakresie metabolizmu adrenalina prowadzi
β1 NA ≥ A
w wyniku pobudzenia adrenoreceptorów β – jak
β2 A > NA to już opisano – do stymulacji cyklazy adenylanowej.
Powstający w wyniku działania tego enzymu z trifo-
β3 NA > A B1
sforanu adenozyny ATP cykliczny monofosforan ade-
nozyny cAMP – aktywuje z jednej strony działanie
kinazy białkowej A, która wyzwala syntezę aktywnej
fosforylazy wątrobowej i mięśniowej z nieaktywnych
substancji prekursorowych. Fosforylazy są z kolei
się efekt zwiększenia częstości pracy serca oraz mi- odpowiedzialne za rozkład glikogenu do glukozo-1-
nutowej objętości wyrzutowej – przez noradrenali- fosforanu oraz jej transformację izomerową do glu-
nę. Ponieważ noradrenalina wykazuje jedynie słabe kozo-6-fosforanu – co zachodzi w wątrobie i mięś-
działanie agonistyczne wobec receptorów β2 w ob- niach szkieletowych. W wątrobie proces defosfory-
rębie mięśni gładkich – jej rozkurczowe działanie lacji prowadzi do powstania glukozy, która trafia
w obrębie przewodu pokarmowego oraz oddechowe- do krwi: wzrasta poziom cukru w surowicy. W obrę-
go jest nieznaczne. Również niewielki jest jej wpływ bie mięśni szkieletowych, gdzie nie występuje gluko-
na zwiększanie poziomu cukru w osoczu. zo-6-fosfataza, glukozo-6-fosforan ulega rozkładowi
glikolitycznemu. Produktem końcowym glikolizy
Adrenalina (epinefryna) – w fizjologicznym stężeniu jest wzrost stężenia kwasu mlekowego we krwi.
– prowadzi do skurczu naczyń krwionośnych skóry, Działanie lipolityczne adrenaliny również polega
śluzówek i narządów trzewnych, powoduje natomiast na pobudzeniu cyklazy adenylanowej z następczym
rozszerzenie naczyń mięśni szkieletowych i serca. tworzeniem cyklicznego AMP. Ten z kolei aktywuje
Ukrwienie mózgu zmienia się jedynie w niewielkim lipazę tkanki tłuszczowej – prowadząc do zwiększenia
stopniu. Ponieważ w sumie pobudzenie receptorów stężenia wolnych kwasów tłuszczowych w osoczu.
β2 prowadzi do zmniejszenia się obwodowego oporu
w obrębie naczyń krwionośnych – co zmniejsza ciś- Wskazania. Noradrenalina – ze względu na swoje
nienie rozkurczowe krwi. Wzrasta natomiast ciśnienie działanie skurczowe w obrębie naczyń krwionośnych
skurczowe – co jest wynikiem zwiększenia minutowej – wskazana jest w przypadku wstrząsu neurogennego
objętości wyrzutowej serca związanej z pobudzeniem i septycznego, a także jako dodatek do preparatów
receptorów β1. Fizjologiczne znaczenie adrenaliny – do znieczulenia miejscowego. W przypadku pierw-
w zakresie układu krążenia – polega na regulacji roz- szego z wymieniowych wskazań najbardziej celowe
kładu krwi w organizmie; podczas gdy noradrenalina jest stosowanie powolnego wlewu dożylnego, gdyż
odpowiedzialna jest za utrzymanie odpowiedniego po (szybkiej) iniekcji leku wzrost ciśnienia krwi
napięcia mięśniówki naczyń i w odpowiedniej sytua- utrzymuje się jedynie przez kilka minut. W przypad-
cji zwiększenie tego napięcia (skurcz). ku podania doustnego – w związku z wysokim efek-
W obrębie serca adrenalina wywołuje zwiększenie tem pierwszego przejścia – noradrenalina jest prawie
siły skurczu i częstości skurczów (dodatnie działanie nieskuteczna.
inotropowe i chronotropowe). Wzrasta również mi- Adrenalina stosowana jest w przypadku wstrzą-
nutowa objętość wyrzutowa serca. Większe dawki su anafilaktycznego i septycznego oraz miejscowo
adrenaliny wyzwalają ponadto heterotopowe wyzwa- do zwężania naczyń krwionośnych. W przypadku
lanie bodźców pobudzających. W wyniku tego dojść zatrzymania pracy serca iniekcji adrenaliny nale-
może nawet do wystąpienia pobudzeń dodatkowych ży dokonywać dożylnie lub dotchawiczo (w wy-
i nawet migotania komór. W przypadku stosowania jątkowych przypadkach również dosercowo!)
adrenaliny należy również uwzględnić to, że zwięk- – po wcześniejszym wdrożeniu podstawowych czyn-
sza ona zużycie tlenu przez mięsień sercowy i nawet ności takich jak sztuczne oddychanie i masaż serca
mimo rozszerzenia naczyń wieńcowych – może oraz bez ich przerywania w trakcie przeprowadza-
wywołać napad dusznicy bolesnej. Na mięśniówkę nia iniekcji.
MUTSCHLER-2009.indd 359 2010-01-07 22:12:37
360 Układ nerwowy
Objawy niepożądane i przeciwwskazania. Jako Typowa dawka początkowa wynosi 2,5 mg hydro-
objawy niepożądane mogą wystąpić: stany lękowe, chlorku midodryny przyjmowanego 2–3 razy dzien-
uczucie osłabienia i drżenia mięśniowe (tremor), jak nie. W zależności od reakcji pacjenta dawka może
również – przede wszystkim – zaburzenia sercowe być powoli – w odstępach trzydniowych – zwiększo-
pod postacią arytmii i napadów dusznicy bolesnej. na. Maksymalna dawka dzienna wynosi 30 mg.
Szczególnie wrażliwi są pacjenci z nadczynnością Jako działania niepożądane mogą wystąpić m.in.:
tarczycy. W ich przypadku noradrenalina i adrenali- uczucie swędzenia (świąd), uczucie zimna, zatrzyma-
na są przeciwwskazane – podobnie jak w przypadku nie moczu, komorowe zaburzenia rytmu i dolegliwo-
miażdżycy naczyń wieńcowych i mózgu, ciężkiego ści podobne do dusznicy bolesnej.
nadciśnienia, znieczulenia ogólnego przeprowadza- U pacjentów z nadczynnością tarczycy, guzem
nego z wykorzystaniem chlorowcopochodnych wę- chromochłonnym oraz łagodnym przerostem gruczo-
glowodorów lub eteru, a także po wcześniejszym łu krokowego z zaleganiem moczu – stosowanie α-
podaniu preparatów naparstnicy. sympatykomimetyków jest przeciwwskazane.
Agoniści receptorów α-adrenergicznych do sto-
sowania miejscowego. Fenylefryna, a w szczegól-
1.13.2. Dopamina ności substytuowana imidazolina (zob. tab. 1.13-3)
stosowane są miejscowo w obrzęku śluzówek w przy-
Dopamina – stosowana głównie w leczeniu wstrząsu padku niespecyficznego i alergicznego zapalenia
– została omówiona w rozdziale B 4.2.6. spojówek, zatok oraz nosa i gardła. U noworodków
i małych dzieci wymienione środki mogą być stoso-
wane wyłącznie pod postacią silnie rozcieńczonych
1.13.3. Agoniści receptorów kropli, a nie na przykład pod postacią aerozolu, gdyż
adrenergicznych w określonych okolicznościach w wyniku (nadmier-
nego) wchłaniania może dojść do zaburzeń oddycha-
nia i stanów śpiączkowych. W związku z ryzykiem
1.13.3.1. Agoniści receptorów
ujawnienia się niepożądanych objawów systemo-
α-adrenergicznych
wych wskazane jest również ostrożne dawkowanie
w przypadku osób nadwrażliwych (np. z nadczynnoś-
Agoniści receptorów α-adrenergicznych pobudzają
cią tarczycy i z nadciśnieniem tętniczym).
współczulne receptory α. Stosowane są do uzyskania
systemowego (ogólnego, uogólnionego) lub miejsco-
wego efektu zwężenia naczyń. Obecnie stosowane
terapeutycznie substancje pobudzają zarówno recep- 1.13.3.2. Agoniści receptorów
tory adrenergiczne α1, jak i α2. α- i β-adrenergicznych
Agoniści receptorów α-adrenergicznych do stoso- Wiele syntetycznych pochodnych noradrenaliny po-
wania ogólnego. Z tej grupy leków stosowana jest woduje pobudzenie receptorów noradrenergicznych
jedynie midodryna. Służy w terapii niskiego ciśnie- zarówno typu α, jak i typu β. Przedstawicielami le-
nia krwi warunkowanego neurogennie. W podobny ków z tej grupy są etylefryna oraz oksylofryna (zob.
sposób jak noradrenalina prowadzi do podwyższenia tabela B 1.13-3). Również po podaniu doustnym pro-
obwodowego oporu naczyniowego – zwiększając wadzą one do długotrwałego podwyższenia ciśnie-
tym samym skurczowe i rozkurczowe ciśnienie krwi. nia krwi (biodostępność – ok. 50%). Podwyższenie
Midodryna jest substancją prekursorową (prolekiem, ciśnienia krwi wiąże się ze skurczem naczyń krwio-
pro-drug), która dopiero w organizmie – poprzez nośnych spowodowanych przez stymulację α-adre-
oderwanie grupy amidowej i oksydatywną dealkila- noreceptorów, jak również z dodatnim działaniem
cję grup metoksylowych – zamienia się we właści- inotropowym i chronotropowym w obrębie serca
wą substancję działającą. Aktywny metabolit osiąga – za co odpowiedzialne jest już pobudzenie recepto-
swoje najwyższe stężenie w osoczu po 1 godzinie rów adrenergicznych β.
od podania midodryny. Okres półtrwania w osoczu Działania niepożądane i przeciwwskazania odpo-
wynosi 3 godziny. Midodryna i jej aktywny metabolit wiadają tym spotykanym przy stosowaniu agonistów
ulegają prawie całkowitemu wydaleniu przez nerki. receptorów α-adrenergicznych.
MUTSCHLER-2009.indd 360 2010-01-07 22:12:37
Układ nerwowy 361
Tabela B 1.13-3. Leki α-sympatykomimetyczne (α-adrenomimetyczne)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
I. Leki α
α-adrenomimetyczne
-adrenomimetyczne stosowane ogólnie
OH midodryna Gutron 0,5 5-30
H3CO NH
NH2
O
OCH3
II. Leki α-adrenomimetyczne do stosowania miejscowego B1
OH fenylefryna Neosynephrin-POS, Otriven 0,3 do worka
Baby, Visadron, Coldrex, spojówkowego,
HO NH CH
3 Febrisan, Gripex, 0,2 donosowo
Neosynephrin, Otrivin,
Theraflu
N nafazolina Privin, Proculin, Siozwo, 0,05-0,2 do worka
Tele-Stulln, Betadrin, spojówkowego,
HN Rhinazin, Cincol, Mibalin, 0,05% jako maść
Oculosan, Rhinex, donosowa
Rhinophenazol, Sulfarinol
CH3 oksymetazolina Nasivin, Vistoxyn, 0,025-0,05 do worka
HO Wick Sinex, Afrin, Acatar, spojówkowego,
N Nasox, Oxalin, 0,05-0,15 donosowo
HN Resoxym
(H3C) 3C CH3
CH3 ksylometazolina Balkis, GeloNasal, Olynth, do 0,3 donosowo
N Otriven, Schnupfen endrine,
Otrivin, Xylogel,
HN Xylometazolin, Xylorhin,
(H3C) 3C CH3 Xylorin
NH N tramazolina Biciron, Ellatun, do 0,13 do worka
Rhinospray spojówkowego,
HN do 0,5 donosowo
N tetryzolina Berberil N, Ophthalmin-N, do 0,07 do worka
Sparsallerg, Strazolin, spojówkowego,
HN Tetrilin, Tatryvil, Visine Yxin, do 0,5 donosowo
Visine, Tyzine
III. Agoniści receptorów adrenergicznych α i β
OH etylefryna Bioflutin, 2,5 50-75
HO NH Cardanat,
C2H5 Effortil,
Efileftin-ratiopharm
OH oksylofryna Carnigen 2,5-4 32-96
NH
CH3
CH3
HO
MUTSCHLER-2009.indd 361 2010-01-07 22:12:37
362 Układ nerwowy
1.13.3.3. Agoniści receptorów cego porodu i przedwczesnej akcji porodowej (przed-
β-adrenergicznych wczesnych skurczów).
Działania niepożądane i przeciwwskazania odpo-
wiadają – szczególnie przy stosowaniu dużych dawek
Pobudzenie receptorów β1 serca zwiększa częstość
– tym obserwowanym w przypadku adrenaliny lub
pracy serca, siłę skurczu i prędkość przewodze-
orcyprenaliny.
nia impulsów. Stymulacja receptorów β2 prowadzi
z kolei do rozkurczu mięśniówki oskrzeli i macicy,
jak również do rozszerzenia naczyń krwionośnych.
Substancje o działaniu β-sympatykomimetycznym 1.13.4. Pośrednie sympatykomimetyki
(β-adrenomimetycznym) mogą w ten sposób pro-
wadzić do bradykardii i zaburzenia przewodnictwa Działanie pośrednio działających sympatykomime-
impulsów, a ponadto mogą być stosowane jako leki tyków (sympatomimetyków, adrenomimetyków;
broncholityczne i tokolityczne. leków o pośrednim działaniu sympatykomimetycz-
nym/adrenomimetycznym) polega na tym, że uwal-
Agoniści receptorów β-adrenergicznych o podob- niają noradrenalinę z pęcherzyków synaptycznych
nym w przybliżeniu działaniu na receptory β1 i β2. współczulnych zakończeń nerwowych i/lub hamują
Orcyprenalina jest jedynym preparatem z tej grupy jej zwrotne wchłanianie (wychwyt) ze szczeliny sy-
leków (zob. tab. 1.13-4). Stosowana bywa w terapii naptycznej do aksoplazmy komórki presynaptycznej.
występujących w przebiegu bradykardii zaburzeń ge- Przez zwiększenie stężenia noradrenaliny w oko-
nerowania i przewodzenia impulsów, jak również jako licy receptorów dochodzi do zwiększenia napięcia
antidotum w przedawkowaniu inhibitorów recepto- w układzie współczulnym. W przypadku wielokrot-
rów β-adrenergicznych. Okres półtrwania w osoczu nego stosowania – szczególnie przy stosowaniu du-
po jednorazowej iniekcji wynosi ok. godziny żych dawek – skuteczność większości substancji
Jako działania niepożądane mogą ujawnić się za- szybko zmniejsza się, gdyż noradrenalina nie może
burzenia rytmu serca, napady dusznicy bolesnej, nud- być zsyntetyzowana na nowo z wystarczającą szyb-
ności, uczucie osłabienia i nadmierne pocenie się. kością, a tym samym coraz mniejsze jej ilości mogą
Podobnie jak w przypadku adrenaliny – syste- być uwolnione (tachyfilaksja).
mowe podanie orcyprenaliny jest przeciwwskazane Do pośrednich związków o działaniu sympaty-
przy nadciśnieniu, chorobie wieńcowej serca, zabu- komimetycznym należą amfetaminy, które zostały
rzeniach rytmu serca z tachykardią, nadczynności opisane w rozdziale B 1.2.4.2.1, a nadto tyramina,
tarczycy oraz znieczuleniu ogólnym za pomocą pre- efedryna, metylosiarczan amezyny (zob. tab. 1.13-5)
paratów halogenowanych. oraz kokaina.
Efedryna – główna substancja pozyskiwana z ziela przęśli
Agoniści receptorów β-adrenergicznych o prze- (Ephedra vulgaris) – stosowana jest zwykle w preparatach
ważającym działaniu na receptory β2. Poprzez złożonych z innymi substancjami w zapaleniu oskrze-
zmiany cząsteczkowe – szczególnie zmiany rodzaju li do miejscowego zwężania naczyń krwionośnych. Poza
grup podstawnych wiążących się z atomem azotu działaniem obwodowym efedryna cechuje się również
ośrodkowym działaniem pobudzającym, gdyż łatwo poko-
pierścienia adrenaliny – udało się stworzyć prepa- nuje barierę krew-mózg. Ze względu na związany z tym
raty β-sympatykomimetyczne (zob. tab. 1.13-4), potencjał uzależniający efedryna powinna być używana
które prowadzą głównie do pobudzenia receptorów do zastosowań systemowych – jeżeli w ogóle – z bardzo
β2. Kardiologiczne działania niepożądane nieselek- dużą ostrożnością.
tywnych β-sympatykomimetyków mogą być w ten
sposób znacząco zmniejszone. Mimo to selektyw- Metylosiarczan amezyny hamuje doneuronalny
ność β2-sympatykomimetyków wobec receptorów wychwyt zwrotny i wewnątrzneuronalny rozpad
β2 nie jest absolutna, lecz jedynie względna. Ozna- noradrenaliny, a ponadto prowadzi do uwolnienia
cza to, że w przypadku stosowania większych da- noradrenaliny z magazynów – choć jego działanie
wek – jak to konieczne jest np. przy stosowaniu β2- jest istotnie słabsze niż wyżej wymienionych sub-
sympatykomimetyków w trakcie tokolizy – należy stancji. Ze względu na opisany profil działania sto-
liczyć się z wyraźnymi działaniami niepożądanymi sowanie metylosiarczanu amezyny nie prowadzi
ze strony serca. do tachyfilaksji. Metylosiarczan amezyny stosowany
β2-sympatykomimetyki służą przede wszystkim jest w przypadku samoistnego i objawowego niskie-
do terapii astmy oskrzelowej. Fenoterol bywa po- go ciśnienia (np. spadków ciśnienia w wyniku znie-
nadto stosowany do tokolizy w przypadku zagrażają- czulenia nadoponowego i rdzeniowego).
MUTSCHLER-2009.indd 362 2010-01-07 22:12:38
Układ nerwowy 363
Tabela B 1.13-4. Leki β-sympatykomimetyczne (β-adrenomimetyczne)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
I. Substancje o równie silnym działaniu na receptory β1 i β2
OH orcyprenalina Alupent, 2,5 0,5-1 doustnie
Astmopent
HO NH CH3
CH3
OH B1
II. Substancje o przeważającym działaniu na receptory β2
OH klenbuterol Spiropent 34 0,02-0,04 doustnie
Cl NH CH3
H3C CH3
H2N
Cl
OH
fenoterol Berotec, 3 20-40 doustnie,
Partusisten, 0,3-0,8 do inhalacji,
HO NH CH3 Fenoterol, 0,7-4 i.v.
Berodual
OH
OH
OH
H formoterol Diffumax Easyhaler, Foradil, 3-5 0,02-0,04
OHC
NH NH CH3 Forastmin, Formotop, do inhalacji
Formoterol-ratiopharm,
HO Fostex, Oxis, Oxidil, Zafiron,
Symbicort Turbuhaler,
OCH3 Oxis Turbuhaler
OH reproterol Bronchospasmin 1,5 2-4 do inhalacji,
HO NH
O 0,9-0,18 i.v.
N CH3
N
OH N N O
CH3
OH salbutamol Apsomol, Bronchospray, 6 15 doustnie,
HOH2C NH CH3 Buventol Easyhaler, 1-2 do inhalacji
Salbutamol-ratiopharm,
H3C CH3 Sultanol, Salbutamol,
HO Salbuhexal, Salbulair,
Steri-Neb Salamol,
Velaspir, Ventolin
OH salmeterol Aeromax, 2-8 0,2-0,4 do inhalacji
HOH2C NH Pulmetrol,
O
Seretide,
HO Serevent
OH terbutalina Aerodur, 3-4 15 doustnie
HO NH CH3 Bricanyl, 1,5-6 do inhalacji
Contimit,
H3C CH3 Terbul,
Asthmoprotect
OH
MUTSCHLER-2009.indd 363 2010-01-07 22:12:38
364 Układ nerwowy
Tabela B 1.13-4. (Kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
II. Substancje o przeważającym działaniu na receptory β2
Cl OH tulobuterol Brelomax 2,5 2 doustnie
NH CH3
H3C CH3
Okres półtrwania w osoczu oceniany jest na 13 1.13.5. Antagoniści receptorów
godzin.
Dawkowanie w hipotonii wynosi 1–3 razy dzien-
adrenergicznych
nie po 10–30 mg doustnie, z kolei w profilaktyce i te-
rapii spadków ciśnienia wynikających ze znieczule- 1.13.5.1. Antagoniści receptorów
nia nadoponowego i rdzeniowego – 5 mg w powolnej α-adrenergicznych
infuzji dożylnej.
Działania niepożądane i przeciwwskazania odpo- 1.13.5.1.1. Alkaloidy sporyszu
wiadają tym występującym podczas stosowania ago-
Do alkaloidów peptydowych sporyszu (secale cor-
nistów α-adrenergicznych.
nutum = przetrwalniki pasożytniczego grzyba buła-
Środek do znieczulenia miejscowego (jak również środek winki czerwonej z rodziny gruzełkowatych – przyp.
uzależniający) – kokaina – prowadzi wprawdzie do uwal- tłum.) należą ergotamina i substancje z grupy ergo-
niania jedynie bardzo niewielkich ilości noradrenaliny, toksyny (ergokrystyna, ergokornina, ergokryptyna),
lecz mimo to zwiększa stężenie katecholaminy w obrębie jak również ich dwuuwodornione pochodne (zob.
receptorów, gdyż blokuje wychwyt zwrotny noradrenaliny
przez błonę presynaptyczną. Efekt działania ulega nasileniu tab. B 1.13-6). Charakteryzują się złożonym zakresem
w przypadku równoczesnego spożycia kokainy z noradre- działania – co wiąże się z faktem, że leki te są częś-
naliną lub adrenaliną. ciowymi agonistami oraz częściowymi antagonistami
Tabela B 1.13-5. Pośrednie sympatykomimetyki (pośrednio działające leki adrenomimetyczne; z wyjątkiem amfetaminy i kokainy)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
benzylosulfonian Regulton, 9-13 10-30 doustnie
H2N OCH3 amezynium Supratonin 5-10 i.v.
+
N H3COSO 3
N
CH3 efedryna Perdiphen, Wick MediNait 6 8
(część składowa),
CH3 Efrinol,
NH
Ephedrinum hydrochloricum
OH
HO tyramina (dość duże ilości
znajdują się w w serach
twardych, czekoladzie,
NH2 śledziach, mięsie
czerwonym – przyp. tłum.)
MUTSCHLER-2009.indd 364 2010-01-07 22:12:38
Układ nerwowy 365
receptorów α-adrenergicznych, dopaminergicznych w spadkach ciśnienia u starszych osób z nadciśnie-
i serotoninergicznych. W przypadku podania do- niem oraz w objawowej terapii zespołu szyjnego.
ustnego biodostępność macierzystych substancji – Jako działania niepożądane mogą ujawnić się
w związku z niepełnym wchłanianiem oraz wysokim w przypadku wszystkich alkaloidów sporyszu: od-
Układ nerwowy
efektem pierwszego przejścia – jest niewielka; jednak ruch wymiotny i wymioty (efekt dopaminergiczny).
dochodzi do powstawania czynnych metabolitów. W trakcie stosowania dihydroergotoksyny obserwo-
Okres półtrwania w osoczu w przypadku ergota- wane były ponadto: dolegliwości żołądkowo-jeli-
miny i dihydroergotaminy wynosi 1,5–2 godz., dihy- towe, suchy nos, niepożądany spadek ciśnienia lub
droergotoksyny (zob. poniżej) – 2–4 godz. (rzadziej) zaburzenia ortostatyczne, bradykardia oraz
dolegliwości dusznicowe. W przypadku długotrwałe-
Ergotamina w związku z działaniem polegającym go stosowania – szczególnie w większych dawkach
na skurczu naczyń stosowana jest w przypadku cięż- – dihydroergotaminy, a przede wszystkim ergotami- B1
kich ostrych ataków migreny. Dihydroergotamina ny istnieje niebezpieczeństwo obwodowych zaburzeń
nadaje się – w związku z tonizującym działaniem ukrwienia (zgorzel).
w obrębie naczyń żylnych – do terapii zaburzeń or- Nieuwodornione alkaloidy sporyszu przeciwwska-
tostatycznych oraz w profilaktyce przeciwzakrzepo- zane są w ciężkich zaburzeniach funkcji wątroby i ne-
wej, szczególnie w okresie przed- i pooperacyjnym rek, nadciśnieniu, chorobach naczyń, a także w czasie
w terapii skojarzonej z heparyną. Dihydroergo- ciąży i okresie karmienia; z kolei uwodornione alka-
kryptyna – jako agoniści receptorów dopaminer- loidy sporyszu – również w czasie ciąży, a dihydro-
gicznych – znajdują zastosowanie w terapii choroby ergotamina – w przebiegu ciężkiej niewydolności
Parkinsona. Mieszanina dihydroergokorniny, -kry- wieńcowej.
styny i -kryptyny – pod nazwą kodergokryny lub Działanie leków przeciwzakrzepowych oraz inhi-
dihydroergotoksyny stosowana bywa w osłabieniu bitorów agregacji płytek ulega nasileniu pod wpły-
sprawności funkcji mózgowych w podeszłym wieku, wem dihydroergotoksyny. Związki makrolidowe i te-
Tabela B 1.13-6. Alkaloidy sporyszu typu peptydowego
OH
R1 O N
H
O NH N
O
O R2
N
CH3
H
HN
R1 R2
Grupa ergotaminy
ergotamina H3C
CH2
Grupa ergokrystyny CH3
ergokrystyna CH
H3C CH2
CH3 CH3
ergokryptyna CH
H3C H3C CH2
CH3 CH3
ergokornina CH CH
H3C H3C
MUTSCHLER-2009.indd 365 2010-01-07 22:12:39
366 Układ nerwowy
tracykliny nasilają z kolei skurcz naczyń wywołany Fenoksybenzaminę można stosować w neuro-
dihydroergotaminą. gennych zaburzeniach oddawania moczu, w których
wzmożone napięcie zwieracza cewki moczowej
Alkaloidy sporyszu typu ergometryny, które nie jest wynikiem nadmiernej stymulacji receptorów α.
blokują receptorów α-adrenergicznych, zostaną omó- Ponadto fenoksybenzamina podawana jest do opa-
wione w rozdziale B 2.8.10.6. nowania kryz nadciśnieniowych w przebiegu guza
chromochłonnego – guza rdzenia nadnerczy lub innej
Zatrucie alkaloidami sporyszu. W okresie średniowiecza tkanki chromochłonnej produkującego noradrenalinę
zatrucia sporyszem przez spożywanie zboża zawierającego – głównie profilaktycznie w okresie przedoperacyj-
sporysz były częste (ogień świętego Antoniego), obecnie
spotykane są jednak rzadko (głównie w związku ze spoży- nym tego rodzaju guzów.
waniem zboża z upraw bioekologicznych, tj. prowadzonych Dawkowanie jest indywidualne (średnia dawka
z zastosowaniem programów ochrony roślin i oczyszczania pojedyncza wynosi 25–50 mg).
materiału siewnego – przyp. tłum.). Wśród działań niepożądanych wyróżnia się odru-
Ostre zatrucie charakteryzuje się wymiotami, silnymi chowe tachykardie, zaburzenia ortostatyczne, zwęże-
bólami brzucha, pragnieniem, zaburzeniami czucia – pare-
stezjami w obrębie kończyn („chodzenie mrówek”) i roz- nie źrenic, suchy nos, zaburzenia wytrysku, wymioty
szerzeniem źrenic. W wyniku porażenia ośrodka oddecho- i biegunkę. W eksperymentach na zwierzętach obser-
wego lub zatrzymania pracy serca może dojść do śmierci. wowano działania kancerogenne fenoksybenzaminy.
W przypadku przewlekłego zatrucia (ergotyzmu) wy- Wobec powyższego wskazania do stosowania feno-
różnia się dwie postacie: drgawkową i zgorzelinową. ksybenzaminy są bardzo wąskie.
W drgawkowej postaci występują napady padaczkowe,
zaburzenia ośrodkowego układu nerwowego i zmiana oso- Przeciwwskazaniem są zaburzenia w obrębie na-
bowości. Postać zgorzelinowa charakteryzuje się natomiast czyń mózgowych i niewydolność serca, oraz ciąża
występowaniem niezwykle bolesnych zaburzeń ukrwie- oraz okres karmienia.
nia w obrębie kończyn dolnych, które później prowadzą
do zgorzeli.
Poza środkami zapobiegającymi wchłanianiu w terapii
zatrucia alkaloidami sporyszu stosuje się leki rozszerzające 1.13.5.1.3. Selektywni antagoniści
naczynia krwionośne, oraz diazepam – w przypadku drga- receptorów
wek. W przypadku zagrażającego porażenia oddechowego α1-adrenergicznych
pacjent musi być sztucznie wentylowany.
Preparaty należące do tej grupy lekowej zostały ze-
stawione w tab. B 1.13-7.
1.13.5.1.2. Fenoksybenzamina
Prazosyna, pierwowzór leków z tej grupy, różni się
Fenoksybenzamina jest nieselektywnym antagonistą od wcześniej omówionych nieselektywnych antago-
receptorów α-adrenergicznych. Jako alkilująca po- nistów receptorów α-adrenergicznych tym, że działa
chodna β-chloroetyloaminy prowadzi początkowo właściwie wyłącznie na receptory α1 i z tego też po-
do kompetytywnego hamowania związanego z wy- wodu nie prowadzi do blokowania presynaptycznych
tworzeniem się kowalencyjnego wiązania, później receptorów α2 z następczym zwiększeniem uwalnia-
jednak wywołuje nieodwracalną blokadę recepto- nia noradrenaliny – związanym z zahamowaniem
rów α-adrenergicznych. Stąd też charakteryzuje się mechanizmu ujemnego sprzężenia zwrotnego.
długim czasem działania. Działanie preparatu ulega Prazosyna ulega szybkiemu wchłanianiu. W oso-
przełamaniu/zniesieniu dopiero w wyniku zsynte- czu lek w 95% jest wiązany z białkami. W wątrobie
zowania nowych cząsteczek białkowych receptora. poddana jest procesowi daleko idącej biotransforma-
Leki α-sympatykomimetyczne (α-adrenomimetycz- cji. Okres półtrwania w osoczu wynosi 2,5–4 godz.,
ne) jako antidotum są nieskuteczne. a w związku z długim okresem działania (około 10
godzin) wymaga podawania jedynie dwóch dawek
dziennie. Wydalanie następuje prawie wyłącznie
z żółcią lub też z kałem.
W związku z działaniem polegającym na rozsze-
rzeniu naczyń krwionośnych prazosyna wskazana
jest do stosowania w nadciśnieniu tętniczym – jako
lek drugiego wyboru. Ponadto stosowana jest w cho-
fenoksybenzamina
robie Raynauda.
Ze względu na ryzyko wystąpienia zaburzeń or-
tostatycznych należy na początku terapii zwiększać
MUTSCHLER-2009.indd 366 2010-01-07 22:12:39
Układ nerwowy 367
Tabela B 1.13-7. Antagoniści receptorów α1-adrenergicznych
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
O prazosyna Prazosin-ratiopharm 2,5-4 2-20
N
H3CO N N O
N
H3CO
NH2
B1
O terazosyna Flotrin, 8-14 2-10
Heitrin,
N Tetrablock,
H3CO N N O Tera TAD,
Kornam,
N Setegis,
H3CO Tesin
NH2
O doksazosyna Cardular, Diblocin PP, 22 2-4
Doxazosin-ratiopharm,
O Doxazosin STADA,
N
Apo Doxan, Cardura,
H3CO N N
O Doxagen, DoxaLEK,
N Doxanorm, Doxar,
H3CO Doxaratio, Doxonex,
NH2 Kamiren, Postatic,
Zoxon
CH3 alfuzosyna Alfuzosin HEXAL, 8 5-10
H3CO N N Alfuzosin STADA,
NH O
Urion, UroXatral,
N AlfuLEK, Alfuzostad,
H3CO O Alfugen, Dalfaz
NH2
O bunazosyna Andante 12 6
N C3H7
H3CO N N
N
H3CO
NH2
tamsulosyna Alna Ocas, Omnic Ocas, 15-19 0,4
Prostadil, TADIN, Apo-Tamis,
H2NO2S NH Bazetham, Fokusin, Omnic,
O
Omsalm Prostamnic,
CH3 OC2H5 Proximic, Ranlosin,
H3CO
Symlosin, Tamsec, Tamsudil,
Tamsigen, Tamsulek,
Tamsulosin-ratiopharm,
Tanyz, Uprox, Urostad
OCH3 urapidil Ebrantil, 2-4 30-180
Urapidil-Pharmore
N
N NH O
N
H3C N CH3
O
MUTSCHLER-2009.indd 367 2010-01-07 22:12:39
368 Układ nerwowy
dawkę powoli: pierwsza dawka wieczorem 0,5 mg, Profil działania. Rozróżnia się β-blokery z i bez:
następnie 0,5–1 mg 2(–3) razy dziennie. Średnia
■ selektywności w stosunku do receptorów β1 (tzw.
dawka podtrzymująca wynosi w nadciśnieniu 4–6
kardioselektywności),
mg/dzień.
Działania niepożądane występują przede wszyst- ■ częściowo agonistycznej aktywności (PAA – par-
kim na początku terapii (tzw. efekt pierwszej dawki; tial agonistic activity) odpowiadającej wewnętrznej
first dose effect) jako zaburzenia ortostatyczne. Dal- aktywności sympatykomimetycznej (ISA – intrinsic
szymi objawami niepożądanymi są – podobnie jak sympatomimetic activity),
w przypadku leków przeciwnadciśnieniowych – re-
■ nieswoistego działania na błonę komórkową,
tencja sodu i wody, zawroty głowy, nudności, sen-
ność i brak napędu, ponadto – bóle głowy i (rzadziej) ■ dodatkowego działania wazodylatacyjnego (zob.
tachykardia. tab. B 1.13-8).
W przypadku niewydolności serca i ograniczonej
funkcji wątroby, a także w czasie ciąży i okresie kar- Nieselektywni antagoniści receptorów β-adrener-
mienia – prazosyna jest przeciwwskazana. gicznych. Pierwszym z nieselektywnych β-blokerów
jest propranolol. Poza wskazaniami sercowo-na-
Ze względu na długie okresy półtrwania w osoczu czyniowymi (zob. poniżej), co do których został on
(zob. tab. 1.13-7), takie analogi, jak terazosyna, bu- już w większości zastąpiony przez substancje selek-
nazosyna i doksazosyna mogą być podawane w po- tywne w stosunku do receptorów β1, wykorzystując
jedynczej dawce dziennej. Poza nadciśnieniem tera- stymulację receptorów β2 – okazał się skuteczny
zosyna i doksazosyna są wskazane również do stoso- w samoistnym drżeniu, zaburzeniach lękowych,
wania w łagodnym przeroście gruczołu krokowego. a także w profilaktyce migreny. Ponadto możliwe
Do wyłącznego stosowania w tym właśnie wskazaniu jest wykorzystanie propranololu – wraz z innymi spe-
zostały dopuszczone alfuzosyna i tamsulosyna, któ- cyficznymi środkami terapeutycznymi – w leczeniu
re charakteryzują się szczególnie wysokim powino- nadczynności tarczycy, gdyż w wyniku zablokowa-
wactwem do występujących w gruczole krokowym nia receptorów β2 ulega zahamowaniu przemiana T4
receptorów α1A i dlatego też ich działanie hipotensyj- do T3.
ne jest niewielkie. Innymi nieselektywnymi β-blokerami (zob. tab.
B 1.13-9) są m.in. bupranolol, metypranolol, pen-
Urapidil ma oprócz obwodowego działania anta- butolol, timolol oraz stosowany jako lek antyaryt-
gonistycznego w stosunku do receptorów α1 także miczny (do umiarowiania pracy serca) sotalol.
ośrodkowe działanie obniżające ciśnienie krwi, które
wiąże się z pobudzeniem receptorów 5-HT1A. Blokery receptorów β-adrenergicznych z działaniem
PAA (o częściowo agonistycznej aktywności; zob. tab. B
1.13-9). Gdy w przypadku leku blokującego receptory ad-
1.13.5.2. Antagoniści receptorów renergiczne β podawana jest informacja, że charakteryzuje
się on częściowo agonistyczną aktywnością (PAA – partial
β-adrenergicznych agonistic activity) – to oznacza, że mamy do czynienia
(blokery receptorów z substancją o działaniu dualistycznym (częściowy agonista
β-adrenergicznych, i równocześnie częściowy antagonista) – jednak z przewa-
blokery receptorów β, β-blokery; żającą komponentą antagonistyczną. Do β-blokerów z dzia-
łaniem PAA (typu PAA) należą m.in.: acebutolol, carteolol,
leki β-adrenolityczne) oksyprenolol i pindolol. W przypadku większości wskazań
działanie w rodzaju PAA należy ocenić jako negatywne.
Antagoniści receptorów β-adrenergicznych hamu- Duże badania epidemiologiczne wykazały znaczącą wyż-
ją w sposób kompetytcyjny receptory adrenergicz- szość czystych antagonistów receptorów β-adrenergicznych
ne β. W wyniku blokady receptorów β1 znoszone nad β-blokerami z działaniem PAA. U pacjentów z tenden-
jest dodatnie działanie inotropowe i chronotropowe cją do zaburzeń rytmu z jego zwolnieniem (bradyarytmią)
katecholamin na serce; z kolei blokada receptorów obniżają one, chociaż słabiej, częstotliwość spoczynkową
i mogą w związku z tym – choć wyraźnie rzadziej – prowa-
β2 znosi zwiotczające działanie na mięśnie gładkie. dzić do zagrażających życiu epizodów bradykardii.
Ponadto antagoniści receptorów β-adrenergicznych
hamują – przez blokadę receptorów β – efekty me-
Selektywni antagoniści receptorów β1-adrener-
taboliczne katecholamin (glikogenolizę, lipolizę).
gicznych. Podani w tab.1.13-9 selektywni antagoni-
W przypadku większości wskazań terapeutycznie po-
ści receptorów β1-adrenergicznych:
żądana jest blokada receptorów β1.
■ acebutolol,
MUTSCHLER-2009.indd 368 2010-01-07 22:12:40
Układ nerwowy 369
■ atenolol, w przypadku związków selektywnych w stosunku
do receptorów β1. Z drugiej strony należy jednak
■ betaksolol,
zwrócić uwagę, że podanie selektywnych antagoni-
■ metoprolol, stów receptorów β1-adrenergicznych może prowa-
Układ nerwowy
dzić do pogorszenia przebiegu obturacyjnych scho-
■ bisoprolol,
rzeń płuc.
■ talinolol
Antagoniści receptorów β-adrenergicznych z niespecy-
posiadają taką zaletę, że ich powinowactwo do re- ficznym działaniem błonowym (działanie stabilizujące
błonę). Przez pojęcie nieswoistego działania β-blokera
ceptorów β1 jest większe niż do receptorów β2. Tego na błonę rozumiana jest właściwość miejscowego działania
rodzaju selektywność ma jednak charakter jedynie znieczulającego i stabilizującego błonę komórkową, która
względny, a nie absolutny, a ponadto zanika w przy- jest niezależna od blokującego działania na receptory ad- B1
padku stosowania dużych dawek leków. (Kliniczne renergiczne β. Zwiększa się ona wraz ze wzrostem lipofil-
określenie kardioselektywności nie jest popraw- ności β-blokera. Dla większości wskazań dla β-blokerów
działanie polegające na stabilizowaniu błony jest w przy-
ne, gdyż receptory β1 występują nie tylko w sercu, padku stosowania leków o typowym dawkowaniu mało
ale również w obrębie innych narządów. Skądinąd znaczące. Efekt stabilizowania błony ujawnia się bowiem
jednak gęstość receptorów β1 w sercu jest szczegól- dopiero w przypadku uzyskania w tkankach stężeń, które
nie duża). są znacząco wyższe niż wymagane do uzyskania zabloko-
Pomimo opisanych ograniczeń selektywność wania receptorów adrenergicznych β.
względem receptorów β1 jest cechą pożądaną w od-
niesieniu do większości wskazań. Przede wszystkim Antagoniści receptorów β-adrenergicznych o dzia-
u pacjentów z zaburzoną tolerancją glukozy lub łaniu wazodylatacyjnym. Do leków z tej grupy na-
z cukrzycą – selektywni antagoniści receptorów β1 leżą karwedilol, celiprolol i nebiwolol.
są bardziej korzystni niż związki o charakterze nie- Efekt rozszerzania naczyń krwionośnych karwe-
selektywnym, gdyż w mniejszym stopniu wpływają diololu opiera się na równoczesnym blokowaniu re-
na metabolizm węglowodanów. O ile nieselektywne ceptorów adrenergicznych α1. Celiprolol działa roz-
β-blokery znoszą tokolityczny efekt leków β1-sym- szerzająco na naczynia ze względu na częściowo ago-
patykomimetycznych oraz zmniejszają przepływ nistyczną aktywność wobec receptorów β2. Nebiwo-
krwi w obrębie macicy i łożyska, o tyle efekt toko- lol prowadzi do rozszerzenia naczyń krwionośnych
lityczny nie ma większego znaczenia klinicznego w wyniku (pośredniego) uwalniania tlenku azotu.
Tabela B 1.13-8. Profil działania antagonistów receptorów β-adrenergicznych (leków β-adrenolitycznych)
Nazwa Aktywność częściowo Nieswoiste działanie Selektywność Rozszerzenie
międzynarodowa agonistyczna błonowe względem β1 naczyń
acebutolol + + (+) –
alprenolol + + – –
atenolol – – + –
bisoprolol – – + –
bupranolol – + – –
celiprolol – + – +
karwedilol + – (+) +
metipranolol – – + –
metoprolol – – + –
nebiwolol – – + +
oksprenolol + + – –
pindolol + + – –
propranolol – + – –
timolol – + – –
MUTSCHLER-2009.indd 369 2010-01-07 22:12:40
370 Układ nerwowy
Tabela B 1.13-9. Antagoniści receptorów β-adrenergicznych (leki β-adrenolityczne)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
OH
1
O NH R2
R
H3C CH3
R1 R2
β-adrenergicznych
l. Nieselektywni anatgoniści receptorów β-adrenergicznych bez PAA
propranolol Beta-Tablinen, 3-4 240 doustnie,
Dociton, Obsidan, 1-5 i.v.
H
Propra-ratiopharm,
Propranolol
metypranolol Betamann, 0,1-0,3 do worka
H3C Normaglaucon spojówkowego
H
H3C CH3
O CH3
penbutolol Betapressin 1,5 20-80
CH3
bupranolol Betadrenol 1,5 50-400
Cl
CH3
H3C
tymolol Arutimol, Combigan, Cosopt, 0,25-0,5 do worka
O Cusimolol, DuoTrav, Fotil, spojówkowego
N CH3 Ganfort, Nyolol, Oftan,
N Oftensin, Timolol CV,
S N Timomann, Timoxehal,
Tim-Ophtal, Timoptic,
Xalacom
OH sotalol Biosotal, Darob, Sotabeta, 15 80-160 (-480)
NH
CH(CH3) 2
Sotahexal, SotaHEXAL,
O O Sotalex, Sotalol-ratiopharm
H3C S NH
lI. Nieselektywni anatgoniści receptorów β-adrenergicznych z PAA
karteolol Arteoptic, 5-7 2,5-20
Carteol,
CH3 Endak
NH O
pindolol Galuco-Stulln, 3-4 1 do worka
Visken spojówkowego,
H 5-10 (-15)
NH
oksprenolol Trasicor 1-2 40-80 (-160)
O
CH2
H
MUTSCHLER-2009.indd 370 2010-01-07 22:12:40
Układ nerwowy 371
Tabela B 1.13-9. Antagoniści receptorów β-adrenergicznych (leki β-adrenolityczne) (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
R1 R2
III. Selektywni anatgoniści receptorów β1-adrenergicznych
O acebutolol Prent, 4 200-400
Abutol,
CH3 Sectral
H
C3H7
HN
B1
O
atenolol Atenolol-ratiopharm, 6-10 25-100
AtoHEXAL,
Atenol AL,
Atenolol,
Normocard,
NH2 H Tenormin
O
betaksolol Betabion, 16-20 0,5 do worka
Betoptic, spojówkowego,
Betoptima, 10-20 doustnie
Kerlone,
H Lokren,
O Optibetol
metoprolol Betaloc, 3-5 50-200
Beloc-Zok Herz,
Lopresor,
Metocard,
MetoHEXAL,
O Metoprolol,
CH3 H
Metoprolol ZOT STADA
bisoprolol Antipres, 10-12 2,5-10
Bisocard,
BisoHEXAL, Bisopromerck,
Bisoprolol-ratiopharm,
Bisoprolol STADA,
O Bisoratio, Concor, Corectin,
O CH(CH3) 2 H Coronal
talinolol Cordanum 11 50-200
CH3
HN NH
IV. Anatgoniści receptorów β-adrenergicznych z działaniem wazodylatacyjnym
O celiprolol Celipro Lich, 5-7 200-400
Celiprolol-CT,
CH3 Celiprolol-ratiopharm,
CH3 Selectol,
C2H5 Celipres
HN N
C2H5
O
MUTSCHLER-2009.indd 371 2010-01-07 22:12:41
372 Układ nerwowy
Tabela B 1.13-9. Antagoniści receptorów β-adrenergicznych (leki β-adrenolityczne) (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
IV. Anatgoniści receptorów β
β-adrenergicznych
-adrenergicznych z działaniem wazodylatacyjnym
karwedilol Atram, Avedol, Carvedigamma, 6-10 12,5-50
H3CO Carvedilol HEXAL, Carvedilol-
OH
HN
-ratiopharm, Carvedilol TEVA,
O NH
O Carvetrend, Carvilex, Coryol,
Dilatrend, Hypoten, Querto
Symtrend, Vivacor
F F nebiwolol Nebilet, 10 2,5-5
H H Nebinad,
O NH O Nedal
OH OH
Kliniczne znaczenie tej składowej działania nie zo- silnemu gromadzeniu, natomiast gromadzenie me-
stało do końca poznane. toprololu jest znacząco słabsze. W przypadku ate-
nololu stężenie w mózgu jest wyraźnie niższe niż
Racemiczni i optycznie czynni antagoniści recep- w osoczu. Również w zakresie kinetyki eliminacji
torów β-adrenergicznych. Większość blokerów re- istnieją wyraźne różnice między poszczególnymi
ceptorów β-adrenergicznych dostępna jest w handlu β-blokerami. Lipofilne związki charakteryzują się
pod postacią racematów, mimo że lewoskrętny (S) zwykle krótkimi okresami półtrwania, preparaty hy-
enancjomer wykazuje 100-krotnie silniejsze działa- drofilne mają natomiast długie okresy półtrwania.
nie blokujące na receptory β niż jego prawoskrętna
(R) postać. Działanie polegające na stabilizowaniu Wskazania. β-blokery stosowane są głównie w cho-
błony komórkowej jest natomiast podobne w przy- robie wieńcowej serca, czynnościowych zaburze-
padku obu postaci enancjomerowych. Zaletą czyste- niach serca i układu krążenia, niewydolności serca,
go enancjomeru S – czego jednak nie udało się jed- zaburzeniach rytmu serca i nadciśnieniu. Następny-
noznacznie potwierdzić badaniami klinicznymi – ma mi wskazaniami są guz chromochłonny (pheochro-
być mniejsze obciążenie organizmu tą substancją. mocytoma; w terapii skojarzonej z α-blokerami) i ja-
skra oraz jak to opisano w przypadku propranololu
Kinetyka. Lipofilne β-blokery (np. metoprolol, pro- – w profilaktyce migreny, nadczynności tarczycy
pranolol) są niemal całkowicie i szybko wchłania- i w drżeniu mięśni (tremor).
ne; tymczasem hydrofilne substancje z tej grupy
(np. atenolol, nadolol) ulegają jedynie częściowe- Wyzwolone lękiem objawy wegetatywne przed operacją,
mu wchłanianiu. Mimo to biodostępność związków w przypadku wystąpień publicznych lub egzaminów, w trak-
cie jazdy samochodem itp. wyraźnie zmniejszają się po poda-
hydrofilnych nie jest zwykle niższa niż preparatów niu β-blokerów, przy czym somatyczne objawy lęku są opa-
lipofilnych – co można tłumaczyć różnicami w za- nowywane lepiej niż subiektywne uczucie lęku. W przypad-
kresie efektu pierwszego przejścia (przez wątrobę; ku pacjentów z chorobą Parkinsona – β-blokery mogą być
first pass effect): lipofilni antagoniści receptorów stosowane łącznie z lekami przeciwparkinsonowymi w te-
β-adrenergicznych wykazują bowiem silny efekt rapii drżenia mięśni. Szczególnie dobre wyniki osiąga się
w leczeniu drżenia samoistnego. Nieselektywne β-blokery,
pierwszego przejścia; tymczasem efekt ten w przy- np. propranolol (zob. powyżej), przewyższają w tym wska-
padku związków hydrofilnych albo jest mały, albo zaniu preparaty selektywne w stosunku do receptorów β1,
w ogóle nie występuje. gdyż drżenie mięśniowe powstaje głównie w wyniku pobu-
Zjawisko lipo- i hydrofilności ma również zna- dzenia receptorów β2. W związku z tym przeciwdrżeniowym
czenie dla stężenia β-blokerów w tkankach. W przy- działaniem – β-blokery były lub też są stosowane przez spor-
towców uprawiających strzelectwo. Dlatego znajdują się one
padku lipofilnego propranololu stężenie w płucach na liście zabronionych środków dopingowych.
jest około 60 razy większe niż w osoczu, umiarko-
wanie lipofilnego metoprololu – 17 razy, a hydrofil- Dawkowanie. Średnie i pojedyncze dawki po-
nego atenololu – jedynie 4 razy wyższe w płucach szczególnych β-blokerów podane zostały w tabeli
niż w osoczu. Także w mózgu – propranolol ulega B 1.13-9.
MUTSCHLER-2009.indd 372 2010-01-07 22:12:41
Układ nerwowy 373
Efekt rebound (z odbicia). Ponieważ w wyniku dłu- Interakcje. Znaczącą interakcją β-blokerów jest spo-
żej trwającej terapii za pomocą β-blokerów zwiększa wolnienie zwiększania się poziomu cukru we krwi
się ilość receptorów β, a ponadto zwiększa się uwal- po uprzednim podaniu insuliny lub doustnych środ-
nianie noradrenaliny, w wyniku nagłego odstawienia ków przeciwcukrzycowych. Efekt ten grozi przedłu-
Układ nerwowy
leków należy liczyć się z objawami rebound (wzrost żoną reakcją hipoglikemiczną. Ponadto u pacjentów
ciśnienia krwi, niebezpieczeństwo wyzwolenia napa- nie ujawniają się objawy ostrzegawcze występujące
du dusznicy bolesnej lub nawet zawału serca). Stąd typowo przy pobudzeniu układu współczulnego, gdyż
też kończenie terapii tymi lekami powinno polegać β-blokery tłumią przewodzenie pobudzeń w układzie
na powolnym zmniejszaniu dawki. współczulnym. Szczególnie wyraźna jest ta interak-
cja w przypadku nieselektywnych β-blokerów.
Działania niepożądane. W sytuacji przestrzegania Działanie licznych leków antyarytmicznych ulega
przeciwwskazań i interakcji – β-blokery należą do le- nasileniu pod wpływem β-blokerów; ich działanie B1
ków szczególnie dobrze znoszonych. spowalniające pracę serca nasila się pod wpływem
Wśród niespecyficznych działań niepożądanych środków do znieczulenia ogólnego. Z powodu hamo-
opisano następujące: objawy żołądkowo-jelitowe, wania enzymatycznego – równoczesne podanie pro-
np. dolegliwości w górnej części brzucha, nudności, pranololu i cymetydyny prowadzi niemal do podwo-
wymioty i biegunka, a w dalszej kolejności zabu- jenia stężenie propranololu w osoczu.
rzenia ze strony ośrodkowego układu nerwowego:
zmęczenie, uczucie rozbicia i bóle głowy oraz sto- Zatrucia. W przypadku zatruć z użyciem β-bloke-
sunkowo rzadko – reakcje alergiczne pod postacią rów – siłę skurczów serca można poprawić przez
wyprysków i świądu. podane agonistów receptorów β, a jeżeli działanie
Specyficzne (swoiste) działania niepożądane to nie będzie wystarczające – wskazane jest zastoso-
są wynikiem blokady receptorów β, a wśród nich wanie krótkiego wlewu z glukagonem.
należy wymienić: zwiększenie oporu w drogach od-
dechowych, osłabienie siły skurczu serca (stąd też
wskazane powolne zwiększanie dawki w przypadku
niewydolności serca!), bradykardie, zaburzenia krą- 1.13.6. Leki sympatolityczne
żenia z niskim ciśnieniem krwi, pogorszenia w prze-
biegu zaburzeń krążenia obwodowego, kryzy nadciś-
Poza blokowaniem receptorów współczulnych – na-
nieniowe w guzie chromochłonnym – gdy jednocześ-
pięcie w obrębie układu współczulnego można ob-
nie nie podano preparatów blokujących receptory
niżyć przez ingerencję ośrodkową lub pozazwojowo
α-adrenergiczne, podwyższenie progu pobudliwości
presynaptycznie. Po zmniejszeniu oporów obwodo-
u pacjentów z rozrusznikiem serca, a także nasilenie
wych oraz minutowej objętości wyrzutowej serca
hipoglikemii u małych pacjentów z cukrzycą i skłon-
maleje ciśnienie krwi. Stąd też substancje o działaniu
nością do kwasicy ketonowej. Szczególnie po poda-
antysympatotonicznym (przeciwsympatotonicznym,
niu nieselektywnych β-blokerów bez działania PAA
hamującym układ współczulny) służą głównie jako
opisano ujawnianie się zmian w zakresie lipidów
leki przeciwnadciśnieniowe.
(zwiększenie stężenia VDLD, nieznaczne obniżenie
stężenia HDL). Zgodnie z dotychczasową wiedzą –
opisane zmiany lipidowe nie mają jednak większego
znaczenia klinicznego. 1.13.6.1. Ośrodkowo
i zazwojowo presynaptycznie
Przeciwwskazania. Z wymienionymi niespecyficz- działający agoniści receptorów
nymi działaniami niepożądanymi wiążą się przeciw- α2-adrenergicznych/receptorów
wskazania do stosowania β-blokerów: obturacyjne imidazolinowych
zaburzenia wentylacji, zaburzenia rytmu serca
w przebiegu bradykardii, blok przedsionkowo-ko- Klonidyna, moksonidyna. Klonidyna – będąca po-
morowy II i III stopnia, szok kardiogenny, nasilona chodną imidazolu oraz jej analog – moksonidyna
hipotonia, bradykardia. Jeżeli w przypadku podania już w małych dawkach obniżają ciśnienie krwi, czę-
β-blokerów dojdzie do (zbyt znacznego) spowolnie- stość pracy serca oraz minutową objętość wyrzutową
nia pracy serca – efekt ten można opanować przez serca. Zidentyfikowany mechanizm działania pole-
dożylne podanie leków parasympatykolitycznych, ga na tym, że klonidyna i moksonidyna – w związku
np. atropiny, lub też β1-sympatykomimetycznych, ze swoją lipofilnością – szybko przenikają do ośrod-
np. orcyprenaliny. kowego układu nerwowego. Tam pobudzają post-
MUTSCHLER-2009.indd 373 2010-01-07 22:12:42
374 Układ nerwowy
dyny – w związku z jej większym powinowactwem
Cl do receptorów imidazolinowych niż do receptorów
N NH adrenergicznych α2. Należą tu także zaburzenia orto-
klonidyna statyczne i retencja sodu (obrzęki). W dalszej kolej-
HN
Cl ności – podobnie jak w przypadku stosowania innych
leków sympatolitycznych – może ujawnić się osłabie-
nie libido i potencji. W przypadku nagłego odstawie-
Cl nia leku – szczególnie w sytuacji gdy był on stosowa-
N NH
N moksonidyna
ny w dużych dawkach – istnieje niebezpieczeństwo
HN znacznych skoków ciśnienia (objaw rebound).
H3C N OCH3 W przypadku bradykardii, zaparcia, depresyjnego
obniżenia nastroju i zaburzeń przewodnictwa przed-
sionkowo-komorowego – klonidynę i jej pochodne
wolno stosować jedynie w warunkach intensywnego
synaptyczne receptory α2-adrenergiczne w obrębie nadzoru nad pacjentem.
– będącego ośrodkową strukturą przełączającą dla Działanie leków neuroleptycznych, nasennych
odruchów z baroreceptorów – jądra pasma samot- i alkoholu ulega nasileniu pod wpływem agonistów
nego (nucleus tractus solitarius). Skutkiem tego receptorów α2-adrenergicznych typu klonidyny.
jest osłabienie pobudzeń współczulnych w ośrodku
naczynioruchowym, a napięcie w układzie współ- Metylodopa. W podobny sposób jak klonidyna działa
czulnym ulega obniżeniu. Ponadto dochodzi tak- również metylodopa. Jako aminokwas może przedo-
że do stymulacji obwodowych presynaptycznych stać się – dzięki aktywnemu transportowi – do ośrod-
receptorów α2-adrenergicznych, czego skutkiem kowego układu nerwowego i tam jest w znaczącym
jest zmniejszenie uwalniania noradrenaliny. Jeszcze stopniu metabolizowana do właściwej substancji
większe znaczenie dla efektu działania tych leków działającej – α-metylonoradrenaliny. Maksymalne
polegającego na obniżeniu ciśnienia krwi jest ich do- stężenie w osoczu stwierdza się po 2–6 godzinach.
datkowe działanie agonistyczne na tzw. receptory 20–70% leku ulega wydaleniu w niezmienionej po-
imidazolinowe. Działanie to jest szczególnie nasilo- staci. Pozostała część dawki ulega natomiast metabo-
ne w przypadku moksonidyny. Receptory imidazo- lizowaniu w ścianie jelit oraz w wątrobie.
linowe występują m.in. w obrębie brzuszno-bocznej Metylodopa służy przede wszystkim jako lek prze-
części rdzenia przedłużonego (medulla oblongata) ciwnadciśnieniowy u kobiet w ciąży.
– ważnego ośrodka przełączającego dla impulsów Dawka wynosi początkowo 125–375 mg/dzień
współczulnych. Wiązanie klonidyny i moksonidyny i może być następnie – jeżeli jest to konieczne – stop-
z tymi receptorami powoduje zmniejszenie aktyw- niowo zwiększana (średnia dzienna dawka podtrzy-
ności układu współczulnego. mująca wynosi 500–750 mg; maksymalna dawka
Klonidyna i pochodne klonidyny ulegają pełnemu w ciąży 2000 mg). Wobec względnie krótkiego okre-
i szybkiemu wchłanianiu. Pod względem kinetyki róż- su działania metylodopy dawka dzienna powinna być
nią się one między sobą głównie okresem półtrwania podzielona na liczne dawki pojedyncze.
w osoczu (moksonidyna 2–3 godziny, klonidyna 8–12 Działania niepożądane są podobne do tych spo-
godzin). Wydalanie odbywa się głównie przez nerki. tykanych u wcześniej wymienionych agonistów re-
Głównym wskazaniem do stosowania tych leków ceptorów α2-adrenergicznych. Innymi objawami nie-
jest nadciśnienie tętnicze. Poza ich stosowaniem jako pożądanymi są gorączka polekowa (rzadko), anemia
leków przeciwnadciśnieniowych używa się ich rów- hemolityczna oraz uszkodzenie wątroby.
nież do łagodzenia objawów w przebiegu zespołu od- W guzie chromochłonnym, zaburzeniach funkcji
stawiennego w uzależnieniu od opioidów. wątroby i depresjach – metylodopa jest przeciwwska-
Średnia pojedyncza dawka wynosi w przypad- zana.
ku klonidyny 0,075–0,3 mg, a moksonidyny 0,2–
0,4 mg.
Zależnymi od dawki działaniami niepożądanymi ago- 1.13.6.2. Rezerpina
nistów receptorów α2-adrenergicznych typu klonidy-
ny są: nadmierne uspokojenie (sedacja) oraz zahamo- Alkaloid rauwolfii – rezerpina – zmniejsza zdolność
wanie wydzielania śliny i śluzu (suchość w ustach), gromadzenia katecholamin w pęcherzykach synap-
które to objawy są słabsze w przypadku moksoni- tycznych. Hamuje działanie ATP-azy zależnej od jo-
MUTSCHLER-2009.indd 374 2010-01-07 22:12:42
Układ nerwowy 375
HO NH2 dawkach (0,05–0,1 mg) i to w terapii skojarzonej
z lekami moczopędnymi.
H3C COOH Zależne od dawki działania niepożądane rezerpiny
HO
– w przypadku stosowania małych dawek – są dobrze
Układ nerwowy
metylodopa tolerowane i polegają zasadniczo na objawach wy-
padowych ze strony układu współczulnego bądź też
OH z wtórnymi do nich objawami (względnego) zwięk-
HO NH2 szenia się napięcia w obrębie układu przywspółczul-
nego. I tak przy stosowaniu rezerpiny mogą ujawnić
CH3
HO się – podobnie jak w przypadku klonidyny – zaburze-
nia ortostatyczne, zawroty głowy, bradykardia oraz
α-metyloadrenalina
zaburzenia potencji. W przypadku długotrwałego sto- B1
sowania rezerpiny w dużych dawkach obserwowano
depresyjne obniżenia nastroju (z niebezpieczeństwem
nów magnezu Mg2+, która aktywnie pompuje protony
samobójstwa!) oraz objawy zespołu Parkinsona.
(H+) do wnętrza pęcherzyków i prowadzi do dużego
stężenia protonów w ich wnętrzu. W wyniku zablo-
kowania pompy protonowej przez rezerpinę – zasa-
dowe substancje (jak np. noradrenalina i dopamina)
nie mogą być więcej protonowane wewnątrzpęche- OCH3
rzykowo i dlatego też nie mogą być dalej gromadzo- OCH3
ne we wnętrzu tychże pęcherzyków pod postacią soli. H
O
Po krótkim czasie pęcherzyki przestają więc zawierać N OCH3
jakiekolwiek substancje neuroprzekaźnikowe, gdyż O
OCH3
z jednej strony do pęcherzyków nie przedostaje się NH
H H
COOCH3
dopamina – substancja prekursorowa dla noradre-
naliny, a z drugiej strony – zawarta w pęcherzykach H3CO
noradrenalina zgodnie z gradientem stężeń przenika
rezerpina
do cytoplazmy i mitochondriów, gdzie ulega rozkła-
dowi przez monoaminooksydazę.
U osób z nadciśnieniem rezerpina wywołuje utrzy-
mujące się długo obniżenie wartości ciśnienia krwi. Rezerpina jest przeciwwskazana w przypadku cho-
Ze względu na ujawniające się w trakcie stosowania roby wrzodowej żołądka lub dwunastnicy oraz astmy
dużych dawek znaczne działania niepożądane rezer- oskrzelowej – w związku z przewagą napięcia układu
pina stosowana jest obecnie jedynie w niewielkich przywspółczulnego.
MUTSCHLER-2009.indd 375 2010-01-07 22:12:42
376 Układ nerwowy
1.14. Substancje wpływające
na układ przywspółczulny
W wyniku pobudzenia układu przywspółczulnego receptory cholinergiczne = receptory M-choliner-
(parasympatycznego, cholinergicznego) wyzwalane giczne) rozmieszczone w narządach wykonawczych
są przede wszystkim reakcje o charakterze trofotro- i w ten sposób prowadzi do wywołania określonych
powym – służące restytucji organizmu. W tab.1.14-1. efektów biologicznych.
zestawione zostały skutki aktywacji układu przy-
współczulnego w obrębie różnych narządów. Uwalnianie, gromadzenie i rozkład acetylocho-
liny. Pobudzenie przywspółczulnych włókien ner-
Droga pobudzenia w układzie przywspółczul- wowych prowadzi do uwolnienia acetylocholiny.
nym. Wychodzące z ośrodkowego układu nerwo- Po uwolnieniu szybko ulega szybkiemu rozkładowi
wego włókna nerwowe ciągną się aż do zwojów przez (swoistą) acetylocholinoesterazę (hydrolazę
przywspółczulnych. Tam bodziec nerwowy przeka- acetylocholiny), która jest zlokalizowana w obrębie
zywany jest na neuron pozazwojowy – neuroprze- błony prze- i postsynaptycznej, do nieaktywnej choli-
kaźnikiem uczestniczącym w tym procesie jest ace- ny i kwasu octowego. Cholina za pomocą aktywnego
tylocholina. Pobudzenie neuronu pozazwojowego transportu ulega wchłonięciu zwrotnemu do wnętrza
również prowadzi do uwalniania acetylocholiny aksonu, z kolei kwas octowy ulega odtransportowa-
– tym razem w obrębie przywspółczulnych zakoń- niu wraz z krwią.
czeń nerwowych (zob. ryc. 1.14-1). W wyniku dy- Oprócz związanej z błoną komórkową swoistej
fuzji przez szczelinę synaptyczną acetylocholina acetylocholionesterazy – we krwi i w wątrobie znaj-
pobudza receptory przywspółczulne (muskrynowe duje się jeszcze nieswoista cholinoesteraza (pseudo-
cholinoesteraza, butyrylocholinoesteraza), za pomo-
cą której hydrolizie ulega nie tylko acetylocholina,
ale również inne estry choliny, np. chlorek suksa-
metonium. Funkcja niespecyficznej cholinoesterazy
ośrodkowy układ nerwowy polega prawdopodobnie na zapobieganiu działaniu
acetylocholiny w miejscu odległym od miejsca uwal-
niania.
Receptory acetylocholiny. Acetylocholina dzia-
neuron ła jako neuroprzekaźnik w synapsach ośrodkowego
przedzwojowy
układu nerwowego, w zwojach przywspółczulnych
i przywspółczulnych włóknach pozazwojowych.
acetylocholina
W dalszej kolejności służy również przekazywaniu
impulsów w zwojach współczulnych oraz w obrębie
płytki końcowej (nerwowo-mięśniowej) w mięśniach
zwój
przywspółczulny poprzecznie prążkowanych. Acetylocholina pobudza
przy tym receptory cholinergiczne typu nikotynowego
i muskarynowego.
W przypadku receptorów nikotynowych (recep-
neuron torów N-cholinergicznych) chodzi o kanały jonowe
pozazwojowy sterowane ligandami, które – jak to wyjaśnia ich na-
zwa – są pobudzane nie tylko przez acetylocholinę,
ale również nikotynę. Receptory tego typu występują
acetylocholina w neuronach ośrodkowego układu nerwowego i zwo-
jach oraz w płytce nerwowo-mięśniowej.
mięśnie gładkie skurcz Receptory muskarynowe (receptory M-choliner-
giczne) są natomiast receptorami sprzężonymi z biał-
gruczoły wydzielanie
kiem G, które występują w obrębie ośrodkowego
Ryc. B 1.14-1. Schemat przekaźnictwa w układzie przywspół- układu nerwowego, w zwojach i w synapsach przy-
czulnym. współczulnych i które mogą być pobudzone zarówno
MUTSCHLER-2009.indd 376 2010-01-07 22:12:42
Układ nerwowy 377
Tabela B 1.14-1. Wpływ pobudzenia układu przywspółczulne- Tabela B 1.14-1. (Kontynuacja)
go na różne narządy (zmodyf. według Goodmana i Gilmana)
Narząd Działanie układu
Narząd Działanie układu lub układ przywspółczulnego
Układ nerwowy
lub układ przywspółczulnego Nerki i drogi moczowe
Oczy wydzielanie reniny Ø
mięsień rozwieracz źrenicy Ø mięśnie ściany pęcherza skurcz
(m. dilatator pupillae)
zwieracz wewnętrzny zwiotczenie
mięsień zwieracz źrenicy zwężenie źrenic
(m. sphincter pupillae) Narządy płciowe
mięsień rzęskowy akomodacja do bliży macica Ø B1
Metabolizm
gruczoł łzowy wydzielanie ↑
wątroba (synteza glikogenu)
Serce
węzeł zatokowy częstotliwość serca ↓ komórki tłuszczowe Ø
mięśnie przedsionków kurczliwość ↓ mięśnie szkieletowe Ø
węzeł przedsionkowo-komorowy szybkość Ø brak działania
przewodzenia ↓
mięsień komór Ø przez acetylocholinę, jak i muskarynę. Istnieje przy
Naczynia tym 5 podtypów receptorów. Receptory M1 występują
skóra, błona śluzowa Ø
wyłącznie w strukturach neuronalnych (ośrodkowy
układ nerwowy, zwoje) i uczestniczą one w proce-
mięśnie szkieletowe Ø sach pamięci oraz uczenia się, jak również w prze-
jama brzuszna Ø kazywaniu impulsów w zwojach. Receptory M2 mają
funkcjonalne znaczenie głównie w sercu (obniżenie
naczynia wieńcowe Ø częstości pracy serca), a receptory M3 – w mięśniach
mózg Ø gładkich (skurcz) i gruczołach wydzielniczych (se-
krecja). Receptory M4 zostały stwierdzone między in-
narządy płciowe rozszerzenie naczyń nymi w przodomózgowiu, hipokampie i prążkowiu,
nerki Ø lecz ich znaczenie fizjologiczne jak dotąd nie zostało
jednoznacznie wyjaśnione. Prawdopodobnie uczest-
żyły Ø niczą one w zjawiskach związanych z bólem. W dal-
Przewód pokarmowy szym ciągu nieznane jest również fizjologiczne zna-
gruczoły ślinowe silne wydzielanie śliny surowiczej czenie występujących w ośrodkowym układzie ner-
wowym receptorów M5.
gruczoły trawienne zwiększenie wydzielania
drogi żółciowe skurcz Pre- i postsynaptyczne receptory muskarynowe.
Podobnie jak w przypadku innych synaps – również
pasaż/napięcie ↑
w przypadku przekaźnictwa synaptycznego oprócz
zwieracze zwiotczenie receptorów postsynaptycznych istnieją również re-
Trzustka ceptory presynaptyczne, których pobudzenie prowa-
wydzielanie endokrynne Ø dzi do zahamowania procesu uwalniania acetylocho-
liny. Wszystkie podtypy receptorów muskarynowych
Układ oskrzelowy
zostały stwierdzone również w obszarze presynap-
mięśniówka skurcz tycznym.
gruczoły zwiększenie wydzielania
Działanie acetylocholiny. Po podaniu dożylnym
Skóra
acetylocholiny występują następujące, bardzo krótko
gruczoły potowe Ø
utrzymujące się efekty (zob. tab. 1.14-1):
■ zwalnia się częstość pracy serca,
■ zmniejsza się opór obwodowy naczyń,
MUTSCHLER-2009.indd 377 2010-01-07 22:12:42
378 Układ nerwowy
■ zwiększa się wydzielanie śliny, soku żołądkowego zwiotczeniem naczyń. Ponadto w wyniku stymulacji
oraz śluzu w oskrzelach i potu, presynaptycznych heteroreceptorów acetylocholina
osłabia uwalnianie noradrenaliny.
■ nasila się napięcie mięśni gładkich w obrębie prze-
wodu pokarmowego, odprowadzających dróg mo-
czowych i oskrzeli,
1.14.1. Agoniści receptorów
■ zwężają się źrenice oczu, muskarynowych
■ oko ulega akomodacji do patrzenia na bliską od- (agoniści receptorów
ległość. M-cholinergicznych,
bezpośrednie
Działanie acetylocholiny polega na tym, że po zwią-
zaniu się z odpowiednim receptorem wpływa na prze-
parasympatykomimetyki)
puszczalność błony komórkowej dla jonów sodowych,
potasowych i wapniowych. W komórkach zwojowych Mimo pełnienia rozmaitych funkcji fizjologicznych
oraz motorycznej płytce końcowej acetylocholina acetylocholina nie ma – z powodu szybkiego rozkładu
zwiększa przepuszczalność dla jonów sodu znacząco – żadnego znaczenia terapeutycznego – poza zastoso-
silniej niż dla jonów potasu, czego skutkiem jest depo- waniem jako miotyk, tj. lek do zwężenia źrenic przed
laryzacja. W komórkach rozrusznikowych serca pro- operacjami okulistycznymi. Do celów terapeutycznych
wadzi natomiast do zwiększenia przepuszczalności jo- nadają się natomiast agoniści receptorów muskaryno-
nów potasu i prowadzi tym samym do hiperpolaryza- wych, które podobnie jak acetylocholina pobudzają
cji z następczym zwolnieniem czynności pracy serca. przywspółczulne receptory cholinergiczne, ale są wol-
W przypadku różnych komórek gruczołowych (np. gru- niej od niej inaktywowane (bezpośrednie parasympaty-
czołów ślinowych, rdzenia nadnerczy) i różnych na- komimetyki/cholinomimetyki = bezpośrednio działające
rządów zawierających mięśnie gładkie (np. pęcherz leki parasympatykomimetyczne/cholinomimetyczne, leki
moczowy, mięśniówka oskrzeli) acetylocholina nasila M-cholinomimetyczne; zob. tab. B 1.14-2).
dokomórkowy napływ jonów wapnia, co wzmaga se-
krecję lub też umożliwia skurcz. W obrębie nieuszko- Betanechol działa zasadniczo podobnie do acetylo-
dzonego śródbłonka naczyń krwionośnych acetylocho- choliny, choć jego działanie jest dłuższe, gdyż w or-
lina powoduje uwolnienie tlenku azotu z następczym ganizmie ulega powolnemu rozkładowi. Wskazany
Tabela B 1.14-2. Agoniści receptorów muskarynowych
Wzór strukturalny Nazwa Preparaty handlowe Okres pół- Średnia dawka
międzynarodowa trwania (godz.) dzienna (mg)
O karbachol Carbamann, 1,5-3 do worka
CH3 Carbachol, spojówkowego
+ –
H2N O N CH3 Cl Miostat,
CH3 Isopto-Carbachol
O CH3 betanechol Myocholme-Glenwood brak danych 50-200 doustnie
CH3
+ –
H2N O N CH3 Cl
CH3
H5C2 pilokarpina Pilokarpin ankerpharm, 1 1,5-6 do worka
Pilomann, Pilopos, spojówkowego;
O N Serscarpin, Salagen, 20 doustnie
N Pilocarpinum, Fotil,
O H3C Normoglaucon
CH3 muskaryna
HO O
CH3
+
N CH3
CH3
MUTSCHLER-2009.indd 378 2010-01-07 22:12:43
Układ nerwowy 379
jest do stosowania w przypadku (pooperacyjnej) ato- mimetyczne) wyróżnia się oprócz opisanych w roz-
nii jelit i pęcherza. dziale B 2.5.2 leków przeciwotępiennych:
Jako objawy niepożądane – będące wyrazem
■ pochodne kwasu karbaminowego (grupa fizostyg-
wzmożonego napięcia w układzie przywspółczulnym
miny; zob. tab. B 1.14-3),
Układ nerwowy
i które mogą być łatwo zniesione dzięki dożylnej in-
iekcji atropiny, mogą ujawnić się: zlewne poty, nasi- ■ estry kwasu fosforowego (alkilowane fosforany,
lony ślinotok, nudności, wymioty i biegunka. związki fosforoorganiczne; zob. poniżej).
W przypadku niewydolności serca, dusznicy boles-
nej, astmy oskrzelowej, nadczynności tarczycy (nie- Ich mechanizm działania polega na tym, że wcho-
bezpieczeństwo migotania przedsionków i komór), dzą one w reakcję z centrum esterazowym acetylo-
owrzodzeń peptycznych, hiperaktywnej mięśniówki cholinoesterazy – prowadząc do powstania estrów
pęcherza moczowego i w chorobie Parkinsona – be- karbaminowych lub fosforanowych tego enzymu, B1
tanechol jest przeciwwskazany. czego skutkiem jest zahamowanie rozkładu acetylo-
choliny (zob. ryc. B 1.14-2).
Karbachol – ester kwasu karbamidowego oraz pi-
lokarpina – główny alkaloid liści krzewu potoślinu
(Pilocarpus jaborandi) stosowane są miejscowo jako centrum centrum
leki przeciwko jaskrze. Ponadto pilokarpina – stoso- anionowe esterazowe
wana ogólnie – służy do zwiększenia wydzielania łez
i śliny w zespole Sjögrena oraz do łagodzenia dolegli- CH2
wości w niedoczynności gruczołów ślinowych po na- OH
świetlaniach okolicy głowy i szyi (preparat Salagen;
dawkowanie 4 razy dziennie po 5 mg). CH3 CH3
H3C +
Arekolina – terapeutycznie niestosowany główny alka- N O N
CH3
loid nasienia orzecha areki (Areca catechu) – działa mu- H3C
skarynergicznie, a dodatkowo wykazuje słabe działanie O
nikotynopodobne. Ujawniające się w trakcie żucia betelu
(rozdrobniony orzech) objawy pobudzenia ośrodkowego
układu nerwowego należy wiązać głównie z działaniem zablokowany enzym
arekaidyny – produktu hydrolizy arekoliny.
Muskaryna została po raz pierwszy wyizolowana z mucho-
CH2 CH3
mora czerwonego, gdzie występuje jedynie w znikomych
ilościach. Znacząco większe stężenia muskaryny zawierają O N
CH3
różne gatunki strzępiaków (Inocybe). Również muskaryna
nie znalazła klinicznego zastosowania. Okazała się jednak O
CH3
cenną substancją w farmakologii eksperymentalnej. H3C +
N OH
H3C
1.14.2. Inhibitory cholinoesterazy
(pośrednie Ryc. B 1.14-2. Blokada acetylocholinoesterazy przez neostyg-
parasympatykomimetyki) minę (schemat).
Do utrzymania prawidłowego napięcia mięśni gładkich Podczas gdy hydroliza i związana z nią regeneracja enzy-
i poprzecznie prążkowanych służy acetylocholina, która mu w przypadku pochodnych kwasu karbaminowego prze-
jest wydzielenia w zwojach, neuronach pozazwojowych biega względnie szybko – przykładowo enzym dimetylo-
i motorycznych płytkach końcowych pod wpływem ośrod- karbamoilowany jest regenerowany w czasie połowiczym
kowych pobudzeń, a wkrótce potem ulega procesowi hy- około 25 minut – enzym sfosforylyzowany ulega bardzo
drolizy pod wpływem acetylocholinoesterazy. W wyniku wolnemu rozkładowi. Stąd też pochodne kwasu karbami-
zahamowania aktywności tego enzymu za pomocą inhi- nowego określa się mianem odwracalnych, a estry kwasu
bitorów acetylocholinoesterazy zwiększa się stężenie ace- fosforowego – nieodwracalnych inhibitorów acetylocholi-
tylocholiny – prowadząc do wzmożenia napięcia układu noesterazy.
przywspółczulnego oraz napięcia mięśni poprzecznie prąż-
kowanych.
1.14.2.1 Pochodne kwasu karbaminowego
Wśród inhibitorów cholinoesterazy (pośrednie para-
sympatykomimetyki/cholinomimetyki = pośrednio Poszczególne pochodne kwasu karbaminowego są ze-
działające leki parasympatykomimetyczne/cholino- stawione łącznie w tab. B 1.14-3.
MUTSCHLER-2009.indd 379 2010-01-07 22:12:44
380 Układ nerwowy
Tabela B 1.14-3. Pośrednie parasympatykomimetyki (pochodne kwasu karbaminowego)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
H fizostygmina (eseryna) Physostigminum 0,25 2-10 i.v., i.m.
H3C
N O salicylicum,
H3C N Anticholium
O CH3
N H
CH3
bromek neostygminy NEOSTIG, 0,5 mg Carino, 0,5-1 0,5-2 i.v.
CH3 CH3 Neostigmin DeltaSelect,
+ CH3 – Neostigmin-Rotexmedica,
N O N Br
H3C CH3 Polstigminum
O
bromek pirydostygminy Kalymin, 1,5 30-720 doustnie,
CH3 Mestinon 1,5 i.m., s.c.
–
N O + CH3 Br
H3C N
O
bromek distygminy Ubretid 65-69 5-10 doustnie,
0,5-1 i.m.
O CH3
+ N O
H3C
N + CH3 –
O N N 2 Br
CH3 O
Fizostygmina – główny alkaloid bobu kalabarskiego Bromek distygminy – substancja podwójnie czwar-
(Physostigma venenosum) i główna substancja po- torzędowa – wykazuje działanie podobne do wyżej
chodnych kwasu karbaminowego – obecnie znajduje wymienionych leków.
zastosowanie wyłącznie jako antidotum w zatruciach
związkami o działaniu parasympatykolitycznym
(Anticholum). Ze względu na dobrą przenikalność 1.14.2.2. Estry kwasy fosforowego
do ośrodkowego układu nerwowego – w przypad-
ku zbyt szybko wykonanej iniekcji – może dojść Substancje z tej grupy zostały omówione w rozdziale
do uogólnionych napadów drgawkowych. Ponadto C 3.12.2.
może wystąpić bradykardia.
Bromek neostygminy – jako czwartorzędowy zwią-
zek amoniowy nie jest w stanie przeniknąć bariery
krew-mózg – wskazany jest natomiast do stosowania 1.14.3. Antagoniści receptorów
w przypadku atonii jelit i pęcherza moczowego, ja- M-cholinergicznych
skrze i miastenii (miasthenia gravis pseudoparaliti- (bezpośrednie
ca). Ponadto służy do znoszenia działania stabilizują- parasympatykolityki,
cych środków zwiotczających mięśnie. spazmolityki neurotropowe)
Działania niepożądane i przeciwwskazania odpo-
wiadają tym występującym w trakcie stosowania be-
Antagoniści muskarynowych receptorów choliner-
tanecholu.
gicznych (receptorów M-cholinergicznych; para-
Bromek pirydostygminy jest wprawdzie słabszy niż sympatykolityki, cholinolityki; zob. tab. 1.14-4)
neostygmina, wyróżnia się jednak równomiernym blokują – na skutek kompetycyjnego antagonizmu
ujawnianiem się efektów działania, dłuższym czasem – przekaźnictwo pobudzeń wykorzystujące acetylo-
działania i szerszym zakresem terapeutycznym. cholinę w obrębie receptorów M-cholinergicznych.
MUTSCHLER-2009.indd 380 2010-01-07 22:12:44
Układ nerwowy 381
Tabela B 1.14-4. Antagoniści receptorów M-cholinergicznych
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
Układ nerwowy
I. Alkaloidy belladonny i substancje pokrewne
H3C atropina Atropin EDO, 12-30 0,5-3 doustnie,
N Atropin-POS, i.v., do worka
Atropinsulfat-100 mg, spojówkowego;
H Dysurgal, w zatruciach
CH2OH Atropinum sulfuricum, – w zależności
Reasec, od objawów
O
Tolargin B1
O
bromek BS-ratiopharm 5 30-60 doustnie,
H3C(CH2) 3 + CH3 N-butyloskopolaminy Buscopan, 20-40 (-100)
N Butylscopolamin- i.m., s.c., i.v.
O /Rotexmedica,
H – Spasman scop Buscolysin,
CH2OH Br Scopolan
O
H3C + CH(CH3) 2 bromek ipratropium Atrovent, 2-4 0,6-1,6
N Berodual, do inhalacji,
Itrop, 20-45 doustnie,
Steri-Neb Ipratropium 0,5 i.v.
H –
CH2OH Br
O
H3C skopolamina Boro-Scopol N, 4 1,5 plaster,
N
Scopoderm TTS 0,2 do worka
O spojówkowego
H
CH2OH
O
H3C + CH3 bromek tiotropium Spiriva 120-150 0,018 do inhalacji
N
O
–
H Br
HO S
O S
chlorek trospium Spasmex, 6-18 5-40 doustnie
+ Spasmolyt,
N Spasmo-Urgenin TC,
Trospi
–
H Cl
HO
O
MUTSCHLER-2009.indd 381 2010-01-07 22:12:44
382 Układ nerwowy
Tabela B 1.14-4. Antagoniści receptorów M-cholinergicznych (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna (mg)
II. Leki parasympatykolityczne o różnej budowie chemicznej
bromek glikopyronium Robinul 1 0,2-0,4 i.v., i.m.
HO
–
H3C + O Br
N
H3C
O
tolterodyna Detrusitol, Uroflow 6 2-4 doustnie
OH
(H3C) 2HC
N
CH(CH3) 2
CH3
O daryfenacyna Emselex 12 7,5-15 doustnie
O
NH2
solifenacyna Vesikur, 45-68 5-10 doustnie
Vesicare
O
N O N
N
tropikamid Mydriaticum Stulln, 0,25-1,5 do worka
Mydrum, spojówkowego
CH2OH
Tropicamidum,
N
H5C2
O
Z powodu zwiotczenia/rozkurczu mięśni gładkich moczowodów i pęcherza moczowego (szczególnie
– wyzwolonego działaniem na układ przywspółczul- jeżeli wcześniej była spastycznie skurczona); po-
ny – substancje te zwane są również spazmolitykami przez porażenie mięśnia zwieracza źrenicy (muscu-
neurotropowymi. lus sphincter pupillae) – źrenice ulegają rozszerzeniu
– czego skutkiem jest światłowstręt oraz zwiększenie
Działanie. W wyniku blokady receptorów muskary- ciśnienia śródgałkowego (w wyniku zablokowania
nowych występujących w różnych narządach – ujaw- odpływu cieczy wodnistej); ponadto zaburzeniu ule-
niają się następujące działania: ośrodkowe neurony ga akomodacja – w wyniku porażenia mięśnia rzęs-
ulegają pobudzeniu lub zahamowaniu, przyspieszeniu kowego (musculus ciliaris).
ulega częstość pracy serca (w większych dawkach),
zmniejsza się wydzielanie łez, śliny i potu oraz sekre- Kinetyka. Leki parasympatykolityczne z trzeciorzę-
cja gruczołów układu pokarmowego, zmniejsza się dowym azotem (np. atropina, zob. poniżej) po podaży
wydzielanie śluzu w górnym i dolnym odcinku dróg doustnej ulegają pełnemu i szybkiemu wchłanianiu.
oddechowych. Zwiotczeniu ulega gładka mięśniówka W przypadku czwartorzędowych związków amonio-
oskrzeli, przewodu pokarmowego, dróg żółciowych, wych (np. bromek ipratropium; zob. poniżej) wchła-
MUTSCHLER-2009.indd 382 2010-01-07 22:12:45
Układ nerwowy 383
nianie leków jest natomiast niewielkie. Dla uzyskania ni gładkich przewodu pokarmowego jest działaniem
odpowiedniego działania konieczne jest więc poda- niepożądanym. Wyzwolone działaniem antagonistów
nie leków na drodze pozajelitowej lub też stosowanie receptorów M-cholinergicznych zwiększenie często-
dawek doustnych znacznie większych od dawek po- ści pracy serca jest pożądanym efektem w przypadku
Układ nerwowy
dawanych dożylnie. Zaletą jest, że związki czwarto- pacjentów z bradykardią, a równocześnie objawem
rzędowe nie potrafią przeniknąć bariery krew-mózg niepożądanym u pacjentów z dolegliwościami spa-
i dlatego też – w porównaniu z lekami parasympaty- stycznymi w obrębie górnej części jamy brzusznej.
kolitycznymi z trzeciorzędowym azotem – są ośrod-
kowo nieskuteczne. Przeciwwskazania. W jaskrze i łagodnym przeroście
gruczołu krokowego – leki parasympatykolityczne
Wskazania. Stosowanie antagonistów receptorów są przeciwwskazane. W przypadku miażdżycy naczyń
M-cholinergicznych jest wskazane: w skurczach sercowych powinny być podawane w dawkach, które B1
mięśniówki gładkiej przewodu pokarmowego, dróg jedynie nieznacznie zwiększają częstość pracy serca.
żółciowych i moczowych oraz w obszarze żeńskich
narządów płciowych (np. w spastycznych zaparciach, Interakcje. Amantadyna, chinidyna, disopiramid, leki
skurczu odźwiernika, kolkach w przebiegu kamicy neuroleptyczne i trójcykliczne leki przeciwdepre-
żółciowej i nerkowej, nadpobudliwym pęcherzu mo- syjne nasilają działanie antagonistów receptorów
czowym; ciężkich zaburzeniach miesiączkowania), M-cholinergicznych.
w zaburzeniach rytmu serca z bradykardią (brady-
arytmie), do wyłączenia odruchów z nerwu błędnego,
jak również dla zmniejszenia wydzielania śluzu w dro-
1.14.3.1. Alkaloidy belladonny
gach oddechowych w trakcie znieczulenia ogólnego,
w zespole Parkinsona – w celu zmniejszenia nasile-
i pokrewne aminy trzeciorzędowe
nia objawów pozytywnych oraz przed badaniem dna
oka, a także w zapaleniu tęczówki i ciała rzęskowego Alkaloid estrowy – atropina jest po podaniu doust-
oraz zapaleniu błony naczyniowej oka – jagodówki nym jako amina trzeciorzędowa dobrze wchłaniana
(uveitis) – w celu zwiększenia szerokości źrenic. i szybko ulega rozmieszczeniu po całym organizmie.
Jako preparat doustny stosowana jest przeciwko kol-
Niektóre związki czwartorzędowe są w dalszej ko- kom przewodu pokarmowego oraz dróg żółciowych
lejności stosowane jako leki rozszerzające oskrzela. i moczowych. Jako preparat do podawania dożylnego
Pomimo że substancje wykazujące powinowactwo stosowana jest również w premedykacji przed znie-
do receptorów muskarynowych typu M1 nadają się czuleniem ogólnym oraz w krótkotrwałej terapii bra-
do wykorzystania w terapii choroby wrzodowej – zo- dyarytmii.
stały wyparte przez lepiej działające leki. Antagoni- Zatrucia atropiną występują głównie po spożyciu
ści receptorów M-cholinergicznych służą ponadto pokrzyku wilczej jagody (Atropa belladonna) lub na-
do opanowania przywspółczulnych działań niepożąda- sion bielunia dziędzierzawy (Dathura stramonium),
nych występujących po podaniu morfiny lub też – jak a tylko bardzo rzadko w wyniku przypadkowego
to już podano – w terapii miastenii łącznie z inhibitora- zażycia leków zawierających atropinę (np. kropli
mi cholinoesterazy. Atropina (zob. poniżej) jest ponad- do oczu). Charakterystycznymi objawami zatrucia
to najważniejszym i często ratującym życie antidotum są: zaczerwienienie twarzy, suchość śluzówek, trud-
w zatruciach estrami kwasu fosforowego (związkami ności w połykaniu, przyspieszona akcja serca (puls)
fosfoorganicznymi). i bardzo szeroko otwarte źrenice. W dużych dawkach
ujawniają się dodatkowo hipertermia (w wyniku zaha-
Działania niepożądane. W zależności od wskazań mowania wydzielania potu), stany pobudzenia, oma-
– działania pożądane i niepożądane antagonistów my i drgawki kloniczne, po których następuje stan
receptorów M-cholinergicznych są odmienne. Jeżeli głębokiej utraty przytomności. Może dojść do zejścia
wykorzystuje się te substancje jako leki rozkurczowe śmiertelnego – w wyniku ośrodkowego porażenia od-
(spazmolityczne) – to rozszerzenie źrenic i zmniej- dechu. W przypadku wystarczająco wczesnego pod-
szenie wydzielania śluzu będzie się określało mianem jęcia leczenia rokowanie nawet w przypadku ciężkich
działania niepożądanego. Kiedy jednak stosowane zatruć jest dobre. Terapia składa się z zastosowania
są jako leki rozszerzające źrenicę (mydriatyczne) preparatów zapobiegających wchłanianiu oraz fizy-
– wówczas wywołany objaw mydriazy jest głównym kalnych, niefarmakologicznych metod obniżania cie-
pożądanym efektem, a m.in. występujące po wchło- płoty ciała i sztucznego oddychania w przypadku za-
nięciu leku z worka spojówkowego zwiotczenie mięś- grażającego porażenia oddechu oraz domięśniowego
MUTSCHLER-2009.indd 383 2010-01-07 22:12:46
384 Układ nerwowy
podania salicylanu fizostygminy (zob. powyżej) jako 1.14.3.3. Trzecirzędowe
antidotum, a także podania diazepamu w przypadku parasympatykolityki
stanów pobudzenia i drgawek.
Tropikamid nadaje się do diagnostycznego rozsze-
Skopolamina różni się farmakologicznie od atropi-
rzenia źrenicy oraz do porażenia akomodacji przy
ny jedynie tym, że wykazuje ośrodkowe działanie
badaniu refrakcji.
hamujące. W celach terapeutycznych stosowana
jest w kroplach do oczu, jak również pod postacią
Pirenzepina – antagonista działający głównie na re-
przezskórnych systemów terapeutycznych – jako
ceptory muskarynowe typu M1 – jest omówiona jako
preparat przeciwwymiotny w przebiegu kinetoz, tj.
lek stosowany w terapii choroby wrzodowej.
choroby lokomocyjnej.
Tolterodyna, darifenacyna i solifenacyna są wska-
W przypadku rzadko występujących zatruć skopo-
zane do stosowania – podobnie jak chlorek trospium
laminą ujawniają się oprócz typowych objawów za-
– w zaburzeniach wypróżniania pęcherza moczowe-
blokowania układu przywspółczulnego (rozszerzenie
go, przede wszystkim w nietrzymaniu moczu wywo-
źrenic, suchość śluzówek itp.) – głębokie zaburzenia
łanym naglącym parciem.
świadomości. Śmierć następuje w wyniku ośrodko-
wego porażenia oddechu. Terapia jest podobna do tej Obie na końcu wymienione substancje działające posiadają
po zatruciu atropiną. pewną selektywność względem receptorów typu M3, mimo
to brak jest przekonujących danych, że zwiększa to ich to-
lerancję. (Receptory M3 mięśni gładkich i gruczołów śli-
nowych są identyczne). Również preferencyjna blokada
1.14.3.2. Czwartorzędowe receptora M3 w terapii nadaktywności mięśnia wypieracza
parasympatykolityki moczu wydaje się mało przekonująca, gdyż oprócz recep-
torów M3 także receptory M2 hamują poprzez zmniejszoną
Bromek glikopyronium stosowany jest w aneste- syntezę cAMP rozkurczowy wpływ układu współczulnego
zjologii do zmniejszenia wydzielania śliny, sekrecji – przyczyniając się do skurczu wypieracza.
śluzu w układzie oskrzelowym oraz do zablokowa-
nia sercowych odruchów ze strony nerwu błędnego
i związanej z nimi bradyarytmii przy wprowadzaniu 1.14.3.4. Muskulotropowe i neurotropowo-
do znieczulenia ogólnego.
-muskulotropowe
Chlorek trospium służy w leczeniu nietrzymania
leki spazmolityczne
moczu wywołanego naglącym parciem i odruchowe-
go nietrzymania z nadaktywnością mięśnia wypiera- Niezależnie od unerwienia wegetatywnego – mięś-
cza moczu. nie gładkie mogą ulec zwiotczeniu w wyniku bez-
pośredniego działania na komórki mięśni gładkich.
Bromek butyloskopolaminy wskazany jest w skur- W ten sposób działające substancje określane są mia-
czach mięśni gładkich – przede wszystkim układu nem leków spazmolitycznych muskulotropowych,
pokarmowego i żeńskich narządów płciowych, jak a ze względu na alkaloid opioidowy – papawerynę
również w celu ułatwienia endoskopowego badania – są nazywane również spazmolitykami papaweryno-
żołądka i jelit. podobnymi (typu papaweryny) (zob. tab. B 1.14-5).
Substancje, które posiadają również składową neuro-
Bromek ipratropium i bromek tiotropium są stoso- tropową (antycholinergiczną = cholinolityczną), zali-
wane w przebiegu obturacyjnej choroby płuc, zwłasz- czane są do leków spazmolitycznych neurotropowo-
cza POChP (przewlekłej obturacyjnej choroby płuc). -muskulotropowych.
Bromek ipratropium używany jest ponadto w przy-
padku bradyarytmii. Papaweryna – występująca w 1% w opium – pro-
wadzi do zwiotczenia wszystkich mięśni gładkich,
Zaletą parasympatykolityków czwartorzędowych tzn. działaniem dotknięta jest w podobnym stopniu
w porównaniu z trzeciorzędowymi parasympaty- mięśniówka naczyń krwionośnych jak np. mięśniów-
kolitykami (zob. poniżej) jest to, że nie wywołują ka oskrzeli lub przewodu pokarmowego. Działanie
one żadnych ośrodkowych działań niepożądanych; to jest szczególnie wyraźne w przypadku wcześniej
ich wadą jest natomiast że gorsze wchłanianie przy zwiększonego napięcia mięśniowego. Jej stosowanie
zażyciu doustnym. jest jednak obecnie przestarzałe.
MUTSCHLER-2009.indd 384 2010-01-07 22:12:46
Układ nerwowy 385
Działający podobnie jak papaweryna – hymekro- moczowo-płciowych, mebeweryny – zespół jelita
mon służy w terapii funkcjonalnych dolegliwości drażliwego, a oksybutyniny i propiweryny – zaburze-
gastrycznych. nia opróżniania pęcherza moczowego (nadczynność
mięśnia wypieracza, moczenie nocne, częste odda-
Układ nerwowy
Do leków spazmolitycznych o działaniu neurotropo- wanie moczu).
wo-muskulotropowym należą: danaweryna, mebe- Wśród działań niepożądanych obserwuje się m.in.
weryna, oksybutynina oraz propiweryna. sporadycznie suchość w ustach, zawroty bóle głowy,
Głównym wskazaniem do zastosowania dena- nudności, zgagę i zaparcia, jak również reakcje aler-
weryny są skurcze przewodu pokarmowego i dróg giczne.
B1
Tabela B 1.14-5. Leki spazmolityczne o działaniu muskulotropowym i neurotropowo-muskulotropowym
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
między- półtrwania dawka
narodowa (godz.) dzienna (mg)
H3CO papaweryna Biosmasmil, 0,5-2 –
Papaverinum
N hydrochloricum,
H3CO Spasticol,
OCH3 Spazmogen,
Theopaverin,
Tolargin
OCH3
CH3 denaweryna Spasmalgan 30 20-200
O
N
O CH3
C2H5
O
C2H5
CH3 propiweryna Mictonetten, 14-20 15-45
N Mictonorm
O
O
OC3H7
O
oksybutynina Ditropan, 2 7,5-15
Driptane,
C2H5 Dridase,
O C
OH C N Oxybutynin-ratiopharm,
C2H5 Oxymedin,
Spasyt,
Uroton
O C2H5 mebeweryna Duspatal, brak danych 400
H3CO N Duspatalin,
O Mebemerck
CH3
H3CO OCH3
CH3 hymekromon Cholspasmin forte, 1 1200
Chol-Spasmoletten,
Cholestil
HO O O
MUTSCHLER-2009.indd 385 2010-01-07 22:12:46
MUTSCHLER-2009.indd 386 2010-01-07 22:12:46
Układ hormonalny 387
2. Hormony i leki wpływające
na funkcjonowanie układu
wewnątrzwydzielniczego
Zarówno w organizmach zwierzęcych, jak i ludzkich do potrzeb organizmu wytwarzania hormonów, a tak-
za utrzymanie stałości środowiska wewnętrznego że stałego kontrolowania wydzielania i unieczynnia-
Układ hormonalny
organizmu, poza wegetatywnym układem nerwo- nia. Zarówno nadmierne, jak i zbyt małe wytwarzanie
wym, odpowiada także układ hormonalny. W ukła- hormonów prowadzi do rozwoju chorób, które opi-
dzie nerwowym informacje przenoszone są wzdłuż sano w podrozdziałach omawiających poszczególne
włókien nerwowych łączących się poprzez synapsy gruczoły dokrewne.
przekazujące impuls pomiędzy neuronami na drodze Na podstawie mechanizmu wydzielania oraz za-
chemicznej oraz – rzadziej – elektrycznej. Natomiast kresu przekazywania informacji działanie hormonów
układ hormonalny można porównać z bezprzewodo- można podzielić na:
wym systemem komunikacyjnym (zob. ryc. B 2-1).
Treść informacji w tym przypadku jest zakodowana
■ endokrynne, B2
w strukturze specjalnych substancji wytwarzanych ■ neuroendokrynne,
i wydzielanych przez wyspecjalizowane (wewnątrz-
■ parakrynne,
wydzielnicze) komórki gruczołowe. Substancje te
zwane hormonami wywołują swoiste działanie ■ autokrynne.
w komórkach docelowych.
Podczas gdy układ nerwowy służy przede wszyst- Gdy hormony są wydzielane przez komórki gruczo-
kim do szybkiego i ukierunkowanego przekazywania łowe do krwiobiegu i wywołują działanie daleko
informacji, zadaniem układu hormonalnego jest głów- od miejsca wytworzenia, mówi się o wydzielaniu
nie długotrwałe i globalne sterowanie funkcjami ko- wewnętrznym lub w węższym znaczeniu o działa-
mórek. Zadanie to wymaga ciągłego, dopasowanego niu hormonalnym (endokrynnym). Termin wydziela-
synaptyczne endokrynne neuroendokrynne parakrynne autokrynne
neuroprzekaźnik hormon
Ryc. B 2-1. Przekazywanie sygnałów pomiędzy komórkami oraz autostymaulacja komórek (zmodyf. wg Zieglera).
MUTSCHLER-2009.indd 387 2010-01-07 22:12:46
388 Układ hormonalny
nie neuroendokrynne oznacza uwalnianie hormonu
z zakończeń nerwowych do krwiobiegu. Jeśli hormon
wpływa na sąsiadujące komórki, wtedy mówi się podwzgórze
o wydzielaniu lub działaniu parakrynnym. Efekt au- przysadka
tokrynny polega na regulowaniu własnych funkcji ko-
mórki poprzez uwalniane przez nią substancje czynne
(zob. ryc. B 2-1).
Niektóre hormony wykazują zarówno efekty endokrynne, tarczyca
jak i parakrynne. Przykładem mogą być hormony przed- i przytarczyce
niego płata przysadki, które poza działaniem endokrynnym
mają także działanie parakrynne, tj. wpływają na wydzie- prawy przedsionek
lanie hormonów przez sąsiadujące komórki. Równoczesne serca
działanie parakrynne i autokrynne wykazuje np. wytwarza-
na przez chondrocyty somatomedyna C. Poza tym stwier- nadnercza
dzono, że pewne hormony pełnią również funkcję neuro- trzustka
przekaźników, np. niektóre peptydowe hormony wpływają- nerki
ce na perystaltykę jelit. Złożoność i wzajemne powiązania
pomiędzy różnymi drogami przekazywnia sygnału unie-
możliwiają jednoznaczne rozgraniczenie i zaklasyfikowa-
nie biologicznych substancji przekaźnikowych.
Chemiczna struktura hormonów. Według budowy
chemicznej hormony można podzielić na: gruczoły płciowe
(jądra)
■ hormony peptydowe lub białkowe (hormony pod-
wzgórza, przysadki, przytarczyc, grasicy, wysp
trzustkowych Langerhansa, przewodu pokarmowe- Ryc. B 2-2. Lokalizacja gruczołów dokrewnych (według
go, wątroby, nerek i przedsionków serca), Benninghoffa-Goerttlera).
■ hormony steroidowe (hormony nadnerczowe i hor-
mony płciowe),
aminokwasów. Przykładem takiego hormonu jest będąca
■ analogi hormonów steroidowych (hormony z grupy glikoproteiną erytropoetyna. Przyłączenie reszt glukozo-
witaminy D) oraz wych do łańcucha peptydowego warunkuje jej działanie
hormonalne oraz wydłuża okres półtrwania.
■ hormony będące pochodnymi aminokwasów: tyro-
zyny (hormony rdzenia nadnerczy i gruczołu tar-
Biosyntezę hormonów steroidowych, hormonów
czowego) i tryptofanu (melatonina).
nadnerczy i gruczołu tarczowego oraz retinoidów
opisano w odpowiednich podrozdziałach.
Poza tym do hormonów są zaliczane pochodne reti-
noidów (witaminy A) oraz nienasycone kwasy tłusz-
Miejsca wytwarzania hormonów. Większość hor-
czowe.
monów jest syntetyzowana w gruczołach pochodze-
nia nabłonkowego, które wydzielają je poprzez pory
Synteza hormonów. Wytwarzanie hormonów pepty-
w śródbłonku włośniczek do krwiobiegu i dlatego
dowych i białkowych, tak jak innych białek, zachodzi
są one określane jako gruczoły wewnątrzwydzielni-
na drodze transkrypcji i translacji. Najpierw jest syn-
cze (endokrynne). Hormony są także syntetyzowane
tetyzowana cząsteczka prekursorowa, tzw. prepro-
w wyspecjalizowanych grupach komórek lub poje-
hormon, z którego powstaje prohormon przez od-
dynczych komórkach narządów, które spełniają prze-
szczepienie peptydu częściowego. Preprohormon lub
de wszystkim inne funkcje.
prohormon zawierają jako swoją częściową sekwen-
cję czynny hormon, który później zostaje uwolniony
Do znanych od wielu lat gruczołów wewnątrzwy-
w wyniku biohydrolizy. W niektórych wypadkach
dzielniczych (zob. ryc. B 2-2) należą:
podczas tej modyfikacji potranslacyjnej powstaje kil-
ka hormonalnie aktywnych produktów. ■ podwzgórze,
Poza tym istotne znaczenie dla aktywności hormonów może ■ przysadka,
mieć glikozylacja prohormonów powstałych po połączeniu ■ tarczyca,
MUTSCHLER-2009.indd 388 2010-01-07 22:12:47
Układ hormonalny 389
■ przytarczyce, stępuje według różnych, dłużej trwających rytmów.
W odróżnieniu od tego rytm wydzielania hormonu
■ grasica,
uwalniającego gonadotropinę z podwzgórza jest wy-
■ wyspy trzustkowe Langerhansa, raźnie krótszy (jest on uwalniany mniej więcej co 90
minut – wydzielanie pulsacyjne).
■ nadnercza,
Niektóre horomony są natomiast wydziela-
■ komórki śródmiąższowe (Leydiga) i komórki Serto- ne w sposób ciągły i przez to ich stężenie we krwi
lego w jądrach, jest utrzymywane w dużej mierze na stałym poziomie.
Typowymi przedstawicielami tej grupy są hormony
■ pęcherzyki jajnikowe i ciałka żółte,
Układ hormonalny
tarczycy.
■ łożysko u kobiet ciężarnych.
Transport hormonów. Transport hydrofilnych hor-
Hormony są również syntetyzowane i wydzielane monów jest możliwy w wolnej postaci, natomiast
przez: nadwzgórze, przewód pokarmowy, wątrobę, hormony słabo rozpuszczalne w wodzie występują
nerki i przedsionki serca. we krwi związane ze specyficznymi białkami trans-
W niektórych wypadkach hormony nie są wytwa- portującymi. Na przykład hormony tarczycy są trans-
rzane i wydzielane w tym samym miejscu. Przykłado- portowane przez globulinę wiążącą tyroksynę (TBG)
wo, synteza niektórych hormonów magazynowanych i prealbuminę wiążącą tyroksynę (TBPA), glukokor- B2
i wydzielanych przez przysadkę zachodzi w pobli- tykosteroidy przez transkortynę, a androgeny przez
skim podwzgórzu. białko wiążące androgeny (ABP). Białka transportu-
jące poszczególne hormony omówiono w odpowied-
W przebiegu niektórych chorób może dojść do ektopowe- nich podrozdziałach.
go wytwarzania hormonu, tj. do (niekontrolowanej) synte-
zy hormonu w nieendokrynnych tkankach wskutek zmian
związanych z nowotworzeniem. W trakcie różnicowania Inaktywacja hormonów. Hormony mogą pełnić
się komórek nowotworych nastąpuje wówczas reaktywacja funkcję nośników informacji dzięki temu, że działa-
wyłączonych genów odpowiedzialnych za syntezę hormo- ją one tylko przez ograniczony czas oraz nie kumu-
nów. Przykładem mogą być komórki raka drobnokomórko- lują się w narządach docelowych. Jest to możliwe
wego płuca, które dość często wytwarzają kortykotropinę. dzięki biotransformacji w narządzie efektorowym
Dolegliwości wywołane przez nadmierną syntezę hormo-
nów mogą wyprzedzać pojawienie się objawów związa- lub innych organach, przede wszystkim w wątrobie,
nych z miejscowym wzrostem guza. ale również w płucach lub nerkach. W niektórych wy-
padkach działanie hormonów zostaje zniesione także
przez wychwyt zwrotny do komórek (np. adrenaliny)
Magazynowanie i uwalnianie hormonów. Ana-
lub uwalnianie hormonów antagonistycznych.
logicznie do neuroprzekaźników hormony peptydo-
we są magazynowane w ziarnistościach, z których
Regulacja hormonalna. Reakcje zachodzące za po-
są wydzielane na drodze egzocytozy pod wpływem
średnictwem hormonów przebiegają wielotorowo we-
odpowiedniego bodźca. Hormony gruczołu tarczo-
dług jednolitego schematu, który charakteryzuje się
wego są magazynowane w tzw. koloidzie. Natomiast
trzystopniową strukturą hierarchiczną (zob. ryc. B
hormony steroidowe nie są magazynowane w gru-
2-3 B). Centralnym narządem regulacji jest podwzgó-
czołach i są one zazwyczaj uwalniane do krwiobiegu
rze. Wytworzony tam hormon uwalniający (libery-
bezpośrednio po zsyntetyzowaniu.
na) wywołuje syntezę i uwalnianie innego hormonu
w przysadce. Z kolei ten hormon wywiera wpływ
Wydzielanie hormonów. Niektóre hormony są uwal-
na obwodowy gruczoł wewnątrzwydzielniczy i dla-
niane według potrzeb w zależności od stanu metabo-
tego jest nazywany hormonem gruczołotropowym
licznego organizmu albo w odpowiedzi na określo-
(lub skrótowo tropowym). Hormon tropowy pobudza
ne wewnętrzne lub zewnętrzne bodźce. Zalicza się
w obwodowym gruczole wytwarzanie i wydzielanie
do nich m.in. wazopresynę, insulinę, aldosteron i ad-
hormonu efektorowego, który z krwiobiegiem ulega
renalinę.
dystrybucji w organizmie i wyzwala pożądaną reak-
W przypadku glukokortykosteroidów i hormonu
cję w komórkach posiadających właściwe receptory
wzrostu podstawowe wydzielanie zachodzi według
hormonalne (zob. tab. B 2-1).
rytmu dobowego; oprócz tego dodatkowe ilości hor-
Uwolnione do krwi hormony efektorowe mogą, za-
monów są uwalniane odpowiednio do zapotrzebo-
zwyczaj na zasadzie sprzężenia zwrotnego ujemnego
wania organizmu. Częstotliwość uwalniania innych
(negative feedback), poprzez podwzgórze wpływać
hormonów, np. żeńskich hormonów płciowych, na-
na funkcjonowanie obwodowych gruczołów dokrew-
MUTSCHLER-2009.indd 389 2010-01-07 22:12:47
390 Układ hormonalny
A B
wartość wartość
pożądana pożądana
regulator regulator
podzwgórze podzwgórze
wartość wartość
rzeczywista rzeczywista
liberyna (hormon uwalniający)
receptory, receptory,
np. receptory np. receptory dla
osmotyczne przysadka mózgowa hormonów T3/T4
przysadka mózgowa
sprzężenie zwrotne
sprzężenie zwrotne
hormon efektorowy, hormon tropowy,
np. wazopresyna np. TSH
parametr parametr gruczoł wykonawczy,
podlegający regulacji, organ wykonawczy, podlegający regulacji, np. tarczyca
np. ciśnienie osmotyczne np. nefron np. stężenie T3/T4
hormony efektorowe,
np. T3/T4
tkanki
Ryc. B 2-3. Podstawowe układy regulacji hormonalnej (według Thewsa, Mutschlera, Vaupela).
nych. Wzrost stężenia hormonu prowadzi najczęściej osmotycznego w przedziale pozakomórkowym. Ośrodek
do zmniejszonego wydzielania hormonu uwalniają- regulacji tego układu znajduje się w podwzgórzu, gdzie
jest wytwarzana wazopresyna, która jest przekazywana
cego, natomiast spadek do jego wzmożenia. następnie za pomocą transportu aksonalnego do przy-
Należy jednak zwrócić uwagę na to, że taki sche- sadki mózgowej. Uwalniana z niej wazopresyna do-
mat regulacji gruczołów wewnątrzwydzielniczych ciera z krwią do nefronów (narząd wykonawczy), gdzie
odnosi się jedynie do części hormonów. W niektó- wpływa na resorpcję zwrotną wody i w ten sposób jako
rych przypadkach nie występuje ani trzystopniowa wartość nastawcza reguluje stężenie osmotyczne płynów
pozakomórkowych (wielkość regulowana). Informacja
struktura łańcucha hormonów, ani sprzężenie zwrot- o osmolarności osocza przekazana przez receptory osmo-
ne ujemne. Na przykład wydzielanie insuliny z wysp tyczne powoduje, że podwzgórze (regulator) wzmaga lub
trzustkowych Langerhansa nie jest regulowane przez ogranicza syntezę wazopresyny i w konsekwencji wpły-
układ podwzgórze-przysadka, lecz zależy od stęże- wa to na jej uwalnianie z przysadki mózgowej (ujemne
nia glukozy we krwi. Wydzielanie innych hormo- sprzężenie zwrotne).
Rzadszym zjawiskiem jest spełnianie przez stężenie hor-
nów, np. parathormonu, również jest regulowane monu we krwi funkcji wielkości regulowanej, która ma być
bez udziału układu podwzgórze-przysadka. Jest ono utrzymywana na stałym poziomie. Przykładem takiego zja-
dostosowywane do stężenia substancji, które poprzez wiska jest przedstawiony na ryc. 2-3 B układ kontrolujący
działanie danego hormonu ma być utrzymane na sta- stężenie trijodotyroniny i tyroksyny we krwi. Stężenie hor-
łym poziomie (np. wydzielanie parathormonu jest za- monu we krwi, tj. wielkość regulowana, jest kontrolowana
przez receptory hormonalne. Spadek wartości rzeczywistej
leżne od stężenia jonów wapnia we krwi). w stosunku do wartości pożądanej powoduje uwolnienie
Zgodnie z procesami regulacji funkcji gruczołów przez ośrodek umiejscowiony w podwzgórzu (regulator)
dokrewnych stężenie danego hormonu we krwi może hormonu uwalniającego (TRH), który drogą krwi dociera
stanowić wielkość nastawczą albo jego wielkość re- do części gruczołowej przysadki mózgowej. TRH powo-
gulowaną, czyli podlegającą kontroli. duje wydzielenie hormonu tropowego – tyreotropiny, która
dociera drogą krwi do tarczycy (narządu wykonawczego)
i stymuluje tam uwalnianie hormonów tarczycy. W ten spo-
Funkcję hormonu jako wielkości nastawczej przed- sób następuje korekcja wielkości regulowanej aż do mo-
stawiono na ryc. B 2-3 A na przykładzie układu, które- mentu, kiedy stężenie hormonu będzie znowu odpowiadać
go celem jest utrzymanie na stałym poziomie stężenia wartości należnej.
MUTSCHLER-2009.indd 390 2010-01-07 22:12:47
Układ hormonalny 391
Tabela B 2-1. Hormony efektorowe
Nazwa Skrót Synonimy Budowa Miejsce Główne działania
wydzielania
hormon antydiuretyczny ADH wazopresyna nonapeptyd przysadka mózgowa zatrzymanie wody
oksytocyna nonapeptyd przysadka mózgowa skurcz mięśni macicy,
wytrysk mleka
somatotropina STH hormon białko część gruczołowa pobudzenie wzrostu
Układ hormonalny
(GH) somatotropowy, przysadki mózgowej kości i mięśni;
hormon wzrostu wzmożenie syntezy białek,
nasilenie lipolizy,
zahamowanie wychwytu
glukozy przez komórki
hormon MSH melanotropina polipeptyd część gruczołowa pigmentacja skóry
melanotropowy przysadki mózgowej
prolaktyna PRL laktotropina białko część gruczołowa rozrost gruczołów mlecznych,
(LTH) przysadki mózgowej wydzielanie mleka B2
tyroksyna T4 tetrajodotyronina wzmożenie procesów
pochodne tarczyca przemiany materii,
trijodotyronina T3 liotyronina tyrozyny pobudzenie wzrostu
kalcytonina tyreokalcytonina polipeptyd tarczyca obniżenie stężenia Ca2+,
podwyższenie stężenia
fosforanów
paratyryna PTH parathormon polipeptyd przytarczyce podwyższenie
tężenia Ca2+
somatomedyna IgF-1, insulinopodobny polipeptyd wątroba i in. pobudzenie wzrostu
IgF-2 czynnik wzrostowy
insulina polipeptyd wyspy Langerhansa, zwiększenie wychwytu
komórki B (trzustka) i metabolizmu glukozy
przez komórki, pobudzenie
syntezy glikogenu (obniżenie
stężenia glukozy we krwi)
glukagon HGF czynnik polipeptyd wyspy Langerhansa, wzmożenie glikogenolizy
hiperglikemiczny komórki A (trzustka) i glukoneogenezy
(podwyższenie stężenia
glukozy we krwi)
glzkokortykosteroidy steroidy kora nadnerczy wzmożenie glukoneogenezy,
(kortyzol) (warstwa siatkowata) proteolizy i lipolizy,
hamowanie procesu zapalnego
mineralokortykosteroidy steroidy kora nadnerczy zatrzymanie Na+,
(Aldosteron) (warstwa kłębuszkowa) wydalanie K+,
zatrzymanie wody
androgeny steroidy jądra (komórki rozwój męskich
(testosteron) Leydiga) narządów płciowych,
wzmożenie syntezy białek
estrogeny steroidy jajniki (nabłonek rozwój żeńskich
(estradiol) pęcherzyka), łożysko narządów płciowych,
proliferacja śluzówki macicy
MUTSCHLER-2009.indd 391 2010-01-07 22:12:47
392 Układ hormonalny
Tabela B 2-1. Hormony efektorowe (kontynuacja)
Nazwa Skrót Synonimy Budowa Miejsce Główne
wydzielania działania
progesteron steroid jajniki przemiana śluzówki
(ciałko żółte), macicy na fazę
łożysko wydzielniczą,
podwyższenie temperatury
ciała (0,4°C)
adrenalina (A) epinefryna pochodna tyrozyny rdzeń nadnerczy pobudzenie czynności
serca, stymulacja
glikogenolizy,
stymulacja OUN
noradrenalina (NA) norepinefryna pochodna tyrozyny rdzeń nadnerczy podwyższenie ciśnienia krwi
erytropoetyna glikoproteina nerki tworzenie erytrocytów
przedsionkowy peptyd (ANF atriopeptyd polipeptyd przedsionki serca wzmożenie diurezy
natriuretyczny ANP) i natriurezy
Mechanizm działania hormonów. Fizjologiczne ■ wzmożona lub – rzadziej – zmniejszona synteza bia-
działania hormonów (funkcje koordynacyjne, udział łek wskutek wzajemnego oddziaływania pomiędzy
w zaopatrzeniu w energię, sterowanie wzrostem i róż- hormonem i wewnątrzkomórkowym receptorem
nicowaniem płciowym) są wyzwalane przez pierwot- oraz następczą interakcją z fragmentami DNA.
ne reakcje ze swoistym receptorem i reakcje następ-
cze w komórkach organów wykonawczych. Typowym przykładem hormonu oddziałującego z re-
Obecnie wiadomo, że istnieją przede wszystkim ceptorową kinazą tyrozynową jest insulina.
trzy rodzaje reakcji, za których pośrednictwem moż- Większość hormonów peptydowych i białkowych,
na wpływać pobudzająco lub hamująco na bioche- a także katecholamin wykazuje swoje działanie po-
miczne procesy w komórkach: przez wytworzenie przekaźnika wtórnego.
Do związków mających wpływ na syntezę bia-
■ fosforylacja wewnątrzkomórkowej domeny fosfo-
łek przez wzajemne oddziaływanie z wewnątrz-
rylacyjnej i występująca wskutek tego aktywacja
komórkowym receptorem należą hormony stero-
receptorowej kinazy tyrozynowej z następczymi,
idowe, hormony tarczycy, witamina A oraz wita-
dalszymi wewnątrzkomórkowymi reakcjami fo-
mina D3. Ważnym wstępnym warunkiem wiązania
sforylacji, jak również (częściową) internalizacją
z wewnątrzkomórkowym receptorem jest zdolność
kompleksu hormon–receptor,
hormonu do przenikania przez błony komórkowe.
■ wytworzenie przekaźnika drugiego rzędu (second Zdolność taką posiadają jedynie hormony lipofilne
messenger) w następstwie interakcji hormonu lub przenoszone przez błonę komórkową za pomocą
z obecnym w błonie kompleksem receptor hormo- transportu aktywnego.
nalny – białko wiążące – enzym,
MUTSCHLER-2009.indd 392 2010-01-07 22:12:48
Układ hormonalny 393
2.1. Podwzgórze
W procesach regulacji funkcjonowania organizmu nie hormon uwalniający i hormon hamujący (zob. po-
mających na celu utrzymanie jego homeostazy, służą- niżej) przedostają się do przysadki.
cych rozmnażaniu i zdolności do pracy, pośredniczą, Regio hypothalamica posterior otaczający jądra
jak opisano powyżej, układ wewnątrzwydzielniczy ciał suteczkowatych pozostaje w ścisłym związku
i wegetatywny układ nerwowy. Wymaga to ścisłej z ośrodkami wegetatywnymi w śródmózgowiu i rdze-
koordynacji pracy obu układów, za którą odpowiada niu przedłużonym.
Układ hormonalny
podwzgórze. W podwzgórzu zlokalizowane są nad-
rzędne ośrodki wegetatywne, które z jednej stro-
ny wpływają na aktywność układu współczulnego 2.1.1. Hormony podwzgórza
i przywspółczulnego, a z drugiej strony na uwalnianie
hormonów przez przysadkę mózgową.
Podwzgórze wytwarza hormony oligopeptydowe,
Podwzgórze i przysadka stanowią razem nadrzęd-
które z krwią poprzez układ wrotny dostają się
ną jednostką czynnościową regulacji hormonalnej.
do przedniego płata przysadki i tam, jak wstępnie
opisano, albo powodują uwalnianie odpowiednich B2
Podstawy anatomiczne. Podwzgórze tworzy najniż-
hormonów (hormony uwalniające – liberyny), albo
sze piętro oraz dno międzymózgowia. Dzieli się ono
blokują ich uwalnianie (hormony hamujące uwal-
(zob. ryc. B 2.1-1) na część przednią i środkową za-
nianie – inhibiny). W tab. B 2.1-1 zestawiono hormo-
wierające niewielką ilość włókien mielinowych oraz
ny uwalniające i hormony hamujące uwalnianie.
część tylną z dużą ilością włókien mielinowych.
Wpływ na wydzielanie liberyn lub hormonów ha-
W części przedniej (regio hypothalamica anterior)
mujących uwalnianie oprócz ujemnego lub dodatnie-
znajdują się jądra przykomorowe i jądro nadwzroko-
go sprzężenia zwrotnego wywierają także neurony
we. Ich neurony wytwarzają tzw. neurosekret, który
noradrenergiczne, adrenergiczne i serotoninergiczne,
za pośrednictwem transportu aksonalnego dociera
przede wszystkim śródmózgowia i układu limbiczne-
do tylnego płata przysadki (zob. poniżej).
go. W ten sposób wpływy środowiska zewnętrznego
Jądra w regio hypothalamica intermedia (m.in. ją-
i wewnętrznego mogą być integrowane w system re-
dra guza, jądro lejka) wpływają na wydzielanie hor-
gulacji neuroendokrynnej.
monów przez przysadkę mózgową. Wydzielane przez
W podwzgórzu są ponadto wytwarzane w formie
hormonów prekursorowych hormony tylnego płata
przysadki – oksytocyna i adiuretyna (wazopresyna)
(wazopresyna jako preprowazopresyna i oksytocyna
NP
wazopresyna jako preprooksyfizyna). Jak już wspomniano, są one
oksytocyna jako tzw. neurosekret przekazywane na drodze transpor-
RH
RIH tu aksonalnego do tylnego płata przysadki. Zostały one
NI omówione w rozdziale o hormonach przysadki mózgo-
NSO
skrzyżowanie wej (zob. poniżej).
wzrokowe RH, RIH
lejek przysadki W obu cząsteczkach preprohormonów za peptydem sygna-
(infundibulum) łowym znajduje się odpowiedni hormon nonapeptydowy
tętnica przysadkowa (wazopresyna lub oksytocyna) oraz większy peptyd – neu-
górna (krążenie wrotne)
tętnica rofizyna. Prekursor wazopresyny zawiera dodatkowo gliko-
RH proteinę.
część gruczołowa część nerwowa
przysadki przysadki
2.1.1.1. Zastosowanie hormonów
podwzgórza w diagnostyce
wazopresyna oksytocyna
Niektóre hormony podwzgórza wykorzystuje się
Ryc. B 2.1-1. Schemat układu podwzgórze-przysadka (według w diagnostyce, np.:
Brücka). NI – jądro lejka, NP – jądro przykomorowe, NSO – jądro
nadwzrokowe, RH – hormon uwalniający (liberyna), RIH – hor- ■ tyreoliberynę (protyrelinę) – w diagnostyce zabu-
mon hamujący uwalnianie (inhibina). rzeń funkcji tarczycy,
MUTSCHLER-2009.indd 393 2010-01-07 22:12:48
394 Układ hormonalny
Tabela B 2.1-1. Hormony wpływające na czynność części gruczołowej przysadki mózgowej
A. Hormony uwalniające (liberyny)
Nazwa Skrót Synonimy Powoduje uwalnianie
hormon uwalniający somatotropinę GRH, somatoliberyna, somatotropiny (STH)
GH-RH somatorelina,
hormon uwalniający
hormon wzrostu
hormon uwalniający tyreotropinę TRH tyreoliberyna tyreotropiny (TSH),
prolaktyny (PRL)
hormon uwalniający kortykotropinę CRH kortykoliberyna, kortykotropiny (ACTH)
kortykorelina
hormon uwalniający gonadotropinę GnRH gonadoliberyna, FSH + LH (ICSH)
(LH-RH) gonadorelina
B. Hormony hamujące uwalnianie
Nazwa Skrót Synonimy Hamuje uwalnianie
hormon hamujący GIH somatostatyna somatotropiny (STH)
uwalnianie somatotropiny (GH-IH)
hormon hamujący uwalnianie prolaktyny = PIH prolaktostatyna prolaktyny (PRL)
dopamina (PRL-IH)
■ kortykoliberynę (kortykorelinę) – w diagnostyce Rzadkie działania niepożądane to miejscowe i ukła-
zaburzeń funkcji kory nadnerczy, dowe reakcje alergiczne.
Podanie hormonów podwzgórza jest przeciwwska-
■ gonadoliberynę (gonadorelinę) – w czynnościowej
zane w razie ciężkich reakcji nietolerancji podczas
diagnostyce jajników i jąder,
poprzednio przeprowadzanych badań diagnostycz-
■ somatoliberynę (somatorelinę) – przy podejrzeniu nych z wykorzystaniem tych hormonów.
niedoboru hormonu wzrostu.
Ponieważ hormony podwzgórza należą do hormonów 2.1.1.2. Terapeutyczne zastosowanie
peptydowych muszą być podawane parenteralnie. hormonów podwzgórza
Wyjątkiem jest tripeptyd protyrelina, który może być i ich analogów
również podawany drogą doustną. Okresy półtrwa-
nia hormonów podwzgórza ze względu na szybką
Gonadoliberyna i analogi gonadoliberyny zostaną
hydrolizę są relatywnie krótkie (np. osoczowy okres
omówione w dalszych rozdziałach (rozdz. B 2.8.1).
półtrwania protyreliny wynosi 5 minut). Hormony
podwzgórza są eliminowane przez nerki w postaci
Somatostatyna i analogi somatostatyny. Somato-
aminokwasów.
statyna będąca tetradekapeptydem hamuje (przede
Dawki tyreoliberyny wynoszą 200–400 μg, kortyko-
wszystkim wskutek hamowania cyklazy adenylano-
i gonadoliberyny – 100 μg, a somatoliberyny 50 μg.
wej) nie tylko uwalnianie hormonu wzrostu, ale tak-
że wydzielanie hormonów peptydowych przewodu
pokarmowego, m.in. gastryny, insuliny i glukagonu.
O CONH2
O Wykazano, że jest ona skuteczna w leczeniu krwa-
HN NH wień z przewodu pokarmowego w przebiegu choroby
N
O wrzodowej oraz nadżerkowego zapalenia błony ślu-
N zowej żołądka. Somatostatyna jest także stosowana
HN w profilaktyce powikłań pooperacyjnych po zabie-
gach w obrębie trzustki. Z powodu krótkiego okresu
tyreoliberyna
półtrwania wynoszącego kilka minut musi ona być
podawana w ciągłym wlewie kroplowym (dawko-
MUTSCHLER-2009.indd 394 2010-01-07 22:12:49
Układ hormonalny 395
wanie 3,5 μg/kg/h [maksymalnie 250 μg/h – przyp. akromegalii znalazł zastosowanie w terapii objawo-
tłum]). Z powodu wpływu na czynność wewnątrzwy- wej hormonalnie czynnych guzów przewodu pokar-
dzielniczą trzustki po podaniu somatostatyny może mowego (karcinoidu, VIPoma, glukagonoma). Jego
początkowo dojść do krótkotrwałej hipoglikemii, wpływ na wzrost guza lub przerzutów jest jednak
a po upływie 2–3 godzin do hiperglikemii, dlatego nieudowodniony. Ponieważ oktreotyd hamuje także
należy w krótkich odstępach czasu kontrolować gli- czynność zewnątrzwydzielniczą trzustki, znalazł za-
kemię. stosowanie w przed- i pooperacyjnej profilaktyce za-
palenia trzustki u pacjentów poddawanych zabiegom
Do syntetycznych analogów somatostatyny charakte- operacyjnym trzustki.
Układ hormonalny
ryzujących się wyższą aktywnością i dłuższym okre- Dawkowanie wynosi dla lanreotydu w formie re-
sem półtrwania należą lanreotyd i oktreotyd. Dłuż- tard 60–120 mg i.m. co 28 dni, a dla oktreotydu po-
szy czas działania (okres półtrwania w osoczu obu czątkowo 50–100 μg s.c. od jednego do trzech razy
substancji, z wyjątkiem postaci retard, wynosi 1,5 go- dziennie, a następnie 20–30 mg w formie retard poda-
dziny) jest następstwem zastąpienia L-aminokwasów wanych głęboko domięśniowo co 28 dni.
przez niefizjologiczne D-aminokwasy bądź w przy- Do działań niepożądanych poza wpływem na go-
padku oktreotydu także równoczesnego wbudowania spodarkę węglowodanową (hipo- i hiperglikemia) na-
aminoalkoholu, który hamuje rozkład enzymatyczny leżą: ból w miejscu podania leku, bóle głowy, utrata
okreotydu. Podczas gdy lanreotyd jest stosowany tyl- apetytu, nudności, wymioty, kamica pęcherzyka żół- B2
ko w leczeniu akromegalii, oktreotyd poza leczeniem ciowego, biegunki tłuszczowe.
H2N
HN
O
NH NH
HN NH
O NH O O NH
O CONH2 O NH
S S H3C OH O NH2
NH O O O NH
H3C NH NH OH
O HOOC NH NH
NH2 O O CH3
HO
somatostatyna
OH
HN HN
O O
NH NH
NH NH
O NH O NH2 O NH O NH2
O O NH O O NH
H2N S NH OH H2N S NH CH3
NH S NH S
O CH3 O CH3
O NH O NH
HO OH HO NH2
CH3 CH3 O
oktreotyd
lanreotyd
MUTSCHLER-2009.indd 395 2010-01-07 22:12:49
396 Układ hormonalny
2.2. Przysadka
Anatomia. Przysadka jest małym, ważącym tylko 2.2.1.1. Tyreotropina
około 0,6 g narządem, położonym w niszy w kostnej (hormon tyreotropowy, TSH)
podstawie czaszki, tj. w siodle tureckim (sella turci-
ca). Poprzez lejek jest ona połączona z podwzgórzem
Tyreotropina, składająca się z dwóch podjednostek
(zob. ryc. B 2.1-1).
α i β, jest swoistą gatunkowo glikoproteiną o masie
cząsteczkowej wynoszącej 28 000 Da, przy czym
Można wyróżnić trzy części przysadki:
podjednostka α jest identyczna jak w hormonie foli-
■ płat przedni (= część gruczołowa przysadki) za- kulotropowym i luteinizującym oraz gonadotropinie
wierający odrębne, zróżnicowane histologicznie kosmówkowej (zob. poniżej), natomiast podjednost-
komórki (komórki kwasochłonne, zasadochłonne ka β jest odpowiedzialna za aktywność biologiczną
i barwnikooporne), spośród których komórki kwa- i zdolność wiązania się z receptorem.
sochłonne wytwarzają somatotropinę i prolaktynę,
komórki zasadochłonne – tyreotropinę i gonadotro- Tyreotropina:
pinę oraz komórki barwnikooporne kortykotropinę
■ pobudza wychwyt jodku z krwi do tarczycy,
(zob. poniżej);
■ przyspiesza utlenianie jodków do jodu oraz prze-
■ płat środkowy albo przejściowy (część pośrednia
kształcanie dijodotyrozyny w tyroksynę i trijodoty-
przysadki), który u ludzi występuje tylko w postaci
roninę,
szczątkowej;
■ zwiększa enzymatyczne uwalnianie tyroksyny (lub
■ płat tylny (= część nerwowa przysadki), który po-
trijodotyroniny) z tyreoglobuliny, a następnie uwal-
wstaje z ektodermy nerwowej, co wyjaśnia, dla-
nianie hormonów do krwi,
czego przysadka nerwowa zawiera drogi nerwowe
wywodzące się z ww. okolic jąder podwzgórza. ■ stymuluje w niewielkim stopniu wzrost tarczycy.
Wydzielanie hormonu tyreotropowego zależy od stę-
żenia hormonów tarczycy we krwi: przy wysokim
2.2.1. Hormony części gruczołowej stężeniu hormonów tarczycy wydzielanie tyreotropi-
ny zostaje ograniczone, natomiast przy niskim ulega
przysadki (hormony
pobudzeniu. Ponadto wydzielanie TSH podlega ryt-
przedniego płata przysadki) mowi dobowemu: późnym wieczorem i w nocy stę-
żenie TSH we krwi osiąga najwyższy poziom. Okres
Przedni płat przysadki wytwarza hormony tropowe półtrwania we krwi wynosi około 1 godziny.
i efektorowe hormony peptydowe. Tyreotropina ma znaczenie jedynie diagnostycz-
ne, a nie terapeutyczne. Uzyskany dzięki inżynierii
Do hormonów tropowych przedniego płata przysadki genetycznej preparat handlowy Thyrogen zawiera
należą: tyreotropinę α. Podany przed scyntygrafią całego
ciała wykonywaną z użyciem jodu promieniotwór-
■ tyreotropina,
czego poprzez pobudzanie jodochwytności pozwala
■ kortykotropina, na wykrycie pozostałości tarczycy oraz rozróżnienie
jodochwytnych nowotworów tarczycy u pacjentów
■ gonadotropiny (hormon folikulotropowy i hormon
po tyroidektomii.
luteinizujący).
Thyrogen podaje się domięśniowo, dwukrotnie
w odstępach 24-godzinnych w dawce 0,9 mg. Dru-
Do hormonów efektorowych przedniego płata przy-
ga dawka powinna zostać podana 24 godziny przed
saki należą:
radiojodem.
■ somatotropina, Opisano następujace działania niepożądane: nud-
ności, wymioty, zawroty głowy, gorączkę, objawy
■ melanotropina,
grypopodobne, a także świąd i pokrzywkę w miejscu
■ prolaktyna. podania.
MUTSCHLER-2009.indd 396 2010-01-07 22:12:49
Układ hormonalny 397
2.2.1.2. Kortykotropina W hormonalnej diagnostyce różnicowej niewy-
(ACTH = hormon dolności nadnerczy pochodzenia przysadkowego
adrenokortykotropowy) i pozaprzysadkowego, zamiast już niedostępnego
w handlu ACTH, stosuje się obecnie tetrakozaktyd
(β1–24 kortykotropinę). Podanie tetrakozaktydu po-
Kortykotropina jest polipeptydem zbudowanym z 39
woduje wzrost stężenia kortyzolu przy przysadkowej
aminokwasów (masa cząsteczkowa 4500 Da), który
niewydolności kory nadnerczy i brak wzrostu przy
jest wytwarzany w komórkach proopiomelanokor-
niewydolności pochodzenia pozaprzysadkowego. Te-
tynowych (komórki POMC) w postaci proopiome-
rapeutycznie stosuje się go, choć zdecydowanie rza-
lanokortyny (POMC; ryc. B 2.2-1), będącej równo-
Układ hormonalny
dziej niż w przeszłości, w leczeniu niektórych postaci
cześnie prekursorem β-endorfiny i melanotropiny
padaczki występujących u niemowląt i małych dzie-
(zob. poniżej).
ci, a w szczególności w zespole Westa. Ze względu
na to, że tetrakozaktyd jest polipeptydem, musi być
Kortykotropina:
on podawany pozajelitowo. Po wstrzyknięciu i.v. czas
■ pobudza wytwarzanie i wydzielanie glukokortyko- działania jest bardzo krótki, a po wstrzyknięciu i.m.
steroidów oraz w mniejszym stopniu mineralokor- wynosi także jedynie kilka godzin, ale można go wy-
tykosteroidów i androgenów w korze nadnerczy, dłużyć, podając preparaty dépôt.
Działania niepożądane i przeciwwskazania są B2
■ zmniejsza wskutek tego zawartość cholesterolu
w większości identyczne jak w przypadku zastosowa-
i kwasu askorbinowego w korze nadnerczy.
nia glukokortykosteroidów. Dodatkowym przeciw-
wskazaniem do stosowania tetrakozaktydu jest zespół
Mechanizm działania ACTH jest przede wszystkim
nadnerczowo-płciowy (zob. poniżej).
związany z pobudzaniem receptora połączonego
z białkiem Gs, czego następstwem jest zwiększone
wytwarzanie cAMP.
ACTH nie jest specyficzny gatunkowo i kortyko- 2.2.1.3. Melanotropina
tropiny poszczególnych gatunków zwierząt różnią Melanotropina (MSH; hormon pobudzający melanocyty)
się od siebie tylko nieznacznie. Fragmenty ACTH jest efektorowym hormonem części gruczołowej przysadki.
Ten polipeptyd (zob. ryc. B 2.2-1) pobudza wytwarzanie
zawierające N-końcowe aminokwasy 20–24 wykazu- melaniny, a także rozpraszanie ziarenek pigmentu w skórze
ją jeszcze pełną jego aktywność. Jeżeli ochroni się i w wyniku tego jej pigmentację. Wydaje się jednak, że u ludzi
koniec aminowy i karboksylowy przed rozkładem w warunkach fizjologicznych ma ona podrzędne znaczenie.
enzymatycznym, to nawet jeszcze mniejsze peptydy Jak już wspomniano, melanotropina jest wytwarzana w po-
wykazują prawie pełną aktywność hormonalną. staci proopiomelanokortyny (POMC) i dlatego jej wydziela-
nie jest zawsze skojarzone z wydzielaniem kortykotropiny
Przysadka wydziela w rytmie dobowym około (i β-endorfiny). Ze względu na fakt, że wydzielnie POMC
20 mg ACTH dziennie. Jego osoczowy okres pół- jest zależne od hormonu uwalniającego kortykotropinę
trwania wynosi około 15 minut. (CRH), przy nadmiernym wytwarzaniu CRH, np. w przebiegu
choroby Addisona, występuje zwiększona pigmentacja skóry.
2.2.1.4. Gonadotropiny
PS POMC
Gonadotropiny, glikoproteiny o masie cząsteczko-
ACTH β-LPH
wej 20 000–50 000 Da, sterują czynnością narządów
γ-MSH płciowych u kobiet i mężczyzn. Można je określić
jako nadrzędne hormony płciowe, które nie są swo-
γ-LPH β-Endo
iste płciowo, jednak w dużym stopniu swoiste gatun-
γ-MSH CLIP
kowo. Zostały one dokładniej omówione w rozdziale
dotyczącym hormonów płciowych.
β-MSH Met-Enk
Ryc. B 2.2-1. Struktura proopiomelanokortyny (POMC). 2.2.1.5. Prolaktyna
ACTH = kortykotropina, (PRL; LTH = hormon
MSH = hormon melanotropowy, laktotropowy)
Met-Enk = met-enkefalina,
β-Endo = β-endorfina, Ten zbudowany ze 199 aminokwasów hormon o ma-
β-LPH = β-lipotropina. sie cząsteczkowej wynoszącej 22 500 Da ma budowę
MUTSCHLER-2009.indd 397 2010-01-07 22:12:49
398 Układ hormonalny
chemiczną zbliżoną do somatotropiny (zob. poni- Poza bromokryptyną, która stosowana jest najdłu-
żej). Prolaktyna pobudza wytwarzanie mleka (lak- żej, jako lek hamujący wydzielanie prolaktyny wy-
tację) w gruczołach piersiowych. Przypisywany korzystywane są również kabergolina, metergolina
w przeszłości prolaktynie wpływ na żeńskie gru- i chinagolid. Oprócz leczenia gruczolaka przysadki
czoły płciowe nie został, przynajmniej u ludzi, po- wydzielającego prolaktynę znajdują one zastosowa-
twierdzony. U kobiet osoczowe stężenia prolaktyny nie w hamowaniu laktacji po porodzie i zaprzestaniu
jest około 1,5 razy wyższe niż u mężczyzn. U obu karmienia piersią, oraz w leczeniu mlekotoku i braku
płci wydzielanie prolaktyny podlega wahaniom miesiączki lub niepłodności uwarunkowanych hiper-
w ciągu doby, a maksymalne stężenia prolaktyny prolaktynemią.
jest stwierdzane podczas snu. Mechaniczna stymu- Dawkowanie jest indywidualne. Dziennie podaje
lacja pochwy i szyjki macicy hamuje wzmożone się przeciętnie 5 mg bromokryptyny, 4–8 mg meter-
wydzielanie prolaktyny za pośrednictwem aferen- goliny i 0,05 mg chinagolidu. W celu pierwotnego
tnej impulsacji do podwzgórza. Podczas ciąży ob- odstawienia od piersi stosuje się jednorazową dawkę
serwuje się fizjologiczne zwiększone wydzielanie l mg długo działającej kabergoliny, a w zaburzeniach
prolaktyny. Ponadto drażnienie mechanoreceptorów typu hiperprolaktynemii 0,5–1 mg tygodniowo.
w brodawkach sutkowych przez ssące niemowlę
prowadzi – ponownie za pośrednictwem aferentnej
impulsacji do podwzgórza – do wzrostu uwalniania 2.2.1.6. Somatotropina
prolaktyny. (GH = hormon wzrostu,
Sprzężenie zwrotne regulujące wydzielanie pro- STH = hormon somatotropowy)
laktyny jest oparte na stężeniu tego hormonu – przy
podwyższonym stężeniu prolaktyny w podwzgórzu
Hormon wzrostu jest jednołańcuchowym peptydem
zwiększa się wydzielanie dopaminy, która hamuje
o masie cząsteczkowej 21 500 Da, zbudowanym
uwalnianie prolaktyny. Dopamina może być zatem
ze 191 aminokwasów. Fizjologicznymi bodźcami
uznana za hormon hamujący uwalnianie prolaktyny.
wyzwalającymi wydzielanie somatotropiny są m.in.
Osoczowy okres półtrwania prolaktyny jest krót-
hipoglikemia, wzrost stężenia aminokwasów w oso-
szy niż jedna godzina.
czu oraz wyrzut katecholamin (np. przy znacznym
Prolaktyna nie ma znaczenia terapeutycznego.
Patologiczną hiperprolaktynemię stwierdza się
w przebiegu wielu chorób, np. w przebiegu prolacti-
noma będącego najczęstszym gruczolakiem przed- ujemne sprzężenie zwrotne
podwzgórze
niego płata przysadki. U kobiet objawami hiperpro-
laktynemii są: mlekotok (galaktorea) oraz zaburzenia podwyższenie
miesiączkowania (amenorea – brak miesiączki), na- hormon stężenia glukozy
tomiast u mężczyzn utrata libido i zaburzenia poten- uwalniający i FFA we krwi
somatotropinę somato-
cji. Z tego powodu podwyższone stężenia prolaktyny statyna
są ważną przyczyną niepłodności.
ujemne sprzężenie zwrotne
hamowanie wychwytu
Farmakoterapia prolactinoma i innych zaburzeń glukozy i jej
wydzielania prolaktyny. Jak opisano powyżej, dopa- przysadka wykorzystania przez
komórki mięśniowe
mina pełni funkcję hormonu hamującego uwalnianie
prolaktyny. Z tego powodu agoniści receptorów do-
somatotropina uruchomienie
paminergicznych stosowani przede wszystkim w tera- zapasów tłuszczów
pii choroby Parkinsona (leki te omówiono w rozdzia-
wątroba soamato- wzrost tkanki
le dotyczącym farmakoterapii choroby Parkinsona) mięśniowej
medyna
mogą poprzez pobudzanie receptorów dopaminer-
gicznych skutecznie hamować wydzielanie prolakty- wzrost kości
ny i równocześnie w przypadku gruczolaka przysadki
wydzielającego prolaktynę prowadzić do długotrwa-
wzrost chrząstek
łego znacznego zmniejszenia nasilenia objawów guza
poprzez regresję guza zarówno w przypadku mikro-,
jak i makrogruczolaków. W przypadku mikrogruczo- Ryc. B 2.2-2. Wpływ somatotropiny i somatomedyny C na
laków próba przerwania terapii po 2–3 latach rokuje przemianę materii (według Thewsa, Mutschlera, Vaupela);
powodzenie. FFA = wolne kwasy tłuszczowe.
MUTSCHLER-2009.indd 398 2010-01-07 22:12:50
Układ hormonalny 399
obciążeniu fizycznym organizmu lub w stresie emo- cześnie osiągnięte są fizjologiczne stężenia hormonów
cjonalnym). GH jest wydzielany przede wszystkim tarczycy, kory nadnerczy i hormonów płciowych. Na-
w pierwszej fazie snu głębokiego. Warunkiem jego tomiast w przypadku braku somatotropiny efekt po-
wydzielania jest wzmożone uwalnianie somatolibe- budzający wzrost tych hormonów jest zmniejszony.
ryny z równoczesnym spadkiem wydzielania soma- Okres półtrwania w surowicy po podaniu dożyl-
tostatyny. nym wynosi 20–30 minut.
Somatotropina jest bardzo swoista gatunkowo
i dlatego zwierzęce hormony wzrostu nie wywo- Somatotropinę do leczenia karłowatości przysadkowej po-
łują u ludzi reakcji analogicznej do ludzkiego GH. czątkowo otrzymywano tylko przez izolację z przysadek
ludzkich (ze zwłok). Istniało przy tym niebezpieczeństwo
Układ hormonalny
Zakres działania somatootropiny jest bardzo szero- przeniesienia choroby Creutzfeldta-Jakoba, charakteryzu-
ki, co przedstawiono na ryc. B 2.2-2. W większości jącej się rozległą utratą komórek zwojowych w mózgu, po-
przypadków za efekt działania somatotropiny odpo- stępującym otępieniem i zaburzeniami motorycznymi, które
wiada nie sam GH, ale wytwarzane pod jej wpły- prowadzą w końcowym etapie choroby do zgonu pacjenta.
wem czynniki wzrostu, czyli somatomedyny (IGF, Pierwotnie uznawano, że przyczyną choroby Creutzfeldta-
-Jakoba jest zakażenie wirusami powolnymi, jednak obec-
insulinopodobne czynniki wzrostu). Somatomedyny nie – tak jak w przypadku gąbczastej encefalopatii bydła
są syntetyzowane w wątrobie oraz w chondrocytach. rogatego (= BSE, „choroby szalonych krów”) – uważa się,
Najważniejszym insulinopodobnym czynnikiem że czynnikiem etiologicznym są priony.
wzrostu jest IGF-1 (somatomedyna C), który poprzez B2
zwiększenie syntezy białek we wszystkich komór- Problem ten nie istnieje przy obecnie dostępnych
kach stymuluje podziały komórkowe. Poza tym wy- preparatach somatotropiny otrzymanych metodami
wołuje on krótko trwające działanie insulinopodob- inżynierii genetycznej.
ne (stąd nazwa insulinopodobny czynnik wzrostu 1). Wskazaniami do stosowania somatotropiny są kar-
Podsumowując, można stwierdzić, że somatotropina łowatość przysadkowa (somatotropinowa niedoczyn-
działa z jednej strony jak hormon efektorowy, a z dru- ność przysadki) (zob. poniżej) oraz zaburzenia wzro-
giej strony jak gruczołotropowy. stu u dziewcząt z zespołem Ullricha-Turnera (mono-
somia chromosomu X).
Somatotropina jako hormon efektorowy: Najkorzystniejszy efekt terapeutyczny osiąga się
podając lek we wstrzknięciach wieczornych.
■ uwalnia kwasy tłuszczowe z tkanki tłuszczowej
Ze zrozumiałych względów leczenie ma sens tylko
i tą drogą prowadzi do zmniejszenia zapasów tłusz-
wtedy, gdy nie doszło jeszcze do zarośnięcia przyna-
czów (nasilenie lipolizy i ketogenezy; działanie an-
sadowych chrząstek wzrostowych.
tagonistyczne w stosunku do insuliny),
Z uwagi na opisany wcześniej wpływ somatotro-
■ podwyższa stężenie glukozy we krwi wskutek pobu- piny na metabolizm glukozy przy zwiększonym daw-
dzania glukoneogenezy w wątrobie i zmniejszenia kowaniu somatotropina działa diabetogennie. W na-
wychwytu glukozy oraz jej zużycia w tkance mieś- stępstwie przyspieszonych podziałów komórkowych
niowej oraz tłuszczowej (działanie antagonistyczne nie można wykluczyć działania przyspieszającego
w stosunku do insuliny), wzrost nowotworów. Z tego względu cukrzyca i cho-
roby nowotoworowe są przeciwwskazaniami do sto-
■ zwiększa wchłanianie aminokwasów, co w kon-
sowania somatotropiny.
sekwencji prowadzi do nasilenia syntezy białek
Jednoczesne podanie glukokortykosteroidów osła-
w tkance mięśniowej (efekt zwiększenia masy).
bia działanie somatotropiny.
Jako hormon gruczołotropowy somatotropina – po- Antagoniści somatotropiny. Pegwisomant jest pe-
średnio poprzez wytwarzanie somatomedyn – pobu- gylowanym analogiem somatotropiny o działaniu an-
dza wzrost tkanki chrzestnej, kostnej i mięśniowej: tagonistycznym, który został zarejestrowany do lecze-
wzmożone wytwarzanie IGF-1 w chondrocytach nia akromegalii. Pegwisomant jest wysoce wybiórczy
zwiększa autokrynną i parakrynną aktywność mito- dla receptora hormonu wzrostu i nie reaguje krzyżo-
tyczną tych komórek. U dzieci i młodzieży prowadzi wo z innymi receptorami. Po podaniu podskórnym
do wzrostu kości na długość, natomiast u dorosłych pegwisomant wchłania się powoli, a maksymalne
do nadmiernego pogrubienia i przerostu kości. Wy- stężenie w surowicy jest osiągane po 1,5–3 dniach.
tworzony w wątrobie i uwalniany do krwiobiegu Osoczowy okres półtrwania wynosi 74–172 godziny.
IGF-1 pobudza wzrost masy tkanki mięśniowej. Dawkowanie: początkowo 80 mg s.c. pod kontro-
Pełny efekt działania hormonu wzrostu, a tym sa- lą lekarza doświadczonego w leczeniu akromegalii,
mym somatomedyn rozwija tylko wtedy, gdy jedno- a następnie 10 mg s.c. dziennie.
MUTSCHLER-2009.indd 399 2010-01-07 22:12:50
400 Układ hormonalny
Lek jest zazwyczaj dobrze tolerowany. Do dzia- 15 mg i późnym popołudniem 5 mg kortyzolu, przy
łań niepożądanych należą: reakcje w miejscu podania czym należy pamiętać, że niektórzy chorzy wyma-
leku, świąd, bóle głowy, zaburzenia snu, zaburzenia gają większych, a inni mniejszych dawek. Decyzja
żołądkowo-jelitowe oraz astenia. Istnieje również o wielkości dawek kortyzolu musi opierać się na sta-
ryzyko, że podczas terapii pegwisomantem dojdzie nie klinicznym pacjenta. W sytuacjach związanych
do powiększenia się guza przysadki wydzielającego ze zwiększonym zapotrzebowaniem na glukokorty-
hormon wzrostu. kosteroidy (np. zabiegi operacyjne, wysoka gorącz-
U chorych przyjmujących insulinę lub doustne ka) konieczne jest zwiększenie dawek kortyzolu.
leki hipoglikemizujące w początkowym okresie te- W przypadku braku lub zmniejszenia wydzielania
rapii pegwisomantem może być konieczna redukcja tyreotropiny obserwuje się objawy niedoczynności
dawki leków obniżających poziom glikemii. tarczycy, które są wskazaniem do codziennej doustnej
suplementacji 75–150 μg lewotyroksyny pod kontro-
lą osoczowych stężeń hormonów tarczycy.
2.2.1.7. Zaburzenia czynności Przy niedoborze gonadotropin podaje się gonado-
przysadki gruczołowej tropiny lub hormony płciowe (estrogeny, gestageny,
androgeny).
2.2.1.7.1. Niewydolność przedniego płata Jeśli u osoby dorosłej w następstwie niedoboru
przysadki hormonu wzrostu obserwuje się zaburzenia prze-
miany materii oraz istotne pogorszenie wydolności,
Patofizjologia. Schorzenie to, zwane także hipopitui- wskazane jest codzienne wieczorne podawanie pod-
taryzmem, jest stanem, w którym doszło do zmniej- skórnych iniekcji somatotropiny w dawce początko-
szonego wydzielania hormonów przedniego płata wej 0,15–0,2 mg, którą można stopniowo zwiększać
przysadki. Przyczynami niewydolności przedniego maksymalnie do 1,5 mg/dobę.
płata przysadki mogą być: wady genetyczne, guzy
przysadki lub mózgu, stany zapalne, zmiany nacieko-
we (np. w przebiegu sarkoidozy) i zmiany wsteczne
2.2.1.7.2. Niedobór wzrostu pochodzenia
(np. na skutek zaburzeń ukrwienia przysadki) oraz
przysadkowego
zaburzenia rozwojowe i urazy.
(karłowatość przysadkowa)
Brak wydzielania niektórych lub wszystkich hor-
monów części gruczołowej przysadki jest określany
jako częściowa lub całkowita niewydolność przed- Patofizjologia. Niedobór wzrostu pochodzenia
niego płata przysadki. Kliniczne konsekwencje zale- przysadkowego jest spowodowany niedoborem so-
żą od tego, które hormony są wydzielane w niedosta- matotropiny w okresie wzrastania. W ciężkiej po-
tecznej ilości. staci obserwuje się karłowatość z zachowanymi
W przypadku powolnej progresji obrazu choro- proporcjami ciała. (O karłowatości mówi się, gdy
bowego, np. w przebiegu gruczolaka przysadki lub po zakończeniu okresu wzrastania wysokość chore-
obecnie rzadziej występującej niedokrwiennej mar- go jest mniejsza od 140 cm). Możliwymi przyczyna-
twicy przysadki w następstwie porodu z dużą utratą mi karłowatości są zaburzenia genetyczne (stosun-
krwi (tzw. zespół Sheehana), w pierwszej kolejności kowo rzadko), wady rozwojowe, zakażenia oraz no-
obserwuje się zazwyczaj objawy niedoboru gonado- wotwory ośrodkowego układu nerwowego, a także
tropin. Rozwija się wówczas (wtórny) hipogonadyzm. urazy czaszki i mózgu. Stosunkowo często stwier-
U kobiet obserwuje się brak miesiączki, a u mężczyzn dza się w wywiadzie uraz okołoporodowy. Wyjaśnia
ulegają zmniejszeniu jądra oraz dochodzi do utraty to, dlaczego często nie stwierdza się izolowanego
libido i potencji. Zanika wtórne owłosienie płciowe, niedoboru somatotropiny, lecz uogólnioną niewy-
skóra staje się cienka, biała, pomarszczona i sucha. dolność przedniego płata przysadki z nieznaczny-
Później dodatkowo w następstwie niedoboru tyreo- mi objawami niedoboru pozostałych hormonów.
tropiny i kortykotropiny rozwijają się objawy (wtór- U Pigmejów stwierdzono prawidłowe wytwarzanie
nej) niedoczynności tarczycy i kory nadnerczy. hormonu wzrostu przy niewystarczającej syntezie
IGF-1.
Farmakoterapia. Leczenie niewydolności przednie-
go płata przysadki skierowane jest na uzupełnianie Farmakoterapia. Leczenie niedoboru hormonu
niedoborów hormonalnych. wzrostu u dzieci polega na codziennym podawaniu
Niedobór kortyzolu pochodzenia przysadkowego przed snem iniekcji somatotropiny w dawce 0,025–
wyrównuje się zazwyczaj, podając wcześnie rano 0,04 mg/kg.
MUTSCHLER-2009.indd 400 2010-01-07 22:12:50
Układ hormonalny 401
2.2.1.7.3. Gigantyzm przysadkowy W dalszej kolejności mogą być stosowani agoni-
i akromegalia ści receptorów dopaminergicznych, którzy w przeci-
wieństwie do działania u ludzi zdrowych u chorych
na akromegalię wywołują zmniejszenie wydzielania
Patofizjologia. Nadmierny wzrost pochodzanie przy-
somatotropiny (tzw. paradoksalne hamowanie wy-
sadkowego (gigantyzm przysadkowy), tzn. ogólny
dzielania somatotropiny).
nadmierny wzrost z zachowanymi proporcjami cia-
ła, jest następstwem nadmiernego wytwarzania so-
matotropiny przed zakończeniem okresu wzrastania.
W tym okresie chrząstki przynasadowe nie są jeszcze 2.2.2. Hormony tylnego płata
Układ hormonalny
zarośnięte, czyli kości mogą nadal wzrastać na dłu- przysadki
gość. Akromegalia (schorzenie występujące u doro-
słych), występująca z częstością 40–70 przypadków Z tylnego płata przysadki są wydzielane dwa, synte-
na 1 milion mieszkańcow, jest także następstwem tyzowane w podwzgórzu, cyklonanopeptydy, tj. adiu-
nadmiernej produkcji somatotropiny. W przeciwień- retyna (wazopresyna) i oksytocyna, które choć wyka-
stwie do gigantyzmu przysadkowego, z powodu zują odmienny zakres działania, w budowie chemicz-
zarośnięcia chrząstek przynasadowych, dochodzi nej różnią się jedynie dwoma aminokwasami. Ludzka
do przerostu kości (ich pogrubienia). Ponadto ob- wazopresyna w odróżnieniu od oksytocyny zawiera B2
serwuje się pobudzenie wzrostu skóry i jej przydat- zamiast leucyny argininę, a zamiast izoleucyny feny-
ków oraz częściowo także narządów wewnętrznych loalaninę.
(splaichnomegalia). Z powodu zwiększonej aktyw-
ności osteoblastów, a co za tym idzie – nasilonej prze-
budowy kostnej, stwierdza się zwiększoną aktywność 2.2.2.1. Wazopresyna
fosfatazy alkalicznej w surowicy. Obraz kliniczny (ADH = hormon antydiuretyczny)
akromegalii obejmuje pogrubienie rysów twarzy, i jej analogi
powiększenie twarzoczaszki, rąk i stóp, które nadają
pacjentowi typowy wygląd, a także pogrubienie nosa,
Fizjologiczna rola wazopresyny polega na nasilaniu
warg i języka. W 40% przypadków z powodu anta-
zagęszczania moczu w nerkach. Już jej niewielkie stę-
gonistycznego w stosunku do insuliny działania hor-
żenie zwiększa istotnie przepuszczalność kanalików
monu wzrostu stwierdza się hiperglikemię lub inne
dystalnych i cewek zbiorczych dla wody, w wyniku
(przysadkowe) objawy cukrzycy. U 30% chorych
czego zwiększa się resorpcja wody. Stąd wywodzi się
rozwija się wole tarczycy. Pacjenci skarżą się na bóle
nazwa adiuretyny. W większych stężeniach powoduje
głowy, bóle stawów oraz nadmierną potliwość. Ob-
ona skurcz wszystkich mięśni gładkich (stąd nazwa
serwowane zaburzenia libido i potencji oraz brak
wazopresyna), co prowadzi do:
miesiączki są następstwem pobudzenia receptorów
dla prolaktyny w związku z bardzo wysokim stęże- ■ wzrostu ciśnienia krwi,
niem hormonu wzrostu.
■ nasilenia perystaltyki jelit,
Najczęstszą przyczyną obu schorzeń jest w więk-
szości przypadków gruczolak przysadki wywodzący ■ zwiększenia napięcia mięśniówki w drogach żół-
się z komórek kwasochłonnych wytwarzających hor- ciowych i moczowych.
mon wzrostu.
Efekt antydiuretyczny lub wazokonstrykcyjny
Farmakoterapia. Leczenie akromegalii polega prze- są następstwem działania wazopresyny poprzez róż-
de wszystkim (oprócz radioterapii, która jest mniej ne receptory sprzężone z białkiem G. Receptory V1
skuteczna i bardziej niebezpieczna z uwagi na ryzyko odpowiadają za wpływ wazopresyny na naczynia,
rozwoju niewydolności przysadki) na operacyjnym receptory V2 – na nerki. Pobudzenie receptorów V1
usunięciu guza. W przypadku niewystarczającej sku- powoduje przekazywanie sygnału drogą fosfoinozy-
teczności powyższych metod lub przeciwwskazań tolu, natomiast receptorów V2 aktywuje cyklazę ade-
do ich stosowania, a także w okresie przygotowywa- nylanową (zob. ryc. B 2.2-3).
nia do leczenia operacyjnego zaleca się farmakote-
rapię. W leczeniu stosuje się analogi somatostatyny Ponadto wazopresyna uczestniczy w regulacji uwal-
– oktreotyd i lanreotyd, a gdy są one nietolerowane niania kortykotropiny, skłonności do picia płynów,
lub nie dają wystarczających efektów agonistę recep- a także wpływa na pamięć i procesy uczenia się.
tora dla hormonu wzrostu – pegwisomant. Poza tym za pomocą dotychczas niepoznanego me-
MUTSCHLER-2009.indd 401 2010-01-07 22:12:50
402 Układ hormonalny
wazopresyna jądro przykomorowe
terli- desmo-
presyna (adiuretyna) presyna
jądro nadwzrokowe
osmorecetory
część
śródmiąższowa
aferentne gruczołowa część
osocze
tkanka
drogi przysadki nerwowa
V1 V2 nerwowe przysadki
wazopresyna
Gq Gs oksytocyna (adiuretyna,
ADH)
wypływ mleka H2O
cyklaza
PLC adenylanowa
ATP
PIP2
cAMP↑
IP3, DAG resorpcja
PKA skurcz mięśnia macicy kanalik nerkowy wody
[Ca2+]↑
kanał Ryc. B 2.2-4. Działanie wazopresyny i oksytocyny oraz regulacja
wodny ich wydzielania (według Thewsa, Mutschlera, Vaupela).
–2
działanie antydiuretyczne skurcz
ciowe oraz stres nasilają wydzielanie wazopresyny.
mięśnie gładkie cewka dystalna, szczególnie Wydzielanie wazopresyny mogą również nasilać
naczyń krwionośnych komórki nabłonka
kanalików zbiorczych substancje cholinergiczne, niektóre leki przeciwpa-
daczkowe oraz trójpierścieniowe leki przeciwdepre-
Ryc. 2.2-3. Mechanizm działania wazopresyny i jej analogów. syjne. W przeciwieństwie do nich alkohol, kofeina,
V – receptor dla wazopresyny, IP3 – 1,4,5-trifosfoinozytol, DAG glukokortykosteroidy i fenytoina hamują wydzielanie
– diacyloglicerol, cAMP – cykliczny monofosforan adenozyny, ADH. W związku z opisanymi właściwościami wazo-
PKA – kinaza białkowa A, PIP2 – difosforan fosfatydyloinozytolu, presyna była stosowana przede wszystkim jako śro-
PLC – fosfolipaza C. dek antydiuretyczny w terapii moczówki prostej oraz
ze względu na działanie zwężające naczynia krwio-
nośne w tamowaniu krwawienia z żylaków przełyku.
chanizmu pobudza ona syntezę VIII czynnika krzep-
Zwężenie naczyń trzewnych powoduje obniżenie ciś-
nięcia.
nienie w żyle wrotnej i w wyniku tego także w żyłach
przełyku. Po wprowadzeniu do terapii otrzymanych
W kontroli działania wazopresyny (zob. ryc. B 2.2-4)
przez modyfikację cząsteczki adiuretyny analogów
biorą udział:
wazopresyny, które działają prawie wyłącznie anty-
■ receptory objętościowe rejestrujące objętość krwi diuretycznie lub naczyniozwężająco, zmniejszyło się
w przedsionkach serca i żyłach płucnych, bardzo znaczenie terapeutyczne wazopresyny.
■ receptory osmotyczne znajdujące się głównie
Analogi wazopresyny. Na receptor V2 działa przede
w podwzgórzu, ale także w obszarze krążenia wrot-
wszystkim:
nego podwzgórze-przysadka i w wątrobie, które
nadzorują stężenie osmotyczne osocza, ■ desmopresyna (DDAVP),
■ baroreceptory w zatokach szyjnych i łuku aorty, natomiast selektywność w stosunku do receptorów
które biorą udział w regulacji ciśnienia tętniczego V1 wykazuje
krwi.
■ terlipresyna.
Receptory osmotyczne w podwzgórzu reagują
na wzrost ciśnienia osmotycznego już o 1% i w od-
powiedzi na taki bodziec wywołują wzmożone wy-
dzielanie wazopresyny (zob. ryc. B 2.2-4). Poza tym
bodźcem fizjologicznym także ból, inne bodźce czu-
MUTSCHLER-2009.indd 402 2010-01-07 22:12:50
Układ hormonalny 403
Desmopresyna, której podstawową zaletą w porów- W leczeniu moczówki prostej desmopresynę po-
naniu z wazopresyną jest znaczne wydłużenie okresu daje się dwa razy dziennie donosowo w dawce 10–40
półtrwania (po podaniu doustnym i donosowym oko- μg lub doustnie – w trzech dawkach podzielonych
ło 2 godzin), jest stosowana przede wszystkim w le- – 0,2–1,2 mg/dobę. W hemofilii A desmopresynę po-
czeniu moczówki prostej. Bywa również stosowana, daje się dożylnie w dawce 0,3–0,4 μg/kg m.c.
jako leczenie wspomagające psychoterapię, u cho- Do działań niepożądanych desmopresyny należy
rych z moczeniem nocnym. U pacjentów z hemofilią zatrzymanie wody, które w ciężkich przypadkach
A o nasileniu od lekkiego do średnio ciężkiego sto- może doprowadzić do obrzęku mózgu. Poza tym
suje się desmopresynę do leczenia lub profilaktyki mogą wystąpić nudności, wymioty, kurczowe bóle
Układ hormonalny
krwawień podczas zabiegów operacyjnych. brzucha oraz sporadycznie reakcje nadwrażliwości.
Przeciwwskazaniami do stosowania wazopresyny
są: polidypsja w przebiegu choroby alkoholowej, hi-
ponatremia, niewydolność krążenia oraz ciężkie za-
Cys Tyr Cys Tyr burzenia gospodarki wodno-elektrolitowej.
S Ile S Phe
S Gln S Gln Szczególne powinowactwo do receptorów V1 charak-
Cys Asn Cys Asn teryzuje terlipresynę, która znalazła zastosowanie
w leczeniu krwawień z żylaków przełyku. Terlipre- B2
Pro Leu Gly NH2 Pro Arg Gly NH2
syna jest prolekiem lizynowazopresyny (lipresyny),
która powstaje powoli poprzez odszczepienie trzech
oksytocyna wazopresyna podstawników glicynowych. W wyniku tego czas
(adiuretyna) działania wydłuża się do 3–4 godzin.
Dawkowanie wynosi początkowo 2 mg i.v., a na-
stępnie 1 mg i.v. co 4 godziny.
Gly Do działań niepożądanych należą bradykardia,
Gly skurcz naczyń wieńcowych, wzrost ciśnienia tętni-
O
Gly czego krwi, bóle brzucha, biegunka oraz skurcz mięś-
Tyr Cys Tyr nia macicy.
S Phe S Phe W ciąży terlipresyna jest przeciwwskazana. Na-
leży zachować szczególną ostrożność, podając terli-
S Gln S Gln
presynę chorym na astmę oskrzelową, nadciśnienie,
Cys Asn Cys Asn
zaawansowaną miażdżycę oraz niewydolność nerek.
Pro D-Arg Gly NH2 Pro Lys Gly NH2
desmopresyna terlipresyna
2.2.2.2. Oksytocyna
Oksytocynę opisano w rozdziale B 2.8.10.1.
MUTSCHLER-2009.indd 403 2010-01-07 22:12:51
404 Układ hormonalny
2.3. Tarczyca
2.3.1. Anatomia gruczołu tarczowego ■ L-tyroksynę (lewotyroksynę = T4),
■ w znacznie mniejszej ilości trijodotyroninę (lioty-
U dorosłych ważąca około 20–25 g tarczyca otacza
roninę = T3),
podkowiasto tuż poniżej chrząstki tarczowatej tcha-
wicę dwoma płatami bocznymi i leżącą pomiędzy
oraz hormon biorący udział w regulacji stężenie Ca2+
nimi więzinę (zwaną cieśnią tarczycy) (zob. ryc. B
we krwi
2.3-1). Zazwyczaj prawy płat boczny jest trochę le-
piej wykształcony niż lewy. W 15% przypadków ■ kalcytoninę.
stwierdza się także trzeci płat, tzw. płat piramidowy,
z przodu przed chrząstką tarczowatą. Budowa chemiczna, biosynteza i magazynowa-
Unaczynione przegrody łącznotkankowe dzielą nie. Tyroksyna i trijodotyronina są pochodnymi ami-
tarczycę na różnej wielkości okolice płatowe. Tkan- nokwasu tyrozyny. Podstawowym składnikiem hor-
ka gruczołowa składa się z nieregularnie położonych, monów tarczycy jest niezawierająca jodu tyronina.
oddzielonych od siebie przegrodami łącznotkanko-
wymi, pęcherzyków, których ścianki są utworzone Tyroksyna (T4) to tetrajodotyronina, której czte-
z nabłonka jednowarstwowego. Wewnątrz pęcherzy- ry atomy jodu znajdują się w pozycji 3,3′,5,5′. Tri-
ków znajduje się jednorodna masa, tj. koloid, zawie- jodotyronina (T3) zawiera 3 atomy jodu w pozycji
rający prekursory hormonów tarczycy. 3,3′,5.
W wytwarzaniu i magazynowaniu T3 i T4 biorą udział
komórki nabłonka tarczycy (tyreocyty) i koloid pę-
A B wakuole brzeżne cherzyków. Początkowo jony jodku transportowane
nabłonek pęcherzyków są przez błonę podstawną tyreocytów razem z jona-
mi sodu z krwi wbrew gradientowi stężeń za pomocą
aktywnego kotransportu (jodowanie). W tyreocytach
pod wpływem peroksydazy ulegają one utlenieniu
do elementarnego jodu (zob. ryc. B 2.3-2). Elemen-
tarny jod zostaje następnie wbudowany do reszt
tyrozylowych białka, tzw. niejodowanej tyreoglo-
buliny. W ten sposób powstają odpowiednie mono-
jodo- i dijodozwiązki, które drogą oksydacji zostają
przekształcone w jodowane tyroniny związane z biał-
koloid kami. W tych przemianach bierze udział peroksydaza.
płat piramidowy Syntetyzowana w ten sposób tyreoglobulina zostaje
na drodze egzocytozy wydzielona do koloidu, gdzie
tarczyca odgrywa rolę magazynu hormonów tarczycy. W razie
potrzeby – po ponownym przetransportowaniu drogą
endocytozy z koloidu do tyreocytów – tyreoglobulina
ulega w lizosomach proteolizie i uwalniane są drogą
dyfuzji oba hormony, czyli tyroksyna i trijodotyro-
Ryc. B 2.3-1. Tarczyca (z płatem piramidowym) – widok z przo- nina w stosunku 10:1. Większość zawartej we krwi
du (według Bennighoffa-Goerttlera. T3 nie pochodzi jednak bezpośrednio z tarczycy, lecz
powstaje w tkankach obwodowych w wyniku odjo-
dowania tyroksyny (zob. poniżej).
Mechanizm działania i rola hormonów tarczycy.
2.3.2. Hormony tarczycy Właściwym, aktywnym metabolicznie hormonem
jest trijodotyronina. Po przeniknięciu przez błonę
Tarczyca wytwarza dwa hormony wpływające komórkową wiąże się ona ze swoim receptorem ją-
na przemianę materii: drowym. Aktywny kompleks hormon-receptor wiąże
MUTSCHLER-2009.indd 404 2010-01-07 22:12:52
Układ hormonalny 405
I I
HO I COOH HO I COOH
NH2 NH2
I O H O
I I
tyroksyna (lewotyroksyna,T4) trijodotyronina (liotyronina, T3)
Układ hormonalny
się ze swoistymi sekwencjami DNA i tą drogą wpły- i tworzenie ciepła w całym organizmie (z wyjątkiem
wa na ekspresję genów, stanowiąc ważny regulator mózgu). W wątrobie stwierdza sie nasilenie glikoge-
transkrypcji i wskutek tego wtórnie biosyntezy białek nolizy i glukoneogenezy (efekt antagonistyczny w sto-
w komórkach docelowych (zob. ryc. B 2.3-3). Pod sunku do insuliny). T3 powoduje ponadto zwiększe-
wpływem T3 dochodzi przede wszystkim do wzmo- nie liczby receptorów LDL w błonie komórkowej B2
żonego wytwarzania pompy Na+/K+ ATP-zależnej hepatocytów oraz gęstości receptorów β1 w sercu.
i enzymów mitochondrialnych (głównie enzymów W stężeniach fizjologicznych T3 działa anabolicznie,
biorących udział w metabolizmie węglowodanów natomiast w wyższych stężeniach działa katabolicz-
i tłuszczów). nie, nasilając rozkład białek w mięśniach. Wpływ T3
Tą drogą trijodotyronina powoduje nasilenie prze- na metabolizm tłuszczów jest dwukierunkowy: z jed-
mian energetycznych, zwiększenie zużycia tlenu nej strony wzmaga ona lipolizę w osoczu, a z drugiej
MIT
peroksydaza
– jodowa T3-TG-T4
I
koloid I2
DIT
egzocytoza endocytoza MIT
CI–
T3-TG-T4
DIT
dejody-
tyreocyt naza
prekursor MIT
tyreoglobuliny
tyrozyna proteoliza
DIT
I– Na+ T3 T4
Gs
TSH
T3 T4
I–
TSH
naczynie włosowate białka transportujące
Ryc. B 2.3-2. Schemat biosyntezy, magazynowania i uwalniania hormonów tarczycy (według Thewsa, Mutschlera, Vaupela).
TSH – hormon tyreotropowy, MIT – monojodotyrozyna; DIT – diodotyrozyna, T3 – trijodotyronina; T4 – tyroksyna, TG - tyreoglobulina.
MUTSCHLER-2009.indd 405 2010-01-07 22:12:52
406 Układ hormonalny
T3 T4 Kinetyka. Dziennie tarczyca wydziela ok. 90 μg T4
i 8 μg T3. Po podaniu doustnym tyroksyna wchłania
się w 80% (jednoczesne spożycie pokarmu wyraźnie
zmniejsza odsetek wchłoniętej tyroksyny!), a trijo-
T3
dotyronina w 90–100%. Krążąca we krwi tyroksyna
(ok. 6–12 μg/100 ml) jest prawie całkowicie związa-
T3
jądro na z białkami. Jedynie 0,05% występuje w postaci
komórkowe wolnej. Mniej więcej 60% T4 wiąże się z globuliną
wiążącą tyroksynę (TBG – thyroxin binding globulin)
i 30% z prealbuminą wiążącą tyroksynę (TBPA; trans-
terytyna) oraz ok. 10% z albuminami. Ilość wolnej
transkrypcja trijodotyroniny, tj. 0,5%, dziesięciokrotnie przewyż-
sza ilość wolnej tyroksyny.
translacja białka Estrogeny podwyższają stężenie TBG w osoczu,
podczas gdy hormony kory nadnerczy i androgeny
obniżają jej stężenie. Wynikająca z tego faktu zmiana
stężenia wolnego, czyli aktywnego hormonu, zostaje
pompa Na+/K+ ATP-zależna, enzymy biorące udział
w metabolizmie węglowodanów, tłuszczów i in. jednak szybko wyrównana przez modyfikację wy-
dzielania tyreotropiny.
Ryc. B 2.3-3. Mechanizm działania hormonów tarczycy. T3 – tri-
W tkankach obwodowych, przede wszystkim
jodotyronina, T4 – tyroksyna.
w wątrobie i nerkach, T4 pod wpływem dejodyna-
zy jodotyroninowej zostaje przekształcona przede
wszystkim w T3 (monoodjodowanie końcowego pod-
pobudza lipogenezę w tkance tłuszczowej i wątrobie,
stawnika fenolowego), a w niewielkim stopniu rów-
przy czym w prawidłowych warunkach przeważa li-
nież w nieaktywną 3,5,5′-trijodotyroninę (rT3 = rever-
poliza.
se T3; monoodjodowanie podstawnika fenylowego
Hormony tarczycy odgrywają istotną rolę w pro-
sąsiadującego z alifatycznym łańcuchem bocznym).
cesie wzrostu i rozwoju organizmu. Fizjologiczne stę-
Dalsze przemiany polegają na ponownym odjodowa-
żenia tych hormonów (w połączeniu z somatotropi-
niu, sprzęganiu z kwasem glukuronowym lub siarcza-
ną) warunkują prawidłowy wzrost na długość, a także
nem, dekarboksylacji i/lub dezaminowaniu. Powstałe
prawidłowy rozwój zawiązków narządów i dojrzewa-
metabolity są nieczynne hormonalnie.
nie organów wewnętrznych, przede wszystkim tkanki
Wydalane z żółcią produkty sprzęgania T3 i T4 ule-
kostnej i mózgu. Hormony tarczycy pobudzają roz-
gają w jelitach w dużej mierze rozszczepieniu i wolne
wój mózgowia poprzez stymulację mielinizacji i two-
hormony są ponownie wchłaniane (krążenie wątro-
rzenia dendrytów.
bowo-jelitowe).
Okres półtrwania tyroksyny w osoczu wynosi ok. 7
Regulacja stężenia hormonów tarczycy. Wydziela-
dni, natomiast trijodotyroniny tylko 1–2 dni.
nie hormonów tarczycy jest regulowane przez układ
podwzgórze-przysadka: w zależności od stężenia
Wskazania. Stosowanie hormonów tarczycy
hormonów tarczycy w osoczu wydzielany jest w od-
jest wskazane:
powiedniej ilości hormon uwalniający tyreotropinę,
a pod jego wpływem jest wydzielana tyreotropi- ■ jako leczenie substytucyjne we wszystkich rodza-
na. TSH pobudza następnie tarczycę do uwalniania jach niedoczynności tarczycy (łącznie z chorobą
do krwi T4 oraz – w mniejszym stopniu – T3, które Hashimoto),
zwrotnie wpływają na przysadkę i podwzgórze na za-
■ w celu hamowania działania tyreotropiny w przy-
sadzie ujemnego sprzężenia zwrotnego (ryc. B 2-3 B).
padku wola z eutyreozą, jak również w profilaktyce
Dodatkowo jeszcze inne czynniki, np. stres i tempera-
nawrotu wola po strumektomii,
tura ciała, mogą wpływać modyfikująco na regulację
wydzielania hormonów tarczycy. Zimno, obciążenie ■ w połączeniu z lekami tyreostatycznymi przy le-
psychiczne i fizyczne podwyższają wartość żądaną czeniu nadczynności tarczycy w celu zapobiegania
układu regulacji (wymagane stężenie hormonów tar- rozwojowi jatrogennej niedoczynności tarczycy.
czycy), natomiast ciepło i stan spoczynku obniżają
tę wartość. Somatostatyna, dopamina i glukokortyko- Lekiem z wyboru – z powodu wyrównanego stę-
steroidy hamują uwalnianie tyreotropiny. żenia hormonu wskutek długiego czasu działania
MUTSCHLER-2009.indd 406 2010-01-07 22:12:53
Układ hormonalny 407
– jest lewotyroksyna? Obecnie nie ma żadnych wska- 2.3.3.1. Wole
zań do użycia T3 lub kombinacji T3 i T4.
Patofizjologia. Termin wole oznacza powiększenie
Dawkowanie. Dawki są dobierane indywidualnie
objętości tarczycy w wyniku zwiększenia liczby pę-
pod odpowiednią kontrolą. Z powodu dużego ry-
cherzyków (rozrost tarczycy). Wtórnie do rozrostu
zyka kumulacji leku terapię rozpoczyna się od ma-
może dojść do zaburzenia funkcji pęcherzyków oraz
łych dawek, które następnie stopniowo się zwiększa.
pojawienia się zwapnień i martwicy. Według cha-
Przeciętna dzienna dawka podtrzymująca w terapii
rakteru rozrostu tarczycy wyróżnia się wole proste
substytucyjnej hormonami tarczycy, a także w tera-
i guzkowe. Na podstawie typu wola nie można jednak
Układ hormonalny
pii supresyjnej wola z eutyreozą wynosi 0,1–0,2 mg
wnioskować o czynności tarczycy: wole pojawia się
tyroksyny. W profilaktyce nawrotów wola z eutyreo-
przy prawidłowej funkcji tarczycy (wole obojętne),
zą po strumektomii wystarczająca jest dawka 0,025–
jak również przy jej niedoczynności lub nadczynno-
0,1 mg T4. Leczenie musi zazwyczaj być prowadzone
ści (zob. poniżej).
do końca życia.
Podczas ciąży zapotrzebowanie na hormony tar-
Wole obojętne, tzn. wole cechujące się prawidłowymi
czycy zwiększa się o ok. 40%.
stężeniami T3 i T4 (wcześniej zwane wolem łagodnym),
Działania niepożądane. Jeśli dawkowanie jest pra-
jest najczęstszym schorzeniem tarczycy w Niemczech B2
oraz w Polsce. Jego główną przyczyną jest występują-
widłowe, nie powinny pojawić się działania niepo-
cy powszechnie niedobór jodu w pożywieniu (wole en-
żądane. Przedawkowanie prowadzi do wystąpienia
demiczne). U ponad 10% ludności Niemiec stwierdza
objawów nadczynności tarczycy (zob. poniżej).
się powiększenie tarczycy w następstwie niedoboru
jodu. Dopóki rozrost tkanki tarczycy oraz optymalne
Przeciwwskazania. Względnymi przeciwwskaza-
wykorzystanie dostarczanego jodu – rozpatrując to ca-
niami są dusznica bolesna, zawał mięśnia sercowego,
łościowo – kompensuje niedostateczne zaopatrzenie
zapalenie mięśnia sercowego i zaburzenia rytmu ser-
w jod, nie rozwijają się objawy niedoboru hormonów
ca z tachykardią.
tarczycy, a jedynie powstanie wola. Poza wzmożo-
nym wydzielaniem tyreotropiny za proces tworzenia
Interakcje. Żywice jonowymienne, np. cholestyrami-
wola odpowiada również obecność wielu czynników
na, zmniejszają lub całkowicie hamują wchłanianie
wzrostu, np. czynnika wzrostowego naskórka (EGF)
hormonów tarczycy z przewodu pokrmowego. Związ-
lub IGF-1, których wytwarzanie jest wzmożone przy
ki zawierające glin lub żelazo również zmniejszają
wewnątrztarczycowym niedoborze jodu. Jeżeli jednak
wchłanianie tyroksyny. Glukokortykosteroidy, pro-
mechanizmy kompensacyjne nie pozwalają na wyrów-
pylotiouracyl oraz beta-blokery hamują obwodową
nanie niedoboru, rozwija się (jawna klinicznie) hipo-
konwersję T4 do T3. Estrogeny zmniejszają osoczowe
tyreoza (zob. poniżej). Długo utrzymujące się wole
stężenie wolnej tyroksyny. Barbiturany i inne leki in-
obojętne z towarzyszącym długotrwałym niedoborem
dukujące enzymy mogą zwiększyć klirens wątrobo-
jodu stwarza ryzyko powstania tzw. zimnych i gorą-
wy tyroksyny. Hormony tarczycy nasilają działanie
cych guzków będących następstwem współistnienia
doustnych leków przeciwzakrzepowych będących
tzw. mikroheterogenności prawidłowych tyreocytów,
pochodnymi hydroksykumaryny oraz zmniejszają
oraz przewlekłego bodźca wzrostowego uwarunkowa-
lub nasilają działanie hipoglikemizujące doustnych
nego niedoborem jodu.
leków przeciwcukrzycowych.
Mikroheterogenność tyreocytów oznacza, że już w warun-
kach fizjologicznych w następstwie somatycznych mutacji
dochodzi do powstania różnych klonów tyreocytów, róż-
2.3.3. Zaburzenia funkcji tarczycy niących się od siebie pod względem aktywności (m.in. wy-
chwytu jodu, syntezy i uwalniania hormonów, wrażliwości
na TSH), jak również reakcji na niedobór jodu.
Schorzenia tarczycy charakteryzują się zmianami
wielkości gruczołu tarczowego i/lub jego czynności.
Jako guzki zimne (w scyntygramie tarczycy) określa
Można je podzielić na:
się guzkowate obszary w tarczycy charakteryzujące
■ powiększenie tarczycy (wole), się zmniejszoną jodochwytnością (obszary hormo-
nalnie nieczynne). Ich znaczenie kliniczne wynika
■ niedoczynność tarczycy (hipotyreozę),
przede wszystkim z tego, że mogą one ulegać złośli-
■ nadczynność tarczycy (hipertyreozę). wemu zwyrodnieniu.
MUTSCHLER-2009.indd 407 2010-01-07 22:12:53
408 Układ hormonalny
Profilaktyka i farmakoterapia wola obojętnego. u kobiety ciężarnej lub stosowania przez nią leków
Działania profilaktyczne mające zapobiegać rozwo- hamujących czynność tarczycy.
jowi wola obojętnego polegają na zapewnieniu od- Jeśli tarczyca matki podczas ciąży wytwarza hor-
powiedniej podaży jodu i powinny być prowadzone mony w wystarczającym stopniu, zapotrzebowanie
od wieku młodzieńczego. Dzienne zapotrzebowanie płodu może być pokryte i dzięki temu po porodzie
na jod wynosi 150–200 μg, a u kobiet w ciąży wzrasta nie obserwuje się u noworodka żadnych wyraźnych
do 230–260 μg. W przypadku niedostatecznej podaży objawów niedoczynności tarczycy, chociaż on sam
jodu z pożywieniem i wodą pitną wskazane jest pro- w życiu płodowym nie był w stanie syntetyzować
filaktyczne spożywanie 5 g jodowanej soli kuchennej wystarczającej ilości hormonów tarczycy. Jednak
dziennie, co odpowiada 100 μg jodu. Alternatywą po porodzie, jeśli nie zastosuje się terapii substy-
jest przyjmowanie tabletek z jodkiem potasu, także tucyjnej hormonami tarczycy, stopniowo pojawia
w dawce 100 μg jodu dziennie. się opóźnienie rozwoju fizycznego i umysłowego,
adynamia, zaparcia i suchość skóry. Budowa ciała
Celem farmakoterapii wola prostego jest zmniejsze- pozostaje masywna i niezgrabna (niedobór wzrostu
nie objętości wola oraz utrzymanie osiągniętego celu z nieprawidłowymi proporcjami ciała). Dłonie i stopy
terapii (tzw. profilaktyka wtórna). Zastosowanie znaj- wyglądają jak łapy u zwierząt, wargi są pogrubiałe,
dują jodki lub terapia kobinowana jodkami i tyrok- język jest powiększony, twarz pomarszczona, a ciś-
syną (w stosunku 2:1–1:1). Zazwyczaj w ciągu 6–12 nienie krwi i napięcie mięśniowe są obniżone. Jeżeli
miesięcy udaje się uzyskać zmniejszenie objętości jednak możliwie szybko po porodzie zostaną włączo-
tarczycy o 25–40%. Szanse powodzenia są większe ne do terapii odpowiednie dawki hormonów tarczycy,
u młodych chorych oraz w przypadku wola o wielkim dzieci rozwijają się prawidłowo.
lub średnim rozmiarze. U dzieci i młodzieży stosu- Nie dotyczy to jednak sytuacji, w których rów-
je się zazwyczaj monoterapię jodkami, podczas gdy nież u matki podczas ciąży występował niedobór
u osób po 40 r.ż. i kobiet w ciąży zazwyczaj stosu- hormonów tarczycy. W takim wypadku nawet przy
je się terapię kombinowaną. Dzienna dawka jodków zastosowaniu bezpośrednio po porodzie pełne-
stosowana w leczeniu wola wynosi 200–300 μg. go substytucyjnego leczenia hormonami tarczycy
nie jest już możliwe osiągniecie przez dziecko pra-
Przy bardzo dużym wolu, które zaburza funkcję okolicz- widłowego rozwoju umysłowego. Dzieci pozostają
nych narządów, konieczne jest leczenie operacyjne, a przy niedorozwinięte umysłowo, ponieważ odpowiednie
przeciwwskazaniach do operacji – radioterapia (zob. poni-
żej). stężenie hormonow tarczycy w okresie prenatalnym
jest niezbędne do prawidłowego rozwoju funkcji
mózgu.
W przeszłości szczególnie często występowały
2.3.3.2. Niedoczynność tarczycy tego rodzaju przypadki na terenach (np. w dolinach
górskich), gdzie woda pitna zawierała zbyt małą ilość
Patofizjologia. Termin niedoczynność tarczycy ozna- jodu, wskutek czego całkowita jego ilość przyjmo-
cza niedostateczne wytwarzanie lub wydzielanie albo wana z pożywieniem była niedostateczna. Od czasu,
(rzadko) działanie hormonów tarczycy w stopniu nie- gdy wprowadzono w profilaktyczne jodowanie soli
wystarczającym. Odpowiednio do przyczyn lub cza- kuchennej, prawie nie pojawiają się na tych terenach
su wystąpienia schorzenia rozróżnia się następujące nowe zachorowania.
jego postacie:
Dzieci z objawami ciężkiego niedoboru hormonów tarczy-
■ niedoczynność tarczycy noworodków, cy, zwłaszcza takie z objawami upośledzenia umysłowego,
nazywano dawniej kretynami, a odpowiedni obraz klinicz-
■ niedoczynność tarczycy nabytą po urodzeniu się, ny choroby kretynizmem. Obecnie nie należy już używać
■ obwodową oporność na hormony (bardzo rzadko). tego terminu, ponieważ ciężki niedobór hormonów tarczy-
cy nie stanowi jednolitej jednostki chorobowej, lecz można
go podzielić na postacie z (długo) istniejącym niedoborem
Niedoczynność tarczycy noworodków (wrodzo- jodu i inne z genetycznie uwarunkowanym nieprawidło-
na) jest następstwem aplazji lub hipoplazji tarczy- wym wykorzystaniem jodu.
cy, genetycznie uwarunkowanych zaburzeń syntezy
hormonów lub niewystarczającego wydzielania. Niedoczynność tarczycy nabytą po urodzeniu, bio-
Ta postać niedoczynności tarczycy może być także rąc pod uwagę patogenezę można podzielić na pier-
następstwem znacznego ciągłego niedoboru jodu wotną i wtórną.
MUTSCHLER-2009.indd 408 2010-01-07 22:12:53
Układ hormonalny 409
Pierwotna niedoczynność tarczycy, której przy- 2.3.3.3. Nadczynność tarczycy
czyną są zaburzenia dotyczące bezposrednio gru-
czołu tarczowego, może być wywołana przez stały 2.3.3.3.1. Patofizjologia
niedobór jodu, procesy zapalne (np. przewlekłe za-
palenie tarczycy, inaczej choroba Hashimoto), nowo-
W przebiegu nadczynności tarczycy obserwuje się
twory tarczycy, leczenie jodem radioaktywnym lub
zwiększone wydzielanie tyroksyny i trijodotyroniny.
podawanie tyreostatyków (zob. poniżej). Stężenia T3
Biorąc pod uwagę różne przyczyny, należy rozróż-
i T4 są obniżone. Wskutek tego na drodze sprzężenia
niać:
zwrotnego dochodzi do wzrostu stężenia TSH (zob.
Układ hormonalny
tab. B 2.3-1). ■ nadczynność tarczycy o podłożu nieimmunologicz-
nym,
Tabela B 2.3-1. Zmiany stężeń tyreotropiny (TSH), trijodotyro- ■ nadczynność tarczycy o podłożu immunologicz-
niny (T3) i tyroksyny (T4) w przebiegu różnych schorzeń tarczycy nym.
(n = stężenie prawidłowe)
W przypadku nadczynności tarczycy o podło-
Choroba tarczycy TSH T3, T4 żu nieimmunologicznym stwierdza się jedno- lub
wieloogniskową czynnościową autonomię tarczycy B2
pierwotna niedoczynność tarczycy ↑ ↓ (guzek autonomiczny lub mnogie gorące guzki).
wtórna niedoczynność tarczycy ↓ ↓ Jest to odgraniczony, podobny do nowotworowego
rozrost części tkanki tarczycy, która już nie podlega
wole obojętne ↑ n
regulacji podwzgórzowo-przysadkowej, lecz w spo-
choroba Basedowa ↓ ↑
sób autonomiczny wytwarza i wydziela do krwiobie-
guzek autonomiczny ↓ ↑ gu zwiększoną ilość hormonów tarczycy. Nie stwier-
wtórna nadczynność tarczycy ↑ ↑ dza się objawów ocznych (zob. poniżej). (Poza tym
występuje także rozproszona postać autonomii czyn-
nościowej).
Innymi możliwymi przyczynami hipertyreozy
Wtórna niedoczynność tarczycy jest następstwem o podłożu nieimmunologicznym są stany zapalne,
zaburzeń funkcji podwzgórza albo przysadki, w któ- nowotwory, wzmożone uwalnianie TSH lub wy-
rych przebiegu obniżone jest zarówno stężenie TSH, chwyt dużych dawek jodu, który może ujawnić utajo-
jak i T3 oraz T4 w osoczu (zob. tab. B 2.3-1). ną tkankę autonomiczną, a także podanie zbyt dużych
Pełny obraz kliniczny niedoczynności tarczycy dawek hormonów tarczycy.
występującej u dorosłych, z powodu obserwowanego
specyficznego pogrubienia i obrzęku skóry w następ- Nadczynność tarczycy o podłożu immunologicz-
stwie odkładania się glikozoaminoglikanów określa nym cechuje się uogólnionym powiększeniem tar-
się jako obrzęk śluzowaty. Wskutek zmniejszonej czycy (tzw. wole toksyczne). Do tej grupy zalicza się
podstawowej przemiany materii dochodzi do obniże- przede wszystkim te typy choroby Basedowa, któ-
nia temperatury ciała oraz zmniejszenia aktywności rych podłoże stanowią reakcje autoimmunologiczne.
psychicznej, czemu towarzyszy spowolnienie mowy Czynnikiem wywołującym nadczynność tarczycy
i chrypka. U chorych stwierdza się często nadwraż- w przebiegu choroby Basedowa są wytwarzane przez
liwość na zimno, łatwą męczliwość, bradykardię, limfocyty B immunoglobuliny klasy IgG, stymulują-
zaparcia i nadwagę. Włosy i paznokcie są łamliwe, ce czynność gruczołu tarczowego (TSI). Te działają-
a ponadto występuje utrata libido i zaburzenia po- ce agonistycznie i prowadzące do rozrostu tarczycy
tencji. autoprzeciwciała (przeciwciała przeciw receptorom
TSH, anty-TSHR) wypierają tyreotropinę z jej miejsc
Podłożem obwodowej oporności na hormony tar- wiązania się w błonie komórkowej tyreocytów i wy-
czycy są genetycznie uwarunkowane wady recepto- wołują długotrwałą stymulację gruczołu tarczowego.
rów hormonów tarczycy. W następstwie zwiększonego uwalniania T3 i T4, od-
wrotnie niż przy hipertyreozach (wtórnych nadczyn-
Farmakoterpia. Leczenie niedoczynności tarczycy nościach tarczycy) wywołanych oddziaływaniem
polega na trwającej całe życie substytucji hormonów podwzgórza i przysadki, obserwuje się obniżone stę-
tarczycy, a w szczególności tyroksyny. żenie TSH (zob. tab. B 2.3-1).
MUTSCHLER-2009.indd 409 2010-01-07 22:12:53
410 Układ hormonalny
W przebiegu wszystkich hipertyreoz dochodzi dowanie jodu w prekursory hormonów tarczycy
do zwiększenia podstawowej przemiany materii, (środki hamujące jodowanie tyrozyny);
w wyniku czego stwierdza się podwyższoną tempe-
■ jony jodku i jod-jodek potasowy – krótkotrwale ha-
raturę ciała, zwiększoną pojemność minutową serca,
mują uwalnianie hormonów tarczycy.
przyspieszenie akcji serca i wzmożoną pobudliwość,
a mimo dobrego apetytu dochodzi do utraty masy
ciała. Chorzy skarżą się na kołatanie serca, nietole-
2.3.3.3.2.1. Nadchlorany
rowanie wysokich temperatur, biegunkę i nadmierną
potliwość. Wskutek zwiększonej stymulacji OUN Jony nadchloranowe hamują na zasadzie antagonizmu
obserwuje się niepokój, bezsenność i drżenia drob- kompetycyjnego wychwyt jonów jodkowych przez tarczycę.
nofaliste. Wadą terapii nadchloranem jest to, że wskutek wzajemnego
W przebiegu klasycznej choroby Basedowa poza hamowania kompetycyjnego i długiego działania utrzymu-
wyżej wymienionymi objawami często stwierdza jącego się przez kilka tygodni, bezpośrednio po odstawie-
niu nadchloranów nie można przeprowadzić przedopera-
się orbitopatię tarczycową, najczęściej w postaci cyjnego leczenia jodem oraz terapii jodem radioaktywnym
obustronnego wytrzeszczu (exophthalmus). Jednak (zob. poniżej).
u 30% chorych z nadczynnością tarczycy o podłożu Nadchlorany są wskazane przede wszystkim, gdy ko-
immunologicznym nie stwierdza się tego objawu. nieczne jest wykonanie badania obrazowego z podaniem jo-
Przyczyna orbitopatii tarczycowej, będącej następ- dowego środka kontrastowego u chorego ze zwiększonym
ryzykiem rozwoju przełomu tarczycowego w wyniku wy-
stwem nagromadzenia glikozoaminoglikanów w tkan- chwytu jodu przez tarczycę. W takiej sytuacji nadchlorany
ce pozagałkowej, nacieku limfocytów i obrzęku tkan- podaje się w terapii kombinowanej z pochodnymi tiomocz-
ki pozagałkowej, nie została jeszcze jednoznacznie nika (zob. poniżej) i w ten sposób zapobiega wychwytowi
wyjaśniona. Pewne jest jej autoimmunologiczne pod- jodu oraz jego wykorzystaniu przez tarczycę. W przeszłości
łoże. Nie stwierdzono jednak bezpośredniej korelacji stosowano nadchlorany także w razie nietolerancji pochod-
nych tiomocznika, przy czym obecnie w takich przypad-
pomiędzy objawami ocznymi i stężeniem przeciwciał kach stosuje się przede wszystkim radioterapię.
anty-TSHR we krwi. Przy podawaniu jodowego środka kontrastowego stosu-
je się dawkę 1 g nadchloranu (razem z 15–30 mg karbima-
Przełom tarczycowy jest nacięższą ostrą postacią zolu – zob. poniżej) dziennie przez 8–10 dni, a przy prze-
nadczynności tarczycy. Może rozwinąć się niespo- wlekłym leczeniu choroby Basedowa 300 mg dziennie.
Działaniami niepożądanymi nadchloranów są zaburze-
dziewanie i gwałtownie w ciągu kilku godzin lub dni. nia żołądkowo-jelitowe. Natomiast do poważniejszych,
W następstwie ciężkich zaburzeń przemiany materii rzadko występujących działań niepożądanych zalicza się
rozwijają się tachykardia, zaburzenia rytmu serca, agranulocytozę, niedokrwistosć aplastyczną i zespół ne-
wysoka gorączka, nasilona biegunka z wymiotami frotyczny. Powikłania te pojawiają się przeważnie dopiero
oraz zaburzenia świadomości ze śpiączką włącznie. po upływie kilku miesięcy od rozpoczęcia terapii.
Nadchlorany są przeciwwskazane przy dużym wolu
guzkowym, przy wolu zamostkowym, w chorobie Basedo-
W przypadku wszystkich postaci hipertyreozy należy wa oraz w czasie ciąży lub karmienia piersią.
pamiętać o tym, że wraz z wiekiem objawy stają się Substancje zawierające jod ograniczają działanie jonów
coraz bardziej nietypowe i mniej nasilone. W pode- nadchloranowych.
szłym wieku zazwyczaj występują tachykardia i często
nastrój depresyjny.
2.3.3.3.2.2. Pochodne tiouracylu
i merkaptoimidazolu
2.3.3.3.2. Tyreostatyki Charakterystyczną cechą budowy wszystkich tyreo-
statyków z tej grupy jest ugrupowanie zawierające
Tyreostatyki hamują syntezę i uwalnianie hormonów tiomocznik. Z pochodnych tiouracylu stosuje się
z tarczycy i z tego powodu znajdują zastosowanie jeszcze tylko propylotiouracyl. Takie samo działanie
w terapii nadczynności tego gruczołu. Na podsta- pod względem jakościowym, ale ilościowo znacznie
wie mechanizmu działania można je podzielić na na- silniejsze, wykazują pochodne merkaptoimidazolu,
stępujące grupy: tj. karbimazol i jego aktywny metabolit tiamazol.
■ nadchlorany – blokują wychwyt jodku przez tarczy-
Właściwości farmakologiczne i mechanizm dzia-
cę (środki hamujące jodowanie tyreoglobuliny);
łania. Substancje te, jak wyżej wspomniano, hamują
■ pochodne tiouracylu i merkaptoimidazolu – bloku- peroksydazę. W wyniku tego dochodzi do hamowa-
ją przekształcanie jodku w jod i wskutek tego wbu- nia przekształcania jodku w jod, wbudowywania jodu
MUTSCHLER-2009.indd 410 2010-01-07 22:12:54
Układ hormonalny 411
do ugrupowania tyrozyny oraz łączenia jodotyrozyn Obecnie zaleca się przede wszystkim terapię małymi
w jodotyroniny (zob. ryc. B 2.3-4). Propylotioura- dawkami z powodu mniejszego ryzyka pojawienia
cyl zmniejsza także obwodową konwersję T4 do T3 się ciężkich działań niepożądanych (zob. poniżej).
w wyniku odjodowania, aczkolwiek kliniczne zna-
czenie tego efektu stanowi kwestię sporną. Istnieją Farmakokinetyka. Pochodne tiouracylu i merkapto-
również dane świadczące o działaniu immunosupre- imidazolu dobrze wchłaniają się z przewodu pokar-
syjnym pochodnych tiomocznika, które jest istotne mowego. Prolek karbimazol w całości ulega biotrans-
przede wszystkim u pacjentów z chorobą Basedowa. formacji do postaci czynnej tiamazolu.
Wskutek wymienionych działań: Osoczowy okres półtrwania propylotiouracy-
Układ hormonalny
lu wynosi 1,5–2 godz., a karbimazolu i tiamazolu
■ stopniowo zmniejsza się zawartość hormonów
6–13 godzin. Ponieważ tiamazol jest magazynowany
w tarczycy,
w tarczycy, jego działanie trwa znacznie dłużej, tj.
■ obniża się stężenie hormonów tarczycy we krwi. około 36–72 godzin. Ważne reakcje biotransforma-
cji to S-oksydacja i sprzęganie z kwasem glukurono-
Efekt działania pochodnych tiomocznika widoczny wym. Wszystkie trzy związki wydalane są z moczem
jest dopiero po upływie 1–2 tygodni, ponieważ po- i żółcią.
czątkowo tarczyca dysponuje jeszcze wystarczającą
ilością tyreoglobuliny. Wskazania. Tyreostatyki będące pochodnymi tio- B2
Występujący ewentualnie wytrzeszcz nie ustępuje mocznika są wskazane w terapii nadczynności tar-
w trakcie terapii pochodnymi tiomocznika, a może na- czycy, a w szczególności w leczeniu choroby Base-
wet ulec nasileniu. Podobnie jak w przypadku innych dowa.
tyreostatyków, z powodu zwiększonego wydzielania
hormonu tyreotropowego istnieje niebezpieczeństwo Dawkowanie. Początkowe dobowe dawki propylo-
działania wolotwórczego (tworzenie wola). Można tiouracylu wynoszą 150–400 mg, karbimazolu 15–60
tego uniknąć, stosując odpowiedni schemat terapeu- mg i tiamazolu 10–40 mg. Po osiągnięciu eutyreozy
tyczny. Istnieją przy tym następujące podstawowe rozpoczyna się podawanie dawek podtrzymujących:
możliwości postępowania: 50–150 mg propylotiouracylu, 5–20 mg karbimazo-
lu i 2,5–10 mg tiamazolu dziennie. Po roku terapii
■ podawanie tyreostatyków w małych dawkach po-
można podjąć próbę odstawienia pochodnych tio-
woduje jedynie częściowe hamowanie syntezy hor-
mocznika.
monów, tzn. zapobiega wystąpieniu stanu hipoty-
reozy,
■ przy całkowitym blokowaniu wytwarzania hormo-
nów tarczycy przez tyreostatyki podawane w du- NH2
tiomocznik
żych dawkach stosuje się dodatkowo tyroksynę. S NH2
C3H7
peroksydaza
HN propylotiouracyl
S NH O
jodki jod
N
pochodne tiomocznika
HS N tiamazol
CH3
peroksydaza O
H5C2O N
karbimazol
S N
jodotyrozyna T3 T4 CH3
Ryc. B 2.3-4. Mechanizm działania pochodnych tiomocznika.
MUTSCHLER-2009.indd 411 2010-01-07 22:12:54
412 Układ hormonalny
Działania niepożądane. Działania niepożądane 2.3.3.3.2.4. Jod promieniotwórczy
(w pewnym stopniu zależne od dawki) to nudności,
zaburzenia powonienia, a także reakcje alergiczne
Jod promieniotwórczy (131I), który charakteryzuje się
(m.in. rumień, pokrzywka) oraz częściowo zagra-
okresem półtrwania wynoszącym 8 dni i ulega roz-
żające życiu działanie mielotoksyczne (leukopenia,
padowi, emitując promienie β i γ, jest gromadzony
trombocytopenia, < 1% agranulocytoza). Ryzyko
w tarczycy w taki sam sposób jak jod nieradioak-
zahamowania czynności szpiku jest najmniejsze przy
tywny. Jego promieniowanie radioaktywne, przede
podawaniu małych dawek pochodnych merkaptoimi-
wszystkim promienie β, może być wykorzystane
dazolu. Ponieważ rozwój agranulocytozy przebiega
w celu zniszczenia tkanki tarczycy i wskutek tego
bardzo szybko, chorzy muszą zostać poinformowa-
zmniejszenia wytwarzania hormonów tarczycy. Z po-
ni o możliwości wystąpienia określonych objawów
wodu niewielkiej penetracji promieniowania β jego
agranulocytozy (zapalenie jamy ustnej, zapalenie
działanie dotyczy prawie wyłącznie tkanki tarczycy
gardła, zapalenie migdałków, gorączka) i koniecz-
i tylko w znikomym stopniu wpływa na sąsiadujące
ności natychmiastowego przerwania terapii w razie
narządy. Efektów terapeutycznych można oczekiwać
ich wystąpienia.
dopiero po upływie 2–4 miesięcy.
Tyreostatyki przenikają przez barierę łożyskową.
Oprócz złośliwych nowotworów tarczycy wskaza-
Z powodu niebezpieczeństwa wystąpienia hipoty-
niami do zastosowania jodu radioaktywnego jest nad-
reozy u dziecka – w czasie ciąży i karmienia piersią
czynność tarczycy (autonomia czynnościowa, nawrót
tyreostatyki muszą być podawane w możliwie naj-
choroby Basedowa, nietolerancja pochodnych tio-
mniejszej dawce.
mocznika) oraz wole obojętne wymagające terapii.
Dawkowanie jest ustalane indywidualne, przy
Przeciwwskazania. Jako przeciwwskazania do sto-
czym średnia dawka w leczeniu autonomii czynnoś-
sowania należy wymienić wole zamostkowe z powo-
ciowej wynosi 200 Gy.
du niebezpieczeństwa ucisku na tchawicę oraz duży
Zaletą terapii jodem promieniotwórczym jest
wytrzeszcz oczu.
względnie duża skuteczność (ok. 90%), brak miejsco-
wych działań niepożądanych oraz mały odsetek na-
Interakcje. Jodki upośledzają działanie pochodnych
wrotów. Niekorzystnymi cechami tej terapii jest długi
tiomocznika.
okres oczekiwania na wystąpienie efektu terapeutycz-
nego, w czasie którego trzeba wielokrotnie stosować
przerywane leczenie tyreostatyczne, a przede wszyst-
2.3.3.3.2.3. Jodki i jod kim ryzyko rozwoju po wielu latach hipotyreozy (tzw.
późnej niedoczynności tarczycy).
Jodki i jod w dużych dawkach działają krótkotrwale, Ogólnie można stwierdzić, że ryzyko terapii jo-
jak tyreostatyki, wywołując przejściowe zmniejszenie dem radioaktywnym jest jednak niewielkie, także
aktywności enzymów proteolitycznych uwalniających nieznaczne jest niebezpieczeństwo powstania raka
hormony tarczycy z tyreoglobuliny. W następstwie tarczycy lub białaczki czy uszkodzenia gonad. Dlate-
tego zwiększa się gromadzenie hormonów tarczycy go obecnie, inaczej niż w przeszłości, leczenie jodem
w koloidzie oraz zmniejsza się wielkość i ukrwienie radioaktywnym jest stosowane również u młodszych
wola nadczynnego, co ułatwia przeprowadzenie za- pacjentów (od 25 r.ż.).
biegu strumektomii. Przeciwwskazaniami do terapii jodem radioak-
Obecnie rzadko istnieją wskazania do przedope- tywnym są ciąża, okres karmienia piersią i okres
racyjnego podania dużych dawek jodu u chorych wzrostu.
z wolem. W pierwszej kolejności należy podać ty-
reostatyki z grupy pochodnych tiomocznika (nie Jod promieniotwórczy w małych dawkach jest stosowany
nadchlorany!) w celu uzyskania eutyreozy. Następ- w czynnościowej diagnostyce tarczycy (test z jodem radio-
aktywnym).
nie przez 1–2 tygodnie podaje się 10 mg jodku/dobę.
Po takim leczeniu przygotowawczym (tzw. terapii
Plummera) należy wykonać strumektomię, ponie-
2.3.3.3.3. Zasady leczenia nadczynności
waż w przeciwnym razie może dojść do pogorszenia
tarczycy
stanu pacjenta.
Celem leczenia nadczynności tarczycy jest zlikwido-
wanie objawów oraz zmniejszenie powikłań (np. cho-
roby niedokrwiennej serca). Jest to możliwe przez:
MUTSCHLER-2009.indd 412 2010-01-07 22:12:54
Układ hormonalny 413
■ (opisane powyżej) stosowanie tyreostatyków, kosteroidy w celu zahamowania procesu zapalnego
(początkowo przez 1–2 tygodnie prednizolon w daw-
■ terapię jodem radioaktywnym,
ce 1–2 mg/kg m.c., potem dawkę dobową należy
■ operacyjne usunięcie nadczynnej tkanki gruczołu zmniejszać o 5 mg tygodniowo; typowa dawka pod-
tarczowego. trzymująca wynosi 20 mg dziennie przez 6 miesięcy;
jedynie przy objęciu procesem chorobowym nerwu
Podczas gdy tyreostatyki tylko hamują funkcję tar- wzrokowego i zagrażającej utracie wzroku na począt-
czycy, pozostałe metody długotrwale usuwają tkankę ku leczenia podaje się metyloprednizolon w dawce
gruczołu i są w przeciwieństwie do terapii konser- 500 mg dziennie przez 3 dni).
Układ hormonalny
watywnej (farmakologicznej) metodami leczenia ra- Rutynowe stosowanie innych leków immunosu-
dykalnego. Farmakoterapia jest szczególnie zalecana presyjnych nie jest wskazane, jest ono zastrzeżone
w chorobie Basedowa, która charakteryzuje się skłon- dla wyspecjalizowanych ośrodków.
nością do spontanicznych remisji i w której przebiegu
często po prawie roku terapii tyreostatykami nie do- Przełom tarczycowy. Aby wyrównać związaną
chodzi do nawrotu choroby. z przełomem tarczycowym utratę płynów i soli, poda-
W przypadku nadczynności tarczycy o podłożu je się pod ścisłą kontrolą wlewy dożylne roztworów
nieimmunologicznym za pomocą farmakoterapii glukozy i elektrolitów. Muszą również zostać wyrów-
nie da się jednak uzyskać długotrwałych efektów. nane zaburzenia stężenia potasu w surowicy. Hiper- B2
W tych sytuacjach oraz w przypadku nawrotowej termię leczy się metodami fizycznymi (worki z lodem,
choroby Basedowa wskazane jest leczenie radykalne. zimne okłady).
Ale również u tych chorych, aby zmniejszyć ryzy- Jeśli nie przeprowadzi się w trybie nagłym ope-
ko zabiegu operacyjnego lub w trakcie oczekiwania racji tarczycy, będącej leczeniem z wyboru, lub gdy
na efekt terapii jodem promieniotwórczym, celowe nie jest możliwe jej wykonanie, w celu zahamowania
jest przejściowe stosowanie tyreostatyków. syntezy hormonów tarczycy podaje się tyreostatyki,
np. tiamazol w dawce 160–240 mg/dobę w ciągłym
Orbitopatia tarczycowa. U chorych z orbitopa- wlewie dożylnym. Za pomocą plazmaferezy można
tią tarczycową poza normalizacją funkcji tarczycy usunąć krążące we krwi hormony tarczycy zwią-
może być konieczne stosowanie środków ogólnych zane z białkami osocza. Z uwagi na często współ-
(np. przyciemniane szkła okularów, sztuczne łzy, istniejącą niewydolność kory nadnerczy korzystne
wysokie ułożenie głowy podczas snu, abstynencja ni- jest podanie dużych dawek glukokortykosteroidów,
kotynowa), napromienianie tkanek pozagałkowych, np. 100–200 mg prednizolonu dziennie i.v. W celu
a w wyjątkowych przypadkach także leczenie chi- zahamowania obwodowego działania hormonów
rurgiczne (operacje odbarczające, zmniejszenie ilości tarczycy podaje się leki blokujące receptory β-ad-
tkanki tłuszczowej w oczodole i in.). W aktywnym renergiczne, np. propranolol w dawce 40 mg 3 razy
okresie choroby stosuje się dodatkowo glukokorty- dziennie.
MUTSCHLER-2009.indd 413 2010-01-07 22:12:55
414 Układ hormonalny
2.4. Hormony tarczycy, przytarczyc
i nerek wpływające na homeostazę wapniową
W regulacji gospodarki wapniowej biorą udział głów- (Pi) na poziomie ≈ 1,3 mmol/1. Mniej więcej połowa
nie trzy hormony: wapnia występuje w postaci związanej, druga połowa
zaś w postaci wolnej (zjonizowanej).
■ paratyryna (parathormon) syntezowana w przytar-
Osoczowy okres półtrwania parathormonu wynosi
czycach,
ok. 4 minut.
■ pochodząca z tarczycy kalcytonina, Parathormon ma następujące punkty uchwytu
(zob. ryc. B 2.4-1):
■ kalcytriol wytwarzany z 25(OH)D3 w nerkach.
W nerkach PTH zwiększa:
Narządami efektorowymi dla powyższych hormonów ■ wchłanianie zwrotne Ca2+ i Mg2+ w kanalikach dal-
są kości, jelita i nerki. Celem regulacji gospodarki szych,
wapniowej jest utrzymanie stałego stężenia jonów
■ wydalanie HPO2– –
4 i H2PO4 w kanalikach bliższych,
wapnia (≈ 2,5 mmol/1) w przestrzeni pozakomórko-
wej. ■ syntezę kalcytriolu, czyli czynnej formy witaminy
D3, poprzez pobudzanie 1-hydroksylazy.
2.4.1. Anatomia przytarczyc Znaczna część efektów działania parathormonu
jest osiągana pośrednio poprzez wzmożone wytwa-
rzanie kalcytriolu.
Przytarczyce (glandulae parathyreoideae, gruczoły
przytarczyczne) mające wielkość ziaren soczewicy
W kościach PTH z jednej strony stymuluje kościotwo-
i łączny ciężar ok. 150 mg są położone na tylnej po-
rzenie, a z drugiej resorpcję kości. Nasilenie resorp-
wierzchni tarczycy. Zazwyczaj znajdują się tam czte-
cji kostnej jest następstwem aktywacji osteoklastów.
ry przytarczyce – dwie górne i dwie dolne.
Efekt osteolityczny opiera się przede wszystkim
Przytarczyce są otoczone delikatną torebką łącz-
notkankową i składają się z komórek nabłonkowych,
które na podłożu tkanki tłuszczowej i tkanki łącznej
tworzą pasma nabłonkowe. Gruczoły przenika gęsta
sieć naczyń włosowatych. W tkance nabłonkowej
parathormon
przytarczyc można wyróżnić:
■ małe ciemne komórki główne,
■ duże przezroczyste jak woda komórki główne, absorpcja Ca2+
w jelicie
■ komórki eozynochłonne.
[Ca2+]e
Parathormon jest wytwarzany w komórkach głów- mobilizacja
nych. Ca2+
kalcytonina
2.4.2. Parathormon (PTH, paratyryna)
cewkowa resorpcja Ca2+
kalcytriol
Wytwarzany w przytarczycach parathormon jest
istotnym dla życia polipeptydem składającym się z 84
aminokwasów o masie cząsteczkowej 9500 Da, któ-
wiązanie kalcytriolu
rego niektóre fragmenty wykazują także aktywność
hormonalną.
Hormon ten – za pośrednictwem cAMP – utrzymu- Ryc. B 2.4-1. Regulacja gospodarki wapniowej i fosforowej
je całkowite stężenie wapnia w osoczu na poziomie przez parathormon, kalcytoninę i witaminę D3 (według Thewsa,
2,25–2,5 mmol/1 oraz fosforanów nieorganicznych Mutschlera, Vaupela).
MUTSCHLER-2009.indd 414 2010-01-07 22:12:55
Układ hormonalny 415
na uwalnianiu interleukiny-6 (IL-6). Równocześnie się on (napadowymi) skurczami mięśni poprzecznie
PTH pobudza proliferację osteoblastów i uwalnianie prążkowanych oraz parestezjami.
przez nie cytokin oraz reguluje uwalnianie czynników Typowe napady tężyczkowe dotyczą kończyn.
pobudzających wzrost kości, a w szczególności IGF-1. Objawiają się one tonicznymi skurczami mięśni dłoni
Tą drogą hamowana jest również apoptoza osteo- i stóp (np. tzw. ręka położnika); mówi się o kurczach
blastów. Podsumowując, można stwierdzić, że PTH nadgarskowo-stopnych. Wyraz twarzy jest napięty.
przesuwa równowagę na korzyść kościotworzenia. U dzieci kurcz głośni oznacza stan ostrego zagroże-
nia życia.
W jelitach parathormon – pośrednio poprzez wzmo- Mogą także wystąpić następujące dodatkowe ob-
Układ hormonalny
żone wytwarzanie kalcytriolu – powoduje zwiększe- jawy (głównie w przypadku postaci pierwotnych):
nie wchłaniania jonów wapnia i fosforanowych. zaburzenia troficzne narządów pochodzenia ektoder-
malnego (sucha i krucha skóra, wypadanie włosów,
Sumaryczne działanie parathormonu polega zatem łamliwe paznokcie), zagęszczenie struktury tkanki
na szybkim zwiększeniu stężenia wapnia zjonizowane- kostnej z powodu zmniejszonej przebudowy kości
go we krwi i zmniejszeniu w takim samym stopniu stę- oraz ogniskowe zwapnienia wewnątrzczaszkowe.
żenia fosforanów. Czynnikiem regulującym wydzie-
lanie parathormonu jest stężenie Ca2+ w przestrzeni Farmakoterapia. W leczeniu stosuje się cholekal-
pozakomórkowej: niskie stężenie Ca2+ wywołuje cyferol (witaminę D3) lub dihydrotachysterol, które B2
wzrost wydzielania PTH, natomiast wysokie stężenie wywołują taki sam efekt jak parathormon, lecz są tań-
Ca2+ powoduje hamowanie tego wydzielania za po- sze i można je podać doustnie. Za pomocą obu tych
średnictwem czujnika Ca2+ (receptora Ca2+) w błonie substancji możliwe jest prowadzenie wystarczającej
komórek głównych. Nie jest znana nadrzędna regula- terapii substytucyjnej niedoboru parathormonu. Za-
cja hormonalna wydzielania PTH. letą dihydrotachysterolu w porównaniu z witaminą
Poza leczeniem osteoporozy parathormon nie ma D3 jest jego krótszy okres półtrwania i dzięki temu
znaczenia terapeutycznego. Zamiast niego stosuje się łatwiejsze sterowanie jego działaniem (t1/2 witaminy
witaminę D3 lub dihydrotachysterol (zob. poniżej). D3 wynosi 4–5 dni, a dihydrotachysterolu 16–18 go-
Fragment parathormonu teriparatyd jest stosowa- dzin).
ny w leczeniu osteoporozy.
H3C
CH(CH3) 2
2.4.3. Zaburzenia funkcji przytarczyc CH3
CH3
H CH3
2.4.3.1. Niedoczynność przytarczyc
OH
Patofizjologia. Obraz kliniczny niedoczynności
przytarczyc jest spowodowany niedoborem parat- dihydrotachysterol
hormonu. Przyczyną rzadko występującej postaci
pierwotnej są zaburzenia wrodzone (aplazja lub hipo-
plazja przytarczyc) bądź choroba autoimmunologicz-
na prowadząca do uszkodzenia przytarczyc. Indywidualne dawkowanie można ustalić jedynie
Znacznie częściej występująca postać jatrogenna na podstawie ciągłego monitorowania stężenia wap-
jest następstwem operacji tarczycy, w czasie któ- nia we krwi. Należy znaleźć dawkę pośrednią między
rych częściowo lub całkowicie usunięto przytarczyce zbyt małą ze zwiększoną skłonnością do skurczów
wraz z tkankami tarczycy albo uszkodzono naczynia a przedawkowaniem pociągającym za sobą niebez-
krwionośne zaopatrujące przytarczyce. pieczeństwo uogólnionych zwapnień. Przeciętna
Niedobór parathormonu prowadzi do hipokalce- dawka dihydrotachysterolu wynosi 0,5–1,5 mg/dobę.
mii i hiperfosfatemii. Konsekwencją hipokalcemii
jest ułatwiona aktywacja (dostępnych) kanałów so-
dowych we włóknach nerwowych i wskutek tego 2.4.3.2. Nadczynność przytarczyc
nadmierna pobudliwość całego układu nerwowego.
Powstały zespół objawów klinicznych określa się Patofizjologia. Jako nadczynność przytarczyc okre-
mianem tężyczki – w tym przypadku jako tężycz- śla się wszystkie formy nadczynności gruczołów
ki spowodowanej brakiem przytarczyc – a objawia przytarczycznych z podwyższonym stężeniem PTH.
MUTSCHLER-2009.indd 415 2010-01-07 22:12:55
416 Układ hormonalny
Przyczyną pierwotnej (autonomicznej) formy nad- rozpocząć jej substytucję. Jeżeli przyczyną nadczyn-
czynności przytarczyc, której towarzyszą hiperkalce- ności przytarczyc jest niewydolność, najważniejszy-
mia i hipofosfatemia, jest w 80% gruczolak przytar- mi elementami terapii są: ograniczenie ilości dostar-
czyc, a w 20% przerost przytarczyc. czanych fosforanów do 600 mg/dobę przez spożywa-
Objawy składające się na obraz kliniczny wyni- nie pokarmów zawierających mało fosforanów oraz
kają przede wszystkim ze stopnia i czasu trwania hi- podawanie węglanu wapnia (2–3 g/dobę). Działa on
perkalcemii (zespół hiperkalcemiczny). W lżejszych z jednej strony jako środek wiążący fosforany w wy-
postaciach przebieg jest bezobjawowy, a przy nasi- niku tworzenia niewchłaniającego się fosforanu wap-
lonej hiperkalcemii poza adynamią, nykturią i po- nia, a jednocześnie powoduje zwiększenie ilości do-
liurią obserwuje się często kolkę nerkową wskutek starczanych jonów wapnia.
tworzenia się złogów z fosforanu wapnia, natomiast Środki wiążące fosforany zawierające glin,
rzadziej wapnicę nerek lub schorzenia kości. Poza np. wodorotlenek glinu, nie powinny być stosowane
tym w przebiegu nadczynności przytarczyc zwiększa z powodu niebezpieczeństwa wywołania przez glin
się częstość owrzodzeń żołądka oraz zapaleń trzust- osteopatii i encefalopatii.
ki w następstwie wywołanego przez hiperkalcemię
zwiększonego wydzielania gastryny. Sewelamer jest niewchłaniającym się z jelit polime-
Przy wtórnej (regulacyjnej) nadczynności przytar- rem, który stanowi alternatywę dla węglanu wapnia
czyc organizm poprzez wzmożone wydzielanie parat- i jako (organiczny) środek wiążący fosforany może
hormonu próbuje wyrównać wywołane innymi scho- być podawany z każdym posiłkiem w dawce 0,8–1,6 g.
rzeniami podwyższone stężenie fosforanów i obniżo-
ne stężenie wapnia we krwi. Do schorzeń mogących Jeżeli za pomocą wymienionych środków nie uda się
wywołać wtórną nadczynność przytarczyc należy osiągnąć normalizacji stężenia Ca2+ we krwi, wska-
krzywica, będąca następstwem braku odpowiedniej zane jest stosowanie pochodnych witaminy D hydrok-
ilości witaminy D niezbędnej do wytwarzania odpo- sylowanych w pozycji 1, przede wszystkim kalcytrio-
wiedniej ilości 1,25-dihydroksy-witaminy D3, oraz lu. U chorych dializowanych celowe jest rozważenie
przewlekła niewydolność nerek ze zmniejszonym zastąpienia kalcytriolu parikalcytolem będącym po-
wydalaniem fosforanów i zmniejszoną hydroksylacją chodną witaminy D.
25-hydroksy-witaminy D3 do 1,25-dihydroksy-wita-
miny D3. Lek ten podaje się w postaci bolusa przez cewnik central-
ny podczas dializy, nie częściej jednak niż co drugi dzień.
Średni osoczowy okres półtrwania u chorych dializowanych
O trzeciorzędowej nadczynności mówi się wówczas, gdy wynosi ok. 15 godzin. Dawka początkowa wynosi 0,04–0,1
po trwającej wiele lat wtórnej postaci dochodzi do autono- μg/kg. Kolejne dawki są ustalane na podstawie stężenia
mizacji przytarczyc. PTH.
Objawy nadczynności przytarczyc rozwijają się rów- Do najczęstszych działań niepożądanych należą hiper-
nież w przebiegu zespołów paraneoplastycznych, np. przy kalcemia, hiperfosfatemia, świąd i zaburzenia smaku.
drobnokomórkowym, wytwarzającym parathormon raku Parikalcytol jest przeciwwskazany u chorych z hiperkal-
oskrzeli lub przy innych złośliwych nowotworach, wydzie- cemią i zatruciem witaminą D.
lających białko podobne czynnościowo do parathormonu.
Inną możliwością terapeutyczną u dializowanych
Leczenie. Objawowa pierwotna nadczynność przy- pacjentów z wtórną nadczynnością przytarczyc
tarczyc wymaga zazwyczaj operacyjnego usunięcia jest zastosowanie cynakalcetu. Jest to lek o działa-
guza. Leczenie farmakologiczne stosowane w cięż- niu kalcymimetycznym, który zwiększa wrażliwość
kiej hiperkalcemii polega na podawaniu środków receptorów Ca2+ komórek głównych przytarczyc
obniżających stężenie Ca2+. Podaje się wówczas kil- na wapń pozakomórkowy i tą drogą zmniejsza wy-
ka litrów soli, co daje duże zwiększenie natriurezy. dzielanie PTH. Może być podawany również w te-
Efekt ten można nasilić, podając furosemid w dawce rapii kombinowanej z witaminą D i środkami wią-
do 100 mg/godzinę. Kolejnym ważnym etapem tera- żącymi fosforany.
pii jest zahamowanie aktywności osteoblastów przez
podawanie bisfosfonianów do momentu normalizacji Po podaniu doustnym cynakalcet osiąga maksymalne stęże-
stężenia wapnia w osoczu. Diuretyki tiazydowe, któ- nie w surowicy po 2–6 godzinach. Równoczesne podawanie
re powodują retencję Ca2+, oraz glikozydy nasercowe pokarmów zwiększa jego biodostępność. Biotransformacja
zachodzi głównie w CYP3A4 i CYP1A2. Osoczowy okres
są u tych chorych przeciwwskazane. półtrwania wynosi 30–40 godzin. Zalecana dawka począt-
W przypadku wtórnej nadczynności przytarczyc kowa wynosi 30 mg dziennie w dawce jednorazowej. Może
ważne jest leczenie przyczyny, która ją wywołuje. być ona następnie stopniowo zwiększana co 2–4 tygodnie
Jeżeli przyczyną jest niedobór witaminy D, należy maksymalnie do dawki dobowej 180 mg.
MUTSCHLER-2009.indd 416 2010-01-07 22:12:55
Układ hormonalny 417
W nerkach kalcytonina wywołuje nieznaczny
CH3 wzrost wydalania wapnia i fosforanów, a także sodu,
H3C CH3 potasu i magnezu. W ten sposób parathormon i kal-
CH3 cytonina działają synergistycznie na zmniejszenie
H3C OH
zwrotnego wchłaniania fosforanów w nerkach.
Niezależnie od efektu hormonalnego kalcytonina
H wykazuje także działanie analgetyczne.
Uwalnianie kalcytoniny zależy od stężenia jonów
wapnia w przestrzeni pozakomórkowej: wzrost stę-
Układ hormonalny
żenia wapnia powoduje wzmożenie wydzielania tego
HO OH hormonu, a jego spadek – hamowanie.
Po przyjęciu pożywienia uwalnianie kalcytoniny
parikalcytol jest pobudzane przez hormony przewodu pokarmo-
wego, np. gastrynę i cholecystokininę. Dzięki temu
dostarczone wraz z pożywieniem jony wapnia zostają
szybko wbudowane do magazynów w tkance kostnej,
tak że nie dochodzi do wzrostu stężenia jonów wap-
F3C
NH
niowych we krwi. To z kolei oznacza, że nie dochodzi B2
CH3 do zmniejszenia stężenia parathormonu, czego na-
stępstwem byłoby szybkie wydalanie wchłoniętych
cynakalcet jonów wapnia przez nerki (tzw. chroniące wapń dzia-
łanie kalcytoniny).
Osoczowy okres półtrwania kalcytoniny wynosi
ok. 45 minut.
Opisano następujące działania niepożądane: nudności, Zastosowanie terapeutyczne. W lecznictwie stoso-
wymioty, anoreksję, zawroty głowy, parestezje, zwiewne
wykwity skórne (wysypka), bóle mięśni, hipokalcemię wana jest przede wszystkim syntetyczna kalcytonina
oraz obniżenie stężenia testosteronu. Z powodu ryzyka hi- łososiowa, która działa dłużej i silniej od kalcytoni-
pokalcemii wskazane jest monitorowanie stężenia wapnia ny ludzkiej. Wadą kalcytoniny łososiowej jest ewen-
w surowicy. tualne wytwarzanie przeciwciał neutralizujących,
Należy zachować szczególną ostrożność przy podawa- wskutek czego częściej dochodzi do powstania
niu cynakalcetu łącznie z lekami silnie hamującymi lub in-
dukującymi CYP3A4 i/lub CYP1A2. oporności lekowej niż w przypadku stosowania biał-
ka ludzkiego.
Podawanie kalcytoniny jest wskazane w (ciężkiej)
hiperkalcemii, chorobie Pageta (osteodystrophia de-
2.4.4. Kalcytonina (tyreokalcytonina) formans), II i III stadium zespołu Sudecka, osteopo-
rozie, a także przy dolegliwościach bólowych spo-
Kalcytonina jest hormonem peptydowym, składa- wodowanych osteoporozą lub przerzutami do kości.
jącym się z 32 aminokwasów (masa cząsteczkowa Kalcytoninę stosuje się także w objawowym leczeniu
3500 Da). Potrzebny jest cały łańcuch peptydowy, osteolizy spowodowanej chorobą nowotworową.
aby kalcytonina wykazywała biologiczne działanie. Jako polipeptyd kalcytonina musi być podawana
Synteza kalcytoniny odbywa się w komórkach C tar- pozajelitowo (s.c., głęboko domięśniowo, i.v. lub do-
czycy; takie komórki stwierdza się także w przytar- nosowo).
czycach i trzustce. Zwierzęce kalcytoniny są również Dawkowanie – pod ścisłą kontrolą stężenia wapnia
skuteczne u ludzi (zob. poniżej). we krwi – w ciężkiej hiperkalcemii wynosi l IU/kg
W warunkach fizjologicznych kalcytonina działa m.c. (wlew dożylny), w chorobie Pageta początko-
częściowo antagonistycznie w stosunku do parathor- wo i po wystąpieniu efektu terapeutycznego 100 IU
monu. Hamuje ona uwalnianie wapnia i fosforanów 1–3 razy tygodniowo, w zespole Sudecka 50–100
z tkanki kostnej i jednocześnie wzmaga ich wbudo- IU/dobę przez 2–6 tygodni, w osteoporozie 50–100
wywanie w tkankę kostną oraz powoduje szybkie IU/dobę przez 4–8 tygodni jako terapia przejściowa,
obniżenie stężenia jonów wapnia we krwi (zob. ryc. a w leczeniu bólów kostnych spowodowanych choro-
B 2.4-1). Efekt ten jest częściowo uwarunkowany bą nowotworową 100–200 IU co 6 godz.
zmniejszaniem aktywności osteoklastów przez kal- 100 IU odpowiada 25 μg syntetycznej kalcytoniny
cytoninę. łososiowej.
MUTSCHLER-2009.indd 417 2010-01-07 22:12:55
418 Układ hormonalny
Działania niepożądane to przejściowe, stosunkowo W przypadku osteoporozy starczej (osteoporo-
rzadko pojawiające się nudności, nagłe zaczerwienie- za typu II), w której przebiegu masa kostna kości
nie twarzy i dolegliwości żołądkowo-jelitowe. Stoso- gąbczastej i zbitej ulega w takim samym stopniu
wanie kalcytoniny łososiowej wiąże się także z ryzy- zmniejszeniu, istotny czynnik patogenetyczny oprócz
kiem rozwoju miejscowej lub uogólnionej reakcji aler- predyspozycji genetycznej stanowi także obniżone
gicznej. Należy unikać zbyt dużych spadków stężenia stężenie kalcytriolu w osoczu. Pacjenci w podeszłym
jonów wapnia we krwi, ponieważ może to spowodo- wieku często przyjmują zbyt małe ilości witaminy D
wać rozwój wtórnej nadczynności przytarczyc. będącej prekursorem kalcytriolu. W tej grupie wieko-
wej także endogenna synteza witaminy D jest zmniej-
szona z powodu niedostatecznego czasu przebywania
w miejscach nasłonecznionych. Poza tym w starszym
2.4.5. Osteoporoza
wieku zmniejsza się prawdopodobnie zdolność wy-
twarzania kalcytriolu z 25-hydroksycholekalcyferolu
2.4.5.1. Patofizjologia (kalcyfediolu) przez nerki.
Osteporoza jest schorzeniem kości (osteopatią) uwa- Osteoporoza wtórna. Ta postać osteoporozy, stano-
runkowanym zaburzeniami przemiany materii tkanki wiąca tylko około 5% zachorowań, jest spowodowa-
kostnej, które prowadzi do uogólnionego lub (znacz- na m.in. przez zaburzenia endokrynologiczne (np. hi-
nie rzadziej) miejscowego zmniejszenia masy kostnej perkortyzolemię, hipertyreozę), unieruchomienie, te-
przypadającej na jednostkę objętości (gęstość mine- rapie glukokortykosteroidami lub cytostatykami oraz
ralna kości) oraz do zaburzenia mikroarchitektury niedożywienie. W odróżnieniu od pierwotnej osteo-
tkanki kostnej. Wskutek zmian strukturalnych i czyn- porozy jest ona stwierdzana częściej u mężczyzn niż
nościowych dochodzi często do niskoenergetycznych u kobiet.
złamań kości (przede wszystkim kręgów, szyjki kości
udowej oraz kości przedramienia). W przeciwień-
stwie do krzywicy i osteomalacji nie występują zabu- 2.4.5.2. Leki stosowane
rzenia mineralizacji. w terapii osteoporozy
Osteoporoza jest najczęstszym schorzeniem kości
Zastosowanie soli wapnia, witaminy D3, kalcytoniny
(często występowania w Niemczech wynosi 4–6 mi-
oraz estrogenów w profilaktyce i terapii osteoporozy
lionów, z tego 80% zachorowań stwierdza się u ko-
omówiono w rozdziałach B 7.2.2.4.2, B 10.1.1.2, B
biet). Wyróżnia się postać pierwotną i wtórną.
2.4.4. i B 2.8.8.
Osteoporoza pierwotna. Bardzo rzadko pojawiają-
ca się osteoporoza młodzieńcza rozwija się w okresie
dojrzewania i ustępuje spontanicznie. Jej przyczyna 2.4.5.2.1. Bisfosfoniany
nie jest znana.
Szczególne znaczenie – nie tylko z powodu często- Bisfosfoniany, charakteryzujące się podwójnym ugru-
ści występowania – ma występująca u kobiet osteo- powaniem P-C-P, są stabilnymi analogami pirofosfo-
poroza pomenopauzalna (osteoporoza typu I). Wyni- ranu i w wielu przypadkach wpływają na metabolizm
kający z zaniku czynności jajników spadek stężenia wapnia jak pirofosforany. Podczas gdy pirofosforany
estrogenów we krwi powoduje w następstwie zmniej- nie mogą być stosowane w terapii, ponieważ ulegają
szonego wydzielania kalcytoniny i uwrażliwienia szybkiej hydrolizie katalizowanej przez tkankowe fo-
osteoklastów na parathormon wzmożoną osteolizę. sfatazy, w wyniku której są przekształcane do fosfo-
Konsekwencją tego jest wzrost stężenia jonów wap- ranów, ich bardziej stabilne pod względem biotrans-
nia w przestrzeni pozakomórkowej. To zaś stanowi formacji analogi mogą być wykorzystywane.
przyczynę zmniejszonego wytwarzania kalcytriolu Na podstawie budowy chemicznej można bisfo-
prowadzącego do zmniejszenia wchłaniania wapnia sfoniany podzielić na (zob. tab. B 2.4-1):
z przewodu pokarmowego, a także zwiększenia jego
■ bisfosfoniany niezawierające podstawników zasa-
wydalania z moczem. W następstwie tych zjawisk
dowych (chlorowcowane, alkilo- i arylobisfosfo-
dochodzi do przesunięcia stanu równowagi między
niany; klodronian, etydronian, tiludronian),
kościotworzeniem a resorpcją kości na korzyść roz-
kładu kości. Zmiany w głównej mierze dotyczą istoty ■ aminobisfosfoniany (aminoalkilobisfosfoniany; alen-
gąbczastej kości. dronian, pamidronian),
MUTSCHLER-2009.indd 418 2010-01-07 22:12:56
Układ hormonalny 419
Tabela B 2.4-1. Bisfosfoniany
Budowa chemiczna Nazwa Preparaty handlowe
międzynarodowa
OH R1 OH
O P P O
OH R2 OH
R1 R2
Układ hormonalny
klodronian Bonefos, Clodron Beta, Clodeon HEXAL,
Cl Cl Ostac [w Polsce: Bonefos,
Sindronat – przyp. tłum.]
etydronian Didronel, Diphos, Etidronat 200 JENAPHARM,
CH3 OH Etidron HEXAL [w Polsce: Difosfen,
Ostedron, Ostedron PAG – przyp. tłum.]
S tiludronian Skelid
H B2
Cl
alendronian Alendron beta, Alendron-HEXAL,
CH2 OH FOSAMAX [w Polsce: Ostolek, Alenato,
NH2
Fosamax, Lindron, Osalen, Ostenil,
Rekostin, Ostemax, Alendromax,
Alendronat-rathipharm– przyp. tłum.]
CH2 NH2 pamidronian Aredia, Pamindronat Gry, Pamindorn HEXAL,
OH Pamifos i in. [w Polsce: Aredia, Pamindronat
medac, Pamifos, Pamisol, Pamitor – przyp.tłum.]
CH3 ibandronian Bondronat, Bonviva
CH2 N OH
(CH2) 4CH3
CH2 risedronian Actonel [w Polsce: Actonel, Norifas,
N
OH Rizendros – przyp. tłum.]
zolendronian Zometa, Aclasta
CH2
N N OH
■ aminobisfosfoniany zawierające podstawnik azoto- mogą one jednak upośledzać prawidłowe wbudowy-
wy (ibandronian), wanie Ca2+ do kości i w ten sposób mineralizację ma-
cierzy tkanki kostnej.
■ bisfosfoniany zawierające zasadową grupę hetero- Pomimo że profil działania wszystkich bisfosfo-
cykliczną (risedronian, zolendronian). nianów jest właściwie taki sam, poszczególne związ-
ki różnią się nie tylko pod względem właściwości
Dzięki skutecznemu hamowaniu aktywności osteokla- farmakokinetycznych (zob. poniżej), lecz także siłą
stów bisfosfoniany blokują uwalnianie wapnia z tkan- działania i stosunkiem hamowania resorpcji kości
ki kostnej oraz resorpcję kości. Poza tym zapobiegają do hamowania mineralizacji. Podczas gdy np. dla al-
one adhezji komórek nowotworowych do macierzy kilobisfosfonianu etydronianu ustalono, że stosunek
kości, a tym samym spowodowaną przez nowotwór hamowania resorpcji do hamowania mineralizacji
osteolizę oraz hiperkalcemię. Bisfosfoniany zapobie- wynosi 1:1, w przypadku aminobisfosfonianu alen-
gają również wapnieniu tkanek miękkich w przebiegu dronianu efektywna terapeutycznie dawka hamująca
hiperfosfatemii (i związanemu z hiperfosfatemią wy- resorpcję kości jest 6000 razy mniejsza od najmniej-
trącaniu się fosforanu wapniowego). Z drugiej strony szej dawki hamującej mineralizację. Przyczyną tej
MUTSCHLER-2009.indd 419 2010-01-07 22:12:56
420 Układ hormonalny
różnicy jest fakt, że osteoklasty razem z kwasem sol- go lub 300 mg/dobę i.v. przez 5 (–7) dni, lub 800–
nym powodują enzymatyczną resorpcję tkanki kost- 1600 (–3200) mg/dobę. doustnie przez 6 miesięcy;
nej i dlatego bisfosfoniany z podstawnikami zasado-
■ etydronian (etydronian disodowy) – w leczeniu
wymi gromadzą się w kwaśnym środowisku zatoki
osteoporozy 400 mg/dobę doustnie przez 14 dni
resorpcyjnej.
(powtarzanie leczenia co trzy miesiące), w lecze-
niu choroby Pageta 5–10 mg/kg m.c./dobę doustnie
Mechanizm działania. Mechanizm działania na po-
przez maksymalnie 6 miesięcy;
ziomie molekularnym jest tylko częściowo wyjaś-
niony. Bisfosfoniany niezawierające podstawników ■ ibandronian – w leczeniu osteoporozy 150 mg
zasadowych są w osteoblastach wbudowywane za doustnie raz w miesiącu lub 3 mg i.v. cztery razy
pomocą transferazy do ATP i w ten sposób wywołu- w roku, w przypadku nowotworów złośliwych
ją działanie cytotoksyczne. W przypadku bisfosfo- z zajęciem kości doustnie 50 mg dziennie lub jed-
nianów zawierających azot stwierdzono, że hamują norazowo 6 mg w powolnym wlewie w odstępach
one syntazę pirofosforanu farnezylu, a tym samym 3–4 tygodni;
przyłączanie reszt farnezylowych (tzw. prenylację)
■ pamidronian – w leczeniu choroby Pageta 30 mg/
do białka G (m.in. Ras, Rho), istotnego dla funkcjo-
dobę przez trzy dni, w leczeniu hiperkalcemii spo-
nowania osteoblastów.
wodowanej nowotworem jednorazowo 60–90 mg,
w obu przypadkach w powolnym wlewie dożylnym
Skuteczność. Skuteczność bisfosfonianów zosta-
w 0,9-proc. roztworze chlorku sodowego;
ła wykazana w wielu badaniach z podwójnie ślepą
próbą. Np. u kobiet w okresie pomenopauzalnym ■ risedronian (risedronian disodowy) – w leczeniu
z co najmniej jednym złamaniem trzonu kręgu na po- osteoporozy 5 mg dziennie lub 35 mg tygodniowo,
czątku badania stosowanie przez trzy lata alendro- w leczeniu choroby Pageta 30 mg dziennie przez
nianu spowodowało zmniejszenie o 50% częstości dwa miesiące;
występowania złamań kręgów, biodra i kości przed-
■ tiludronian – 400 mg/dobę doustnie przez trzy mie-
ramienia w porównaniu z placebo.
siące.
Właściwości farmakokinetyczne. Po podaniu do- ■ zolendronian – 4 mg w powolnym wlewie co 3–4
ustnym bisfosfoniany źle wchłaniają się z przewo- tygodnie.
du pokarmowego (klodronian wchłania się w 4–5%,
etydronian w < 10%, tiludronian w 2–3%, a związki Stosując bisfosfoniany w celu zapobiegania rozwojowi za-
zawierające azot w 0,5–2%). Równoczesne przyjmo- palenia lub nadżerek w przełyku, lek należy przyjmować
w pozycji z uniesionym pionowo tułowiem na czczo ok. 30
wanie pokarmów na skutek zawartego w nich wapnia minut przed śniadaniem i popijać go szklanką wody. Po-
zmniejsza ich biodostępność. Podczas, gdy osoczowy nadto pacjenta należy poinformować, że po przyjęciu leku
okres półtrwania jest stosunkowo krótki (zazwyczaj nie powinien się ponownie kłaść przez 30 minut (w przy-
kilka godzin), okres półtrwania w tkance kostnej padku ibandronianu przez 60 minut).
wynosi od kilku miesięcy do kilku lat. Bisfosfoniany
są wydalane głównie przez nerki. Działania niepożądane. Ogólnie bisfosfoniany
są dobrze tolerowane. Jako działania niepożądane
Wskazania. Z powodu opisanego działania stosowa- mogą pojawić się zaburzenia żołądkowo-jelitowe
nie bisfosfonianów jest wskazane w leczeniu osteopo- (szczególnie po podaniu doustnym), alergiczne
rozy i choroby Pageta, w terapii paliatywnej osteolizy odczyny skórne, bóle kości i mięśni, objawy gry-
i hiperkalcemii wywołanej przez nowotwór, a także popodobne, hipokalcemia i hipofosfatemia oraz
w leczeniu bólów będących następstwem przerzutów wrzody trawienne. Przy podawaniu dożylnym ist-
nowotworowych do kości. Wskazania do stosowania nieje niebezpieczeństwo przejściowej proteinurii.
poszczególnych substancji nieznacznie się różnią. Etydronian powoduje opóźnienie mineralizacji
nowo powstałej tkanki kostnej w stopniu zależnym
Dawkowanie. Dawkowanie poszczególnych leków od dawki. Przy niewłaściwym przyjmowaniu bisfo-
jest następujące: sfonianów może dojść do rozwoju zapalenia prze-
łyku oraz nadżerki i owrzodzenia błony śluzowej
■ alendronian – doustnie 10 mg dziennie lub 70 mg
przełyku. Do innych działań niepożądanych należą
tygodniowo jako leczenie przewlekłe;
stosunkowo rzadkie zaburzenia ze strony ośrodko-
■ klodronian – jednorazowo 1500 mg w powolnym wego układu nerwowego (splątania, halucynacje,
wlewie i.v. w 0,9-proc. roztworze chlorku sodowe- zaburzenia widzenia).
MUTSCHLER-2009.indd 420 2010-01-07 22:12:56
Układ hormonalny 421
Przeciwwskazania. U chorych z niewydolnością ne- Stosowanie w leczeniu osteoporozy fluorków
rek, a także u kobiet w ciąży i w okresie karmienia w dawkach terapeutycznych jest przeciwwskazane
piersią bisfosfoniany są przeciwwskazane. Ciężkie, w przypadku ciężkich zaburzeń czynności wątro-
ostre stany zapalne przewodu pokarmowego wy- by i nerek, osteomalacji, ostrego zapalenia stawów,
kluczają podawanie doustne. Poza tym etydronianu u dzieci i młodzieży w okresie wzrostu, a także u ko-
nie wolno stosować u chorych z osteomalacją. biet w wieku rozrodczym.
Jednoczesne przyjmowanie preparatów zawierają-
Interakcje. Sole wapnia, żelaza i magnezu zmniej- cych glin, wapń lub magnez zmniejsza wchłanianie
szają wchłanianie bisfosfonianów z przewodu pokar- fluorku sodu, natomiast nie wpływa na wchłanianie-
Układ hormonalny
mowego. fluorofosforanu sodu.
Stosowanie fluorków w profilaktyce próchnicy
opisano w rozdz. B 10.2.
2.4.5.2.2. Fluorki
Fluorki, które zostaną wbudowane do cząsteczki apa- 2.4.5.2.3. Parathormon, teryparatyd
tytu zamiast grupy hydroksylowej, pobudzają proli-
ferację osteoblastów, jak również syntezę macierzy Długo niestosowany w lecznictwie parathormon zo-
zewnątrzkomórkowej i tą drogą nasilają kościotwo- stał w 2006 roku zarejestrowany do terapii osteoporo- B2
rzenie. Ich działanie przynajmniej częściowo wynika zy pomenopauzalnej u pacjentek z wysokim ryzykiem
z hamowania fosfatazy fosfotyrozylowej w osteo- załamań. Jego osoczowy okres półtrwania wynosi 1,5
blastach, w wyniku czego zwiększa się aktywność godziny. Jest on podawany raz dziennie podskórnie
kinazy białkowej (p44 MAPK) uczynnianej przez (dawka 100 μg).
mitogen.
Temu korzystnemu efektowi przeciwstawia się, Teryparatyd jest otrzymywanym metodami inżynie-
szczególnie przy stosowaniu dużych dawek, działa- rii genetycznej fragmentem parathormonu składają-
nie niepożądane polegające na nieregularnej i opóź- cym się z aminokwasów 1–34, który jest stosowany
nionej mineralizacji macierzy kostnej oraz zmianie w terapii objawowej osteoporozy pomenopauzalnej
struktury kryształów apatytów: tworzą się kości łam- u kobiet oraz pierwotnej lub hipogonadalnej osteopo-
liwe, wytrzymujące mniejsze obciążenie. rozy u mężczyzn przy współistnieniu wysokiego ry-
Z tego powodu leczenie osteoporozy fluorkami zyka złamań. Osoczowy okres półtrwania wynosi ok.
było (i jest) kwestią sporną, i to nie tylko z powodu 1 godziny, a zalecane dawkowanie 20 μg s.c. dziennie
nieprzestrzegania wąskiego zakresu stężenia terapeu- przez maksymalnie 18 miesięcy (z powodu potencjal-
tycznego fluorków, stosowania zbyt dużych dawek nego ryzyka rozwoju mięsaka kości).
oraz zbyt długiego czasu trwania terapii. Przy właś- Łącząc się z receptorem dla parathormonu, tery-
ciwym stosowaniu fluorki zmniejszają ryzyko wystą- paratyd i parathormon aktywują różne przekaźniki
pienia złamań kręgów, lecz nie zmniejszają niebez- wtórne i tą drogą pobudzają osteoblasty do kościo-
pieczeństwa złamań kości w obrębie kończyn. tworzenia.
Aby uniknąć maksymalnych stężeń w osoczu, Najczęstszymi działaniami niepożądanymi są poza
przekraczających zakres terapeutyczny 90–190 ng/ml, ryzykiem hiperkalcemii nudności, bóle głowy i koń-
powinno się stosować preparaty typu retard. Praw- czyn, oraz zawroty głowy.
dopodobnie korzystne jest także prowadzenie tera- Oba leki są przeciwwskazane u chorych z hiperkal-
pii przerywanej. Stosowane są preparayty zawiera- cemią, ciężką niewydolnością nerek, pierwotną nad-
jące fluorofosforan sodu (Na2PO3F) i fluorek sodu czynnością przytarczyc i chorobą Pageta, jak również
(NaF). po przebytym napromienianiu szkieletu.
Obecnie najczęściej stosowane dawki wynoszą W trakcie leczenia teryparatyd zwiększa stężenie
15–25 mg biodostępnego fluorku dziennie (25 mg glikozydów naparstnicy w osoczu.
fluorku sodu odpowiada 11,3 mg F–), podawane mak-
symalnie przez okres 3 lat. Jednocześnie konieczne
jest dostarczanie wystarczającej ilości wapnia i wi- 2.4.5.2.4. Ranelinian strontu
taminy D3.
Obserwowane działania niepożądane to dolegli- Nowym lekiem stosowanym w leczeniu osteoporozy
wości żołądkowe, jak również obrzęki i bóle stawów jest ranelinian strontu. Składa się on z dwóch jonów
skokowych, kolanowych i biodrowych z możliwymi (stabilnego, nieradioaktywnego) strontu oraz jednej
mikrozłamaniami lub bez nich. cząsteczki kwasu ranelinowego (kwas czterokarbok-
MUTSCHLER-2009.indd 421 2010-01-07 22:12:56
422 Układ hormonalny
Wczesne, regularne stosowanie substytucyjnego
Brak!!! leczenia estrogenami umożliwia obniżenie o mniej
ranelinian strontu więcej 50% ryzyka wystąpienia osteoporozy u ko-
biet w okresie pomenopauzalnym, jednak wskazania
do stosowania estrogenów sa ograniczone z uwagi
na zwiększone ryzyko rozwoju raka piersi oraz wy-
stąpienia incydentów zakrzepowo-zatorowych. Moż-
liwe jest rozważenie terapii modulatorami receptora
estrogenowego.
Należy unikać palenia papierosów i spożywania
dużych dawek alkoholu.
Podobnie jak w profilaktyce osteoporozy, różwnież
w jej leczeniu, którego celem jest poprawa stabilności
kości oraz zapobieganie (nowym) złamaniom, należy
sylowy). Ranelinian strontu nasila kościotworzenie
pamiętać o dostarczeniu wystarczającej ilości wapnia
poprzez zwiększenie ilości preosteoblastów, jak rów-
i witaminy D3 w ramach terapii podstawowej. Jeżeli
nież powoduje nasilenie syntezy kolagenu i równo-
zaburzona jest przemiana aktywująca witaminę D3
czesne hamowanie resorpcji kości poprzez zmniej-
do hormonu-witaminy D3, np. w przebiegu chorób
szenie różnicowania osteoklastów i tą drogą ich ak-
nerek lub wątroby, zamiast witaminy D3 musi być
tywności resorpcyjnej. Struktura kryształów apatytu
podawany alfakalcydol lub kalcytriol.
w kościach pozostaje niezmieniona.
U kobiet po menopauzie, jeżeli nie ma przeciw-
Znaczna polarność cząsteczki ranelinianu stronu
wskazań, po ocenie indywidualnego stosunku korzy-
zmniejsza jego wchłanianie z przewodu pokarmowe-
ści do ryzyka można zastosować w terapii estrogeny
go (na czczo biodostępność wynosi 25%; równoczes-
lub modulator receptora estrogenowego.
ne spożycie pokarmu znacznie ją obniża). Objętość
W przypadku bisfosfonianów dostępnych jest naj-
dystrybucji oraz wiązanie z białkami osocza również
więcej danych wskazujących na hamowanie przez
są niskie. W związku z tym efektywny okres półtrwa-
nie resorpcji kości oraz zwiększanie masy kości
nia wynosi 60 godzin. Wydalanie zachodzi przez ner-
w obrębie kręgosłupa i bioder, a także zmniejszania
ki i z kałem.
częstości złamań. Są one dzisiaj terapią z wyboru
Dawkowanie wynosi 2 g dziennie w jednej dawce
w leczeniu osteoporozy.
przed snem.
Fluorki, jak opisano powyżej, wprawdzie działają
Do działań niepożądanych należą bóle głowy,
anabolicznie na kości, jednak jakość nowo powstałej
nudności, biegunki, reakcje skórne i in.
kości nie odpowiada normalnym kościom, i dlatego
Ranelinian strontu może zmniejszać wchłanianie
ich zastosowanie jest ograniczone do specjalnych
tetracyklin i fluorochinolonów.
przypadków.
Teryparatyd jest co najmniej tak skuteczny jak
bisfosfoniany, lecz jego podstawową wadą jest ko-
2.4.5.3. Zasady prowadzenia profilaktyki nieczność codziennego wykonywania iniekcji, a po-
i leczenia osteoporozy nadto terapia nie może trwać dłużej niż 18 miesięcy
i jest bardzo droga.
Zapobieganie rozwojowi osteoporozy polega na regu- Kalcytonina w aerozolu donosowym, dopuszczona
larnej aktywności fizycznej, dostarczaniu przynajmniej w celu zmniejszenia ryzyka złamania kręgów u ko-
1000 mg (lepiej 2000 mg) wapnia dziennie przez spo- biet z osteoporozą pomenopauzalną, nie należy jed-
żywanie pokarmów bogatych w wapń (przede wszyst- nak do leków pierwszego rzutu.
kim mleka lub jego przetworów) bądź przyjmowanie Chociaż w przypadku ranelinianu strontu potrzeb-
preparatów zawierających wapń oraz na dodatko- ne są dalsze dane w celu dokonania ostatecznej oceny,
wym podawaniu 500 jednostek witaminy D3 – przede jest on już dziś pełnowartościowym lekiem w terapii
wszystkim u pacjentów w starszym wieku. osteoporozy.
MUTSCHLER-2009.indd 422 2010-01-07 22:12:57
Układ hormonalny 423
2.5. Grasica
2.5.1. Anatomia grasicy
Grasica jest płaskim, podłużnym narządem poło- 2.5.2. „Hormony grasicy”
żonym w śródpiersiu nad osierdziem, za mostkiem (czynniki grasicze)
i przed głównymi żyłami. Narząd ten jest największy
Układ hormonalny
u dzieci; u noworodków waży około 12 g, natomiast Grasica wytwarza szereg peptydów o masie cząsteczkowej
w 2–3 roku życia ok. 35 g. Taki ciężar utrzymuje się od 1200 Da do 14000 Da. Ich struktura i funkcja nie zo-
aż do okresu dojrzewania, a potem następuje stopnio- stały jeszcze w pełni poznane i dlatego czasami określa się
wy fizjologiczny zanik grasicy i ulega ona przekształ- je nie jako hormony, lecz jako czynniki. Do grupy tymozyn
mieszaniny różnych hormonów grasicy – należy tzw. frakcja
ceniu w tkankę tłuszczową. 5, z której m.in. syntetycznie w czystej postaci wyizolowano
tymozynę α1. Innymi hormonami grasicy są: tymopoetyna,
U dzieci grasica otoczona jest torebką i wykazuje wyraź- tymopentyna (fragment tymopoetyny). tymulina (facteur thy-
ną strukturę płatową, w której można wyróżnić położone mique serigue, nonapeptyd) i tymostymulina (TP-1).
w środku części rdzenne i na obwodzie części korowe. Pod- Hormony grasicy, jak wyżej wspomniano, wpływają B2
stawowy szkielet miąższu grasicy stanowią rozgałęzione ko- na dojrzewanie limfocytów T. W badaniach na zwierzętach
mórki siatkowate, tworzące gąbczaste utkanie siateczkowa- wykazano także, że nasilają one proliferację tkanki limfa-
te. W przestrzeni międzykomórkowej znajdują się małe lim- tycznej, wzmagają regenerację tkanki limfatycznej uszko-
focyty. Komórki siateczki często układają się w grupy ściśle dzonej przez promieniowanie jonizujące, przyspieszają
przylegające do siebie, tworząc w ten sposób tzw. ciałka odrzucanie przeszczepów skóry oraz poprawiają ogólną
Hassala, których funkcja nie została jeszcze poznana. odporność.
U ludzi próbowano i nadal próbuje się stosować hormo-
ny grasicy w terapii wielu schorzeń, np. reumatoidalnego
Grasica ma szczególne znaczenie dla odporności zapalenia stawów, twardziny układowej, przewlekłego za-
immunologicznej, przede wszystkim dla dojrzewania palenia wątroby, niedoborów odporności i nowotworów
limfocytów T . złośliwych. Uzyskane wyniki nie są jednoznaczne.
MUTSCHLER-2009.indd 423 2010-01-07 22:12:57
424 Układ hormonalny
2.6. Wyspy trzustki
2.6.1. Anatomia wysp trzustki Dopiero w 1921 r. Banting i Best rozwiązali ten problem.
Oparli się oni przy tym na dwóch roboczych hipotezach:
Trzustka łączy w sobie dwa oddzielne układy czyn- ■ substancja czynna jest wytwarzana w wyspach trzustko-
nościowe: wych Langerhansa.
■ w komórkach nabłonkowych gruczołów trzustko- ■ otrzymane w sposób tradycyjny wyciągi z trzustki nie wyka-
wych jest wytwarzany sok trzustkowy, który przez zują odpowiedniego działania, dlatego że insulina – tak na-
przewód wyprowadzający jest wydzielany do jelita zwano tę substancję czynną – ulega zniszczeniu przez pro-
teolityczne enzymy trzustkowe w trakcie ich sporządzania.
cienkiego (funkcja zewnątrzwydzielnicza),
■ specjalne grupy komórek, tzw. wyspy trzustkowe Podwiązywali oni przewód wyprowadzający trzustki
u zwierząt doświadczalnych, wskutek czego ulegała znisz-
Langerhansa, wytwarzają hormony, które są uwal- czeniu wydzielnicza tkanka gruczołowa, natomiast wy-
niane bezpośrednio do krwiobiegu (funkcja we- spy trzustkowe Langerhansa pozostawały niezmienione.
wnątrzwydzielnicza). Z pozostałej tkanki trzustki udało się im otrzymać wyciągi
ze stosunkowo dużą zawartością substancji czynnej. Ko-
Wyspy trzustkowe Langerhansa są grupami komó- lejnym decydującym krokiem było stwierdzenie, że jako
materiał wyjściowy można także wykorzystać prawidłowe
rek o średnicy 75–300 μm, które są rozsiane (w for- trzustki, pod warunkiem że ekstrakcję przeprowadzi się
mie wysp) w zewnątrzwydzielniczej tkance trzustki na zimno z wykorzystaniem alkoholu zawierającego kwas
(przede wszystkim w ogonie trzustki) i są zaopatry- solny, co zapobiega enzymatycznemu rozkładowi insuliny.
wane licznymi naczyniami mającymi szerokie świat-
ło. Łącznie te grupy komórek są często określane W 1926 r. Abel otrzymał insulinę w formie krystalicznej,
w 1954 r. Sanger ustalił sekwencję aminokwasową hete-
jako wyspy trzustkowe Langerhansa, co ma podkre- rocyklicznego peptydu, a wiele zespołów badawczych
ślić ich morfologiczną i czynnościową niezależność. (Katsoyannis, Zahn) opisało jego syntezę ze związków
Orientacyjnie stanowią one 2–3% tkanki trzustki. prostych.
Główną masę komórek wysp (ok. 70%) stanowią
słabo barwiące się komórki β wytwarzające insulinę W 1979 r. udało się w wyniku transferu genów dokonać
syntezy insuliny w bakteriach E. coli.
i amylinę. Drugie pod względem masy sumarycznej
(ok. 20%) są zawierające liczne ziarnistości komórki
Budowa chemiczna, synteza, magazynowanie
α, które są odpowiedzialne za wytwarzanie glukago-
i rozkład. Insulina jest polipeptydem (masa cząstecz-
nu. Pozostałe 10% stanowią komórki D wytwarzające
kowa ok. 5800 Da) składającym się z dwóch łańcu-
somatostatynę oraz dodatkowy typ komórek (komór-
chów polipeptydowych, łańcucha A złożonego z 21
ki PP, komórki F) wytwarzających polipeptyd trzust-
aminokwasów i łańcucha B z 30 aminokwasów, które
kowy, który hamuje wytwarzanie soku trzustkowego
są ze sobą połączone 2 mostkami dwusiarczkowymi
i perystaltykę jelit.
(ryc. B 2.6-1).
Pierwszym produktem translacji powstającym na ryboso-
mach komórek B jest składająca się tylko z jednego łańcu-
2.6.2. Hormony trzustki cha preproinsulina. W siateczce cytoplazmatycznej zostaje
z niej utworzona proinsulina, w której łańcuch A i B in-
2.6.2.1. Insulina suliny są połączone przez tzw. polipeptyd C, wykazujący
strukturalną homologię z somatomedynami. Odszczepienie
Badania nad insuliną są pasjonującym rozdziałem w histo- polipeptydu C odbywa się w aparacie Golgiego. Powstała
rii biochemii i farmakoterapii. w taki sposób insulina jest w formie heksameru – związa-
na z cynkiem – magazynowana w pęcherzykach, z których
W 1869 r. Langerhans odkrył nazwane później jego nazwi- w razie potrzeby zostaje uwolniona (razem z polipeptydem
skiem grupy komórek w trzustce. C) na drodze egzocytozy.
W 1889 r. Mering i Minkowski wykazali, że u psów po usu-
nięciu trzustki pojawia się stan chorobowy odpowiadają- W osoczu znaczna część insuliny pozostaje w formie
cy obrazowi klinicznemu cukrzycy oraz że wszczepienie niezwiązanej. Osoczowy okres półtrwania wynosi
tkanki trzustki pod skórę powoduje ustąpienie wywoła-
nych objawów. Nie udało im się jednak otrzymać wyciągu poniżej 10 min, a połowiczy czas działania – ok. 40
z trzustki, który pozwoliłby na wyleczenie tych zwierząt min. Rozkład insuliny odbywa się przede wszystkim
doświadczalnych. w wątrobie i nerkach.
MUTSCHLER-2009.indd 424 2010-01-07 22:12:57
Układ hormonalny 425
NH2 NH2 NH2 NH2
Gly – Ile – Val – Glu – Glu – Cys – S – S– Cys – Ser – Leu – Tyr – Glu – Leu – Glu – Asp – Tyr – Cys – Asp
Cys A10 S
NH2 NH2 S A8 – Ser S
Phe – Val – Asp – Glu – His – Leu S Val – Glu – Ala – Leu – Tyr – Leu – Val – Cys – Gly
Cys Leu Glu
Układ hormonalny
Gly – Ser – His Arg
B30 – Lys – Pro – Thr – Tyr – Phe – Phe – Gly
Ryc. B 2.6-1. Sekwencja aminokwasowa insuliny ludzkiej, wieprzowej i wołowej. Człowiek: A8 = Thr; A10 = Ile; B30 : Thr. Świnia: A8 =
Thr; A10 = Ile; B30 = Ala. Bydło: A8 = Ala; A10 = Va; B30 = Ala.
B2
Trzustka zawiera w sumie mniej więcej 80 IU jest wzrost stężenia glukozy we krwi (zob. ryc. 2.6-2).
(zob. poniżej) insuliny, z czego dziennie wydzielana Poza tym wydzielanie insuliny wywołuje także
jest mniej więcej połowa. wzrost stężenia w osoczu różnych aminokwasów
Insulina ludzka, insuliny wołowe i wieprzowe pod (np. argininy, lizyny), wolnych kwasów tłuszczo-
względem sekwencji aminokwasowej różnią się od sie- wych i niektórych hormonów przewodu pokarmowe-
bie tylko w nieznacznym stopniu (zob. ryc. B 2.6-1), go – w szczególności tzw. inkretyn. Ilość wydzielanej
natomiast ich biologiczne działanie jest w identyczne. insuliny jest modulowana przez autonomiczny układ
nerwowy: impulsacja z części przywspółczulnej i po-
Kontrola i mechanizm uwalniania insuliny. Właś- budzenie receptorów β2 części współczulnej wzma-
ciwym bodźcem powodującym uwolnienie insuliny gają uwalnianie, natomiast pobudzenie receptorów α2
glukoza
GLUT2
gluko- komórka β
kinaza
diazoksyd
(hiperpolaryzacja)
glukozo-6-fosforan
ADP
K+
mitochondrium
pochodne sulfonylomocznika,
ATP glinidy
[Ca2+]↑ uwolnienie insuliny
Ca2+ depolaryzaja na skutek
zablokowania kanałów K+
Ryc. B 2.6-2. Stymulacja egzocytozy insuliny przez pochodne sulfonylomocznika i glinidy w wyniku wzrostu stężenia wapnia śród-
komórkowego, zamknięcia kanałów potasowych za pośrednictwem ATP. Hamowanie uwalniania insuliny przez diazoksyd. GLUT2
– białko transportujące glukozę 2.
MUTSCHLER-2009.indd 425 2010-01-07 22:12:57
426 Układ hormonalny
hamuje wydzielanie. Glukagon (w dużych dawkach) Działanie insuliny. Insulina jest ważnym dla życia,
wzmaga uwalnianie insuliny, a somatostatyna i adre- zwiększającym wzrost, hormonem anabolicznym. Ma
nalina je hamują. ona następujące właściwości:
Uwolnienie insuliny na skutek wzrostu stężenia
glukozy we krwi jest następstwem wzmożonego utle- ■ poprawia wychwyt glukozy i aminokwasów przez
niania glukozy w komórkach β i następczego zwięk- komórki większości tkanek,
szonego wytwarzania ATP. W odpowiedzi na zwięk-
■ wzmaga oksydacyjny rozkład glukozy,
szenie ilości ATP zamknięte zostają kanały potasowe
zależne od ATP. To z kolei prowadzi do zmniejsze- ■ zwiększa glikogenogenezę w wątrobie i mięśniach
nia przepuszczalności błony komórkowej dla potasu oraz zapobiega glikogenolizie,
i wskutek tego do spadku potencjału spoczynkowego
■ wzmaga wytwarzanie tłuszczów z glukozy,
błony komórkowej z –65 do –30 mV. To prowadzi
do aktywacji zależnych od napięcia kanałów wap- ■ hamuje przekształcanie białek w glukozę.
niowych, przez które jony wapnia napływają z prze-
strzeni zewnątrzkomórkowej, co uruchamia proces Wszystkie te procesy prowadzą do obniżenia stężenia
egzocytozy. glukozy we krwi.
insulina
glukoza
receptor insulinowy
GLUT4
P P
P P
endocytoza aktywacja
kompleksu domeny β aktywacja białka transkrypcja
insulina-receptor kinazy tyrozynowej RAS genów
fosforylacja IRS
przemieszczenie białek
transportujących aktywacja kinaz
glukozę i fosfataz
zwiększenie syntezy zwiększenie wychwytu poprawa wykorzystania zmniejszenie
glikogenu, białek glukozy glukozy glukoneogenezy
i triglicerydów
obniżenie stężenia glukozy
we krwi
Ryc. B 2.6-3. Mechanizm działania insuliny (schemat uproszczony) (według Ranga, Dale’a, Rittera). IRS – substrat dla receptora in-
sulinowego.
MUTSCHLER-2009.indd 426 2010-01-07 22:12:58
Układ hormonalny 427
W tkance tłuszczowej insulina poprzez aktywację ła, można zwiększyć liczbę receptorów, co zmniejsza
osoczowych lipaz lipoproteinowych zwiększa wnika- insulinooporność lub doprowadza do jej ustąpienia.
nie wolnych kwasów tłuszczowych do komórek, gdzie
są one następnie odkładane w postaci triglicerydów Zastosowanie lecznicze. Zastosowanie lecznicze in-
(tłuszcz zapasowy). Synteza triglicerydów wzrasta suliny omówiono w rozdziale B 2.6.5.2.1.
także wskutek nasilenia przemiany glukozy do acety-
lo-CoA. Poza tym insulina przeciwdziała mobilizacji
i rozkładowi tłuszczów. Hamuje ona także lipolizę 2.6.2.2. Glukagon
w wątrobie.
Układ hormonalny
Innymi efektami działania insuliny są: nasile- Budowa chemiczna, synteza, magazynowanie
nie dokomórkowego wnikania jonów potasu oraz i rozkład. Wytwarzany w komórkach α wysp trzust-
zmniejszenie katabolicznego działania glukokortyko- kowych Langerhansa glukagon, podobnie jak insuli-
steroidów i hormonów tarczycy. na, jest polipeptydem (masa cząsteczkowa 3500 Da).
Składa się on tylko z jednego łańcucha zbudowane-
go z 29 aminokwasów. Sekwencja aminokwasowa
Mechanizm działania. Wzajemne oddziaływanie
glukagonu wykazuje znaczną homologię z sekre-
między hormonem i receptorem błonowym pro-
tyną i VIP. Osoczowy okres półtrwania glukagonu,
wadzi do autofosforylacji wewnątrzkomórkowych
w związku z szybką hydrolizą w wątrobie, nerkach B2
struktur receptora. W wyniku tego receptor nabie-
i krwi, wynosi jedynie kilka minut.
ra właściwości aktywnej kinazy tyrozynowej (znie-
sienie przestrzennej inhibicji domeny kinazy tyro-
Mechanizm i kontrola uwalniania. Uwalnianie
zynowej w podjednostce β receptora przez zmianę
glukagonu z komórek α wywołują: hipoglikemia
konformacyjną podjednostki α). Następująca potem
(np. w następstwie głodu), wzrost stężenia w osoczu
fosforylacja różnych substratów (m.in. substratu
aminokwasów wykorzystywanych do glukoneoge-
receptora insulinowego l i 2) powoduje aktywację
nezy, a także intensywny wysiłek fizyczny, stres, po-
wielu enzymów biorących udział w przemianie ma-
drażnienie nerwu błędnego i pobudzenie receptorów
terii w komórkach efektorowych (zob. ryc. B 2.6-3).
β-adrenergicznych. Hamujący wpływ na wydzielanie
Ponadto w komórkach mięśni i tkanki tłuszczowej
glukagonu wywierają hiperglikemia, agoniści recep-
kompleks insulina-receptor insulinowy powoduje
torów α-adrenergicznych, insulina, inkretyna i soma-
przemieszczenie preformowanych białek transpor-
tostatyna.
tujących glukozę (GLUT) z pęcherzyków cytopla-
zmatycznych do błony komórkowej. Dzięki zależnej
Działanie. Glukagon bierze udział w regulacji stęże-
od GLUT ułatwionej dyfuzji dochodzi do znacznego
nia glukozy we krwi oraz wielu procesach metabo-
przyspieszenia transportu glukozy z przestrzeni ze-
licznych w wątrobie, działając antagonistyczne w sto-
wnątrzkomórkowej do wewnątrzkomórkowej. Poza
sunku do insuliny. Powoduje on mobilizację rezerw
tym wskutek aktywacji pompy Na+/K+ zależnej
energii organizmu w stanach zwiększonego zapotrze-
od ATP następuje wzrost dokomórkowego wnika-
bowania na energię i zagrażającej hipoglikemii.
nia jonów potasu. W końcowym efekcie powyższe
Podobnie jak adrenalina, powoduje on wzmożenie
procesy prowadzą do opisanego już spadku stęże-
glikogenolizy w wątrobie, ale – w przeciwieństwie
nia glukozy we krwi na skutek poprawy przemian
do adrenaliny – nie wywołuje on takiego efektu w ko-
glukozy oraz zmniejszenia glukoneogenezy, jak
mórkach mięśni. Ponadto w wątrobie zwiększa on
również nasilenia syntezy glikogenu, białek i trigli-
glukoneogenezę z aminokwasów i mleczanu, co pro-
cerydów.
wadzi do wzrostu stężenia glukozy we krwi.
Na metabolizm tłuszczów glukagon wpływa dwu-
Gęstość receptorów insulinowych. U ludzi z pra- kierunkowo: z jednej strony zwiększa utlenianie
widłową przemianą materii występuje nadmiar re- kwasów tłuszczowych i tworzenie ciał ketonowych
ceptorów insulinowych, dzięki czemu do wywołania w wątrobie, z drugiej strony natomiast wzmaga lipo-
maksymalnego efektu wystarczy pobudzenie tylko lizę w tkance tłuszczowej i wątrobie.
ich części. Podwyższone stężenie insuliny prowadzi Wpływ glukagonu na metabolizm białek polega
w następstwie internalizacji receptorów do spadku na zwiększeniu proteolizy, która dostarcza aminokwa-
ich liczby (down-regulation). Nadwaga także pro- sów do glukoneogenezy.
wadzi do zmniejszenia gęstości receptorów insulino-
wych. Z drugiej strony, za pomocą diety redukcyjnej Mechanizm działania. Efekt działania glukagonu
i będącego jej konsekwencją zmniejszenia masy cia- jest następstwem reakcji między hormonem i sprzę-
MUTSCHLER-2009.indd 427 2010-01-07 22:12:58
428 Układ hormonalny
żonym z białkiem G receptorem błonowym, co powo- węglowodanów, zmian intensywności utleniania glu-
duje aktywację cyklazy adenylanowej i w rezultacie kozy, która podczas wysiłku fizycznego może zwięk-
indukcję tworzenia cAMP. Proces ten poprzez fosfo- szyć się kilkakrotnie, oraz stale występujących od-
rylację enzymów wywołuje następnie właściwe efek- chyleń od wartości pożądanej układ regulacji jest za-
ty metaboliczne. zwyczaj w stanie szybko i na dłuższy czas przywrócić
prawidłowe stężenie glukozy. Jedynie w razie przyję-
Zastosowanie lecznicze. W porównaniu z insuli- cia większej ilości węglowodanów może się pojawić
ną znaczenie terapeutyczne glukagonu jest znacznie przejściowy wzrost stężenia cukru we krwi (hipergli-
mniejsze. Poza stanami hipoglikemicznymi znalazł kemia poposiłkowa, zob. ryc. B 2.6-4).
on zastosowanie w hamowaniu perystaltyki podczas
badania przewodu pokarmowego, przy stanach skur- Można zatem stwierdzić, że stężenie glukozy we krwi
czowych zwieracza wpustu oraz przy zatruciach leka- stanowi wypadkową zachodzących w organizmie
mi blokującymi receptory β-adrenergiczne. procesów dostarczających i zużywających glukozę
(zob. ryc. B 2.6-5). Przez aktywację lub hamowanie
poszczególnych procesów regulowanych głównie
2.6.3. Podstawy fizjologiczne regulacji hormonalnie układ jest w stanie utrzymać pożądaną
stężenia glukozy we krwi wartość glikemii we krwi.
Szczególnie ważną funkcję pełni, jak opisano wcześ-
W warunkach fizjologicznych stężenie glukozy
niej, insulina. Żołądkowy peptyd hamujący (GIP)
we krwi na czczo jest utrzymywane na poziomie
oraz glukagonopodobny peptyd-1 (GLP-1) zwiększa-
3,05–5,55 mmol/1 (55–100 mg/dl). Pomimo zmienia-
ją uwalnianie insuliny; somatostatyna, przeciwnie,
jącej się ilości przyjmowanych wraz z pożywieniem
hamuje jej uwalnianie.
Adrenalina, glukagon i somatotropina uczestniczą
także w regulacji stężenia glukozy we krwi. Adrena-
lina i glukagon powodują przede wszystkim szybkie
Ś O DP K PK
SP SP SP sen
stężenie insuliny w osoczu (μE/ml)
70 insulina,
60 somatostatyna
50 somato-
statyna
40 glukoneogeneza glukagon,
(wątroba) katecholaminy,
30 T3, T4
20
10
0
glikemia
insulina 3,05-5,55 mmol/l glukokortyko-
(0,55-1,00 g/l) steroidy
8
(mmol/l)
glikemia
6
4
2
wykorzystanie glukozy
0 IGF-1 (mięśnie, tkanka tłuszczowa) hormon
7 11 15 19 23 3 7 wzrostu
godziny
Ś – śniadanie K – kolacja
GIP
O – obiad PK – późna kolacja peptyd podobny do glukagonu-1
DP – dodatkowy posiłek SP – spacer (1 godzina)
Ryc. B 2.6-5. Schematycznie przedstawiona regulacja hormo-
Ryc. B 2.6-4. Dobowy profil glikemii i stężenia insuliny w osoczu nalna glikemii. GIP – żołądkowy peptyd hamujący, IGF-1 – insu-
u osoby z prawidłową masą ciała (wg Molnara). linopodobny czynnik wzrostu 1
MUTSCHLER-2009.indd 428 2010-01-07 22:12:58
Układ hormonalny 429
uwalnianie glukozy z nagromadzonych zapasów, Objawy autonomiczne, pojawiające się ze szcze-
a somatotropina hamuje (długotrwale) dokomórkowe gólną intensywnością po szybkim spadku stężenia
wnikanie glukozy. Wydzielanie tych trzech antagoni- glukozy we krwi u pacjentów z hiperglikemią, to nie-
stów insuliny wzrasta w odpowiedzi na spadek stę- pokój, uczucie lęku, kołatanie serca, nudności, dresz-
żenia glukozy we krwi poniżej pożądanej wartości, cze, wilczy głód i pocenie się. Przy leczeniu cukrzycy
co prowadzi do wzrostu jej stężenia. Należy jednak pełnią one funkcję ostrzegawczą.
pamiętać o tym, że adrenalina i glukagon działają Objawy neuroglikopenii, rozwijające się przy po-
antagonistycznie do insuliny tylko w aspekcie wpły- wolnym obniżaniu stężenia glukozy we krwi, to osła-
wu na stężenie glukozy we krwi; w rzeczywistości, bienie, zaburzenia snu i myślenia, splątanie, zabu-
Układ hormonalny
w fizjologicznych warunkach oba te hormony dzia- rzenia widzenia, zawroty głowy i kurcze mięśnio-
łają raczej synergistycznie z insuliną, gdyż poprzez we. Bardzo niskie stężenia glukozy we krwi (< 1,95
uwalnianie glukozy stwarzają one warunki do wzmo- mmol/l lub < 35 mg/dl), których przyczyną jest za-
żonego jej wykorzystania. zwyczaj przedawkowanie insuliny lub wyspiak, pro-
Inkretyna pobudza zależną od glukozy sekrecję wadzą do wstrząsu hipoglikemicznego (śpiączki hi-
insuliny. poglikemicznej).
W odniesieniu do glukokortykosteroidów oraz T3
i T4 nie udało się wykazać bezpośredniego ich udzia- Leczenie. W leczeniu hipoglikemii zastosowanie
łu w procesie regulacji stężenia glukozy we krwi; znajdują: B2
substancje te działają przede wszystkim permisyw-
■ glukagon (zob. poniżej),
nie, tzn. poprzez wzmożenie efektów wywieranych
przez adrenalinę czy glukagon. ■ diazoksyd.
Przeciętna dawka glukagonu wynosi 0,5–1 mg dzien-
nie s.c., i.m. lub i.v. Do działań niepożądanych należą
2.6.4. Hipoglikemia sporadycznie obserwowane nudności i wymioty oraz
reakcje nadwrażliwości. Indometacyna osłabia efekt
Patofizjologia. O hipoglikemii mówimy gdy stęże- działania glukagonu, natomiast efekt przeciwkrzepli-
nie cukru we krwi – oznaczane metodą enzymatyczną wy warfaryny ulega nasileniu pod wpływem gluka-
– spadnie poniżej 2,5 mmol/l (45 mg/dl). gonu. U chorych z guzem chromochłonnym stosowa-
Zgodnie z etiologią wyróżnia się hipoglikemię nie glukagonu jest przeciwwskazane.
egzogenną i endogenną. Hipoglikemia egzogenna Diazoksyd (poza obniżaniem ciśnienia krwi)
może rozwinąć się m.in. w następstwie powstrzy- zwiększa stężenie glukozy we krwi poprzez hamo-
mywania się od jedzenia, niewłaściwego dawkowa- wanie wydzielania insuliny oraz zwiększanie synte-
nia insuliny lub doustnych leków przeciwcukrzyco- zy glukozy w wątrobie. Mechanizm działania – tak
wych oraz nadużycia alkoholu (hamowanie gluko- jak w przypadku rozszerzania naczyń krwionośnych
neogenezy). – jest związany z otwarciem kanałów potasowych
Hipoglikemia endogenna może być spowodowana (kanały potasowe zależne od ATP) prowadzącym
obecnością wyspiaka (insulinoma) wytwarzającego do depolaryzacji komórek wysp trzustki, a w konse-
insulinę lub pozatrzustkowego nowotworu wytwa- kwencji do zahamowania uwalniania insuliny (zob.
rzającego produkty rozkładu tryptofanu, które zwięk- ryc. B 2.6-2).
szają zużycie glukozy w tkankach obwodowych. Jako Dawkowanie ustala się indywidualnie (dawka po-
inne przyczyny bierze się pod uwagę ciężkie schorze- czątkowa 5 mg/kg/dobę).
nia wątroby z zaburzeniami glukoneogenezy, wro- W schorzeniach kardiologicznych diazoksyd
dzone choroby metaboliczne z defektami enzymów nie jest już stosowany.
uczestniczących w glikogenolizie, niedoczynność
przysadki i kory nadnerczy oraz wzmożone zużycie
glukozy spowodowane ciężką pracą fizyczną lub glu-
kozurią.
2.6.5. Cukrzyca
Objawy hipoglikemii można podzielić na dwie
grupy: objawy autonomiczne uwarunkowane akty- 2.6.5.1. Patofizjologia
wacją autonomicznego układu nerwowego (przede
wszystkim części współczulnej) oraz objawy neuro- Termin cukrzyca oznacza zespół chorobowy cha-
glukopeniczne spowodowane niedostatecznym za- rakteryzujący się (bezwzględnym lub względnym)
opatrzeniem mózgu w glukozę. niedoborem insuliny ze współistniejącą przewle-
MUTSCHLER-2009.indd 429 2010-01-07 22:12:59
430 Układ hormonalny
kłą hiperglikemią (stężenie glukozy we krwi pełnej W cukrzycy typu I (cukrzycy insulinozależnej)
na czczo > 6,15 mmol/l lub > 110 mg/dl) i będącymi szybko nasila się niedobór insuliny, aż do całkowi-
tego następstwem zaburzeniami przemiany materii tego braku wydzielania insuliny. Dlatego niezbędne
oraz uszkodzeniem narządów (powikłania późne). jest leczenie substytucyjne insuliną.
Bezwzględny niedobór insuliny stwierdzamy, gdy W cukrzycy typu II progresja zaburzeń jest znacz-
trzustka wskutek zniszczenia komórek wysp Langer- nie wolniejsza. Zazwyczaj występuje tylko względny
hansa nie jest już w stanie wydzielać insuliny. niedobór insuliny. Uwzględniając masę ciała, wyróż-
O względnym niedoborze insuliny mówimy, gdy nia się następujące podtypy: z masą ciała od niedo-
wytwarzanie insuliny jest zbyt małe w stosunku wagi do prawidłowej (typ IIa) oraz z nadwagą (typ
do potrzeb, działanie insuliny na komórki docelowe IIb). 80% chorych na cukrzycę pierwotną typu II ma
jest osłabione przez przeciwciała przeciwinsulinowe, nadwagę!
liczba receptorów insulinowych w narządach efek-
torowych jest zmniejszona lub występuje defekt re- W rozwoju obu typów cukrzycy biorą udział zarów-
ceptora insulinowego albo sprzężonego z receptorem no czynniki genetyczne, jak i egzogenne. Określone
wewnątrzkomórkowego przekazywania sygnałów antygeny HLA (np. DR3, DR4) stanowią genetycz-
(defekt postreceptorowy). ną predyspozycję do rozwoju cukrzycy typu I (inne
Cukrzyca jest najczęstszym i zarazem najistotniej- antygeny HLA, np. DR2, mają wpływ ochronny).
szym pod względem swoich następstw zaburzeniem W przypadku predyspozycji genetycznej decydujące
przemiany materii. W Niemczech (i w Polsce) choru- znaczenie dla pojawienia się cukrzycy typu I mają
je na nią 5–8% ludności i obserwuje się istotną ten- procesy immunologiczne. W wyniku zachodzącej
dencję wzrostową (!). Jeszcze częściej stwierdza się za pośrednictwem limfocytów T reakcji autoimmu-
nieprawidłową tolerancję glukozy (stan przedcukrzy- nologicznej wywołującej limfocytarne (autoimmu-
cowy), która dotyczy 16% populacji. nologiczne) zapalenie wysp Langerhansa dochodzi
do wybiórczego uszkodzenia komórek β i przez
Typy cukrzycy. Na podstawie etiologii, patogenezy to do stopniowo rozwijającego się przez wiele lat za-
i objawów klinicznych wyodrębniono różne typy przestania wytwarzania i uwalniania insuliny. Uważa
cukrzycy (zob. tab. B 2.6-1). Spośród różnych typów się, że czynnikami egzogennymi mogącymi wywołać
cukrzycy około 90% stanowi cukrzyca typu II. reakcję autoimmunologiczną są zakażenia wirusowe
(np. wirusami Coxsackie lub świnki) albo szkodliwe
czynniki środowiskowe. Już podczas fazy wstępnej,
tzn. jeszcze przed wystąpieniem objawów choroby,
Tabela B 2.6-1. Etiologiczna klasyfikacja cukrzycy (zmodyf. we- można stwierdzić przeciwciała przeciw komórkom β,
dług WHO, 1997)
insulinie i białku wysp trzustkowych – dekarboksy-
lazie glutaminianowej, która pod względem budowy
Typ cukrzycy Dawniej stosowane nazwy jest bardzo podobna do białek wirusów Coxsackie.
I. Cukrzyca typu l
W przypadku cukrzycy typu II prawdopodobień-
A: uwarunkowana czynnikami cukrzyca młodzieńcza;
immunologicznymi, cukrzyca insulinozależna stwo dziedziczenia (inaczej niż według wcześniej-
B: idiopatyczna (IDDM) szych poglądów) jest wyraźnie większe niż w typie
II. Cukrzyca typu II I, jednak sposób przekazywania potomstwu cech
genetycznych nie został dokładnie wyjaśniony.
typ IIa bez otyłości; cukrzyca wieku starczego;
typ IIb z otyłością cukrzyca insulinoniezależna Nie stwierdzono związku z układem HLA. Pewne
(NIDDM) jest to, że dziedziczenie to ma charakter wieloge-
III. Inne swoiste typy cukrzycy nowy. Decydującą rolę jako czynniki wyzwalające
m.in. będące następstwem:
odgrywające nadmierne odżywianie i nadwaga (oty-
– wad genetycznych, łość), a także brak aktywności fizycznej.
– schorzeń zewnątrzwydzielniczej
części trzustki,
– endokrynopatii, Wstępną fazą pełnoobjawowej cukrzycy typu II jest tzw.
– działania leków i środków zespół metaboliczny (zespół poprzedzający cukrzy-
chemicznych
cę pierwotną typu II, zespół X) charakteryzujący się
IV. Cukrzyca w przebiegu ciąży otyłością, hiperglikemią, insulinoopornością, nadciś-
cukrzyca w przebiegu ciąży cukrzyca ciężarnych nieniem, dyslipidemią, a także trombofilią. W Polsce
stwierdza się go u 5–9 milionów ludzi. Z tego powodu
zespół metaboliczny ma szczególnie duże znaczenie.
MUTSCHLER-2009.indd 430 2010-01-07 22:12:59
Układ hormonalny 431
masy w bardzo wysokim stopniu biorą udział w meta-
bolizmie glukozy, nie są w stanie przyjąć wystarcza-
otyłość bez cukrzycy
jącej ilości glukozy do swoich komórek. Znaczenie
patogenetyczne ma prawdopodobnie także wydzie-
1,5 lany razem z insuliną amyloidowy polipeptyd wysp
trzustkowych amylina, blokujący działanie insuliny
w wątrobie i mięśniach szkieletowych.
U mających nadwagę chorych na cukrzycę typu
Immunoreaktywność insuliny
otyłość z cukrzycą II tkanki obwodowe wykazują znacznie zmniejszoną
Układ hormonalny
1,0 wrażliwość na insulinę i dlatego na początku choroby
stwierdza się zwiększone wydzielanie insuliny, mają-
prawidłowa masa ciała bez cukrzycy ce na celu utrzymanie prawidłowego stężenia glukozy
we krwi (zob. ryc. B 2.6-6). W późniejszych stadiach
z powodu zwiększenia zapotrzebowania czynnościo-
0,5 wego może jednak ulec wyczerpaniu zdolność komó-
rek β trzustki do pełnienia swojej funkcji. Stężenie
insuliny w osoczu obniża się wówczas do wartości
prawidłowa masa ciała z cukrzycą prawidłowych i jednocześnie wyraźnie wzrasta stę- B2
żenie glukozy we krwi.
0 15 30 45 60 90 120 150 180 Na ryc. B 2.6-7 przedstawiono schematycznie po-
czas (min) glądy na temat patogenezy cukrzycy typu II.
Ryc. B 2.6-6. Zmiany stężenia insuliny w osoczu po doustnym W grupie III w tab. B 2.6-1 zestawiono inne formy
podaniu 100 g glukozy (według Bagdade’a i in.). Tylko u chorych cukrzycy o różnej etiologii.
na cukrzycę z prawidłową masą ciała występuje zmniejszone wy-
dzielanie insuliny w porównaniu z osobami zdrowymi. U otyłych O cukrzycy w przebiegu ciąży (cukrzyca ciężarnych, grupa
chorych na cukrzycę wydzielanie insuliny jest zwiększone w po- IV) mówimy, gdy u kobiety stwierdzi się po raz pierwszy
równaniu z osobami zdrowymi. hiperglikemię podczas ciąży. U mniej więcej 1–3% ciężar-
nych należy się liczyć z pojawieniem cukrzycy w przebiegu
Stwierdzany w pełnoobjawowej cukrzycy typu II ciąży. Jeżeli nie rozpocznie się leczenia, zwiększa się ry-
zyko wystąpienia stanu przedrzucawkowego, wielowodzia,
(względny) niedobór insuliny jest spowodowany porodów przedwczesnych, zwiększonej masy ciała nowo-
zarówno zaburzeniem wydzielania insuliny, jak i in- rodka po urodzeniu się oraz zwiększonej okołoporodowej
sulinoopornością komórek docelowych. Często do- chorobowości i śmiertelności wśród noworodków. Z tego
chodzi do zaburzenia przekształcania proinsuliny powodu jest wskazane wczesne rozpoznanie i leczenie cuk-
w insulinę oraz do pulsacyjnego uwalniania insuliny. rzycy ciężarnych.
Także tkanki wrażliwe na insulinę (wątroba, mięśnie
i tkanka tłuszczowa) reagują w mniejszym stopniu Zaburzenia przemiany materii wywołane niedo-
na insulinę. Pomimo hiperglikemii w wątrobie nie borem insuliny. Charakterystyczne dla niedoboru
jest hamowana glukoneogeneza i uwalnianie glukozy insuliny zaburzenia metabolizmu węglowodanów,
do krwi; mięśnie szkieletowe, które z powodu dużej tłuszczów i białek przedstawiono w tab. B 2.6-2. Wy-
Tabela B 2.6-2. Następstwa cukrzycy
Zaburzenia prowadzą do z następującymi objawami
metabolizmu węglowodanów wskutek upośledzonego hiperglikemii, poliurii, polidypsji,
zużytkowania glukozy, wzmożonej glukoneogenezy glikozurii świądu narządów płciowych, odwodnienia
i glikogenolizy
metabolizmu tłuszczów wskutek hamowania syntezy lipidów, hiperlipidemii, nudności, wzbudzenia odruchu
zwiększonej lipolizy i wzmożonego tworzenia ciał ketonowych hiperketonemii, ketonurii, wymiotnego, zmniejszenia masy ciała,
kwasicy ketonowej zapachu acetonu w wydychanym powietrzu
metabolizmu białek wskutek wzmożonego rozkładu białek, hiperglikemii, glikozurii, aminoacydurii, adynamii,
zwiększonej glukoneogenezy, zmniejszonej syntezy białek wzrostu stężenia produktów przemian zmniejszenia masy ciała,
azotowych we krwi zaniku mięśni
MUTSCHLER-2009.indd 431 2010-01-07 22:12:59
432 Układ hormonalny
W najcięższej postaci ostrych zaburzeń przemia-
ny materii w przebiegu cukrzycy, czyli w śpiączce
czynniki cukrzycowej (coma diabeticum), stwierdza się eks-
środowiskowe uwarunkowania
(niedostateczna genetyczne tremalne zaburzenia zużytkowania glukozy. Wyróżnia
aktywność fizyczna, się dwie postacie śpiączki cukrzycowej:
nieprawidłowe
odżywianie, otyłość) wiek ■ znacznie częstszą (ok. 80–90% przypadków)
śpiączkę z kwasicą ketonową, występującą przede
wszystkim w przebiegu cukrzycy typu I,
insulinooporność hiperinsulinemia,
normoglikemia ■ śpiączkę hiperosmolarną w przebiegu cukrzycy
typu II.
nieprawidłowa W przypadku klasycznej śpiączki z kwasicą ketonową
gluko- uszkodzenie śpiączka rozwija się w następstwie tworzenia dużych ilości
tolerancja toksyczność komórek β
glukozy ciał ketonowych i następczej kwasicy ketonowej. Wydala-
nie kwasów w postaci soli sodowych i potasowych prowa-
dzi do utraty Na+ i K+. Ponadto utrata glukozy z moczem
wywołuje diurezę osmotyczną prowadzącą do utraty wody.
Śpiączka z kwasicą ketonową charakteryzuje się zatem wy-
hipoinsulinemia, stępowaniem kwasicy, zaburzeń elektrolitowych, odwod-
cukrzyca hiperglikemia
typu II nienia oraz zmniejszeniem ukrwienia mózgu z hipoksją.
W przypadku śpiączki hiperosmolarnej nie rozwija się
kwasica ketonowa, ponieważ dostępna ilość insuliny wy-
Ryc. B 2.6-7. Patogeneza cukrzycy typu II (schemat). starcza do tego, żeby zapobiec tworzeniu ciał ketonowych
w wątrobie. Stężenie glukozy we krwi przekracza 6 g/l
i jest wyraźnie wyższe niż w przebiegu śpiączki z kwasicą
ketonową.
nikające ze zmniejszonego dokomórkowego wnikania
Śpiączka cukrzycowa, w szczególności jej postać
glukozy upośledzone zużytkowanie glukozy w ko-
hiperosmolarna, powstaje zazwyczaj stopniowo
mórkach oraz wzmożenie glukoneogenezy prowadzą
i jest poprzedzona stadium objawów prodromalnych
do hiperglikemii, a i po przekroczeniu progu nerko-
(stan przedśpiączkowy), któremu towarzyszą utrata
wego dla glukozy – do glikozurii.
apetytu, nudności, wymioty, osłabienie siły mięśnio-
wej, senność oraz nadmierne pragnienie, polidypsja
Wzmożona lipoliza w tkance tłuszczowej oraz w wą-
i poliuria. Podczas śpiączki skóra, śluzówki i język
trobie i mięśniach powoduje wzrost stężenia wol-
są suche, gałki oczne miękkie, a oddech pogłębio-
nych kwasów tłuszczowych w osoczu. W ciężkich
ny i spowolniony. W śpiączce w przebiegu kwasicy
przypadkach w wyniku zwiększonego rozkładu
ketonowej wydychane przez pacjenta powietrze ma
kwasów tłuszczowych i ograniczonego utleniania
zapach acetonu.
acetylo-CoA w cyklu kwasu cytrynowego pojawiają
się we krwi ciała ketonowe (kwasica ketonowa). Wy-
twarzana jest nadmierna ilość acetoacetylo-CoA. Po- Późne powikłania cukrzycy. W nieleczonej cukrzy-
przez stadium pośrednie β-hydroksy-β-metylo-gluta- cy typu I przewidywana długość życia jest znacznie
rylo-CoA powstaje acetooctan, który w następstwie skrócona. Leczenie insuliną prowadzi – zależnie
redukcji zostaje przekształcony w β-hydroksymaślan, od tego, jak dobrze zostanie wyregulowany meta-
a wskutek dekarboksylacji w aceton. (Acetooctan, bolizm glukozy – do wyraźnego przedłużenia życia,
β-hydroksymaślan i aceton są określane – niewłaściwie aczkolwiek w następstwie tzw. późnych powikłań
pod względem chemicznym – jako tzw. ciała ketono- cukrzycy dochodzi do zgonu wcześniej niż u ludzi
we). W następstwie zwiększonej dostępności kwasów niechorujących na cukrzycę. Ponadto choroby na-
tłuszczowych w wątrobie wzrasta również tworzenie czyniowe i zaburzenia ze strony układu nerwowego
lipoprotein bogatych w triglicerydy, a to prowadzi wpływają niekorzystnie na jakość życia. Dotyczy
do zwiększenia zawartości lipoprotein we krwi. to także do chorych na cukrzycę typu II.
Poza tym obserwuje się nasilenie rozkładu białek, Ważną przyczyną tych powikłań jest hiperglikemia.
przede wszystkim w tkance mięśniowej. Rozkład tzw. Glukoza, szczególnie w wyższym stężeniu, może
ketogennych aminokwasów, tj. aminokwasów mogą- wejść w nieenzymatyczną reakcję z szeregiem endo-
cych ulec przekształceniu w ciała ketonowe, wzmaga gennych białek (tzw. glikozylację nieenzymatyczną).
dodatkowo tworzenie ciał ketonowych. Prowadzi to do zmiany struktury i funkcji tych bia-
MUTSCHLER-2009.indd 432 2010-01-07 22:13:00
Układ hormonalny 433
przedłużająca się
hiperglikemia
nieenzymatyczna
dyslipoproteinemia glikolizacja gromadzenie sorbitolu
białek
Układ hormonalny
pogrubienie błony osmotyczne uszkodzenie komórek,
miażdżyca tętnic podstawnej i błony wewnętrznej pęcznienie komórek
w naczyniach końcowych
(niespecyficzna) (specyficzna)
makroangiopatia mikroangiopatia
B2
zaburzenia ukrwienia stwardnienie kłębuszków retinopatia, polineuropatia
mózgu, serca nadciśnienie nerkowych, ślepota
i tkanek obwodowych niewydolność nerek zaćma
Ryc. B 2.6-8. Schemat patogenezy późnych powikłań cukrzycy.
łek. Oprócz białek osocza krwi i tkanek zmiany doty- często dochodzi do zawałów mięśnia sercowego, rozwoju
czą również hemoglobiny. Wytworzona zostaje m.in. miażdżycy zarostowej tętnic oraz udarów mózgu. Z powo-
HbA1c, której stężenie stanowi ważny parametr wy- du współistniejącej neuropatii w wielu wypadkach chorzy
nie odczuwają bólu typowego dla zaburzeń ukrwienia,
korzystywany do kontroli terapii: im niższe jest stę- np. w przebiegu choroby niedokrwiennej serca (nieme nie-
żenie HbA1c, tym lepsza kontrola cukrzycy u pacjenta dokrwienie!).
(pożądana wartość < 6,5%).
Neuropatię, która, podobnie jak osmotyczne uszkodzenie
Na ryc. B 2.6-8 przedstawiono schemat patogenezy póź- komórek, jest następstwem mikroangiopatii, stwierdza się
nych powikłań cukrzycy. u 60–90% chorych na cukrzycę. Najczęściej rozwija się
Cukrzycowe powikłania naczyniowe można podzielić polineuropatia obwodowa z dominującymi zaburzeniami
na swoistą dla cukrzycy mikroangiopatię oraz nieswoistą czucia (m.in. drętwienie, parestezje, nocne kurcze mięśni
makroangiopatię. łydek, zanik odruchów). Rzadziej występują zaburzenia
motoryczne (np. osłabienie mięśni, niedowłady) lub neuro-
Mikroangiopatia cukrzycowa obejmuje przede wszyst- patia autonomicznego układu nerwowego z zaburzeniami
kim naczynia w nerkach i siatkówce oka. Nefropatia cuk- perystaltyki przewodu pokarmowego (opóźnione opróżnia-
rzycowa charakteryzuje się proteinurią, nadciśnieniem i po- nie żołądka, zaparcia), ortostatycznymi spadkami ciśnienia,
stępującą niewydolnością nerek w następstwie stwardnie- zaburzeniami rytmu serca, impotencją.
nia kłębuszków nerkowych. Stała się ona jedną z głównych
przyczyn niewydolności nerek wymagającej dializoterapii. Tak zwana stopa cukrzycowa jest innym późnym po-
Objawem uszkodzenia siatkówki (retinopatii) są mikrotęt- wikłaniem cukrzycy, w którego przebiegu stwierdza się
niaki i zamknięcie światła naczyń włosowatych. Uwarun- atroficzne, wrzodziejące i zgorzelinowe zmiany skórne
kowane hipoksją tworzenie nowych naczyń w siatkówce z wtórnymi powikłaniami (np. nadkażenie, zapalenie ko-
wiąże się, z powodu ich kruchości, z niebezpieczeństwem ści i szpiku). U 60% pacjentów przyczyną stopy cukrzyco-
krwawień i następczego powstania ślepoty. wej jest zakażenie, u mniej więcej 25% zaburzenia neuro-
Makroangiopatia cukrzycowa odpowiada miażdżycy wegetatywno-niedokrwienne, a u pozostałych zaburzenia
tętnic u ludzi niechorujących na cukrzycę. Ma ona jednak makroangiopatyczno-niedokrwienne, które stosunkowo
większe nasilenie, pojawia się wcześniej i częściej niż często wymagają amputacji kończyny objętej procesem
zmiany występujące w populacji ogólnej. Jako czynnik patologicznym.
predysponujący, przede wszystkim u chorych na cukrzycę
typu II, należy wymienić opisany wcześniej zespół metabo- Następstwem uszkodzenia naczyń i neuropatii autonomicz-
liczny. Z powodu makroangiopatii u chorych na cukrzycę nego układu nerwowego mogą być także inne powikłania
MUTSCHLER-2009.indd 433 2010-01-07 22:13:00
434 Układ hormonalny
cukrzycy w postaci różnorodnych chorób skóry (np. wypry- niono miejscami aminokwasy w pozycji 28 i 29 (Pro-
ski, zakażenia bakteryjne i grzybicze). Lys na Lys-Pro), w insulinie aspart – także w łańcuchu
B – prolina 28 została zastąpiona kwasem asparagi-
nowym, natomiast insulinę glulizynową otrzymano,
2.6.5.2. Leki przeciwcukrzycowe zamieniając kwas asparaginowy w pozycji 3 lizyną,
a lizynę w pozycji 29 kwasem glutaminowym. W wy-
2.6.5.2.1. Insulina niku wprowadzenia tych zmian znacznie zmniejszo-
no, w porównaniu z niezmienioną insuliną ludzką,
tworzenie się heksamerów i w rezultacie znacznie
Obecnie, w przeciwieństwie do czasów wcześniej-
zwiększyła się szybkość wchłaniania z tkanki pod-
szch, gdy stosowano głównie insuliny zwierzęce,
skórnej. Czas upływający do osiągnięcia maksymal-
w leczeniu cukrzycy stosuje się niemal wyłacznie
nego stężenia w osoczu jest mniej więcej o połowę
insulinę ludzką – szczególnie tą otrzymywaną za po-
krótszy niż w przypadku insuliny ludzkiej. Dzięki
mocą inżynierii genetycznej z wykorzystaniem pałe-
szybkiemu początkowi działania te trzy substan-
czek okrężnicy, jak również insuliny (analogi insuli-
cje można podawać bezpośrednio przed posiłkami
ny) modyfikowane metodami inżynierii genetycznej.
i nie jest już konieczne przestrzeganie odstępu czasu
(Bez otrzymywania insuliny metodami inżynierii ge-
między wstrzyknięciem a jedzeniem, co jest wyma-
netycznej nie można by pokryć stale wzrastającego
gane przy innych preparatach insuliny.
zapotrzebowania na insulinę).
Analogi insuliny (rekombinowane analogi insuli-
Insuliny z opóźnionym działaniem cechują się nato-
ny ludzkiej) zostały opracowane w celu otrzymania
miast tym, że za pomocą metod galenowych lub częś-
preparatów mających konkretne właściwości farma-
ciowo syntetycznej modyfikacji uzyskano zmniejsze-
kokinetyczne (szczególnie szybki początek działania,
nie szybkości ich wchłaniania i wskutek tego dłuższy
oraz krótki lub długi czas działania).
czas trwania działania.
Na podstawie początkowej siły działania, czasu
Galenowe metody otrzymywania insulin o opóź-
upływającego do osiągnięcia maksymalnej siły dzia-
nionym działaniu polegają wiązaniu insuliny z za-
łania oraz czasu działania można podzielić preparaty
sadowymi białkami. Do przygotowania tzw. insulin
insulin na (zob. tab. B 2.6-3):
NPH (NPH – neutral protamine hagedom, insulina
■ insuliny krótko działające (insuliny „stare”, insuli- izofanowa) jako białko zasadowe wykorzystywana
ny normalne, insuliny do podawania w bolusie), jest protamina. Insuliny NPH charakteryzują się tym,
że można je mieszać z insuliną normalną, co daje
■ insuliny o szczególnie szybkim początku działania
możliwość tworzenia indywidualnego składu wstrzy-
(i krótkim działaniu),
kiwanego roztworu.
■ insuliny o opóźnionym działaniu (insuliny podsta-
wowe, preparaty insuliny typu dépôt). Zastąpienie asparaginy w pozycji 21 glicyną i wy-
dłużenie łańcucha B na C-końcu o dwie cząsteczki
Insuliny o opóźnionym działaniu można dodatkowo asparaginy doprowadziło do uzyskania długo działa-
podzielić na insuliny o pośrednim czasie działania jącego analogu insuliny ludzkiej – insuliny glarginy,
(< 24 godziny) i insuliny długo działające (24–36 która podawana w postaci przezroczystego roztwo-
godzin). ru bez żadnych dodatków opóźniających uwalnianie
działa równomiernie przez 24 godz.
Terminem insulina normalna (stara) określa się pre- W przypadku insuliny detemir wydłużenie czasu
paraty insuliny zawierające rozpuszczoną insulinę działania uzyskano dzięki następującym modyfika-
bez dodatków opóźniających wchłanianie. Charak- cjom łańcucha B insuliny ludzkiej: połączeniu lizy-
teryzują się one szybkim początkiem działania (po ny-29 z kwasem mirystynowym oraz usunięciu treo-
10–30 minutach), wystąpieniem maksymalnej siły niny z pozycji 30.
działania po upływie 1–3 godzin od wstrzyknięcia W tab. B 2.6-3 przedstawiono preparaty insulin.
i działaniem trwającym 5–8 godzin.
Insulina do inhalacji. Podczas gdy wszystkie do-
Do insulin o szczególnie szybkim początku dzia- tychczas stosowane prepararty insuliny, jak opisano
łania należą otrzymywane na drodze inżynierii ge- powyżej, muszą być podawane w iniekcjach, prepa-
netycznej analogi insuliny ludzkiej: insulina lispro, rat Exubera był próbą stworzenia insuliny ludzkiej
insulina aspart i insulina glulizyna. W przypadku podawanej wziewnie. Z uwagi na niską względną
insuliny lispro w łańcuchu B insuliny ludzkiej zamie- biodostępność (ok. 10% w porównaniu z podaniem
MUTSCHLER-2009.indd 434 2010-01-07 22:13:00
Układ hormonalny 435
Tabela B 2.6-3. Preparaty insulin transport jonów potasu z przestrzeni zewnątrzkomórkowej
do wnętrza komórek). Współistniejącą kwasicę (wartość
pH < 7,1) należy wyrównać, podając dożylnie roztwór wo-
Preparaty dorowęglanu sodu.
handlowe
Działania niepożądane. W przypadku każdej terapii
I. Insuliny krótko działające
insuliną istnieje ryzyko hipoglikemii wywołanej prze-
roztwór insuliny ludzkiej Actrapid, dawkowaniem. Doświadczony chory na cukrzycę,
Berlinsulin H Normal,
Huminsulin Normal, który wcześnie rozpozna pierwsze objawy nadmier-
Insuman Rapid nego obniżenia się stężenia glukozy we krwi, może
Układ hormonalny
II. Insuliny o bardzo szybkim i krótkim czasie działania wyrównać przedawkowanie insuliny przez szybkie
NovoRapid
dostarczenie pożywienia bogatego w węglowodany.
insulina aspart,
insulina glulizyna, Apidra W ciężkiej hipoglikemii konieczne jest pozajelitowe
insulina lispro HUMALOG, podanie glukozy.
Liprolog
Innymi działaniami niepożądanymi insulin są reak-
III. Insuliny o pośrednim czasie działania cje alergiczne, które jednak od czasu wprowadzenia
insulina ludzka + Actraphane, insulin ludzkich i analogów insulin ludzkich pojawia-
siarczan protaminy Berlinsulin H Basal
Huminsulin Basal,
ją się znacznie rzadziej. Reakcje alergiczne należy B2
Insuman Basal odróżniać od insulinooporności, która także jest wy-
wołana tworzeniem przeciwciał przeciw insulinie.
dwufazowa insulina aspart, NovoMix W takich przypadkach zmiana stosowanego prepara-
dwufazowa insulina lispro Humalog Mix,
Liprolog Mix tu insuliny może okazać się korzystna, a u niektórych
pacjentów konieczne jest ponadto podawanie bardzo
IV. Insuliny długo działające
dużych dawek insuliny w celu przezwyciężenia insu-
insulina detemir, Levemir linooporności.
insulina glargina Lantus
Do miejscowych działań niepożądanych należy li-
podystrofia w miejscu podania leku.
Interakcje. Chloropromazyna, glukokortykosteroidy,
podskórnym insuliny ludzkiej), znaczne wahania
pochodne kwasu nikotynowego, leki natriuretyczne,
glikemii przy współistniejących schorzeniach dróg
hormony gruczołu tarczowego i sympatykomimetyki
oddechowych, np. przy ostrym zapaleniu oskrze-
zmniejszają spadek glikemii wywołany przez insuli-
li, oraz konieczność modyfikowania dawkowania
nę, natomiast nieselektywne β1-blokery i cytostaty-
u chorych z upośledzoną funkcją oddechową prepa-
ki typu cyklofosfamidu nasilają go. Nieselektywne
rat został wycofany z rynku.
β-blokery dodatkowo maskują objawy hipoglikemii.
Dawkowanie. Dawkowanie insulin, jak wspomniano,
jest indywidualne i dostosowane do czasu spożywa-
nia posiłków oraz ilości przyjmowanego pożywienia. 2.6.5.2.2. Doustne leki przeciwcukrzycowe
W przypadku (pełnoobjawowej) cukrzycy typu I prze-
ciętne dawkowanie (s.c.) u chorych w okresie wzrostu Wstrzyknięcia insuliny, zwłaszcza przed wprowadze-
wynosi 0,8–1 IU/kg m.c., natomiast u dorosłych 30–70 niem stosowanych obecnie powszechnie przyborów
IU dziennie. Średnie dobowe zapotrzebowanie na in- pomagających je wykonać, stanowiły duże obciążenie
sulinę u chorych na cukrzycę typu II wynosi 30–45 IU. dla chorych na cukrzycę, dlatego doustne leki prze-
Insulinę podaje się zazwyczaj podskórnie, przy czym ciwcukrzycowe (pochodne sulfonylomocznika i bigu-
miejsce wstrzyknięcia należy zmieniać między wybra- anidy) uznano za istotny postęp w terapii cukrzycy.
nymi okolicami ciała (brzuch, udo). Biorąc pod uwagę zdobyte doświadczenie, powinno
się je, przede wszystkim pochodne sulfonylomoczni-
W leczeniu śpiączki cukrzycowej (cotma hyperglycaemi- ka (zob. poniżej), stosować jednak bardziej ostrożnie
cum) przez pierwsze 4 godziny podaje się insulinę normal- niż dotychczas. Stosowanie tych leków jest wskazane
ną (krystaliczną) we wlewie dożylnym w dawce 4 (–12) IU/ jedynie wtedy, gdy:
godzinę, kontrolując co godzinę glikemię, oraz równocześ-
nie uzupełnia niedobory płynów, podając 0,9-proc. roztwór ■ wykluczono cukrzycę typu I,
chlorku sodu z prędkością 500 ml na godzinę. Aby uniknąć
hipokaliemii, należy dodatkowo podawać chlorek potasu ■ leczenie dietetyczne i redukcja masy ciała okazały
(od 5 do maksymalnie 20 mEq K+). (Insulina powoduje się niewystarczające,
MUTSCHLER-2009.indd 435 2010-01-07 22:13:00
436 Układ hormonalny
■ nie ma wskazań do podawania insuliny zamiast do- do dawki trzy razy dziennie po 100 mg (maksymalnie
ustnych leków przeciwcukrzycowych, np. w kwasicy 3 × 200 mg).
ketonowej. Działania niepożądane – wzdęcia lub biegunka –
są spowodowane tym, że wskutek hamowania gluko-
Niestety, kryteria te często nie są przestrzegane. zydazy poli- i disacharydy są częściowo rozkładane
W związku z tym jeszcze raz usilnie zwraca się uwagę dopiero w okrężnicy (poddawane fermentacji) przez
na to, że chorych na cukrzycę typu IIb można leczyć bakterie jelitowe z wydzieleniem gazu. Tak jak wspo-
z mniejszym ryzykiem i niejednokrotnie z zadowala- mniano, stopniowe zwiększanie dawkowania może
jącym efektem za pomocą konsekwentnie przestrze- wyraźnie zmniejszyć nasilenie tych działań niepożą-
ganej diety i redukcji masy ciała. danych. Bardzo rzadko obserwuje się wzrost aktyw-
ności enzymów wątrobowych, który jest prawdopo-
dobnie spowodowany przez metabolity akarbozy.
Stosowanie akarbozy jest przeciwwskazane w przy-
2.6.5.2.2.1. Inhibitory α-glukozydazy padku chorób jelit z zaburzeniami trawienia i wchła-
niania, jak również w przypadku schorzeń, których
Inhibitory α-glukozydazy hamują, jak to wynika przebieg może się pogorszyć w wyniku zwiększone-
z ich nazwy, enzymatyczne rozszczepianie oligo- go wytwarzania gazów w jelitach (np. owrzodzenia
i disacharydów przez glukozydazę w rąbku szczo- i zwężenia jelit).
teczkowym jelita cienkiego. W rezultacie opóźnia się Ponieważ akarboza opóźnia wchłanianie węglowo-
wchłanianie węglowodanów, co pozwala uniknąć hi- danów, przy hipoglikemii wywołanej terapią kombino-
perglikemii poposiłkowej charakterystycznej przede waną akarbozą i insuliną lub akarbozą i pochodnymi
wszystkim dla chorych na cukrzycę typu II. W nie- sulfonylomocznika należy podawać glukozę, której
wielkim stopniu ulega także obniżeniu podstawowy wchłanianie – w przeciwieństwie do sacharozy –
poziom glikemii i w sumie udaje się uzyskać niższe nie zostaje upośledzone przez inhibicję α-glukozydazy.
przeciętne wartości stężenia glukozy we krwi. Stęże-
nie hemoglobiny glikozylowanej obniża się o 0,7–1% Miglitol. Innym inhibitorem glukozydazy o podob-
(działanie słabsze niż metforminy czy insulinotropo- nych do akarbozy właściwościach farmakodynamicz-
wych leków przeciwcukrzycowych). Równolegle nych jest miglitol będący wielohydroksylowaną po-
z tym obniża się hipertriglicerydemia. chodną piperydyny. W przeciwieństwie do akarbozy
miglitol wchłania się w 60–90% i zostaje wydalony
Akarboza. Akarboza, będąca pseudotetrasachary- w postaci niezmienionej przez nerki. Okres półtrwa-
dem, jest najdłużej stosowanym w terapii inhibitorem nia wynosi 2–3 godz.
glukozydazy i jej powinowactwo do tego enzymu
jest około 15 000 razy większe niż naturalnego sub-
stratu, czyli sacharozy.
Po podaniu doustnym akarboza wchłania się tylko CH2OH OH CH3 OH
w nieznacznym stopniu (< 2%). W dalszych odcinkach
O OH NH OH
jelita jest ona hydrolizowana przez bakterie jelitowe O O
i enzymy trawienne. Większość produktów jej rozkła- O
HO OH O OH OH
du zostaje wydalona z kałem, natomiast ok. 20% ulega
OH CH2OH OH CH2OH
wchłonięciu. Powstały w wyniku odszczepienia czą-
steczki glukozy pseudotrisacharyd wykazuje jeszcze
akarboza
ok. jednej trzeciej aktywności substancji macierzystej.
Czas trwania działania wynosi 4–6 godz.
Akarboza jest stosowana w monoterapii lub terapii
OH
kombinowanej z innymi doustnymi lekami przeciw-
cukrzycowymi u pacjentów z cukrzycą typu IIa i IIb. HOH2C N
W połączeniu z insuliną można ją podawać także
u chorych na cukrzycę typu I. HO OH
Aby zmniejszyć działania niepożądane (zob. ni- OH
żej), dawkowanie należy zwiększać powoli, podając
początkowo jeden lub maksymalnie dwa razy dzien- miglitol
nie po 50 mg bezpośrednio przed posiłkiem. Następ-
nie można dawkę zwiększać co tydzień o 50 mg, aż
MUTSCHLER-2009.indd 436 2010-01-07 22:13:01
Układ hormonalny 437
Terapię rozpoczyna się od dawki 3 razy 50 mg nia. Poza tym metformina obniża stężenie triglicery-
dziennie. Zależnie od uzyskanego efektu i tolerowa- dów i cholesterolu we frakcji LDL i VLDL w osoczu,
nia leku można w okresie kilku tygodni zwiększyć a także wzrost cholesterolu HDL.
dawkę do 3 razy po 100 mg dziennie. Mechanizm działania metforminy na poziomie
Działania niepożądane, przeciwwskazania i inter- molekularnym nie został jeszcze w pełni wyjaśniony.
akcje są takie jak w przypadku akarbozy. Wydaje się potwierdzone, że powoduje ona silniej-
sze wiązanie się insuliny z jej receptorami, a ponadto
wpływa na procesy będące następstwem wzajemnego
2.6.5.2.2.2. Metformina oddziaływania między insuliną i receptorem (efekty
Układ hormonalny
poreceptorowe). W ten sposób metformina, podobnie
Spośród biguanidów wcześniej wprowadzonych jak insulina, wywołuje przemieszczenie białek trans-
do terapii jako doustne leki przeciwcukrzycowe sto- portujących glukozę i zwiększoną aktywność białek
sowana jest już tylko metformina, której podawanie transportowych w błonie komórkowej. Wydaje się,
wiąże się z mniejszym ryzykiem wystąpienia kwa- że działa ona podobna jak inkretyna.
sicy mleczanowej (zob. poniżej) niż w przypadku Tylko w stężeniach toksycznych spowodowanych
innych biguanidów. Metformina ma duże znaczenie przedawkowaniem lub niedostatecznym wydalaniem
między innymi dlatego, że w odróżnieniu od pochod- nerkowym w przebiegu niewydolności nerek metfor-
nych sulfonylomocznika (zob. poniżej) nie powoduje mina blokuje łańcuch oddechowy w nabłonku jelit B2
uwalniania insuliny. W długookresowych badaniach, i w wątrobie zmniejszając wytwarzanie ATP. Na-
przede wszystkim w Prospektywnym Badaniu dot. stępstwem tego jest nasilenie beztlenowego rozkła-
Cukrzycy w Wielkiej Brytanii (UKPDS), stwierdzo- du glukozy. Ponadto z powodu wzmożonej glikolizy
no dużą skuteczność ze zmniejszeniem się o 36% powstaje większa ilość kwasu pirogronowego, który
śmiertelności w porównaniu z konwencjonalnym le- następnie wskutek zaburzenia jego rozkładu tlenowe-
czeniem dietą. go jest redukowany do kwasu mlekowego. Ten z ko-
lei nie może zostać zużytkowany, tak jak zazwyczaj,
Efekt i mechanizm działania. U chorych na cukrzy- na drodze glukoneogenezy. W konsekwencji pojawia
cę, ale nie u ludzi z prawidłową przemianą materii, się niebezpieczeństwo rozwoju zagrażającej życiu
metformina po podaniu doustnym w sposób zależny kwasicy mleczanowej.
od dawki obniża stężenie glukozy we krwi w wyni-
ku zmniejszonej syntezy glukozy w wątrobie (wsku- Farmakokinetyka. Po podaniu doustnym metfor-
tek hamowania glikogenolizy i glukoneogenezy), mina wchłania się dobrze, jej biodostępność wynosi
a także lepszego zużytkowania glukozy w tkankach 50–60%, a okres półtrwania – ok. 3 godzin. Substan-
obwodowych. Stężenie HbAlc obniża się o 1–2%. Jak cja czynna gromadzi się w przewodzie pokarmowym,
wspomniano, metformina nie powoduje uwalniania wątrobie i nerkach. Nie wiąże się ona z białkami
insuliny z komórek β. Dzięki temu, poza rzadkimi i jest wydalana przez nerki w postaci niezmienionej.
przypadkami (np. jednoczesnego picia dużych ilości
alkoholu), nie trzeba obawiać się hipoglikemii. Jed- Wskazania i dawkowanie. Metformina jest wskaza-
nak metformina, podobnie jak pochodne sulfonylo- na w leczeniu cukrzycy typu II, kiedy dieta, reduk-
mocznika, wykazuje skuteczność jedynie przy zacho- cja masy ciała i aktywność fizyczna nie wystarczają
wanym przynajmniej resztkowym wytwarzaniu insu- do normalizacji stężenia glukozy we krwi. Ponieważ
liny. Metformina nie powoduje przyrostu masy ciała, metformina nie wywołuje hiperinsulinizmu i ułatwia
a przestrzeganie zaleceń dietetycznych jest ułatwione zmniejszenie masy ciała, jest szczególnie polecana
z powodu zmniejszonego w pewnym stopniu łaknie- u chorych na cukrzycę z nadwagą (cukrzycę typu
IIb). Poza monoterapią metformina może być także
stosowana w terapii kombinowanej z innymi doust-
nymi lekami przeciwcukrzycowymi lub z insuliną.
CH3
Początkowa dawka wynosi jeden raz dziennie 500
lub 850 mg rano, następnie dwa razy dziennie (rano
N NH NH2
H3C i wieczorem) po 850 mg przy posiłku. Maksymalna
NH NH dawka dobowa wynosi 2 g.
metformina Działania niepożądane. W zakresie działań niepo-
żądanych należy szczególnie zwrócić uwagę na to,
że chociaż metformina stosunkowo często wywołuje
MUTSCHLER-2009.indd 437 2010-01-07 22:13:01
438 Układ hormonalny
zaburzenia żołądkowo-jelitowe, mogą one być także 2.6.5.2.2.3.1. Pochodne sulfonylomocznika
objawem rozpoczynającej się kwasicy mleczanowej.
Do innych działań niepożądanych należą meta- W 1942 r. zaobserwowano, że niektóre pochodne sulfona-
liczny smak w ustach i (rzadko) alergie skórne, jak midów obniżają stężenie glukozy we krwi u pacjentów cho-
rych na dur brzuszny. W 1955 r. wprowadzono do terapii
również w pojedynczych wypadkach zaburzenia he- pierwszy doustny lek przeciwcukrzycowy należący do gru-
matopoezy (niedokrwistość megaloblastyczna spo- py sulfonamidów – karbutamid, który wykazywał jeszcze
wodowana hamowaniem wchłaniania witaminy B12 wszystkie właściwości bakteriostatyczne sulfonamidów.
i kwasu foliowego).
Przez podstawienie grupy aminowej grupą metylową
w pierścieniu aromatycznym usunięto niepotrzebne dzia-
Przeciwwskazania. Stosowanie metforminy jest łanie bakteriostatyczne karbutamidu bez utraty działania
przeciwwskazane w niewydolności nerek, śpiączce hipoglikemizującego. Wkrótce po tolbutamidzie poja-
cukrzycowej lub cukrzycowym stanie przedśpiącz- wiło się wiele preparatów o analogicznej strukturze (zob.
kowym, kwasicy ketonowej, niewydolności odde- tab. B 2.6-4), wśród których związki wprowadzone póź-
chowej, ciężkim uszkodzeniu układu sercowo-naczy- niej do terapii okazały się skuteczne już w ilościach rzędu
miligramów. Po dołączeniu do grupy sufonylomoczniko-
niowego lub wątroby, alkoholizmie, w stanach szcze- wej podstawnika lipofilnego uzykano tzw. pochodne sul-
gólnego obciążenia organizmu (zakażenia, zabiegi fonylomocznika 2 generacji charakteryzujące się większą
operacyjne), gorszym stanie ogólnym oraz w czasie aktywnością.
ciąży i karmienia piersią.
Farmakokinetyka. Po doustnym podaniu pochodne
Interakcje. Tak jak w przypadku stosowania innych
sulfonylomocznika wchłaniają się zazwyczaj szybko
leków przeciwcukrzycowych, działanie obniżające
i dobrze, z wyjątkiem glibenklamidu, a ich biodo-
stężenie glukozy we krwi osłabiają glukokortykoste-
stępność zależnie od preparatu galenowego waha się
roidy, natriuretyki, hormony tarczycy i sympatyko-
od 50 do 90%. W osoczu pochodne sulfonylomoczni-
mimetyki.
ka wiążą się w znacznym stopniu z białkami (np. gli-
benklamid i glimepiryd w 99%). Pod względem meta-
bolizmu poszczególne substancje różnią się w sposób
2.6.5.2.2.3. Insulinotropowe doustne
bardziej istotny. Na przykład grupa metylowa tolbu-
leki przeciwcukrzycowe
tamidu szybko ulega utlenieniu do grupy karboksy-
lowej. Powstały w ten sposób kwas karboksylowy
Insulinotropowe doustne leki przeciwcukrzycowe,
nie działa hipoglikemizująco. W glibenklamidzie gru-
do których należą pochodne sulfonylomocznika i glini-
pa cykloheksylowa ulega hydroksylacji, a glimepiryd
dy, powodują uwalnianie insuliny z komórek β trzust-
ulega przekształceniu do pochodnej hydroksylowej
ki, zwiększając wrażliwość tych komórek na bodźce
i karboksylowej.
fizjologiczne. To oznacza, że są one skuteczne tylko
Okres półtrwania wynosi 3–4 godzny w przy-
wtedy, gdy zachowane jest przynajmniej częściowo
padku glikwidonu, około 5 godzin dla tolbutamidu,
wytwarzanie insuliny. Poza tym, jeśli trzustka wydzie-
5–8 godzin dla glimepirydu, 8 godzin dla glibor-
liła nagromadzoną insulinę, to do ponownego jej uwal-
nurydu i 5–9 godzin dla glibenklamidu. Ponieważ
niania pod wpływem przeciwcukrzycowego leku insu-
po maksymalnej stymulacji komórki β trzustki, jak
linotropowego może dojść dopiero po upływie pewne-
wspomniano, stają się wrażliwe na bodźce dopiero
go okresu latencji. Klinicznie istotne dodatkowe efekty
po upływie stosunkowo długiego czasu, okres pół-
pozatrzustkowe wywoływane przez tę grupę leków
trwania pochodnych sulfonylomocznika nie ma takie-
nie są dostatecznie udokumentowane.
go samego znaczenia jak w przypadku innych leków.
Mechanizm działania. Insulinotropowe leki prze-
ciwcukrzycowe blokują kanały potasowe w ko- Dawkowanie. Typowe dawki dobowe poszczegól-
mórkach β (kanały K+ zależne od ATP). Prowadzi nych leków podano w tabeli B 2.6-4. Podczas gdy
to do zmniejszenia przepuszczalności błony komór- glibenklamid i tolbutamid podawane są tylko raz
kowej dla jonów potasu i w konsekwencji do zmniej- na dobę, glibornuryd podaje się 1–2 razy dziennie,
szenia potencjału spoczynkowego błony komórko- a glikwidon 2–3 razy dziennie (zgodnie z rejstracją
wej. To powoduje otwarcie zależnych od napięcia w Polsce: tolbutamid i glibenklamid przyjmuje się
kanałów wapniowych, prowadząc do zwiększenia w 2–3 dawkach, glikwidon w 1–3 dawkach, glime-
wewnątrzkomórkowego stężenia jonów wapnia rid w 1 dawce, a glikliazyd w 2 dawkach (wyjątkiem
i wskutek tego do wzmożonej egzocytozy insuliny jest postać MR podawana 1 raz dziennie) – przyp.
(zob. ryc. B 2.6-2). tłum.).
MUTSCHLER-2009.indd 438 2010-01-07 22:13:01
Układ hormonalny 439
Tabela B 2.6-4. Doustne leki przeciwcukrzycowe z grupy pochodnych sulfonylomocznika
Wzór strukturalny Nazwa Preparaty handlow Czas pół- Dawka
międzynarodowa (zarejestrowne trwania dobowa
nazwy handlowe) (h) (mg)
NH2 karbutamid 500-1500
NH NH
H3C(CH2) 3 S
O O O
Układ hormonalny
CH3 tolbutamid Orabet [w Polsce: 5 500-1500
Diabetol –przyp.tłum.]
NH NH
H3C(CH2) 3 S
O O O
Cl glibenklamid Euglucon N, Glibenclami- 5-9 3,5 - 10,5
-ratiopharm, Gliben
HEXAL, Maninil i in.
NH [w Polsce: Euclamin –
NH NH O OCH3
przyp.tłum.] B2
S
O O O
glimepiryd Amaryl, Glimepirid AL., 5-8 1-6
CH3
Glimepirid-CT, Glimepirid-
N
-ratipharm i in. [w Polsce:
NH C2H5 Amaryl, Amyx,Apo-Glim,
NH NH O O Avaron, Betaglid, Diaril,
S Glemid, Glibetic, Glibezid,
H3C O O O Glidiamid i in. –
przyp.tłum.]
H3C CH3 glikwidon Glurenorm 3-4 15-120
O
N
OCH3
NH NH O
S
O O O
Działania niepożądane. Możliwe działania niepo- kich zaburzeniach czynności nerek oraz we wszyst-
żądane to dolegliwości żołądkowo-jelitowe, a także kich stanach dekompensacji metabolicznej, m.in.
reakcje alergiczne, rzadko także leukopenia i trombo- w przebiegu chorób infekcyjnych, wywołanych ope-
cytopenia. Tak jak po wstrzyknięciach insuliny, może racją lub innymi sytuacjami wymagającymi szczegól-
rozwinąć się ciężka hipoglikemia. Takie niebezpie- nie dobrej kontroli cukrzycy. Przejście na podawanie
czeństwo wiąże się przede wszystkim ze stosowaniem insuliny jest także konieczne w czasie ciąży i karmie-
preparatów działających silnie i długotrwale (stosun- nia piersią, ponieważ wtedy bardzo ważna jest dobra
kowo częste są hipoglikemie w początkowym okre- kontrola glikemii, a pobudzanie uwalniania insuliny
sie terapii glibenklamidem) oraz dotyczy pacjentów u płodu mogłoby wywołać ujemne skutki.
z niewydolnością nerek i nadużywających alkoholu.
Wadą tych leków jest wywołany przez wydzieloną in- Interakcje. Pochodne kumaryny, leki blokujące re-
sulinę wzmożony apetyt, który utrudnia zmniejszenie ceptory β-adrenergiczne, chloramfenikol, cytostatyki
masy ciała. typu cyklofosfamidu, fenylobutazon, salicylany, sul-
fonamidy i tetracykliny nasilają działanie hipoglike-
Przeciwwskazania. Stosowanie pochodnych sulfo- mizujące pochodnych sulfonylomocznika, natomiast
nylomocznika jest przeciwwskazane przy cukrzycy glukokortykosteroidy, leki natriuretyczne, hormony
typu I, znacznej acetonurii, w cukrzycowym stanie gruczołu tarczowego i sympatykomimetyki zmniej-
przedśpiączkowym i śpiączce cukrzycowej, w cięż- szają to działanie.
MUTSCHLER-2009.indd 439 2010-01-07 22:13:01
440 Układ hormonalny
2.6.5.2.2.3.2. Glinidy Wskazania, działania niepożądane i przeciwwska-
zania są podobne jak w przypadku pochodnych sul-
fonylomocznika.
Oba glinidy, nateglinid i repaglinid różnią się wpraw-
Repaglinid wchodzi w interakcje farmakokinetycz-
dzie budową chemiczną od pochodnych sulfony-
ne szczególnie z substancjami czynnymi, które także
lomocznika, lecz mechanizm działania obu grup
są biotransformowane przez CYP3A4 (np. erytro-
jest w dużym stopniu podobny.
mycyna, ketokonazol). Inne interakcje są takie same
Farmakokinetyczne właściwości determinują
jak w przypadku stosowania pochodnych sulfonylo-
wyjątkowość i związane z tym również zalety gli-
mocznika.
nidów. Wchłaniają się one bardzo szybko z prze-
wodu pokarmowego (tmaks wynosi ok. 30 minut,
biodostępność nateglinidu 75%, a repaglinidu 60%)
2.6.5.2.2.4. Leki uwrażliwiające
i podobnie szybko ulegają eliminacji (t1/2 wynosi
na działanie insuliny (glitazony)
dla nateglinidu 1,5 godziny, a dla repaglinidu ok.
l godziny; biotransformacja zachodzi głównie przy
Należące do tiazolidydendionów glitazony (pioglita-
udziale CYP3A4 i sprzęgania z kwasem glukurono-
zon i rosiglitazon) są grupą doustnych leków hipogli-
wym; nateglinid wydalany jest przez nerki i z kałem,
kemizujących o mechanizmie działania różniącym się
a repaglinid w ponad 90% z żółcią). Dzięki temu gli-
od mechanizmów dotychczas wymienionych leków
nidy można przyjmować tuż przed posiłkami i w ten
przeciwcukrzycowych (zob. ryc. B 2.6-9). Najważ-
sposób w dużej mierze dostosować uwalnianie in-
niejszym z tych mechanizmów jest stymulacja pod-
suliny do fizjologicznego zapotrzebowania. Znacz-
typu γ peroksysomalnych receptorów aktywowanych
nie obniżone zostają poposiłkowe stężenia glukozy
przez proliferator (PPARγ – peroxisomal proliferator
we krwi, natomiast jej stężenie na czczo prawie się
activated receptor γ). Należące razem z receptorami
nie zmniejsza. W porównaniu z pochodnymi sul-
dla retinoidów do receptorów wewnątrzkomórko-
fonylomocznika niebezpieczeństwo hipoglikemii
wych receptory PPARγ poprzez interakcje z DNA
– także w nocy – jest mniejsze.
zwiększają wytwarzanie białek, które przyspieszają
Dawkowanie powinno być dostosowane do warto-
różnicowanie komórek tłuszczowych i przyczyniają
ści HbA1c i dostarczonej ilości węglowodanów, a wy-
nosi przy każdym głównym posiłku 60–120 (–180)
mg w przypadku nateglinidu i 0,5–4 mg dla repaglini- glitazon
du (maksymalna dzienna dawka wynosi 16 mg).
retinoidy
PPARγ + PXR
CH(CH3) 2
jądro
NH komórkowe PPARγ PXR
O
HO O
transkrypcja
nateglinid
translacja
białko
(H3C) 2HC COOH
O
NH OC2H5
różnicowanie lipo- wchłanianie glukoneo-
N komórek geneza↑ ↑ glukozy i kwasów geneza ↓
tłuszczowych tłuszczowych
do komórki↑
repaglinid
Ryc. B 2.6-9. Schemat działania glitazonów. PPARγ – peroxiso-
mal; proliferator activated receptor γ; RXR – receptor X dla reti-
noidów.
MUTSCHLER-2009.indd 440 2010-01-07 22:13:02
Układ hormonalny 441
O O
H5C2 CH3
N
NH NH
S N N S
O O
O O
pioglitazon rosiglitazon
Układ hormonalny
się do utrzymania homeostazy glukozowej i lipido- 2.6.5.3. Leki przeciwcukrzycowe
wej: w następstwie wzmożonej aktywności i zwięk- o zwiększonej sile imitujące
szonego przemieszczania się białek transportujących B2
działanie inkretyny
glukozę zwiększa się wychwyt glukozy przez komór-
ki, zmniejsza glukoneogeneza w wątrobie, natomiast
Do inkretyn należy poza hamującym peptydem żo-
zwiększa glikoliza.
łądkowym (GIP – gastric inhibitory peptide) prze-
Następstwem tych działań jest obniżenie glikemii
de wszystkim glukagonopodobny peptyd-1 (GLP-1
na czczo we krwi oraz stężenia HbA1c. Zmniejsza
– glukagon-like peptide 1). Ten wydzielany w niedo-
się również stężenie triglicerydów, wolnych kwasów
statecznej ilości w przebiegu cukrzycy hormon pobu-
tłuszczowych i peptydu C w surowicy, natomiast
dza zależne od glukozy wydzielanie insuliny, obniża
wzrasta stężenie frakcji HDL cholesterolu.
stężenie glukozy we krwi, zmniejsza apetyt oraz po-
Glitazony w związku z opisanymi powyżej właś-
budza przemianę materii. Pomimo tych obiecujących
ciwościami zmniejszają insulinooporność i dlatego
właściwości niestety z powodu krótkiego czasu dzia-
są wskazane w leczeniu cukrzycy typu II w monote-
łania nie może być on stosowany jako lek. Jednak,
rapii lub w połączeniu z innym lekiem przeciwcuk-
jak wykazały badania prowadzone w ostatnich latach,
rzycowym.
zasadę jego działania można wykorzystać w terapii
cukrzycy typu II za pomocą:
Oba związki dobrze wchłaniają się z przewodu po-
karmowego, prawie w całości wiążą się z białkami ■ substancji o działaniu zbliżonym do inkretyn,
osocza (99%) i niemal w całości ulegają metaboli- ale o dłuższym niż GLP-1 czasie działania,
zmowi w wątrobie. Metabolity są wydalane zarówno
przez nerki, jak i z żółcią. Osoczowy okres półtrwania ■ substacji hamujących enzymy, które będą blokować
pioglitazonu wynosi 3–7 godzin, a rosiglitazonu 3–4 rozkład GLP-1.
godziny.
Dawkowanie pioglitazonu wynosi 15–30 (–45) mg Eksenatyd. W gruczołach ślinowych północnoame-
dziennie w dawce jednorazowej, a rosiglitazonu 4–8 rykańskiej jaszczurki – helodermy arizońskiej (He-
mg dziennie w dawce jednorazowej. loderma suspectum) znaleziono składający się z 39
Obserwowane działania niepożądane to zatrzyma- aminokwasów polipeptyd – eksendynę-4, która cha-
nie płynów w organizmie, przyrost masy ciała, nie- rakteryzuje się właściwościami farmakodynamicz-
dokrwistość, zaburzenia przemiany tłuszczów oraz nymi porównywalnymi z GLP-1 oraz zgodnie z tym
funkcji wątroby, a także bóle głowy, nadmierny ape- wiąże się z tym samym miejscem na receptorze,
tyt oraz wzdęcia. Kwestią sporną jest to, czy rosiglita- ale ma istotnie dłuższy osoczowy okres półtrwania
zon zwiększa ryzyko zawału mięśnia sercowego. (2,4 godziny). Białko to otrzymane syntetycznie, pod
Stosowanie glitazonów jest przeciwwskazane nazwą międzynarodową eksenatyd, zostało wprowa-
u pacjentów z chorobami wątroby i niewydolnością dzone na rynek jako pierwsza substancja działająca
krążenia. podobnie do inkretyn. Dotychczas eksenatyd został
Działanie doustnych leków antykoncepcyjnych dopuszczony do stosowania u chorych z niewystar-
może być osłabione przy przyjmowaniu pioglitazo- czającą odpowiedzią na terapię metforminą i/lub po-
nu; efektu takiego nie wywołuje rosiglitazon. chodnymi sulfonylomocznika.
MUTSCHLER-2009.indd 441 2010-01-07 22:13:02
442 Układ hormonalny
Dawkowanie. Lek podawany jest podskórnie dwa 2.6.6. Zasady leczenia cukrzycy
razy dziennie w dawce 5 μg.
Do działań niepożądanych należą zaburzenia
Leczenie cukrzycy polega na stosowaniu diety i opi-
żołądkowo-jelitowe (nudności, dyspepsja, biegun-
sanych wyżej leków przeciwcukrzycowych. Celem
ka) oraz ze strony ośrodkowego układu nerwowego
leczenia jest:
(m.in. bóle głowy, nerwowość).
Eksenatyd zmniejsza biodostępność lowastatyny ■ spowodowanie ustąpienia objawów z zachowaniem
i paracetamolu. odpowiedniej jakości życia, a także unikniecie
Z powodu braku wystarczających danych ekse- w możliwie dużym zakresie działań niepożądanych
natyd nie powinien być stosowany u kobiet w ciąży (przede wszystkim hiperglikemii),
i w okresie karmienia piersią.
■ profilaktyka lub zmniejszenie późnych powikłań
i dzięki temu
Sitagliptyna. Sitagliptyna jest pierwszym przedsta-
wicielem drugiej grupy wymienionych wyżej nowych ■ wydłużenie życia chorych.
leków przeciwcukrzycowych. Hamując peptydazę di-
peptydylową-4 (DPP-4), zapobiega ona rozkładowi Oznacza to konieczość zmniejszenia glukoneogene-
GLP-1 i tą drogą wydłuża ona jego działanie (inhibi- zy, obniżenia stężenia glukozy we krwi, zmniejszenia
tor peptydazy dipeptydylowej-4), nie powodując przy insulinooporności oraz obniżenia stężenia insuliny
tym przyrostu masy ciała. w surowicy.
Sitagliptyna szybko wchłania się z przewodu po-
karmowego, a jej biodostępność wynosi niemal 90%.
U każdego chorego na cukrzycę jest zasadniczo po-
Prawie 80% leku jest wydalane przez nerki w postaci
trzebne i racjonalnie uzasadnione stosowanie środ-
niezmienionej. Osoczowy okres półtrwania wynosi
ków dietetycznych, które obok edukacji stanowią
około 12 godzin.
podstawę każdej terapii przeciwcukrzycowej.
Wskazania. Sitagliptyna jest stosowana w lecze-
Przy cukrzycy typu I (masa ciała chorych jest za-
niu cukrzycy typu II w połączeniu z metforminą lub
zwyczaj w zakresie od prawidłowej do niedowagi)
glitazonem, kiedy środki niefarmakologiczne (ruch,
należy dostosować dostarczanie pożywienia do poda-
dieta) oraz inne leki przeciwcukrzycowe okazały się
wania insuliny (zob. poniżej) i odwrotnie, by stężenie
nieskuteczne.
glukozy we krwi u pacjenta pozostawało w prawid-
Dawkowanie. Sitagliptynę podaje się doustnie
łowym zakresie. W przeciwieństwie do tego u mają-
w dawce 10 mg dziennie.
cych niejednokrotnie nadwagę chorych na cukrzycę
Działania niepożądane. W trakcie terapii mogą się
typu II – mimo dość częstych niepowodzeń – należy
pojawić, jak przy eksenatydzie, bóle, a rzadziej także
nieustannie podejmować próbę redukcji masy ciała
zawroty głowy, zaparcia i in.
(aż do jej normalizacji), ponieważ po osiągnięciu tego
Badania na zwierzętach wskazują możliwą tok-
celu w wielu wypadkach nie jest już potrzebna tera-
syczność płodową i dlatego sitagliptyna nie powinna
pia farmakologiczna. Zależnie od masy ciała, wieku
być stosowana w ciąży. Stosowanie w okresie kar-
i aktywności fizycznej dzienna podaż kalorii w poży-
miania piersią nie jest zalecane.
wieniu powinna wynosić 1000–1800 kcal (50–60%
w postaci węglowodanów, ok. 30% tłuszczów i 10–
15% białek). Ponadto należy zwracać uwagę na to,
aby pacjenci przyjmowali wystarczającą ilość błonni-
ka pokarmowego. Ważne jest, aby zwracać uwagę pa-
cjentów na to, że regularny wysiłek fizyczny prowadzi
do zmniejszenia insulinooporności. Tzw. słodziki (np.
F F sorbitol) oraz fruktoza nie mają przewagi nad sacha-
NH2 O
N
rozą i dlatego, tak jak inne tzw. produkty dietetyczne
F N dla chorych na cukrzycę, nie powinny być spożywane
N
N w nadmiarze.
CF3
Insulinoterapia jest niezbędna w leczeniu cukrzycy
sitagliptyna
typu I. Może być także konieczna w leczeniu chorych
na cukrzycę typu II, gdy leczenie dietetyczne i/lub
innymi lekami przeciwcukrzycowymi okaże się nie-
MUTSCHLER-2009.indd 442 2010-01-07 22:13:03
Układ hormonalny 443
skuteczne, a zwłaszcza, gdy przestanie być skuteczne ności intensywnej współpracy pacjenta stosuje się
(tzw. nieskuteczność wtórna). ją tylko w wybranych wypadkach, np. przy bardzo
Insuliny szybko działające są wskazane w przypad- wysokich porannych wartościach glikemii lub przy
ku śpiączki cukrzycowej lub stanu przedśpiączkowe- bardzo trudnej kontroli cukrzycy u pacjenta.
go, zagrażającej kwasicy metabolicznej, ciężkich in-
fekcji, hiperglikemii u kobiety w ciąży, jak również Akarboza jest stosowana w monoterapii i w terapii
u chorych z cukrzycą de novo. kombinowanej z innymi lekami przeciwcukrzycowy-
Insuliny o pośrednim czasie działania są obecnie mi u pacjentów z cukrzycą typu IIa i IIb. W połącze-
stosowane zazwyczaj w połączeniu z insulinami krót- niu z insuliną może być ona także stosowana pomoc-
Układ hormonalny
ko działającymi. niczo w cukrzycy typu I.
Na podstawie sposobu podawania insuliny można
wyróżnić trzy formy terapii insulinowej: Metformina jest lekiem pierwszego rzutu w tera-
pii chorych z cukrzycą typu IIb, jeżeli istnieje ko-
■ konwencjonalną terapię insulinową,
nieczność farmakoterapii i brak jest przeciwwskazań
■ intensywną terapię insulinową, do jej stosowania.
■ terapię z wykorzystaniem pomp insulinowych.
B2
Przy konwencjonalnej terapii insulinowej mniej
więcej 30 minut przed śniadaniem i kolacją podaje Terapia podstawowa:
się ustaloną ilość mieszanki insulinowej (stosunek zmiana nawyków żywieniowych, zmniejszenie masy ciała,
edukacja, zwiększenia aktywności fizycznej.
porannej dawki insuliny do wieczornej zazwyczaj Wartość docelowa: HbA1C < 6,5
wynosi 2:1 lub 3:2). Z powodu początkowo wysokie-
go i następnie długo utrzymującego się podstawowe-
go stężenia insuliny pacjent musi ścisle przestrzegać Jeżeli po 3 miesiącach HbA1C > 7%:
wyznaczonego czasu iniekcji insuliny oraz przyjęcia doustne leki przeciwcukrzycowe
posiłku, a także wyliczonej ilości przyjmowanych
węglowodanów. Konieczne jest przyjmowanie dodat-
kowych posiłków pomiędzy posiłkami głównymi. nadwaga prawidłowa masa ciała
Intensywna terapia insulinowa (terapia dawką
podstawową i bolusami) ma na celu zaspokojenie monoterapia metforminą
zapotrzebowania podstawowego przez jedno-, dwu- lub α-GI; monoterapia SM
przy przeciwwskazaniach lub glinidem
lub (rzadziej) trzykrotne w ciągu dnia wstrzyknięcie do metforminy
preparatu insuliny o pośrednim czasie działania (ok.
50% całkowitego zapotrzebowania) oraz lepsze niż
w przypadku konwencjonalnej terapii dopasowanie Jeżeli po 3 miesiącach HbA1C > 7% :
dołączenie drugiego doustnego leku przeciwcukrzycowego
dostarczania insuliny przez dodatkowe stosowanie
insuliny z szybkim początkiem działania przed każ-
dym głównym posiłkiem lub podczas niego. Warun- metformina SM + α-GI lub metformina;
kiem jest dokonywanie przez pacjenta regularnych, + α-GI, glitazon, jeżeli metformina jest
kilkakrotnych w ciągu doby oznaczeń glikemii oraz SM lub glinid przeciwwskazana: glitazon
stosowanie dawek insuliny każdorazowo dostosowa-
nych do wartości glikemii i ilości spożywanego poży-
Jeżeli po 3 miesiącach HbA1C > 7%:
wienia. Specjalne systemy wstrzykiwania, tzw. peny, terapia kombinowana doustnymi lekami przeciwcukrzycowymi i insuliną
ułatwiają wykonywanie częstych wstrzyknięć.
Jeżeli efekt jest niewystarczający:
Terapia z wykorzystaniem przenośnej pompy insu-
linowej, za pomocą której dostarczana jest w sposób (mono)terapia insuliną: połączenie
ciągły dawka podstawowa oraz w sposób przerywa- insuliny krótko i długo działającej
ny dodatkowe dawki insuliny przy każdym posiłku,
stanowi specjalną formę bardziej intensywnej terapii
insulinowej. Z powodu dużej intensywności opieki Ryc. B 2.6-10. Schemat terapii cukrzycy typu II. α-GI – inhibitor
i związanych z tym dużych kosztów oraz koniecz- α-glukozydazy, SM – pochodne sulfonylomocznika.
MUTSCHLER-2009.indd 443 2010-01-07 22:13:03
444 Układ hormonalny
Insulinotropowe leki przeciwcukrzycowe są wska- Tabela B 2.6-5. Kryteria dobrej kontroli przemiany materii
zane w terapii chorych na cukrzycę typu IIa oraz u chorych na cukrzycę (zmodyf. według Griesa i Zieglera)
u niektórych chorych w późnym stadium cukrzycy
typu IIb – w tej drugiej grupie zazwyczaj w połącze- dobra kontrola
niu z innymi lekami hipoglikemizującymi, a w koń-
glikemia (mg/dl)
cowym stadium z insuliną, gdy chorzy z obu grup na czczo 80-110
nie wydzielają dostatecznej ilości insuliny. We wczes- poposiłkowa 80-160
hemoglobina glikozylowana (HbA1c) < 6,5 %
nych stadiach cukrzycy typu IIb insulinotropowe leki
przeciwcukrzycowe nie powinny być stosowane, glukoza w moczu nieobecna
aceton w moczu nieobecny
gdyż, jak opisano wcześniej, nadwaga jest przyczyną
insulinooporności, którą te leki mogą nasilać. hipoglikemie
lekkie rzadko
ciężkie nigdy
Leki uwrażliwiające na insulinę (glitazony) są prze-
znaczone do leczenia w monoterapii lub terapii kom- cholesterol LDL (mg/dl) < 100
triglicerydy (mg/dl) < 150
binowanej chorych z cukrzycą typu IIa i IIb. BMI (kg/m2) < 25
Kryteria właściwej kontroli cukrzycy podano w tab.
B 2.6-5. Kontrola polega przede wszystkim na regu-
larnym oznaczaniu glikemii na czczo, badaniu moczu
na obecność glukozy i ciał ketonowych oraz oznacza-
tętniczego (docelowe wartości ciśnienia wynoszą: dla
niu stężenia hemoglobiny glikozylowanej (HbA1c)
ciśnienia skurczowego – 120 mm Hg, a dla ciśnie-
we krwi. Pomiar stężenia glukozy we krwi pozwala
nia rozkurczowego – 75–80 mm Hg) oraz obniżyć
określić jedynie aktualną kontrolę glikemii, nato-
stężenie lipidów w osoczu (do stężenia LDL < 100
miast stopień glikozylacji hemoglobiny umożliwia
mg/dl).
kontrolę stężenia cukru we krwi w okresie ostatnich
10 tygodni, tzn. w okresie odpowiadającym długości
Aby uniknąć rozwoju stopy cukrzycowej, konieczne
życia erytrocytów.
są regularne badania stóp, ich pielęgnacja, odpo-
Najważniejszą rolę w zapobieganiu późnym po-
wiednie obuwie i ochrona przed urazami.
wikłaniom cukrzycy odgrywają działania profilak-
W terapii polineuropatii cukrzycowej można sto-
tyczne polegające na stałym i w miarę możliwości
sować kwas α-liponowy (kwas tiooktanowy) i ben-
całkowitym wyrównaniu poziomu glukozy we krwi.
fotiaminę. Poza tym w leczeniu bólów neuropatycz-
Działania te są tym ważniejsze, im większa jest prze-
nych podaje się dodatkowo lek przeciwdepresyjny
widywana długość życia pacjenta. Przy tego rodzaju
(np. amitryptylinę), karbamazepinę, gabapentynę lub
terapii, jeśli pacjenci nie są wystarczająco przeszko-
pregabalinę. Leczenie farmakologiczne cukrzycowe-
leni i nie przeprowadzają regularnie samokontroli
go porażenia żołądka polega na podaniu leku proki-
stężenia glukozy we krwi, występuje jednak znacznie
netycznego, np. metoklopramidu.
zwiększone ryzyko wystąpienia hipoglikemii.
Przede wszystkim u chorych na cukrzycę typu II
Na ryc. B 2.6-10 przedstawiono schematycznie kolej-
oraz u pacjentów z zespołem metabolicznym należy
ne stopnie terapii cukrzycy typu II.
poza tym prowadzić skuteczne leczenie nadciśnienia
MUTSCHLER-2009.indd 444 2010-01-07 22:13:03
Układ hormonalny 445
2.7. Hormony nadnerczy
2.7.1. Budowa i fizjologia nadnerczy
torebka
2.7.1.1. Anatomia nadnerczy warstwa kłębkowata
Nadnercza są parzystymi gruczołami, luźno osadzo-
Układ hormonalny
nymi na górnych biegunach nerek o łącznej masie ok.
10 g. Składają się one z dwóch różnych pod wzglę-
dem ontogenetycznym i czynnościowym części: kory warstwa pasmowata
nadnerczy, stanowiącej 80–90% masy całego narzą-
du, oraz rdzenia nadnerczy.
W korze nadnerczy pod względem histologicznym
rozróżnia się trzy warstwy (zob. ryc. B 2.7-1), które
przechodzą w siebie bez wyraźnego odgraniczenia:
B2
■ warstwę kłębkowatą,
■ warstwę pasmowatą, warstwa siatkowata
■ warstwę siatkowatą.
Dla kory nadnerczy charakterystyczna jest duża za-
wartość lipidów, cholesterolu i witaminy C, które rdzeń nadnerczy
biorą udział w syntezie hormonów.
Ryc. B 2.7-1. Obraz histologiczny ludzkiego nadnercza (według
Buchera).
2.7.1.2. Hormony kory nadnerczy
W korze nadnerczy z cholesterolu w wyniku rozkładu
łańcuchów bocznych – poprzez progesteron jako sta-
dium pośrednie – są wytwarzane: podwzgórze
■ w warstwie pasmowatej i siatkowatej glukokorty-
kosteroidy, wpływające zwłaszcza na metabolizm hormon uwalniający
węglowodanów, tłuszczów i białek, kortykotropinę
ujemne sprzężenie zwrotne
■ w warstwie kłębkowatej mineralokortykosteroidy,
działające przede wszystkim na gospodarkę wod-
no-elektrolitową, przysadka
■ w warstwie siatkowatej – w niewielkich ilościach
kortykotropina
– androgeny.
Hormony kory nadnerczy, wpływając na metabolizm kora nadnerczy
węglowodanów, tłuszczów i białek oraz na gospo-
darkę wodno-elektrolitową, umożliwiają zachowanie
równowagi biologicznej – homeostazy. Warunkują kortyzol
one zdolność organizmu do reagowania na obcią-
żenia wewnętrzne i zewnętrzne (stres). Nieleczona
niewydolność kory nadnerczy prowadzi w krótkim mięśnie wątroba
(rozkład białek) (glikoneogeneza)
czasie do zgonu.
Budowa chemiczna i biosynteza. Glukokortyko- Ryc. B 2.7-2. Wpływ kortyzolu na przemianę materii oraz regu-
steroidy i mineralokortykosteroidy są C21-steroida- lacja jego wydzielania.
MUTSCHLER-2009.indd 445 2010-01-07 22:13:04
446 Układ hormonalny
H3C O CH3 HO O
CH(CH3) 2 CH2OH
CH3 CH3 O
CH3 H CH3 H CH3 H
H H H H H H
HO HO O
cholesterol pregnenol aldosteron
O CH3 O CH2OH O CH2OH
CH3 droga syntezy CH3 CH3
HO
mineralokorty-
CH3 H kosteroidów CH3 H CH3 H
H H H H H H
O O O
progesteron kortekson kortykosteron
(deoksykortykosteron)
droga syntezy
glukokortykosteroidów
O CH3 O CH2OH O CH2OH
CH3 CH3 CH3
OH OH HO OH
CH3 H CH3 H CH3 H
H H H H H H
O O O
17α-hydroksyprogesteron 17α-hydroksykortekson kortyzol
(hydrokortyzon)
Ryc. B 2.7-3. Biosynteza glukokortykosteroidów i mineralokortykosteroidów.
mi (pochodnymi pregnanu). W strukturze cząsteczek zować hydroksylacji związków hydroksylowanych przy
obu grup hormonów występuje: C21. W aldosteronie, najważniejszym mineralokortykoste-
roidzie, grupa hydroksylowa przy C11 i grupa aldehydowa
■ łańcuch boczny ketolowy znajdujący się w pozycji przy C18 tworzą półacetal.
β przy C17,
■ α-, β-nienasycony keton przy pierścieniu A. Mechanizm działania. Tak jak inne hormony stery-
dowe (zob. poniżej), gluko- i mineralokortykosteroi-
Glukokortykosteroidy posiadają dodatkowo grupę dy działają poprzez receptory wewnątrzkomórkowe.
17α-hydroksylową. Z receptorem dla glukokortykosteroidów wiąże się
tylko kortyzol i inne glukokortykosteroidy, natomiast
Synteza hormonów kory nadnerczy (zob. ryc. B 2.7-2) receptor dla mineralokortykosteroidów pobudzają
podlega trójstopniowej regulacji na zasadzie sprzężenia w równej mierze gluko- i mineralokortykosteroidy.
zwrotnego. Ich biostynteza (zob. ryc. B 2.7-3) rozpoczyna Droga przekazywania sygnału związana z działa-
się od oksydacji cholesterolu, a z wytworzeniem substancji
pośredniej pregnenolonu jest syntetyzowany progesteron. niem glukokortykosteroidów jest opisana w rozdziale
Z niego powstają glukokortykosteroidy w wyniku pier- B 13.4.2, a związana z mineralokortykosteroidami
wotnej hydroksylacji przy C17 i późniejszej hydroksylacji w rozdziale B 2.7.4.
przy C21 oraz C11. W przypadku mineralokortykosteroidów
pierwsza hydroksylacja następuje, odmiennie, przy C21, Selektywność działań wywoływanych przez receptory glu-
co powoduje, że 17-hydroksylaza nie jest w stanie katali- kokortykosteroidowe, tzn. wyodrębnienie działania prze-
MUTSCHLER-2009.indd 446 2010-01-07 22:13:05
Układ hormonalny 447
ciwzapalnego, antyproliferacyjnego i immunosupresyjne- ■ w wyniku ujemnego sprzężenia zwrotnego zmniejsza
go, może być osiągnięta tylko częściowo (w przypadku wydzielanie ACTH w przednim płacie przysadki.
glukokortykosteroidów miejscowych typu prednikarbatu
i furoinianu mometazonu). Działania przeciwzapalnego
i immunosupresyjnego przy tylko niewielkim wpływie Wyższe stężenia kortyzolu będące następstwem wzmo-
na przemianę materii można spodziwać się po odkryciu żonego uwalniania kortyzolu w sytuacjach stresowych
selektywnych aktywatorów receptora glukokortykostero- (zob. poniżej) lub leczniczego podawania glukokorty-
idowego (SEGRA), które w pierwszej kolejności hamują kosteroidów w większych dawkach oprócz opisanych
ekspresję docelowych genów, tzn. działają silniej transre-
powyżej działań dodatkowo wywołują następujące
presyjnie niż transaktywizująco.
efekty:
Układ hormonalny
Fizjologicznie aldosteron wywiera w dużym stop- ■ hamowanie podziałów fibroblastów (działanie an-
niu selektywny wpływ na gospodarkę elektrolitową, typroliferacyjne) oraz rozkładu i syntezy kolagenu,
co jest spowodowane wytwarzaniem specjalnego izo-
■ hamowanie reakcji zapalnych wskutek hamowania
enzymu dehydrogenazy 11β-hydroksysterydowej (typ
odporności swoistej i nieswoistej (działanie prze-
II) w nerkach, która inaktywuje glukokortykosteroidy,
ciwzapalne),
katalizując ich oksydację do 11-ketosterydów. W prze-
ciwieństwie do glukokortykosteroidów aldosteron ■ hamowanie proliferacji limfocytów T (działanie im-
dzięki posiadanej strukturze półacetalu grypy 11β-hy- munosupresyjne), B2
droksylowej nie stanowi substratu dla tego enzymu.
■ poprawę mikrokrążenia podczas wstrząsu w wyni-
Inny, częściej występujący w organizmie izoenzym dehydro- ku poprawy reaktywności naczyń na katecholami-
genazy 11β-hydroksysteroidowej (typ I) znajduje się głównie ny,
w wątrobie. W przeciwieństwie do działającego utleniająco
typu II typ I enzymu katalizuje reakcję redukcji. Typ I odpo- ■ wzrost liczby trombocytów we krwi,
wiada za odtwarzanie najsilniej działającego fizjologicznego ■ zmniejszenie wydzielania gonadotropin w przed-
glukokortykosteroidu – kortyzolu – z jego utlenionej postaci
(kortyzonu). Wysoka aktywność dehydrogenazy 11β-hy- nim płacie przysadki oraz upośledzenie czynności
droksysterydowej typu I w wątrobie wyjaśnia także, dlacze- gonad,
go kortyzol i kortyzon lub prednizolon i prednizon po po-
daniu doustnym – inaczej niż przy stosowaniu miejscowym ■ wzmożenie pobudliwości mózgu i obniżenie progu
na skórę lub dostawowo, gdzie nie stwierdzono tego enzymu drgawkowego,
– działają tak samo skutecznie.
■ psychotropowe działanie euforyzujące, ale także
działanie depresyjne.
2.7.1.3. Znaczenie fizjologiczne Glukokortykosteroidy są niezbędne do pokonywania
hormonów kory nadnerczy ciężkich sytuacji stresowych (np. zakażeń, urazów,
operacji), ponieważ z jednej strony wzmagają dzia-
łanie katecholamin, a z drugiej zmniejszają uszko-
Najważniejszym z punktu widzenia fizjologii gluko-
dzenia organizmu poprzez hamowanie nadmiernego
kortykosteroidem jest kortyzol (= hydrokortyzon).
wytwarzania cytokin (zob. poniżej).
Z tego powodu fizjologiczne efekty działania glukokor-
tykosteroidów są przede wszystkim efektami działania
Regulacja stężenia glukokortykosteroidów. U zdro-
kortyzolu. Kortyzol wywołuje różnorodne efekty za-
wego dorosłego człowieka pod wpływem kortyko-
leżnie od szybkości wydzielania lub stężenia tkanko-
tropiny kora nadnerczy wydziela mniej więcej 15–
wego, przy czym nie są one od siebie wyraźnie odgra-
60 mg kortyzolu i 1–2 mg kortykosteronu dziennie.
niczone. W fizjologicznych stężeniach kortyzol:
W stresie wydzielanie dobowe kortyzolu może się
■ wzmaga glukoneogenezę w wyniku zwiększonego zwiększyć do 240 mg.
rozkładu białek (działanie kataboliczne) i w ten Uwalnianie glukokortykosteroidów podlega regu-
sposób zwiększa stężenie glukozy we krwi oraz syn- lacji podwzgórzowo-przysadkowej (zob. ryc. B 2.7-2).
tezę glikogenu w wątrobie, Przy spadku stężenia wolnego kortyzolu w podwzgó-
rzu uwalniana jest kortykoliberyna, która powoduje
■ nasila działanie lipolityczne katecholamin,
wydzielanie kortykotropiny w przysadce. Ta z kolei
■ wywołuje zatrzymanie sodu oraz wzmożone wyda- pobudza syntezę i uwalnianie glukokortykosteroidów
lanie potasu (działanie mineralokortykosteroido- w nadnerczach. Wydzielanie glukokortykosteroidów
we) oraz wapnia przez nerki (działanie antagoni- nie jest jednak stałe, lecz podlega rytmowi dobowe-
styczne w stosunku do witaminy D), mu z maksymalnym stężeniem kortyzolu w osoczu
MUTSCHLER-2009.indd 447 2010-01-07 22:13:05
448 Układ hormonalny
powtarzających się sytuacjach nadmiernego obciąże-
nia organizmu nasilenie reakcji stresowej zmniejsza
się – występuje fizjologiczna adaptacja.
200
Farmakokinetyka. We krwi ok. 90% kortyzolu wiąże
stężenie kortyzolu w osoczu
150 się z transkortyną (CBG = globuliną wiążącą korty-
kosteroidy) – α-glikoproteiną o masie cząsteczkowej
100 52 000 Da. Jeżeli stężenie kortyzolu w osoczu prze-
kracza 200 μg/l, z powodu wyczerpania zdolności
50 wiązania transkortyny nasila się niespecyficzne wią-
zanie kortyzolu z albuminami. Wolny kortyzol szyb-
ko ulega inaktywacji, przede wszystkim w wątrobie,
0 4 8 12 16 20 24 godziny
m.in. w wyniku redukcji (α-, β-nienasyconych grup
ketonowych) do dihydrokortyzolu lub tetrahydrokor-
tyzolu (2 stereoizomery, 5α- oraz 5β-tetrahydrokor-
Ryc. B 2.7-4. Zależność między stężeniem kortyzolu w osoczu a
tyzol; zob. ryc. B 2.7-5), które następnie są sprzęga-
porą dnia (dobowy rytm wydzielania kortyzolu).
ne z aktywnym kwasem glukuronowym. Produkty
sprzęgania są wydalane z moczem.
rano między godz. 6 a 9 i minimalnym stężeniem oko- Osoczowy okres półtrwania kortyzolu wynosi u lu-
ło północy (zob. ryc. B 2.7-4). dzi 1,7 godziny.
Sytuacje stresowe (zob. powyżej) pociągają
za sobą znaczne zwiększenie syntezy glukokortyko-
steroidów. Może ono być interpretowane jako zmiana
2.7.1.4. Znaczenie fizjologiczne
wartości żądanej w układzie regulacji wydzielania
glukokortykosteroidów. W stresie dochodzi również
mineralokortykosteroidów
do zwiększenia aktywności układu sympatycznego
i w rezultacie do wzmożonego wydzielania hormo- Oprócz najważniejszego pod względem fizjologicz-
nów rdzenia nadnerczy. Przy długo trwających lub nym mineralokortykosteroidu – aldosteronu – z kory
O O
CH2OH CH2OH
CH3 CH3
HO OH HO OH
CH3 H HSD CH3 H
H H H H
O O
H
kortyzol 5α/5β-dihydrokortyzol
(hydrokortyzon)
HSD
O
CH2OH
CH3
HO OH
CH3 H O
CH2OH
CH3
H H HO OH
HOOC O O UGT CH3 H
H
H H
HO OH
HO
OH H
glukuronian tetrahydrokortyzolu 5α/5β-tetrahydrokortyzol
Ryc. B 2.7-5. Biotransformacja kortyzolu do nieaktywnych metabolitów. HSD – dehydrogenaza hydroksysteroidów, UGT – glukuro-
nylotransferaza.
MUTSCHLER-2009.indd 448 2010-01-07 22:13:05
Układ hormonalny 449
O HO O
CH2OH CH2OH
OHC O
HO
CH3 H CH3 H
H H H H
O O
Układ hormonalny
aldosteron (postać otwarta) aldosteron (postać półacetalowa)
O O
CH2OH CH2OH
CH3 CH3
HO
CH3 H CH3 H
B2
H H H H
O O
kortekson kortykosteron
nadnerczy jest wydzielany także jego prekursor kor- wzmożone wydzielanie kortykotropiny może jednak
tekson (11-deoksykortykosteron (DOC)). Pod wzglę- spowodować wzmożoną syntezę aldosteronu.
dem jakościowym jego działanie jest takie samo jak
aldosteronu, jednak ilościowo jest znacznie słabsze
(ok. 1/30). 2.7.1.5. Znaczenie fizjologiczne
androgenów nadnerczowych
Regulacja stężenia mineralokortykosteroidów. Fi-
zjologicznie wydzielanie mineralokortykosteroidów
Nadnercza wytwarzają w pierwszej kolejności bar-
z kory nadnerczy jest regulowane przede wszystkim
dzo słabo działający dehydroepiandrosteron (DHEA).
przez zmianę objętości krwi krążącej, zmianę ukrwie-
Ten androgen odgrywa istotną rolę u kobiet, u któ-
nia nerek oraz stężenia jonów sodu w kanalikach
rych jako neurosteroid wpływa pozytywnie na ogól-
dystalnych nerek. W odpowiedzi na niedobór Na+
ne samopoczucie. U mężczyzn działanie androgenów
we krwi, spadek ukrwienia nerek i zmniejszone stęże-
nadnerczowych, w przeciwieństwie do tych syntezo-
nie jonów sodu w plamce gęstej komórki przykłębko-
wanych w jądrach, ma znikome znaczenie. (Działanie
we w nerkach wydzielają reninę, która jest proteazą
androgenów opisano w rozdziale B 2.8.2.3.).
powodującą powstanie angiotensyny I z białka pre-
kursorowego angiotensynogenu. Następnie z deka-
peptydu angiotensyny I enzym konwertujący angio-
tensynę (ACE) wytwarza oktapeptyd angiotensynę II. 2.7.2. Zaburzenia czynności
Angiotensyna II oraz powstająca przez odszczepienie kory nadnerczy
kolejnych aminokwasów z angiotensyny II angioten-
syna III pobudzają syntezę aldosteronu (układ renina- Niedoczynność kory nadnerczy. Przy niedoczyn-
angiotensyna-aldosteron). ności kory nadnerczy stwierdza się niedobór hormo-
Przy utracie krwi uwalnianie aldosteronu może nów kory nadnerczy. Postać pierwotna, wywołana
wywołać również pobudzenie receptorów objętościo- uszkodzeniem samych nadnerczy, jest nazywana
wych. W prawidłowych warunkach kortykotropina chorobą Addisona. Do (objawowego) wypadnięcia
w znikomym stopniu wpływa na wytwarzanie i uwal- funkcji dochodzi dopiero wtedy, gdy zniszczeniu
nianie aldosteronu. W warunkach patologicznych ulegnie ok. 90% kory nadnerczy. Najczęstszą przy-
MUTSCHLER-2009.indd 449 2010-01-07 22:13:06
450 Układ hormonalny
Tabela B 2.7-1. Następstwa (pierwotnej) niewydolności kory nadnerczy (zmodyf. według Siegenthalera)
Niedobór w następstwie prowadzi do
mineralokortykosteroidów hiponatremii i hipochloremii zmęczenia, osłabienia, nudności, wymiotów,
hiperkaliemii kurczów mięśni, niedowładów, arytmii,
kwasicy hiperwentylacji, zaburzeń świadomości,
śpiączki
odwodnienia zewnątrzkomórkowego z hipotonią tachykardii, hipotonii, skłonności
do zapaści ortostatycznych,
wewnątrzkomórkowej hydratacji bólów głowy, apatii, splątania
glukokortykosteroidów zaburzeń metabolizmu węglowodanów uczucia głodu, dysfunkcji mózgowej,
(hipoglikemii) lęku, potów, nudności,
tachykardii, zaburzeń świadomości, śpiączki,
zaburzeń metabolizmu białek zmniejszenia masy ciała,
i tłuszczów niedokrwistości normocytowej, leukopenii,
eozynofilii, limfocytozy
zaburzeń hematopoezy zmniejszonego wytwarzania kwasu solnego
zmniejszonej stymulacji błony śluzowej żołądka lub jego braku
wzmożonego uwalniania MSH
zwiększonej pigmentacji skóry
zaburzeń ośrodkowego układu nerwowego i błon śluzowych, zaburzeń psychicznych
androgenów katabolizmu osłabienia, zaniku mięśni, impotencji
czyną (100 przypadków na milion mieszkańców) stanowią duże zagrożenie dla pacjentów, ponieważ
tego rzadkiego schorzenia są reakcje autoimmu- dodatkowa utrata wody i elektrolitów (np. z powodu
nologiczne (tzw. idiopatyczna niewydolność kory biegunki z wymiotami), jeśli nie poda się dożylnego
nadnerczy obejmująca ok. 50% przypadków nie- wlewu elektrolitów, szybko doprowadza do ciężkie-
doczynności kory nadnerczy). Innymi przyczynami go, zagrażającego życiu odwodnienia.
mogą być: gruźlica kory nadnerczy (rzadka w kra- Wtórna postać niewydolności kory nadnerczy
jach z dobrą opieką medyczną), krwawienia do kory jest następstwem zmniejszonego wydzielania ACTH
nadnerczy, przerzuty nowotworowe, AIDS, a także spowodowanego niewydolnością podwzgórza i/lub
działania niepożądane niektórych leków (substan- przedniego płata przysadki, np. w przebiegu nowo-
cji hamujących steroidogenezę, np. ketokonazolu, tworu. Ponieważ wytwarzanie mineralokortykosteroi-
etomidatu, aminoglutetymidu). Wskutek niedoboru dów jest w znacznej mierze niezależne od ACTH, przy
niezbędnych do życia mineralo- i glukokortykoste- wtórnej niewydolności kory nadnerczy nie stwierdza
roidów pojawiają się liczne objawy, które zestawio- się zaburzeń elektrolitowych. W przeciwieństwie
no w tab. B 2.7-1. do postaci pierwotnej nie obserwuje się zwiększo-
nego, a zmniejszone odkładanie melaniny, ponieważ
Zaburzenia gospodarki elektrolitowej objawiają się zostaje wytworzona mniejsza ilość POMC, a w wyni-
hiponatremią, hipochloremią, hiperkaliemią i hipo- ku tego także MSH i ACTH. Obraz chorobowy deter-
wolemią, w których następstwie pacjenci mogą być minuje przede wszystkim niedobór glukokortykoste-
zmęczeni, apatyczni, z zaburzeniami świadomości, roidów (niedobór kortyzolu).
hipotonią i tachykardią. Stale utrzymująca się hipo-
glikemia wywołuje m.in. lęk, zaburzenia czynności W diagnostyce niewydolności kory nadnerczy oznacza się
mózgu oraz nudności. Poza tym pacjenci skarżą się stężenie kortyzolu w surowicy po podaniu ACTH. Różni-
cowanie postaci pierwotnej i wtórnej opiera się na oznacze-
na napady potów, kołatanie serca, zmniejszenie masy niu osoczowego stężenia ACTH.
ciała i osłabienie siły mięśniowej.
Dopiero w zaawansowanym stadium stwierdza się Zespół nadnerczowo-płciowy. W zespole nadnerczowo-
typowe brunatne, szarobrunatne przebarwienia skóry, płciowym obraz chorobowy uwarunkowany jest nadmierną
spowodowane wzmożonym wydzielaniem proopio- syntezą androgenów w korze nadnerczy i może on występo-
wać w dwóch postaciach: wrodzonej i nabytej.
melanokortyny (POMC), a przez to MSH i ACTH, Przyczyną wrodzonego zespołu nadnerczowo-płciowe-
w następstwie niedoboru kortyzolu we krwi. Zakaże- go są defekty enzymów kory nadnerczy, wśród których naj-
nia, a w szczególności infekcje żołądkowo-jelitowe, częstszy jest defekt 21β-hydroksylazy. Poza tym znaczenie
MUTSCHLER-2009.indd 450 2010-01-07 22:13:06
Układ hormonalny 451
Tabela B 2.7-2. Następstwa nadmiernego wytwarzania kortyzolu w przebiegu zespołu Cushinga (zmodif. według Siegenthalera)
Zaburzenie prowadzi do
metabolizmu węglowodanów metabolizmu diabetogennego, hiperglikemii i kwasicy ketonowej, cukrzycy posterydowej,
poliurii, polidypsji
wodno-elekrolitowe nadciśnienia
metabolizmu białek adynamii, zaniku mięśni
Układ hormonalny
metabolizmu tłuszczów twarzy „księżyc w pełni”, otłuszczenia tułowia, hipercholesterolemii, miażdżycy
hematopoezy poliglobulii, leukocytozy, limfo- i eozynopenii, trombocytozy
układu mezenchymalnego rozstępów skórnych (sinoczerwone rozstępy na skórze brzucha), osłabienia reakcji immunologicznych
ośrodkowego układu nerwowego zaburzeń psychicznych z agresją i/lub nastrojem depresyjnym
B2
mają jeszcze defekty 11β-hydroksylazy i 3β-dehydroge- ne objawy zestawiono w tab. B 2.7-2. W przypadkach
nazy. W następstwie tych zaburzeń enzymatycznych kora nieleczonych średni czas przeżycia wynosi 5 lat.
nadnerczy nie jest w stanie w stopniu dostatecznym wy-
twarzać gluko- i mineralokortykosteroidów. Niskie stężenie
kortyzolu na skutek sprzężenia zwrotnego z podwzgórzem Hiperaldosteronizm. Hiperaldosteronizm jest spo-
lub przysadką powoduje wzrost wytwarzania ACTH i – na wodowany wzmożonym wytwarzaniem aldosteronu.
skutek rozrostu kory nadnerczy – androgenów. Tak jak przy hiperkortyzolizmie, wyróżnia się postać
U dziewcząt stwierdza się objawy wirylizacji (m.in. pierwotną (= nadnerczową) i wtórną (= pozanadner-
maskulinizacja zewnętrznych narządów płciowych, mę- czową).
ska budowa ciała i typ owłosienia). U chłopców dochodzi
do przedwczesnego wykształcenia drugorzędowych cech Przyczyną pierwotnego hiperaldosteronizmu (ze-
płciowych (pseudopubertas praecox) bez odpowiedniego społu Conna) jest w 75% przypadków gruczolak
rozwoju gruczołów płciowych, ponieważ podwyższone stę- kory nadnerczy wytwarzający aldosteron, a w po-
żenie androgenów hamuje wydzielanie gonadotropin. Brak zostałych 25% rozrost kory nadnerczy o nieznanej
jest również spermatogenezy. U obu płci po zwracającym etiologii (hiperaldosteronizm idiopatyczny). Do naj-
uwagę szybkim wzroście w pierwszych latach życia nastę-
puje wczesne zatrzymanie wzrostu wskutek przedwczesne- ważniejszych objawów należą (uwarunkowane utratą
go skostnienia chrząstek nasadowych kości długich. potasu) osłabienie siły mięśniowej, adynamia, zmę-
Nabyty zespół nadnerczowo-płciowy jest spowodowany czenie, poliuria, nykturia i parestezje oraz (uwarun-
nowotworami kory nadnerczy (gruczolaki, rak), które wy- kowane hiperwolemią) nadciśnienie, bóle głowy i ob-
twarzają nadmierne ilości androgenów. jawy tężyczkowe.
Hiperaldosteronizm wtórny w ścisłym znacze-
Zespół Cushinga (hiperkortyzolizm). Zespół Cu- niu nie jest chorobą układu wewnątrzwydzielnicze-
shinga jest następstwem nadmiernego wytwarzania go. Wzmożone wytwarzanie aldosteronu jest w tym
glukokortykosteroidów, przede wszystkim kortyzolu. przypadku uwarunkowane aktywacją układu renina-
Przyczyną nadnerczowego pierwotnego zespołu Cus- -angiotensyna-aldosteron w następstwie zmniejszo-
hinga są łagodne lub złośliwe nowotwory kory nad- nej objętości osocza lub obniżonego przepływu oso-
nerczy (15% przypadków zespołu Cushinga). Przy- cza przez nerki (np. w przebiegu zespołu nefrotyczne-
czyną częstszej wtórnej postaci są głównie mikro- go, marskości wątroby, a także podawania wysokich
gruczolaki przysadki wydzielające nadmiar ACTH dawek tiazydów lub diuretyków pętlowych).
(70%) lub nowotwory ektopowo wydzielające ACTH
(15%). Najczęściej stwierdza się jednak jatrogenny Przyczynami zespołu rzekomego nadmiaru mineralokor-
zespół Cushinga będący następstwem długotrwałego tykosteroidów są defekty receptora dla mineralokortyko-
podawania dużych dawek glukokortykosteroidów. steroidów oraz dehydrogenazy 11β-hydroksysteroidowej.
Defekty te mogą być spowodowane przez:
Dla zespołu Cushinga charakterystyczna jest okrąg-
ła, ciemnoczerwona twarz ze zubożoną mimiką („księ- ■ mutację genu kodującego receptor dla mineralokortyko-
życ w pełni”), a także otłuszczenie tułowia. Inne waż- steroidów,
MUTSCHLER-2009.indd 451 2010-01-07 22:13:06
452 Układ hormonalny
■ wrodzony defekt dehydrogenazy 11β-hydroksysteroidowej
(typu II) lub 21 grupa estrowa
O C H2O H
■ hamowanie dehydrogenazy 11β-hydroksysteroidowej
przez kwas glicyretynowy zawarty w wyciągu z lukrecji. CH3 grupa estrowa
HO 17
OH
11
Niezależnie od obecności hormonu mające defekty recep- 1 CH3 16
tory są stale uaktywnione. W przypadku stanów wymie- CH3/OH
2 9 podstawnik
nionych jako dwa ostatnie wskutek brakującej inaktywa- chlorowcowy
cji kortyzolu, którego stężenie w wolnej postaci w oso- 3
6 podstawnik
czu przekracza stukrotnie stężenie aldosteronu, dochodzi O chlorowcowy/CH3
do ciągłej aktywacji receptora dla mineralokortykostero-
idów. Objawy nadmiaru mineralokortykosteroidów poja-
wiają się jednak dopiero po przyjęciu nadmiernych ilości Podstawnik Działanie
wyciągu z lukrecji.
C-6 chlorowcowy ↑↑ wiązania do GR i MR
lub metylowy
C-9 chlorowcowy ↑↑ wiązania do GR i MR
2.7.3. Glukokortykosteroidy
Δ1,2 podwójne ↑ wiązania do GR i ↓ wiązania do MR
jako środki lecznicze wiązanie
Zależnie od dawki lub stężenia glukokortykosteroidy C-16 metylowy brak wiązania z MR
lub hydroksylowy
wywołują różne efekty. Poza terapią substytucyjną
stosowaną w przypadku (stosunkowo rzadkiej) nie- C-17 estrowy ↑ powinowactwo do GR
wydolności kory nadnerczy (zob. poniżej) znaczenie
C-17/ estrowy ↑ resorpcji ze skóry
terapeutyczne glukortykosteroidów jest związane C-21 i płuc
z ich działaniem:
■ przeciwzapalnym, Ryc. B 2.7-6. Powiązania pomiędzy budową chemiczną i działa-
niem pochodnych glukokortykosteroidowych. ↑ – zwiększenie;
■ przeciwalergicznym,
↑ ↑ – znaczne zwiększenie; ↓ – zmniejszenie działania. GR – re-
■ immunosupresyjnym. ceptor glukokortykosteroidowy, MR – recceptor mineralokorty-
kosteroidowy.
Przez częściowo syntetyczną modyfikację natural-
nych hormonów (zob. ryc. B 2.7-6) próbowano uzy-
skać bardziej skuteczne związki. Najważniejszym tywy dalszego względnego zwiększenia selektywno-
krokiem było wprowadzenie metodą dehydrogenacji ści (SEGRA).
mikrobiologicznej podwójnego wiązania między C1 W tab. B 2.7-3 zestawiono preparaty handlowe
i C2. Orzymany tą drogą z kortyzolu prednizolon glukokortykosteroidów do stosowania ogólnego. Hy-
oraz z kortyzonu prednizon wykazują prawie cztery drofilne estry proleków (np. dihydrofosforany) pred-
razy silniejsze działanie przeciwzapalne niż kortyzol nizolonu i kortyzolu mogą być podawane w stanach
i jednocześnie wpływają słabiej na gospodarkę wod- nagłych w iniekcjach dożylnych.
no-elektrolitową. Dalsze zwiększenie siły działania
osiągnięto przez wprowadzenie grup metylowych Farmakokinetyka. Glukokortykosteroidy dobrze
i hydroksylowych lub atomu chlorowca w różnych wchłaniają się z przewodu pokarmowego. Kortyzol
miejscach pierścieni sterydowych. Szczególnie efek- i prednizolon są względnie szybko eliminowane, na-
tywne okazało się wprowadzenie atomu fluoru w po- tomiast chlorowcowane pochodne kortyzolu są wol-
zycji 9α. Podstawienie w pozycji C-16 grupy hydrok- niej metabolizowane.
sylowej lub metylowej eliminuje powinowactwo
do receptora dla mineralokortykosteroidów. Wskazania. Podczas stosowania glukokortykostero-
Wymienione pochodne kortyzolu działają wpraw- idów należy ściśle rozróżnić terapię substytucyjną
dzie silniej i jedynie w niewielkim stopniu wpływają stosowaną przy niewydolności kory nadnerczy i po-
na gospodarkę sodową i potasową, jednak nie spełni- dawanie ich z powodu posiadanych przez nie właś-
ły one oczekiwań dotyczących zmniejszenia innych ciwości przeciwzapalnych, przeciwalergicznych i im-
działań niepożądanych: równolegle z działaniem munosupresyjnych (tzw. terapia farmakodynamicz-
przeciwzapalnym wzrasta także np. wpływ na meta- na). W leczeniu substytucyjnym lekiem z wyboru
bolizm glukozy. Ostatnio pojawiły się nowe perspek- jest kortyzol (zob. rozdz. B 2.7.5).
MUTSCHLER-2009.indd 452 2010-01-07 22:13:07
Układ hormonalny 453
Tabela B 2.7-3. Glukokortykosteroidy do stosowania układowego
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna* (mg)
Niefluorowane glukokortykosteroidy
O kortyzon Cortison CIBA 1,5 10-40
CH2OH
CH3
O OH
Układ hormonalny
CH3 H
H H
O
O kortyzol Hydrocortison GALEN, 1,5 10-40
CH2OH (hydrokortyzon) Hydrocortison Joechst,
CH3
HO OH Hydrocortison JENAPHARM,
CH3 H Hydrocutan
[w Polsce: Hydrocortisonum,
H H Iwostin HC – przyp. tłum.] B2
O
O prednizolon Decortin H, Predni H, 3 5-15
CH2OH Prednisolon JENAPHARM,
CH3
HO OH Prednisolon-ratiopharm
CH3 H [w Polsce: Encortolon,
Fenicort – przyp. tłum.]
H H
O
O prednizon Decortin, Prednison GALEN, 3 5-15
CH2OH Prednison HEXAL, Prednison-
CH3
O OH -ratiopharm
CH3 H
[w Polsce: Encorton – przyp. tłum.]
H H
O
O metyloprednizolon Methyloprednisol JENAPHARM, 2-3 4-12
CH2OH Metypred GALEN, Urbason
CH3
HO OH [w Polsce: Metypred,
CH3 H Medrol – przyp. tłum.]
H H
O
CH3
O kloprednol Syntestan 3 1,25-12,5
CH2OH
CH3
HO OH
CH3 H
H H
O
Cl
* bardzo zróżnicowane w zależności od wskazań
Pomimo tegp, że glukokortykosteroidy działają tylko gicznych. Terapię glukokortykosteroidami stosuje się
objawowo, a nie przyczynowo, często pozwalają one w następujących sytuacjach:
na uzyskanie bardzo dobrego efektu terapeutyczne-
■ w chorobach skóry (m.in. wyprysku, łuszczycy, ru-
go podczas leczenia wielu schorzeń nieendokrynolo-
mieniu guzowatym, rumieniu popromiennym),
MUTSCHLER-2009.indd 453 2010-01-07 22:13:07
454 Układ hormonalny
Tabela B 2.7-3. Glokokortykosteroidy do stosowania układowego (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dzienna* (mg)
Niefluorowane glukokortykosteroidy
O deflazakort Calcort 2 6-18
O
O CH3
CH3 N CH3
HO
CH3 H O
H
H H
O
Fluorowane glikokortykosterydy
O betametazon Celestamine N 7 0,5-2
CH2OH
CH3 [w Polsce: Daivobet,
HO OH Diprophos, Kuterid,
CH3 H CH3 Celestone – przyp. tłum.]
F H
O
O deksametazon Dexamethason GALEN, 36 8-40
CH2OH Dexamethason JENAPHARM,
CH3
HO OH Dexamethason-ratiopharm,
Fortecortin
CH3 H CH3 [w Polsce: Dexaferee,
Dexamethason, Dexapolocort,
F H Pabi-Dexamethason,
O Dexaven – przyp. tłum.]
O fluokortolon Ultralan 1,5 2
CH2OH
CH3
HO H
CH3 H CH3
H H
O
F
O triamcynolon Delphicort, Volon 5 4-48
CH2OH
CH3 [w Polsce: Triacomb,
HO OH Polcortolon – przyp. tłum.]
CH3 H OH
F H
O
Estry proleków do podawania pozajelitowego
O
CH2OPO3H2 dihydrofosforan Celestan 7 8-40
CH3 betametazonu [w Polsce: Celestone, (aktywniejszy
HO OH
Diprophos – przyp. tłum.] metabolit)
CH3 H CH3
F H
O
O dihydrofosforan Hefasolon 40-200 i.v.
CH2OPO3H2 3
CH3 prednizolonu (aktywniejszy
HO OH
metabolit)
CH3 H
H H
O
MUTSCHLER-2009.indd 454 2010-01-07 22:13:07
Układ hormonalny 455
■ w schorzeniach reumatycznych (zwłaszcza w fazie pacjenta i okresowo modyfikować ją zgodnie z ak-
ostrej gorączki reumatycznej z zapaleniem serca, tywnością choroby,
w reumatoidalnym zapaleniu stawów, w polimial-
■ nawet bardzo duże pojedyncze dawki glukokorty-
gii reumatycznej i układowych chorobach tkanki
kosteroidów nie wywołują zazwyczaj żadnych nie-
łącznej),
bezpiecznych działań niepożądanych,
■ w reakcjach alergicznych (np. obrzęku naczynioru-
■ także terapia trwająca kilka dni nie powoduje za-
chowym, alergicznym nieżycie nosa, alergicznym
zwyczaj żadnych poważnych działań niepożąda-
zapaleniu spojówek, ukąszeniach owadów, po-
nych, chyba że stosuje się bardzo duże dawki,
krzywce, wstrząsie anafilaktycznym),
Układ hormonalny
■ w miarę możliwości glukokortykosteroidy należy
■ w chorobach nerek (głównie w zespole nefrotycz-
podawać w jednej dawce rano, gdyż takie dawko-
nym),
wanie w mniejszym stopniu zaburza rytm dobowy
■ w schorzeniach hematologicznych (np. niedokrwi- wydzielania kortyzolu, a tym samym w mniejszym
stościach hemolitycznych, plamicy małopłytko- stopniu hamuje funkcję kory nadnerczy i zmniej-
wej), sza ryzyko atrofii kory nadnerczy. Przy stosowaniu
preparatów dépôt nie jest możliwe osiągnięcie ta-
■ w chorobach płuc (np. przewlekłej obturacyjnej
chorobie płuc – POChP, sarkoidozie, toksycznym
kiego dopasowania do rytmu dobowego i dlatego B2
nie powinno się ich, a także długo działających
obrzęku płuc),
glukokortykosteroidów (np. deksametazonu), sto-
■ w chorobach żołądka i jelit (np. chorobie Crohna), sować w długotrwałej terapii,
■ w wymiotach wywołanych cytostatykami (w połą- ■ u pacjentów, u których jedna dawka dziennie
czeniu z antagonistą 5-HT3), nie pozwala na osiągnięcie efektu terapeutycznego,
wskazane jest podawanie drugiej dawki wczesnym
■ w chorobach wątroby (np. w przewlekłym agre-
popołudniem lub wczesnym wieczorem,
sywnym nieinfekcyjnym zapaleniu wątroby),
■ kończąc długo trwające leczenie glukokortyko-
■ w nowotworach złośliwych, przede wszystkim
steroidami, należy dawki zmniejszać stopniowo,
nowotworach układowych (białaczkach, ziarnicy,
ponieważ w przeciwnym razie istnieje niebezpie-
mięsaku limfatycznym),
czeństwo powikłań będących następstwem atrofii
■ w chorobach układu nerwowego (np. napadach kory nadnerczy. Występują one przede wszystkim
zgięciowych typu Salaam), w sytuacjach stresowych, kiedy istnieje zwiększo-
ne zapotrzebowanie na kortyzol. W takich sytua-
■ w obrzęku mózgu (przede wszystkim przy obrzęku
cjach należy odpowiednio dostosować dawkowa-
mózgu spowodowanym guzem),
nie kortyzolu.
■ w ciężkim wstrząsie,
Dawkowanie. Dawkowanie dostosowuje się
■ w transplantologii.
z uwzględnieniem ww. zasad do uzyskanej odpo-
wiedzi na leczenie oraz ciężkości choroby. Na przy-
Przy współistnieniu choroby infekcyjnej glukokor-
kład początkowe dawki prednizolonu mogą wynosić
tykosteroidy wolno stosować tylko w połączeniu
30–1000 mg, natomiast doustne dawki podtrzymują-
z lekami skierowanymi przeciwko czynnikom in-
ce 5–15 mg dziennie. W ostrych stanach zagrożenia
fekcyjnym. W tych przypadkach podaje się je w celu
życia podaje się duże dawki glukokortykosteroidów
zapobiegania uwarunkowanym zakażeniem, niepo-
we wstrzyknięciach lub wlewach dożylnych.
żądanym reakcjom mezenchymalnym, takim jak wy-
twarzanie wysięku.
Działania niepożądane. Działania niepożądane glu-
kokortykosteroidów wynikają z ich profilu działania.
Zasady stosowania. Glukokortykosteroidy należy
Przy długo trwającej terapii wraz ze zwiększeniem
stosować tylko w przypadku istnienia ściśle określo-
dawki wzrasta ich częstość oraz nasilenie. Objawy
nych wskazań po dokładnym rozważeniu możliwych
zespołu Cushinga (zob. powyżej) pojawiają się, gdy
działań niepożądanych. Powinno się przy tym zwra-
zostanie przekroczony – indywidualnie bardzo różny
cać uwagę na następujące kwestie:
– próg Cushinga. Działania niepożądane występujące
■ dawkę potrzebną do osiągnięcia efektu terapeu- przy podawaniu ≤ 7,5 mg prednizolonu diennie mają
tycznego należy zawsze ustalać indywidualnie dla zazwyczaj małe znaczenie.
MUTSCHLER-2009.indd 455 2010-01-07 22:13:08
456 Układ hormonalny
W związku z działaniem immunosupresyjnym dów nasercowych. Wskutek indukowania enzymów
wzrasta ryzyko infekcji oraz reaktywacji utajonych barbiturany, fenytoina i rifampicyna osłabiają działa-
zakażeń. Przy jednoczesnym stosowaniu niesteroido- nie glukokortykosteroidów.
wych leków przeciwzapalnych mogą ponownie poja-
wić się owrzodzenia w żołądku i jelitach, jednak same Przeciwwskazania. Względnymi przeciwwskaza-
glukokortykosteroidy nie wywołują owrzodzeń. Go- niami, przy których należy dokładnie przeanalizować
jenie się ran jest opóźnione. Z powodu występowa- możliwe korzyści i zagrożenia, są: grzybice układo-
nia efektów katabolicznych lub antyproliferacyjnych we, amebiaza, owrzodzenia żołądka i jelit, ciężka
może się pojawić atrofia mięśni, skóry (tworzenie osteoporoza, psychozy, stadium wiremii w przebiegu
się rozstępów) i tkanki tłuszczowej. U dzieci obser- różnych chorób wirusowych (np. wywołanych wiru-
wuje się zahamowanie wzrostu. Przy długotrwałym sem ospy wietrznej i półpaśca), a także jaskra. Przy
stosowaniu wskutek rozpuszczania się mezenchy- niedoborze kortyzolu powinno się jednak zawsze
malnej macierzy kostnej i częściowo również w wy- przeprowadzić terapię substytucyjną.
niku działania antagonistycznego do witaminy D,
jak również hamowania kościotworzenia w następ-
stwie zmniejszenia ekspresji genu odpowiedzialnego 2.7.4. Mineralokortykosteroidy
za kodowanie prokolagenu I istnieje znaczne ryzyko jako środki lecznicze
pojawienia się osteoporozy. Na to ryzyko są narażeni
w sposób szczególny pacjenci z ograniczoną aktyw-
Mineralokortykosteroidy biorą udział w regulacji
nością ruchową.
gospodarki wodno-elektrolitowej. Po połączeniu
Z powodu wpływu wywieranego na metabolizm
ze swoim receptorem wewnątrzkomórkowym nasila-
glukozy utajona cukrzyca może przejść w postać
ją one wiązanie pompy Na+/K+ zależnej od ATP oraz
objawową (działanie diabetogenne). Z kolei wpływ
syntezę kanałów potasowych i sodowych w cewkach
na metabolizm lipidów przy długo trwającym poda-
dalszych oraz kanalikach zbiorczych nerek (zob. ryc.
waniu dużych dawek prowadzi do opisanej zmiany
B 2.7-7). Prowadzi to do zwiększenia wchłaniania
rozmieszczenia tkanki tłuszczowej.
zwrotnego sodu oraz wydalania potasu i jonów wodo-
Przy stosowaniu związków, których część zakre-
rowych. Równoczesnie dochodzi do retencji wody.
su działania odpowiada mineralokortykosteroidom,
Podobny wpływ wywierają mineralokortykostero-
może wystąpić zatrzymanie sodu i wody w organi-
idy na transport jonów i wody w jelitach oraz ślinian-
zmie oraz wzmożona utrata potasu.
kach i gruczołach potowych.
Z powodu wpływu na czynność ośrodkowego ukła-
du nerwowego istnieje ryzyko wystąpienia zaburzeń
Przez wprowadzenie fluoru w pozycji 9α kortyzolu
snu, zahamowania napędu lub zmian psychicznych.
lub prednizolonu można otrzymać silnie działające
Długo trwająca terapia może, szczególnie u dzieci,
mineralokortykosterydy. Obecnie w handlu jest do-
wywołać wzrost ciśnienia śródczaszkowego i objawy
stępna wyłącznie jedna substancja czynna z tej grupy,
rzekomego guza mózgu.
a mianowicie pochodna kortyzolu – fludrokortyzon.
Z tym wiąże się również spostrzeżenie, że gluko-
Fludrokortyzon, którego okres półtrwania wynosi
kortykosteroidy powodują wzrost ciśnienia śródgał-
około 1 godziny, jest wydalany głównie przez nerki.
kowego i wskutek tego mogą doprowadzić do jaskry,
W porównaniu z glukokortykosteroidami fludro-
a w konsekwencji także do ślepoty. Taki sam wpływ
kortyzon ma niewielkie znaczenie terapeutyczne.
na ciśnienie śródgałkowe wywierają glukokortykoste-
Przy niewydolności kory nadnerczy można go poda-
roidy zarówno stosowane miejscowo na gałkę oczną,
jak i stosowane ogólnie. Poza tym należy wspomnieć
o zwiększonym ryzyku wystąpienia zakrzepicy.
O
Interakcje. Glukokortykosteroidy osłabiają działa- CH2OH
CH3
O OH
nie leków przeciwzakrzepowych i doustnych leków
przeciwcukrzycowych. Przy jednoczesnym stosowa- CH3 H
niu glukokortykosteroidów i niesteroidowych leków H H
przeciwzapalnych, zwłaszcza pochodnych kwasu sa- O
licylowego, wzrasta ryzyko krwawienia do przewodu
pokarmowego. Związana ze stosowaniem glukokor-
fludrokortyzon
tykosteroidów utrata potasu nasila działania glikozy-
MUTSCHLER-2009.indd 456 2010-01-07 22:13:08
Układ hormonalny 457
światło cewki tkanka śródmiąższowa
okołocewkowa
mineralo-
+ kortykosteroid
MR MR
K+
jądro
komórkowe MR
Na+
Układ hormonalny
transkrypcja ATP
3 Na+
+
2K
translacja
białka indukowane przez aldosteron
B2
Ryc. B 2.7-7. Mechanizm działania mineralokortykosteroidów (na przykładzie cewki dalszej nerki). MR – receptor dla mineralokor-
tykosteroidów.
wać jedynie łącznie z glukokortykosteroidami (zob. Kortyzol podaje się w dawce 20–25 mg dziennie
poniżej). Poza tym fludrokortyzon stosuje się przy w pierwotnej niewydolności kory nadnerczy i 15–
ciężkich, opornych na leczenie, zaburzeniach krąże- 20 mg w postaci wtórnej, a dawka dobowa powinna
nia z hipotonią. być tak podzielona, aby chory otrzymywał 2/3 dawki
W terapii substytucyjnej stosuje się fludrokortyzon rano i 1/3 wczesnym popołudniem. Pacjenci z choro-
w dawce 0,l mg, natomiast w leczeniu hipotonii do- bą Addisona powinni dodatkowo otrzymywać 0,05–
ustnie 0,1–0,3 mg dziennie. 0,2 mg fludrokortyzonu dziennie.
Działaniami niepożądanymi pojawiającymi się W sytuacjach stresowych o umiarkowanym nasi-
przy leczeniu hipotonii oraz po przedawkowaniu leniu (np. przy znacznym obciążeniu fizycznym bądź
w terapii substytucyjnej są obrzęki oraz utrata pota- psychicznym organizmu lub infekcji grypopodobnej)
su. dawkę należy przejściowo podwoić. W sytuacjach
Nadciśnienie tętnicze, niewydolność krążenia, mar- stresowych o dużym nasileniu konieczne jest lecze-
skość wątroby i zespół nerczycowy są przeciwwskaza- nie dużymi dawkami glukokortykosteroidów. Przy
niami do stosowania fludrokortyzonu. dawce dobowej hydrokortyzonu ponad 50 mg można
przerwać podawanie fludrokortyzonu.
2.7.5. Zasady leczenia Zespół Cushinga. Leczenie nadnerczowej i wtórnej
zaburzeń funkcji nadnerczy postaci polega na usunięciu produkującej hormony
tkanki. Leczenie farmakologiczne (np. ketokonazo-
Niewydolność kory nadnerczy. W celu uniknięcia lem w dawce 600–1000 mg) jest wskazane tylko jako
zagrażającego życiu przełomu nadnerczowego oraz przygotowanie do zabiegu operacyjnego lub u cho-
poprawienia wydolności fizycznej w leczeniu choro- rych, którzy nie mogą być operowani.
by Addisona (pierwotnej niewydolności nadnerczy)
wskazane jest równoczesne podawanie glukokortyko- Hiperaldosteronizm. Jednostronny gruczolak kory
steroidów (kortyzonu lub kortyzolu) i mineralokorty- nadnerczy produkujący aldosteron może być leczony
kosteroidów. We wtórnej niedoczynności kory nadner- operacyjnie. Jako leczenie przygotowujące, którego
czy, z uwagi na zachowane podstawowe wydzielanie celem jest normalizacja stężenia potasu we krwi, sto-
aldosteronu, zazwyczaj wystarczające jest podawanie suje się antagonistów aldosteronu, np. spironolakton
tylko glukokortykosteroidów. w dawce 200–300 mg dziennie. Pooperacyjny hipo-
MUTSCHLER-2009.indd 457 2010-01-07 22:13:08
458 Układ hormonalny
aldosteronizm wymaga leczenia fludrokortyzonem zapalne oraz nadmierna proliferacja tkanek. Gluko-
w dawce 0,05–0,2 mg. kortykosteroidy powodują zmniejszenie nasilenia
objawów tych schorzeń dzięki wykazywanemu przez
Zespół nadnerczowo-płciowy. Leczenie opiera się nie działaniu przeciwzapalnemu, antyproliferacyjne-
na substytucji kortyzolu, która powoduje normaliza- mu i immunosupresyjnemu. Poza tym glukokorty-
cję stężenia androgenów. kosteroidy są również często stosowane jako specy-
ficzne skuteczne środki lecznicze będące leczeniem
z wyboru, co przedstawiono sumarycznie we wspo-
2.7.6. Glukokortykosteroidy mnianym rozdziale.
w leczeniu immunosupresyjnym
i w terapii chorób zapalnych
2.7.3. Hormony rdzenia nadnerczy
Jak napisano w rozdziale B 2.7.3, glukokortykostero-
idy znajdują zastosowanie w terapii wielu schorzeń, Hormony rdzenia nadnerczy – noradrenalina i adre-
u podstawy których leżą reakcje immunologiczne lub nalina – zostały omówione w rozdziale B 1.13.
MUTSCHLER-2009.indd 458 2010-01-07 22:13:09
Układ hormonalny 459
2.8. Hormony płciowe i wywodzące się z nich leki
Efektorowe hormony płciowe, tak jak glukokortyko- nadoliberyna, gonadorelina, GnRH) powstaje ze zbu-
steroidy, są syntezowane z cholesterolu. U mężczyzn dowanego z 92 aminokwasów prekursora, a proces
jądra wydzielają androgen – testosteron. Najważniej- ten podlega neuronalnej kontroli podwzgórza. Jest on
szymi żeńskimi hormonami płciowymi są syntezo- uwalniany pulsacyjnie, u mężczyzn co 120 minut,
wane przede wszystkim w jajnikach estrogeny i ges- a u kobiet co 90 minut w fazie folikularnej i rzadziej
tagen – progesteron, który powstaje w ciałku żółtym w fazie lutealnej cyklu miesięcznego (zob. rozdz.
Układ hormonalny
i (u kobiet w ciąży) w łożysku. 2.8.4).
Nadrzędne hormony podwzgórza (hormon uwalnia- Wydzielanie pulsacyjne jest niezwykle ważne dla
jący gonadotropiny, GnRH) i przysadki (gonadotro- działania fizjologicznego GnRH. Na skutek aktywacji
piny) regulują syntezę tych hormonów sterydowych. receptorów dla GnRH w przysadce, które są powią-
Obie gonadotropiny (hormon folikulotropowy – FSH zane z białkiem Gq, wzrasta stężenie IP3– lub Ca2+,
i hormon luteinizujący – LH), a także GnRH są wy- co prowadzi do syntezy i uwalniania FSH lub LH.
dzielane zarówno u kobiet, jak i u mężczyzn. (Znaczenie receptorów dla GnRH znajdujących się
w jajnikach, jądrach i nowotworach nie zostało wy- B2
jaśnione.)
2.8.1. Regulacja syntezy hormonów Gonadotropiny. Termin gonadotropiny oznacza hor-
płciowych; budowa i funkcja mony wpływające na gonady. Poza powstającymi
hormonu uwalniającego w przysadce hormonem luteinizującym (LH) i foli-
gonadotropiny i gonadotropin kulotropowym (FSH) do gonadotropin należy także
(ludzka) gonadotropina kosmówkowa (HCG), która
niezależnie od GnRH powstaje w czasie ciąży w ło-
Synteza hormonów płciowych, tak jak hormonów tar-
czycy i glukokortykosteroidów, podlega trójstopniowej
regulacji na zasadzie sprzężenia zwrotnego (zob. ryc.
podwzgórze
B 2.8-1). Powstający w podwzgórzu hormon uwalnia-
jący gonadotropiny (GnRH) jest uwalniany z przerwa-
mi (tzw. uwalnianie pulsacyjne) – w nocy i wcześnie
rano częstość pulsów się zwiększa. W przednim pła- gonadoliberyna
(GnRH)
cie przysadki GnRH pobudza syntezę i wydzielanie
hormonu folikulotropowego (FSH) i hormonu luteini-
zującego (LH). One zaś inicjują w jądrach lub jajni-
przedni płat
kach syntezę testosteronu, estrogenów i progesteronu. przysadki
W związku z pobudzaniem przez hormon luteinizujący
syntezy testosteronu w komórkach śródmiąższowych
(komórkach Leydiga) bywa on też nazywany hormo-
LH
nem pobudzającym komórki śródmiąższowe (ICSH).
Testosteron i progesteron, jak również estrogeny FSH
w niskich stężeniach, hamują wydzielanie hormonów
nadrzędnych – przede wszystkim GnRH. Ujemne
sprzężenie zwrotne powiaząne z syntezą FSH jest na- gonady
silane przez inhibiny produkowane przez komórki
warstwy ziarnistej jajników oraz komórki Sertolego
w jądrach i należące do czynników wzrostu klasy
hormony płciowe inhibina
„białek indukujących syntezę kości i chrząstki”. Wy-
sokie stężenia estrogenów dla odmiany pobudzają
na zasadzie dodatniego sprzężenia zwrotnego wy- wysokie stężenie estrogenów
dzielanie FSH (zob. rozdz. B 2.8.4).
Ryc. B 2.8-1. Regulacja syntezy hormonów płaciowych. GnRH
Hormon uwalniający gonadotropiny. Będący de- – hormon uwalniający gonadotropiny, LH – hormon luteinizują-
kapeptydem hormon uwalniający gonadotropiny (go- cy, FSH – hormon folikulotropowy.
MUTSCHLER-2009.indd 459 2010-01-07 22:13:09
460 Układ hormonalny
żysku. Działanie HCG jest bardzo podobne do LH i gonadotropiny oraz ich pochodne znajdują zastoso-
– oba hormony pobudzają receptor dla LH, podczas wanie jako środki terapeutyczne.
gdy FSH jest jedynym hormonem posiadającym zdol-
ność pobudzania receptora dla FSH.
Gonadotropiny mają także zbliżoną budowę che- 2.8.1.1. Zastosowanie hormonu
miczną. Są one dużymi heterodimerycznymi gliko- uwalniającego gonadotropiny
proteinami składającymi się z identycznych podjed- i jego analogów w lecznictwie
nostek α (takich samych jak w TSH) oraz warunkują-
cych działanie podjednostek β. Obecność komponen- Za pomocą hormonu uwalniającego gonadotropiny
ty cukrowej jest niezbędna do aktywacji kieszonki (gonadoreliny) oraz antagonistów GnRH (zob. tab. B
sygnałowej i utrzymania odpowiedniej szybkości 2.8-1) można zwiększać lub zmniejszać wydzielanie
rozkładu. HCG działa najdłużej. gonadotropin. Wzrost uzyskuje się, podając je jedno-
U mężczyzn LH pobudza syntezę androgenów, razowo lub w sposób pulsacyjny. Podawanie ciągłe,
a w szczególności testosteronu, w komórkach Ley- szczególnie silniej i dłużej działających zmodyfikowa-
diga, a FSH syntezę białek w komórkach Sertolego nych postaci hormonu, powoduje początkowo wzrost
(zob. poniżej). U kobiet FSH pobudza wzrost jajni- wydzielania FSH i LH, a następnie zablokowanie re-
ków, dojrzewanie pęcherzyków, ekspresję receptorów ceptorów dla GnRH (down-regulation) w przysadce,
dla LH w komórkach osłonki i komórkach warstwy które w ciągu 2–3 tygodni prowadzi do zahamowa-
ziarnistej oraz syntezę aromatazy i produkcję estroge- nia wydzielania gonadotropin. W następstwie gona-
nów w komórkach ziarnistych. LH zwiększa syntezę dy przestają syntezować hormony płciowe. Szybkie
androgenów w komórkach osłonki, wywołuje owu- zahamowanie wydzielania gonadotropin powodują
lację oraz warunkuje przemianę resztki pęcherzyka antagoniści GnRH.
w ciałko żółte (zob. rozdz. B 2.8.4).
W związku z tym wielokierunkowym działaniem Hormon uwalniający gonadotropiny. Będąc de-
zarówno hormon uwalniający gonadotropiny, jak kapeptydem GnRH ulega strawieniu w przewodzie
Tabela B 2.8-1. Hormon uwalniający gonadotropiny (gonadorelina) i jego analogi
Nazwa Nazwa handlowa Wskazania Okres Średnie
międzynarodowa półtrwania dawkowanie
gonadorelina Lutrelef, niewydolność jajników, < 10 min 60-240 g dziennie i.v.,
Kryptocur wnętrostwo zależne 1,2 mg dziennie
od podwzgórza donosowo
Analogi GnRH, agoniści
buserelina Suprecur endometrioza, 1-2 godz. 0,9 mg dziennie
zapłodnienie in vitro donosowo
goserelina Zoladex endometrioza, 2,3 godz. 3,6 mg 28-dniowy
mięśniaki macicy implant podskórny
leuprorelina Enantone-Gyn, Trenantone- Gyn endometrioza 3 godz. 3,75 mg/miesiąc s.c.
[w Polsce: Eligard, Lucrin depot lub i.m.
– przyp. tłum.]
nafarelina Synarela [w Polace: endometrioza, 4 godz. 0,4-0,8 mg dziennie
Synarel – przyp. tłum.] zapłodnienie in vitro donosowo
triptorelina Decapeptyl Gyn endometrioza, 20 min 3,75 mg co 28 dni s.c.
[w Polsce: Decapeptyl, mięśniaki macicy lub i.m.
Diphereline – przyp. tłum.]
Analogi GnRH, antagoniści
cetroreliks Cetrotide zapłodnienie in vitro 12-30 godz. 3 mg dziennie s.c.
ganireliks Orgalutran zapobieganie przedwczesnemu 13 godz. 0,25 mg dziennie s.c.
wzrostowi stężenia LH
przy zapłodnieniu in vitro
MUTSCHLER-2009.indd 460 2010-01-07 22:13:09
Układ hormonalny 461
podstawienie D-aminokwasów
zwiększa aktywność i czas działania
gonadorelina pGlu His Trp Ser Tyr Gly Leu Arg Pro Gly-NH2
wiązanie z receptorem wiązanie z receptorem
i aktywacja, podstawienie
D-aminokwasów powoduje
działanie antagonistyczne
Układ hormonalny
Agoniści
leuprorelina pGlu His Trp Ser Tyr D-Leu Leu Arg Pro-NH-CH2-CH3
goserelina pGlu His Trp Ser Tyr tetrabutylo-D-Ser Leu Arg Pro-NH-NH-CO-NH2
buserelina pGlu His Trp Ser Tyr tetrabutylo-D-Ser Leu Arg Pro-NH-CH2-CH3
nafarelina pGlu His Trp Ser Tyr 3-(2-naftylo)-D-Ala Leu Arg Pro Gly-NH2 B2
triptorelina pGlu His Trp Ser Tyr D-Trp Leu Arg Pro Gly-NH2
Antagoniści
cetroreliks N-acetylo-2-(2-naftylo)-D-Ala 4-chloro-D-Phe 3-(3-pirydylo)-D-Ala Ser Tyr
N5-karbamylo-D-Orn Leu Arg Pro D-Ala-NH2
ganireliks N-acetylo-2-(2-naftylo)-D-Ala 4-chloro-D-Phe 3-(3-pirydylo)-D-Ala Ser Tyr
N’-(N,N-dietyloamidyno)-D-Lys Leu N’-(N,N-dietyloamidyno)-D-Lys Pro D-Ala-NH2
Ryc. B 2.8-2. Struktura hormonu uwalniającego gonadotropinę (gonadoreliny) i jej analogów; pGlu – kwas pyroglutaminowy.
pokarmowym, jest jednak dobrze wchłaniany po po- sów w pozycji 6 i 10; wprowadzenie niefizjologicz-
daniu donosowym. W surowicy ulega on szybkiej hy- nego D-aminokwasu w pozycji 6 zwiększa stabilność
drolizie, a okres półtrwania wynosi 4 minuty. analogów (zob. ryc. B 2.8-2).
GnRH ma zastosowanie diagnostyczne i terapeu- Wskazania do ich stosowania wynikają przede
tyczne. Zarówno u kobiet, jak i u mężczyzn jest wy- wszystkim z pełnego zahamowania syntezy hormo-
korzystywany w diagnostyce różnicowej hipogona- nów płciowych oraz całkowitego zablokowania re-
dyzmu. Oznacza się wówczas osoczowe stężenie LH ceptorów w przysadce. Dzięki temu można przeciw-
przed i po podaniu dożylnym 100 μg GnRH. działać wzrostowi nowotworów hormonozależnych.
W zaburzeniach płodności spowodowanych nie- Agoniści GnRH są stosowani w terapii paliatywnej
doborem GnRH terapia pulsacyjna GnRH może rozsianego raka prostaty lub sutka. Ponadto stałe
przynieść powodzenie. Za pomocą pompy infuzyj- podawanie GnRH jest wskazane w leczeniu endo-
nej podaje się 5–20 μg leku w odstępach co 90 minut metriozy i przedwczesnego dojrzewania (pubertas
u kobiet i co 120 minut u mężczyzn. Innym wskaza- praecox), a także w celu przedoperacyjnego zmniej-
niem do stosowania GnRH jest wnętrostwo – wów- szenia mięśniaków macicy. Kolejnym wskazaniem
czas podaje się 1,2 mg dziennie donosowo przez 4 jest kierowanie dojrzewaniem pęcherzyków w trak-
tygodnie. Jeżeli nie spowoduje to przemieszczenia cie wspomagania rozrodu (zapłodnienie in vitro
jąder, konieczne jest leczenie operacyjne. – zob. poniżej).
Do działań niepożądanych należą ciąże mnogie Do działań niepożądanych należą zaburzenia
i zespół hiperstymulacji jajników u kobiet, aczkol- związane z klimakterium, zaburzenia naczynioru-
wiek pojawiają się one rzadziej niż przy leczeniu go- chowe, spadek libido) oraz obrzęki. Długotrwałe
nadotropinami. stosowanie GnRH może doprowadzić do osteoporo-
zy, która, przy braku wskazań związanych z zagro-
Agoniści GnRH. Działające silniej i dłużej analogi żeniem życia, jest przeciwwskazaniem do ich sto-
GnRH otrzymuje się poprzez wymianę aminokwa- sowania.
MUTSCHLER-2009.indd 461 2010-01-07 22:13:10
462 Układ hormonalny
Antagoniści GnRH. Antagonistów GnRH, cetroreliks kosmówkowej (HCG). Początkowo gonadotropiny
i ganireliks, otrzymano przez znaczną modyfikację były uzyskiwane poprzez ekstrakcję z moczu. Mocz
struktury GnRH, a przede wszystkim zamianę ami- kobiet w wieku menopauzalnym zawiera FSH i LH
nokwasów w pozycjach 1–3 na formy prawoskrętne w nienaruszonej formie (ludzka gonadotropina me-
(D-formy) aminokwasów. Spowodowało to dalsze nopauzalna, menotropina), a HCG jest także w for-
wydłużenie okresu półtrwania, który w przypadku mie aktywnej wydalana przez nerki. Jednak uzyska-
cetroreliksu wynosi ok. 12 godzin. Od agonistów ne ekstrakty zawierają bardzo małe ilości substancji
antagoniści GnRH różnią się szybkim spadkiem syn- czynnych, a więc zawartość leków była niewielka.
tezy gonadotropin i hormonów płciowych bez przej- Obecnie gonadotropiny uzyskuje się metodami
ściowego wzrostu ich stężenia. inżynierii genetycznej poprzez wywołanie ekspresji
Antagoniści GnRH są obecnie zarejestrowani w komórkach ssaków. Dostępne są dwie proteiny
do stosowania przy wspomaganiu rozrodu. Szybki spa- FSH (folitropina α i β) różniące się nieznacznie gli-
dek stężenia hormonów skraca czas terapii gonadotro- kozylacją, a co za tym idzie – także siłą działania.
pinami potrzebny do uzyskania nadających się do za- Jako glikoproteiny gonadotropiny muszą być po-
płodnienia komórek jajowych. Chociaż dzięki temu dawane pozajelitowo. W organizmie są potem długo
zmniejszono ryzyko zwiazane z zapłodnieniem in vitro, wykrywalne.
nadal nie można pewnie ocenić jego bezpieczeństwa. Ze względu na zdolność do pobudzania dojrzewa-
Działania niepożądane odpowiadają tym wymie- nia pęcherzyków FSH i HCG są wskazane w lecze-
nionym przy agonistach GnRH. niu niepłodności żeńskiej. Dawkowanie jest ustalane
W USA preparat o przedłużonym działaniu zawie- indywidualnie na podstawie ocenianego ultrasono-
rający abareliks został zarejestrowany w terapii pa- graficznie wzrostu pęcherzyków jajnikowych. Owu-
liatywnej raka prostaty. lację dojrzałych pęcherzyków w celu umożliwienia
naturalnej koncepcji można wywołać, podając HCG
lub LH – komórka jajowa zostaje uwolniona ok. 36
2.8.1.2. Gonadotropiny stosowane godzin po iniekcji. Alternatywnie mogą te substancje
w lecznictwie być wykorzystywane przy zapłodnieniu in vitro. Aby
wywołać dojrzewanie wielu pęcherzyków, konieczne
jest podawanie dużych dawek FSH lub menotropiny.
Poza gonadoreliną i jej analogami również gonado-
Przed operacyjnym pobraniem komórek jajowych,
tropiny znalazły zastosowanie jako środki leczni-
dla uzyskania ich pełnej dojrzałości zostaje podana
cze (zob. tab. B 2.8-2). Dotyczy to obu hormonów
HCG (aby zapobiec przedwczesnemu jajeczkowaniu
przysadkowych (FSH i LH), a także gonadotropiny
łącznie z agonistą lub antagonistą GnRH).
Tabela B 2.8-2. Gonadotropiny
Nazwa Nazwa handlowa Wskazania Okres Średnie
międzynarodowa półtrwania dawkowanie
(godz.)
folitropina α (rekombinowana) GONAL-F wspomaganie rozrodu 24 indywidualnie w zależności
u mężczyzn i kobiet od płci, wskazań i działania
folitropina β (rekombinowana) Puregon wspomaganie rozrodu 40
u mężczyzn i kobiet
menotropina Menogon HP [w Polsce:
(wyciąg zawierający FSH i LH) Menopur – przyp. tłum.] niepłodność u kobiet 75-150 IU
lutropina α (rekombinowana) Luveris niepłodność u kobiet 12 75 IU
gonadotropina kosmówkowa Choragon, Predalon [w Polsce: wywołanie owulacji, 30 dwa razy w tygodniu
(wyciąg) Biogonadyl, Pregnyl, wnętrostwo 5000-10000 IU,
Choragon – przyp. tłum.] jednorazowo 1500 IU
gonadotropina kosmówkowa Ovitrelle brak owulacji, wywołanie 30 250 μg
(rekombinowana) mnogiej owulacji
MUTSCHLER-2009.indd 462 2010-01-07 22:13:10
Układ hormonalny 463
W przypadku niepłodności męskiej spowodo-
wanej niedoczynnością przysadki stosuje się wielo- spermatocyt II rzędu
miesięczną terapię FSH (150 IU 3 razy w tygodniu)
i HCG (1500-6000 IU raz w tygodniu). spermatocyt I rzędu prespermatydy
W leczeniu wnętrostwa chłopcy otrzymują za- spermatogonium
spermatogonium w fazie podziału
leżnie od wieku 500–2000 IU HCG raz w tygodniu
przez 5 tygodni. Jeżeli nie uzyska się pożądanego
efektu, kurację można powtórzyć jeden raz. Przy bra-
ku odpowiedzi klinicznej na kolejną terapię wskaza-
Układ hormonalny
ne jest leczenie operacyjne.
Poza tym u mężczyzn HCG jest wykorzystywana
w diagnostyce różnicowej hipogonadyzmu. Przy pra-
widłowej funkcji gonad po podaniu domięśniowym
5000 IU obserwuje się wzrost syntezy testosteronu.
Oprócz tego, z uwagi na obecność HCG w moczu,
wkrótce po zapłodnieniu jest ona wykorzystywana błona podstawna komórki
w testach ciążowych. We krwi β-HCG jest wykrywa- plemniki Sertolego
ne już 10 dni po zapłodnieniu, a w moczu po 2 tygo-
spermatydy B2
dniach od poczęcia. Proste dostępne zestawy testo- Ryc. B 2.8-3. Przekrój kanalika jądrowego (według Buchera).
we zawierają przeciwciała przeciw podjednostce β
HCG.
Do mających znaczenie kliniczne, ważnych dzia- miniferi contortii, ryc. B 2.8-3), osadzonych w strukturze
łań niepożądanych leczenia niepłodności gonado- drobnowłókienkowej tkanki łącznej. Kanaliki mają śred-
nicę 200–300 μm, a ich łączna długość wynosi 250–300
tropinami należy hiperstymulacja jajników i dlatego metrów. Ich ścianę tworzą cylindryczne komórki (komórki
ponad 10% ciąż kończy się porodem bliźniaków. Ob- podporowe, zwane inaczej komórkami Sertolego), pomię-
jawami hiperstymulacji jajników mogą być groźne dzy którymi znajdują się komórki biorące udział w powsta-
obrzęki brzucha i klatki piersiowej, ostra niewydol- waniu nasienia.
ność oddechowa i oliguria. Ryzyko wystąpienia tych
powikłań jest niższe przy podawaniu krócej działają- W okresie dojrzewania w kanalikach nasiennych
cego rekombinowango LH niż przy stosowaniu dłu- rozpoczyna się wytwarzanie nasienia. W przeciwień-
żej działającej HCG. stwie do kobiet, u których komórki jajowe tworzą się
już w życiu płodowym, u mężczyzn proces spermio-
genezy zachodzi stale dopiero w okresie dojrzałości
2.8.2. Męskie hormony płciowe płciowej. Pomiędzy kanalikami nasiennymi przy na-
i ich analogi czyniach krwionośnych tkanki łącznej śródmiąższo-
wej znajdują się grupy komórek ułożonych nabłon-
kowato jedna obok drugiej, określane jako komórki
2.8.2.1. Podstawy anatomiczne
śródmiąższowe jądra lub komórki Leydiga. W nich
i fizjologiczne
pod wpływem LH (= ICSH) są wytwarzane męskie
hormony płciowe (androgeny) oraz niewielkie ilości
Jądra są miejscem tworzenia plemników oraz synte-
żeńskich hormonów płciowych.
zy męskich hormonów płciowych. Poza tym powstają
w nich – w niewielkich ilościach – także żeńskie hor-
mony płciowe. Dojrzałe płciowo jądra są parzystym
narządem o kształcie elipsoidalnym, którego wymiar 2.8.2.2. Budowa chemiczna,
podłużny wynosi ok. 5 cm. Do tylnego brzegu jądra, biosynteza i rozkład
do tzw. śródjądrza, wnikają lub z niego wychodzą
naczynia, nerwy i nasieniowód w powrózku nasien- Naturalne androgeny są C19-steroidami. Najważniej-
nym. szym męskim hormonem płciowym syntetyzowanym
w komórkach śródmiąższowych jądra (komórkach
Jądra otoczone są zbitą torebką łącznotkankową, od której Leydiga) jest testosteron. W niektórych narządach
w głąb miąższu w kierunku śródjądrza odchodzą promieni-
ście przegródki jądra. Te przegrody dzielą jądra na 200–250 efektorowych, np. zewnętrznych narządach płcio-
stożkowatych płacików (lobuli testis). Płaciki jądra zawie- wych, gruczole krokowym, komórkach łojowych
rają wiele poskręcanych kanalików nasiennych (tubuli se- wytwarzających, a także mieszkach włosowych, te-
MUTSCHLER-2009.indd 463 2010-01-07 22:13:11
464 Układ hormonalny
stosteron ulega przekształceniu do 5α-dihydrotesto- Biosynteza. Tak jak inne hormony steroidowe, an-
steronu, charakteryzującego się wyraźnie większym drogeny są syntetyzowane z cholesterolu poprzez
powinowactwem do receptora androgenów niż testo- substancję pośrednią – pregnenolon. Innym związ-
steron (zob. poniżej). kiem pośrednim jest androstendion (zob. ryc. B 2.8-
W warunkach fizjologicznych u mężczyzn, 4). Zarówno testosteron, jak androstandion mogą być
ale nie u kobiet, androgeny syntetyzowane w korze metabolizowane do 5α-dihydrotestosteronu (andro-
nadnerczy (np. dehydroepiandrosteron – DHEA) stanolonu, 5α-DHT).
mają znaczenie drugorzędne.
H3C O CH3
CH3 CH(CH3) 2 CH3
CH3 H CH3 H
H H H H O
CYP CH3
HO HO 17
D pregnenolon
-HS CH3 H
cholesterol 3β
H H
O CH3 HO
CH3
D prasteron (DHEA)
-HS
CH3 H 3β
H H
C YP
17
O CH3 O CH3 O
progesteron CH3 H H
CYP19
H H H H
O HO
androstendion estron
17β-HSD 17β-HSD
CH3 OH
CH3 OH CH3 OH
CH3 H
CH3 H H
CYP19
H H 5α-reduktaza H H H H
O
H O HO
androstanolon testosteron estradiol
(5α-DHT)
17β-HSD
CH3 O
CH3 OH
CH3 H
H OH
H H
O H H
H
HO
androstandion estriol
Ryc. B 2.8-4. Biosynteza hormonów płciowych. CYP – cytochrom P-450, 3β-HSD – dehydrogenaza 3β-hydrosteroidowa, DHEA – de-
hydroepiandrosteron, 5α-DHT - 5α-dihydrotestosteron.
MUTSCHLER-2009.indd 464 2010-01-07 22:13:11
Układ hormonalny 465
Farmakokinetyka. U dojrzałego płciowo mężczyzny Mechanizm działania. Podobnie jak przy innych
powstaje ok. 7 mg testosteronu dziennie. Nie jest on hormonach steroidowych, działanie androgenów za-
gromadzony w jądrach, a najwyższe jego stężenia chodzi za pośrednictwem receptorów wewnątrzko-
występują przed południem. Po podaniu doustnym mórkowych. Największe powinowactwo do receptora
testosteron w związku z efektem pierwszego przej- androgenowego, a przede wszystkim najdłuższy czas
ścia nie wywołuje niemal żadnego efektu, możliwe wiązania się z tym receptorem i w wyniku tego najsil-
jest jednak jego podawanie transdermalne. niejsze działanie androgenne charakteryzuje 5α-dihy-
W osoczu testosteron wiąże się w 98% z globuli- drotestosteron. Inne androgeny, przede wszystkim te
ną wiążącą hormony płciowe (SHGB). Biologicznie wytwarzane w korze nadnerczy, wykazują jedynie
Układ hormonalny
aktywny jest tylko wolny testosteron, a jego stężenie niewielkie powinowactwo do receptora androgeno-
obniża się wraz z wiekiem wskutek zwiększonej syn- wego.
tezy SHGB.
Osoczowy okres półtrwania wynosi ok. 10 min.
Metabolizm testosteronu zachodzi głównie w wątro- 2.8.2.4. Androgeny stosowane
bie. Ważne metabolity przedstawiono na ryc. B 2.8-4. w lecznictwie
Niewielka część androgenów ulega także przekształ-
ceniu w estrogeny. Testosteron jest wydalany głównie Testosteron. Dzięki dobrej biodostępności (ok. 10%)
przez nerki w postaci glukuronianów lub półestrów możliwe jest podawanie przezskórne testosteronu B2
kwasu siarkowego. w postaci hydrożeli.
Pochodne testosteronu. Aby otrzymać androge-
2.8.2.3. Działanie androgenów ny dłużej działające lub nadające się do stosowania
doustnego, stworzono estry testosteronu (heptanian
Testosteron (lub jego pochodna 5α-dihydrotestoste-
i undecylan). Po podaniu domięśniowym olejowego
ron):
rozworu undecylanu testosteronu czas działania wy-
■ w okresie płodowym pobudza rozwój męskich na- dłuża się do 3 miesięcy. Możliwe jest również poda-
rządów płciowych, a w okresie dojrzewania po- wanie doustne. Substancje lipofilne nie podlegają wą-
wstawanie drugorzędowych męskich cech płcio- trobowemu efektowi pierwszego przejścia, ponieważ
wych (działanie androgenne), poprzez naczynia limfatyczne dostają się bezpośred-
nio do dużego krążenia. Aby zapewnić wystarczają-
■ utrzymuje u mężczyzn prawidłową czynność narzą-
ce wchłanianie (7%), ważne jest przyjmowanie leku
dów płciowych (razem z FSH reguluje powstawa-
razem z posiłkiem.
nie nasienia, natomiast w dużych stężeniach hamu-
je spermiogenezę),
Wskazania. U mężczyzn androgeny są wskazane jako
■ zwiększa libido oraz potencję, terapia substytucyjna przy niedoborze androgenów
(hipogonadyzmie). W trakcie terapii wzrasta napęd,
■ współdeterminuje psychiczne zachowanie się męż-
siła mięśniowa i libido, a także poprawia się ogólne
czyzny,
samopoczucie.
■ wzmaga syntezę białek i kwasów nukleinowych
(działanie anaboliczne), U kobiet androgeny mogą być stosowane w leczeniu
nieoperacyjnego raka sutka. Przy tych wskazaniach
■ pobudza erytropoezę,
z powodu lepszej tolerancji podaje się jednak przede
■ wzmaga wytwarzanie łoju, wszystkim antyestrogeny i pochodne GnRH.
■ prowadzi – przy określonych uwarunkowaniach
Dawkowanie. Dawkowanie zależy w dużym stop-
genetycznych – do utraty włosów (łysienia andro-
niu od stosowanej substancji, formy jej podawania
gennego),
i wskazania do jej stosowania. Przeciętne dawkowa-
■ nasila przebudowę kości oraz przyspiesza zakoń- nie podano w tab. B 2.8-3.
czenie wzrostu kości na długość w okresie dojrze-
wania. Działania niepożądane. Działania niepożądane
w ścisłym znaczeniu występują rzadko. Wirylizacja
U kobiet wzmożone wytwarzanie androgenów w nad- u kobiet lub atrofia męskich i żeńskich gruczołów
nerczach lub jajnikach, a także dłużej trwające lecze- płciowych – według definicji – nie są działaniami
nie testosteronem prowadzi do wirylizacji. niepożądanymi, lecz objawami działania hormonu.
MUTSCHLER-2009.indd 465 2010-01-07 22:13:12
466 Układ hormonalny
Tabela B 2.8-3. Androgeny
Budowa chemiczna Nazwa Nazwa handlowa Okres Średnie
międzynarodowa półtrwania dawkowanie
(mg)
Androgeny
CH3 OR
CH3 H
H H
O
R
testosteron Androtop Gel, STRIANT, Testim 0,5-2 godz. 25-50
H Gel, Testogel [w Polsce: dziennie
Androtop, Omnadren, Tostran transdermalnie
– przyp. tłum.]
O heptanian testosteronu Testosteron-Depot GALEN, 8 dni 250
CH3
Testosteron-Depot (testosteron co 2-3 tygodnie i.m.
JENAPHARM, Estosteron- jak aktywny
-Depot Rotexmedica, metabolit
Testoviron-Depot zob. powyżej)
[w Polsce: Testosteronum
prolongatum – przyp. tłum.]
O undecylan testosteronu Andriol, Nebido roztwór do 2-3 godzin., doustnie 40-120
wstrzyknięć [w Polsce: Nebido, 8 dni dziennie, 1000
CH3
Undestor – przyp. tłum.] co 10-14 dni i.m.
Antagoniści receptora androgenowego
octan cyproteronu Androcur, Cyproteronacetat 2 dni doustnie 50-300
CH3 COCH3 dura, Cyproteronacetat-GRY, dziennie
OCOCH3
H Virilit [w Polsce: Climen
CH3 H – przyp. tłum.]
H
H H
O
Cl
Inhibitory 5α-reduktazy
O finasteryd PROPECIA, PROSCAR [w Polsce: 5-6 godz. doustnie 1-5
NH Ambulase, Androstatin, dziennie
CH3 Antiprost, Apo-Fina, Aprost,
C(CH3) 3
Finamed, Finaran, Finaride,
CH3 H Finaster, Finasterid-STADA,
Finasterid-ratiopharm, Finpros,
H H Fintral, Finxta, Lifin, Penester,
O NH Propecia, Proscar, Symasteride
H – przyp. tłum.]
CF3 dutasteryd Avodart 3-5 tygodni doustnie
0,5 dziennie
O NH
CH3
F3C
CH3 H
H H
O NH
H
MUTSCHLER-2009.indd 466 2010-01-07 22:13:12
Układ hormonalny 467
Przy leczeniu dużymi dawkami pojawia się spo- dotyczącego receptorów androgenowych. Hamują
radycznie żółtaczka cholestatyczna. W razie długo one libido i spermiogenezę. Pierwszym wprowadzo-
trwającej terapii istnieje niebezpieczeństwo zatrzy- nym do terapii antyandrogenem jest octan cyprote-
mania jonów sodu, potasu, wapnia, chlorków i fosfo- ronu, pochodna progesteronu, która oprócz działania
ranów oraz wody w organizmie. antyandrogennego wykazuje także silne właściwości
gestagenne. W wyniku działania gestagennego octan
Interakcje. Androgeny nasilają działanie przeciw- cyproteronu hamuje uwalnianie LH i przez to rów-
krzepliwe pochodnych kumaryny. nież wytwarzanie testosteronu. Octan cyproteronu
jest stosowany:
Układ hormonalny
Przeciwwskazania. Stosowanie androgenów jest prze-
■ w raku gruczołu krokowego,
ciwwskazane u mężczyzn z rakiem gruczołu krokowe-
go lub piersi oraz u kobiet podczas ciąży. ■ u mężczyzn z nieprawidłowym lub wzmożonym
popędem płciowym.
2.8.2.5. Środki anaboliczne Stosowanie octanu cyproteronu u kobiet jest wskaza-
Przez wprowadzanie zmian w cząsteczce testosteronu pró- ne w leczeniu:
bowano zwiększyć efekt anaboliczny androgenów i jed-
nocześnie zmniejszyć działanie androgenne, aby w ten
■ hirsutyzmu lub łysienia typu androgennego, B2
sposób otrzymać substancje czynne wykazujące głównie ■ ciężkich postaci trądziku pospolitego.
właściwości anaboliczne, tzn. zwiększające syntezę bia-
łek w organizmie. Początkowe nadzieje na uzyskanie ta- Aby zapobiec zajściu w ciążę, podaje się octan cypro-
kiego zróżnicowania działania spełniły się jednak tylko teronu u kobiet z trądzikiem w stałej kombinacji z ety-
częściowo. nyloestradiolem. Poza tym z powodu gestagennego
Działania niepożądane środków anabolicznych odpo-
wiadają działaniom występującym w przypadku androge- działania stosuje się go w hormonalnej terapii zastęp-
nów. Należy przy tym szczególnie zwrócić uwagę, że u ko- czej w okresie menopauzy (zob. rozdz. B 2.8.8).
biet pierwszymi objawami rozpoczynającej się wirylizacji Działanie rozpoczyna się między pierwszym
mogą być nieodwracalne zmiany głosu (mutacja głosu!) a trzecim tygodniem stosowania preparatu, a po jego
oraz pojawienie się męskiego typu owłosienia ciała. Innym odstawieniu zanika całkowicie (jest odwracalne)
typowym działaniem niepożądanym jest trądzik. U dzieci
ulega przyspieszeniu dojrzewanie tkanki kostnej, natomiast (normalizacja spermiogenezy następuje po 4–6 mie-
u chłopców dochodzi do przedwczesnego dojrzewania. siącach).
Przeciwwskazania są takie same jak w przypadku an- U mężczyzn dawkowanie wynosi 100–200 mg,
drogenów. a w przypadku trądziku – 2 mg dziennie.
Stosowanie (niewłaściwe) środków anabolicznych u wy- Jako działania niepożądane na początku leczenia
czynowych sportowców w celu zwiększenia masy mięśnio-
wej (doping) jest zabronione, lecz nadal rozpowszechnione. obserwowano zahamowanie napędu a w niektórych
Również kulturyści dość często nadużywają tych środków. przypadkach ginekomastię. Przy podawaniu dużych
Ponieważ takie stosowanie nie jest prowadzone pod kontro- dawek prawie zawsze występujący spadek libido
lą lekarza, dość częste jest podawanie bardzo dużych dawek stanowi działanie pożądane w przypadku podawania
(przekraczających nawet sto razy typowe dawki) oraz łącze- octanu cyproteronu w celu zmniejszenia nieprawid-
nie lub podawanie po sobie różnych substancji czynnych
(również związków, które nie są przeznaczone do stosowa- łowej lub chorobliwie wzmożonej aktywności seksu-
nia u ludzi). Nie jest dokładnie znane ryzyko z tym związane. alnej, natomiast przy wszystkich innych wskazaniach
Znanym działaniem niepożądanym jest zmniejszenie wiel- jest to działanie niepożądane.
kości jąder, które po odstawieniu preparatów może utrzy- Z powodu działania gestagennego przeciwwskaza-
mywać się przez wiele miesięcy. W przypadku stosowania nia do stosowania antyandrogenów są takie same jak
alkilowanych androgenów mogą dodatkowo wystąpić cięż-
kie zaburzenia czynności wątroby, wzrost liczby płytek krwi, w przypadku gestagenów.
spadek stężenia cholesterolu HDL, a także wzrost stężenia
cholesterolu LDL w osoczu oraz zaburzenia psychiczne. W zaawansowanej postaci raka gruczołu krokowego
zastosowanie znalazły związki działające wyłącznie
antyandrogennie: flutamid i bikalutamid, które opisa-
no w rozdziale B 12.6.4.
2.8.2.6. Antagoniści receptorów
androgenowych (antyandrogeny)
2.8.2.7. Inhibitory 5α-reduktazy
Antagoniści receptorów androgenowych (zob. tab. B
2.8-3) znoszą działanie testosteronu oraz 5α-dihydro- 5α-dihydrotestosteron, jak wspomniano wyżej, działa
testosteronu w wyniku antagonizmu kompetycyjnego wyraźnie silniej niż testosteron na wiele narządów,
MUTSCHLER-2009.indd 467 2010-01-07 22:13:12
468 Układ hormonalny
m.in. gruczoł krokowy i mieszki włosowe, z tego pęcherzyk
względu można uzyskać w tych narządach działa- pierwszorzędowy
nie antyandrogenne w wyniku hamowania enzymu pęcherzyk drugorzędowy
5α-reduktazy (zob. ryc. B 2.8-4). Enzym ten występuje
w postaci dwóch izoenzymów (typ l i 2). W gruczole
krokowym i fibroblastach brodawki włosa dominuje
typ 2, natomiast w nieowłosionej skórze i sebocytach pęcherzyk trzeciorzędowy
typ l. Inhibitory 5α-reduktazy nie upośledzają czyn- komórki osłonki
ności narządów, w których efekty androgenne wywo- wewnętrznej
łuje sam testosteron (np. mięśnie, OUN).
Pierwszym wprowadzonym do terapii inhibitorem
5α-reduktazy (hamującym głównie typ 2 enzymu)
był finasteryd – zastosowany w leczeniu łagodnego
przerostu gruczołu krokowego (BPH). Dutasteryd ciałko żółte
hamuje w równym stopniu obie izoformy 5α-reduk-
tazy.
Z powodu oddziaływania na przemiany metabo-
liczne androgenów finasteryd, poza leczeniem BPH Ryc. 2. 8-5. Schemat faz cyklu miesiączkowego i tworzenia ciał-
w dawce l mg dziennie, nadaje się również do terapii ka żółtego (według Fricka i in.).
łysienia androgennego u mężczyzn. Terapia ta hamuje
postęp łysienia. pęcherzyka Graafa dochodzi do pobudzenia wzrostu
także bardzo wielu (ok. 1000!) pęcherzyków towa-
rzyszących, jednakże ulegają one przedwczesnemu
2.8.3. Żeńskie hormony płciowe zanikowi, tzn. bez pęknięcia (alresia folliculi ovaria-
lis).
2.8.3.1. Podstawy anatomiczne Jajniki są jednocześnie miejscem wytwarzania żeń-
i fizjologiczne skich hormonów płciowych: estrogenów i gestagenu
– progesteronu. Jajniki syntetyzują także niewielkie
ilości androgenów.
Jajniki są parzystym narządem wielkości śliwki o cię-
żarze ok. 10 g, położonym przy bocznej ścianie mied-
nicy mniejszej. W łącznotkankowym zrębie jajników
(stroma ovarii) znajdują się tzw. pęcherzyki jajniko- 2.8.3.2. Budowa chemiczna, biosynteza
we, składające się z komórki jajowej i otaczającego i rozkład estrogenów
ją nabłonka pęcherzykowego. Na podstawie stadium oraz progesteronu
rozwoju wyróżnia się pierwszo-, drugo- i trzeciorzę-
dowe pęcherzyki jajnikowe (Graafa) (zob. ryc. B 2.8- Estrogeny. Biologicznie najbardziej aktywnym hor-
5). W chwili narodzin każdy z obu jajników zawiera monem estrogenowym syntezowanym przez nabłonek
około 500 000 pierwszorzędowych pęcherzyków pęcherzykowy jest estradiol. Poza tym syntezowane
jajnikowych, z których w okresie dojrzewania zacho- są estron i estriol. W niewielkich ilościach estrogeny
wanych jest jeszcze mniej więcej 200 000. Spośród powstają także w jądrach, nadnerczach, gruczołach
nich – w każdym jajniku – dojrzewa tylko około 250. piersiowych, mózgu i kościach, w znikomej ilości
Podczas dojrzewania komórki jajowej aż do stadium są także syntezowane w tkance tłuszczowej.
CH3 OH CH3 O CH3 OH
H H H OH
H H H H H H
HO HO HO
estradiol estron estriol
MUTSCHLER-2009.indd 468 2010-01-07 22:13:12
Układ hormonalny 469
LH FSH
LH FSH
Gs Gs
AC AC
ATP cAMP/PKA ATP cAMP/PKA
Układ hormonalny
cholesterol androstendion
CYP19
testosteron testosteron estradiol
komórka osłonki komórka ziarnista
Ryc. B 2.8-6. Synteza estrogenów w pęcherzykach jajnikowych. Komórki osłonki pod wpływem LH syntezują testosteron, który
następnie w pobliskich komórkach ziarnistych ulega za pomocą aromatazy (CYP19) przekształceniu w estradiol. Ta przemiana jest B2
pobudzana przez FSH. Receptory dla gonadotropin sąpowiazane z białkiem G (GS) i ich stymulacja powoduje wzrost stężenia cAMP
oraz wzrost aktywności kinazy proteinowej A (PKA).
Od innych hormonów steroidowych estrogeny zasadniczo genów. Po menopauzie synteza ich spada do 5–10 μg
różnią się posiadaniem pierścienia aromatycznego A. Przy dziennie.
fenolowej grupie hydroksylowej w pozycji 3 jest możliwe Ponadto jajniki uwalniają niewielkie ilości wolne-
tworzenie się soli i w wyniku tego chemiczne oddzielenie
estrogenów od pozostałych hormonów płciowych. W ten go testosteronu. Poza tym – szczególnie po okresie
sposób w 1929 r. dwóch badaczy, Doisy i Butenandt, jedno- menopauzy – estrogeny powstają także z syntezowa-
cześnie i niezależnie od siebie wyizolowało w formie kry- nego w nadnerczach dehydroepiandrosteronu.
stalicznej pierwszy hormon steroidowy – estron. Estradiol ulega przekształceniu do słabiej działają-
cych estrogenów: estronu i estriolu, a następnie ule-
Synteza żeńskich hormonów płciowych (zob. ryc. ga sprzęganiu z aktywnym kwasem glukuronowym
2.8-4 i ryc. B 2.8-6) zachodzi w komórkach osłonki i siarczanami. Sprzężone estrogeny ulegają częścio-
i komórkach ziarnistych jajników. Pod wpływem hor- wo wydaleniu z żółcią i wchłanianiu zwrotnemu w je-
monu luteinizującego (LH) komórki osłonki nabłonka licie grubym. Po ponownym wchłonięciu estrogeny
pęcherzykowego syntezują testosteron, który w po- podlegają krążeniu wątrobowemu. Poza tym estradiol
bliskich komórkach ziarnistych pod wpływem FSH po podaniu doustnym na skutek efektu pierwszego
ulega za pomocą aromatazy (CYP19) przekształceniu przejścia traci > 90% swojej aktywności. Osoczowy
w estradiol. Także ciałko żółte, które powstaje z resz- okres półtrwania wynosi ok. 50 minut.
tek pęcherzyka, syntezuje estrogeny i progesteron.
U kobiet w wieku rozrodczym dziennie wydziela- Progesteron. Progesteron jest jedynym fizjologicz-
ne jest (w zależności od fazy cyklu) 25–100 μg estro- nym gestagenem, który jako pochodna pregnanu wy-
H3C H3C
OH OH
CH3 CH3
CH3 H CH3 H
H H H H
HO HO
H H
pregnandiol allopregnandiol
MUTSCHLER-2009.indd 469 2010-01-07 22:13:13
470 Układ hormonalny
kazuje duże chemiczne pokrewieństwo do korteksanu mineralokortykosteroidowym i wzmożonym wy-
(różni się od niego tylko brakiem grupy hydroksylo- twarzaniem angiotensynogenu) w organizmie,
wej przy C21; zob. tab. B 2.8-5). Poza jajnikami pro-
■ nasilają syntezę receptorów serotoninowych,
gesteron jest syntetyzowany z cholesterolu w łożysku,
korze nadnerczy i jądrach. Odgrywa on ważną rolę ■ wywierają działanie pobudzające.
jako substancja pośrednia w syntezie hormonów kory
nadnerczy – androgenów i estrogenów. Głównymi Wydaje się, że estrogeny mają drugorzędne znacze-
źródłami progesteronu we krwi są ciałko żółte i łoży- nie dla zachowań seksualnych kobiet.
sko. W drugiej połowie cyklu miesiączkowego ciałko
żółte wydziela około 20 mg progesteronu dziennie. Progesteron może z jednej strony znosić działanie
Podczas ciąży ilość wytwarzanego w łożysku proge- estrogenów, a z drugiej je nasilać.
steronu może wzrosnąć aż do 250 mg dziennie.
Podobnie jak estradiol, po podaniu pozajelitowym Wywołana przez estrogeny wzmożona synteza białek
progesteron działa jedynie krótko, natomiast po po- transportujących (np. globuliny wiążącej hormony
daniu doustnym słabo z powodu efektu pierwszego płciowe i transkortyny) zwiększa wiązanie z nimi
przejścia. Osoczowy okres półtrwania wynosi około hormonów i w wyniku tego dochodzi do słabsze-
5 minut. Progesteron ulega przede wszystkim hydro- go działania androgenów i glukokortykosteroidów.
ksylacji w wątrobie. Jego główny metabolit pregnan- Estrogeny oddziałują także na przemianę materii
diol jest wydalany z moczem w postaci glukuronianu. zachodzącą w wątrobie, nasilając wytwarzanie cho-
Poza tym powstaje allopregnandiol. lesterolu HDL i zmniejszając syntezę cholesterolu
LDL, a także zwiększając wytwarzanie czynników
krzepnięcia (fibrynogenu, czynników VII, VIII, X
2.8.3.3. Działanie estrogenów i XII) przy równoczesnym zmniejszeniu syntezy bia-
łek przeciwkrzepliwych (białko C i S, antytrombina).
Estrogeny, szczególnie w dużych dawkach, nasilają
Estrogeny, tj. przede wszystkim estradiol, są czynni-
krzepnięcie krwi.
kami wzrostowymi, które działają głównie na narządy
płciowe, ale także na inne narządy.
Mechanizm działania. Podobnie jak inne hormony
Estrogeny:
steroidowe, estrogeny działają za pośrednictwem
■ pobudzają wzrost żeńskich narządów płciowych, receptorów wewnątrzkomórkowych. Do łączenia
determinują żeńskie wtórne cechy płciowe, zwięk- z receptorem estrogenowym niezbędna jest obecność
szają podskórne magazynowanie tłuszczów (z wy- pierścienia A oraz grupy hydroksylowej przy węglu
tworzeniem typowej żeńskiej budowy ciała), C-3. Podtypy ERα i ERβ są aktywowanymi ligandem
czynnikami transkrypcyjnymi, które różnią się dome-
■ zwiększają ilość receptorów dla progesteronu
ną wiążącą ligand i domeną transaktywizującą. ERα
i tą drogą umożliwiają działanie gestagenów,
występuje przede wszystkim w macicy, jajnikach,
■ wywołują, razem z gestagenami, cykliczne zmiany gruczołach sutkowych i podwzgórzu, a ERβ w jaj-
błony śluzowej macicy oraz lepkości wydzieliny nikach i tkance kostnej, jak również w gruczole kro-
szyjkowej, kowym, płucach, naczyniach i mózgu. Jeżeli recep-
tory ERα i ERβ występują razem na danej komórce,
■ wpływają na procesy przemiany materii zacho-
to ERβ hamuje większość efektów wywoływanych
dzące w wątrobie (przede wszystkim syntezę bia-
przez ERα.
łek osocza, zob. poniżej), w jelitach (zwiększają
W formie nieaktywnej receptory estrogenowe, tak
wchłanianie wapnia) i w tkance kostnej (pobudzają
jak inne receptory wewnątrzkomórkowe, są stabili-
wbudowywanie wapnia do kości, nasilają wzrost
zowane przez białka szoku cieplnego (HSP). Kom-
kości oraz zarastanie chrząstek przynasadowych
pleksy te rozpadają się pod wpływem nagromadzenia
w okresie dojrzewania),
estrogenów, a aktywowane dzięki temu receptory łą-
■ hamują proliferację sebocytów (zmniejszają pro- czą się jako dimery (ERα-ERα, ERα-ERβ, ERβ-ERβ)
dukcję łoju), z fragmentami odpowiadającymi na estrogeny (ERE)
w regionie promotorowym docelowego genu, wcho-
■ obniżają opór naczyń obwodowych (m.in. poprzez
dzą w interakcje z koaktywatorem lub korepresorem
zwiększenie syntezy NO),
i tą drogą wpływają na ekspresję docelowych genów.
■ stosowane w dużych dawkach powodują zatrzyma- Efekt nasilający lub zmniejszający ekspresję genu
nie chlorku sodu i wody (spowodowane działaniem zależy od obecności w komórce docelowej koakty-
MUTSCHLER-2009.indd 470 2010-01-07 22:13:13
Układ hormonalny 471
watorów lub korepresorów oraz podtypu receptora z aktywnym kwasem glukuronowym i aktywnymi
estrogenowego. Za wywołany efekt odpowiada tak- siarczanami. Metabolitem, który jeszcze znajduje za-
że powinowactwo poszczególnych ligandów do obu stosowanie teraputyczne i powstaje głównie lokalnie
podtypów receptora ER (na przykład fitoestrogeny w rejonie narządów płciowych, jest estriol.
wiążą się w pierwszej kolejności z ERβ) oraz zmiany
konformacji zachodzące pod wpływem poszczegól- Farmakokinetyka estrogenów podawanych do-
nych ligandów (zob. SERM). ustnie i w formie dépôt. Z powodu krótko trwające-
Poza tym, jak w przypadku glukokortykosteroi- go działania estrogenów po podaniu pozajelitowym
dów, obserwuje się wzajemne oddziaływanie z in- i słabego działania po podaniu doustnym opracowa-
Układ hormonalny
nymi czynnikami transkrypcyjnymi (np. AP-1, SP-1, no pochodne estradiolu, które po podaniu doustnym
NF-κB), co umożliwia wpływ estrogenów na ekspre- działają dłużej lub lepiej.
sję genów niemających w regionie promotorowym
fragmentów ERE. Walerianian estradiolu cechuje się dłuższym cza-
Poza receptorami wewnątrzkomórkowymi stwier- sem działania, a podaniu domięśniowym jest po-
dzono w komórkach podlegających regulacji estroge- woli wchłaniany i rozkładany. Można go również
nowej także receptory błonowe połączone z białkiem stosować doustnie, ale wówczas jego biodostępność
G, które odpowiadają za szybkie niegenomowe dzia- jest również mała.
łanie estrogenów. Sygnał jest wówczas przekazywa- Jeżeli przy C17 wprowadzi się grupę etynylową, B2
ny za pomocą kanałów jonowych oraz kinazy MAP. to otrzymuje się etynyloestradiol, który jest wolniej
inaktywowany w wątrobie i dzięki temu po podaniu
doustnym działa skutecznie (biodostępność 25–65%)
2.8.3.4. Środki terapeutyczne zawierające Etynyloestradiol i jego 3-metyloeter, czyli prolek
estrogeny i antyestrogeny mestranol, należą do najczęściej stosowanych estro-
genów doustnych, jednak tylko przed wystąpieniem
2.8.3.4.1. Estrogeny o działaniu nieswoistym menopauzy.
narządowo Sprzężone estrogeny, które często stosuje się do-
ustnie w okresie pomenopauzalnym, wchłaniają się
po dekoniugacji w jelicie grubym.
Jako leki o działaniu estrogenowym (zob. tab. 2.8-4)
poza ludzkimi estrogenami (estradiolem i estronem) Doustnie stosowane są także estrogeny otrzymywane cał-
zastosowanie znalazły: kowicie syntetycznie (np. dietylostylbestrol), które nie po-
siadają struktury sterydowej i wykazują tylko dalekie podo-
■ estry estradiolu, bieństwo do naturalnych hormonów. Wykazują one właści-
■ koniugowane estrogeny wyizolowane z moczu wości farmakologiczne zbliżone do estradiolu i podobnie
jak on, wiążą się z receptorem estrogenowym. Ze względu
klaczy (koński estrogen; siarczan estronu, ekwilin na opisaną grupę przypadków wystąpienia raka narządów
i dihydroekwilin), płciowych u kobiet, których matki w celu podtrzymania
ciąży przy zagrażającym poronieniu otrzymywały diety-
■ pochodne estradiolu (etynyloestradiol, mestranol). lstylobestrol przez dłuższy czas w dużych dawkach, wyco-
fano ten preparaty z użycia.
Różnią się one między sobą częściowo pod wzglę-
dem farmakodynamiki, a przede wszystkim właści-
Wskazania. Stosowanie estrogenów jest wskazane
wości farmakokinetycznych.
w przypadku:
Działanie. O tym, że estron działa słabiej niż estra- ■ hipoplazji macicy i jej następstw (np. zaburzeń
diol, wspomniano już wcześniej. Etynyloestradiol miesiączkowania),
(zob. powyżej) działa silniej niż estradiol na ośrod-
■ niedoboru estrogenów spowodowanego niewydol-
kowe receptory estrogenowe i dzięki temu szczegól-
nością jajników, przede wszystkim po ich usunię-
nie skutecznie hamuje dojrzewanie komórki jajowej
ciu lub kastracji radiologicznej oraz podczas kli-
i wywołaną gonadotropinami owulację.
makterium lub w okresie pomenopauzalnym, jeżeli
występują uciążliwe objawy,
Farmakokinetyka ludzkich estrogenów. Po poda-
niu pozajelitowym w iniekcji osoczowy okres pół- ■ pierwotnego i wtórnego braku miesiączki, przy
trwania estradiolu wynosi około 50 minut. Estradiol czym są one wówczas stosowane w sposób dosto-
i estron są metabolizowane w wątrobie, gdzie pod- sowany do cyklu miesiączkowego i w połączeniu
legają hydroksylacji, dehydrogenacji oraz sprzęganiu z gestagenami,
MUTSCHLER-2009.indd 471 2010-01-07 22:13:13
472 Układ hormonalny
Tabela B.2.8-4. Estrogeny i leki o działaniu estrogenowym
Budowa chemiczna Nazwa Nazwa handlowa Okres Średnie
międzynarodowa półtrwania dawkowanie
(godz.) (mg)
Naturalne estrogeny i ich pochodne
estradiol, TTS Cutanum, Dermestril, 1 0,8-1,6
(transdermalny) Estraderm TTS, Estradot co 3-4 dni
CH3 OH
H estradiol, doustny Estrifam, Estronorm, 1 dziennie
Femoston mono
H H
HO estradiol, donosowy AERODIOL 0,3 dziennie
O
walerianian estradiolu Estradiol JENAPHARM, ok. 13 2 dziennie
CH3
Gynokadin, Merimono,
CH3 O
Progynova [w Polsce:
H Progynova – przyp. tłum.]
H H
HO
CH3 O koniugowane: koniugowane końskie Climopax mono, 0,3-1,25 dziennie
R= siarczan lub estrogeny Presomen
glukuronian (np. ekwilin)
H H ekwilin:
RO R=H
CH3 OH estriol Estriol JENAPHARM, 3 2-4 dziennie
OeKolp, Ovestin, Synpause E
H OH [w Polsce: Oekolp,
Ortho-Gynest, Ovestin
H H – przyp. tłum.]
HO
Syntetyczne estrogeny
CH3 OH etynyloestradiol Ethinyloestradiol 13 0,025-0,1 dziennie
C CH JENAPHARM,
H składnik antykoncepcji
hormonalnej
H H
HO
CH3 OH mestranol Esticia 50 0,05 dziennie
C CH
H
H H
H3CO
Estrogeny swoiste narządowo
CH3 OH tybolon (STEAR) Liviella 6 2,5 dziennie
C CH [w Polsce: Livial,
H Ladybon – przyp. tłum.]
H H
O CH3
S OH raloksyfen (SERM) Evista, Optruma 28 56 dziennie
HO [w Polsce: Evista
– przyp. tłum.]
O
N
O
MUTSCHLER-2009.indd 472 2010-01-07 22:13:14
Układ hormonalny 473
■ pierwotnego (hamowanie laktacji) i wtórnego modulatory receptorów estrogenowych (SERM), któ-
(zmniejszanie laktacji) odstawienia od piersi. re od ww. leków różnią się tym, że wywołują jedynie
część działania estrogenowego, natomiast hamują
Dawkowanie. Dawkowanie ustala się indywidualnie inne ich efekty.
w zależności od wskazań. W terapii substytucyjnej Do tej grupy należy raloksyfen (zob. tab. 2.8-4),
estrogenami, np. w okresie przed- i pomenopauzal- który względnie selektywnie hamuje resorpcję tkanki
nym lub po usunięciu jajników, estradiol można po- kostnej oraz jest stosowany przy rozsianym raku sut-
dawać doustnie (np. 1–2 mg walerianianu estradiolu ka obok tamoksyfenu i toramifenu. Zasadniczo także
dziennie), a także w znacznie mniejszych dawkach estrogeny będące składnikami preparatów roślinnych
Układ hormonalny
(25–100 μg dziennie) – z powodu ominięcia efektu (glikozydy genisteiny i daidzeiny pochodzące z ziaren
pierwszego przejścia – przezskórnie. soi, kumesterol z koniczyny czerwonej i lucerny, fla-
wonoidy chmielu i in.) powinny być zaklasyfikowa-
Działania niepożądane. Estrogeny – w sposób za- ne jako SERM. Jednak w tych przypadkach aktywne
leżny od dawki – zwiększają ryzyko wystąpienia incy- substancje nie zostały dokładnie opisane, ich skład-
dentów zakrzepowo-zatorowych. Z tego powodu przy niki są intensywnie metabolizowane i modyfikowane
zapaleniu żył lub zwiększonym ryzyku wystąpienia w świetle jelita oraz w wątrobie, stosownie do tego,
zakrzepicy należy je natychmiast odstawić. Przy także profil działania nie został jasno zdefiniowany
długo trwającym podawaniu estrogenów dochodzi a skuteczność pozostaje mocno dyskusyjna. B2
do atrofii jajników wskutek hamowania wydzielania
gonadotropin. Inne działania niepożądane to odczu- Naturalne estrogeny i ich pochodne w takim samym
cie bólu i obrzmienie piersi, zwiększenie masy cia- stopniu aktywują wszystkie geny regulowane estro-
ła, nudności, zatrzymanie sodu w z tworzeniem się genami (a antagoniści receptorów estrogenowych
obrzęków (w następstwie działania mineralokortyko- hamują te funkcje), natomiast selektywne modulato-
steroidowego i wzmożonego wytwarzania angioten- ry receptorów estrogenowych pobudzają tylko cześć
synogenu), a także hiperpigmentacja skóry. tych genów. Jest to prawdopodobnie związane z:
Planując leczenie, należy wziąć pod uwagę zwięk-
■ różnym powinowactwem do obu podtypów recepto-
szenie częstości występowania raka endometrium
rów estrogenowych (α i β) lub
w przypadku podawania samych estrogenów w okre-
sie pomenopauzalnym. ■ zróżnicowaną strukturą kompleksów ligand-recep-
tor estrogenowy, szczególnie z różnicami w struk-
Interakcje. Induktory enzymatyczne, zwłaszcza bar- turze domeny transaktywizującej podczas wiązania
biturany, karbamazepina, rifampicyna oraz wyciąg estrogenu i SERM.
z dziurawca osłabiają działanie estrogenów. Przy
jednoczesnym podaniu estrogenów z lekami prze- Kompleksy, składające się z SERM i receptora estro-
ciwcukrzycowymi występuje zmniejszona tolerancja genowego, wiążą się w różnym stopniu z elementa-
glukozy. mi odpowiedzi na estrogeny (ERE). Jeden z genów
aktywowanych przez raloksyfen koduje przykładowo
Przeciwwskazania. Stosowanie estrogenów jest prze- transformujący czynnik wzrostu β (TGF-β) – substan-
ciwwskazane w przypadku hormonozależnych nowo- cję silnie hamującą czynność osteoklastów i w ten
tworów macicy i gruczołu sutkowego, endometriozy, sposób resorpcję tkanki kostnej.
ciężkich zaburzeń funkcji wątroby, idiopatycznej Po podaniu doustnym raloksyfen wprawdzie dobrze
żółtaczki ciężarnych i ciężkiego świądu ciężarnych wchłania się z przewodu pokarmowego, ale wsku-
w wywiadzie, hiperbilirubinemii, chorób zakrzepo- tek efektu pierwszego przejścia jego biodostępność
wo-zatorowych, ciężkiej cukrzycy oraz z trudem da- wynosi jedynie ok. 2%. Substancja czynna jest wy-
jącego się opanować nadciśnienia lub hiperlipidemii. dzielana z żółcią w postaci glukuronianu i po roz-
szczepieniu w dalszych odcinkach jelita wchłania
się ponownie. W następstwie krążenia jelitowo-wą-
2.8.3.4.2. Selektywne modulatory trobowego osoczowy okres półtrwania wynoszący 28
receptorów estrogenowych (SERM) godz. jest stosunkowo długi.
Raloksyfen jest wskazany w profilaktyce osteopo-
Poza opisanymi wyżej działającymi agonistycznie rozy w okresie pomenopauzalnym.
estrogenami pochodzenia naturalnego lub syntetycz- Dawkowanie wynosi 60 mg jeden raz dziennie.
nego oraz antyestrogenami opisanymi w podrozdzia- Przy stosowaniu raloksyfenu może się zwiększyć
le 2.8.3.4.4 (zob. poniżej), istnieją tzw. selektywne częstość występowania uderzeń gorąca. Innym dzia-
MUTSCHLER-2009.indd 473 2010-01-07 22:13:14
474 Układ hormonalny
łaniem niepożądanym – podobnie jak w przypadku Po podaniu doustnym tybolon w dużym stopniu
innych estrogenów – jest zakrzepica żył głębokich. wchłania się z przewodu pokarmowego, a następnie
Z tego powodu stosowanie raloksyfenu jest prze- jest wydalany z żółcią w postaci skoniugowanej.
ciwwskazane w przypadku choroby zakrzepowo-za- Tybolon jest wskazany w zespole niedoboru estro-
torowej. genów, a dawka dobowa wynosi 2,5 mg.
Do częstych działań niepożądanych należą nasile-
nie krwawienia po zranieniach, nadmierne owłosie-
2.8.3.4.3. Tybolon nie i przyrost masy ciała. Wpływ tybolonu na ryzyko
rozwoju raka piersi nie został dokładnie oceniony.
Tybolon (zob. tab. 2.8-4), pochodna 19-nortestostero-
nu, jest przekształcany do metabolitów wykazujących
działanie estrogenowe (3α- i 3β-hydroksytybolon), 2.8.3.4.4. Antyestrogeny
gestagenowe i androgenne (izomer Δ4) (zob. ryc. B
2.8-7). 3-hydroksytybolon hamuje, jako wyraz dzia- Antyestrogeny są substancjami, które znoszą działa-
łania estrogenowego, np. resorpcję kości. Natomiast nia estrogenów. Są to zazwyczaj częściowi antagoni-
w przeciwieństwie do estrogenów nie powoduje on ści, tzn. związki będące już tylko słabymi agonistami
wzrostu gruczołów sutkowych, ponieważ w tkankach receptorów estrogenowych. Do tej grupy należy po-
gruczołu sutkowego sulfotransferazy przekształcają chodna stilbenu – klomifen. Wywołuje on wzmożone
aktywne metabolity na drodze koniugacji z siarcza- uwalnianie gonadoliberyny i w wyniku tego zwięk-
nami w związki nieaktywne. Synteza sulfotransferaz szone wydzielanie gonadotropin. U kobiet z cyklami
w kościach jest mniej nasilona lub są w nich produ- bezowulacyjnymi może to pozwolić na wywołanie
kowane sulfatazy, które na drodze hydrolizy ponow- owulacji. Skutecznego działania można oczekiwać je-
nie aktywują wcześniej skoniugowane z siarczanami dynie w przypadku niewydolności jajników z prawid-
metabolity. Dlatego ten działający selektywnie lek łowym stężeniem gonadotropin, tzn. przy niezmienio-
jest określany jako tkankowo selektywny regulator nej czynności układu podwzgórze-przysadka.
o aktywności estrogenowej (STEAR).
CH3 OH sulfonylotransferaza
C CH pula
H siarczanów
H H
sulfataza
HO CH3
H SD
3α- 3α-hydroksytybolon
CH3 OH CH3 OH
C CH C CH
izomeraza
H H
H H H H
O CH3 O CH3
tybolon 3β izomer Δ4
-H
SD
CH3 OH
C CH
H
H H
HO CH3
3β-hydroksytybolon
Ryc. B 2.8-7. Tkankowospecyficzny metabolizm tybolonu. HSD – dehydrogenaza hydroksysteroidowa.
MUTSCHLER-2009.indd 474 2010-01-07 22:13:14
Układ hormonalny 475
Stwierdzono, że mechanizm działania klomifenu ■ zwiększa lepkość śluzu szyjkowego,
polega na wywoływaniu w podwzgórzu zaniku efektu
■ hamuje uwalnianie LH w przysadce i przez to owu-
ujemnego sprzężenia zwrotnego estrogenów poprzez
lację,
blokadę ich receptorów.
Po podaniu doustnym klomifen wchłania się do- ■ pobudza tworzenie gruczołów w sutkach,
brze. Z powodu krążenia jelitowo-wątrobowego klo-
■ podnosi spoczynkową temperaturę ciała (mniej
mifenu osoczowy okres półtrwania wynosi 5 dni.
więcej o 0,5 C°),
Stosowanie klomifenu jest wskazane w celu wy-
zwolenia owulacji u kobiet z niespełnionym pragnie- ■ jako tzw. hormon ciążowy jest niezbędny do utrzy-
Układ hormonalny
niem zajścia w ciążę z powodu cykli bezowulacyj- mania ciąży (np. dzięki hamowaniu kurczliwości
nych. macicy i krwawień miesiączkowych, zwiększeniu
Dawkowanie wynosi 50 mg dziennie, lek podaje wentylacji u ciężarnych, co prowadzi do lepszego
się w kolejnych dniach od 5 do 9 dnia cyklu. Czas zaopatrzenia płodu w tlen),
trwania leczenia nie powinien przekraczać sześciu
■ wzmaga działanie estrogenów na układ szkieleto-
cykli miesiączkowych. Stosowanie klomifenu powin-
wy.
no być prowadzone pod stałym nadzorem lekarza.
Działania niepożądane to uderzenia gorąca, brak
apetytu, bóle głowy, zaburzenia widzenia i odczucie
W dużych dawkach progesteron działa także katabo- B2
licznie.
napiętych gruczołów sutkowych. Rzadko obserwo-
wano powstanie torbielowatych zmian w jajnikach.
Mechanizm działania. Podobnie jak inne hormony
Poza tym może wzrosnąć częstość występowania
steroidowe, progesteron wzmaga ekspresję genów do-
ciąż mnogich.
celowych. Progesteron łączy się z dwoma podtypami
Stosowanie klomifenu jest przeciwwskazane u ko-
receptorów (receptor progesteronowy α i β) będący-
biet w ciąży, przy ciężkich chorobach wątroby oraz
mi wariantami jednego genu różniącymi się N-koń-
niewyjaśnionych krwawieniach z macicy.
cem. Do wiązania z receptorem niezbędna jest grupa
Czystym antyestrogenem, który znalazł zasto-
ketonowa w pozycji C3. Estrogeny, jak wspomniano,
wanie w terapii raka sutka, jest fulwestrant. W raku
pobudzają tworzenie tych receptorów.
sutka stosowane są także inhibitory aromatazy, które
zmniejszają syntezę estrogenów.
2.8.3.6. Gestageny i antygestageny
jako środki lecznicze
2.8.3.6.1. Gestageny
Progesteron, tak jak estradiol, po podaniu pozajelito-
wym działa krótko, a po podaniu doustnym wywołuje
(H5C2) 2N Cl niewielkie działanie z uwagi na efekt pierwszego przej-
O ścia. Z tego powodu stworzono, poprzez chemiczną
modyfikację, pochodne progesteronu, które mogą być
klomifen
stosowane jako leki (zob. tab. B 2.8-5). Poza opisanym
działaniem gestagenowym, polegającym na pobudza-
niu receptorów progesteronowych, stosowane w lecz-
nictwie gestageny wykazują powinowactwo do recep-
2.8.3.5. Działanie gestagenów torów androgenowych lub mineralokortykosteroido-
(hormonów ciałka żółtego, wych, pobudzając lub hamując te receptory. Z tego
hormonów ciążowych) powodu charakteryzują się one specyficznymi profila-
mi działania (zob. tab. B 2.8-6), co należy uwzględnić,
wybierając gestagen dla konkretnej pacjentki.
Progesteron aktywny tylko u płci żeńskiej:
■ zmniejsza ilość receptorów estrogenowych,
Przedłużenie czasu działania uzyskuje się dzięki hy-
■ hamuje uwarunkowaną działaniem estrogenów droksylacji w pozycji 17, a następnie estryfikacji,
proliferację śluzówki macicy oraz pobudza rozwój natomiast siłę działania zwiększa się przez wprowa-
wydzielniczego endometrium, dzenie wiązania podwójnego między C6 a C7 oraz
MUTSCHLER-2009.indd 475 2010-01-07 22:13:15
476 Układ hormonalny
Tabela B 2.8-5. Gestageny
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnie
międzynarodowa półtrwania dawki
(godz.) dzienne (mg)
Grupa progesteronu
O progesteron Utrogest 0,1 200-300
CH3 CH3 [w Polsce: Luteina,
Progesteronum
CH3 H – przyp. tłum.]
H H
O
O octan chlormadinonu Chlormadinon 36 2-4
CH3 JENAPHARM
CH3
O CH3
CH3 H
O
H H
O
Cl
O octan Clinofem, Depo-Clinovir, 30-60 300-1000
CH3
CH3 medroksyprogesteronu MPA-beta, MPA HEXAL
O CH3 [w Polsce: Provera,
CH3 H Depo-Provera – przyp. tłum.]
O
H H
O
CH3
O dydrogesteron Duphaston 36 10-20
CH3 [w Polsce: Femoston,
CH3
Duphaston – przyp. tłum.]
CH3 H
H H
O
O medrogeston Prothil 13 5-20
CH3
CH3
CH3
CH3 H
H H
O
CH3
O octan megestrolu Megastat 15-30 160
CH3 [w Polsce: Cachexan,
CH3
O CH3 Megace, Gestron, Megalia,
CH3 H Megesin – przyp. tłum.]
O
H H
O
CH3
MUTSCHLER-2009.indd 476 2010-01-07 22:13:15
Układ hormonalny 477
Tabela B 2.8-5. Gestageny (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnie
międzynarodowa półtrwania dawkowanie
(godz.) dzienne (mg)
Pochodne nortestosteronu
O octan noretysteronu Gestakadin, Norethisteron 4-13 1-2 dziennie
JENAPHARM, Primolut-Nor-5
CH3 O CH3 [w Polsce: Primolut-Nor
– przyp. tłum.]
Układ hormonalny
C CH
H H
H H
O
H3C OH lewonorgestrel Duofem, Levogynon, 26 1,5 dziennie
C CH Microlut
[w Polsce: Escapelle, Mirena,
H H Postinor – przyp. tłum.]
H H B2
O
H3C OH etonogestrel Implanon 30 68 co 3 lata
H2C
[w Polsce: Nuva-Ring
C CH – przyp. tłum.]
H H
H H
O
dienogest składnik preparatów 9 2 dziennie
CH3 OH złożonych
H CN
H
O
O drospirenon składnik preparatów 27 3 dziennie
złożonych
O
CH3
CH3 H H
H H H
O H
H
H3C gestoden składnik preparatów 12-15 0,075 dziennie
OH złożonych
C CH
H H
H H
O
O norgestymat składnik preparatów 45 0,25 dziennie
złożonych
H3C O CH3
C CH
H H
H H
HO
N
MUTSCHLER-2009.indd 477 2010-01-07 22:13:15
478 Układ hormonalny
Tabela B 2.8-5. Gestageny (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Średnie
międzynarodowa półtrwania dawkowanie
(godz.) dzienne (mg)
Pochodne nortestosteronu (kontynuacja)
H3C OH norelgestormin składnik preparatów 28 0,15 dziennie
złożonych
C CH
H H
H H
HO
N
Proleki pochodnych nortestosteronu
lynestrenol Orgametril 16 10 dni
CH3 OH
C CH
H H
H H
H3C OH desogestrel Cerazette 30 0,075 dziennie
H2C C CH [w Polsce: Azalia,
Cerazette – przyp. tłum.]
H H
H H
reszty metylowej lub chlorowcowej w pozycji 6. Sto- ■ antykoncepcji,
sowane doustnie pochodne 17α-hydroksyprogestero-
■ hormonalnej terapii zastępczej w okresie pomeno-
nu – octan megestrolu, octan chlormadinonu i przede
pauzalnym,
wszystkim octan cyproteronu – wykazują dodatko-
wo właściwości przeciwandrogenowe. Drospirenon ■ w celu hamowania za pośrednictwem jajników po-
działa zarówno antyandrogenowo, jak i antyminera- porodowych krwawień z macicy,
lokortykosteroidowo.
Stosowane doustnie gestageny otrzymuje się rów- a także w leczeniu:
nież przez wprowadzenie grupy etynylowej przy
■ zaburzeń miesiączkowania,
C17 testosteronu lub 19-nortestosteronu. W wyniku
tego powstają związki wykazujące głównie właści- ■ endometriozy oraz
wości gestagenów, ale często także androgenowe.
■ zaawansowanych postaci raka macicy, sutka i ne-
Do tej grupy substancji należą noretysteron, norge-
rek.
strel czy lewonorgestrel i gestoden. Odpowiednie
3-deoksy pochodne, lynestrenol i dezogestrel, są pro-
Gestageny można także stosować w celu hamowania
lekami noretysteronu i etonogestrelu. (Ugrupowanie
miesiączkowania (np. u kobiet uprawiających sport).
3-ketonowe jest niezbędne do wywołania efektu ges-
Częste w przeszłości podawanie gestagenów przy za-
tagenowego). Szczególnie wyraźne resztkowe działa-
grażającym lub nawykowym poronieniu obecnie nie
nie androgenowe wykazuje danazol.
jest praktykowane.
Dienogest i dydrogesteron działają jak czyste ges-
tageny, tzn. nie wykazują one żadnego istotnego po-
Dawkowanie. Dawki ustala się indywidualnie.
winowactwa do receptora androgenowego.
Działania niepożądane. Przy stosowaniu małych
Wskazania. Ze względu na wpływ na endometrium dawek w sposób dopasowany do cyklu działania nie-
i jajniki gestageny (często w połączeniu z estrogena- pożądane występują rzadko. W razie dłuższego stoso-
mi) są stosowane w: wania gestagenów oprócz hamowania owulacji mogą
MUTSCHLER-2009.indd 478 2010-01-07 22:13:16
Układ hormonalny 479
Tabela B 2.8-6. Działania dodatkowe gestagenów
Gestagen Działanie Działanie Działanie Działanie antymineralo-
antyestrogenowe androgenowe antyandrogenowe kortykosteroidowe
progesteron + – (+) +
octan medroksyprogesteronu + (+) – –
octan chlormadinonu + – + –
octan noretysteronu + + – –
Układ hormonalny
dydrogesteron + – – (+)
medrogeston + – – –
etonogestrel + + – –
lewonorgestrel + + – –
dienogest + – + –
drospirenon + – + +
+ dodatkowe działanie; (+) słabe dodatkowe działanie; – brak dodatkowego działania
B2
się pojawić zaburzenia psychiczne oraz somatyczne, z podstawnikiem przy C11. Przy stosowaniu mife-
np. spadek libido, bóle głowy, nudności, wymioty pristonu podczas ciąży dochodzi w ciągu kilku go-
i bóle wywołane napięciem gruczołów sutkowych. dzin do zwyrodnienia śluzówki macicy i zaburzenia
Zwiększenie masy ciała może wynikać z pobudzania czynności łożyska. Podawanie mifepristonu w pierw-
receptorów mineralokortykosteroidowych. Pochodne szych tygodniach ciąży prowadzi u większości kobiet
testosteronu lub 19-nortestosteronu mogą dodatkowo do poronienia.
wywołać objawy wirylizacji. Z tego powodu mifepriston jest środkiem wyko-
rzystywanym do wywoływania poronienia we wczes-
Interakcje. Induktory enzymatyczne, np. barbiturany, nym okresie ciąży (do 49 dnia). Długi okres półtrwa-
karbamazepina, fenytoina, rifampicyna lub wyciąg nia powoduje, że wystarcza jednorazowe podanie
z dziurawca, przyspieszają biotransformację gestage- 600 mg mifepristonu. Aby jednak uzyskać pewne
nów i w wyniku tego zmniejszają działanie zapobie- działanie, po upływie 48 godz. podaje się dodatkowo
gające zajściu w ciążę. pochodną prostaglandyn (np. sulproston lub mizo-
prostol).
Przeciwwskazania. Stosowanie gestagenów posia- Działania niepożądane to ciężkie krwawienia,
dających właściwości androgenowe jest przeciw- nudności, wymioty, silne bóle brzucha i uczucie zmę-
wskazane w ciąży, ponieważ istnieje niebezpieczeń- czenia.
stwo maskulinizacji płodów żeńskich. Analogicznie
nie wolno stosować w ciąży gestagenów z działaniem
antyandrogenowym z powodu niebezpieczeństwa
feminizacji męskich płodów. Stosowanie wszyst-
kich gestagenów jest przeciwwskazane przy cięż-
kich uszkodzeniach wątroby, hiperbilirubinemii oraz (H3C) 2N
w przypadku choroby zakrzepowo-zatorowej. CH3 OH C CH3
C
H
2.8.3.6.2. Antygestageny H
O
Antygestageny jako antagoniści receptorów proge-
steronu hamują prawidłowy cykl rozwojowy błony mifepriston
śluzowej macicy. Jedyną obecnie dostępną substancją
tego typu jest mifepriston, pochodna noretysteronu
MUTSCHLER-2009.indd 479 2010-01-07 22:13:16
480 Układ hormonalny
2.8.4. Cykl jajnikowy i maciczny ny, jak i zmiana odstępów między pulsami prowadzi
do zmniejszonego uwalniania FSH i LH z przysadki.
Należy także pamiętać o tym, że cykl miesiączkowy
Cykl jajnikowy. W powtarzającym się co miesiąc cy-
nie podlega wyłącznie regulacji hormonalnej, lecz
klu u dojrzałej płciowo kobiety biorą udział podwzgó-
jest sprzężony ze złożonymi czynnościami ośrodko-
rze, przysadka, jajniki i macica. Hormon uwalniający
wego układu nerwowego. Oznacza to, że na płodność
gonadotropinę powoduje uwalnianie hormonu foliku-
mogą wywierać wpływ także czynniki psychiczne.
lotropowego (FSH, folitropiny) i hormonu luteinizują-
cego (LH, lutropiny) z przysadki (zob. ryc. B 2.8-1).
Cykliczne zmiany macicy (cykl miesiączkowy, cykl
Pod wpływem działania FSH dojrzewa, jak wspomnia-
maciczny). Trwający średnio 28 dni cykl miesiączko-
no, grupa pęcherzyków jajnikowych, które wraz z po-
wy z pojawiającymi się okresowymi zmianami w en-
stępującym dojrzewaniem wytwarzają coraz więcej
dometrium dzieli się na trzy fazy:
estrogenów, zwłaszcza estradiolu (zob. ryc. B 2.8-8).
Estrogeny powodują wzmożone tworzenie receptorów ■ fazę złuszczania i procesów naprawczych (1–4
FSH na powierzchni pęcherzyków jajnikowych, któ- dzień),
rych pobudzanie wzmaga dojrzewanie pęcherzyków.
■ fazę proliferacyjną (5–14 dzień),
W końcu pęcherzyk posiadający największą liczbę
receptorów zostaje wybrany do pełnego dojrzewania: ■ fazę wydzielniczą (15–28 dzień).
zwiększona synteza estrogenów oraz inhibina wydzie-
lana przez dominujące pęcherzyki powodują ustanie Inwolucja ciałka żółtego wraz ze spadkiem stężenia
wytwarzania FSH, w wyniku czego zmniejsza się progesteronu na końcu cyklu prowadzi zazwyczaj
stymulacja pęcherzyków towarzyszących. Poza tym w 26 i 27 dniu cyklu do wzmożonego wytwarzania
wskutek zmniejszonej aktywności aromatazy w pę- prostaglandyn. Wywołuje to skurcz spiralnie prze-
cherzykach towarzyszących gromadzą się androgeny, biegających tętnic w błonie śluzowej macicy. Począt-
które doprowadzają do ich inwolucji.
Interesujące jest to, że w środku cyklu wraz
ze wzrastającym stężeniem estrogenów nie zmniej-
A
sza się, lecz wzrasta wydzielanie GnRH, LH i później
hormon
także FSH; tzn. w tej fazie jest hamowane ujemne folikulotropowy hormon
sprzężenie zwrotne, a pojawia się dodatnie sprzęże- luteinizujący
nie zwrotne ze szczytem wydzielania LH wywołują-
cym jajeczkowanie (owulację, tj. pęknięcie dojrzałe- owulacja
go pęcherzyka z wydaleniem komórki jajowej). 5 10 15 20 25
Po owulacji pęknięty pęcherzyk ulega przekształ-
ceniu w ciałko żółte, które rozpoczyna wytwarzanie B
progesteronu, równocześnie zmniejsza się stęże-
nie estradiolu. Pod wpływem nasilonego ujemnego
sprzężenia zwrotnego stężenie LH i FSH obniża się C faza pęcherzykowa faza lutealna
z powrotem do wartości podstawowych.
progesteron
Hormonalny mechanizm sprzężenia zwrotnego za- estradiol
pobiega wystąpieniu kolejnej owulacji w drugiej po-
łowie cyklu miesiączkowego, a także w czasie ciąży
i w ten sposób ponownemu zapłodnieniu.
5 10 15 20 25
Decydujące znaczenie, a dla prawidłowego prze-
biegu cyklu miesiączkowego, a przez to dla płodności
D błona śluzowa macicy miesiączka
kobiety ma wspomniane wczesniej pulsacyjne uwal-
nianie GnRH. W pierwszej połowie cyklu (aż do owu-
lacji), tzn. pod wpływem estrogenów, odstęp pomiędzy
pulsami wynosi ok. 90 minut, natomiast w drugiej po- 5 10 15 20 25 dzień
cyklu
lowie cyklu już głównie pod wpływem gestagenów –
2–4 godz. Na podstawie badań na zwierzętach, ale tak- Ryc. B 2.8-8. Cykliczne zmiany stężenia gonadotropin (A) i za-
że (leczniczego) stosowania GnRH u kobiet, np. przy leżne od tego działania wpływające na czynnościowy stan pę-
leczeniu bezpłodności, wykazano, że zarówno ciągłe cherzyków (B), stężenie hormonów płciowych (C) oraz prolifera-
dostarczanie hormonu uwalniającego gonadotropi- cję i różnicowanie błony śłuzowej macicy (D).
MUTSCHLER-2009.indd 480 2010-01-07 22:13:17
Układ hormonalny 481
kowo powoduje to zmniejszone ukrwienie i później w endometrium przy udziale enzymów proteolitycz-
jej niedokrwienne uszkodzenie. Jeżeli podczas słab- nych (zagnieżdżenie) i przekształca się w zarodek.
nącego skurczu tętnic wpłynie do nich ponownie Zarodek jest odżywiany przez łożysko, które jedno-
duża ilość krwi, to w obrębie tych tętnic dochodzi cześnie przejmuje funkcję regulacji hormonalnej.
do krwawień do otaczających tkanek lub jamy maci- Funkcję tę pełnią dwa hormony gonadotropina łoży-
cy. W końcu uszkodzenie śluzowki prowadzi do wy- skowa (HCG – ludzka gonadotropina kosmówkowa)
dalenia jej fragmentów zmieszanych z krwią (mie- wytwarzana głównie w czasie pierwszych miesięcy
siączka, menstruacja). ciąży oraz mammotropina łożyskowa (CS – somato-
Przyjmuje się, że pierwszym dniem nowego cyklu mammotropina łożyskowa) wpływająca m.in. na gru-
Układ hormonalny
miesiączkowego jest dzień, w którym rozpoczyna czoły sutkowe (zob poniżej).
się krwawienie miesiączkowe. Podczas fazy złusz- Pod wpływem hormonów łożyska dochodzi do po-
czania krzepnięcie krwi jest miejscowo zmniejszo- większenia ciałka żółtego, które w pierwszych mie-
ne. Przy jej końcu następuje regeneracja nabłonka siącach ciąży zajmuje znaczną część jajnika Stężenia
i tkanki łącznej oraz zostaje zamknięta rana (faza estrogenów i progesteronu pozostają wysokie. Dzięki
naprawcza). temu endometrium nie zostaje wydalone, nie pojawia
W czasie rozpoczynającej się potem fazy proli- się krwawienie miesięczne. Pod koniec pierwszego
feracyjnej dochodzi do odbudowy błony śluzowej trymestru ciąży następuje inwolucja ciałka żółte-
macicy. Pod wpływem zwiększonego stężenia estro- go. Od tego momentu łożysko przejmuje wytwarza- B2
genów powstają nowe naczynia, które wnikają w ślu- nie estrogenów i progesteronu, które są potrzebne
zówkę oraz wraz ze wzrastającą grubością endome- do utrzymania ciąży.
trium wytwarzane są ponownie wydłużone gruczoły, Różne czynniki hormonalne są uważane za wyzwa-
mające później nieco kręty przebieg. Struktura tkanki lające proces porodu, m.in. spadek stężenia progeste-
staje się luźniejsza. ronu i uwarunkowany tym wzrost stosunku estroge-
Po pęknięciu pęcherzyka rozpoczyna się faza wy- nów do gestagenów, wzmożone tworzenie połączeń
dzielnicza. Równolegle ze wzrastającym stężeniem szczelinowatych (gap junctions) między komórkami
progesteronu błona śluzowa macicy przekształca mięśniowymi macicy pod wpływem estrogenów,
się w nabłonek wykazujący zdolność wydzielania zwiększone uwalnianie oksytocyny oraz wzmożona
i nadający się do zagnieżdżenia zarodka. Pod koniec synteza receptorów dla oksytocyny i prostaglandyn.
fazy wydzielniczej gruczoły są silnie skręcone i za- Dotychczas nie wyjaśniono, który z wymienionych
wierają śluzową wydzielinę; struktura tkanki łącz- czynników odgrywa decydującą rolę.
nej jest rozpulchniona. Błona śluzowa jest wówczas Laktacja (wytwarzanie i wydzielanie mleka przez
silnie ukrwiona i osiąga grubość 6–8 mm. Komórki gruczoły piersiowe) podlega również regulacji hor-
tkanki łącznej powiększają się i zawierają duże ilości monalnej. Podczas ciąży pod wpływem estrogenów
glikogenu. Pod koniec fazy wydzielniczej nabłonek i progesteronu oraz współdziałania mammotropiny
powierzchniowy posiada miejscami urzęsienie. Jeśli łożyskowej są tworzone dystalne pęcherzyki gruczo-
nie dochodzi do zapłodnienia, inwolucja ciałka żółte- łowe i płaciki gruczołu sutkowego. Rozpoczynające
go wyzwala następne krwawienie miesiączkowe. się po porodzie wytwarzanie mleka jest pobudzane
Śluzówka szyjki macicy tylko w niewielkim zakresie przez prolaktynę. Wydzielanie mleka, wyzwalane bez-
bierze udział w cyklicznych zmianach. Znaczenie ma pośrednio przez bodziec w postaci ssania, jest możli-
jednak to, że w przebiegu cyklu ulega zmianie kon- we przy udziale oksytocyny.
systencja śluzu szyjkowego. Tuż przed owulacja pod
wpływem estrogenów śluz szyjkowy ulega upłynnie-
niu, „daje się prząść”, tzn. można go rozciągać w dłu- 2.8.6. Schorzenia
gie nitki i plemniki z łatwością go przenikają. Pod i zaburzenia ginekologiczne
wpływem progesteronu w 2 fazie cyklu śluz staje się
ponownie lepki i utrudnia przechodzenie plemników.
Dysmenorrhoea. Termin miesiączkowanie bolesne
oznacza, że u kobiety podczas miesiączki poważ-
nie pogarsza się samopoczucie. Wyróżnia się postać
2.8.5. Hormonalna regulacja ciąży, pierwotną i wtórną. W postaci pierwotnej krwawie-
porodu i laktacji nia miesięczne są bolesne od samego początku, na-
tomiast przy wtórnym bolesnym miesiączkowaniu
Jeżeli komórka jajowa zostanie zapłodniona, to bla- bolesność występuje dopiero w późniejszych latach.
stocysta po początkowych podziałach „zakopuje się” Miejscowe dolegliwości to rwące bóle w okolicy lę-
MUTSCHLER-2009.indd 481 2010-01-07 22:13:17
482 Układ hormonalny
dźwiowej lub w podbrzuszu, które mogą nasilić się ■ objawów naczynioruchowych (uderzenia gorąca,
i przejść w ciężkie kolki. Występuje także ogólne złe tachykardia) i innych objawów wegetatywnych (za-
samopoczucie, brak apetytu, bóle głowy, drażliwość, wroty głowy, pocenie się),
kołatanie serca.
■ zazwyczaj odwracalnych objawów psychicznych
Przyczynami bolesnego miesiączkowania mogą
(uczucie lęku, nastrój depresyjny),
być: endometrioza, stany zapalne lub guzy macicy
(mechaniczne bolesne miesiączkowanie), przedmie- ■ zaburzeń metabolicznych (osteoporoza, hiperli-
siączkowe rozluźnienie mięśni miednicy (bolesne poproteinemia, atrofia skóry i błon śluzowych
miesiączkowanie uwarunkowane zmianami statycz- np. w obrębie narządów płciowych).
nymi) lub czynniki psychiczne, np. niespełnione
pragnienie urodzenia dziecka (psychicznie uwarun-
kowane bolesne miesiączkowanie). 2.8.7. Hormonalne środki
antykoncepcyjne
Amenorrhoea. Termin brak miesiączki oznacza nie-
wystąpienie krwawienia miesięcznego. Pierwotny Możliwość zapobiegania ciąży istniała także przed
brak miesiączki to brak rozpoczęcia miesiączkowa- wprowadzeniem hormonalnych środków antykon-
nia (pierwszej miesiączki) u kobiety, która ukończyła cepcyjnych. W ich przypadku nowość stanowi to,
18 rok życia. O wtórnym braku miesiączki mówi się, że zamiast używania miejscowych mechanicznych
gdy po początkowym rozpoczęciu miesiączkowania lub plemnikobójczych środków w celu zapobiegania
miesiączka nie występuje przez dłuższy czas. Powin- zajściu w ciążę zaczęto stosować żeńskie hormony
no się rozróżniać brak miesiączki fizjologiczny i wy- płciowe, podawane zazwyczaj doustnie, częściowo
wołany zaburzeniami czynnościowymi. Fizjologiczny również domacicznie lub domięśniowo. Z powodu
brak miesiączki występuje podczas ciąży, połogu dużej niezawodności hormonalna antykoncepcja stała
i laktacji. się jedną z najważniejszych metod kontroli zachodze-
Najczęstszą przyczyną czynnościowego braku nia w ciążę (zob. tab. B 2.8-7 i ryc. B. 2.8-9). Prepara-
miesiączki jest niewydolność jajników. Może to być ty te zawierają kombinację estrogenów i gestagenów
zaburzenie zarówno układu podwzgórze-przysadka, lub tylko gestageny.
jak i samych jajników. Najważniejszą przyczyną jaj-
nikową braku miesiączki jest nadmierna jajnikowa Metoda jednofazowa. W przypadku metody jedno-
produkcja androgenów z powstaniem policyklicz- fazowej przez 21 dni przyjmuje się kombinację estro-
nych jajników, jak również trądziku, hirsutyzmu i ły- genu z gestagenem (tzw. klasyczny preparat złożony),
sienia. Kobiety są wówczas często niepłodne. po czym następuje 7 dni przerwy. 3–4 dni po odsta-
wieniu preparatu występuje krwawienie (krwawienie
Klimakterium. W okresie klimakterium, tzw. okresie z odstawienia hormonów), które odpowiada mniej
przekwitania, czyli fazie przejściowej między pełną więcej krwawieniu miesiączkowemu. Popecane są re-
dojrzałością płciową a starością, gruczoły płciowe paraty zawierające małe dawki estrogenów (< 50 μg,
wygaszają swoją czynność. Następuje to przeciętnie tzw. „mikropigułki”).
w wieku 51±3 lat. Najpierw krwawienia miesięczne
stają się nieregularne i mniej obfite, nie występują Metoda dwufazowa. W przypadku metody dwufa-
owulacje i tworzenie ciałek żółtych. Wraz ze spad- zowej w pierwszej fazie cyklu podaje się tylko estro-
kiem stężenia estrogenów i gestagenów na okres kil- geny lub estrogeny razem z małą dawką gestagenu,
ku lat wzrasta znacznie wydzielanie gonadotropin. natomiast w drugiej fazie typową kombinację estro-
Termin menopauza oznacza, czas wystąpienia ostat- genu z gestagenem. W pierwszym przypadku mówi
niej miesiączki, okres przedmenopauzalny to okres się o preparatach sekwencyjnych, a w drugim o dwu-
trzech lat poprzedzających wystąpienie menopauzy, stopniowych.
a okres pomenopauzalny to okres siedmiu lat nastę-
pujących po menopauzie. Metoda trójfazowa. W przypadku tej metody prepa-
Chociaż utrata funkcji przez żeńskie gruczoły raty są jeszcze bardziej niż przy metodzie dwufazo-
płciowe jest procesem fizjologicznym, u około 2/3 wej dostosowane do cyklu miesiączkowego. Odpo-
kobiet pojawiają się tzw. klimakteryczne objawy wy- wiednio do zmian stężenia hormonów w przebiegu
padowe. Są one uwarunkowane przede wszystkim cyklu tabletki przeznaczone do przyjmowania w cza-
zmniejszeniem stężenia estrogenów Przyjmują one sie pierwszych sześciu dni po rozpoczęciu krwawie-
postać: nia miesiączkowego zawierają małą dawkę estrogenu
MUTSCHLER-2009.indd 482 2010-01-07 22:13:17
Układ hormonalny 483
Tabela B 2.8-7. Doustne środki antykoncepcyjne
Estrogen (μg)* Gestagen (mg)* Preparaty handlowe
Preparaty zawierające gestageny
– lewonorgestrel (0,03), minipigułka Microlut, 28 mini
– desogestrel (0,075) Cerazette
Układ hormonalny
– etonogestrel (68 mg); implant podskórny Implanon
– octan medroksyprogesteronu (150 mg); i.m. Depo-Clinovir
– enantan noretysteronu (200 mg); i.m. Noristerat
Preparaty jednofazowe
etynyloestradiol (20-35); noretysteron (0,5-1) Conceplan M, EVE20
minipigułka
norestymat (0,25) Cilest
B2
lewonorgestrel (0,1-0,15) Femigoa, Leios, Microgynon, Miranova
lynestrenol (0,75) Ovoresta M
desogestrel (0,15) LAMUNA, Lovelle, Marvelon
dienogest (2) Valette
gestoden (0,075) Femovan, Minulet
octan chlormadinonu (2) Belara
drospirenon (3) Petibelle, Yasmin
etynyloestradiol (50) lewonorgestrel (0,125) Gravistat
Preparaty dwufazowe
etynyloestradiol (40-50) desogestrel (0,025/0,125) Biviol, Oviol
octan chlormadinonu (1/2) Neo-Eunomin
Preparaty trójfazowe
etynyloestradiol lewonorgestrel (0,05/0,075/0,125) NovaStep, Trigoa, Triquilar, Trisiston
(30-40-30/35-30-30)
noretysteron (0,5/0,75/1) Synphasec, Trinovum
desogestrel (0,05/0,1/0,15) Novial
norgestymat (0,18/0,215/0,25) Pramino
Preparaty transdermalne
etynyloestradiol (600/tydzień) norelgestromin (6/tydzień) EVRA
Krążki dopochwowe
etynyloestradiol (2700/3 tygodnie) etonogestrel (11,7/ 3 tygodnie) NuvaRing
Antykoncepcja ratunkowa (po stosunku)
– lewonorgestrel (1,5 jednorazowo) Duofem, Levogynon
* Dawki dzienne, chyba że podawany inaczej
MUTSCHLER-2009.indd 483 2010-01-07 22:13:18
484 Układ hormonalny
czyste preparaty gestagenowe:
minipigułki, implanty
podskórne
preparaty trójfazowe
preparaty dwufazowe
preparaty sekwencyjne
preparaty jednofazowe
(doustne, transdermalne)
0 5 10 15 20 25 30 35 40
dni cyklu
miesiączka przerwa w podawaniu
z krwawieniem z odstawienia
estrogen gestagen
Ryc. B 2.8-9. Schemat składu i sposobu podawania środków antykoncepcyjnych.
i gestagenu, przeznaczone do przyjmowania w czasie podawane są w sposób ciągły. Na przykład Microlut
następnych 5 dni większą dawkę estrogenu i gesta- zawiera lewonorgestrel (dawkowanie 0,03 mg).
genu, natomiast na okres pozostałych 10 dni dawki W przypadku tzw. zastrzyku trzymiesięcznego po-
estrogenu są zmniejszone do wartości początkowej daje się 150 mg octanu medroksyprogesteronu lub
i ponownie jest zwiększona dawka gestagenu. 200 mg enantanu noretysteronu domięśniowo co 3
miesiące. Implanty podskórne zawierające etonoge-
System transdermalny. Dostępne są już plastry za- strel uwalniają substancję czynną w ten sposób, że za-
wierające etynyloestradiol i norelgestromin (aktyw- pewniają one ochronę antykoncepcyjną przez 3 lata.
ny metabolit norgestimatu), które dziennie uwalniają
ilość hormonów (20 μg etynyloestradiolu i 150 μg Antykoncepcja wewnątrzmaciczna. Poza wymie-
gestagenu), która zabezpiecza przed zajściem w cią- nionymi preparatami jako środki antykoncepcyjne
żę. Plastry zmnienia się co 7 dni, a po 21 dniach, jak stosuje się także spirale wewnątrzmaciczne zawiera-
w przypadku antykoncepcji doustnej, następuje 7 dni jące gestageny, które uwalniają przez dłuższy okres
przerwy. Skuteczność jest porównywalna z doustny- małe ilości gestagenów. Mirena jest preparatem za-
mi środkami antykoncepcyjnymi. wierającym lewonorgestrel, który zgodnie z nowy-
mi danymi charakteryzuje się znacznym ryzykiem
Pierścień dopochwowy. Pierścień dopochwowy za- niektórych powikłań (perforacja macicy, zwiększona
wierający etynyloestradiol i etonogestrel, który nale- częstość raka piersi, ciąża ektopowa). W starszych
ży założyć przed upływem 5. dnia cyklu i pozostawić systemach środkiem antykoncepcyjnym była miedź
w pochwie na 21 dni, działa równie skutecznie jak do- elementarna.
ustne preparaty jednofazowe. Uwalniana ilość hormo-
nów (15 μg etynyloestradiolu i 120 μg etonogestrelu Środki antykoncepcyjne stosowane po stosunku
dziennie) jest wystarczająca przy stabilnych cyklach. płciowym. W przypadku metod antykoncepcyjnych
stosowanych po stosunku płciowym („pigułka po”),
Preparaty gestagenowe. Antykoncepcja jest tak- przyjmuje się dwukrotnie (750 μg) lub jednorazo-
że możliwa przy podawaniu tylko gestagenów (zob. wo (1500 μg) preparat zawierający lewonorgestrel
tab. B 2.8-7). Termin minipigułka oznacza prepara- po 12 godzniach, ale nie później niż po 72 godzinach,
ty zawierające gestageny w małych dawkach, które od stosunku. Metoda ta nie nadaje się jednak do sto-
MUTSCHLER-2009.indd 484 2010-01-07 22:13:18
Układ hormonalny 485
sowania rutynowego – użycie jej jest ograniczone dobowej 30 μg etynyloestradiolu. Przy podawaniu
do wyjątkowych przypadków. doustnym skuteczna w zapobieganiu plamieniom
śródcyklicznym, tzn. niestabilności cyklu, jest prze-
Mechanizm działania i skuteczność. Preparaty jed- ciętna dawka 20 μg etynyloestradiolu. U kobiet przyj-
nofazowe (doustne, transdermalne, dopochwowe): mujących przez dłuższy czas hormonalne środki an-
tykoncepcyjne może rozwinąć się nadciśnienie. Pale-
■ hamują owulację w wyniku efektu przeciwgonado-
nie papierosów oraz zaawansowany wiek zwiększają
tropowego,
ryzyko powikłań sercowo-naczyniowych.
■ zapobiegają, jeżeli pomimo ich stosowania doj- Działań niepożądanych związanych z efektem an-
Układ hormonalny
dzie do owulacji, zagnieżdżeniu się zapłodnionej drogennym (łojotok, trądzik) można uniknąć, stosu-
komórki jajowej nawet po owulacji (nie dochodzi jąc preparaty zawierające gestageny niewykazujące
do pełnego wydzielniczego przekształcenia się en- powinowactwa do receptorów androgenowych lub
dometrium), o działaniu antyandrogenowym. Przyrost masy cia-
ła jest, np. przy stosowaniu drospirenonu, niewielki
■ hamują zdolność penetracji plemników w wyniku
(zob. tab. B 2.8-6). Zazwyczaj działania niepożądane
zwiększenia lepkości śluzu szyjkowego.
występują w większym zakresie tylko przy pierw-
szym zastosowaniu preparatu. Dotyczy to także nie-
Działanie preparatów sekwencyjnych polega głównie
prawidłowości cyklu. B2
na hamowaniu owulacji. Ze względu na to, że nie wy-
Innym działaniem niepożądanym hormonalnych
kazują oddziaływania na śluz szyjkowy, nie są one
środków antykoncepcyjnych jest zmniejszenie tole-
tak skuteczne jak (klasyczne) preparaty złożone. Pre-
rancji glukozy. Wymienione preparaty nie wykazu-
paraty dwu- i trójfazowe pod względem skuteczności
ją działania kancerogennego. Jednak podobnie jak
odpowiadają natomiast w przybliżeniu preparatom
wszystkich leków zawierających estrogeny, prepara-
jednofazowym. Dotyczy to także podawanych poza-
tów tych nie wolno stosować u pacjentek z nowotwo-
jelitowo gestagenów i gestagenów podawanych do-
rem zależnym od estrogenów z powodu ich działania
ustnie w dużych dawkach.
ułatwiającego wzrost tych nowotworów.
Działanie antykoncepcyjne minipigułki polega
Przy stosowaniu doustnym preparatów zawierają-
głównie na zwiększeniu lepkości śluzu szyjkowe-
cych tylko gestageny rzadziej występują ogólne dzia-
go, natomiast nie jest hamowana owulacja. W wy-
łania niepożądane. Często jednak dochodzi do zabu-
niku tego wskaźnik nieskuteczności (zob. poniżej)
rzenia stabilności cyklu. Mogą wystąpić dodatkowe
jest znacznie wyższy niż w przypadku preparatów
krwawienia pomiędzy krwawieniami miesiączkowy-
jedno- lub dwufazowych.
mi, odchylenia pod względem nasilenia miesiączki
Środki wewnątrzmaciczne zapobiegają zapłodnie-
czy czasu jej trwania. Może się też zdarzyć, że mie-
niu także przez zagęszczenie śluzu szyjkowego, a po-
siączka nie wystąpi. Przy środkach wewnątrzmacicz-
nadto zaburzają one strukturę endometrium. Działają
nych także dochodzi do zmniejszenia się nasilenia
miejscowo, a ich skuteczność jest duża.
miesiączki, a czasem do przerwania miesiączkowania.
Środki antykoncepcyjne stosowane po stosunku
płciowym działają także przede wszystkim hamująco
Po stosowaniu postkoitalnych środków antykoncep-
na owulację i zagnieżdżenie jaja płodowego. Wskaź-
cyjnych jako działanie niepożądane często pojawia-
nik nieskuteczności wynosi ok. 3%.
ją się nudności i wymioty, bóle żołądkowo-jelitowe,
Wskaźnik Pearla, według definicji, oznacza liczbę ciąż a także krwawienia niezależne od czasem opóźnionej
(„odsetek niepowodzeń”) wśród 100 kobiet stosujących miesiączki. Innymi częstymi działaniami niepożąda-
daną metodę antykoncepcyjną w ciągu jednego roku. nymi są bóle i zawroty głowy.
W badaniu obejmującym > 20 000 kobiet, które
Działania niepożądane. Zaburzenia samopoczucia przyjmowały doustne środki antykoncepcyjne (część
(osłabienie libido, zmęczenie, nerwowość, nudności, z nich ponad 25 lat) wykazano, że całkowita śmiertel-
wymioty, odczucie napięcia w gruczołach sutkowych ność w tej grupie nie była większa niż w grupie kobiet
występują bardzo rzadko przy stosowaniu nowoczes- niestosujących antykoncepcji hormonalnej. Analiza
nych preparatów kombinowanych estrogenowo-ges- regularnie przeprowadzanych w Niemczech ankiet
tagenowych zawierających < 50 μg etynyloestradiolu. dotyczących zdrowia wykazała, że kobiety przyjmu-
Przede wszystkim jednak wprowadzenie ich ograni- jące doustne środki antykoncepcyjne charakteryzują
czyło zależne od dawki ryzyko powikłań zakrzepo- się lepszym stanem zdrowia, szczególnie pod wzglę-
wych, które istotnie wzrasta po przekroczeniu dawki dem poziomu żelaza, niż dobrana grupa kontrolna.
MUTSCHLER-2009.indd 485 2010-01-07 22:13:19
486 Układ hormonalny
Tabela B 2.8-8. Środki antykoncepcyjne
Metoda Zalety Ważne działania Skuteczność jest Uwagi
niepożądane zmniejszana przez…
doustne środki anty- odwracalne, pewne często zakrzepica, szczególnie induktory enzymatyczne, przeciwwskazane
koncepcyjne; szczególnie (wskaźnik Pearla 0,1-0,9), przy nadciśnieniu i paleniu np. karbamazepinę, fenobarbital, przy chorobach sercowo-
< 50 μg etynyloestradiolu dobra kontrola cyklu tytoniu (ryzyko bardzo wzrasta primidon, fenytoinę, -naczyniowych oraz ich
(mikropigułki) przy 50 μg etynyloestradiolu) rifampicynę, wyciąg czynnikach ryzyka,
z dziurawca; także a także raku sutka
cholestyraminę, penicyliny przed mniej niż 5 laty
o szerokim zakresie
i tetracykliny
plastry hormonalne stałe niskie stężenie często zakrzepica, jak mikropigułka jak mikropigułka
hormonów, zmieniane podrażnienie skóry poza penicylinami
co tydzień, dobra przez plaster o szerokim zakresie
kontrola cyklu
(wskaźnik Pearla 0,9)
pierścień dopochwowy zakładany raz w miesiącu, często zakrzepica, jak mikropigułka jak mikropigułka
dobra kontrola cyklu trądzik poza penicylinami
(wskaźnik Pearla 0,65) i tetracyklinami
minipigułki małe dawki gestagenów; ryzyko zakrzepicy nieznacznie ograniczona skuteczność szczególnie dla kobiet
może być stosowana podwyższone, (wskaźnik Pearla 0,5-3), nietolerujących
przy karmieniu piersią zła kontrola cyklu szczególnie gdy błąd czasu estrogenów
i przy przeciwwskazaniach podania > 3 godz.
dla estrogenów
gestageny odwracalne i pewne zwiększone ryzyko zakrzepicy,
(duże dawki) (wskaźnik Pearla 0,14); zaburzenia krwawienia,
można stosować w czasie trądzik
karmienia piersią
gestageny zapobiegają ciąży przez nie zwiększają ryzyka alternatywa, gdy nie jest
w formie dépôt 3 miesiące zakrzepicy, spadek gęstości możliwe stosowanie
(zastrzyki trzymiesięczne) (wskaźnik Pearla 0,88), mineralnej kości przy octanie innych metod
brak problemów medroksyprogesteronu
z przestrzeganiem
zaleceń, brak miesiączki
i związanych
z nią dolegliwości
hormonalne implanty pewna ochrona przez zwięszone ryzyko zakrzepicy,
podskórne 3 lata często krwawienia śródcykliczne,
(wskaźnik Pearla < 0,1) trądzik
spirala hormonalna odwracalna, pewna ryzyko zakrzepicy nieznacznie zwiększone ryzyko raka
ochrona przez 5 lat podwyższone, sutka, ciąży ektopowej
(wskaźnik Pearla 0,16) krwawienia śródcykliczne
spirala zawierająca miedź ochrona przez 5 lat upławy, zapalenie przydatków, tylko czasowo,
bez zaburzania czynności endometrioza, gdy niemożliwe jest
układu hormonalnego skąpe miesiączkowanie stosowanie doustnych
(wskaźnik Pearla 0,9-3) środków antykoncepcyjnych
pigułka po stosunku działa po odbyciu stosunku często nudności, wymioty, jak mikropigułka przeciwwskazane
bez zabezpieczenia zaburzenia cyklu, przy migrenie,
w ciągu następnych krwawienia śródcykliczne zapaleniu wątroby,
72 godzin chorobach tętnic,
ostrej porfirii
MUTSCHLER-2009.indd 486 2010-01-07 22:13:19
Układ hormonalny 487
Interakcje. Działanie hormonalnych środków an- dzie moczowo-płciowym (zob. rozdz. B 2.8.6). Poza
tykoncepcyjnych staje się niepewne przy jedno- tym długotrwałe stosowanie estrogenów zapobiega
czesnym podawaniu induktorów enzymatycznych, resorpcji tkanki kostnej. Ryzyko rozwoju raka en-
np. barbituranów, karbamazepiny, fenytoiny, rifampi- dometrium można zmniejszyć, stosując estrogeny
cyny lub wyciągu z dziurawca. Skuteczność ochrony w połączeniu z gestagenami. Taka terapia kombino-
przed zajściem w ciążę mogą również zmniejszyć wana jest wskazana u wszystkich kobiet mających
antybiotyki o szerokim zakresie działania, np. amino- macicę, tylko u pacjentek po histerektomii można
penicyliny i tetracykliny, w następstwie przerwania stosować substytucyjnie estrogeny w monoterapii.
krążenia jelitowo-wątrobowego żeńskich hormonów Jednak (długotrwała) hormonalna terapia zastępcza
Układ hormonalny
płciowych z powodu braku rozprzęgania metabolitów budzi wiele kontrowersji. Duże badania (np. HERS,
fazy II przez bakterie jelitowe. Dotyczy to głównie WHO, Million-Women) wykazały bowiem wyraźnie
wszystkich preparatów stosowanych w małych daw- związane z nią ryzyko. Powikłania sercowo-naczy-
kach. W wyniku zmniejszonej tolerancji na glukozę niowe, a w szczególności udary mózgu i choroba
może wzrosnąć zapotrzebowanie na leki przeciwcuk- niedokrwienna serca oraz zatorowość płucna, wystę-
rzycowe. pują istotnie częściej u kobiet otrzymujących terapię
kombinowanę estrogen/gestagen (0,625 mg koniugo-
Przeciwwskazania. Świeża i podawana w wywiadzie wanych estrogenów z 2,5 mg octanu medroksyproge-
zakrzepica (w przypadku preparatów zawierających steronu). Monoterapia estrogenami zwiększa ryzyko B2
gestageny w monoterapii tylko świeża zakrzepica), sercowo-naczyniowe w mniejszym stopniu. Poza tym
zapalne i zwyrodnieniowe choroby naczyń, ciężkie u kobiet otrzymujących terapię kombinowaną estro-
zaburzenia czynności wątroby, żółtaczka i świąd gen/gestagen lub tybolon częściej rozpoznawany
ciężarnych w wywiadzie oraz hiperbilirubinemia jest rak piersi. Badanie WHO zostało z tego powodu
są przeciwwskazaniami do stosowania hormonal- przerwane.
nych środków antykoncepcyjnych. W przypadku sto- Biorąc pod uwagę przedstawione wyniki, hor-
sowania hormonalnych środków antykoncepcyjnych monalną terapię zastępczą w okresie klimakterium
w razie pojawienia się po raz pierwszy migrenowych lub pomenopauzalnym należy stosować tylko przy
bólów głowy lub ostrych zaburzeń widzenia (podej- wyraźnych objawach i po dokładnym rozważe-
rzenie powikłań zakrzepowo-zatorowych), cholesta- niu związanych z nią korzyści i ryzyka. Estrogeny
zy i dużego wzrostu ciśnienia krwi, a także przed za- powinny być podawane w możliwie jak najmniej-
biegami operacyjnymi należy natychmiast odstawić szej dawce. Monoterapia estrogenami jest u kobiet
te preparaty. po histerektomii możliwa, ale we wszystkich innych
Preparaty zawierające estrogeny nie powinny być przypadkach wskazana jest terapia kombinowana
przyjmowane w czasie karmienia piersią, gdyż estro- z gestagenem.
geny hamują wytwarzanie mleka.
W tabeli B 2.8-8 zestawiono zalecenia dotyczące Z grupy estrogenów stosuje się (zob. tab. B 2.8-9):
stosowania środków antykoncepcyjnych.
■ estradiol i walerianian estrogenu (2 mg dziennie),
■ koniugowane estrogeny (0,6 mg dziennie),
2.8.8. Hormonalna terapia zastępcza ■ raloksyfen,
w okresie pomenopauzalnym ■ tybolon.
Znaczne zmniejszenie wytwarzania estrogenów Należy przy tym pamiętać, że cykliczne stosowanie
u kobiet po zakończeniu okresu płodności prowadzi gestagenów jest związane z występowaniem krwa-
do dolegliwości, które częściowo nie tylko przeszka- wień z odstawienia. Można ich uniknąć, stosując te-
dzają w funkcjonowaniu, ale także stwarzają zagro- rapię ciągłą, ale niestety zwieksza się wówczas ryzy-
żenie życia. Wraz ze wzrostem długości życia okres ko rozwoju raka endometrium.
pomenopauzalny stanowi znaczną część życia kobiet,
co przyczynia się do dużego wzrostu znaczenia pro- Miejscowe stosowanie estriolu (zob. tab. B 2.8-4)
filaktyki chorób układu sercowo-naczyniowego i os- może zmniejszyć nasilenie objawów z okolicy po-
teoporozy. chwy (świądu, uczucia suchości).
Suplementacja estrogenów zmniejsza nasilenie
zaburzeń wazomotorycznych (uderzeń gorąca), ob- Na razie brak jest dla fitoestrogenów badań klinicz-
jawów psychicznych i zmian atroficznych w ukła- nych, które spełniałyby wymogi stawiane badaniom
MUTSCHLER-2009.indd 487 2010-01-07 22:13:20
488 Układ hormonalny
Tabela B 2.8-9. Środki stosowane w hormonalnej terapii zastępczej
Estrogen (mg) Gestagen (mg) Preparaty handlowe
Estrogeny
estardiol, doustnie (1) – Estrifam, Estronorm
walerianian estradiolu, doustnie (2) Estradiol JENAPHARM,
– Gynokadin, Merimono, Progynova
estradiol, transdermalnie (0,78-1,56) Dermestril, Estrabeta, Estraderm,
– Estradot
estrogeny koniugowane, doustnie – Climopax mono, Presomen
(0,3-1,25)
Preparaty złożone do stosowania doustnego
estradiol (1-2) octan noretysteronu (0,5-1,0) Activelle, Clionara, Gynamon, Kliogest N
octan medroksyprogesteronu (2,5-5) Indivina, Osmil
dydrogesteron (5-10) Femoston
drospirenon (2) Angeliq
walerianian estradiolu (1-2) noretysteron (0,7) Mericomb, Merigest
octan medroksyprogesteronu (10) Gianda, Indivina, Procyclo
lewonorgestrel (0,075-0,15) Klimonorm, Östronara
norgestrel (0,5) Cyclo-Progynova
dienogest (2) Climodien, Lafamme
octan cyproteronu (1) Climen
estriol (2) leworgestrel (0,25) CycloÖstrogynal
estrogeny koniugowane (0,3-1,25) medrogeston (5) Presomen compositum
octan medroksyprogesteronu (2,5-5) Climopax
Preparaty złożone do stosowania transdermalnego
estradiol (4,3/0,5) octan noretysteronu (4,8) Estalis sequi
estradiol (1,5) lewonorgestrel (1,5) Fem7 Combi
łania owulacji klomifen lub gonadotropiny. Jednak,
oceniającym skuteczność i bezpieczeństwo leków,
jak wspomniano wcześniej, terapia ta wiąże się
i dlatego nie można wyciągnąć żadnych wniosków
z dużym ryzykiem ciąż mnogich i hiperstymula-
dotyczących ich skuteczności i ryzyka związanego
cji janików. Przy braku pragnienia zajścia w ciążę
z ich stosowaniem.
podstawą do rozpoczęcia leczenia jest brak wydziel-
niczej przebudowy endometrium. Aby zapobiegać
gruczołowej hiperplazji endometrium, wskazane
2.8.9. Farmakoterapia zaburzeń jest wówczas cykliczne podawanie gestagenów lub
miesiączkowania środków antykoncepcyjnych z komponentą antyan-
drogenową.
W terapii zespołu policyklicznych jajników u kobiet Amenorrhoea u kobiet przed 45 r.ż. wymaga lecze-
pragnących zajść w ciążę stosuje się w celu wywo- nia. Także w tym przypadku wskazane jest cykliczne
MUTSCHLER-2009.indd 488 2010-01-07 22:13:20
Układ hormonalny 489
podawanie gestagenów (w monoterapii lub w połą-
czeniu z estrogenami). ■ w czasie porodu przy słabej czynności porodowej,
Objawy bolesnego miesiączkowania można złago-
■ po cięciu cesarskim w celu wywołania skurczu ma-
dzić podając niesteroidowe leki przeciwzapalne lub
cicy,
doustne środki antykoncepcyjne.
■ w okresie poporodowym – zazwyczaj w połączeniu
z metyloergometryną – w celu odklejenia łożyska,
2.8.10. Związki wpływające na macicę zmniejszenia utraty krwi oraz profilaktyki lub le-
czenia atonii macicy,
Układ hormonalny
2.8.10.1. Oksytocyna ■ przy słabym wytrysku mleka i uwarunkowanych
tym trudnościach w karmieniu piersią.
Hormon tylnego płata przysadki, oksytocyna (oxitoci-
num; zob. ryc. B 2.8-10) jest związkiem, który fizjolo- Dawkowanie wynosi:
gicznie powoduje skurcze macicy. Mięśniówka maci-
■ w celu rozpoczęcia porodu i stymulacji czynności
cy – niezależnie od unerwienia – zostaje bezpośrednio
porodowej – rozwór oksytocyny w glukozie o stę-
pobudzona. Jej wrażliwość na oksytocynę jest bar-
żeniu 1 IU/100 ml podaje się dożylnie z szybkością
dzo różna i zależy od wielu czynników, ale przede
8–40 kropli/min, B2
wszystkim od stosunku estrogenów do gestagenów.
Estrogeny zwiększają pobudliwość i spontaniczną ■ w okresie poporodowym – 3–6 IU i.m. lub i.v.,
aktywność macicy, natomiast gestageny powodu-
■ przy zastoju mleka – 4 IU donosowo przed każdym
ją, że macica staje się niewrażliwa na oksytocynę.
karmieniem piersią.
Tym można również wyjaśnić niewielką skuteczność
oksytocyny w początkowym okresie ciąży, kiedy
stosunek estrogenów do gestagenów jest niski. Pod
koniec ciąży łożysko wytwarza coraz większe ilości
estrogenów, które uwrażliwiają mięśniówkę macicy
na oksytocynę. Poza tym rozciągnięcie ściany maci-
cy spowodowane szybkim wzrostem płodu prowadzi
odruchowo do wzmożonego uwalniania oksytocyny.
oksytocyna
W fizjologicznych dawkach oksytocyna wyzwa-
la rytmiczne skurcze macicy. Ciągły skurcz (tetanus
uteri) występuje jedynie przy dużych dawkach lub Cys Tyr
Ile
po wstrzyknięciu oksytocyny do macicy. S
S
Gln
Oksytocyna wywołuje nie tylko skurcze mięśniówki ma- Asn
Cys
cicy, lecz także mięśni gładkich w gruczołach mlecznych.
Powoduje to wyciśnięcie mleka z odcinków wydzielni- Pro
czych do większych przewodów wyprowadzających. Wy-
dzielanie oksytocyny z przysadki następuje odruchowo, Leu Gly NH2
w czasie kiedy dziecko ssie. Jej działanie jest wyjątkowo
swoiste: wystarcza już 0,01 IU (l IU = 0,002 mg oksyto- karbetocyna
cyny). atosiban
CH2 OCH3 CH2 CH3
CO
Oksytocyna ulega inaktywacji w nerkach i wą- CH2 Mpa
Tyr Tyr
CH2 Ile
trobie, natomiast podczas karmienia piersią również S
w gruczole sutkowym. Okres półtrwania wynosi tyl- S Ile S Thr
ko kilka minut. U kobiet ciężarnych wykryto poza Asn
Cys Asn Gln Cys
tym w osoczu enzym rozkładający oksytocynę – ok-
sytocynazę. Pro Pro
Leu Gly NH2 Orn Gly NH2
Stosowanie oksytocyny jest wskazane:
■ w celu rozpoczęcia porodu przy przedwczesnym
pęknięciu pęcherza płodowego, stanie przedrzu- Ryc. B 2.8-10. Struktura oksytocyny, karbetocyny i atosibanu.
cawkowym, rzucawce oraz ciąży przenoszonej, Mpa – kwas merkaptopropionowy.
MUTSCHLER-2009.indd 489 2010-01-07 22:13:20
490 Układ hormonalny
W przypadku rozpoczynania porodu i leczenia słabej 2.8.10.4. Pochodne prostaglandyn
czynności porodowej stosowany wówczas przede
wszystkim ciągły wlew kroplowy umożliwia dokład-
Innymi fizjologicznymi substancjami wywołujący-
ne regulowanie czynności porodowej.
mi skurcze macicy są prostaglandyny, które oprócz
Jako działanie niepożądane może wystąpić zbyt
wpływu na narządy rozrodcze oddziałują także
silna i wskutek tego bardzo bolesna czynność poro-
na inne organy.
dowa. W szczególności przy przedawkowaniu ist-
Tak więc powodują one wzmożenie, np. w próbie
nieje niebezpieczeństwo powstania ciągłego skurczu
u zwierząt, uwalniania gonadoliberyn oraz zmieniają
macicy. Inne działania niepożądane to spadek ciśnie-
motorykę jajowodów, ułatwiając zapłodnienie ko-
nia krwi, nudności i wymioty.
mórki jajowej: proksymalne odcinki jajowodu ulega-
Stosowanie oksytocyny jest przeciwwskazane przy
ją zwężeniu, natomiast dystalne unieruchomieniu.
skurczach tonicznych macicy, w ciężkim zatruciu
U kobiet niebędących w ciąży prostaglandyny,
ciążowym, mechanicznych przeszkodach utrudniają-
przede wszystkim prostaglandyna F2α, uczestniczą
cych poród, zagrażającym pęknięciu macicy i przed-
w wyzwalaniu krwawienia miesiączkowego. Bolesne
wczesnym odklejeniu się łożyska.
miesiączkowanie – przynajmniej częściowo – wywo-
Z powodu wzmożonych skurczów macicy
łuje wzmożone wytwarzanie prostaglandyn.
nie jest możliwe jednoczesne podanie prostaglandyn
W czasie ciąży około terminu porodu wzrasta wy-
(zob. poniżej).
raźnie synteza prostaglandyn.
Z tego powodu, że prostaglandyny wywołują skur-
cze macicy, stosuje się je w celu wywołania poronie-
2.8.10.2. Karbetocyna nia, przy poronieniu chybionym (missed abortion),
zaśniadzie graniastym, w celu rozpoczęcia porodu
W celu wywołania skurczu macicy po cięciu cesar- i w okresie poporodowym.
skim stosuje się karbetocynę, będącą długo działają- W czasie porodu w wyniku działania powodują-
cym agonistą oksytocynowym. Okres półtrwania wy- cego zwiotczenie szyjki macicy i przez to szybsze
nosi ok. 40 minut. Zazwyczaj wystarcza jednorazowe otwarcie ujścia macicy, przyspieszony zostaje przede
podanie dożylne 100 μg. Efekt działania jest porów- wszystkim okres rozwierania. Dostępne są następu-
nywalny z uzyskiwanym podczas wielogodzinnego jące substancje czynne: prostaglandyna E2, a także
wlewu oksytocyny. pochodne prostaglandyny E2 i E1 – sulproston oraz
Karbetocyna nie może być stosowana podczas gemeprost (zob. tab. B 3.3-1).
skurczów porodowych ani do ich wywoływania. Za- Dawkowanie prostaglandyn jest zawsze indywi-
sadniczo należy ją stosować ostrożnie przy migre- dualne. W celu wywołania poronienia są potrzebne
nie, astmie i chorobach sercowo-naczyniowych oraz znacznie większe dawki niż do rozpoczęcia porodu
w innych sytuacjach, w których szybkie zwiększenie pod koniec ciąży.
objętości płynu zewnątrzkomórkowego może być Przy stosowaniu ich jako środka do wywołania
nadmiernym obciążeniem dla organizmu. poronienia podaje się je głównie do jamy macicy
(pozaowodniowo), natomiast z powodu złego tolero-
wania rzadziej dożylnie. W celu rozpoczęcia porodu
2.8.10.3. Atosiban potrzebne są mniejsze dawki i dlatego wówczas sub-
stancje te podaje się często dożylnie. Prostaglandynę
E2 można także stosować dopochwowo.
Atosiban jest antagonistą oksytocyny stosowanym
Działania niepożądane to zależne od dawki nud-
do zahamowania zagrażającego porodu przedwczes-
ności, wymioty, biegunka, uderzenia gorąca i bóle
nego. Lek podaje się we wlewie dożylnym (dawko-
głowy. Poza tym obserwuje się napady astmy, kur-
wanie: 6,75 mg w bolusie, a następnie we wlewie
cze mięśniowe i napady padaczkowe. Przedawkowa-
ciągłym z szybkością 300 μg/min przez 3 godziny,
nie może wywołać tężcowy skurcz mięśnia macicy.
a potem 100 μg/min). Osoczowy okres półtrwania
Przy astmie, jaskrze, kurczach mięśniowych, a także
wynosi 1,7 godziny.
ostrych zaburzeniach żołądkowo-jelitowych pros-
Częstymi działaniami niepożądanymi są nudności,
taglandyny można stosować, jeśli w ogóle, jedynie
bóle i zawroty głowy, a rzadko gorączka i wysypka.
z zachowaniem dużej ostrożności.
Przeciwwskazaniami są: przedwczesne pękniecie
pęcherza płodowego, jak również stan matki lub pło-
du, w którym podtrzymywanie ciąży niesie ze sobą
istotne ryzyko.
MUTSCHLER-2009.indd 490 2010-01-07 22:13:20
Układ hormonalny 491
2.8.10.5. Alkaloidy sporyszu 2.8.10.6. Tokolityki
Tokolityki (środki hamujące czynność skurczową
Alkaloidy sporyszu, podobnie jak oksytocyna, działają
macicy) są związkami stosowanymi do zwiotczenia
bezpośrednio na mięśniówkę macicy i w małych daw-
macicy przy zagrażającym porodzie przedwczesnym,
kach powodują jej rytmiczne skurcze. Niebezpieczeń-
przy zabiegach operacyjnych podczas ciąży (np. ope-
stwo wywołania ciągłego skurczu macicy jest jednak
racji zakładania pierścienia na szyjce macicy, wyni-
znacznie większe niż przy podawaniu oksytocyny.
cowania mięśniaka), przedwcześnie rozpoczynającej
W położnictwie i to wyłącznie w okresie poporo-
się lub patologicznie wzmożonej czynności porodo-
dowym stosuje się pochodną ergometryny – mety-
Układ hormonalny
wej, nagłych przypadkach położniczych (np. cięcie
loergometrynę, ponieważ w przeciwieństwie do al-
cesarskie), a także przy zmianie z zewnątrz z poło-
kaloidów z grupy ergotaminy i ergotoksyny niemal
żenia podłużnego miednicowego płodu na położenie
nie powoduje ona skurczu naczyń krwionośnych
podłużne główkowe. Lekami pierwszego rzutu są
i blokowania receptorów α-adrenergicznych.
β2-sympatykomimetyki, obecnie terapeutycznie wy-
Stosowanie alkaloidów sporyszu jest wskazane
korzystuje się już tylko fenoterol. Poza tym tokolizę
w razie:
można prowadzić podając jony magnezu lub antago-
■ opóźnionego odklejania się łożyska, nistę oksytocyny (zob. rozdz. B 2.8.10.3).
B2
■ krwawienia po odklejeniu się łożyska,
■ zastoju odchodów połogowych,
OH
■ słabego zwijania się macicy w połogu.
HO NH
Dawkowanie wynosi 0,1–0,2 mg doustnie lub poza- CH3
OH
jelitowo.
OH
Ogólnie metyloergometryna jest dobrze tolerowa-
na. W dużych dawkach może wywołać nudności, wy- fenoterol
mioty i bóle w podbrzuszu. Sporadycznie donoszono
również o występowaniu zawrotów i bólów głowy,
tachy- lub bradykardii oraz wysypek.
Stosowanie metyloergometryny jest przeciwwska-
Dawkowanie fenoterolu wynosi 0,5–3 μg/min.
zane w okresie rozwarcia i wydalania płodu, przed
Na początku leczenia należy ściśle kontrolować pa-
urodzeniem główki, a także przy słabej czynności
cjentkę (kontrola tętna, ciśnienia krwi i wydalania
porodowej.
moczu).
Z powodu konieczności podawania dużych da-
wek, mimo względnej β2-selektywności, należy
wziąć pod uwagę możliwość wystąpienia kardiolo-
gicznych działań niepożądanych (tachykardii, komo-
O NH CH2OH rowych zaburzeń rytmu, dolegliwości o charakterze
CH2R
dławicy piersiowej). Jednoczesne stosowanie leków
selektywnie blokujących receptory β1-adrenergiczne
N
CH3
(np. atenololu lub metoprololu) może spowodować
H ustąpienie tych działań niepożądanych lub przynaj-
mniej je zmniejszyć.
HN Tokolityki osłabiają działanie hipoglikemiczne le-
ków przeciwcukrzycowych.
Kardiomiopatia przerostowa, zwężenie zastawek
R
serca, tachykardia, tyreotoksykoza i nadciśnienie
H ergometryna płucne są przeciwwskazaniami do stosowania toko-
lityków tego typu.
CH3 metyloergometryna
Jony magnezu w celu uzyskania tokolizy muszą być
podawane we wlewie w dużych dawkach (początko-
wo 4–6 g; dawka podtrzymująca 2–4 g co godzinę).
MUTSCHLER-2009.indd 491 2010-01-07 22:13:21
492 Układ hormonalny
Do działań niepożądanych należą kołatanie serca, β-sympatykomimetyków i soli magnezu istotnie się
bóle i zawroty głowy oraz nudności. Zmniejszenie zmniejsza. Jednak ten czas może wystarczyć na uzy-
odruchów głębokich, zmiany w EKG aż do asysto- skanie dojrzałości płuc dziecka dzięki podaniu gluko-
lii oraz depresja ośrodka oddechowego są objawami kortykosteroidów oraz przeniesieniu kobiety ciężar-
przedawkowania. nej do ośrodka posiadającego oddział intensywnej te-
Często jednak dzięki tokolizie można opóźnić za- rapii noworodków, co zdecydowanie zwiększa szanse
grażający poród o 24–48 godzin; potem skuteczność dziecka na przeżycie.
MUTSCHLER-2009.indd 492 2010-01-07 22:13:21
Mediatory (autokoidy, hormony parakrynne) 493
3. Mediatory
(autakoidy, hormony parakrynne)
Mediatory (autakoidy) to substancje wydzielane przez ■ czynnik aktywujący płytki (PAF),
określone komórki lub grupy komórek (hormony tkan-
■ kininy.
kowe), które wpływają na sąsiednie komórki, tzn. wy-
wołują efekt parakrynny. Do mediatorów należą:
■ histamina, Histamina i serotonina pełnią poza tym funkcję neu-
roprzekaźników.
■ serotonina,
Cytokiny, chemokiny i interferony (zob. rozdz. B
■ produkty przemian kwasu arachidonowego (pros- 13.3.1) nie są zaliczane do mediatorów, chociaż wy-
taglandyny, tromboksan A2, prostacyklina, leuko- wołują efekt parakrynny.
trieny),
Mediatory
3.1. Histamina
B3
Synteza, uwalnianie i rozkład. Histamina powsta- py należą: morfina, radiologiczne środki kontrastowe
je we wszystkich tkankach ludzkiego organizmu i preparaty krwiozastępcze (np. HES – skrobia hy-
jako produkt dekarboksylacji aminokwasu histy- droksyetylowana). Innym związkiem uwalniającym
dyny. Najwyższe stężenia stwierdza się w płucach, histaminę jest mastoparan, będący składnikiem jadu
skórze i przewodzie pokarmowym. W mastocytach osy, który aktywując białko G, w sposób niezależny
i granulocytach zasadochłonnych histamina jest ma- od IgE prowadzi do uwolnienia histaminy.
gazynowana w formie dodatniego jonu związanego
z anionami, np. heparyną lub proteoglikanami. Uwal- Receptory histaminowe i mechanizm działania
nianie histaminy z tych magazynów następuje na sku- histaminy. Histamina łączy się z czterema różnymi
tek uszkodzenia komórek (np. podczas zapalenia), związanymi z białkiem G receptorami: H1, H2, H3
IgE-zależnej reakcji nadwrażliwości oraz w sposób i H4 (zob. ryc. B 3.1-1).
IgE-niezależny w następstwie działania różnych
związków chemicznych, tzw. substancji uwalniają- Stymulacja receptora H1 poprzez aktywację fosfolipa-
cych histaminę (zob. poniżej).Wolna histamina ulega zy C powoduje rozszerzenie tętniczek, a w konsekwen-
szybko rozkładowi na drodze dezaminacji oksyda- cji spadek ciśnienia tętniczego krwi. Ponadto tą samą
tywnej pod wpływem histaminazy, a także na drodze drogą histamina, powodując skurcz komórek śród-
metylacji grupy NH i utlenienia powstałych metabo- błonka drobnych naczyń żylnych, zwiększa przepusz-
litów za pomocą monoaminooksydazy. czalność naczyń włosowatych, co prowadzi do przeni-
kania białek osocza, wody i komórkowych składników
Substancje uwalniające histaminę. Niektóre leki krwi do otaczających tkanek. Rozszerzenie naczyń
mogą powodować uwolnienie histaminy. Do tej gru- krwionośnych i zwiększona przepuszczalność naczyń
włosowatych prowadzą do rozwoju wstrząsu anafilak-
tycznego (zob. poniżej). Ponadto histamina ułatwia mi-
grację leukocytów przez tworzenie białek adhezyjnych
N na powierzchni komórek śródbłonka, co z kolei nasila
HN
NH2 reakcję zapalną. Stymulując zakończenia nerwowe, hi-
stamina wywołuje świąd. Poza tym kurczy ona mięśnie
histamina gładkie oskrzeli i przewodu pokarmowego. W OUN re-
ceptory H1 biorą udział w utrzymaniu stanu czuwania
oraz uczestniczą w regulacji przyjmowania pokarmów.
MUTSCHLER-2009.indd 493 2010-01-07 22:13:21
494 Mediatory (autokoidy, hormony tkankowe)
alergen NO
A
histamina światło naczynia
H1
Gq Ca2+↑ CaM eNOS
IP3
ER IP3R
mastocyt
(komórka tuczna) komórka śródbłonka
NO
sGC cGMP↑ PKG rozszerzenie
mięśniówka naczyń naczyń
(mięśnie gładkie)
B alergen histamina
H1
Gq
Ca2+↑ CaM MLCK
IP3
ER skurcz
IP3R
mastocyt mięśniówka oskrzeli
(komórka tuczna) (mięśnie gładkie)
C
Gs
K+
H2
AC
PKA↓ nie
cAMP pobudze
PKA
Gq
neuron histaminergiczny histamina
H1
DAG
H3
cAMP↓ PKC
Gi
cyklaza K+
adenylanowa K+
ATP
Na+
neuron postsynaptyczny
glutaminian
PKA↓
receptor
NMDA
neuron glutaminergiczny
H3
cAMP↓ Na+, Ca2+
Gi
pobudzen
ie
cyklaza
adenylanowa
Ryc. B 3.1-1. Przykłady działania histaminy. (A) Histamina uwolniona z mastocytów, pobudzając syntezę NO, rozszerza naczynia
krwionośne. Histaminergiczna stymulacja śródbłonka powoduje zmianę przepuszczalności naczyń włosowatych i prowadzi do po-
wstania wysięku. (B) W mięśniówce gładkiej oskrzeli w przeciwieństwie do mięśniówki naczyń pobudzenie receptorów H1 wywołuje
skurcz. (C) W OUN znajdują się receptory H1, H2 i H3. Aktywacja receptora H1 prowadzi do pobudzenia pompy Na+/K+ zależnej od
ATP oraz hamowania kanałów K+. W obu przypadkach, podobnie jak po aktywacji receptora H2, który pobudza cyklazę adenylanową
i zwiększa śródkomórkowe stężenia cAMP, dochodzi do pobudzenia neuronu i przekazywania impulsu. Receptory H3, będąc recep-
torami o właściwościach auto- i heterohamujacych, blokują wydzielanie histaminy i innych neuroprzekaźników. CaM – kalmomo-
dulina, DAG – diacyloglicerol, IP3R – receptor IP3, MLCK – kinaza łańcuchów lekkich miozyny, PKA – kinaza białkowa A, PKG – kinaza
białkowa G, PKC – kinaza białkowa C, sGC – rozpuszczalna cyklaza guanylanowa.
MUTSCHLER-2009.indd 494 2010-01-07 22:13:21
Mediatory (autokoidy, hormony parakrynne) 495
Pobudzenie receptorów H2 poprzez aktywację cy- padowe zaczerwienienie skóry (flush), nasiloną po-
klazy adenylanowej przyspiesza akcję serca, zwięk- krzywkę i obrzęki o ciężkim przebiegu, a także spazm
sza kurczliwość mięśnia sercowego oraz nasila wy- oskrzelowy.
dzielanie gruczołów, szczególnie gruczołów błony Histamina nie ma znaczenia terapeutycznego. Wy-
śluzowej żołądka. korzystywana jest jednak jako pozytywna kontrola
Receptory H3 położone są presynaptycznie. Jako w śródskórnych testach alergicznych.
autoreceptory hamują one uwalnianie histaminy,
a jako heteroreceptory regulują uwalnianie innych
neuroprzekaźników. Uwalnianie histaminy z komó-
rek tucznych żołądka oraz serotoniny z komórek en- 3.1.1. Leki przeciwhistaminowe H1
terochromochłonnych podlega podobnej regulacji.
Receptory H4 stwierdzono przede wszystkim Leki przeciwhistaminowe I generacji (por. tab. B
na limfocytach T, granulocytach zasadochłonnych 3.1-1) działają nie tylko na obwodowe, lecz także
i komórkach tucznych i na tej podstawie wysunięto na ośrodkowe receptory H1 oraz blokują receptory
przypuszczenie o ich roli prozapalnej. Poza tym ist- muskarynowe. Leki przeciwhistaminowe II genera-
nieją przesłanki wskazujące na rolę receptorów H4 cji, dzięki niewielkiej lipofilności, działają niemal
w różnicowaniu komórek macierzystych szpiku. An- selektywnie na obwodowe receptory H1. Nowszymi
tagoniści receptorów H4 mogliby stać się nową klasą przedstawicielami leków przeciwhistaminowych dru-
Mediatory
leków stosowanych w terapii reumatoidalnego zapa- giej generacji są lewocetyryzyna, desloratadyna i fek-
lenia stawów, wrzodziejącego zapalenia jelit, astmy sofenadyna, które przez niektórych autorów są okre-
oskrzelowej i przewlekłego świądu. Pobudzenie re- ślane mianem leków przeciwhistaminowych trzeciej
ceptora H4 przez białko Gi powoduje hamowanie cy- generacji. Lewocetyryzyna jest aktywnym enancjo-
klazy adenylanowej, a przez białko Gβ,γ pobudzenie merem cetyryzyny; desloratadyna i feksofenadyna
fosfolipazy C. są dla odmiany aktywnymi metabolitami loratadyny B3
i terfenadyny. Zaletą feksofenadyny w porównaniu
Patofizjologiczne znaczenie histaminy. Histamina z terfenadyną jest mniejsza liczba działań niepożą-
uwolniona w skórze, np. pod wpływem ukłucia przez danych ze strony układu sercowo-naczyniowego.
owada lub kontakt z pokrzywą, powoduje powstanie Natomiast lewocetyryzyna i desloratadyna nie mają
bolesnego zaczerwienienia na skutek rozszerzenia żadnej przewagi nad cetyryzyną i loratadyną.
naczyń oraz swędzącego bąbla w wyniku wzrostu
przepuszczalności naczyń włosowatych. Działanie. Leki przeciwhistaminowe H1 blokują kom-
Histamina odgrywa szczególną rolę w reakcji aler- petycyjnie działanie histaminy na receptor H1. Środki
gicznej typu natychmiastowego. Uwolnienie histami- należące do I generacji istotnie częściej w porów-
ny może być przyczyną pokrzywki alergicznej lub naniu z pozostałymi wykazują działanie sedacyjne
obrzęku Quinckego, a w ciężkich przypadkach także z uwagi na blokowanie ośrodkowych receptorów H1
wstrząsu anafilaktycznego. Także w przebiegu wstrzą- (zob. zastosowanie leków przeciwhistaminowych H1
su endotoksycznego, zapalenia i oparzenia stwierdza jako leków nasennych) i z tego powodu nie powin-
się degranulację mastocytów i zwiększone stężenie ny być zalecane kierowcom. Działanie zwiększające
histaminy we krwi. Osoby z genetycznie uwarun- wydzielanie oraz inne efekty pobudzenia receptorów
kowaną predyspozycją do wytwarzania przeciwciał H2 nie są hamowane przez przedstawicieli tej grupy.
w klasie IgE skierowanych przeciw powszechnym Poza działaniem związanym z blokowaniem recep-
i zazwyczaj tolerowanym antygenom (atopia) mogą torów H1 niektóre leki przeciwhistaminowe, np. pro-
chorować (równocześnie lub sekwencyjnie) na ato- metazyna lub difenhydramina, łącznie z działaniem
powe zapalenie skóry, sezonowe (katar sienny) lub antycholinergicznym w dużych dawkach działają tak-
stałe (całoroczne) zapalenie błony śluzowej nosa oraz że miejscowo znieczulająco. Niektóre związki z tej
astmę oskrzelową. grupy, np. azelastyna, cetyryzyna i ketotifen, stabili-
Znaczna ilość egzogennej histaminy, na przykład zują błonę komórkową mastocytów. Cyproheptadyna,
pochodzącej z zepsutej ryby, może wywołać duszność, która pobudza także receptory serotoninowe, zostanie
spadek ciśnienia tętniczego krwi, zaczerwienienie skó- omówiona w rozdziale B 3.2.2.
ry, nudności, wymioty, bóle głowy i biegunkę.
Guz wywodzący się z komórek tucznych (masto- Farmakokinetyka. Po podaniu doustnym leki prze-
cytoma) oraz zwiększona ilość mastocytów we krwi ciwhistaminowe H1 wchłaniają się dobrze i szybko.
lub tkankach (mastocytoza) mogą w następstwie Większość z nich podlega intensywnym procesom
zwiększonego uwalniania histaminy powodować na- biotransformacji w wątrobie i jest wydalana w formie
MUTSCHLER-2009.indd 495 2010-01-07 22:13:22
496 Mediatory (autokoidy, hormony tkankowe)
Tabela B 3.1-1. Leki przeciwhistaminowe H1
Wzór strukturalny Nazwa Preparaty handlowe Okres Dawka
międzynarodowa półtrwania dzienna
(godz.) (mg)
Leki przeciwhistaminowe H1 pierwszej generacji: pochodne etylenodiaminy
prometazyna Atosil, 10-14 25-100 doustnie
CH3 Promethazin-neuraxpharm,
Proneurin, Prothazin
N [w Polsce: Dophergan,
N CH3
Polfergan – przyp. tłum.]
S CH3
Leki przeciwhistaminowe H1 pierwszej generacji: pochodne kolaminy
difenhydramina Dolestan, 4 50-150 doustnie
Emesan,
O CH3 Mordorm,
N Sediat
CH3 [w Polsce:
Betadrin – przyp. tłum]
dimenhydrynat Reisetabletten-ratiopharm, 4 50-300 doustnie
(8-chlorteofilinat Superpep, Vomacur,
difenhydraminy) Vomex A
[w Polsce:
Aviomarin - przyp. tłum.]
chlorfenoksamina Systral miejscowo żel
Cl 1,5-proc.
O CH3
N
CH3 CH3
doksylamina Gittalun, 10 25-50 doustnie
Hoggar Night,
Sedaplus
[w Polsce:
O
N CH3 Tabcin Impact – przyp. tłum.]
CH3 CH3
N
Leki przeciwhistaminowe H1 pierwszej generacji: pochodne propylaminy
dekschlorfenyramina Polaronil 17-30 6-8 doustnie
Cl
CH3
N
CH3
MUTSCHLER-2009.indd 496 2010-01-07 22:13:22
Mediatory (autokoidy, hormony parakrynne) 497
Tabela B 3.1-1. Leki przeciwhistaminowe H1 (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Dawka
międzynarodowa półtrwania dzienna
(godz.)
Leki przeciwhistaminowe H1 pierwszej generacji: pochodne pozostałe
CH3 bampina Soventol miejscowo żel
N 2-proc.
N
Cl klemastyna Tavegil 35-40 1-2 mg doustnie,
[w Polsce: miejscowo żel
CH3 Clemastinum – przyp. tlum.] 0,03-proc.
O N
CH3
Mediatory
CH3 dimetynden Fenistil 6 3-6 mg dousnie,
N miejscowo żel
0,1-proc.
CH3
N
H3C B3
CH3 ketotifen Ketof, 20 1-2 mg doustnie
N Ketotifen STADA,
Zaditen
[w Polsce: Ketotifen,
Zaditen – przyp. tłum.]
O S
OC2H5 emedastyna Emadine 2-6 0,1 mg – krople
do oczu
N CH3
N
N
N
Leki przeciwhistaminowe H1 drugiej generacji: pochodne etylenodiaminy
Cl O COOH cetyryzyna Cetirizin HEXAL, Cetirizin STADA, 7 10 mg doustnie
N Reactine, Zyrtec [w Polsce:
N Acer, Allertec, Zyrtec
– przyp. tłum.]
lewocetyryzyna XUSAL [w Polsce: Xyzal 7 5 mg doustnie
– przyp. tłum.]
Leki przeciwhistaminowe H1 drugiej generacji: pochodne pozostałe
Cl azelastyna Allergodil 20 4 mg doustnie,
0,03 mg w kroplach
do oczu,
0,3 mg donosowo
N
N
N CH3
O
MUTSCHLER-2009.indd 497 2010-01-07 22:13:23
498 Mediatory (autokoidy, hormony tkankowe)
Tabela B 3.1-1. Leki przeciwhistaminowe H1 (kontynuacja)
Wzór strukturalny Nazwa Preparaty handlowe Okres Dawka
międzynarodowa półtrwania dzienna
(godz.) (mg)
Leki przeciwhistaminowe H1 drugiej generacji: pozostałe (kontynuacja)
feksofenadyna Telfast 11-15 120-180 doustnie
OH [w Polsce:
N
Telfas, Fexofast
– przyp. tłum.]
OH
H3C CH3
COOH
CH3 lewokabastyna Livocab 0,1-0,2 w kroplach
[w Polsce: do oczu,
NC COOH Histimet – przyp. tłum.] 0,5-1 donosowo
N
COOC2H5 loratadyna Lisino, 3-20 10 doustnie
Cl N Lobeta,
Loratidin AD,
Loratidin STADA
[w Polsce:
N Claritine, Aleric, Flonidam,
Loram – przyp. tłum.]
NH desloratadyna Aerius 27 5 doustnie
Cl
F mizolastyna Mizollen, 13 10 doustnie
CH3
Zolim
N NH O
N N N
metabolitów. W formie niezmienionej wydalane są ce- obrzęku Quinckego, chorobie posurowiczej, aler-
tyryzyna i feksofenadyna. W przypadku niektórych no- gii na leki oraz po ukąszeniach owadów. Substancje
wych związków szybka przemiana do hydrofilnych, o silnym działaniu sedatywnym, np. difenhydramina
ale wciąż wykazujących wpływ na receptory H1 me- lub doksylamina, mogą być także zalecane jako środ-
tabolitów ogranicza możliwość ich przenikania przez ki nasenne. Natomiast te o silnym działaniu antycho-
barierę krew-mózg. Inne, np. cetyryzyna i fenoksyfena- linergicznym (np. difenhydramina i jej sole z 8-chlo-
dyna, są tylko w niewielkim stopniu lipofilne i dzięki roteofiliną, dimenhydrynat) są stosowane jako leki
temu wykazują nieznaczną penetrację do OUN. przeciwwymiotne w kinetozach.
Wskazania. Antagoniści receptorów histamino- Drogi podania. Leki z tej grupy mogą być podawane
wych H1 są wskazani we wszystkich schorzeniach, miejscowo lub doustnie. W przypadku ciężkiej reak-
w których istotną rolę odgrywa uwolnienie histami- cji alergicznej, a w szczególności w rozpoczynającym
ny, np. w pokrzywce, alergicznym zapaleniu błony się wstrząsie, należy rozważyć podanie domięśniowe
śluzowej nosa i alergicznym zapaleniu spojówek, lub dożylne.
MUTSCHLER-2009.indd 498 2010-01-07 22:13:23
Mediatory (autokoidy, hormony parakrynne) 499
Działania niepożądane. Najczęstszym działaniem niewydolności krążenia. W sposób szczególny nara-
niepożądanym leków przeciwhistaminowych H1 żone są dzieci. W leczeniu zatrucia podaje się środki
pierwszej generacji jest ich wpływ na ośrodkowy opóźniające wchłanianie z przewodu pokarmowego,
układ nerwowy. Ze względu na hamowanie ośrodko- w przypadku drgawek diazepam, a w razie niewy-
we układu nerwowego leki te wydłużają czas reakcji dolności oddechowej wskazane jest zastosowanie
i dlatego, jak już wspomniano, ograniczają zdolność oddechu wspomaganego. Jako odtrutkę w stosunku
do prowadzenia pojazdów mechanicznych. Do in- do działania cholinolitycznego można podać fizostyg-
nych działań niepożądanych należą zaburzenia żo- minę.
łądkowo-jelitowe, suchość w jamie ustnej, zaburze-
nia mikcji oraz zaburzenia rytmu serca. Po podaniu
miejscowym do worka spojówkowego lub na błonę 3.1.1.1. Zasady postępowania
śluzową nosa może pojawić się przejściowe podraż- w alergicznym nieżycie nosa
nienie (np. pieczenie). i zapaleniu spojówek
Znaczne upośledzenie czynności wątroby lub rów-
noczesne podawanie substancji hamujących CYP3A4
Nowsze leki przeciwhistaminowe H1 są chętniej niż
(np. makrolidów lub azolowych związków przeciw-
starsze stosowane w leczeniu alergicznego nieżytu
grzybicze) może być przyczyną przedawkowania
nosa i zapalenia spojówek. Charakteryzują się one
i zwiększenia stężenia niektórych leków przeciwhi-
dużą skutecznością w hamowaniu objawów sezono-
Mediatory
staminowych (np. terfenadyny) we krwi, co w kon-
wego i całorocznego alergicznego zapalenia błony
sekwencji może prowadzić do wydłużenia odcinka
śluzowej nosa i spojówek, wykazują mniejszą sku-
QT i zagrażających życiu tachyarytmii komorowych
teczność w usuwaniu uczucia zatkania nosa. Syste-
(torsades de pointes).
matyczne przewlekłe przyjmowanie jest skuteczniej-
sze niż podawanie ich doraźnie. Jeżeli po terfenady- B3
Środki ostrożności i przeciwwskazania. Stosowa-
nie wystąpią powikłania kardiologiczne (zaburzenia
nie leków przeciwhistaminowych H1, szczególnie
rytmu serca, torsades de pointes), należy ją zastąpić
tych ulegających biotransformacji w wątrobie, u pa-
bezpieczniejszym preparatem. Równoczesne syste-
cjentów z chorobami serca lub zaburzeniami czynno-
matyczne miejscowe stosowanie glukokortykostero-
ści wątroby wiąże się z istotnym ryzykiem rozwoju
idów prowadzi do ustąpienia wszystkich objawów
powikłań.
ze strony nosa, w tym lepszego opanowania uczucia
Leki przeciwhistaminowe H1 wykazujące aktyw-
jego niedrożności niż przy monoterapii lekami prze-
ność antycholinergiczną są przeciwwskazane u cho-
ciwhistaminowymi. Terapia kombinowana obniża
rych z jaskrą z wąskim kątem przesączania.
także stężenie mediatorów zapalenia. Skuteczność
miejscowych glukokortykosteroidów w opanowy-
Interakcje. Leki przeciwhistaminowe H1 działające
waniu objawów ocznych jest niestety mniejsza. Pa-
sedatywnie nasilają działanie leków przeciwbólo-
cjenta należy poinformować o opóźnionym działaniu
wych, nasennych, narkotycznych, psychotropowych
glukokortykosteroidów (początek działania pojawia
działających hamująco na ośrodkowy układ nerwowy
się po godzinie, a czasem po jednym dniu) i ich pra-
oraz alkoholu. Antagoniści H1 wykazujący działanie
widłowym dawkowaniu oraz zapewnić o bezpieczeń-
antymuskarynowe nasilają działanie antycholiner-
stwie nowych donosowych glukokortykosteroidów
giczne leków parasympatykolitycznych i niektórych
i możliwości redukcji dawki po opanowaniu obja-
przeciwdepresyjnych.
wów. W początkowym okresie terapii i przy bardzo
O wzajemnym oddziaływaniu z substancjami ha-
nasilonych objawach można przez dwa tygodnie sto-
mującymi CYP3A4 wspomniano wcześniej.
sować podwojone dawki. Jeżeli powyższe leczenie
nie jest skuteczne, należy rozważyć dołączenie kro-
Zatrucia. W dawkach toksycznych działające ośrod-
moglikanu lub nedokromilu.
kowo leki przeciwhistaminowe H1 z komponentą
miejscowo znieczulającą lub cholinolityczną mogą
wywołać pobudzenie, drgawki toniczno-kloniczne,
rozszerzenie źrenicy, zaburzenia akomodacji i mikcji 3.1.2. Leki przeciwhistaminowe H2
oraz tachykardię i bóle stenokardialne. Zgon nastę-
puje w wyniku depresji ośrodka oddechowego lub Zostały one omówione w rozdziale B 6.2.2.2.
MUTSCHLER-2009.indd 499 2010-01-07 22:13:24
500 Mediatory (autokoidy, hormony tkankowe)
3.2. Serotonina
Biosynteza, rozkład i występowanie. Serotonina jelita cienkiego. Rozciąganie ścian jelita pod wpły-
(5-hydroksytryptamina, 5-HT) występuje w wielu wem wzrostu ciśnienia w jego świetle prowadzi
tkankach roślinnych i zwierzęcych. W organizmie po- do uwalniania 5-HT. Ponadto komórki enterochłonne
wstaje z należącego do tzw. aminokwasów niezbęd- przekazują serotoninę płytkom krwi, gdy przepływają
nych – tryptofanu, który ulega hydroksylacji do 5-hy- one przez naczynia jelitowe. Biorąc udział w pierwot-
droksytryptofanu, a ostatecznie dekarboksylacji (zob. nej hemostazie uwolniona z trombocytów serotonina
ryc. B 3.2-1). Ostatni etap biosyntezy zachodzi pod powoduje miejscowy skurcz naczyń krwionośnych.
wpływem dopa-dekarboksylazy, tj. enzymu, który
odpowiada także za przekształcenie L-dopy w dopa- Działanie fizjologiczne serotoniny i receptory se-
minę. Na początku głównej drogi rozkładu 5-HT ule- rotoninowe. Serotonina wywołuje wiele różnych
ga przemianie w aldehyd 5-hydroksyindolylooctowy działań. Zwiększa ona kurczliwość mięśniówki je-
pod wpływem monoaminooksydazy A. Następnie al- lit, obkurcza duże tętnice i poszerza tętniczki, nasila
dehyd zostaje przez oksydazę aldehydową utleniony agregację płytek krwi, powoduje pobudzenie nocy-
do kwasu 5-hydroksyindolylooctowego, który zosta- ceptorów bólowych i odgrywa istotną rolę w ośrod-
je wydalony przez nerki. kowym układzie nerwowym. Stwierdzone podczas
W organizmach ssaków 5-HT występuje zarów- badań na zwierzętach działanie kurczące oskrzela
no w komórkach nerwowych, jak i nieneuronalnych. oraz mięsień macicy u ludzi jest bardzo słabe.
Serotonina zmagazynowana w neuronach, które znaj- Przyczyną tak wielu efektów działania serotoniny
dują się w tylnej części mózgu, przede wszystkim jest znaczna ilość różniących się między sobą recep-
w jądrach szwu, a także w całym przewodzie pokar- torów serotoninowych (zob. tab. B 3.2-1). Dla żad-
mowym, pełni rolę neuroprzekaźnika. Serotonina po- nego innego neuroprzekaźnika nie zidentyfikowano
zaneuronalna powstaje i jest zmagazynowana w du- dotychczas tak wielu różnych receptorów. Podzie-
żych ilościach w komórkach enterochromochłonnych lono je na 7 typów (od receptora 5-HT1 do 5-HT7),
COOH COOH
NH2 NH2 NH2
Trp-H HO OH-Trp-DC HO
NH NH NH
tryptofan 5-hydroksytryptofan serotonina (5-HT)
COOH MAO
HO ADH
CHO
NH
HO
kwas 5-hydroksyindolo-
octowy NH
H
Alk-D
OH
aldehyd 5-hydroksyindolo-
octowy
HO
NH
alkohol 5-hydroksyindoloetylowy
Ryc. B 3.2-1. Biosynteza i biotransformacja serotoniny. ADH – dehydrogenaza aldehydowa, Alk-DH – dehydrogenaza alkoholowa,
OH-Trp-DC – dekarboksylaza hydroksytryptofanu, Trp-H – hydroksylaza tryptofanu.
MUTSCHLER-2009.indd 500 2010-01-07 22:13:24
Mediatory (autokoidy, hormony parakrynne) 501
Tabela B 3.2-1. Receptory serotoninowe – podtypy i ich działanie
Receptor Podtyp Lokalizacja Efekt wywołany
(przekaźnik) stymulacją
5-HT1 5-HT1A OUN lęk, spadek ciśnienia tętniczego krwi, regulacja snu
(białko G, i łaknienia, autoreceptory
↓cAMP) 5-HT1B OUN hamowanie uwalniania GABA i glutaminianu (heteroreceptory),
skurcz naczyń krwionośnych (np. naczyń wieńcowych)
5-HT1D naczynia opon mózgowych skurcz
OUN pobudzenie motoryczne, regulacja zachowania
przedsionek serca autoreceptor
5-HT2 5-HT2A OUN pobudzenie neuronów
(białko G, mięśnie gładkie uwolnienie endoteliny, skurcz dużych naczyń
↑IP3/DAG)
płytki krwi agregacja
5-HT2B naczynia rozszerzenie naczyń krwionośnych w następstwie uwolnienia NO
5-HT2C OUN regulacja łaknienia, regulacja snu, lęk,
zachowania stresowe
Mediatory
5-HT3 – OUN regulacja zachowania, lęk
(zależny włókna aferentne nerwu błędnego, nudności, wymioty
od liganda układ nerwowy przewodu pokamowego pobudzenie aktywności neuronalnej
kanał jonowy)
area postrema nudności, wymioty
włókna bólowe ból B3
5-HT4 – okrężnica (mięśnie, przyspieszenie motoryki, uwolnienie acetylocholiny
(białko G, neurony)
↑cAMP) serce tachykardia
OUN uwolnienie dopaminy
a dla wielu z nich znane są jeszcze podtypy. Znacze- dzięki powstaniu trifosforanu inozytolu i diacylogli-
nie receptorów typów 1-4 zostało poznane, natomiast cerolu prowadzą do uwolnienia jonów Ca2+ z maga-
fizjologiczna i terapeutyczna rola znajdujących się zynów wewnątrzkomórkowych. Receptory te znajdu-
w ośrodkowym układzie nerwowym receptorów typu ją się także na płytkach krwi, a ich pobudzenie nasila
5-7 nie została jeszcze wyjaśniona. Receptor 5-HT7 ich agregację. Poza tym część działań dietyloamidu
bierze udział w regulacji temperatury ciała. kwasu lizergowego (LSD) jest związana z pobudze-
Poza receptorem 5-HT3, będącym kanałem jono- niem receptorów 5-HT2A.
wym regulowanym ligandem, wszystkie pozostałe Stymulacja neuronalnych receptorów 5-HT3 wpły-
receptory serotoninowe należą do receptorów powią- wa na pobudzenie tych neuronów i w konsekwencji
zanych z białkiem G. powoduje uwolnienie neuroprzekaźników (np. nora-
Pobudzenie receptorów 5-HT1, które jako presy- drenaliny, substancji P). W ten sposób receptory 5-HT3
naptyczne receptory hamujące są rozpowszechnione uczestniczą w powstaniu wielu reakcji odruchowych.
w ośrodkowym układzie nerwowym, blokuje cyklazę Znajdują się one m.in. w ośrodku wymiotnym (area
adenylanową, a tym samym syntezę cAMP. Stymu- postrema) w rdzeniu przedłużonym oraz (presynap-
lacja receptorów 5-HT1B rozszerza duże naczynia tycznie) we włóknach aferentnych nerwu błędnego,
krwionośne, a kurczy naczynia opon mózgowych a wyniku tego w sąsiedztwie komórek enerochromo-
i naczynia wieńcowe. Podobnie pobudzenie znajdują- chłonnych (komórki ECL) jelit. Pobudzając te recepto-
cych się w naczyniach opon mózgowych receptorów ry, serotonina, która jest uwalniana w dużych ilościach
5-HT1D powoduje zwężenie naczyń, a równocześnie przez komórki ECL w następstwie chemioterapii lub
hamuje uwalnianie białek prozapalnych. radioterapii, wywołuje nasilone nudności, a częściowo
Działanie kurczące na naczynia krwionośne także bardzo ciężkie wymioty. Także wolne zakończe-
jest dodatkowo nasilane przez obwodowe receptory nia włókien bólowych zawierają receptory 5-HT3 i dla-
5-HT2A, które aktywują fosfolipazę C, a tym samym tego serotonina, drażniąc nocyceptory, wywołuje ból.
MUTSCHLER-2009.indd 501 2010-01-07 22:13:24
502 Mediatory (autokoidy, hormony tkankowe)
Receptory 5-HT4 znajdują się w ośrodkowym uwalnianie serotoniny i wystąpienie charakterystycz-
układzie nerwowym i w przewodzie pokarmowym. nego zespołu objawów (zespół rakowiaka). Do typo-
Ich pobudzenie powoduje na drodze bezpośredniej wych objawów należą: biegunka z kolkowymi bó-
oraz pośredniej (poprzez uwalnianie acetylocholiny) lami brzucha, spazm oskrzelowy, oliguria, obrzęki
pobudzenie perystaltyki jelit. i zwłóknienie wsierdzia ze zmianami na zastawkach,
a także napadowe zaczerwienienie (flush) twarzy,
Aspekty patofizjologiczne. Istotne znaczenie pato- szyi i górnej części tułowia.
fizjologiczne ma serotonina w przypadku migreny.
Poza tym przypuszcza się, że uczestniczy ona w wy- Znaczenie terapeutyczne agonistów i antagoni-
woływaniu niektórych postaci zaburzeń rytmu, w na- stów receptorów serotoninowych. Agoniści recep-
głym zatrzymaniu krążenia, w zespole nagłej śmierci tora 5-HT1 znajdują zastosowanie jako leki obniżają-
niemowląt, w zaburzeniach lękowych i depresyjnych, ce ciśnienie tętnicze krwi – urapidil oraz przeciwlę-
a także w regulacji łaknienia. kowe – buspiron. Agoniści 5-HT1B/5-HT1D (triptany)
Rakowiak to rzadko występujący rozrost nowo- są szczególnie przydatni w terapii ostrych napadów
tworowy komórek enterochromochłonnych przewo- migreny. Stosowana w profilaktyce migreny dihy-
du pokarmowego, który powoduje m.in. nadmierne droergotamina ma działanie agonistyczne i antago-
Tabela B 3.2-2. Antagoniści receptora 5-HT3 (setrony)
Wzór strukturalny Nazwa Preparaty handlowe Okres Dawka
międzynarodowa półtrwania dzienna
(godz.) (mg)
H dolasteron Anemet 7-9 100-200
N
O
N
H
O
O
H
CH3 ondasetron Axosetron, 3 8-16
N cellondan,
Ondasetron-ratiopharm,
Zofran
CH3 [w Polsce:
Atossa, Emetron,
O N Zofran – przyp. tłum.]
N
H tropisetron Navoban 8 5
N CH3
N
O
O
CH3 granisetron Granisetron beta, 9 2
Granisetron HEXAL,
N CH3
N Granisetron STADA
N
NH
O
palonosetron Aloxi 40 0,25
H
O N
MUTSCHLER-2009.indd 502 2010-01-07 22:13:25
Mediatory (autokoidy, hormony parakrynne) 503
nistyczne na podtypy receptora 5-HT2. W leczeniu lekiem przeciwhistaminowym H1, cyproheptadyna
anoreksji wykorzystywany jest antagonista 5-HT2 wykazuje także działanie sedatywne.
– cyproheptadyna (zob. poniżej). Wpływ antagoni-
styczny na receptory 5-HT2 jest prawdopodobnie
odpowiedzialny za działanie przeciwpsychotyczne 3.2.3. Selektywni antagoniści
atypowych neuroleptyków, np. klozapiny. Antagoni- receptorów 5-HT3
ści receptora 5-HT3 odgrywają istotną rolę w leczeniu
wymiotów wywołanych chemio- i radioterapią. Inny- Do tej grupy należą setrony (zob. tab. B 3.2-2): do-
mi lekami wpływającymi na układ serotoninergiczny lasteron, granisetron, ondasetron, palonosetron
są należące do leków przeciwdepresyjnych inhibitory i tropisetron. Są one, jak wspomniano, stosowane
zwrotnego wychwytu serotoniny. w leczeniu wymiotów wywołanych emetogennymi
cytostatykami lub radioterapią oraz wymiotów po-
operacyjnych.
3.2.1. Triptany i alkaloidy sporyszu Skuteczność we wczesnej fazie wymiotów (w cią-
gu pierwszych 24 godzin od podania np. cisplatyny),
jednak nie w późnej (po 2–5 dniach), przewyższa
Te grupy leków zostały omówione w rozdziałach B
inne leki przeciwwymiotne.
1.4.7.1 i B 1.12.5.1.
Metoklopramid w dużych dawkach, poza recep-
Mediatory
torami dopaminergicznymi, blokuje także receptory
5-HT3 w ośrodku wymiotnym (area postrema) oraz
3.2.2. Antagoniści receptorów 5-HT2 w jelitach. Przy powtarzanych cyklach chemioterapii
cisplatyną działa on jednak słabiej niż inni antagoni-
Antagonistą receptorów 5-HT2A i 5-HT2B jest wspo- ści receptorów 5-HT3.
mniana powyżej cyproheptadyna, którą stosuje się Antagoniści 5-HT3 w przeciwieństwie do le- B3
jako lek zwiększający apetyt u chorych wyniszczo- ków przeciwhistaminowych H1 i cholinolitycznych
nych, np. w przebiegu choroby nowotworowej lub nie są skuteczni w leczeniu nudności i wymiotów
AIDS. Będąc równocześnie działającym ośrodkowo w przebiegu kinetoz.
MUTSCHLER-2009.indd 503 2010-01-07 22:13:25
504 Mediatory (autokoidy, hormony tkankowe)
3.3. Eikozanoidy (prostaglandyny, tromboksan A2,
prostacyklina, leukotrieny)
Kolejną ważną grupą mediatorów są eikozanoidy. działają częściowo synergistycznie, a częściowo an-
Biorą one udział w licznych procesach fizjologicznych tagonistycznie (ryc. B 3.3-1) – jest spowodowany
i patologicznych, a w szczególności w reakcjach za- tym, że zazwyczaj w jednym narządzie stwierdza się
palnych. W przeciwieństwie do serotoniny i histaminy różne typy receptorów dla prostaglandyn, równocześ-
eikozanoidy nie są magazynowane w ziarnistościach nie powstają różne prostanoidy, a ich receptory cha-
wewnątrzkomórkowych, a powstają w odpowiedzi rakteryzują się ograniczoną swoistością.
na różne bodźce z prekursora, którym jest kwas ara-
chidonowy (kwas all-cis-5,8,11,14-eikozatetraeno-
wy). W postaci wolnej występuje on tylko w niewiel-
kich ilościach, większość kwasu arachidonowego 3.3.1.1. Prostaglandyny
jest wbudowana do fosfolipidów bony komórkowej.
Pod wpływem różnych czynników, a w szczególno- Nazwa prostaglandyny (PG) wywodzi się stąd,
ści bodźców uszkadzających komórkę, kwas arachi- że po raz pierwszy zostały one wyizolowane z prosta-
donowy zostaje uwolniony przy udziale fosfolipazy ty. Jednak są one syntezowane nie tylko w gruczole
A2, a następnie na drodze przemian oksydacyjnych krokowym, ale w wielu różnych narządach. Znacze-
powstają z niego następujące produkty: nie fizjologiczne i patologiczne mają PGD2, PGE2
i PGF2α. Uwalnianie prostaglandyn jest wyzwalane
■ prostaglandyny (PGD, PGE, PGF), prostacyklina
na drodze neuronalnej, pod wpływem różnych me-
(PGI) i tromboksan (TXA) na szlaku cyklooksyg-
diatorów (np. histaminy) oraz przez hormony prze-
enazy,
wodu pokarmowego (np. gastrynę). Uwalnianie pro-
■ leukotrieny (LTB, LTC i LTD) na szlaku lipoksyge- staglandyn pobudza także noradrenalina, natomiast
nazy (zob. poniżej). prostaglandyny hamują jej uwalnianie z zakończeń
nerwowych.
Działanie. Wielokierunkowe działanie fizjologiczne
3.3.1. Substancje i patologiczne prostaglandyn u ludzi przedstawiono
szlaku cyklooksygenazy na ryc. B 3.3-1. Oprócz opisanego w rozdziale B
1.5.1.2 udziału prostaglandyn w patofizjologii bólu
Cyklooksygenaza (COX, występująca w dwóch i reakcji zapalnej znaczenie terapeutyczne ma także
izoformach: COX-1 i COX-2) katalizuje przemianę wpływ PGE2 na wydzielanie śluzu i kwasu solnego
kwasu arachidonowego do nadtlenku prostaglandy- w żołądku, działanie antyagregacyjne i rozszerzające
nowego (PGG2), a następnie przekształca go w PGH2. naczynia krwionośne PGI2 i PGE2 oraz wywoływa-
Z PGH2 w wielu tkankach powstają prostaglandyny nie skurczu mięśnia macicy przez PGE2 i PGF2α.
(PG), w płytkach krwi – tromboksan A2 (TXA2),
a w komórkach śródbłonka naczyń – prostacyklina Metabolizm. Inaktywacja prostaglandyn zachodzi
(PGI2) (zob. ryc. B 3.3-1). bardzo szybko pod wpływem różnych enzymów
wewnątrzkomórkowych, a w szczególności dehy-
Receptory dla prostanoidów. Nazywane prostano- drogenazy 15-hydroksyprostaglandyn i Δ13-reduk-
idami substancje powstające na szlaku cyklooksyge- tazy. Najwyższą aktywność dehydrogenazy prosta-
nazy łączą się ze specyficznymi receptorami połączo- glandyn stwierdzono w płucach, mleku i nerkach,
nymi z białkiem G. Pobudzenie receptorów dla PGD2 a najwyższą aktywność reduktazy w tkance tłusz-
i PGI2 aktywuje cyklazę adenylanową, podczas gdy czowej. Już po jednorazowym przejściu przez płuca
pobudzenie receptorów EP – w zależności od pod- nie stwierdza się prostaglandyn we krwi. Osoczowy
typu – może powodować wzrost lub spadek stężenia okres półtrwania nie przekracza 1 minuty. Nieaktyw-
cAMP. Pobudzenie receptorów dla TXA2 i PGF2α ne pierwotne metabolity są następnie rozkładane, jak
prowadzi do powstania trifosforanu inozytolu (IP3) inne kwasy tłuszczowe, na drodze β-oksydacji.
oraz diacyloglicerolu, a w konsekwencji do wzro-
stu stężenia wewnątrzkomórkowego Ca2+. Złożony Zastosowanie terapeutyczne. Ze względu na wpływ
obraz działania prostaglandyn – różne substancje na mięsień macicy oraz mięśniówkę naczyń naturalni
MUTSCHLER-2009.indd 504 2010-01-07 22:13:25
Mediatory (autokoidy, hormony parakrynne) 505
lipidy błony komórkowej
PLA2
kwas arachidonowy
COX-1 COX-2
PGG2
COX-1 COX-2
PGH2
PGI2 TXA2 PGF2α PGD2 PGE2
Mediatory
IP-R TP-R FP-R DP-R EP1-R EP2-R EP3-R EP4-R
Gs Gq Gq Gs Gq Gs Gi Gs
B3
cAMP IP3/Ca2+ IP3/Ca2+ cAMP IP3/Ca2+ cAMP cAMP cAMP
rozszerzenie agregacja skurczy rozszerzenie skurcz mięśni relaksacja hamowanie zwiększone
naczyń płytek krwi, mięśnia naczyń oskrzeli mięśni wydzielania wydzielanie
krwionośnych, skurcz naczyń macicy krwionośnych, i przewodu oskrzeli, kwasu solnego śluzu
hamowanie krwionośnych, hamowanie pokarmowego przewodu w żołądku, w żołądku,
agregacji płytek skurcz oskrzeli agregacji płytek pokarmowego zwiększenie zapobiega
krwi, hamowanie krwi, relaksacja i naczyń siły skurczu zamknięciu
uwalniania reniny mięśni przewodu mięśnia macicy przewodu
pokarmowego w czasie ciąży Botalla
i mięśnia macicy
Ryc. B 3.3-1. Synteza, receptory i fizjologiczne działanie prostaglandyn. Lipidy błony komórkowej są przekształcane przez fosfoli-
pazę A do kwasu arachidonowego, który jest substratem dla cyklooksygenazy 1 i 2. Pod ich wpływem powstają prekursory prosta-
glandyn PGG2 i PGH2, z których na drodze enzymatycznej (za pomocą syntaz) powstają aktywne fizjologicznie prostaglandyny. PLA2
– fosfolipaza A2; IP-R, TP-R, FP-R, DP-R, EP-R – receptory dla prostanoidów.
i syntetyczni przedstawiciele grup PGE i PGF znaj- Dinoprost, gemeprost i sulproston są wykorzy-
dują zastosowanie jako środki lecznicze. stywane, jak wspomniano, ze względu na wywoły-
Alprostadil ze względu na wywoływane rozsze- wany przez nie skurcz mięśnia macicy do indukowa-
rzenie naczyń jest stosowany w zaburzeniach erek- nia poronienia we wczesnej ciąży. W trakcie porodu
cji oraz w ciężkich postaciach choroby zarostowej przyspieszają one otwarcie kanału szyjki macicy,
tętnic. Podaje się go miejscowo w iniekcjach do ciał a podane pod koniec porodu zapobiegają atonii ma-
jamistych prącia lub ogólnoustrojowo we wlewie cicy. Podaje się je dożylnie lub zewnątrzowodniowo.
dożylnym. Kolejnym wskazaniem do jego stosowa- Gemeprost można podawać miejscowo w postaci
nia jest zapobieganie zamknięciu przewodu tętni- globulek dopochwowych.
czego Botalla w celu zwiększenia przepływu przez Latanoprost, trawoprost i bimatoprost po poda-
krążenie płucne u noworodków z wrodzonymi wa- niu miejscowym zmniejszają ciśnienie śródgałkowe
dami serca. i dzięki temu znalazły zastosowanie w leczeniu jaskry.
MUTSCHLER-2009.indd 505 2010-01-07 22:13:26
506 Mediatory (autokoidy, hormony tkankowe)
Tabela B 3.3-1. Prostaglandyny i ich pochodne (poza okulistyką)
Wzór strukturalny Nazwa Preparaty handlowe Wskazania Okres Dawkowanie
między- półtrwania
narodowa (godz.)
alprostadil CAVERJECT, zaburzenia 0,1-0,2 10-40 μg
O
VIRIDAL erekcji do ciał
COOH jamistych
Minprog utrzymanie początkowo
CH3
drożności 3-6 μg/kg/godz.
HO OH
przewodu
tętniczego Botalla
MUSE zaburzenia 250-1000 μg
erekcji do cewki
Prostavasin zaburzenia moczowej
krążenia
obwodowego 20-40 (-80) μg
we wlewie/
/dzień
O gemeprost Cergem rozwarcie szyjki 0,75-1 1-5 mg
COOCH3 macicy
w zabiegach
H3C CH3 ginekologicznych
CH3
i aborcji
HO OH
O dinoprost MINIPROSTIN E2, indukcja porodu 0,01 1-2 mg
Prepidil,
COOH Propess
CH3
HO OH
O sulproston Nalador indukcja 2 do 1 mg
NHSO 2CH3 poronienia,
atoniczne
O krwawienie
O
HO po porodzie
OH lub aborcji
O mizoprostol składnik Arthotecu profilaktyka 0,5 0,2-0,6 mg/dzień
COOCH3 choroby
wrzodowej
HO CH3 przy podawaniu
CH3
NLPZ
HO
Mizoprostol, pochodna prostaglandyny E1, dzię-
burzeń erekcji alprostadilem może być przyczyną
ki wpływowi na wydzielanie śluzu i kwasu solnego
priapizmu.
w żołądku jest stosowany w profilaktyce choroby
wrzodowej u chorych otrzymujących niesteroidowe
Interakcje. Mifepriston może nasilić wczesnoporon-
leki przeciwzapalne.
ne działanie prostaglandyn.
Działania niepożądane. Jako działania niepożąda-
ne mogą pojawić się nudności, biegunka, napadowe
zaczerwienienie skóry, bóle głowy i nasilone bóle 3.3.1.2. Prostacyklina i jej pochodne
podbrzusza. Są one silniejsze przy podaniu ogól-
nym niż miejscowym. W miejscu podania prosta- Prostacyklina (PGI2), która powstaje przy udziale
glandyny mogą wywołać bolesność. Leczenie za- syntazy prostacykliny w komórkach śródbłonka na-
MUTSCHLER-2009.indd 506 2010-01-07 22:13:26
Mediatory (autokoidy, hormony parakrynne) 507
czyń, powoduje rozszerzenie naczyń i hamuje agrega- Lipooksygenaza jest enzymem znajdującym się
cję płytek krwi. Efekty te są następstwem pobudzenia w cytoplazmie leukocytów, płytek krwi, komórek
receptora IP i aktywacji cyklazy adenylanowej (zob. tucznych i płuc. Poszczególne izoformy lipooksyge-
ryc. B 3.3-1). Prostacyklina szybko ulega biotransfor- nazy występujące w różnych tkankach różnią się mię-
macji – czas półtrwania wynosi 3 minuty. dzy sobą punktem uchwytu kwasu arachidonowego,
Stabilnym syntetycznym analogiem prostacykliny co w konsekwencji prowadzi do powstania po wpro-
jest iloprost. Tak jak alprostadil, podawany pozaje- wadzeniu grupy hydroperoksydowej różnych postaci
litowo, jest stosowany w leczeniu zaburzeń ukrwie- kwasu hydroperoksyeikozatetraenowego (HPETE),
nia tętniczego oraz w celu zapobiegania zamknię- który następnie pod wpływem peroksydazy jest prze-
ciu przewodu tętniczego Botalla. Podanie wziewne kształcany do kwasu hydroksyeikozatetraenowego
jest skuteczne w pierwotnym nadciśnieniu płucnym. i leukotrienów (LT) (zob. ryc. B 3.3-2).
Działania niepożądane są zbliżone do przedstawio- Nazwa leukotrieny wywodzi się stąd, że tego ro-
nych dla pochodnych prostaglandyny E. dzaju związki zawierające trzy skoniugowane wiąza-
nia podwójne wyizolowano po raz pierwszy z leuko-
cytów. Tak jak w przypadku prostaglandyn, u ludzi
3.3.1.3. Tromboksan A2 dominują produkty utleniania kwasu arachidonowego
zawierające dodatkowe (czwarte) wiązanie podwójne
– na co wskazuje indeks 4 stosowany w skrótach.
Przy udziale syntazy tromboksanu w płytkach krwi
Mediatory
Szczególną rolę odgrywa znajdująca się w leuko-
powstaje tromboksan A2 (TxA2), który stymulując
cytach 5-lipooksygenaza, która po pobudzeniu ko-
receptory TP (zob. ryc. B 3.3-1), pobudza agregację
mórki wiąże się z obecnym w błonie komórkowym
trombocytów, a w konsekwencji tworzenie skrzepu
białkiem aktywującym 5-lipooksygenazę (FLAP), co
płytkowego. Poza tym wykazuje on działanie naczy-
zapoczątkowuje syntezę leukotrienów przez produkt
nioskurczowe. Jest on w tym względzie antagonistą B3
pośredni 5-HPETE.
prostacykliny. Jest uwalniany przede wszystkim pod-
Z 5-HPETE powstaje nie tylko 5-HETE, ale tak-
czas adhezji trombocytów do uszkodzonego śród-
że 5,6-epoksyd (LTA4), który (w leukocytach) ulega
błonka naczyń.
hydrolizie enzymatycznej do LTB4 lub przy udziale
Substancje hamujące agregację płytek krwi opisa-
S-transferazy glutationu (w eozynofilach i bazofi-
no w rozdziale B 4.1.4.2.
lach) zostaje przekształcony do LTC4. Z LTC4, który
należy do leukotrienów cysteinylowych (peptydoleu-
kotrienów skoniugowanych z cysteiną), poprzez od-
3.3.1.4. Inhibitory cyklooksygenazy szczepienie kwasu glutaminowego powstaje LTD4,
a z niego po odszczepieniu glicyny LTE4.
Opisane powyżej różne drogi przemiany kwasu ara- Leukotrieny cysteinylowe (LTC4, LTD4, LTE4)
chidonowego czynią zrozumiałym fakt, że wpływ tworzą tzw. wolno działającą substancję anafilaktycz-
farmakologiczny na te procesy ma dużą wartość tera- ną (slow-reacting substance of anaphylaxis, SRS-A,
peutyczną. Działanie niesteroidowych leków przeciw- zob. poniżej).
zapalnych (NLPZ) opiera się w dużej mierze na wpły- W analogiczny sposób powstają w trombocytach
wie na kaskadę kwasu arachidonowego poprzez za pomocą 12-lipooksygenazy i w granulocytach przy
hamowanie aktywności cyklooksygenazy. Jest zro- udziale 15-lipooksygenazay 12-HPETE i 15-HPETE,
zumiałe, ze zahamowanie cyklooksygenazy nasila które zostają następnie przekształcone do odpowied-
syntezę innych pochodnych kwasu arachidonowego nich HETE. 15-HETE jest w końcowym etapie prze-
(zob. poniżej) i tą drogą prowadzi do rozwoju działań kształcany do tzw. lipoksyny.
niepożądanych (np. rozwoju tzw. astmy aspirynowej
na skutek stosowania NLPZ). Receptory leukotrienowe. Tak jak w przypadku
prostaglandyn, zidentyfikowano wiele receptorów
leukotrienowych. Pobudzenie receptora dla LTB4 ak-
tywuje fosfolipazę C i tą drogą prowadzi do powsta-
3.3.2. Substancje nia trifosforanu inozytolu (IP3) i wzrostu stężenia
szlaku lipooksygenazy wapnia wewnątrzkomórkowego w leukocytach. Na-
stępstwem tego jest nasilona chemotaksja granulocy-
Oprócz przemian pod wpływem cyklooksygenazy, tów obojętnochłonnych i makrofagów, synteza białek
kwas arachidonowy może być utleniany pod wpły- adhezyjnych i wolnych rodników tlenowych, a także
wem lipooksygenazy (LOX) do hydroperoksydów. uwolnienie enzymów lizosomalnych. Istnieje jeszcze
MUTSCHLER-2009.indd 507 2010-01-07 22:13:27
508 Mediatory (autokoidy, hormony tkankowe)
FLAP
PLA2 5-LOX
kwas arachidonowy
leukocyt, 5-HPETE
mastocyt,
trombocyt
leukotrien A4
leukotrien B4 leukotrien C4
leukotrien D4 leukotrien E4
leukotrien B4 leukotrien C4
BLT
leukocyt Gq montelukast CysLT
Gq
IP3/Ca2+
IP3/Ca2+
chemotaksja
mięśnie oskrzeli skurcz
eozynofile migracja
komórki gruczołowe wydzielanie śluzu
Ryc. B 3.3-2. Leukocyty, mastocyty, trombocyty i komórki nowotworowe są w stanie wytwarzać leukotrieny. Z fosfolipidów błony
komórkowej za pomocą fosfolipazy A2 (PLA2) jest syntezowany kwas arachidonowy, który pod wpływem 5-lipooksygenazy (5-LOX)
jest przekształcany do kwasu hydroperoksyeikozatetraenowy (5-HPETE). Aktywność 5-LOX jest regulowana przez FLAP (białko ak-
tywujące 5-lipoksygenazę). 5-HPETE zostaje przekształcony w leukotrien A4, z którego powstają leukotrieny B4 i C4. Specjalne białka
transportowe odpowiadają za wydzielanie leukotrienów z komórek. Leukotrien B4, aktywując receptor BLT, wpływa na chemotaksję
komórek zapalnych, natomiast leukotrien C4 może ulec dalszemu przekształceniu do leukotrienów D4 i E4. Trzy ostatnie leukotrieny
zawierają w swej cząsteczce cysteinę (= leukotrieny cysteinylowe) i wiążą się z receptorem CysLT1, którego pobudzenie powoduje
skurcz oskrzeli, produkcję śluzu przez komórki gruczołowe i migrację eozynofili. Receptor BLT – receptor dla leukotrienów B4.
co najmniej jeden receptor leukotrienowy (receptor
patofizjologii istotny jest ich udział w procesach za-
CysLT1), za pomocą którego leukotrieny cysteiny-
palnych związany z syntezą wolnych rodników tleno-
lowe wywołują skurcz oskrzeli i nadmierną sekrecję
wych i uwalnianiem enzymów lizosomalnych, które
śluzu w drzewie oskrzelowym. Tą samą drogą leuko-
powodują depolimeryzację związków macierzy tkan-
trieny wywołują skurcz naczyń wieńcowych.
ki łącznej (np. kolagenu, kwasu hialuronowego), de-
naturację enzymów, uszkodzenie błon komórkowych
Znaczenie fizjologiczne i patofizjologiczne.
i zwiększenie przepuszczalności naczyń. Przypusz-
5-HETE, 12-HETE, a w szczególności LTB4, dzia-
cza się, że LTB4 uczestniczy w rozwoju nieswoistych
łają chemotaktycznie na leukocyty, fibroblasty i ke-
zapaleń jelit (wrzodziejącego zapalenia jelita grube-
ratynocyty. Z tego względu odgrywają one istotną
go i choroby Crohna), reumatoidalnego zapalenia sta-
rolę w procesie gojenia się ran. Z punktu widzenia
wów i łuszczycy.
MUTSCHLER-2009.indd 508 2010-01-07 22:13:27
Mediatory (autokoidy, hormony parakrynne) 509
LTC4, LTD4 i LTE4 uczestniczą dla odmiany 3.3.2.2. Inhibitory lipoksygenazy
w rozwoju astmy oskrzelowej. Wywołują one bardzo
silny skurcz oskrzeli (ok. 1000 razy silniejszy niż hi-
Wpływ hamujący na lipooksygenazę wykazano
stamina!), jednak ich działanie rozpoczyna się póź-
w przypadku mesalazyny, cenionej w terapii wrzo-
niej niż innych mediatorów (stąd określenie „wolno
dziejącego zapalenia jelita grubego. Odpowiada on
działająca substancja anafilaktyczna”; slow-reacting
przynajmniej częściowo za działanie terapeutyczne
substance of anaphylaxis, SRS-A). Poza tym opisano
mesalazyny oraz jej prekursorów: olsalazyny i sulfa-
działanie naczynioskurczowe leukotrienów.
salazyny. Skuteczność sulfasalazyny jako leku pod-
stawowego w reumatoidalnym zapaleniu stawów
3.3.2.1. Antagonisci leukotrienów może także wynikać z tego punktu uchwytu. Wyni-
ki badań z inhibitorami 5-lipooksygenazy w terapii
Antagonistów receptorów leukotrienowych stosowa- astmy są jak na razie rozczarowujące (np. zileuton
nych w leczeniu astmy oskrzelowej, takich jak mon- nie został w Niemczech zarejestrowany).
telukast, opisano w rozdziale B 5.3.2.2.1.3.
Mediatory
3.4. Czynnik aktywujący płytki
Nazwa czynnika aktywującego płytki wywodzi się z kwasem arachidonowym jest magazynowany jako
z faktu, że został on po raz pierwszy stwierdzony prekursor PAF. Poza płytkami krwi PAF mogą synte- B3
w trombocytach. zować komórki zapalne.
Do powstania PAF konieczna jest aktywacja fosfo-
lipazy C, np. przez trombinę lub fibrynogen i wapń. Znaczenie fizjologiczne i patofizjologiczne. Istot-
PAF nie tylko uczestniczy w agregacji trombocytów, ny z punku widzenia fizjologii jest udział PAF w he-
ale także prowadzi do skurczu oskrzeli, spadku ciś- mostazie. Bierze on udział w patofizjologii tworzenia
nienia tętniczego krwi i wzrostu przepuszczalności zakrzepów. Poza tym w związku z działaniem bron-
naczyń oraz małopłytkowości i leukopenii. Jego chospastycznym i chemotaktycznym współuczestni-
działanie wynika z wiązania się z receptorem bło- cy on w rozwoju astmy oskrzelowej. Prawdopodobny
nowym i aktywacji fosfolipazy C. W surowicy lub jest udział PAF w rozwoju zapalnych i alergicznych
w nieaktywowanych komórkach PAF jest inaktywo- chorób skóry.
wany na drodze deacetylacji, a po ponownej acylacji
3.5. Kininy
Do kinin, biologicznie aktywnych peptydów, należą i krwi obwodowej w ciągu kilku minut pod wpływem
nonapeptyd bradykinina oraz dekapeptyd kalidy- specyficznej peptydazy (kininazy). Kininaza I od-
na. Kininy są w osoczu odszczepiane przez proteazę szczepia jeden, a kininaza II (tak samo jak w przy-
serynową, tzw. kalikreinę, od α2-globuliny – kinino- padku enzymu konwertującego angiotensynę, ACE)
genu. Nieaktywny prekursor kalikreiny, prekalikrei- dwa aminokwasy od C-końca peptydu.
na (kalikreinogen), powstaje w różnych narządach
i tkankach, np. w trzustce i osoczu krwi. Aktywacja Działanie. Kininy są bardzo aktywnymi związkami.
osoczowej prekalikreiny zachodzi pod wpływem Zwiększają one pojemność minutową serca, powodu-
czynnika Hagemana (czynnik XII układu krzepnię- ją rozszerzenie obwodowych naczyń krwionośnych
cia). Po enzymatycznym podziale kininogenu przez i tą drogą obniżenie ciśnienia tętniczego krwi. Poza
kalikreinę osoczową powstaje bradykinina, a po za- tym zwiększają przepuszczalność naczyń włosowa-
działaniu enzymu trzustkowego – kalidyna. Rozkład tych, prowadząc do powstania obrzęków, mają silne
biologicznie aktywnych kinin zachodzi w tkankach działanie bronchospastyczne oraz są w stanie spowo-
MUTSCHLER-2009.indd 509 2010-01-07 22:13:27
510 Mediatory (autokoidy, hormony tkankowe)
dować zarówno skurcz, jak i relaksację mięśniówki nej na chemiczne i termiczne bodźce nocyceptywne.
gładkiej jelit. Kininy są także mediatorami pobudzającymi recep-
Ich działanie jest następstwem pobudzenia re- tory bólowe, a wywołując tą drogą odczucia bólowe,
ceptorów bradykininowych, których znane są dwa współuczestniczą w powstaniu typowych miejsco-
podtypy: B1 i B2; za wystąpienie opisanych powyżej wych objawów zapalenia (przekrwienie, obrzęk i ból).
działań odpowiada pobudzenie receptora B2. Oba re- Poza tym przypuszcza się, że uwolniona z uszkodzo-
ceptory są związane z białkiem G, a przekaźnikami nych tkanek kalikreina razem z innymi czynnikami
wtórnymi są trifosforan inozytolu i diacyloglicerol. prowadzi do rozwoju wstrząsu w przebiegu ciężkiego
Aktywacja fosfolipazy A2 i następcza nasilona syn- zapalenia trzustki. Z punku widzenia farmakokine-
teza prostaglandyny E2 lub prostacykliny prowa- tyki istotny jest udział bradykininy w wywoływaniu
dzi do rozszerzenia naczyń krwionośnych i skurczu kaszlu w trakcie terapii inhibitorami ACE.
mięśni gładkich przewodu pokarmowego.
Aprotynina. Aprotynina jest inhibitorem kalikreiny
Znaczenie fizjologiczne i patofizjologiczne. Re- i innych proteaz (np. trypsyny, chymotrypsyny, pla-
ceptory B2, a tym samym prawdopodobnie także zminy). Jest ona składającym się z 58 aminokwasów
bradykinina są odpowiedzialne za rozwój prawidło- peptydem, którego zastosowanie omówiono w roz-
wo funkcjonujących nerek, natomiast receptory B1 dziale B 4.1.4.5.
odgrywają istotną rolę w inicjacji odpowiedzi zapal-
MUTSCHLER-2009.indd 510 2010-01-07 22:13:28
Krew 511
4. Serce i układ krążenia
Serce i układ krążenia stanowią funkcjonalną ca- ■ serca jako pompy,
łość składającą się z:
■ systemu regulacji nerwowej, hormonalnej oraz
■ krwi jako środka transportu, śródbłonkowej (miejscowej).
■ naczyń jako dróg transportu,
4.1. Krew
Z czynnościowego punktu widzenia krew można gotrwałym intensywnym poceniu się lub wskutek
określić jako płynną tkankę. Składa się ona z komó- niskiej podaży płynów) lub zwiększeniu (hiperwole-
Serce i układ krążenia
rek (krwinek) oraz osocza, które jest płynem zawie- mia u niemowląt, kobiet w ciąży lub u ludzi żyjących
rającym białka i elektrolity. Wszystkie komórkowe wysoko nad poziomem morza).
składniki krwi powstają ze wspólnych komórek pre-
kursorowych, tzw. pluripotencjalnych komórek ma- Utrata krwi. Utrata krwi (wskutek zewnętrznego
cierzystych (zob. ryc. B 4.1-1). bądź wewnętrznego krwawienia) o objętości do 500
(–800) ml nie prowadzi u zdrowego dorosłego czło-
Zadania krwi. Głównym zadaniem krwi, której wieka do istotnych zaburzeń funkcjonowania układu
skład i objętość ulega jedynie niewielkim wahaniom, sercowo-naczyniowego. Przy utracie krwi większej
jest transport substancji. Tlen pobrany w płucach niż 30% prawidłowej objętości występują objawy
jest dostarczany przez erytrocyty do wszystkich tka- wstrząsu hipowolemicznego. Nagła utrata więcej niż
B4
nek organizmu, z kolei ditlenek węgla transportowany 50% całkowitej objętości krwi w przypadku braku
jest z tkanek do płuc. Równocześnie z krwią do ko- odpowiedniego postępowania leczniczego jest śmier-
mórek dostarczane są składniki odżywcze, mineral- telna.
ne, hormony i inne mediatory, a także leki i ich me- Narządy, które są niewystarczająco zaopatrywane
tabolity, a produkty metabolizmu wszystkich tych w krew lub w ogóle pozbawione ukrwienia, szybko
związków są następnie z komórek usuwane. Krew przestają funkcjonować w sposób prawidłowy.
odgrywa również decydującą rolę w utrzymywaniu Po niewielkiej utracie krwi (np. 500 ml podczas
pH organizmu, dzięki różnym systemom buforują- oddawania krwi) prawidłowa ilość krwi zostaje
cym (bufor białkowy, fosforanowy i wodorowęgla- przywrócona w ciągu kilku godzin dzięki zastąpie-
nowy). Kolejnym zadaniem krwi jest termoregulacja niu utraconego osocza płynem śródmiąższowym.
organizmu dzięki odprowadzaniu ciepła powstałe- Regeneracja utraconych krwinek trwa znacznie dłu-
go w procesach przemiany materii na powierzchnię żej – ich odtworzenie następuje dopiero po tygodniu.
ciała. Oprócz tego krew uczestniczy w dużej mierze Potrzebne do tego żelazo jest uwalniane z zapasów,
w obronie organizmu przed wnikającymi do środka które z kolei odtwarzane są najwcześniej po ośmiu
ciałami obcymi oraz zarazkami. tygodniach.
Objętość krwi. Całkowita ilość krwi to 7–8% masy
ciała. U dorosłych odpowiada to 4–6 l całkowitej ob- 4.1.1. Składniki komórkowe krwi
jętości krwi (normowolemia). Z tego na układ tętniczy
(wysokociśnieniowy) przypada około 15%, a na układ
4.1.1.1. Erytrocyty
niskociśnieniowy – naczynia włosowate, żyły i krąże-
nie płucne – około 85%. Krew składa się w 44% ze składników komórkowych.
Objętość krwi może ulec zmniejszeniu dzięki Największą ich część stanowią krwinki czerwone
mechanizmom fizjologicznym (hipowolemia po dłu- (erytrocyty) – pozbawione jąder, dwuwklęsłe dyski
MUTSCHLER-2009.indd 511 2010-01-07 22:13:28
512 Krew
IL-1, IL-2,
IL-4, IL-6
limfocyt T
IL-7
limfoidalna komórka
macierzysta IL-2, IL-15 komórka NK
IL-6, IL-1, IL-2,
IL-7 IL-4, IL-6 antygen komórka
plazmatyczna
IL-1
IL-6 limfocyt B
SCF
pluripotencjalna komórka EPO EPO
macierzysta erytrocyt
IL-3
CFU-E
IL-3, IL-4 komórka
dendrytyczna
SCF
IL-1
IL-3 GM-CSF
monocyt
IL-6 M-CSF
IL-3
GM-CSF granulocyt
CFU-GM obojętnochłonny
G-CSF
szpikowa komórka
macierzysta
GM-CSF granulocyt
IL-3, IL-5 kwasochłonny
CFU-Eo
IL-3, IL-4 granulocyt
zasadochłonny
CFU-Bas
GM-CSF TPO
trombocyt
IL-3, IL-6
CFU-Mega megakariocyt
Ryc. B 4.1-1. Uproszczone przedstawienie hematopoezy z wybranymi czynnikami regulującymi wzrost. Strzałki oznaczają kierunek
różnicowania się. CFU – zdeterminowane komórki macierzyste (colony forming units), IL interleukiny, SCF – czynnik komórek pnia
(stem cell factor), CSF – czynnik wzrostowy kolonii (colony stimulating factor), GM – granulocyty i monocyty, EPO – erytropoetyna,
TPO – trombopoetyna.
MUTSCHLER-2009.indd 512 2010-01-07 22:13:28
Krew 513
o średnicy około 7,5 μm (normocyty) i grubości od 1 zwierzęce (np. jad węży) i bakteryjne, konflikty sero-
μm w części środkowej do 2 μm w części obwodo- logiczne (nieprawidłowości przy przetoczeniu krwi)
wej. Zawierają one odpowiedzialną za przenoszenie oraz czynniki mechaniczne i termiczne. Wskutek
tlenu hemoglobinę (zob. poniżej). W 1 μl krwi męż- tego nieprzepuszczalna wcześniej dla światła krew
czyzny stwierdza się średnio 5,2 miliona erytrocytów, staje się przejrzysta i słabo wybarwiona.
a u kobiety 4,6 miliona erytrocytów.
Wytwarzanie, czas życia i rozpad. Po urodzeniu ery- 4.1.1.1.1. Hemoglobina
trocyty wytwarzane są w czerwonym szpiku kostnym
(zob. ryc. B 4.1-1). Powstają z komórki macierzy- Około 30% zawartości erytrocytu stanowi czerwo-
stej, która różnicuje się następnie do proerytroblastu. ny barwnik krwi – hemoglobina. Służy ona przede
Po wchłonięciu żelaza i syntezie hemoglobiny prze- wszystkim jako substancja transportująca tlen z płuc
kształca się on do makroblastu, z którego, po zagęsz- do tkanek oraz ditlenek węgla z tkanek do płuc.
czeniu i zmniejszeniu jądra, powstaje normoblast. Oprócz tego pełni ona funkcję buforu. Zawartość he-
Pyknotyczne jądro zostaje następnie usunięte, a po- moglobiny u mężczyzny wynosi średnio 130–160 g/l
wstały w ten sposób erytrocyt przechodzi do krwi krwi.
krążącej. Nie w pełni rozwinięte erytrocyty zawie-
rają jeszcze podobne do siateczki struktury i dlate- Budowa cząsteczki hemoglobiny. Mająca w przy-
go nazywane są retikulocytami. Każde wzmożenie bliżeniu kształt kuli hemoglobina jest chromoprotei-
erytropoezy prowadzi w rezultacie do zwiększenia ną (zob. ryc. B 4.1-2), składającą się z czterech łań-
Serce i układ krążenia
frakcji retikulocytów w krwi krążącej. Niedobór tle- cuchów polipeptydowych, z których każdy zawiera
nu w tkankach będący silnym bodźcem do tworzenia w swojej cząsteczce barwnik określany jako hem.
erytrocytów prowadzi do uwolnienia z nerek erytro- Masa cząsteczkowa hemoglobiny wynosi 64 500 Da.
poetyny, która wraz z krwią dociera do czerwonego Hemoglobina dorosłych (HbA) składa się z ułożo-
szpiku kostnego, gdzie pobudza różnicowanie się ko- nych w sposób symetryczny dwóch łańcuchów po-
mórek macierzystych. lipeptydowych α zawierających 141 aminokwasów
Czas życia krążących we krwi erytrocytów wy- oraz dwóch łańcuchów polipeptydowych β zawiera-
nosi przeciętnie 110–120 dni. Następnie są one (lub jących 146 aminokwasów.
ich fragmenty) fagocytowane przez komórki układu Część hemowa zawiera strukturę protoporfiryno-
siateczkowo-histiocytarnego w śledzionie, wątrobie wą z dwuwartościowym atomem żelaza w swoim cen-
B4
i szpiku kostnym. Oznacza to, że w ciągu 24 godzin trum. Struktura protoporfirynowa składa się z czte-
około 0,8% wszystkich erytrocytów ulega rozkładowi rech pierścieni pirolowych połączonych mostkami
i jest zastępowanych nowo wytworzonymi. W każdej metinowymi i posiadających różnorakie łańcuchy
minucie powstaje więc 160 milionów nowych czerwo- boczne.
nych ciałek krwi.
Wiązanie tlenu przez hemoglobinę. Tlen przeno-
Hematokryt. Hematokryt jest to stosunek objętości szony jest przez krew w znaczącej większości w połą-
erytrocytów do całkowitej objętości krwi. Wynosi on czeniu wiązaniem koordynacyjnym z dwuwartościo-
u mężczyzny średnio 0,47, a u kobiety 0,42. Oznacza wym atomem żelaza grupy hemowej. W wyniku tego
to, że prawie połowę objętości krwi stanowią krwinki dochodzi do powstania oksyhemoglobiny. Ponieważ
czerwone.
Do podwyższenia wartości hematokrytu dochodzi
wskutek niedoboru tlenu podczas pobytu na dużych
wysokościach nad poziomem morza, a także u nowo- CH2
CH3
rodków. U małych dzieci z kolei wartość hematokry-
H3C
tu jest często niższa. CH2
N N
2+
Hemoliza. W roztworach hipo- i hipertonicznych Fe
N N
oraz w obecności substancji powierzchniowo czyn-
nych lub lipofilnych rozpuszczalników (eter, chloro- H3C CH3
form i in.) dochodzi do uwolnienia się hemoglobiny HOOC COOH
z erytrocytów. Tego typu uszkodzenie erytrocytu
określa się mianem hemolizy. Hemolizę mogą powo-
dować także niektóre leki (np. metylodopa), toksyny Ryc. B 4.1-2. Budowa hemu.
MUTSCHLER-2009.indd 513 2010-01-07 22:13:28
514 Krew
hemoglobina jest tetramerem, jej 1 mol może przy- Oprócz głównych grup układu AB0 rozróżnić można także
łączyć maksymalnie 4 mole O2. O tym, jaki odsetek podgrupy, np. A1 i A2.
hemoglobiny występować będzie pod postacią oksy-
hemoglobiny decyduje stężenie rozpuszczonego O2, Kiedy dojdzie do połączenia erytrocytów określonej
a także powinowactwo O2 do hemoglobiny (tzw. na- grupy krwi w układzie AB0 z osoczem zawierają-
sycenie O2). cym skierowane przeciw nim przeciwciała, dochodzi
Powinowactwo O2 do hemoglobiny zależy od wie- do aglutynacji: erytrocyty zlepiają się ze sobą, a na-
lu czynników, a w szczególności od wartości pH, stępnie ulegają hemolizie. Stąd w przypadku transfuzji
ciśnienia parcjalnego CO2, stężenia kwasu 2,3-difo- niezgodnej grupowo krwi może dojść do ciężkich po-
sfoglicerynowego wewnątrz erytrocytu oraz od tem- wikłań (żółtaczka hemolityczna, wstrząs hemolitycz-
peratury. ny), zwłaszcza jeśli osocze biorcy zawiera przeciw-
Zmniejszenie wartości pH, podwyższenie ciśnie- ciała przeciw erytrocytom dawcy. W sytuacji odwrot-
nia parcjalnego CO2, zwiększenie stężenia kwasu nej, tzn. kiedy to krew dawcy zawiera przeciwciała
2,3-difosfoglicerynowego oraz podwyższenie tem- przeciw erytrocytom biorcy, reakcja ta, ze względu
peratury prowadzą do zmniejszenia powinowactwa na duże rozcieńczenie przeciwciał w krwiobiegu, ma
tlenu do hemoglobiny. I odwrotnie – zwiększenie lżejszy przebieg.
wartości pH, obniżenie ciśnienia parcjalnego CO2 lub
obniżenie temperatury prowadzą do wzrostu powino- Układ Rh. Oprócz układu AB0 duże znaczenie kli-
wactwa tlenu do hemoglobiny (przesunięcie krzywej niczne ma również układ antygenowy Rh, nazwany
wiązania O2 w lewo). tak, ponieważ po raz pierwszy stwierdzono go u małp
z gatunku rezus (rhesus = Rh). Jest to skomplikowa-
Wiązanie ditlenku węgla przez hemoglobinę. Di- ny układ z licznymi antygenami, spośród których naj-
tlenek węgla transportowany jest przez krew z tkanek większe znaczenie ma dziedziczony w dominujący
do płuc zarówno w formie rozpuszczonej we krwi, sposób antygen D. Osoby, które posiadają ten anty-
jak i związanej chemicznie. Wiąże się on w sposób gen – czyli około 85% populacji Europy – określa się
odwracalny przede wszystkim w postaci wodoro- mianem osób Rh-dodatnich, a osoby, które go nie po-
węglanu w erytrocytach i osoczu, a w mniejszym siadają, jako osoby Rh-ujemne. Jeżeli erytrocyty Rh+
stopniu poprzez przyłączenie się do wolnych grup pojawią się we krwi kobiety Rh– na przykład wsku-
aminowych hemoglobiny, w wyniku czego powstaje tek przetoczenia krwi lub po porodzie dziecka Rh+,
karbaminohemoglobina. to dochodzi u niej do wytworzenia się przeciwciał
przeciw antygenowi D. Przy kolejnej ciąży istnieje
ryzyko, że poziom przeciwciał anty-Rh+ będzie tak
wysoki, że u płodu Rh+ dojść może do zagrażającej
4.1.1.1.2. Grupy krwi życiu hemolizy erytrocytów. Przeciwciała, które prze-
nikają przez barierę łożyskową, uszkadzają bowiem
Ludzkie erytrocyty posiadają na swojej błonie dużą układ erytropoetyczny płodu oraz prowadzą do he-
ilość specyficznych, genetycznie zdeterminowanych molizy jego erytrocytów (erytroblastoza, żółtaczka
struktur o właściwościach antygenowych. Przeciw hemolityczna noworodków). U nowo narodzonego
wielu z tych antygenów mogą być wytwarzane prze- dziecka z erytroblastozą terapią z wyboru jest natych-
ciwciała, przy czym wobec własnych antygenów wy- miastowa transfuzja wymienna krwi.
stępuje w normalnych warunkach tolerancja. Dzięki W celu profilaktyki uczulenia na antygen Rh,
różnym właściwościom antygenowym błony ko- a co za tym idzie – erytroblastozy, matkom z grupą
mórkowej erytrocytów wyróżnić można różne grupy krwi Rh– wstrzykuje się immunoglobulinę anty-D.
krwi. Do chwili obecnej poznano ponad 30 układów Standardowa dawka to 300 mg i.m. w 28 tygodniu
grupowych krwi, spośród których największe znacze- ciąży oraz po inwazyjnych zabiegach (np. amniocen-
nie kliniczne mają układy AB0 oraz Rh. tezie) i urodzeniu dziecka Rh+.
Układ AB0. Antygeny tego układu to specyficzne cu-
kry, które są za pomocą enzymów związane z biał- 4.1.1.1.3. Metabolizm żelaza
kami lub lipidami błony komórkowej erytrocytów.
Rozróżnia się antygeny A, B i AB, a u ludzi z grupą Całkowita zawartość żelaza w organizmie dorosłego
krwi 0 brakuje enzymu odpowiedzialnego za tworze- człowieka wynosi 3,5–5 g. Z tego w hemoglobinie
nie antygenów grupowych krwi bądź też nie funkcjo- znajduje się 65–70%, w mioglobinie ok. 4%, w szpi-
nuje on prawidłowo (stąd określenie układ AB0) ku kostnym 2,5%, w enzymach zawierających żelazo
MUTSCHLER-2009.indd 514 2010-01-07 22:13:28
Krew 515
0,1%, a tzw. żelazo zmagazynowane (w ferrytynie żelaza przekazywana do krwi, co znaczy, że system
i hemosyderynie, zob. poniżej) stanowi prawie 25%. transportujący żelazo dostosowuje się każdorazowo
Dzienna utrata żelaza u mężczyzny wynosi ok. 1 mg, do zapotrzebowania. Jednakże, inaczej niż uważano
a u dojrzałej płciowo kobiety około 2 mg. Straty te w przeszłości, system ten nie jest w stanie chronić
wynikają ze złuszczania się komórek – szczególnie organizmu przed nadmierną podażą żelaza.
w przewodzie pokarmowym – a także z utraty krwi
podczas miesiączki. Transport, wykorzystanie i magazynowanie żela-
Wiarygodnym wskaźnikiem zawartości żelaza za. Żelazo po uprzednim utlenieniu do postaci trój-
w organizmie jest stężenie ferrytyny w surowicy krwi wartościowej wiąże się we krwi z białkiem trans-
(zob. poniżej). Prawidłowa wartość wynosi 100– portującym – transferyną i jest dalej transportowane
250 μg/l. Wartości poniżej 12 μg/l oznaczają niedo- jako kompleks transferyna-żelazo. Dopiero od tego
bór żelaza. momentu możliwe jest wykorzystanie żelaza przez
potrzebujące komórki dzięki obecności na ich po-
Wchłanianie żelaza. Z żelaza dostarczanego wraz wierzchni odpowiednich receptorów dla transferyny.
z pożywieniem dziennie wchłania się 10–12 mg, Maksymalnie transferyna może związać 14 mg żela-
jeśli nie występuje jego niedobór (zob. poniżej).
Z tego 10–15% wchłania się w dwunastnicy oraz enterocytarne przyswajanie żelaza
górnym odcinku jelita czczego (zob. ryc. B 4.1-3).
W ten sposób w pełni wyrównywana jest fizjolo- światło krew
jelita enterocyt
giczna utrata żelaza.
Serce i układ krążenia
W pokarmach pochodzenia roślinnego obecne ferroportyna
są pewne aniony (np. fosforany, fityniany), z którymi DMT1
żelazo w nich zawarte tworzy słabo rozpuszczalne sole Fe2+ Fe2+ Fe2+
i z tego powodu wchłania się znacznie gorzej niż że-
lazo zawarte w pokarmach pochodzenia zwierzęcego.
ferrytyna
Żelazo wchłania się głównie w postaci dwuwar- Dcytb
tościowej, ponieważ przy wartościach pH panujących Fe3+ hepcydyna
w górnej części jelita cienkiego (pH 5–7) trójwartoś-
ciowe żelazo tworzy słabo wchłaniające się wodoro-
tlenki. Oprócz tego białko przenośnikowe – transpor- enterocyt B4
ter metali dwuwartościowych (divalent metal trans-
porter 1, DMT1) – jest specyficzne względem żelaza
dwuwartościowego. Trójwartościowe żelazo może
jednak przejść w dwuwartościowe dzięki reduktazie
żelaza (Dcytb), a następnie zostać wchłonięte za po- wchłanianie
średnictwem DMT1. Transfer cząsteczek żelaza przez w jelicie 1–2 mg
komórki enterocytów jest do chwili obecnej słabo
poznany. Transport żelaza przez błonę podstawną
10 – armem
szp
z pok daż
ik ko
12 m
enterocytu do krwi zachodzi dzięki białku transpor-
po
stny
tującemu z rodziny SLC, ferroportynie (SLC40A1).
Nowo odkryta proteina syntetyzowana w wątrobie żelazo w osoczu dobowa
– hepcydyna – jest w stanie wpływać na ferropor- wymiana
tynę, a co za tym idzie – regulować przechodzenie 3 mg 20–30 mg
dobowa
żelaza do krążenia (zob. ryc. B 4.1-3). Za wchłania-
zmaga ina, en
wymiana
mioglo
nie i możliwość wykorzystania żelaza odpowiedzial- 2 mg
żela owa
y yt
zyn zy
b
zo, n
troc
ny jest więc system, który jest w stanie dopasować
e
er y
poziom przyswajania żelaza do potrzeb organizmu: my ,
z wchłoniętego żelaza do krążenia przechodzi okre-
ślona część, a reszta magazynowana jest w komórkach utrata
1 mg
śluzowych jako ferrytyna błony śluzowej. Ferrytyna 2 mg
powstaje wskutek połączenia się żelaza z białkiem
o masie 450 kD zwanym apoferrytyną. Cząsteczka
apoferrytyny może związać do 4500 cząsteczek że- Ryc. B 4.1-3. Schematyczne przedstawienie metabolizmu że-
laza. W przypadku niedoboru żelaza wzrasta część laza.
MUTSCHLER-2009.indd 515 2010-01-07 22:13:28
516 Krew
Tab. B 4.1-1. Dzienne zapotrzebowanie na żelazo (w mg) w różnych okresach życia (według Beckera)
Całkowite dzienne Dzienne wydalanie Dodatkowe dzienne zapotrzebowanie
zapotrzebowanie żelaza na żelazo spowodowane
na żelazo wzrostem miesiączką ciążą
Dorośli mężczyźni 1,0 1,0
Miesiączkujące kobiety 1,8 1,0 0,8
Kobiety po menopauzie 1,0 1,0
Dzieci 1,2 0,2 1,0
Mężczyźni w okresie dojrzewania 2,0 1,0 1,0
Kobiety w okresie dojrzewania 2,8 1,0 1,0 0,8
Kobiety ciężarne 3,7 1,0 2,7
za. W prawidłowych warunkach w osoczu w posta- żelaza. Dlatego, zwłaszcza w drugiej połowie ciąży,
ci związanej z transferyną występuje jedynie około wskazane jest podawanie (po odpowiedniej kontroli
4 mg żelaza. Całkowita zdolność wiązania wykorzy- laboratoryjnej) dodatkowych 50 mg żelaza dziennie.
stywana jest zaledwie w jednej trzeciej. W tab. B 4.1-1 podano wartości dziennego zapo-
Większa część żelaza wykorzystywana jest do trzebowania na żelazo w różnych okresach życia.
syntezy hemoglobiny. Dodatkowym źródłem żelaza
jest żelazo pochodzące z codziennego rozpadu ery-
trocytów, w ilości 20–30 mg. 4.1.1.2. Leukocyty
Żelazo jest ponadto potrzebne w postaci tzw. żela-
za czynnościowego jako składnik mioglobiny i enzy-
W przeciwieństwie do erytrocytów leukocyty zawie-
mów zawierających żelazo.
rają jądro komórkowe. Ich ilość nie jest tak samo
Oprócz komórek śluzowych żelazo magazynowa-
stała jak erytrocytów. Średnia liczba leukocytów wy-
ne jest przede wszystkim w wątrobie, śledzionie oraz
nosi 6000/μl krwi, ale wartość prawidłowa mieści się
w szpiku kostnym w postaci opisanej wyżej rozpusz-
w zakresie 4000–10 000/μl. W przypadku wartości
czalnej ferrytyny, skąd w razie potrzeby może zostać
powyżej 10 000/μl mówi się o leukocytozie, a w przy-
szybko zmobilizowane. Przy zbyt dużej podaży nad-
padku mniejszej niż 4000/μl o leukopenii.
miar żelaza magazynowany jest jako nierozpuszczal-
We wzorze krwinek rozróżnia się następujące białe
na hemosyderyna, która powstaje wskutek degradacji
ciałka krwi:
ferrytyny (agregacji wielu cząsteczek ferrytyny za
pomocą części zawierających żelazo wraz z częścio- ■ granulocyty, które ze względu na różne wybarwia-
wym rozkładem otoczki białkowej). nie się za pomocą barwników kwaso- i zasado-
chłonnych dzieli się na obojętnochłonne, kwaso-
Zapotrzebowanie na żelazo. Zapotrzebowanie chłonne i zasadochłonne,
człowieka na żelazo odpowiada jego dziennej utracie
■ monocyty,
– odpowiednio 1 mg u mężczyzn i 2 mg u miesiącz-
kujących kobiet. Fizjologicznie zwiększone zapotrze- ■ limfocyty.
bowanie na żelazo występuje w okresie wzrostu oraz
u kobiet w ciąży. W przeliczeniu na masę ciała u dzie- Z tego 55–70% stanowią granulocyty obojętnochłon-
ci zapotrzebowanie to jest większe, a u dorastającej ne, 2–4% kwasochłonne, 0–1% zasadochłonne, 25–
młodzieży znacznie większe niż u dorosłych. Podczas 40% limfocyty, a 2–6% monocyty.
ciąży płód pobiera z organizmu około 400 mg żelaza, Wszystkie leukocyty mają zdolność do ruchu
z czego około 100 mg zawiera łożysko. Jeśli weźmie pełzakowatego i mogą po adhezji do śródbłonka na-
się pod uwagę utratę krwi podczas porodu, to w czasie czyniowego opuszczać światło naczyń włosowatych
ciąży konieczna jest dodatkowa podaż około 800 mg (leukodiapedeza).
MUTSCHLER-2009.indd 516 2010-01-07 22:13:28
Krew 517
4.1.1.3. Granulocyty reakcjach nadwrażliwości, a także w przypadku hi-
perlipoproteinemii oraz w przebiegu przewlekłej bia-
łaczki szpikowej i czerwienicy prawdziwej.
Granulocyty powstają z komórek macierzystych
Komórki tuczne stanowią komórki efektorowe re-
w szpiku kostnym i rozwijają się początkowo przez
akcji alergicznych typu natychmiastowego. Ponadto
podziały komórkowe, a następnie przez dojrzewa-
biorą one udział w innych rodzajach reakcji zapal-
nie. Ich przeciętna długość życia wynosi 8–14 dni,
nych, ponieważ po ich aktywacji posiadają zdolność
z czego we krwi krążącej przebywają tylko około
uwalniania histaminy i serotoniny, a także leukotrie-
14 godzin. Dlatego w prawidłowo funkcjonują-
nów oraz innych substancji chemotaktycznych.
cym szpiku kostnym są one tworzone nieustannie.
Oprócz tego znajduje się tam wystarczający ich za-
pas, który w razie konieczności może zostać natych-
miast wykorzystany. Wpływ czynników wzrostu 4.1.1.4. Monocyty
(G-CSF i GM-CSF) na tworzenie się granulocytów
został w rozdz. 13.1.1. Monocyty o średnicy 12–20 μm stanowią największe
komórki krwi obwodowej. Podobnie jak granulocyty,
Granulocyty obojętnochłonne. Granulocyty obo- są one wytwarzane w szpiku kostnym. Są one w du-
jętnochłonne, zwane też leukocytami o polimorficz- żym stopniu zdolne do ruchu pełzakowatego i dzię-
nym jądrze albo mikrofagami, są odpowiedzialne ki temu mogą przemieszczać się do różnych tkanek,
przede wszystkim za odporność nieswoistą. Nadają gdzie różnicują się w makrofagi. Wraz z limfocytami
Serce i układ krążenia
się one do tej roli szczególnie ze względu na umie- uczestniczą one w mechanizmach odporności nieswo-
jętność samodzielnego poruszania się oraz dużą za- istej, a przede wszystkim swoistej, dzięki zdolności
wartość enzymów proteolitycznych, a także zdolność rozpoznawania i prezentacji antygenów. Monocyty,
do wytwarzania aktywnych związków tlenu i fagocy- a także makrofagi wydzielają zarówno spontanicz-
tozy. Oprócz tego są one zdolne do przeżycia w tkan- nie jak i wskutek aktywacji liczne substancje o bar-
kach słabo zaopatrzonych w tlen dzięki umiejętności dzo różnym działaniu biologicznym. Należą do nich
uzyskiwania energii w procesie glikolizy. Ich liczba enzymy (np. kolagenaza, elastaza) i białka, które
wzrasta przede wszystkim na początku ostrej reakcji uczestniczą w odporności swoistej i nieswoistej (lizo-
zapalnej. zym, składniki układu dopełniacza, interferon, inter-
Ropa składa się w większości z obumarłych granu- leukina-1). Układ monocytów-makrofagów odgrywa B4
locytów obojętnochłonnych oraz ich fragmentów. ponadto ważną rolę w powstawaniu miażdżycy.
Granulocyty kwasochłonne. Nieco większe w po-
równaniu z granulocytami obojętnochłonnymi gra- 4.1.1.5. Limfocyty
nulocyty kwasochłonne również posiadają zdolność
do fagocytozy. Zawierają one duże ziarnistości kwa-
Limfocyty powstają nie tylko w szpiku kostnym,
sochłonne, w których magazynowane są liczne enzy-
ale także w tkankach limfatycznych (węzły chłon-
my i inne substancje. Ilość granulocytów obojętno-
ne, śledziona, migdałki itp.). Pełnią one decydującą
chłonnych we krwi podlega dobowym wahaniom od-
funkcję w mechanizmach swoistej odpowiedzi immu-
wrotnie do poziomu uwalnianych glukokortykostero-
nologicznej organizmu:
idów. Oznacza to, że rano i po południu stwierdza się
mniejższą ich liczbę niż w nocy. ■ limfocyty B po przekształceniu się w komórki pla-
Do wzrostu ilości granulocytów kwasochłonnych zmatyczne produkują przeciwciała odpowiedzialne
(eozynofilia) dochodzi podczas chorób alergicznych, za odporność humoralną,
np. w astmie alergicznej, oraz podczas zakażenia pa-
■ limfocyty T są odpowiedzialne za komórkowe re-
sożytniczego szczególnie robakami.
akcje odpornościowe przeciw obcym tkankom lub
chorobowo zmienionym komórkom organizmu.
Granulocyty zasadochłonne. Komórki tuczne.
Granulocyty zasadochłonne występujące we krwi
Liczba limfocytów wzrasta w przypadku różnych
obwodowej nazywane są także komórkami tucznymi
chorób wirusowych (np. różyczki, wirusowego zapa-
krwi, analogicznie do komórek tucznych w tkankach.
lenia wątroby, wirusowego zapalenia płuc), a także
Zawierają one heparynę, histaminę i serotoninę,
w przypadku przewlekłych reakcji zapalnych.
a także wiele enzymów proteolitycznych (m.in. pro-
We krwi krążącej znajduje się mniej niż 1% lim-
teaz). Wzrost liczby bazofili zachodzi niekiedy przy
focytów, ponieważ większość z nich znajduje się
MUTSCHLER-2009.indd 517 2010-01-07 22:13:28
518 Krew
w tkankach. Ze względu na czas przeżycia można kami osoczowymi dochodzi do rozpoczęcia procesu
wyróżnić wśród nich dwie grupy: krótko żyjące lim- krzepnięcia krwi,
focyty z czasem życia wynoszącym maksymalnie
■ wzmacniają obkurczanie się (retrakcję) skrzepu.
8 dni oraz limfocyty długo żyjące, które mogą przeżyć
kilkaset dni, a prawdopodobnie nawet kilka lat. Analogicznie do wzrostu liczby erytrocytów pod wpływem
erytropoetyny w przypadku trombocytów endogennym
związkiem przyspieszającym rozwój komórek prekursoro-
wych trombocytów – megakariocytów – jest trombopoety-
4.1.1.6. Trombocyty na (TPO), wytwarzana głównie przez wątrobę, nerki i szpik
kostny (zob. ryc. B 4.1-1).
Pozbawione jąder trombocyty (płytki krwi) mają Obecnie w trakcie badań klinicznych są dwie odmia-
kształt dysków o średnicy 1,5–4 μm i grubości 0,5–2 ny otrzymanej metodami genetycznymi trombopoetyny.
μm. Podobnie jak erytrocyty i granulocyty, powstają Rekombinowana ludzka trombopoetyna (rhTPO) zawiera
w szpiku kostnym. Ich tworzenie polega na oddzie- wszystkie 353 aminokwasy. Ponadto badana jest mocno
skrócona odmiana, która ogranicza się jedynie do regionu
leniu się z cytoplazmy megakariocytów (komórek rozpoznawanego przez receptor związanego z glikolem
olbrzymich szpiku kostnego). Ponieważ trombocyty polietylenowym (PEG-rhMGDF – PEG-conjugated re-
są bardzo wrażliwe, ich liczbę ustalić można tylko combinant human megakaryocyte growth and development
dzięki szczególnie ostrożnym metodom pomiaro- factor). TPO powinna być stosowana w przypadku trombo-
wym. U zdrowego człowieka ich liczba we krwi cytopenii, m.in. w trakcie leczenia nowotworów. Ponieważ
trombocyty mogą wychwytywać TPO i w ten sposób wpły-
waha się pomiędzy 160 000 a 300 000/μl. Czas życia wać na jej stężenie obwodowe, kluczowe znaczenie ma
płytek krwi wynosi 1–2 tygodnie. Ich rozpad zacho- czas podawania. (Przy dużej liczbie trombocytów wychwy-
dzi przede wszystkim w układzie siateczkowo-histio- tują one większą ilość TPO i w związku z tym nie wywiera
cytarnym. ona wystarczającego działania). Kwestia ta jest szczególnie
Płytki krwi zawierają liczne ziarnistości, w których istotna w przypadku podawania preparatów płytek krwi
od dawców w ramach leczenia trombocytopenii. Oprócz
znajdują się enzymy hydrolityczne, czynniki krzep- wysokich kosztów oraz możliwych reakcji immunolo-
nięcia krwi oraz czynnik płytkowy 4, który hamuje gicznych istnieje także niebezpieczeństwo, że przetoczone
działanie heparyny. W ich błonie zmagazynowane trombocyty wychwycą TPO i w związku z tym zmniejszą
są kompleksy fosfolipidowe, eksponowane podczas jej wpływ na dojrzewanie megakariocytów w organizmie
aktywacji płytek (tzw. mechanizm flip-flop), a także biorcy. Dotychczasowe badania dotyczące rekombinowanej
TPO są raczej rozczarowujące i dlatego dalszy rozwój idzie
osoczowe czynniki krzepnięcia. raczej w kierunku niskocząsteczkowych analogów TPO.
Na powierzchni trombocytów znajduje się wiele
receptorów odgrywających istotną rolę w procesie W USA dopuszczono rekombinowaną ludzką interleu-
adhezji do struktur podśródbłonkowych (czynnik von kinę-11 o nazwie oprelvekin do leczenia trombocytopenii
Willebrandta – vWF; kolagen, fibronektyna), a także spowodowanej chemioterapią.
tworzenia sieci podczas agregacji trombocytów (re- Zwiększona liczba trombocytów, podobnie jak trombocy-
ceptor GP-IIb/IIIa). Są one ważnym punktem uchwy- topenia, może być leczona. Lekiem z wyboru u młodszych
tu dla substancji farmakologicznych wpływających pacjentów jest interferon α, a u starszych hydroksymocznik
na funkcje płytek. (anagrelid). Dawkowanie początkowe anagrelidu wynosi 1
Trombocyty są niezbędne do zatrzymania krwawie- mg dziennie, dawkę podtrzymującą ustala się indywidualnie
w zależności od liczby płytek krwi. Lek ten jest metaboli-
nia oraz krzepnięcia krwi (zob. poniżej). Spełniają zowany przez cytochrom CYP1A2, a jego okres półtrwania
one przy tym następujące funkcje: wynosi 1,3 godziny. Stosowany jest u pacjentów z pierwot-
ną trombocytemią, którzy nie tolerują wymienionych leków
■ uszczelniają ubytki w naczyniach włosowatych bądź u chorych są one nieskuteczne. Mechanizm działania
również nieuszkodzonych tkanek, tego leku nie jest w pełni poznany. Wykazano, że anagrelid
hamuje dojrzewanie megakariocytów.
■ w razie uszkodzenia naczynia tworzą jego mecha-
niczne zamknięcie (czop trombocytowy),
■ uwalniane z trombocytów mediatory, a w szcze- N NH
gólności tromboksan A2, prowadzą do szybkiego O
N
skurczu naczyń w uszkodzonym układzie naczy- Cl
niowym, Cl
■ po aktywacji trombocytów dzięki uwolnieniu za- anagrelid
wartych w nich czynników w połączeniu z czynni-
MUTSCHLER-2009.indd 518 2010-01-07 22:13:28
Krew 519
4.1.2. Niedokrwistości 4.1.2.1. Niedokrwistości
i środki stosowane z niedoboru żelaza
w niedokrwistościach
Jak już wspomniano, w prawidłowych warunkach
zapotrzebowanie na żelazo jest bardzo niewielkie,
Termin niedokrwistość (anemia) nie jest do końca
ponieważ organizm użytkuje ponownie całe żelazo
właściwy, ponieważ w większości przypadków stan
uwalniane ze związków. Pomimo tego najczęstszą po-
ten nie oznacza niedoboru krwi, a wyłącznie niedobór
stacią niedokrwistości jest niedokrwistość z niedobo-
hemoglobiny lub erytrocytów. Niedokrwistość rozpo-
ru żelaza. W tym rodzaju niedokrwistości całkowita
znaje się, kiedy zawartość hemoglobiny u mężczyzny
ilość hemoglobiny jest obniżona bardziej niż liczba
wynosi mniej niż 130 g/l, a u kobiety mniej niż 120
erytrocytów, więc zawartość hemoglobiny w erytro-
g/l. W celu dokładnego zdiagnozowania rodzaju nie-
cytach jest niższa od prawidłowej. Dlatego też ten ro-
dokrwistości muszą zostać również zbadane ilość,
dzaj niedokrwistości nazywa się niedokrwistością nie-
wielkość i rodzaj erytrocytów oraz wysokość hema-
dobarwliwą. Do przyczyn tego rodzaju anemii należą:
tokrytu. Niekiedy w celu ustalenia dokładnego rozpo-
znania wymagana jest również punkcja szpiku kost- ■ zwiększone zapotrzebowanie na żelazo (np. w okre-
nego. Jeżeli zawartość hemoglobiny w erytrocycie sie wzrostu lub ciąży),
jest prawidłowa, mamy do czynienia z niedokrwistoś-
■ zwiększona utrata żelaza (np. przy przedłużających
cią normochromiczną. W przypadku zmniejszonego
się lub nasilonych krwawieniach miesiączkowych
stężenia hemoglobiny mówi się o niedokrwistości hi-
Serce i układ krążenia
bądź w przypadku krwawień w obrębie przewodu
pochromicznej (niedobarwliwej), a przy podwyższo-
pokarmowego związanych z rozwojem nowotwo-
nym jej stężeniu o niedokrwistości hiperchromicznej
ru),
(nadbarwliwej).
■ niedostateczna podaż żelaza (np. z powodu niewy-
Do przyczyn niedokrwistości zaliczamy: starczającej zawartości żelaza w pożywieniu wy-
stępującej przeważnie przy diecie mlecznej w okre-
■ zaburzenia syntezy hemoglobiny (niedokrwistości
sie dziecięcym lub z powodu zaburzeń wchłaniania
z niedoboru żelaza, niedokrwistość syderoachre-
żelaza w chorobach przewodu pokarmowego).
styczna),
■ upośledzenie erytropoezy (anemia aplastyczna, Ponieważ przyczyną tego typu niedokrwistości
B4
niedokrwistości z niedoboru erytropoetyny = nie- jest niedobór żelaza, leczenie polega na doustnym,
dokrwistość nerkowa, niedokrwistość makrocy- a w wyjątkowym przypadku parenteralnym, poda-
tarna = niedokrwistość z niedoboru witaminy B12 waniu soli żelaza. Ze względu na słabe wchłanianie
i kwasu foliowego, niedokrwistości wskutek terapii żelaza trójwartościowego w leczeniu doustnym po-
cytostatykami i in.), winny być stosowane jedynie preparaty zawierające
żelazo dwuwartościowe.
■ przyspieszony rozpad erytrocytów (anemie hemo-
lityczne),
Preparaty żelaza. Dostępne na rynku doustne prepa-
■ ostre i przewlekłe krwawienia. raty żelaza dwuwartościowego często zawierają sta-
bilizatory, np. kwas askorbinowy, zapobiegające jego
Ogólne objawy wszystkich rodzajów niedokrwisto- utlenieniu się do żelaza trójwartościowego. Dodatek
ści wynikają z niedoboru hemoglobiny. Szczególną kwasów organicznych, np. kwasu bursztynowego
uwagę zwraca blada skóra i śluzówki. Przy dużym nie zwiększa zdolności wchłaniania dostarczonego
obciążeniu wysiłkiem fizycznym wskutek konieczno- żelaza.
ści zwiększenia dopływu krwi dochodzi do znaczne-
go zwiększenia akcji serca. Inne objawy to duszność, Pozajelitowe stosowanie żelaza jest niebezpieczne
zwroty głowy, mroczki przed oczami i tendencja z powodu ryzyka wystąpienia ostrego zatrucia że-
do łatwego męczenia się. lazem oraz uszkodzenia ściany naczynia w miejscu
wstrzyknięcia i dlatego powinno być zarezerwo-
Za pomocą terapii farmakologicznej leczy się przede wane wyłącznie do przypadków zapalnych chorób
wszystkim niedokrwistości z niedoboru żelaza oraz jelit (np. wrzodziejącego zapalenia jelita grubego),
erytropoetyny, a także niedokrwistości megalobla- które mogą się zaostrzyć wskutek podawania żela-
styczne, które omówiono poniżej. za doustnie, oraz w sytuacji, kiedy doustne prepa-
MUTSCHLER-2009.indd 519 2010-01-07 22:13:29
520 Krew
Tab. B 4.1-2. Doustne i pozajelitowe preparaty żelaza do 50% pacjentów) w postaci nudności, wymiotów,
biegunki, ale także zaparć i drgawek. Przyjmowanie
Sól żelaza Preparat handlowy preparatu wraz z posiłkiem lub wkrótce po nim
zmniejsza działania niepożądane, ale może powodo-
Doustnie wać większe wahania ilości wchłoniętego żelaza.
Preparaty żelaza w postaci tabletek musują-
asparginian żelaza (II) Inzelloval
cych są często lepiej tolerowane. Mogą one jednak
fumaran żelaza (II) Ferrum Hausmann Retardkapseln, w przypadku dłuższego podawania powodować
Rulofer N uszkodzenia szkliwa zębów wskutek obniżania war-
glukonian żelaza (II) Eisen-Sandoz, Ferrum Verla, tości pH.
Lösferron, Rulofer G, Vitaferro Przy pozajelitowym podawaniu preparatów żelaza
glicynosiarczan Ferro sanol poza wspomnianym uszkodzeniem ściany naczynia
żelaza (II) może dojść do nudności i wymiotów, a także dole-
gliwości ze strony mięśni i stawów oraz reakcji aler-
bursztynian żelaza (II) Ferrlecit 2 gicznych. Szczególnie w przypadku przedawkowania
i związanego z tym przekroczenia zdolności wiązania
siarczan żelaza (II) Eisendragees-ratiopharm, Eryfer,
Haemoprotect, Vitaferro żelaza przez osoczową transferynę istnieje niebezpie-
czeństwo dużego spadku ciśnienia krwi (wstrząs).
Pozajelitowo
Przy przewlekłym podawaniu nadmiernych ilości
glukonian sodowo- Ferrlecit żelaza jest ono odkładane w układzie siateczkowo-
-żelazowy (III)
śródbłonkowym (hemosyderoza).
Leki zmniejszające kwaśność soku żołądkowego
chlorek żelaza (II) Tracutil
(antacida), szczególnie te zawierające jony magnezu,
chlorek żelaza (III) Addel N, Tracitrans plus wapnia lub glinu, a także kolestyramina upośledza-
ją wchłanianie żelaza. Związki żelaza mogą osłabiać
kompleks wodorotlenku Venofer Ampullen
żelaza (III) i sacharozy wchłanianie niektórych tetracyklin i inhibitorów gy-
razy poprzez tworzenie trudno rozpuszczalnych kom-
kompleks wodorotlenku CosmoFer pleksów.
żelaza (III) i dekstranu
Zatrucie żelazem. Związki żelaza przyjęte w dużej
ilości mogą spowodować śmierć przede wszystkim
u dzieci, rzadko u dorosłych (2000 przypadków rocz-
raty żelaza są bardzo źle tolerowane. Do podawania nie w USA). Dawka śmiertelna siarczanu żelaza (II)
ogólnego stosuje się trójwartościowe sole żelaza dla dzieci w wieku 1–3 lat wynosi ok. 2 g.
w związkach kompleksowych (np. glukonian sodowo- W przypadku zatrucia ostrego, po upływie mniej
-żelazowy (III)) (zob. tab. B 4.1-2). więcej 30–120 minut od przyjęcia, wskutek krwo-
Preparaty zawierające połączenie żelaza z innymi tocznego zapalenia żołądka i jelit pojawiają się
substancjami, np. witaminami, nie zwiększają tera- silne wymioty, bóle żołądka i biegunka. Następnie
peutycznej wartości żelaza i dlatego nie powinny być dochodzi do rozwoju ciężkiego wstrząsu z powo-
stosowane. du silnego rozszerzenia się naczyń krwionośnych.
Przy ustalaniu dawkowania należy uwzględnić Niejednokrotnie może to prowadzić do zgonu.
oprócz ostrego niedoboru żelaza także niedobór żela- U pacjentów, którzy przeżyją obserwuje się często
za zapasowego. Do obliczenia całkowitej dawki żela- po upływie około jednej doby ponowny duży spadek
za można użyć następującego prostego wzoru: ciśnienia krwi, a także drgawki i niekiedy toksycz-
ne zapalenie wątroby. Zatrucie żelazem jest więc
niedobór hemoglobiny w g/dl × 250 = dawka całko- nagłym wypadkiem, którego śmiertelność można
wita w mg. zmniejszyć dzięki możliwie natychmiastowej inter-
wencji terapeutycznej.
W przypadku drogi doustnej podaje się dziennie Leczenie polega przede wszystkim na doustnym
100–300 mg żelaza, a w przypadku drogi parenteral- i pozajelitowym podawaniu deferoksaminy, produktu
nej 20–100 mg. przemiany materii bakterii Actinomyces, który wiąże
Do działań niepożądanych obserwowanych przy się z żelazem, tworząc kompleksy. Deferoksamina
podawaniu żelaza drogą doustną należą przede podana doustnie zmniejsza dalsze wchłanianie żelaza,
wszystkim zaburzenia żołądkowo-jelitowe (nawet a podana pozajelitowo wiąże wchłonięte żelazo, któ-
MUTSCHLER-2009.indd 520 2010-01-07 22:13:29
Krew 521
re jest dzięki temu względnie szybko usuwane przez Ekspresję genu EPO reguluje czynnik induko-
nerki. Korzystne jest przy tym to, że deferoksamina wany hipoksją (HIF-1). Erytropoetyna rozpoczy-
wiąże żelazo z ferrytyny i transferyny, a nie z hemo- na swoje działanie po związaniu się z receptorem
globiny czy cytochromów. na powierzchni komórek prekursorowych erytro-
Dawkowanie wynosi 5–10 g doustnie i równocześ- cytów (CFU-E; zob. ryc. B 4.1-1) i pobudza w ten
nie 1–2 g pozajelitowo. sposób kinazę tyrozynową (JAK2; kinaza Janusowa).
U pacjentów, u których nie rozwinęły się objawy Ta z kolei aktywuje szlak kinaz Ras/MAP, a także
wstrząsu, można także jako środek zmniejszający czynniki transkrypcyjne typu STAT (signal transdu-
wchłanianie zastosować płukanie żołądka za pomocą cer and activator of transcription).
1-proc. roztworu wodorowęglanu sodu (dzięki czemu Jeżeli wskutek choroby nerek (niewydolności)
tworzą się nierozpuszczalne sole węglanu żelaza). hormon ten nie może być wytwarzany i uwalniany
w odpowiedniej ilości, dochodzi do rozwoju nie-
Przedawkowanie. Deferasirox używany jest do le- dokrwistości. Można ją leczyć przez podawanie re-
czenia przewlekłego przedawkowania żelaza wsku- kombinowanej erytropoetyny (epoetyny), co w ciągu
tek częstych przetoczeń (więcej niż 7 ml/kg/mie- 2–6 tygodni prowadzi do wzrostu liczby erytrocytów.
siąc koncentratu krwinek czerwonych) u pacjentów Docelowa wartość hematokrytu powinna wynosić
z beta-talasemią większą oraz w innych stanach nad- 30–35% (nie należy przekraczać wartości 35% z po-
miernej podaży żelaza wskutek częstych przetoczeń. wodu ryzyka wystąpienia ciężkich działań niepożą-
Deferasirox działa jako związek chelatujący, który danych).
jest selektywny względem żelaza trójwartościowego Oprócz leczenia niedokrwistości nerkowej erytro-
Serce i układ krążenia
i wiąże się z nim w stosunku 2:1. Przez to zwiększa poetynę podaje się również w przypadku niedokrwi-
się wydalanie związanego żelaza z kałem. Dzięki ni- stości spowodowanych stosowaniem chemioterapii
skiemu powinowactwu do cynku i miedzi deferasirox czy też chorobami wirusowymi i nowotworowymi,
nie wpływa na poziom tych metali w surowicy krwi. jak również w ramach przygotowania do oddania
Związek ten jest metabolizowany przez UGT1A1 krwi do autotransfuzji. W leczeniu stosuje się różne
i transportowany na zewnątrz komórki za pomocą postacie rekombinowanej erytropoetyny, takie jak:
ABCC2.
■ epoetyna alfa,
Dawka początkowa wynosi 20 mg/kg masy ciała.
■ epoetyna beta.
B4
Okres półtrwania w osoczu wynosi 4–12 godzin.
N
Dawkowanie wynosi zwykle 150–300 jednostek/
kg i.v. lub s.c. trzy razy w tygodniu.
OH N N OH
Darbepoetyna alfa jest kolejną rekombinowaną ery-
tropoetyną z wyraźnie dłuższym okresem półtrwania
(ok. 25 godzin), co wynika z tego, że ma większą
COOH część węglowodanową, a co za tym idzie, większą
masę cząsteczkową. Lek ten może być podawany raz
deferasirox w tygodniu.
Działania niepożądane preparatów erytropoetyno-
wych mogą przypominać objawy grypy oraz podwyż-
szonego ciśnienia krwi (zwłaszcza jeśli podawane
są w sposób przewlekły w ramach terapii niewydol-
4.1.2.2. Niedokrwistość ności nerek). Może również dojść do zamknięcia się
z niedoboru erytropoetyny przetoki tętniczo-żylnej u pacjentów dializowanych.
(niedokrwistość nerkowa) Obserwowane były też ponadto pojedyncze przypad-
ki trombocytozy, zakrzepicy, wybiórczej aplazji czer-
Dla erytropoezy, która zachodzi w czerwonym szpi- wonokrwinkowej (PRCA – pure red cell aplasia),
ku kostnym, jak wynika z ryc. B 4.1-1, niezbędna zmian skórnych oraz zaburzeń ośrodkowego układu
jest erytropoetyna (EPO), która wytwarzana jest prze- nerwowego, m.in. drgawki.
de wszystkim w komórkach przykłębuszkowych dy-
stalnych kanalików nerkowych (masa cząsteczkowa Epoetyna jest przeciwwskazana w ciężkich przypad-
34 000 Da). kach nadciśnienia opornego na leczenie.
MUTSCHLER-2009.indd 521 2010-01-07 22:13:29
522 Krew
Podawanie epoetyny może zwiększyć liczbę erytrocytów, Ponieważ dzienne zapotrzebowanie na witaminę
a co za tym idzie – zdolność przenoszenia tlenu, również B12 wynosi zaledwie 1 μg, a w wątrobie znajduje
u osób z normalnymi wartościami hematokrytu. Dlatego
się zapas 1000–2000 μg, od momentu zaprzestania
epoetyna jest nadużywana przez sportowców wyczyno-
wych jako „doping za pomocą substancji endogennych”. wchłaniania witaminy B12 do rozwinięcia się nie-
Ponieważ do niedawna trudno było odróżnić egzogenne dokrwistości megaloblastycznej upływa 2–5 lat.
rekombinowane epoetyny od erytropoetyny endogennej, Choroba ta charakteryzuje się bladością, lekko żół-
za wyraźny wskaźnik użycia epoetyny uznawano wartości tym zabarwieniem skóry, zapaleniem języka i bie-
hematokrytu powyżej 50%. Obecnie nowe metody analizy
gunką, a nieleczona prowadzi do śmierci. Dodatkowo
pozwalają jednoznacznie odróżnić je od siebie.
w 20–30% przypadków obserwuje się ubytkowe ob-
jawy neurologiczne spowodowane uszkodzeniem
komórek nerwowych rdzenia kręgowego (zob. poni-
4.1.2.3. Niedokrwistości makrocytarne
żej) z towarzyszącymi parestezjami i niedowładami
mięśni.
Niedokrwistości makrocytarne (megaloblastyczne)
polegają na zaburzeniach rozwoju erytrocytów wsku-
Witamina B12 (cyjanokobalamina) należy do grupy
tek niedoboru witaminy B12 lub kwasu foliowego.
korynoidów zawierających w miejscu centralnym
Obie te witaminy uczestniczą w syntezie kwasów
atom kobaltu. Cząsteczka podstawowa dla struktury
dezoksyrybonukleinowych w niedojrzałych komór-
korynoidów – koryna różni się od porfiryn brakiem
kach szpiku kostnego i wpływają na podziały ko-
grupy metinowej między pierścieniami A i D. Innym
mórkowe. Ich niedobór prowadzi w szpiku kostnym
ważnym elementem budowy cyjanokobalaminy
do opóźnienia podziałów komórek macierzystych
jest nukleotyd, zawierający zamiast zasady puryno-
erytrocytów. W związku z tym nie wykształcają się
wej lub pirymidynowej 5,6-dimetylobenzimidazol.
z nich prawidłowe erytrocyty (normocyty), tylko po-
wstają tzw. megalocyty, które wskutek braku wielu
Po wyizolowaniu i odkryciu budowy cyjanokobala-
podziałów komórkowych przechodzą w zmniejszo-
miny okazało się, że jest ona produktem powstałym
nej liczbie do krwi obwodowej. Megalocyty zawie-
wskutek sztucznej obróbki. Właściwe substancje
rają w porównaniu z normocytami zbyt dużą ilość
czynne, 5′-dezoksy-adenozylo-kobalamina oraz me-
hemoglobiny i dlatego ten typ anemii określa się jako
tylokobalamina, powstają w organizmie z witaminy
nadbarwliwą niedokrwistość makrocytarną.
B12 przez zastąpienie grupy CN resztą 5′-dezoksy-
adenozynową bądź metylową. Utworzone w ten spo-
sób koenzymy witaminy B12 biorą udział w:
4.1.2.3.1. Niedokrwistość złośliwa
(niedokrwistość
z niedoboru witaminy B12)
O
Najważniejszą postacią niedokrwistości makrocytar-
nej jest niedokrwistość złośliwa (choroba Addisona- CN
H2N CH3 O
-Biermera, anaemia perniciosa), która powstaje O
CH3
wskutek długotrwałego niedoboru witaminy B12. NH2
Warunkiem wystąpienia tego typu niedokrwistości H2N H3C N N
NH2
z niedoboru witaminy B12 jest niezdolność błony ślu- O H3C Co+
H CH3 O
zowej żołądka do produkcji soku żołądkowego (achy- H2N N N
CH3
lia gastrica) w zapaleniu żołądka typu A. Dochodzi
wtedy do zaprzestania wytwarzania przez komórki CH3
H3C NH2
okładzinowe błony śluzowej żołądka tzw. czynnika O
wewnętrznego (IF – intrinsic factor) – mukoproteiny HN N CH3
koniecznej do prawidłowego wchłaniania witaminy -
O
O OH
B12, czyli czynnika zewnętrznego. Kompleks utwo- H3C N CH3
O P O
rzony przez witaminę B12 i czynnik wewnętrzny O O
jest wskutek endocytozy wchłaniany przez komórki
HOH2C
błony śluzowej. Tam czynnik wewnętrzny zastępo-
wany jest białkiem transportującym transkobalaminą
II, a nowo powstały kompleks jest na drodze egzocy- witamina B12
tozy wydalany z komórki.
MUTSCHLER-2009.indd 522 2010-01-07 22:13:29
Krew 523
■ biosyntezie zasad purynowych i pirymidynowych, 4.1.2.3.2. Niedokrwistość
z niedoboru kwasu foliowego
■ redukcji rybonukleotydotrifosforanów do 2-dez-
oksyrybonukleotydotrifosforanów,
Oprócz niedokrwistości złośliwej związanej z niedo-
■ przekształceniu metylomalonylo-CoA w sukcy-
borem witaminy B12 znanych jest wiele niedokrwi-
nylo-CoA, a co za tym idzie – w syntezie kwasów
stości makrocytarnych, które wynikają z niedoboru
tłuszczowych,
kwasu foliowego. W przypadku tego typu niedokrwi-
■ syntezie metioniny z homocysteiny, stości skutecznym leczeniem jest podawanie kwasu
foliowego.
■ tworzeniu osłonek mielinowych układu nerwowe-
go.
Kwas foliowy należy, podobnie jak witamina B12,
do witamin z grupy B. W cząsteczce kwasu foliowe-
Oprócz tego witamina B12 jest odpowiedzialna
go kwas glutaminowy jest połączony wiązaniem pep-
za prawidłowe odzyskiwanie kwasu tetrahydrofolio-
tydowym z kwasem p-aminobenzoesowym, którego
wego z kwasu N-metylotetrahydrofoliowego (stąd
azot podstawiony jest ugrupowaniem 2-amino-4-hy-
wtórny niedobór kwasu foliowego przy niedokrwi-
droksy-6-metylopterydynowym. W organizmie kwas
stości złośliwej). Niedobór nukleotydów wyjaśnia
foliowy ulega redukcji z udziałem kwasu askorbino-
też, dlaczego w przypadku niedoboru witaminy B12
wego do kwasu tetrahydrofoliowego, który służy jako
zaburzona jest erytropoeza, a dodatkowo wskutek
przenośnik dla grup metylowych („aktywny formal-
niewystarczającej syntezy lipidów tworzą się jedynie
dehyd”) oraz formylowych („aktywny kwas mrów-
Serce i układ krążenia
wadliwe osłonki mielinowe, w wyniku czego oprócz
kowy”; kwas N10-formylotetrahydrofoliowy), czyli
niedokrwistości mogą pojawić się ciężkie ubytkowe
grup jednowęglowych.
objawy neurologiczne (powrózkowe zwyrodnienie
rdzenia). Oprócz tego dochodzi do atrofii błony ślu-
„Aktywny formaldehyd” powstaje w ten sposób,
zowej przewodu pokarmowego.
że przy przemianie seryny w glicynę odszczepiony
Przyczynowe leczenie tych schorzeń polega
formaldehyd zostaje przyłączony do kwasu N10-for-
na pozajelitowym podawaniu preparatów witaminy
mylotetrahydrofoliowego. W przypadku „aktywnego
B12 (podawanie doustne nie jest zalecane, nawet je-
kwasu mrówkowego” grupa hydroksymetylowa zo-
żeli preparat zawiera dodatek czynnika wewnętrz-
nego, ponieważ w dużym odsetku przypadków za-
staje dzięki NADP+ (fosforanu dwunukleotydu niko- B4
tynoamidoadeninowego) utleniona do grupy formy-
palenia błony śluzowej żołądka typu A wytwarzane
lowej.
są przeciwciała przeciw czynnikowi wewnętrzne-
mu zarówno pochodzenia ludzkiego, jak i zwierzę-
Dzięki „aktywnemu formaldehydowi” z deoksyury-
cego).
dyno-5′-fosforanu powstaje deoksytymidyno-5′-fo-
Dawkowanie w ciągu pierwszych dwóch tygodni
sforan, ważny składnik budowy kwasu dezoksyrybo-
wynosi 100 μg dziennie, a po osiągnięciu prawidło-
nukleinowego.
wych wartości hematokrytu 100 μg dwa razy w tygo-
„Aktywny kwas mrówkowy” jest niezbędny
dniu. W przypadku leczenia przewlekłego wystarcza
do syntezy nukleotydów purynowych. Podczas synte-
dawka 100 μg raz w miesiącu. Szczególnie wskaza-
zy pierścienia pirymidynowego oraz imidazolowego
na jest hydroksykobalamina, która wiąże się silniej
za każdym razem wbudowywana jest grupa jednowę-
z białkami niż cyjanokobalamina i stanowi naturalną
glowa.
formę zapasową.
Nie istnieje niebezpieczeństwo przedawkowania
witaminy B12 nawet przy stosowaniu dużych dawek.
Pomimo to zaprzestano stosowanego zwykle w prze- O COOH
szłości podawania w początkowej fazie leczenia 1000 OH NH
μg na dobę, ponieważ większa część leku była sto- N
sunkowo szybko wydalana z moczem. N NH COOH
H2N N N
Skuteczność witaminy B12 w przypadku wielu in-
nych wymienianych dla niej wskazań (np. zapalenia kwas foliowy
nerwów, migreny, półpaśca, uszkodzenia miąższu
wątroby) jest wątpliwa lub nieudowodniona.
MUTSCHLER-2009.indd 523 2010-01-07 22:13:29
524 Krew
O COOH O COOH
OH NH OH NH
NH NH
N N COOH N N COOH
H2N N NH OH H2N N NH H O
„aktywny formaldehyd“ „aktywny kwas mrówkowy“
Chociaż kwas foliowy syntezowany jest przez wej). Ponadto dochodzi do biegunek i zmniejszenia
bakterie jelitowe w jelicie grubym, można wywołać masy ciała. W przypadku niedoboru kwasu folio-
eksperymentalnie niedobór kwasu foliowego przez wego we wczesnym okresie ciąży dochodzi u płodu
zmniejszanie jego ilości w pożywieniu. Przemawia do rozszczepów, szczególnie do rozszczepu kręgosłu-
to za tym, że kwas foliowy wytwarzany przez bakterie pa (spina bifida).
słabo wchłania się ze światła jelita grubego. Natomiast W leczeniu wymienionych postaci niedokrwistości
kwas foliowy dostarczany wraz z pożywieniem re- stosuje się preparaty kwasu foliowego drogą doustną
sorbowany jest w jelicie cienkim na drodze trans- w dawce 10–15 mg dziennie, a drogą pozajelitową
portu aktywnego. Warunkiem jest jednak uprzednie 1–5 mg dziennie. Podawanie pozajelitowe jest wska-
rozszczepienie obecnych w pożywieniu koniugatów zane tylko w przypadku zespołu złego wchłaniania
kwasu foliowego z licznymi cząsteczkami kwasu glu- lub po resekcji górnej części jelita cienkiego.
taminowego (poliglutaminianów) dzięki zawartym Dzięki leczeniu za pomocą kwasu foliowego do-
m.in. w komórkach śluzowych rąbka szczoteczkowe- chodzi do normalizacji obrazu krwi także w przy-
go enzymom dekoniugującym. Wewnątrz komórek padku niedokrwistości złośliwej. Mimo to nie po-
kwas foliowy jest ponownie magazynowany w posta- winno się go stosować w monoterapii, ponieważ
ci poliglutaminianu. Metylowane w pozycji 5 analogi nie spowoduje on wyleczenia uszkodzeń układu
kwasu tetrahydrofoliowego muszą przed utworze- nerwowego.
niem poliglutaminianu ulec demetylacji z udziałem Nie są znane objawy przedawkowania kwasu fo-
witaminy B12. liowego, jednak duże jego dawki mogą znacznie
Wynosząca ok. 15 mg zawartość zmagazynowane- osłabić, a nawet całkiem znieść przeciwpadaczkowe
go prawidłowo w organizmie kwasu foliowego po- działanie barbituranów, prymidonu i fenytoiny.
krywa zapotrzebowanie na 3–4 miesiące.
Przy prawidłowym odżywianiu niedobór kwa-
su foliowego występuje rzadko. Dochodzi do niego
jedynie w przypadku zaburzeń trawienia, przy upo- 4.1.3. Osocze krwi, surowica krwi
śledzonym wchłanianiu lub stosowaniu antymetabo-
litów kwasu foliowego oraz niekiedy w wyniku le- Osocze jest to płynna część krwi. Składa się w 90%
czenia pirymetaminą, lekami przeciwpadaczkowymi z wody, w 7–8% z białek, a oprócz tego zawiera sole,
(barbituranami, prymidonem, fenytoiną), a także przy węglowodany, lipidy, aminokwasy i in. Dzięki do-
podawaniu doustnych środków antykoncepcyjnych brze przepuszczalnym dla wody i elektrolitów ścia-
wskutek zahamowania reduktazy dihydrofoliano- nom naczyń włosowatych między osoczem a płynem
wej lub dekoniugacji poliglutaminianów. Niedobór wewnątrzkomórkowym dochodzi do ciągłej wymia-
kwasu foliowego występuje ponadto u alkoholików ny substancji.
(wskutek zaburzeń wchłaniania oraz bezpośredniego Termin surowica krwi oznacza przezroczysty płyn
działania przeciwfolianowego etanolu) oraz u kobiet pozostający podczas krzepnięcia po oddzieleniu się
w ciąży. Dlatego w ciąży wskazane jest profilaktyczne skrzepu. Od osocza różni się głównie tym, że nie za-
podawanie kwasu foliowego. wiera czynnika krzepnięcia krwi – fibrynogenu.
Niedobór kwasu foliowego objawia się zaburze-
niami podziałów komórek, głównie układu krwio- Białka krwi. Największą frakcję substancji roz-
twórczego (w obrazie krwi nie można odróżnić puszczonych w osoczu krwi stanowią białka, które
niedokrwistości makrocytarnej spowodowanej nie- są syntezowane głównie w wątrobie. Prawidłowe
doborem kwasu foliowego od niedokrwistości złośli- stężenie białka całkowitego w osoczu krwi wynosi
MUTSCHLER-2009.indd 524 2010-01-07 22:13:29
Krew 525
65–80 g/l. Stężenie molarne wynosi około 1 mmol/l. Oprócz wymienionych cząsteczek białkowych
Dzięki elektroforezie białka osocza można rozdzielić w osoczu krwi znajdują się również różne enzymy,
na frakcje albumin oraz globulin α1, α2, β, i γ (fibry- np. esterazy, amidazy, oraz czynniki grupowe krwi.
nogen znajduje się między frakcją β- i γ-globulin). W przypadku zniszczeniu komórek organizmu enzy-
Albuminy stanowią około 56% białek osocza, α1-glo- my w nich zawarte przedostają się do krwi. W taki
buliny ok. 4%, α2, β, i γ-globuliny zaś odpowiednio właśnie sposób dochodzi do podwyższenia zawarto-
ok. 10%, 12% i 18%. ści określonych transaminaz w surowicy krwi w przy-
Elektroforeza białek osocza lub surowicy krwi padku zawału serca lub zapalenia wątroby. Dlatego
jest badaniem ważnym z klinicznego punktu widze- oznaczanie zawartości enzymów w surowicy krwi
nia, ponieważ wiele schorzeń prowadzi do zmian stało się ważnym badaniem diagnostycznym.
obrazu białek krwi. Za pomocą tzw. immunoelektro-
forezy, czyli połączenia elektroforezy osocza i immu- Elektrolity osocza. Najważniejsze elektrolity wystę-
noprecypitacji, można przeprowadzić dokładniejszą pujące w osoczu i ich średnie stężenia przedstawione
analizę. zostały w tab. B 4.1-3.
Podobnie jak komórki krwi, białka osocza pod-
legają nieustannym procesom syntezy i rozkładu, Dla wszystkich stężeń jonów istnieją określone prze-
a po rozpadzie na oligopeptydy lub aminokwasy działy prawidłowe, które wynoszą dla Na+ – kationu
są wchłaniane przez komórki, które wykorzystują o najwyższym stężeniu w osoczu – 135–145 mmol/l,
je w procesach syntezy białek. Stanowią one rezerwę a dla Cl– – najważniejszego pod względem ilościo-
białkową, która jest w razie potrzeby bardzo szyb- wym anionu osocza – między 98 a 106 mmol/l. (W ta-
Serce i układ krążenia
ko dostępna. Oprócz tego służą do swoistego i nie- beli wartości te są dodatkowo podane w mEq/l w celu
swoistego transportu licznych substancji, a ponadto pokazania, że osocze jest neutralne pod względem
pełnią funkcję bufora krwi. Dodatkowo odgrywają ładunku). Do grupy nieopisanej szczegółowo należą
ważną rolę w regulacji podziału wody między oso- kwasy organiczne osocza, m.in. kwas mlekowy, kwas
czem a przestrzenią wewnątrzkomórkową, ponieważ cytrynowy, kwas pirogronowy i aminokwasy.
jako białka nie przechodzą przez ściany naczyń wło- Ciśnienie osmotyczne osocza w wysokości 7,3 atm
sowatych i dzięki temu, wywierając ciśnienie koloi- utrzymywane jest dzięki dobrze rozwiniętym me-
doosmotyczne (onkotyczne), zatrzymują wodę w ło- chanizmom osmoregulacji, za którą odpowiedzialne
żysku naczyniowym. Najważniejszą rolę grają w tym
przypadku albuminy (ok. 80%). Albuminy są oprócz
B4
Tab. B 4.1-3. Stężenia elektrolitów w osoczu krwi
tego odpowiedzialne za wiązanie się leków z białkami
krwi. mEq/l mmol/l mg/dl
Globuliny spełniają liczne funkcje biologiczne. Wiele kationy
α- i β-globulin pełni swoiste funkcje transportujące. sód 142 142 327
I tak z grupy α1-globulin transkobalamina transportuje potas 5 5 20
witaminę B12, a transkortyna – kortyzol. Inna α1-glo-
bulina, protrombina, odgrywa kluczową rolę w proce- wapń 5 2,5 10
sie krzepnięcia krwi. Ponadto α2-globulina, angioten- magnez 2 1 2
synogen, jest nieaktywnym prekursorem silnego czyn-
nika kurczącego naczynia – angiotensyny II. razem 154 150,5
Natomiast α2-haptoglobina posiada zdolność wią- aniony
zania w osoczu uwolnionej w procesie hemolizy he- chlorki 102 102 362
moglobiny.
diwęglany* 27 27 165
Transferyna, będąca β-globuliną, jest odpowiedzial-
na za transport trójwartościowego żelaza w osoczu. fosforany 2 1 10
Z kolei γ-globuliny są glikoproteinami, które pod-
siarczany 1 0,5 5
czas elektroforezy wędrują najwolniej. Ze względu
na to, że uczestniczą one w reakcjach odpornościo- kwasy organiczne 6 6 21
wych, nazywane są także immunoglobulinami (Ig). białko 16 1 7200
Jeżeli chodzi o właściwości koloidoosmotyczne,
to globuliny ze względu na dużą masę cząsteczko- razem 154 137,5
wą oraz stosunkowo niewielką ilość w organizmie * we krwi żylnej z wysokim parcjalnym ciśnieniem CO2
nie odgrywają tu dużej roli.
MUTSCHLER-2009.indd 525 2010-01-07 22:13:30
526 Krew
są w 96% nieorganiczne elektrolity. Wzajemny stosu- naczyniowego i powoduje zmniejszenie ilości krwi
nek jonów oraz wartość pH osocza są również utrzy- żylnej powracającej do serca, a co za tym idzie – po-
mywane na stałym poziomie dzięki wyspecjalizowa- jemności minutowej serca. W ciężkich przypadkach
nym mechanizmom regulacyjnym. może dojść do wstrząsu.
Inne składniki osocza. Oprócz wymienionych skład- Z tego względu w przypadku utraty krwi lub osocza
ników w osoczu znajdują się również inne substancje, (np. przy urazach lub poparzeniach) metodą lecze-
które w obrębie swoich fizjologicznych zakresów stę- nia jest centralne podanie odpowiednich preparatów
żeń nie mają istotnego wpływu na fizykochemiczne w celu wypełnienia łożyska naczyniowego. Utrata
właściwości osocza i dlatego są czasami nazywane krwi nie musi przy tym być koniecznie leczona
składnikami transportowanymi przez osocze. Należą za pomocą transfuzji krwi. Wskazania do przetocze-
do nich: nia krwi należy ustalać na podstawie indywidual-
nej oceny stanu pacjenta. Nie ma też sensu ustana-
■ składniki odżywcze, np. glukoza,
wiać sztywnych granic dla takiego postępowania.
■ witaminy i pierwiastki śladowe, Przetoczenie jest konieczne tylko wtedy, kiedy utrata
krwi sięga jednej trzeciej całkowitej objętości krwi
■ zawierające azot produkty metabolizmu białek i pu-
w organizmie. W przypadku niewystarczającego wy-
ryn, które mają być wydalone (mocznik, kwas mo-
pełnienia łożyska naczyniowego używane są obecnie
czowy, kreatynina i in.),
zazwyczaj pozbawione krwinek roztwory koloidalne
■ hormony. pochodzące z organizmu pacjenta lub obcego.
Podawanie wyłącznie roztworów soli (krystalo-
Wartości stężeń tych składników osocza zostały po- idów), np. roztworu Ringera, w leczeniu hipowole-
dane w tab. B 4.1-4. mii jest skuteczne tylko na krótką metę ze względu
na krótki czas utrzymywania się tego typu roztworów
Tab. B 4.1-4. Stężenia innych ważnych składników ludzkiego w łożysku naczyniowym. Dlatego roztwory soli sto-
osocza (wartości prawidłowe) suje się często z roztworami koloidalnymi.
g/l Istnieją cztery rodzaje koloidalnych płynów zastępu-
jących osocze: albuminy, dekstrany, skrobia hydro-
glukoza 0,6 – 1,0 ksyetylowana oraz pochodne żelatyny. W przypadku
kwas mlekowy 0,09 – 0,16 tych substancji mamy do czynienia z dwoma prob-
kwas pirogronowy 0,005 – 0,017
lemami: po pierwsze, płyny zastępcze osocza poda-
związki występujące w moczu wane są również w leczeniu schorzeń związanych
mocznik 0,2 – 0,6 z ubytkiem śródbłonka i występującym wskutek tego
kwas moczowy 0,034 – 0,07 przeciekiem kapilarnym (capillary leak). Istnieje
0,024 – 0,057 wtedy możliwość, że płyn koloidalny przedostanie
kreatynina 0,004 – 0,012 się do przestrzeni międzykomórkowej i będzie wią-
kreatyna 0,002 – 0,005
zał płyn poza naczyniami. Po drugie, nie udało się
całkowite lipidy 3,05 – 8,8 do tej pory uzyskać roztworów koloidalnych zdol-
całkowity cholesterol 1,0 – 2,5 nych do przenoszenia tlenu, w związku z czym moż-
fosfolipidy 1,25 – 2,3 liwości zastąpienia krwi przy dużej utracie za pomocą
triglicerydy 0,5 – 2,0 płynów zastępczych osocza są ograniczone.
wolne kwasy tłuszczowe 0,08 – 0,12
bilirubina 0,002 – 0,01
4.1.3.1.1. Homologiczne preparaty osocza
Wśród endogennych składników osocza skutecznymi
środkami stosowanymi w celu uzupełniania objętości
4.1.3.1. Płyny zastępcze osocza krwi okazały się roztwory ludzkiej albuminy, oraz pa-
steryzowane roztwory białek osocza.
Do prawidłowego funkcjonowania układu krążenia
niezbędne jest wystarczające wypełnienie łożyska Roztwory ludzkiej albuminy są w większości pro-
naczyniowego. Ubytek krążącej krwi. np. wskutek dukowane przez frakcjonowanie alkoholowe metodą
utraty krwi. zmniejsza ciśnienie wypełnienia łożyska Cohna mieszaniny wielu osoczy. Na rynku dostępne
MUTSCHLER-2009.indd 526 2010-01-07 22:13:30
Krew 527
są izotoniczne roztwory 5-proc. oraz hipertonicz- ■ brak wpływu na oznaczanie grup krwi oraz badania
ne roztwory 20- i 25-proc. Ponieważ nie zawierają w diagnostyce laboratoryjnej,
one przeciwciał ani aglutynin można je podawać
■ brak wpływu na krzepnięcie krwi.
bez uprzedniego sprawdzania grupy krwi. Trwałość
wynosi około roku, a w związku z ich stosowaniem
Środki zastępujące osocze powinny ponadto cecho-
nie istnieje ryzyko przeniesienia zapalenia wątroby.
wać się brakiem właściwości antygenowych, być ła-
twe do przygotowania, możliwe do sterylizacji, trwa-
Pasteryzowane roztwory białek osocza niewiele
łe także przy wahaniach temperatury.
różnią się od roztworów ludzkiej albuminy. Oprócz
Warunki takie spełniają w większości następujące
albumin zawierają one niewielkie ilości globulin.
preparaty:
Obydwa rodzaje roztworów wskazane są w przy- ■ dekstran,
padku niewystarczającej objętości łożyska naczynio-
■ skrobia hydroksyetylowana,
wego, hipoalbuminemii, hiperglobulinemii, a także
w celu wykonania normowolemicznej hemodylucji. ■ żelatyna.
Podczas stosowania tych preparatów w rzadkich
przypadkach pojawiają się objawy gorączki oraz re- W przypadku środków zastępujących osocze szcze-
akcji alergicznych. gólnie ważna jest skuteczność w uzupełnianiu obję-
tości łożyska naczyniowego, która zależy od zdolno-
ści wiązania wody oraz czasu przebywania koloidu
Serce i układ krążenia
4.1.3.1.2. Pozaustrojowe środki zastępcze w łożysku żylnym. Jeżeli dany roztwór koloidalny,
osocza dzięki wywołaniu przemieszczenia się płynu z tka-
nek do układu naczyniowego, powiększa objętość
Ze względu na wysoką cenę homologicznych prepa- łożyska naczyniowego o wartość większą niż podana
ratów osocza oraz ograniczoną ich ilość, jaką można dożylnie objętość, to określany jest mianem środka
mieć do dyspozycji, jako płynów zastępczych oso- zwiększającego objętość osocza.
cza używa się często pozaustrojowych roztworów W tab. B 4.1-5 podano najważniejsze właściwości
koloidalnych. Powinny one mieć następujące właś- koloidalnych roztworów zastępczych osocza.
ciwości:
Wszystkie środki zastępcze osocza mogą powodo-
B4
■ dobrą skuteczność w uzupełnianiu objętości łoży-
wać wystąpienie reakcji anafilaktycznych lub rzeko-
ska naczyniowego,
moanafilaktycznych z różną częstością i o różnym
■ wystarczająco długi okres utrzymywania się w ukła- nasileniu w zależności od preparatu. Dlatego należy
dzie naczyniowym, uważnie obserwować pacjentów po rozpoczęciu wle-
wu dożylnego.
■ dobrą tolerancję i biologiczną nieaktywność,
Stosowanie środków zastępczych osocza jest prze-
■ metabolizm w organizmie i/lub możliwość wydale- ciwwskazane w przypadku ciężkiej niewydolności
nia przez nerki, serca, hiperwolemii i przewodnienia.
■ brak wpływu zwiększającego lepkość krwi,
Tab. B 4.1-5. Charakterystyka koloidalnych roztworów osoczozastępczych (według Grubera i Kloseego)
Dekstran 60 Dekstran 40 Żelatyna Skrobia
hydroksyetylowana
średnia masa cząsteczkowa (Da) 60 000 40 000 30 000-50 000 200 000
stężenie (%) 6 10 3,0-5,5 10
efekt objętościowy w zależności od ilości 1,05-krotny 1,5-krotny 0,8-krotny 1,05-krotny
wiązanie wody (ml/g) 25,6 29,2 14,3-39,0 10-14
okres trwania w naczyniach (godz.) 6 3 3 8
MUTSCHLER-2009.indd 527 2010-01-07 22:13:30
528 Krew
4.1.3.1.2.1. Dekstrany 200 000 Da (średniocząsteczkowa skrobia hydroksy-
etylowana) oraz 70 000 (niskocząsteczkowa skrobia
Termin dekstrany oznacza polisacharydy złożone z cząste- hydroksyetylowana).
czek glukozy jako monomerów połączonych ze sobą wią- Roztwory skrobi hydroksyetylowanej odpowiada-
zaniami α-glikozydowymi. Dekstrany są podobne w swojej
budowie chemicznej do skrobi i glikogenu, jednak w dek-
ją pod względem swoich właściwości farmakologicz-
stranach występują głównie wiązania 1,6-glikozydowe, nych roztworom dekstranów. Jednakże ich wpływ
dzięki czemu są one trudniej rozkładane enzymatycznie na krzepnięcie krwi jest słabszy i rzadziej dochodzi
w organizmie. Otrzymuje się je dzięki metodom mikrobio- do wystąpienia ciężkich reakcji anafilaktycznych.
logicznym za pośrednictwem Leuconostoc mesenteroides Wydalanie skrobi hydroksyetylowanej zachodzi
z roztworów zawierających sacharozę. Surowy dekstran ma
zbyt dużą masę cząsteczkową i dlatego musi zostać częś-
głównie za pośrednictwem nerek i dodatkowo z ka-
ciowo zhydrolizowany, a następnie frakcjonowany. Na ryn- łem. Preparaty wysokocząsteczkowe są uprzednio
ku obecne są preparaty wysoko oczyszczonego dekstranu wychwytywane przez układ fagocytów wielojądrza-
o średniej masie cząsteczkowej od 60 000 (dekstran 60) stych i tam rozkładane enzymatycznie.
do 40 000 (dekstran 40, dekstran niskocząsteczkowy).
Preparaty dekstranu mają wyraźną zdolność zwięk-
szania objętości. Działanie zwiększające objętość osocza
jest szczególnie wyraźne w przypadku roztworów hipe- 4.1.3.1.2.3. Żelatyny
ronkotycznych. Dekstran 40 jest oprócz tego w stanie,
szczególnie podczas wstrząsu, poprawić mikrokrążenie W poszukiwaniu substancji koloidalnych, które na-
dzięki rozpuszczaniu agregatów krwinek czerwonych dawałyby się na preparaty zastępcze osocza zbada-
(sludge phenomenon), zmniejszaniu lepkości krwi i zwięk-
szeniu jej stabilności. Inne dekstrany zmniejszają również
no również żelatyny. Pierwsze preparaty miały jed-
w mniejszym stopniu agregację płytek krwi. nak wady: nie można było ich sterylizować, miały
Wydalanie dekstranów zachodzi głównie przez nerki, właściwości antygenowe, w temperaturze pokojowej
przy czym próg nerkowy dla masy cząsteczkowej wynosi zestalały się do postaci żelu i musiały być przed po-
ok. 50 000, w związku z czym dekstrany o większej masie daniem podgrzewane. Dzięki specjalnym metodom
cząsteczkowej nie mogą być odfiltrowane. Dlatego muszą
one zostać enzymatycznie rozłożone przed wydaleniem
obróbki (depolimeryzacji i ponownej polimeryzacji)
przez nerki. udało się te wady wyeliminować. Obecnie w użyciu
Przy podawaniu większych dawek dekstranu (od około są następujące preparaty żelatyn o masie cząsteczko-
1,5 g na kilogram masy ciała) istnieje niebezpieczeństwo wej od 30 000 do 35 000:
zaburzeń hemostazy wskutek agregacji płytek krwi oraz
dodatkowego hamowania czynników krzepnięcia V i VIII. ■ żelatyny połączone mostkami mocznikowymi,
Z tego powodu należy ściśle przestrzegać zaleceń doty-
czących jego dawkowania (np. początkowo maksymalnie ■ modyfikowane płynne żelatyny, polimeryzat skła-
1500 ml dekstranu 40 na dobę). dający się z poddanej rozkładowi żelatyny podsta-
Reakcje anafilaktyczne po podaniu dekstranu obser- wionej resztami sukcynylowymi.
wuje się w 0,03% przypadków. Prawdopodobnie docho-
dzi do nich z powodu uwrażliwienia na dekstrany zawarte
w środkach spożywczych oraz używkach, a także wskutek
W porównaniu z preparatami dekstranów preparaty
krzyżowej antygenowości z antygenami bakteryjnymi. żelatyny mają mniejszą zdolność wiązania wody oraz
krótszy czas utrzymywania się w łożysku naczynio-
wym. W niewielkim stopniu wpływają one na hemo-
stazę. Większa część pochodnych żelatyny jest wy-
4.1.3.1.2.2. Skrobia hydroksyetylowana
dalana z moczem, mniejsza część przez przewód po-
karmowy. Oprócz tego w niewielkim stopniu są one
Rozpuszczalna skrobia nie nadaje się do użycia jako rozkładane enzymatycznie przez peptydazy.
preparat zastępczy osocza, ponieważ jest bardzo W porównaniu z dekstranami po preparatach żela-
szybko rozkładana przez α-amylazę. Jednakże dzię- tyn częściej występują reakcje rzekomoanafilaktycz-
ki wprowadzeniu grup hydroksyetylowych do hy- ne, ale ich przebieg jest z reguły lżejszy.
drolizatu amylopektyny można uzyskać biopolimer
o opóźnionym rozkładzie enzymatycznym, który
może być stosowany jako preparat zastępczy osocza 4.1.4. Hemostaza
(amylopektyna składa się z cząsteczek glukozy po-
wiązanych wiązaniami 1,4-α-glikozydowymi oraz (zatrzymywanie krwawienia)
1,6-α-glikozydowymi, wykazuje więc duże pokre-
wieństwo chemiczne z glikogenem). Prawidłowo funkcjonujący układ hemostazy jest nie-
Na rynku dostępne są preparaty skrobi hy- zbędny do życia, ponieważ w przypadku zaburzeń
droksyetylowanej o średniej masie cząsteczkowej hemostazy już niewielkie urazy mogą prowadzić
MUTSCHLER-2009.indd 528 2010-01-07 22:13:30
Krew 529
do ciężkich krwawień, a z drugiej strony przy nad- Obydwa te mechanizmy wyzwalane są natych-
miernym krzepnięciu zwiększona jest skłonność miast po uszkodzeniu naczynia. Aktywacja układu
do tworzenia zakrzepów i w związku z tym wzrasta zewnątrzpochodnego przebiega szybko (w ciągu
ryzyko zakrzepicy i powstawania zatorów. kilku sekund), natomiast układ wewnątrzpochodny
Ze względu na przebieg hemostazę dzieli się aktywowany jest znacznie wolniej (w ciągu kilku
na dwa połączone ze sobą procesy: minut).
Aktywacja układu zewnątrzpochodnego (zob. ryc.
■ hemostazę pierwotną,
B 4.1-4) rozpoczyna się od uwolnienia czynnika tkan-
■ hemostazę wtórną (krzepnięcie krwi). kowego (tromboplastyny tkankowej) wskutek uszko-
dzenia komórek okołonaczyniowych. Ten czynnik,
Hemostaza pierwotna. Tuż po powstaniu uszkodze- który składa się głównie z bogatych w fosfatydy struk-
nia w naczyniu krwionośnym płytki krwi ulegają ad- tur błonowych, aktywuje następnie osoczowy czynnik
hezji do kolagenowych włókien tkanki łącznej na brze- VII. Ten z kolei powoduje przejście czynnika X w ak-
gach rany i jeżeli uszkodzone naczynie nie jest zbyt tywną postać czynnika Xa, która wraz z czynnikiem
duże, tworzą czop zamykający (odwracalna agrega- V oraz jonami wapnia prowadzi do przekształcenia
cja płytek krwi, czop trombocytowy). Pod wpływem protrombiny w trombinę (czynnik IIa). Protrombina
utworzonej podczas aktywacji układu krzepnięcia powstaje w wątrobie przy udziale witaminy K i nale-
trombiny (zob. poniżej) płytki krwi przekształcają ży do grupy α1-globulin. Powstająca z niej trombina
się nieodwracalnie w jednorodną masę i uwalniają jest endopeptydazą, która prowadzi do powstania fi-
na zewnątrz całą swoją zawartość. Uwolnione media- bryny (zob. poniżej). Oprócz wpływu na tworzenie
Serce i układ krążenia
tory (m.in. serotonina) prowadzą do skurczu naczyń się fibryny trombina może aktywować czynniki V
w uszkodzonym obszarze. Proces hemostazy dodat- i VIII i w ten sposób nasilać własną syntezę.
kowo wspiera zwijanie się oraz sklejanie śródbłonka
naczyń. Podczas łączenia się trombocytów uwalniane W przypadku układu wewnątrzpochodnego reakcja
są zawarte w nich składniki (ADP, czynnik płytkowy zaczyna się aktywacją czynnika Hagemana (czynni-
4) oraz eksponowane są naładowane ujemnie fosfoli- ka XII), która zachodzi na uszkodzonej powierzchni
pidy błony komórkowej trombocytów. ADP i fosfoli- naczynia. Układ ten również polega na kaskadzie
pidy odgrywają ważną rolę podczas krzepnięcia krwi, aktywacji kolejnych czynników, przy czym dodat-
a czynnik płytkowy 4 inaktywuje hamującą krzepnię- kowo niezbędne są jony wapnia i fosfolipoprotei-
cie krwi heparynę. ny uwolnione z trombocytów. Ostatecznie również
B4
Czas upływający od momentu uszkodzenia naczy- w tym przypadku dochodzi do przekształcenia pro-
nia do pierwotnego, niestabilnego jeszcze zamknięcia trombiny w trombinę. Na etapie aktywacji czynnika
uszkodzonego miejsca określany jest mianem czasu X przez układy zewnątrz- i wewnątrzpochodne roz-
krwawienia. Prawidłowo wynosi on 2–3 minuty. poczyna się wspólny dla obu układów ostatni etap
krzepnięcia.
Hemostaza wtórna. Czop trombocytowy nie jest wy-
starczający do trwałego zamknięcia uszkodzonego na- Ze względów dydaktycznych wciąż stosuje się podział
czynia. Niezbędną trwałość zapewnia dopiero utwo- na zewnątrz- i wewnątrzpochodny układ krzepnięcia, cho-
rzenie skrzepu i powstanie sieci fibryny (zob. poniżej). ciaż aktywny czynnik VII (czynnik VIIa) oddziałuje rów-
Mianem hemostazy wtórnej określa się więc proces nież na układ wewnątrzpochodny, w związku z czym in
prowadzący do trwałego mechanicznego zamknięcia vivo oba układy są aktywowane równocześnie.
powstałego ubytku oraz jego pełnej naprawy przez
utworzenie blizny. W procesie tym oprócz płytek Synteza fibryny rozpoczyna się od odszczepienia
krwi uczestniczą liczne czynniki osoczowe i tkan- przez trombinę fibrynopeptydów A i B z powstałego
kowe, które określa się ogólnie mianem czynników w wątrobie fibrynogenu. Fibrynogen jest podłużnym
krzepnięcia krwi. W tab. B 4.1-6 zestawiono różne białkiem (masa cząsteczkowa 340 000 Da), które
czynniki krzepnięcia. składa się z dwóch identycznych podjednostek za-
wierających po trzy łańcuchy polipeptydowe.
Aktywacja krzepnięcia zachodzi na drodze dwóch Monomery fibryny ulegają następnie spontanicznej
mechanizmów: agregacji, tworząc długie włókna. Świeżo utworzona
fibryna jest niestabilna, ponieważ jej poszczególne
■ pozanaczyniowego (układ zewnątrzpochodny),
elementy są połączone ze sobą za pośrednictwem
■ wewnątrznaczyniowego (układ wewnątrzpochod- wiązań niekowalencyjnych. Dopiero pod wpływem
ny). czynnika XIII dochodzi do tworzenia się sieci ko-
MUTSCHLER-2009.indd 529 2010-01-07 22:13:30
530 Krew
Tab. B 4.1-6. Czynniki krzepnięcia krwi
Czynnik Nazwa (synonimy) Miejsce syntezy
I fibrynogen wątroba
II protrombina wątroba
III tromboplastyna tkankowa (trombokinaza tkankowa) komórki tkanek
IV jony wapnia
V (VI*) proakceleryna (ac-globulina) głównie wątroba
VII prokonwertyna wątroba
VIII globulina antyhemofilowa A (kompleks czynnika VIII) wątroba, śledziona,
układ siateczkowo-śródbłonkowy
IX globulina antyhemofilowa B (czynnik Christmasa) wątroba
X czynnik Stuarta-Prowera wątroba
XI PTA (czynnik przeciwhemofilowy C, czynnik Rosenthala) ?
XII czynnik Hagemana ?
XIII czynnik stabilizujący włóknik (FSF, czynnik Laki-Loranda) wątroba
XIV wysokocząsteczkowy kininogen (czynnik Fitzgeralda) różne narządy
XV prekalikreina (czynnik Fletchera) różne narządy
* VI nie jest samodzielnym czynnikiem, tylko prawdopodobnie aktywowanym czynnikiem V
walencyjnych wiązań poprzecznych i podłużnych, Xa, XIa i XIIa hamuje ich aktywność proteolitycz-
co prowadzi do wzmocnienia struktury. Również ną, a co za tym idzie – hamuje kaskadę krzepnięcia
czynnik XIII jest aktywowany przez trombinę. krwi.
Ostatni etap krzepnięcia polega na obkurczaniu
się włókien fibryny (retrakcji). Proces ten zostaje za- Siarczan heparanu występuje na powierzchni ko-
początkowany przez rozpad płytek krwi, a uwolniona mórek śródbłonka i prowadzi do wzmocnienia po-
w wyniku tego ATP-aza (trombostenina) prowadzi wyższego mechanizmu.
do obkurczania się włókien fibryny. Wskutek retrakcji
dochodzi do zbliżenia się brzegów rany, a oprócz tego Białko C i białko S są zależne od witaminy K i po-
zwiększa się wytrzymałość mechaniczna skrzepu. dobnie jak większość czynników krzepnięcia są syn-
tetyzowane w wątrobie. Białko C po aktywacji przez
Fizjologiczne inhibitory krzepnięcia krwi. Oprócz trombinę i związaniu się na powierzchni fosfolipido-
czynników prowadzących do krzepnięcia krwi orga- wej inaktywuje, z udziałem białka S jako kofaktora,
nizm dysponuje różnymi inhibitorami krzepnięcia. czynniki Va oraz VIIIa. Dzięki temu mechanizmowi
Największe znaczenie spośród nich mają: ujemnego sprzężenia zwrotnego dochodzi do zatrzy-
mania procesu krzepnięcia.
■ antytrombina (poprzednio antytrombina III),
■ siarczan heparanu,
Fibrynoliza. Dzięki procesowi fibrynolizy – okre-
■ białko C i białko S. ślanemu często jako „lustrzane odbicie krzepnięcia”
– dochodzi do rozpuszczenia fibryny. Układ fibry-
Antytrombina to wytwarzana w wątrobie α2-glo- nolityczny jest równie ważny jak układ krzepnięcia,
bulina o masie cząsteczkowej 64 000 Da, która po- ponieważ również w warunkach fizjologicznych
przez tworzenie kompleksów z czynnikami IIa, IXa, w krążącej krwi dochodzi do powstawania niewiel-
MUTSCHLER-2009.indd 530 2010-01-07 22:13:30
Krew 531
kiej ilości fibryny, co bez nieustannej fibrynolizy Produkty degradacji fibrynogenu hamują z kolei two-
prowadziłoby do powstawania wewnątrznaczynio- rzenie trombiny, a także polimeryzację monomerów
wych zakrzepów. Układ fibrynolityczny ma również fibryny i w ten sposób blokują krzepnięcie krwi.
za zadanie uwalniać ze złogów fibryny inne układy
przewodowe (przewody wyprowadzające gruczołów, Do aktywatorów układu pozanaczyniowego, tzw. ak-
drogi odprowadzające mocz), a także rozpuszczać tywatorów tkankowych, należą:
niepotrzebną już fibrynę (np. przy zapaleniu włókni-
■ tkankowy aktywator plazminogenu (t-PA – tis-
kowym).
sue plasminogen activator), który wytwarzany
Aktywacja układu fibrynolizy zachodzi podobnie
jest głównie w śródbłonku naczyniowym,
jak w przypadku układu krzepnięcia dzięki układo-
wi zewnątrz- i wewnątrznaczyniowemu za pośredni- ■ urokinaza, która występuje w nerkach, skąd jest
ctwem aktywatorów plazminogenu. Przekształcają wydalana z moczem, a także w innych tkankach.
one nieaktywny prekursor – plazminogen (zob. ryc. B
4.1-4) w endopeptydazę plazminę, która rozszczepia Zanim aktywatory układu wewnątrznaczyniowego
fibrynę i w ten sposób prowadzi do jej rozpuszcze- będą mogły przekształcić plazminogen w plazminę,
nia. Plazmina prowadzi również do rozszczepienia muszą zadziałać na nie proaktywatory. Zalicza się
plazminogenu, a także wpływa na czynniki V i VIII. do nich lizokinazy uwalniane z leukocytów wskutek
Serce i układ krążenia
układ wewnątrzpochodny układ zewnątrzpochodny
kontakt z powierzchnią uszkodzenie tkanki
XIV K
XII XIIa
tromboplastyna
XV tkankowa (III)
XI XIa
Ca2+ B4
IX IXa VIIa VII
Ca2+
VIII Ca2+
X Xa X
PF3
Ca2+ Va V
XIII
protrombina trombina
fosfolipidy
XIIIa
fibrynogen fibryna fibryna produkty rozpadu fibryny
niestab. stab.
plazminogen plazmina
aktywatory osoczowe aktywatory tkankowe α2-antyplasmina
proaktywatory inhibitory aktywatorów (PAI-1)
a = aktywowany czynnik krzepnięcia przekształcanie
kompleks aktywujący protrombinę oddziaływanie
Ryc. B 4.1-4. Przebieg procesu krzepnięcia krwi (u góry) oraz fibrynolizy (u dołu). Nazwy czynników krzepnięcia podano w tab. B
4.1-6.
MUTSCHLER-2009.indd 531 2010-01-07 22:13:31
532 Krew
uszkodzenia tkanek oraz czynnik Hagemana (czyn- zaburzenia czynnościowe płytek krwi są bardzo rzad-
nik XII), który uczestniczy również w aktywacji we- kie. Charakterystyczne dla zaburzeń hemostazy uwa-
wnątrznaczyniowego układu krzepnięcia. Dlatego runkowanych przez płytki krwi są krwawienia typu
ma on znaczenie zarówno w krzepnięciu krwi, wybroczyn (petechiae). W przypadku zmniejszenia
jak i w fibrynolizie. Proaktywatorem egzogennym się liczby płytek krwi poniżej 10 000/μl występują
jest streptokinaza wytwarzana przez paciorkowce ciężkie objawy kliniczne.
β-hemolizujące. Koagulopatie dzieli się na wrodzone i nabyte.
W przypadku koagulopatii wrodzonych uwarunko-
Poziom aktywatorów plazminogenu w osoczu może być wany genetycznie niedobór lub brak dotyczy z reguły
podwyższany przez liczne substancje endogenne oraz leki, jednego czynnika, podczas gdy koagulopatie nabyte
np. hormony kory nadnerczy, testosteron czy doustne leki
przeciwcukrzycowe. cechują się niedoborem kilku czynników krzepnię-
cia.
Najczęstszą postacią wśród koagulopatii wrodzo-
W prawidłowych warunkach w osoczu nie dochodzi
nych jest dziedziczona w sposób dominujący choro-
do układowej aktywacji plazminogenu, a jeśli doj-
ba von Willebranda, która dzieli się na kilka odmian.
dzie do wytworzenia plazminy, jest ona szybko in-
Wszystkie one charakteryzują się niedoborem lub
aktywowana przez α2-antyplazminę. W ten sposób
zaburzeniami czynności czynnika von Willebranda,
nie dochodzi do występowania zwiększonej skłonno-
który jest konieczny do adhezji płytek krwi do ma-
ści do krwawień. Jeżeli jednak dojdzie do wytworze-
cierzy obecnej pod komórkami śródbłonka w uszko-
nia fibryny wewnątrz naczynia, to niewielka część pla-
dzonych naczyniach. Choroba ta występuje z taką
zminogenu wiąże się z nią i dochodzi wtedy do miej-
samą częstością zarówno u kobiet, jak i u mężczyzn.
scowej aktywacji plazminy. Tak związana z fibryną
Zaburzenia w chorobie von Willebranda dotyczą
plazmina jest inaktywowana przez α2-antyplazminę
przede wszystkim hemostazy pierwotnej. Wyniki
wolniej w przeciwieństwie do szybko i nieodwracal-
klasycznego testu krzepnięcia i liczba trombocytów
nie inaktywowanej plazminy uwolnionej z rozłożonej
są prawidłowe, natomiast czas krwawienia jest zwy-
fibryny. Oznacza to, że fibryna nie tylko rozpoczyna
kle wydłużony.
proces fibrynolizy, ale także go ogranicza.
Do koagulopatii wrodzonych należą również he-
W prawidłowych warunkach istnieje równowaga
mofilie. Najczęściej występująca i równocześnie naj-
między procesami tworzenia fibryny a fibrynolizą.
ważniejsza z nich hemofilia A polega na niedoborze
W przypadku jej zaburzenia w zależności od tego,
globuliny antyhemofilowej A (czynnik VIII), a rza-
który proces przeważa, dochodzi albo do tworzenia
dziej występująca hemofilia B na niedoborze globu-
zakrzepów, albo zwiększenia skłonności do krwa-
liny antyhemofilowej B (czynnik Christmasa, czynnik
wień.
IX). Obie te hemofilie są dziedziczone w sposób re-
cesywny sprzężony z chromosomem X. Genotypowo
Zaburzenia hemostazy (skaza krwotoczna). Zabu-
chore, a fenotypowo zdrowe kobiety (posiadające
rzenia hemostazy mogą wynikać:
uszkodzony gen wyłącznie na jednym chromoso-
■ ze zmiany liczby płytek krwi albo z upośledzenia mie X) przenoszą chorobę, która ujawnia się w pełni
ich czynności, u mężczyzn.
Dzięki prawidłowej funkcji płytek krwi mecha-
■ z niedoboru czynników krzepnięcia (koagulopatie),
nizm hemostazy pierwotnej pozostaje w normie.
■ ze zmian naczyniowych. Jednakże z powodu zaburzeń krzepnięcia krwi na-
wet niewielkie urazy mogą prowadzić do ciężkich
Skaza krwotoczna związana ze zmianą liczby płytek krwawień, przede wszystkim w mięśniach i stawach.
krwi polega najczęściej na jej zmniejszeniu (trom- Ponadto istnieje niebezpieczeństwo wystąpienia za-
bocytopenie), natomiast zaburzenia czynnościowe grażającego życiu krwawienia w obrębie przewodu
płytek krwi (trombostenie) występują stosunkowo pokarmowego oraz w mózgu.
rzadko. Uwarunkowane immunologicznie trombo- Koagulopatie nabyte są najczęściej uwarunkowa-
cytopenie powstają w konsekwencji zakażeń (np. ró- ne niedoborem witaminy K, uszkodzeniem miąższu
życzki) oraz po zastosowaniu różnych leków (np. sul- wątroby prowadzącym do zmniejszenia syntezy czyn-
fonamidów, fenylbutazonu), a oprócz tego jako tzw. ników krzepnięcia lub też masywnym wykrzepianiem
trombocytopenie objawowe wskutek toksycznego wewnątrz naczyń (rozsiane krzepnięcie wewnątrzna-
uszkodzenia szpiku kostnego. Zaburzenia czynnoś- czyniowe, zob. poniżej).
ciowe płytek krwi są najczęściej nabyte (np. w prze- W takich przypadkach wydłużaniu ulegają zarów-
biegu zespołu mielodysplastycznego). Wrodzone no czas krwawienia, jak i krzepnięcia.
MUTSCHLER-2009.indd 532 2010-01-07 22:13:31
Krew 533
Rzadko obecnie występujące naczyniowe skazy żeniach, nowotworach złośliwych, a także w czasie
krwotoczne polegają na wrodzonych lub nabytych ciąży lub podczas przyjmowania środków hamują-
zmianach w obrębie ścian naczyń krwionośnych. cych owulację. Poza tym skłonność do krzepnięcia
Przykładem nabytej skazy naczyniowej jest szkorbut rośnie w przypadku podwyższonego poziomu gluko-
występujący w przypadku niedoboru witaminy C. kortykosteroidów we krwi.
Kliniczne następstwa zakrzepicy zależą od jej
Zakrzepica i zatory. Zakrzep jest to powstający umiejscowienia i zakresu. Szczególnie niebezpieczna
wewnątrznaczyniowo w trakcie życia skrzep. Wyróż- jest zakrzepica w tętnicach końcowych, gdyż prowa-
nia się: dzi do martwicy niedokrwiennej.
Jeżeli zakrzep oderwie się i popłynie z biegiem
■ zakrzep biały (płytkowy),
krwi, powstaje skrzep zatorowy (embolus), który
■ zakrzep czerwony. może spowodować zator (embolię), czyli ostre za-
mknięcie światła naczynia.
Zakrzepy białe powstają przede wszystkim w tętni-
cach, kiedy to po agregacji i rozpadzie płytek krwi
między trombocytami odkładają się włókna fibryny. 4.1.4.1. Środki wspomagające
W przypadku zastoju krwi, głównie w krążeniu żyl- hemostazę
nym, a także w zwężonych tętnicach, po powstaniu
zakrzepu białego tworzy się zakrzep czerwony. Środki takie stosowane są w przypadku skaz krwo-
tocznych oraz przy ostrych krwawieniach. Racjonalna
Serce i układ krążenia
Powstawaniu zakrzepów sprzyja: terapia powinna uwzględniać przyczynę powstawania
zaburzenia hemostazy.
■ uszkodzenie ścian naczyń,
■ zwolniony przepływ krwi,
4.1.4.1.1. Grupa witaminy K (filochinony)
■ przyspieszony proces krzepnięcia (nadkrzepli-
wość). Podstawowym elementem budowy wspólnym dla fi-
lochinonów jest 2-metylo-1,4-naftochinon, a różnią
Uszkodzenie ściany naczyń tętniczych występu- się one między sobą jedynie łańcuchem bocznym.
je głównie w postaci zmian arteriosklerotycznych (zob. tab. B 4.1-7). B4
(blaszka miażdżycowa). Fizjologiczną postacią witaminy K jest mena-
Szczególnie ważną rolę w powstawaniu zakrze- chinon (witamina K2). Może on zostać zastąpiony
pów odgrywa zwolniony przepływ krwi lub tworze- fitomenadionem (witamina K1), który występuje
nie się prądów wirowych (stąd częste występowanie w zielonych częściach roślin. Po odszczepieniu resz-
zakrzepów przy dłuższym okresie leżenia w łóżku). ty fitylowej z fitomenadionu otrzymuje się menadion
Nadkrzepliwość występuje w przypadku zwięk- (witamina K3), który jest w organizmie częściowo
szonej nadmiernie liczby płytek krwi oraz po zabie- przekształcany do menachinonu.
gach chirurgicznych, urazach, chorobach związanych Dzienne zapotrzebowanie na witaminę K wynosi
z niedrożnością naczyń tętniczych lub żylnych, zaka- ok. 1 mg.
Tab. B 4.1-7. Witamina K
Wzór strukturalny R Nazwa Preparaty handlowe
międzynarodowa
ftylowy fitomenadion Kanavit, Konakion MM
O (fitonadion, witamina K1)
CH3
difarnezylowy oraz menachinon (witamina K2)
R farnezylo-geranylo-geranylowy
O
–H menadion
witamina K (witamina K3)
MUTSCHLER-2009.indd 533 2010-01-07 22:13:31
534 Krew
Znaczenie fizjologiczne. Witamina K jest koniecz- na, np. w przypadku marskości lub atrofii, preparaty wita-
na do syntezy w wątrobie czynników II, VII, IX miny K są nieskuteczne.
oraz X, a także białek C i S. Działa ona jako koen-
zym w procesie γ-karboksylacji łańcuchów bocznych
zawierających kwas glutaminowy. Powstające w ten 4.1.4.1.2. Preparaty czynnika VIII,
sposób związki γ-karboksyglutamylowe są w sta- czynnika IX i czynnika VIIa
nie wiązać kompleksowo jony wapnia, co prowadzi
Skuteczne leczenie krwawień spowodowanych he-
do zmian konformacyjnych czynników krzepnięcia,
mofilią polega na podaniu świeżego osocza lub – jak
będących warunkiem ich działania. Właściwa postać
w przypadku hemofilii A – globuliny antyhemofilo-
czynna witaminy K jest pochodną hydrochinonu, któ-
wej A, najczęściej w postaci koncentratów czynnika
ra jest utleniana podczas γ-karboksylacji kwasu gluta-
VIII lub też uzyskiwanej metodą inżynierii genetycz-
minowego do 2,3-epoksydu witaminy K, a w reakcji
nej globuliny antyhemofilowej A. Na rynku dostępny
redukcji przez reduktazę epoksydową z NADPH jako
jest również preparat Moroctocog alfa, który jest re-
koenzymem ulega ponownie regeneracji.
kombinowanym czynnikiem VIII zalecanym w kon-
trolowaniu i profilaktyce epizodów krwawień, a także
Niedobór witaminy K. Dopóty witamina K dostar-
w rutynowej oraz przedzabiegowej profilaktyce u do-
czana jest z pożywieniem może być wchłaniana,
rosłych i dzieci chorych na hemofilię.
dlatego niedobór witaminy K jest rzadki. Jej wchła-
Chorym na hemofilię B podaje się koncentrat
nianie jest jednak, podobnie jak w przypadku innych
czynnika IX lub też białko rekombinowane.
witamin rozpuszczalnych w tłuszczach oraz samych
U pacjentów z niedoborem czynnika VII stosuje
tłuszczów, zależne od obecności w świetle jelita kwa-
się preparat czynnika VII.
sów żółciowych. Oznacza to, że w przypadku zmniej-
W przypadku ciężkich krwawień spowodowanych
szenia lub zatrzymania wydzielania żółci witamina K
niedoborem zależnych od witaminy K czynników
się nie wchłania. Może do tego dojść w przypadku
krzepnięcia (II, VII, IX, X), np. przy przedawkowa-
niedrożności lub ciężkiego zapalenia dróg żółcio-
niu fenprokumonu, można stosować preparaty kom-
wych oraz przy żółtaczce mechanicznej.
pleksu protrombiny.
Obserwuje się wtedy niską zawartość protrombiny
W przypadku wrodzonego niedoboru czynnika
we krwi i wynikający z tego wydłużony czas krwa-
XIII (czynnika stabilizującego fibrynę) również moż-
wienia. Również czynniki VII, IX oraz X są wówczas
na prowadzić terapię substytucyjną za pomocą odpo-
wytwarzane w niewystarczającej ilości.
wiedniego preparatu.
Niedobór witaminy K z podwyższonym ryzykiem
Terapia substytucyjna przy użyciu brakujących
krwawienia do mózgu występuje również u noworod-
czynników krzepnięcia będzie skuteczna jedynie
ków (melaena neonatorum) , których matki nie przyj-
wtedy, kiedy wymaga tego stopień ciężkości zabu-
mowały wystarczających ilości witaminy K.
rzeń krzepnięcia. Do komplikacji takiej terapii na-
leży, rzadko występujące, zjawisko tworzenia się
Wskazania. Preparaty witaminy K zawierające fi-
przeciwciał przeciw czynnikowi VIII co prowadzi
tometadion są wskazane we wszystkich postaciach
do pojawienia się hemofilii na skutek wytworzenia
niedoboru witaminy K. Służą one również jako an-
inhibitorów. W takim przypadku można podawać
tidotum przy przedawkowaniu leków przeciwzakrze-
rekombinowany czynnik VIIa, co pozwala ominąć
powych np. fenprokumonu.
zależną od czynnika VIII aktywację układu krzep-
nięcia. Jest to terapia skuteczna, ale niezwykle kosz-
Dawkowanie. U dorosłych w terapii substytucyjnej
towna.
podaje się doustnie 10–20 mg witaminy K1 dziennie.
W przypadku mniej nasilonych krwawień wystar-
Jedynie w przypadku zagrażających życiu krwawień,
czają zwykle środki miejscowe (tamponada ucisko-
np. po przedawkowaniu leków przeciwzakrzepo-
wa, środki opatrunkowe, zastosowanie preparatów
wych, wskazane jest dodatkowe podawanie dożylne
trombiny). Ponadto w mniej ciężkich przypadkach
czynników krzepnięcia (z powodu ryzyka powikłań
można podawać desmopresynę, która nasila uwalnia-
w postaci wstrząsu). Noworodkom podaje się profi-
nie czynnika VIII ze śródbłonka naczyń.
laktycznie 1–2 mg doustnie, a w przypadku istnieją-
cego ryzyka również domięśniowo lub dożylnie.
4.1.4.1.3. Fibrynogen
Witamina K może zwiększać zawartość protrombiny
we krwi jedynie wtedy, kiedy wątroba jest jeszcze w stanie Nagłe stany niedoboru fibryny występują w przebie-
produkować ten związek. Jeżeli jest ona ciężko uszkodzo- gu rozsianego krzepnięcia wewnątrznaczyniowego
MUTSCHLER-2009.indd 534 2010-01-07 22:13:31
Krew 535
(tzw. koagulopatii ze zużycia). Termin ten ozna- bocytów), jak i prostacykliny (hamującej agregację
cza szybkie zużycie krążących we krwi czynników trombocytów).
krzepnięcia wskutek rozległego uszkodzenia ścian Dlatego istnieje niebezpieczeństwo, że korzystny
naczyń, zwolnienia przepływu krwi (np. we wstrzą- efekt hamujący agregację płytek krwi może zostać
sie), podczas pewnych powikłań okołoporodowych przynajmniej częściowo zniesiony przez zmniejsze-
(np. przedwczesnego odklejenia się łożyska, zatoru nie syntezy prostacykliny. Oprócz tego wskutek upo-
wodami płodowymi), a także niekiedy podczas ope- śledzenia czynności trombocytów wzrasta skłonność
racji płuc lub mózgu. Istnieje również hipofibrynoge- do krwawień.
nemia wrodzona. Powstałe wskutek tego krwawienia
mogą zostać zahamowane dzięki dożylnym wlewom Jako inhibitory agregacji trombocytów zastosowanie
fibrynogenu. W przypadku zastosowań miejscowych znajdują:
dostępne są mieszaniny fibrynogenu, trombiny i czyn-
■ inhibitory cyklooksygenazy (np. kwas acetylosali-
nika VIII pod postacią kleju fibrynowego.
cylowy),
■ antagoniści receptorów ADP (tiklopidyna i klopi-
4.1.4.1.4. Hormony kory nadnerczy dogrel),
■ antagoniści receptora glikoproteinowego IIb/IIIa
W przypadku skłonności do krwawień uwarunko-
(GP-IIb/IIIa),
wanej zbyt małą ilością trombocytów (trombocyto-
penia) wskutek obecności przeciwciał (autoimmu- ■ dipirydamol.
Serce i układ krążenia
notrombocytopenia) można stosować glukokortyko-
steroidy. Powodują one, często tylko przejściowo,
wzrost liczby płytek krwi, zmniejszają tworzenie 4.1.4.2.1. Inhibitory cyklooksygenazy
się przeciwciał i przepuszczalność błony naczyń
włosowatych. Nie ma natomiast wskazań do stoso- Kwas acetylosalicylowy jest w dalszym ciągu najczęś-
wania glukokortykosteroidów w przypadku trom- ciej stosowanym inhibitorem agregacji trombocytów.
bocytopenii wynikającej z zaburzeń wytwarzania Jego mechanizm działania polega na acetylacji błony
trombocytów. płytek krwi i białek osocza oraz przede wszystkim
na nieodwracalnym hamowaniu, również wskutek
acetylacji, cyklooksygenazy 1 (COX-1). Przy czym
B4
zaletą kwasu acetylosalicylowego jest to, że hamo-
4.1.4.2. Środki hamujące czynność
wanie agregacji trombocytów trwa znacznie dłużej
trombocytów
niż – zachodzące również za pośrednictwem bloko-
(inhibitory agregacji płytek krwi) wania COX-1 – hamowanie syntezy prostacykliny
w śródbłonku. W przeciwieństwie do wytwarzają-
cych prostacyklinę komórek śródbłonka płytki krwi,
Choroby zakrzepowo-zatorowe są najczęstszą przy- w których została zablokowana COX-1, nie są w sta-
czyną zgonów szczególnie w krajach uprzemysłowio- nie zsyntetyzować nowego enzymu oraz tromboksa-
nych. Dlatego niezwykle duże znaczenie mają sku- nu A2, ponieważ nie posiadają jądra komórkowego.
teczna profilaktyka i leczenie tych schorzeń. W związku z tym w agregacji uczestniczyć mogą wy-
W przypadku zakrzepicy naczyń tętniczych, która łącznie nowo powstałe płytki krwi.
rozwija się najczęściej na podłożu zmian miażdżyco-
wych lub na powierzchni materiałów egzogennych Dodatkowo korzystnym zjawiskiem jest to, że kwas acety-
(np. sztucznych zastawek aortalnych), dochodzi, losalicylowy acetyluje płytkową COX-1 już w żyle wrotnej,
gdzie występuje w wyższym stężeniu, ponieważ nie uległ
przynajmniej początkowo, do rozwoju zakrzepów jeszcze szybkiemu metabolizmowi wskutek deacetylacji.
białych (płytkowych). Dlatego racjonalną metodą W sąsiedztwie komórek śródbłonka naczyń krążenia duże-
leczenia jest z założenia hamowanie czynności trom- go znajduje się już w o wiele niższym stężeniu i dlatego
bocytów w celu zapobiegania powstawaniu zakrze- jego wpływ na syntezę prostacykliny jest mniejszy.
pów i ich powikłań. Aby jednak w pełni zrozumieć
problematykę dotyczącą stosowania odpowiednich Chociaż nie jest w pełni wyjaśnione, jakie dawko-
środków, a w szczególności inhibitorów cyklooksy- wanie kwasu acetylosalicylowego przynosi najlep-
genazy, należy zwrócić uwagę na to, że cyklooksy- sze skutki przy równocześnie możliwie najmniej-
genaza uczestniczy w syntezie w śródbłonku zarów- szych efektach niepożądanych, wydaje się, że dawka
no tromboksanu A2 (wzmagającego agregację trom- 100 mg dziennie jest wystarczająca dla wywołania
MUTSCHLER-2009.indd 535 2010-01-07 22:13:31
536 Krew
hamowania agregacji trombocytów. Jest to znacznie za pośrednictwem kompleksu receptora GP-IIb/IIIa
mniejsza dawka od dawki koniecznej dla wywołania i w ten sposób hamowana jest agregacja trombocy-
efektu przeciwbólowego. tów. Oprócz długotrwałego stosowania w celu hamo-
wania agregacji płytek u pacjentów z nietolerancją
kwasu salicylowego tę grupę leków podaje się w celu
4.1.4.2.2. Antagoniści receptorów ADP zapobiegania tworzeniu się zakrzepów po wszczepie-
niu stentów naczyniowych.
Ta grupa leków blokuje wybiórczo wiązanie się
adenozynodifosforanu (ADP) z jego receptorami Klopidogrel jest prolekiem, który zostaje aktywowa-
na trombocytach (zob. ryc. B 4.1-5). Na powierzch- ny po oksydacji przez cytochrom P-450 3A4, a na-
ni płytek krwi występują dwa receptory purynowe stępnie hydrolizie. Dlatego silne inhibitory CYP3A4
(P2Y1 i P2Y12), obydwa związane z białkiem G (Gq są w stanie zmniejszyć bioaktywację klopidogrelu,
w przypadku P2Y1, a Gi w przypadku P2Y12). Dzięki wpływając tym samym na osłabienie jego działania.
blokadzie tych receptorów nie dochodzi do skutku Okres półtrwania wynosi osiem godzin, ale z powodu
indukowane przez ADP tworzenie sieci trombocytów nieodwracalnej blokady przez bioaktywny metabolit
A trombina
PAF (czynnik ADP (adenozynodifosforan)
aktywujący płytki)
TXA2 (tromboksan A2)
adrenalina
wazopresyna Aktywator Receptor/
punkt uchwytu
PAFR PAR-1
P2Y12 TxA2
α2 Gi Gi ADP P2Y12; P2Y1
Gi Gi Gq V1 adrenalina α2
kolagen
Gq
serotonina Gq kolagen glikoproteina VI
5HT2A
PAF PAFR
GpVI
serotonina 5 HT2A
G
trombocyt trombina PAR-1; PAR-4
tromoksan TxA2
GP-IIb/IIIa wazopresyna V1
B
fibrynogen
aktywowany trombocyt
trombocyt GP IIb
opróżnianie
ziarnistości
GP IIIa
śródbłonek GP Ib
vWF vWF
warstwa podśródbłonkowa
uszkodzenie śródbłonka
Ryc. B 4.1-5. (A) Mechanizm aktywacji płytek oraz (B) agregacji za pośrednictwem receptorów GP-IIb/IIIa. vWF – czynnik von
Willebranda.
MUTSCHLER-2009.indd 536 2010-01-07 22:13:31
Krew 537
receptora P2Y12 musi upłynąć 5–7 dni po odstawie- 4.1.4.2.3. Antagoniści receptora
niu klopidogrelu do przywrócenia czynności płytek glikoproteinowego IIb/IIIa
krwi.
Klopidogrel jest wskazany w prewencji występo-
Ta grupa leków działa na nowej zasadzie – hamo-
wania incydentów zakrzepowych po zawale serca lub
wania ostatecznego etapu agregacji trombocytów,
udarze niedokrwiennym, a także przy obwodowych
wspólnego dla wszystkich szlaków niezależnie
zatorach naczyń tętniczych. Dawkowanie wynosi 75
od czynnika wywołującego (tromboksan, ADP i in.).
mg dziennie. W przypadku ostrego zespołu wieńco-
Na powierzchni płytek krwi znajduje się 30 000–
wego powinien być zastosowany w znacznie więk-
50 000 receptorów GP-IIb/IIIa, które należą do gru-
szej dawce wysycającej (300 mg).
py tzw. integryn (integryna αIIb, β3).Składają się one
Działania niepożądane wynikające z mechanizmu
z podjednostek α i β, które wiążą się ze swoim ligan-
działania obejmują zwiększenie częstości krwawień.
dem (fibrynogenem) dopiero po uprzedniej aktywa-
Dlatego inne leki hamujące hemostazę (np. kwas
cji wskutek zmiany konformacji np. przez trombinę,
acetylosalicylowy, heparyna) można podawać rów-
ADP lub noradrenalinę. Fibrynogen tworzy wtedy
nocześnie wyłącznie w przypadku ściśle określo-
mostki między aktywowanymi trombocytami i pro-
nych wskazań (np. po wszczepieniu stentu). Zmiany
wadzi w ten sposób do utworzenia zakrzepu płyt-
w obrazie krwi (leukopenia) obserwuje się rzadziej
kowego.
niż w przypadku stosowania tiklopidyny (zob. poni-
żej). Działania niepożądane dotyczące przewodu po- Zidentyfikowano dwie sekwencje peptydowe na recepto-
karmowego obserwuje się rzadziej niż w przypadku rze odpowiedzialne za rozpoznawanie liganda. Sekwencja
Serce i układ krążenia
leczenia kwasem acetylosalicylowym. arginina/lizyna/kwas asparginowy jest identyfikowana
w różnych integrynach, podczas gdy sekwencja lizyna/gli-
Tiklopidyna pod względem budowy jest lekiem cyna/kwas asparginowy jest swoista względem wiązania
się z fibrynogenem w receptorze GP-IIb/IIIa. Związki, któ-
ściśle spokrewnionym z klopidogrelem. Substancja re zawierają obie te sekwencje, swoiście hamują agregację
ta również jest podawana jako prolek, który ulega płytek krwi.
bioaktywacji przez CYP3A4 i wiąże się nieodwracal-
nie z receptorem P2Y12. Tiklopidyna jest stosowana
Korzystne efekty tej grupy leków w połączeniu
u dializowanych pacjentów z powikłaniami w zakre-
z kwasem acetylosalicylowym i heparyną potwier-
sie przetoki tętniczo-żylnej, a także w profilaktyce
udarów niedokrwiennych, kiedy nie można stosować
dzono w dużych badaniach klinicznych, szczegól- B4
nie u pacjentów z ostrym zespołem wieńcowym
kwasu acetylosalicylowego z powodu jego nietole-
(np. niestabilną dusznicą bolesną), a także w przy-
rancji przez pacjenta.
padku przezskórnych zabiegów na tętnicach wień-
Dawkowanie wynosi 250–500 mg dziennie.
cowych. Najważniejszym działaniem niepożądanym
Do działań niepożądanych zalicza się zmiany
są krwawienia.
w obrazie krwi (trombocytopenię, leukopenię, tak-
że agranulocytozę), zaburzenia żołądkowo-jelitowe,
pokrzywkę i uszkodzenie wątroby. Najgroźniejszym Abcyksymab to fragment Fab chimerowego mono-
działaniem niepożądanym jest zakrzepowa plamica klonalnego przeciwciała przeciw receptorowi GP-IIb/
małopłytkowa (choroba Moschowitza). Z powodu IIIa, który wiąże się z nim, a także z innymi recepto-
nieco korzystniejszego profilu działań niepożąda- rami (np. witronektyną). Lek ten jest podawany poza-
nych zamiast tiklopidyny obecnie zwykle stosowany jelitowo. Okres półtrwania postaci wolnej w osoczu
jest klopidogrel. wynosi w fazie pierwszej 10 minut, a w fazie drugiej
30 minut. Ponieważ jednak abcyksymab jest wy-
chwytywany przez trombocyty i może być następnie
N z nich ponownie uwalniany, substancja czynna może
tiklopidyna być wykrywana przez ponad tydzień. Dlatego agre-
S Cl gacja płytek jest zahamowana do 24 godzin po zakoń-
czeniu podawania leku.
Dawkowanie wynosi 0,25 mg/kg masy ciała
COOCH3 w dożylnym bolusie 10–60 minut przed rozpoczę-
ciem przezskórnej angioplastyki naczyń wieńcowych
N (PTCA), a następnie w stałym wlewie z prędkością
klopidogrel
S Cl 0,125 μg/kg/min (maksymalnie 10 μg/min) przez 12
godzin.
MUTSCHLER-2009.indd 537 2010-01-07 22:13:31
538 Krew
Tab. B 4.1-8. Właściwości antagonistów receptora GP-IIb/IIIa
Właściwość Abcyksymab Tirofiban Eptifibatyd
swoistość względem GP-IIb/IIIa nie tak tak
powinowactwo do receptora wysokie niskie niskie
okres półtrwania 2 dni 2 godz. 2 godz.
odwracalność (przywrócenie funkcji płytek 2 dni 4–8 godz. 4 godz.
po zakończeniu leczenia)
stwierdzona antygenowość tak nie nie
ryzyko trombocytopenii tak tak nie
Do działań niepożądanych należy trombocytopenia
obserwowana u 2% pacjentów przyjmujących abcyk-
symab. Występuje ona najprawdopodobniej wskutek
tworzenia się epitopów antygenowych po związaniu
O O OH
się przeciwciał na receptorze. O (CH2) 3CH3
HN S
NH O
Eptifibatyd to syntetyczny heptapeptyd będący swo-
istym antagonistą receptora GP-IIb/IIIa, który po- tirofiban
dobnie jak abcyksymab jest podawany dożylnie (180
μg/kg w bolusie a następnie ciągły wlew z szybkością
2,0 μg/kg/min do 72 godzin). Czas działania jest o 6
do 12 godzin krótszy niż abcyksymabu.
Tirofiban to natomiast niepeptydowy, niskocząstecz-
kowy antagonista receptora GP-IIb/IIIa. Również ten
lek podawany jest pozajelitowo (najpierw 0,4 μg/kg/ 4.1.4.2.4. Dipirydamol
min przez 30 minut, następnie dawka podtrzymująca
0,1 μg/kg/min). Okres półtrwania wynosi 2 godzi-
Dipirydamol był początkowo opracowany jako lek
ny. Lek ten jest w większości wydalany przez nerki
stosowany w terapii choroby wieńcowej. Dzięki
w niezmienionej postaci. Czas leczenia powinien być
zdolności do hamowania fosfodiesterazy zawartej
nie krótszy niż 48 godzin i nie dłuższy niż 108 go-
w płytkach krwi prowadzi do gromadzenia w nich
dzin.
cAMP, wiązania wapnia i w rezultacie do zmniejsze-
nia uwalniania mediatorów z płytek krwi. Stosowany
jest głównie w skojarzeniu z kwasem acetylosalicylo-
CONH2 wym dwa razy dziennie po 200 mg.
S
NH2 S NH
HN NH O 4.1.4.3. Leki przeciwzakrzepowe
HN O
N (antykoagulanty)
HOOC
O O
O
Antykoagulanty są stosowane, podobnie jak leki
HN NH NH
NH hamujące agregację trombocytów, w profilaktyce
O i leczeniu procesów zakrzepowo-zatorowych. Są one
ponadto niezbędne przy produkcji preparatów krwi
eptifibatyd
konserwowanej.
Ze względu na mechanizm działania rozróżnia
się:
MUTSCHLER-2009.indd 538 2010-01-07 22:13:31
Krew 539
■ leki przeciwzakrzepowe bezpośrednie, które od- jaśnione. Pozajelitowe podanie heparyny zapobiega
działują bezpośrednio na czynniki krzepnięcia, krzepnięciu krwi poprzez działanie na różne miejsca
układu krzepnięcia. Podstawowy mechanizm działa-
■ leki przeciwzakrzepowe pośrednie, które zmniej-
nia polega na aktywacji antytrombiny, która hamuje
szają produkcję czynników krzepnięcia.
trombinę i inne proteazy serynowe. Aktywacja anty-
trombiny zachodzi dzięki pentasacharydowej struk-
Oprócz tego leki przeciwzakrzepowe podzielić moż-
turze wewnątrz cząsteczki heparyny, która prowadzi
na na te, które hamują procesy krzepnięcia wyłącznie
do zmian konformacji, a w rezultacie do aktywacji
in vitro lub in vivo, oraz na te, które działają zarówno
antytrombiny. Do zahamowania trombiny niezbędne
in vitro, jak i in vivo.
jest wytworzenie trójskładnikowego kompleksu zło-
Podczas stosowania leków przeciwzakrzepowych
żonego z heparyny, antytrombiny i trombiny (zob.
należy również wziąć pod uwagę to, czy chodzi je-
ryc. B 4.1-6).
dynie o częściowe zmniejszenie krzepnięcia krwi,
czy o całkowite zahamowanie procesu krzepnięcia
Do utworzenia takiego kompleksu dochodzi tylko
(np. w przypadku krążenia pozaustrojowego).
wtedy, gdy zastosowana heparyna ma łańcuch zło-
żony z więcej niż 18 monomerów. Do zahamowania
czynników XIa i IXa jest natomiast potrzebne zwią-
4.1.4.3.1. Usunięcie jonów wapnia
zanie heparyny jedynie z antytrombiną, co zachodzi
również w przypadku heparyn o krótszych łańcu-
Jony wapnia są niezbędne do procesu krzepnięcia
chach. Ten mechanizm wyjaśnia, dlaczego heparyna
Serce i układ krążenia
krwi. Dlatego dzięki ich usunięciu można zapobiec
niefrakcjonowana hamuje czynnik Xa i trombinę,
krzepnięciu krwi. Dokonuje się tego za pomocą wią-
podczas gdy heparyny niskocząsteczkowe (low mo-
zania w kompleksach z cytrynianem sodu, bądź też
lecular weight heparins; LMWH) hamują głównie
wytrącania przy użyciu szczawianu sodu lub fluorku
czynnik Xa. Oprócz działania na układ krzepnięcia
sodu.
heparyna ma działanie lipemiczne klarujące osocze
Usuwanie jonów wapnia może być stosowane je-
poprzez uwalnianie ze śródbłonka lipazy lipopro-
dynie in vitro, ponieważ obniżenie poziomu jonów
teinowej, która rozpuszcza chylomikrony. Ponadto
wapnia we krwi in vivo doprowadziłoby do tężyczki.
heparyna przyspiesza rozkład histaminy poprzez
Można jednak użyć do transfuzji krwi cytrynianowej,
wpływ na uwalnianie oksydazy diaminowej, która
pod warunkiem że szybkość przetaczania nie jest zbyt
utlenia histaminę, oraz zmniejsza wytwarzanie al-
B4
duża. W takim przypadku niedobór jonów wapnia zo-
dosteronu.
staje szybko wyrównany dzięki uruchomieniu maga-
Działanie heparyny związane jest z występowa-
zynów wapnia przez parathormon.
niem w jej cząsteczce licznych ładunków ujemnych,
wzrastających wraz ze wzrostem liczby reszt siarcza-
nowych. Dlatego jej działanie może zostać szybko
4.1.4.3.2. Heparyny
zniesione przez polikationy, np. siarczan protaminy
(zob. poniżej), który składa się w dwóch trzecich
Heparyna jest bezpośrednią endogenną substancją z zasadowego aminokwasu – argininy.
przeciwzakrzepową, która występuje w mastocytach Istotną zaletą heparyny jest to, że działa ona na-
i granulocytach zasadochłonnych. To polianionowy tychmiast po podaniu. Podobnie jednak jak więk-
polisacharyd o masie cząsteczkowej 6000–30000 Da szość występujących naturalnie w organizmie sub-
zawierający grupy karboksylowe oraz rodniki siar- stancji o strukturze polimerowej, jest szybko rozkła-
czanowe, które sprawiają, że jest to jeden z najsilniej- dana i dlatego jej działanie utrzymuje się tylko kilka
szych kwasów obecnych w organizmie. W tym poli- godzin.
merze na jedną cząsteczkę kwasu uronowego (kwasu Z powodu dużej masy cząsteczkowej oraz ujemne-
glukuronowego, kwasu iduronowego) przypada jedna go ładunku, który uniemożliwia wchłanianie po po-
cząsteczka glukozaminy, a oba te rodzaje cząsteczek daniu doustnym, heparyna w zastosowaniach ogól-
są częściowo siarczanowane, przy czym liczba i miej- nych musi być podawana parenteralnie (dożylnie,
sce podstawienia reszt siarczanowych są zmienne. podskórnie).
Heparyna jest wytwarzana i magazynowana wraz
z histaminą w ziarnistościach komórek tucznych, któ- Heparyna standardowa (heparyna niefrakcjono-
re występują w szczególnie dużych ilościach w wą- wana, UFH). Heparynę pozyskuje się z błony śluzo-
trobie, płucach i błonie śluzowej jelit. Biologiczne wej świńskich jelit oraz bydlęcych płuc. Produkt nie-
znaczenie endogennej heparyny nie jest do końca wy- frakcjonowany określany jest mianem heparyny stan-
MUTSCHLER-2009.indd 539 2010-01-07 22:13:32
540 Krew
- -
SO3 - SO3 - -
HN CO2 HN CO2 OSO3
-
HO O O3SO O O OCH3
O O
O O
HO HO O HO O NH
- -
- OH - OSO3 OH SO3
SO3 OSO3
sekwencja
pentasacharydowa
czynnik Xa
heparyna
niefrakcjonowana
antytrombina
trombina
sekwencja
pentasacharydowa
heparyna
drobnocząsteczkowa antytrombina
antytrombina
Ryc. B 4.1-6. Mechanizm działania heparyny niefrakcjonowanej i heparyny drobnocząsteczkowej (według Weitza).
dardowej. Ponieważ różne preparaty heparyny różnią Heparyny drobnocząsteczkowe (low molecular
się pomiędzy sobą składem, szczególnie stopniem weight heparins; LMWH). Za pomocą ograniczone-
usiarczanowania oraz masą cząsteczkową, a co za tym go rozkładu można uzyskać z produktu wyjściowego
idzie – skutecznością, dawkowanie określa się w jed- tzw. heparynę drobnocząsteczkową o średniej masie
nostkach międzynarodowych, a nie w miligramach. cząsteczkowej 4000–6000 Da, przy czym w zależno-
1 mg heparyny standardowej odpowiada ok. 170 ści od metody uzyskiwania można otrzymać prepa-
IE. Heparyna niefrakcjonowana jest pod względem raty o różnej masie cząsteczkowej, ale o podobnych
masy cząsteczkowej bardzo heterogenna (od 5 000 właściwościach farmakodynamicznych. Jak opisano
do 30 0000 Da ze średnią wartością 15 000, co od- to wcześniej, LMWH różnią się od heparyny standar-
powiada długości 50 monomerów). Ponieważ dłu- dowej przede wszystkim tym, że ze względu na mniej-
gie łańcuchy heparyny są eliminowane szybciej niż szą długość łańcuchów hamują one głównie czynnik
krótkie, farmakokinetyka różnych frakcji heparyny Xa. Heparyny drobnocząsteczkowe wiążą się też
standardowej jest zróżnicowana. Oprócz tego jedynie w mniejszym stopniu niż heparyna standardowa z biał-
część heparyny (ok. 30%) zawiera strukturę pentasa- kami krwi, trombocytami i śródbłonkiem, a mimo
charydową odpowiedzialną za jej działanie, dlatego to ich okres półtrwania jest wyraźnie dłuższy (2–4 go-
trudno jest określić ogólnie jej wpływ na krzepnięcie. dziny po podaniu dożylnym, 4–6 godzin po podaniu
Z tego powodu konieczne jest częste monitorowanie podskórnym). Względna zawartość odpowiedzialnych
jej działania. za działanie struktur pentasacharydowych jest wyższa
MUTSCHLER-2009.indd 540 2010-01-07 22:13:32
Krew 541
niż w heparynie niefrakcjonowanej. Z tych różnych cząsteczki heparyn drobnocząsteczkowych nie mogą
właściwości wynika lepsza przewidywalność efektu zostać zneutralizowane przy użyciu siarczanu prota-
hamującego krzepnięcie krwi. Dlatego w przypadku miny (zob. poniżej).
stosowania heparyn drobnocząsteczkowych u więk- Fondaparinuks jest obecnie wskazany wyłącznie
szości pacjentów nie jest konieczne regularne moni- u pacjentów, którzy muszą zostać poddani dużym za-
torowanie odpowiednich parametrów (z wyjątkiem biegom ortopedycznym kończyn dolnych.
pacjentów z niewydolnością nerek).
Dawkowanie. Dawkowanie zależy od tego, czy ko-
Fondaparinuks. Wychodząc od odpowiedzialnego nieczne jest uzyskanie pełnego efektu przeciwzakrze-
za wiązanie antytrombiny rejonu heparyny, zsyntety- powego czy tylko zmniejszenie nadkrzepliwości.
zowano pentasacharyd fondaparinuks, który podob- W celu uzyskania pełnego efektu przeciwzakrzepo-
nie jak heparyna drobnocząsteczkowa, umożliwia wego (np. przy krążeniu pozaustrojowym) po począt-
wybiórcze działanie hamujące czynnik Xa bez wpły- kowym podaniu w bolusie 5000–10000 IE. podaje
wu na trombinę. Fondaparinuks, wiążąc się selek- się we wlewie ciągłym 20 000–30 000 IE. heparyny
tywnie z antytrombiną, wzmaga około 300-krotnie standardowej. Działanie heparyny należy monitoro-
jej działanie na czynnik Xa. Lek ten ma zapewniać wać, oznaczając czas trombinowy oraz czas rekalcy-
lepszą kontrolę nad terapią, a oprócz tego z powodu nacji osocza. Natychmiastowo działającym antidotum
braku wpływu na czynnik płytkowy 4 nie powinna jest siarczan protaminy. Jest to silnie zasadowe biał-
występować po nim indukowana heparyną trombocy- ko, które inaktywuje heparynę, tworząc z nią kom-
topenia. Niedawno wykazano co prawda, że wytwa- pleksy. Siarczan protaminy stosuje się przy powikła-
Serce i układ krążenia
rzanie przeciwciał przeciw czynnikowi płytkowemu niach wywołanych heparyną, pod koniec zabiegów
4 zachodzi po podaniu fondaparinuksu z taką samą z wykorzystaniem krążenia pozaustrojowego oraz
częstością jak po podaniu enoksaparyny, jednak ani po zakończeniu transfuzji wymiennej.
razu nie doszło do wystąpienia trombocytopenii. W profilaktyce zakrzepicy (szczególnie po za-
Fondaparinuks jest wydalany w postaci niezmie- biegach) oraz zatorów płucnych stosuje się obecnie
nionej przez nerki. Jego okres półtrwania wynosi 15 przede wszystkim podawane podskórnie małe dawki
godzin, co umożliwia dawkowanie raz na dobę. heparyny. Pacjentowi podaje się 3000–5000 IE. 2–
5 godzin przed zabiegiem oraz 5–10 dni po zabiegu
Wskazania do stosowania heparyn. Zasadniczo w odstępach co 6, 8, a później co 12 godzin. Liczba
wskazania do stosowania heparyny standardowej epizodów zakrzepicy dzięki takiemu dawkowaniu
B4
i drobnocząsteczkowej nie różnią się. Obie są podob- zmniejsza się przy jednocześnie niewielkim wpływie
nie skuteczne w przed- i pooperacyjnej profilaktyce na testy krzepnięcia.
zakrzepicy i zatorów, w leczeniu niestabilnej posta- Dawkowanie fondaparinuksu wynosi 2,5 mg pod-
ci dławicy piersiowej, w ostrej fazie zawału mięśnia skórnie raz na dobę.
sercowego (razem ze środkami fibrynolitycznymi)
w celu redukcji komplikacji zakrzepowo-zatoro- Działania niepożądane. Podczas podawania he-
wych, w rozsianym krzepnięciu wewnątrznaczynio- paryny w dawkach powodujących pełny efekt
wym, a także w krążeniu pozaustrojowym i dializie. przeciwzakrzepowy mogą wystąpić krwawienia
Ze względu na opisane wyżej lepsze właściwości far- np. w obrębie skóry lub błon śluzowych. Kolejnym
makokinetyczne heparyny drobnocząsteczkowe mają istotnym działaniem niepożądanym jest indukowa-
w porównaniu z heparyną standardową przewagę przy na heparyną trombocytopenia. Rozróżnia się dwa
podobnej skuteczności klinicznej. Przede wszystkim jej typy: typ 1 to trombocytopenia nieimmunogenna,
mają one istotnie dłuższy czas działania z możliwoś- która pojawia się wkrótce po rozpoczęciu leczenia
cią podawania raz dziennie, a także możliwość pro- i przebiega bez powikłań, a liczba trombocytów nor-
wadzenia leczenia bez intensywnego monitorowania malizuje się szybko, nawet jeśli nie przerwie się po-
terapii. Dlatego pomimo znacznie wyższych kosztów dawania heparyny. Znacznie większe znaczenie ma
heparyny drobnocząsteczkowe są stosowane coraz trombocytopenia typu II, która rozpoczyna się u le-
częściej. Z drugiej strony heparyny drobnocząstecz- czonych po raz pierwszy pacjentów po trwającym
kowe mają też wady: po pierwsze, monitorowanie te- 5–10 dni okresie utajenia. Polega ona na aktywacji
rapii polega na oznaczaniu aktywności czynnika Xa, płytek krwi za pośrednictwem IgG, których celem
a nie czasu częściowej tromboplastyny po aktywacji jest kompleks heparyny z czynnikiem płytkowym 4.
(APTT, czas koalinowo-kefalinowy), co nie jest moż- Aktywacja płytek wyjaśnia paradoksalny na pierw-
liwe we wszystkich szpitalach, a także w przypad- szy rzut oka efekt w postaci powikłań zakrzepo-
kach nagłych; po drugie, przy przedawkowaniu małe wo-zatorowych mimo trombocytopenii. Ta forma
MUTSCHLER-2009.indd 541 2010-01-07 22:13:32
542 Krew
trombocytopenii indukowanej heparyną utrzymuje 4.1.4.3.3. Heparynoidy
się aż do momentu odstawienia heparyny. Częstość
występowania u pacjentów po dużych zabiegach or-
Termin heparynoidy oznacza preparaty o działaniu
topedycznych na kończynach dolnych wynosi około
podobnym do heparyny, które podawane są pozajeli-
3% u leczonych heparyną standardową i 0,3% u le-
towo i mają duże znaczenie przede wszystkim w le-
czonych heparynami frakcjonowanymi (w innych
czeniu pacjentów, u których wystąpiła trombocytope-
grupach pacjentów trombocytopenia indukowana
nia indukowana heparyną.
heparyną występuje rzadziej). Ze względu na czę-
stość stosowania heparyny trombocytopenia typu
Sól sodowa danaparoidu jest to mieszanka drobno-
II indukowana podawaniem heparyny jest bardzo
cząsteczkowych glikozaminoglikanów, które pozy-
istotnym działaniem niepożądanym. Aby móc leczyć
skiwane są z błony śluzowej świńskich jelit. Preparat
pacjentów, u których w przebiegu terapii heparyną
składa się w 84% z siarczanu heparanu, 12% stano-
rozwinęła się trombocytopenia typu II, opracowano
wi siarczan dermatanu, a 4% siarczan chondroityny.
alternatywnie kilka innych leków (np. hirudynę lub
Mechanizm działania polega na hamowaniu czynnika
danaproid, zob. poniżej).
Xa (siarczan heparanu) oraz trombiny (siarczan der-
Pozostałe działania niepożądane heparyny obej-
matanu). Okres półtrwania wynosi 24 godziny, wyda-
mują reakcje alergiczne (pokrzywkę, nieżyt nosa
lanie zachodzi w 40–50% przez nerki.
i in.), martwicę skóry, odwracalne wypadanie
Danaparoid jest wskazany w profilaktyce choroby
włosów, a przy długotrwałym podawaniu osteo-
zakrzepowej u pacjentów z indukowaną heparyną
porozę. W pojedynczych przypadkach opisywano
trombocytopenią typu II w wywiadzie, jako lek prze-
wystąpienie indukowanego przez heparynę hipo-
ciwzakrzepowy przy objawowej zakrzepicy i współ-
aldosteronizmu. Po odstawieniu heparyny może
istniejącej indukowanej heparyną trombocytopenii
dojść do wystąpienia efektu z odbicia. U pacjentów
typu II, a także w profilaktyce zakrzepicy w przypad-
z paraproteinemiami istnieje niebezpieczeństwo
ku nietolerancji heparyny.
wywołania zmian lepkości krwi wskutek tworzenia
Dawkowanie w profilaktyce zakrzepicy żylnej wy-
się trudno rozpuszczalnych kompleksów heparyny
nosi 750 jednostek anty-Xa dwa razy dziennie przez
z białkami.
7–10 dni.
Przeciwwskazania. Stosowanie heparyny jest prze-
Polisiarczan pentosanu jest to podobny do hepary-
ciwwskazane przy podejrzeniu lub stwierdzeniu
ny drobnocząsteczkowy polisacharyd pochodzenia
zwiększonej skłonności do krwawień, owrzodzeniach
roślinnego (1,4-β-D-ksylan-2,3-dihydrosiarczan) se-
przewodu pokarmowego, zagrażającym poronieniu,
lektywnie hamujący czynnik Xa. Podobnie jak dana-
a także w przypadku ciężkich schorzeń wątroby, ne-
paroid, wskazany jest on w profilaktyce zakrzepicy
rek i trzustki.
przed i po dużych zabiegach chirurgicznych, jeśli
nie można zastosować heparyny.
Interakcje. Równoczesne podawanie środków ha-
W tym celu 1–2 godziny przed zabiegiem należy
mujących agregację trombocytów, a także niektó-
podać w iniekcji 50 mg, a następnie 50 mg podskór-
rych penicylin i cefalosporyn zwiększa ryzyko wy-
nie co 12 godzin co najmniej przez 7 dni.
stąpienia krwawienia. Działanie heparyny zmniej-
szają leki przeciwhistaminowe, glikozydy naparst- Dodatkowo istnieje wiele syntetycznie uzyskiwanych he-
nicy i tetracykliny. parynoidów do zastosowań miejscowych, które powstają
przez podstawienie odpowiednich polisacharydów resztami
Preparaty heparyny do stosowania miejscowego. kwasu siarkowego. Ich zastosowanie jest podobne jak sto-
Na rynku dostępne są liczne preparaty zawierające sowanej miejscowo heparyny i również sporna jest ich sku-
teczność.
heparynę do stosowania miejscowego w przypadku
zamkniętych urazów sportowych i powypadkowych,
podbiegnięć krwawych, zespołu żylakowatego, za-
krzepicy naczyń powierzchownych oraz zakrzepo- 4.1.4.3.4. Hirudyna, pochodne hirudyny
wego zapalenia żył. Skuteczność tych preparatów i inne inhibitory trombiny
w profilaktyce zakrzepicy naczyń powierzchownych
oraz w leczeniu chorób żył jest bardzo sporna. Nieco Wydzielina gruczołów pijawki (Hirudo medicinalis)
lepsze rezultaty są w przypadku stosowania w lecze- zawiera hirudynę, substancję o działaniu silnie hamu-
niu powierzchownych podbiegnięć krwawych oraz jącym krzepnięcie krwi. Jest to polipeptyd składający
tępych urazów. się z 65 aminokwasów stabilizowanych trzema most-
MUTSCHLER-2009.indd 542 2010-01-07 22:13:32
Krew 543
kami dwusiarczkowymi, który w przeciwieństwie aminokwasów, z których pierwsze cztery wiążą się
do heparyny jest bezpośrednim inhibitorem trombiny z centrum katalitycznym trombiny.
(hamuje również trombinę związaną z fibryną), to zna- Lek ten jest wydalany przez nerki, a jego okres
czy działa bez pośrednictwa antytrombiny. Z powodu półtrwania wynosi 25 minut, przy czym jego dzia-
trudności z uzyskaniem wystarczającej ilości hirudy- łanie zmniejsza się również dlatego, że trombina
ny z pijawek badania kliniczne z jej udziałem były jest w stanie powoli rozkładać biwalirudynę katali-
możliwe dopiero po uzyskaniu możliwości jej syn- tycznie. Lek ten może być używany jako alternatywa
tezy metodami inżynierii genetycznej. W niektórych dla heparyny w przypadku przezskórnych zabiegów
państwach w lecznictwie stosowana jest sama hirudy- na tętnicach wieńcowych.
na, natomiast w Niemczech stosuje się trzy pochodne Jeśli chodzi o dawkowanie, to podaje się początko-
hirudyny – lepirudynę, desirudynę i biwalirudynę. wo dożylny bolus 0,75 mg/kg biwalirudyny, a później
w postaci następujących po sobie infuzji w dawce
Lepirudyna jest to wytwarzana przez komórki 1,75 mg/kg/godzinę co najmniej przez okres trwania
drożdży hirudyna zmodyfikowana w dwóch miej- zabiegu.
scach, która blokuje zarówno centrum katalityczne,
jak i miejsca rozpoznawania substratów trombiny. W trakcie badań jest wiele drobnocząsteczkowych inhibi-
Okres półtrwania w osoczu wynosi około 2 godzin. torów trombiny. Dotychczasowe doświadczenia kliniczne
są ograniczone, a pierwsza nadająca się do zastosowania
Wydalanie zachodzi głównie przez nerki, dlate- substancja z tej grupy, ksymelagatran, musiała zostać wy-
go należy mieć szczególne baczenie w przypadku cofana z rynku z powodu działań niepożądanych (uszko-
osłabienia funkcji nerek. Lepirudyna jest szczegól- dzenie wątroby).
Serce i układ krążenia
nie wskazana u pacjentów z indukowaną heparyną
trombocytopenią typu II, którzy wymagają dalsze-
go pozajelitowego podawania leków hamujących 4.1.4.3.5. Drotrekogina alfa
krzepnięcie. Oprócz tego jest stosowana u pacjen-
tów z niestabilną dusznicą bolesną, zawałem mięś- Drotrekogina alfa jest to rekombinowane ludzkie ak-
nia sercowego i innymi chorobami zakrzepowo-za- tywowane białko C. Jak napisano wyżej, białko C ha-
torowymi. muje krzepnięcie poprzez inaktywację czynników Va
Typowe dawkowanie wynosi początkowo 0,4 mg/ i VIIIa. Drotrekogina alfa jest wskazana w leczeniu
kg w dożylnym bolusie, a następnie 0,15 mg/kg/go- pacjentów z ciężką sepsą i niewydolnością wielona-
dzinę we wlewie ciągłym. Cztery godziny po rozpo- rządową. Leczenie należy rozpocząć w ciągu 24 go-
B4
częciu infuzji należy dostosować dawkowanie w za- dzin od wystąpienia sepsy w dawce 24 μg/kg/godz.
leżności od wyniku oznaczenia czasu aktywowanej w ciągłym wlewie dożylnym przez ponad 96 godzin.
częściowo tromboplastyny (APTT).
Najpoważniejszym działaniem niepożądanym
są krwawienia (na lepirudynę nie ma antidotum). 4.1.4.3.6. Antagoniści witaminy K
Oprócz tego obserwowano również w rzadkich przy- (pochodne 4-hydroksykumaryny)
padkach występowanie reakcji alergicznych.
W roku 1944 Link wyizolował ze zgniłego ziela siekiernicy
Desirudyna jest również rekombinowaną pochodną 3,3′-metyleno-bis-4-hydroksykumarynę (dikumarol), a na-
stępnie określił jej budowę i udowodnił, że jest ona przy-
hirudyny, różniącą się od niej dwoma aminokwasa- czyną choroby bydła występującej od lat dwudziestych XX
mi. Używana jest w profilaktyce zakrzepicy żył głę- wieku w północnych stanach USA i w Kanadzie. U zwie-
bokich kończyn dolnych po operacjach wszczepiania rząt nią dotkniętych występowała zwiększona skłonność
endoprotez stawu biodrowego i kolanowego. Jej okres do krwawień, z powodu których większość z nich ginęła.
półtrwania wynosi 2–3 godziny, z czego 40–50% wy-
dalane jest w postaci niezmienionej przez nerki.
Dawkowanie wynosi 15 mg podskórnie dwa razy
dziennie przez 9–12 tygodni po zabiegu. O OH OH
W porównaniu z heparyną ryzyko wystąpienia za- CH3
krzepicy jest znacząco mniejsze.
O OO O
Biwalirudyna, podobnie jak inne pochodne hirudy- O
ny, jest bezpośrednim inhibitorem trombiny o profilu witamina K3 dikumarol
działania porównywalnym do lepirudyny. Pod wzglę-
dem budowy jest to polipeptyd złożony z dwudziestu
MUTSCHLER-2009.indd 543 2010-01-07 22:13:32
544 Krew
Porównanie wzoru strukturalnego dikumarolu i wi- od witaminy K γ-karboksylacji kwasu glutamino-
taminy K3 ujawnia chemiczne pokrewieństwo tych wego znajdującego się w prekursorach czynników
związków. krzepnięcia (a co za tym idzie – m.in. przekształcenie
Dikumarol i jego analogi, będąc antagonistami wi- dekarboksyprotrombiny w protrombinę). Blokują one
taminy K, hamują syntezę prawidłowo funkcjonują- bowiem poprzez hamowanie reduktazy epoksydowej
cej protrombiny oraz czynników VII, IX i X w wątro- konieczną dla prawidłowego procesu dekarboksylacji
bie. Są to więc środki przeciwzakrzepowe działające regenerację hydrochinonu witaminy K z epoksydu wi-
pośrednio. taminy K (zob. ryc. B 4.1-7). Dotyczy to czynników
krzepnięcia II, VII, IX i X, a także białka C i biał-
W leczeniu stosuje się: ka S.
■ fenprokumon,
Mechanizm ten wyjaśnia, dlaczego działanie tych le-
■ warfarynę – w Europie Zachodniej (w Polsce naj- ków nie występuje od razu, lecz dopiero po pewnym
bardziej popularny pozostaje acenokumarol). czasie. Dopiero bowiem kiedy stężenie obecnych
wcześniej we krwi czynników krzepnięcia obniży
Mechanizm działania. Punktem uchwytu dla anta- się poniżej poziomu krytycznego, może ujawnić się
gonistów witaminy K jest potranslacyjna obróbka ich zmniejszone wytwarzanie lub brak wytwarzania
czynników krzepnięcia. Zapobiegają one zależnej w wątrobie. Przy czym wartości okresów półtrwania
czynnik okres półtrwania
(godz.)
wątroba
II 60
VII 4
białko S IX
IX 24
białko C
II
X 48
X
VII białko C 6
białko S 10
O
O
NH
NH
kwas glutaminowy
COOH
kwas γ-karbo-
COOH O2 CO2
ksyglutaminowy COOH
OH
O
CH3
CH3
O
R
R
OH
O
witamina K epoksyd witaminy K
reduktaza witaminy K reduktaza epoksydu
witaminy K
O
CH3
R
(chinon) O
fenprokumon, fenprokumon,
warfaryna warfaryna
Ryc. B 4.1-7. Mechanizm działania antagonistów witaminy K (według Ranga, Dale’a i Rittera).
MUTSCHLER-2009.indd 544 2010-01-07 22:13:32
Krew 545
Wskazania. Antagoniści witaminy K, podobnie jak
OH C2H5
heparyna, są wskazani w profilaktyce i leczeniu cho-
roby zakrzepowo-zatorowej przede wszystkim w te-
rapii długoterminowej.
O O
Rozpoczynając leczenie antagonistami witaminy K, należy
fenprokumon zawsze dodatkowo podać pozajelitowo lek przeciwzakrze-
powy, np. heparynę drobnocząsteczkową. Oprócz bowiem
prozakrzepowo działających czynników II, VII, IX i X
O zależne od witaminy K są również działające przeciwza-
krzepowo białko C i białko S. Przede wszystkim białko
OH CH3 C ma o wiele krótszy okres półtrwania (kilka godzin) niż
np. czynnik X (kilka dni). Dlatego niedługo po rozpoczę-
ciu terapii za pomocą antagonistów witaminy K docho-
O O dzi do niedoboru białka C, podczas gdy ilość czynnika X
i czynnika II jest jeszcze prawidłowa. W tej prozakrzepowej
warfaryna fazie leczenia ryzyko zamknięcia światła naczynia jest wy-
raźnie podwyższone, jeżeli układ krzepnięcia nie zostanie
zahamowany przy użyciu heparyny. U pacjentów z ciężkim
wrodzonym niedoborem białka C, którzy są szczególnie
narażeni po rozpoczęciu leczenia antagonistami witami-
dla różnych czynników krzepnięcia są bardzo różne. ny K, można prowadzić substytucję przy użyciu białka C
Oprócz czynników krzepnięcia wiele innych białek oczyszczonego za pośrednictwem monoklonalnych mysich
Serce i układ krążenia
potrzebuje do normalnego działania procesu karbok- przeciwciał.
sylacji.
Dawkowanie. Dawkowanie antagonistów witaminy
Farmakokinetyka. Po podaniu doustnym antago- K prowadzi się indywidualnie (typowa dawka pod-
niści witaminy K wchłaniają się dobrze. Istotny trzymująca fenprokumonu wynosi 1,5–6 mg/dzień,
jest ich wysoki stopień wiązania się z białkami osocza warfaryny 5–15 mg/dzień). Dawniej leczenie było
(fenprokumon ponad 99%, warfaryna ok. 90%). monitorowane przez mierzenie w regularnych odstę-
Warfaryna, której okres półtrwania wynosi 40 go- pach czasu (początkowo codziennie, później co dwa,
dzin, cechuje się średnio długim czasem działania. trzy tygodnie) tzw. wskaźnika Quicka (wskaźnik pro-
Podawana jest w postaci mieszaniny racemicznej, trombinowy), którego wartość powinna wynosić 20–
B4
przy czym enancjomer S ma o wiele silniejsze działa- 30% wartości prawidłowej. Ponieważ pomiędzy la-
nie przeciwzakrzepowe niż enancjomer R. Warfaryna boratoriami występowały duże różnice w oznaczaniu
jest metabolizowana przez CYP2C9, który wykazuje wskaźnika Quicka, obecnie metoda ta została zastą-
genetyczny polimorfizm. Z licznych badań wynika, piana przez pomiar tzw. międzynarodowego czynnika
że pacjenci z uwarunkowaną dziedzicznie zmniejszo- znormalizowanego INR (International Normalized
ną aktywnością CYP2C9 wymagają niższych dawek Ratio). W przypadku INR uwzględnia się różnice
warfaryny, gdyż w przypadku podawania im dawek w aparaturze i odczynnikach, dlatego wyniki tego
standardowych istnieje wyższe ryzyko wystąpienia badania można ze sobą porównywać. Wartość INR
krwawień. Oprócz tego opisane są uwarunkowane nie powinna przekraczać 4,5 (wartość prawidłowa
dziedzicznie różne warianty reduktazy epoksydowej wynosi 1).
witaminy K (VKORC1) (zob. ryc. B 4.1-7), co pro-
wadzi do konieczności bardzo zindywidualizowane- Działania niepożądane, przeciwwskazania. Działa-
go podejścia do pacjenta w przypadku terapii antago- nia niepożądane i przeciwwskazania są podobne
nistami witaminy K. jak w przypadku heparyny. Oprócz tego przede
Fenprokumon z okresem półtrwania wynoszącym wszystkim u kobiet po porodzie oraz podczas kli-
150 godzin jest lekiem długo działającym. Należy makterium może dojść do wystąpienia martwicy
przy tym uwzględnić, że u pacjentów występują skóry (najczęściej na przyśrodkowych powierzch-
duże wahania szybkości jego eliminacji, które można niach ud, brzuchu, w okolicy gruczołu piersiowe-
przynajmniej częściowo wyjaśnić przez indywidual- go), szczególnie w przypadku podania dużych da-
ne różnice w metabolizmie tego leku. Fenprokumon wek wstępnych. W przeciwieństwie do heparyny
w porównaniu z warfaryną jest w mniejszym stopniu pochodnych kumaryny nie można podawać w cza-
metabolizowany przez CYP2C9, a w większym przez sie ciąży i karmienia piersią, ponieważ przechodzą
CYP3A4. Częściowo jest wydalany przez nerki w po- one przez ścianę łożyska i mogą też przedostać się
staci niezmienionej. do mleka matki.
MUTSCHLER-2009.indd 545 2010-01-07 22:13:32
546 Krew
Witamina K jako antidotum. Za pomocą dużych rów plazminogenu (fibrynolityki pośrednie) w dużej
dawek witaminy K można zniwelować działanie części udaje się doprowadzić do fibrynolizy i dzięki
pochodnych dikumarolu. Należy jednak liczyć się, temu rozpuścić skrzeplinę, szczególnie jeżeli terapię
podobnie jak w przypadku działania antagonistów rozpocznie się możliwie szybko po utworzeniu za-
witaminy K, z opóźnieniem początku jej działania, krzepu.
ponieważ synteza wystarczającej ilości czynników
krzepnięcia wymaga określonego czasu (6–12 go- W terapii zastosowanie znajdują następujące związki
dzin). Jeżeli konieczne jest natychmiastowe przywró- endogenne:
cenie zdolności krzepnięcia krwi, należy, jak opisano
■ urokinaza,
wyżej, podać czynniki krzepnięcia, a w przypadku
ciężkich krwawień przetoczyć krew. ■ tkankowy aktywator plazminogenu lub jego po-
chodne – retaplaza i tenekteplaza
Interakcje. Podanie antagonistów witaminy K rów-
oraz związek egzogenny
nocześnie z innymi środkami farmakologicznymi
może, z powodu wąskiego indeksu terapeutycznego ■ streptokinaza.
pochodnych kumaryny, doprowadzić do niebezpiecz-
nych zmian ich działania. Najważniejsze z tych inter- Leczenie środkami fibrynolitycznymi musi być prowa-
akcji zestawiono w tab. B 4.1-9. dzone w warunkach szpitalnych, ponieważ ze wzglę-
du na niebezpieczeństwo wystąpienia krwawienia
pacjent musi być ściśle monitorowany. Podczas fibry-
Tab. 4.1-9. Interakcje z antagonistami witaminy K
nolizy fizjologicznej uwolniona do krążenia plazmina
jest szybko inaktywowana przez inhibitory (antypla-
Nasilenie działania Osłabienie działania zmina) i w związku z tym nie dochodzi do uogólnionej
amiodaron barbiturany
proteolizy. W przypadku terapeutycznego stosowania
aktywatorów plazminogenu, ze względu na koniecz-
allopurinol fenytoina ne w leczeniu duże dawki, może dojść do przekro-
cymetydyna karbamazepina czenia zdolności działania inhibitorów. Wskutek tego
dochodzi do rozkładu z użyciem plazminy nie tylko
fenylobutazon i pochodne rifampicyna fibryny, ale także fibrynogenu oraz innych czynników
kwas acetylosalicylowy (duże dawki) gryzeofulwina krzepnięcia, a w szczególności czynnika V i czynnika
VIII (zob. poniżej). Wiele danych wskazuje jednak
metronidazol, sulfonamidy, cholestyramina na to, że terapia lityczna przed rozpoczęciem leczenia
cefalosporyny, ciprofloksacyna
szpitalnego (fibrynoliza przedszpitalna) może prowa-
ketokonazol i inne imidazolowe estrogeny dzić do lepszych prognoz leczenia.
leki przeciwgrzybicze
hormony tarczycy 6-merkaptopuryna Urokinaza była pierwotnie izolowania z kultur komó-
5-fluorouracyl preparaty zawierające
rek nerkowych lub moczu, a obecnie jest częściowo
dziurawiec uzyskiwana za pomocą technik inżynierii genetycz-
nej. Jest to enzym proteolityczny zbudowany z dwóch
fluwastatyna artykuły spożywcze
zawierające witaminę K łańcuchów, który występuje w organizmie w dwóch
formach. Pierwsza z nich o masie cząsteczkowej
fibraty
54 000 Da może wskutek proteolizy przejść w drugą
formę o masie cząsteczkowej 33 000 Da. Obie for-
my są w stanie rozkładać plazminogen do plazminy.
Aktywacja plazminogenu polega na rozerwaniu wią-
zania między argininą a waliną.
Dawkowanie standardowe wynosi początkowo
4.1.4.4. Fibrynolityki działające
3500–4500 IE/kg m.c., a następnie taka sama dawka
pośrednio (trombolityki) na godzinę w ciągłym wlewie kroplowym.
W USA urokinaza została wycofana z rynku.
Za pomocą wymienionych leków przeciwzakrzepo-
wych nie można rozpuścić zakrzepu już powstałego Tkankowy aktywator plazminogenu (tissue plas-
w świetle naczynia. Natomiast za pomocą aktywato- minogen activator = t-Pa) zbudowany jest z łańcucha
MUTSCHLER-2009.indd 546 2010-01-07 22:13:33
Krew 547
527 aminokwasów (masa cząsteczkowa ok. 65 000 matycznej. Dopiero kompleks utworzony ze strepto-
Da), a produkowany przez komórki śródbłonka jako kinazy i plazminogenu działa poprzez zmianę kon-
fizjologiczny aktywator plazminogenu. Otrzymywany formacji na wolny, niezwiązany ze streptokinazą
jest metodami inżynierii genetycznej (alteplaza; plazminogen.
rekombinowany ludzki aktywator plazminogenu = Dawkowanie streptokinazy w przypadku tzw. dłu-
rt-Pa). rt-Pa w przeciwieństwie do innych aktywato- gotrwałej fibrynolizy w terapii obwodowych zatorów
rów plazminogenu uzyskuje pełną aktywność enzy- naczyniowych wynosi na początku 250 000 IE przez
matyczną dopiero po związaniu się z fibryną. rt-Pa 30 minut, a następnie 100 000 IE na godzinę; w przy-
związany z fibryną ma istotnie wyższe powinowactwo padku ostrego zawału mięśnia sercowego 1,5 miliona
do plazminogenu niż niezwiązany, przez co wykazuje jednostek w ciągu godziny. Dawki przy rozpoczęciu
wysoką selektywność działania na zakrzepy. Pomimo leczenia streptokinazą są tak duże dlatego, że pra-
to również w przypadku jego stosowania może dojść wie wszyscy pacjenci wskutek uprzednich infekcji
do wystąpienia uogólnionej aktywacji układu fibryno- paciorkowcowych mają przeciwciała przeciw strep-
litycznego (stężenie w preparatach stosowanych w le- tokinazie i trzeba najpierw przełamać ich działanie
czeniu przekraczają fizjologiczne od 5 do 10 ng/ml, hamujące.
czyli około 1000 krotnie). Białko to ma krótki okres
półtrwania (5–10 minut), który wynika z klirensu Wskazania. Terapia fibrynolityczna jest wskazana
wątrobowego oraz endocytozy przez komórki śród- w leczeniu świeżego zawału mięśnia sercowego, kie-
błonka. Dodatkowo t-Pa jest bardzo szybko inakty- dy nie ma możliwości natychmiastowego wykonania
wowany przez inhibitor aktywatora plazminogenu 1 przezskórnej angioplastyki wieńcowej, w przypadku
Serce i układ krążenia
(PAI-1). ostrego zamknięcia tętnic w kończynach, świeżej za-
W przypadku zawału mięśnia sercowego podaje krzepicy żył miednicy i kończyn, zatorów płucnych
się początkowo 15 mg w bolusie i.v., a następnie 50 oraz przy tworzeniu się zakrzepów w przetokach tęt-
mg przez 30 minut i w końcu 35 mg przez 60 minut niczo-żylnych u pacjentów dializowanych.
(dawka całkowita 100 mg). U pacjentów z zawałem mięśnia sercowego dzię-
ki takiemu leczeniu można w 60–80% przypadków
Reteplaza jest rekombinowanym analogiem ak- doprowadzić do rekanalizacji zamkniętego naczynia
tywatora plazminogenu. Pod względem budowy wieńcowego, jednak terapia fibrynolityczna ma sens
jest to złożona z 355 aminokwasów część t-PA, która jedynie wtedy, gdy przeprowadzi się ją najpóźniej
ma dłuższy okres półtrwania niż alteplaza. Reteplaza w ciągu 6 godzin od wystąpienia zawału. Szczególnie
B4
jest w większości wydalana przez nerki. Dawkowanie dobre rezulataty osiąga się przy równoczesnym stoso-
wynosi dwa razy po 560 mg w odstępie 30 minut waniu ze środkiem fibrynolitycznym kwasu acetylo-
w bolusie. salicylowego w celu hamowania agregacji trombocy-
tów. Obserwuje się wtedy przede wszystkim zmniej-
Tenekteplaza jest również rekombinowanym akty- szoną częstość ponownego zamknięcia się naczynia
watorem plazminogenu, utworzonym przez mutacje wieńcowego.
punktowe w t-PA. Dzięki tym zmianom struktural- U pacjentów z udarem niedokrwiennym mózgu
nym tenekteplaza ma dłuższy okres półtrwania niż można prowadzić leczenie za pomocą alteplazy po-
t-PA, a także w mniejszym stopniu podlega inaktywacji dawanej ogólnie w odpowiednio wyposażonych od-
przez inhibitor aktywatora plazminogenu 1 (PAI-1). działach (stroke units).
Tenekteplaza podawana jest w pojedynczym bolusie,
a jej dawkowanie zależy od masy ciała i wynosi mak- Działania niepożądane. Najważniejsze działania
symalnie 50 mg. (Pacjenci o masie ciała poniżej 60 niepożądane to powikłania w postaci krwawień (z
kg otrzymują maksymalnie 30 mg). miejsc wkłucia, krwiaki, krwiomocz, krwawienia
z przewodu pokarmowego, płuc, mózgu). W przy-
Pomimo lepszej farmakokinetyki i wolniejszej inak- padku zbyt silnego zahamowania układu krzepnięcia
tywacji reteplazy i tenekteplazy ich skuteczność kli- działanie środków fibrynolitycznych można znieść
niczna równa jest skuteczności t-PA. za pomocą aprotyniny (zob. poniżej).
Przy podawaniu streptokinazy z powodu jej eg-
Streptokinaza (masa cząsteczkowa 47 000 Da) jest zogennego pochodzenia należy liczyć się również
wytwarzana przez paciorkowce β-hemolizujące. Po- z możliwością wystąpienia reakcji alergicznych (go-
mimo że końcówka -aza wskazuje, że jest to enzym, rączki, zwężenia oskrzeli, wysypki, zaczerwienienia,
streptokinaza nie wykazuje żadnej aktywności enzy- bólu stawów, wstrząsu).
MUTSCHLER-2009.indd 547 2010-01-07 22:13:33
548 Krew
Przeciwwskazania. Przeciwwskazania są takie same uczestniczy lizyna. Z tego powodu blokują one prze-
jak w przypadku leczenia lekami przeciwzakrzepo- kształcenie się plazminogenu w plazminę. Ponieważ
wymi. działanie samej plazminy nie zostaje zniesione, musi
minąć około dwóch godzin od momentu podania
do wystąpienia pożądanego działania. Przy okre-
sie półtrwania wynoszącym dwie godziny działanie
4.1.4.5. Środki przeciwfibrynolityczne to utrzymuje się 4–6 godzin. Środki te wydalane
są w przeważającej części przez nerki.
Do nadmiernej fibrynolizy (hiperfibrynolizy) docho-
dzi w wielu patologicznych stanach, przede wszystkim Dawkowanie wynosi:
w przebiegu różnych postaci wstrząsu, po zabiegach
■ kwas traneksamowy – 250–500 mg powoli i.v.,
w obrębie układu moczowo-płciowego (uwolnienie
a następnie 125 mg/godzinę w ciągłym wlewie
urokinazy), w przypadku białaczek, nowotworów,
kroplowym,
marskości wątroby i in. Również w trakcie leczenia
fibrynolitycznego istnieje możliwość wystąpienia hi- ■ kwas p-aminometylobenzoesowy – 50–150 (–300)
perfibrynolizy, a co za tym idzie – niebezpieczeństwo mg/dzień powoli dożylnie lub w ciągłym wlewie
krwawień. kroplowym.
W tego typu przypadkach można podawać pod ści-
słą kontrolą układu krzepnięcia inhibitory fibrynolizy W lżejszych przypadkach można te leki podawać
(środki przeciwfibrynolityczne). również doustnie.
Do działań niepożądanych zaliczają się zaburzenia
Aprotynina. Za pomocą aprotyniny można uzyskać ortostatyczne, a także nudności, wymioty i biegunka.
natychmiastowe zahamowanie fibrynolizy, ponieważ W przypadku ciężkich zaburzeń czynności nerek
jako inhibitor proteaz serynowych hamuje ona nie tyl- leki te są przeciwwskazane ze względu na niebez-
ko tworzenie się plazminy, lecz także wpływa hamu- pieczeństwo kumulacji w organizmie. W przypad-
jąco na jej działanie. Aprotynina jest to polipeptyd ku skłonności do zakrzepów i podczas ciąży można
złożony z 58 aminokwasów, początkowo uzyskiwany je podawać wyłącznie w ściśle określonych wskaza-
z płuc bydlęcych, a obecnie metodami inżynierii ge- niach.
netycznej. Używana jest w stanach hiperfibrynolizy,
we wstrząsie krwotocznym, a także w przypadku po-
wikłań krwotocznych powstałych wskutek leczenia
trombolitycznego. Ponieważ aprotynina inaktywu-
je proteazy serynowe, jest również używana w celu
zmniejszania utraty krwi u pacjentów wymagających NH2
krążenia pozaustrojowego podczas zabiegów wszcze- H2N COOH
pienia by-passów wieńcowych. Z powodu ciężkich
lizyna
działań niepożądanych ze strony układu sercowo-na-
czyniowego (zawałów serca, udarów mózgu) zosta-
ła dopuszczona do leczenia czasowo.
Drobnocząsteczkowe środki przeciwfibrynoli- H2N
tyczne. Oprócz aprotyniny w leczeniu stosuje się COOH
drobnocząsteczkowe środki przeciwfibrynolityczne,
które pod względem budowy są podobne do lizyny. kwas p-aminometylobenzoesowy
Należą do nich:
■ kwas traneksamowy,
H2N
■ kwas p-aminometylobenzoesowy.
COOH
Substancje te blokują miejsce wiązania się z lizyną
kwas traneksamowy
w cząsteczce plazminogenu i w ten sposób hamują
jego wiązanie się z fibryną. Dzięki temu hamują nie-
odwracalnie fizjologiczne aktywatory plazminogenu,
a przez to rozkład wiązań peptydowych, w których
MUTSCHLER-2009.indd 548 2010-01-07 22:13:33
Krew 549
blaszka miażdżycowa
pęknięcie
inhibitory agregacji adhezja, aktywacja czynników leki przeciwzakrzepowe
trombocytów aktywacja, krzepnięcia (np. antagoniści
(np. kwas acetylosalicylowy, agregacja wit. K,
inhibitory GP-IIb/IIIa) trombocytów heparyna)
zakrzep agregaty fibryny produkty rozpadu fibryny
leki przeciwfibrynolityczne
(np. kwas traneksamowy)
plazminogen plazmina
Serce i układ krążenia
leki fibrynolityczne
(np. urokinaza, kompleks
streptokinaza-
-plazminogen,
alteplaza)
śródbłonek naczyniowy
Ryc. B 4.1-8. Możliwości interwencji farmakologicznych w celu zapobiegania tworzeniu się zakrzepów oraz ich zmniejszania.
B4
MUTSCHLER-2009.indd 549 2010-01-07 22:13:33
550 Układ naczyniowy i krążenie
4.2. Układ naczyniowy i krążenie
4.2.1. Podstawy anatomiczne Warstwa środkowa składa się z mięśni gładkich
i w zależności od typu naczynia może mieć różną bu-
i fizjologiczne dowę.
Duże tętnice typu sprężystego znajdujące się w oko-
Rodzaje naczyń krwionośnych. Naczynia, którymi
licy serca charakteryzują się gęstą siecią włókien sprę-
krew płynie z lewej części serca na obwód, nazywane
żystych, dzięki czemu mają dużą zdolność do rozcią-
są tętnicami. Naczynia, którymi krew płynie z obwo-
gania się. W tętnicach bardziej oddalonych od serca
du do prawej części serca, nazywane są żyłami. W za-
zmniejsza się ilość włókien sprężystych, a wzrasta
leżności od budowy i funkcji rozróżnia się:
liczba włókien mięśni gładkich (typu mięśniowego).
■ tętnice typu sprężystego (aorta, tętnica płucna), któ- Warstwa środkowa żył jest cienka i luźna, a wystę-
re zapewniają ciągły przepływ krwi dzięki temu, pujące w niej komórki mięśni gładkich są przedzie-
że podczas każdego skurczu serca część objętości lone występującymi w dużej ilości włóknami spręży-
krwi jest pochłaniana przez rozszerzające się ścia- stymi i kolagenowymi tkanki łącznej.
ny tętnic, a w fazie rozkurczu ich elastyczne ściany
obkurczają się, wypychając zawartą w nich krew Przydanka łączy naczynie z otoczeniem za pomo-
na obwód (tzw. naczynia powietrzni). cą włókien sprężystych i kolagenowych. Obecne
są w niej również komórki mięśni gładkich.
■ tętnice typu mięśniowego oraz tętniczki (arteriole),
które regulują opór naczyniowy, a co za tym idzie
Ściana naczynia włosowatego zbudowana jest jedy-
– ciśnienie krwi oraz dowóz krwi do zaopatrywa-
nie z cienkiej warstwy komórek śródbłonka otoczo-
nych przez siebie organów (naczynia oporowe),
nego rozciągliwą błoną podstawną.
■ naczynia włosowate (kapilary), w których dochodzi
do wymiany substancji pomiędzy krwią a tkankami Zastawki żylne. W żyłach duże znaczenie mają za-
(naczynia wymiany), stawki żylne o kształcie kieszonek. Występują z reguły
parami poniżej połączenia z inną żyłą. Mają one za za-
■ małe żyły i żyłki, które decydują o ilości krwi
danie zapobiegać wstecznemu przepływowi krwi oraz
w tkankach (naczynia pojemnościowe),
ułatwiać przepływ krwi w kierunku serca następujący
■ duże żyły (np. venae cavae), w których gromadzi dzięki uciskowi z zewnątrz (pompa mięśniowa).
się krew zmierzająca do serca (naczynia zbiorcze).
Krążenie duże i małe (płucne). Krew żylna płynie
Duże znaczenie mają również naczynia zwierające, z obwodu przez vena cava superior i inferior (żyłę
regulujące wielkość przepływu krwi przez naczynia główną górną i dolną) do prawej części serca, skąd
włosowate oraz zespolenia tętniczo żylne (shunts), przez arteria pulmonalis (tętnicę płucną) jest pompo-
które łączą naczynia krążenia żylnego i tętniczego wana do płuc. W kapilarach płucnych ditlenek węgla
z pominięciem kapilar, dzięki czemu umożliwiają przechodzi do powietrza wydychanego, a tlen pobie-
czasowe wyłączenie z krążenia obszaru naczyń wło- rany jest z powietrza wdychanego. Krew przez venae
sowatych. pulmonales (żyły płucne) przechodzi do lewej części
serca, gdzie kończy się krążenie małe (płucne) (zob.
Budowa ściany naczynia. Ściana naczynia krwio- ryc. B 4.2-1).
nośnego zbudowana jest z trzech warstw. Są to: Z lewej części serca krew płynie przez aortę (tęt-
nicę główną), która rozgałęzia się wkrótce po wyjściu
■ warstwa wewnętrzna (intima),
z serca na duże tętnice. Te rozgałęziają się coraz bar-
■ warstwa środkowa (media), dziej, tworząc następnie tętniczki, a w końcu liczne
naczynia włosowate.
■ warstwa zewnętrzna, przydanka (adventitia).
W naczyniach włosowatych substancje odżywcze
przechodzą do tkanek, a produkty przemiany materii
Warstwa wewnętrzna utworzona jest z komórek są z nich usuwane. Tlen wymieniany jest na ditlenek
śródbłonka otoczonych włóknami kolagenowymi węgla. Wskutek tego powstaje krew żylna. Płynie
i sprężystymi. ona następnie przez małe żyły i zbiera się w więk-
MUTSCHLER-2009.indd 550 2010-01-07 22:13:33
Układ naczyniowy i krążenie 551
Układ ciśnienia wysokiego i niskiego. Układ krą-
żenia z anatomicznego i czynnościowego punktu
15% mózg widzenia dzieli się na układ wysokociśnieniowy i ni-
skociśnieniowy. Do układu wysokociśnieniowego za-
licza się tętnice i tętniczki (arteriole) krążenia dużego,
do układu niskociśnieniowego naczynia włosowate,
małe żyły i żyły dużego układu krążenia oraz całe krą-
płuca żenie płucne. Termin krążenie końcowe (mikrokrąże-
oskrzela
nie) określa wszystkie naczynia krwionośne o średni-
100% cy mniejszej niż 30–50 μm, czyli tętniczki, naczynia
włosowate i żyłki.
aorta
Ciśnienie tętnicze. Pulsacyjny napływ krwi do aor-
ty prowadzi do wspomnianych zmian objętości oraz
wahań ciśnienia krwi, które rozprzestrzeniają się
jako fala tętna po całym układzie naczyń tętniczych.
Zmiany ciśnienia występujące w obrębie całego
5% naczynia wieńcowe układu tętniczego umożliwiają pomiar jednego z naj-
ważniejszych parametrów krążenia krwi – ciśnienia
tętniczego. Maksymalna wartość fali ciśnienia krwi
Serce i układ krążenia
28% 7% podczas skurczu określana jest mianem ciśnienia
skurczowego, wartość minimalna podczas rozkurczu
trzewia określana jest mianem ciśnienia rozkurczowego,
21% a różnica między ciśnieniem skurczowym a rozkur-
czowym nazywana jest amplitudą ciśnienia krwi.
U zdrowej dorosłej osoby skurczowe ciśnienie krwi
mierzone na ramieniu wynosi prawidłowo 110–
17% mięśnie szkieletowe 130 mm Hg, a rozkurczowe 70–85 mm Hg.
Innym charakterystycznym parametrem układu
krążenia jest średnie ciśnienie tętnicze, które liczone
B4
jest jako wartość średnia ciśnienia w układzie tętni-
23% nerki czym w określonym przedziale czasu. W tętnicach
położonych bliżej serca wartość ta leży pośrodku
wartości ciśnienia skurczowego i rozkurczowego,
a w tętnicach obwodowych jest nieco bliższa ciśnie-
12% skóra i in. niu rozkurczowemu.
Regulacja ciśnienia krwi. Średnie ciśnienie tętnicze
Ryc. B 4.2-1. Schemat krążenia krwi wraz z procentowym udzia-
wynika z pojemności minutowej serca oraz oporu ob-
łem ukrwienia narządów w warunkach spoczynkowych i przy
neutralnej temperaturze.
wodowych naczyń tętniczych. Wartość ciśnienia krwi
może się więc zmieniać wskutek zmian pojemności
minutowej serca i/lub oporu obwodowego.
Na ryc. B 4.2-2 przedstawiono schemat regulacji
szych żyłach. Krew płynąca z głowy i gardła spływa ciśnienia krwi w formie schematu kołowego, w któ-
do vena cava superior. Krew z większej części prze- rym wielkością regulowaną jest średnie ciśnienie tęt-
wodu pokarmowego, śledziony i trzustki zbiera się nicze krwi.
w żyle wrotnej (vena porta), a następnie przepływa
przez wątrobę i z żyłami wątrobowymi wpada do żyły Rzeczywista wartość średniego ciśnienia rejestrowa-
głównej dolnej (układ krążenia wrotnego). na przez baroreceptory rozmieszczone w łuku aorty
Dolne żyły główne przyjmują krew płynącą z koń- oraz w rozwidleniu tętnicy szyjnej (sinus aorticus)
czyn dolnych i miednicy. Wraz z wpłynięciem do pra- jest przekazywana w postaci impulsu nerwowego
wej części serca kończy się układ krążenia dużego, do ośrodka krążenia w ośrodkowym układzie ner-
a rozpoczyna na nowo krążenie płucne. wowym. Tam dochodzi do porównania z założoną
MUTSCHLER-2009.indd 551 2010-01-07 22:13:33
552 Układ naczyniowy i krążenie
ciśnienie
pożądane
regulator
neurony regulujące
układu krążenia
medulla oblongata
ciśnienie
rzeczywiste
układ
receptory sterujący
baro- hormonalny
receptory i wegetatywny
układ regulowany
wartość regulowana serce
ciśnienie tętnicze CO
krwi elementy
regulujące
mięśnie gładkie naczyń
TPR
czynniki zaburzające,
np. utrata krwi
Ryc. B 4.2-2. Schemat krótkoterminowej regulacji ciśnienia krwi. CO – pojemność minutowa serca (cardiac output), TVR – całkowity
opór naczyniowy (total vascular resistance).
docelową wartością ciśnienia tętniczego. Istniejące sodowych we krwi prowadzi do uwalniania dużej ilo-
odchylenia są korygowane przez zmiany pojemności ści reniny i wskutek tego do wzrostu syntezy angio-
minutowej serca lub oporu naczyń obwodowych. tensyny II, która powoduje skurcz naczyń oporowych
Ośrodki kontroli układu krążenia mieszczą i wzrost oporu obwodowego oraz prowadzi, podob-
się w rdzeniu przedłużonym (medulla oblongata) nie jak angiotensyna III, do uwalniania aldosteronu
oraz w sąsiadujących z nim rejonach pnia mózgu. z kory nadnerczy, w wyniku czego dochodzi do nasi-
Rozróżnia się przy tym umieszczony obustronnie lenia resorpcji zwrotnej jonów sodowych do osocza
ośrodek pobudzający (naczynioruchowy) oraz po- krwi. Podwyższone stężenie jonów sodu zwiększa
środkowo i głębiej położony ośrodek hamujący. wrażliwość naczyń na katecholaminy, a zwłaszcza
Pobudzenie ośrodka naczynioruchowego powodu- na noradrenalinę. Oznacza to, że zwiększenie stęże-
je podwyższenie napięcia układu współczulnego nia jonów sodu nasila efekt presyjny.
i wynikające z tego podwyższenie pojemności mi-
nutowej serca oraz oporu obwodowego. Rezultatem Miejscowa regulacja krążenia krwi. Miejscowa re-
jest podwyższenie ciśnienia krwi. Pobudzenie gulacja krążenia krwi w narządach zachodzi przede
ośrodka hamującego powoduje natomiast spadek wszystkim za pośrednictwem substancji, które uczest-
ciśnienia krwi. niczą w komórkowej przemianie materii. Miejscowe
Oprócz ośrodków umieszczonych w pniu mózgu rozszerzenie naczyń krwionośnych jest powodowane
dodatkowo wpływ na układ krążenia mają również przez wzrost miejscowego ciśnienia parcjalnego CO2,
inne rejony mózgu, a zwłaszcza podwzgórze. Dzięki silne zmniejszenie parcjalnego ciśnienia O2, obniże-
regulacji podwzgórza układ sercowo-naczyniowy nie pH oraz wzrost stężenia adenozynotrifosforanu,
przystosowuje się do uogólnionych reakcji wegeta- adenozynomonofosforanu, adenozyny oraz jonów
tywnych (wzrost ciśnienia w stanie ergotropowym, potasu. Siła działania tych bodźców w różnych orga-
obniżenie ciśnienia w stanie trofotropowym). W re- nach może być odmienna. I tak na przykład wzrost
gulacji ciśnienia krwi uczestniczy oprócz tego układ ukrwienia mózgu wynika przede wszystkim z miej-
renina-angiotensyna-aldosteron. Zmniejszenie prze- scowego zwiększenia się ciśnienia parcjalnego CO2
pływu krwi przez nerki lub obniżenie stężenia jonów (hiperkapnia).
MUTSCHLER-2009.indd 552 2010-01-07 22:13:33
Układ naczyniowy i krążenie 553
Niektóre narządy dzięki zjawisku autoregulacji dzi również śródbłonkowy czynnik hiperpolaryzujący
miogennej są zdolne do zwężania naczynia w przy- (EDHF; endothelium derived hyperpolarizing fac-
padku podwyższenia ciśnienia krwi oraz rozsze- tor), za który uważa się kwasy epoksyeikozatrieno-
rzania naczyń w przypadku spadku ciśnienia krwi. we (EET, zob. ryc. B 4.2-3). Oprócz tego śródbłonek
Szczególnie wyraźnie widoczna jest ta reakcja w na- syntezuje również endotelinę 1 – jedną z substancji
czyniach doprowadzających nerki. najsilniej obkurczających naczynia.
Funkcja regulacyjna śródbłonka. Od dawna wia- Endoteliny, pośród których rozróżnia się typy od 1 do 3,
domo, że śródbłonek naczyniowy nie jest jedynie są to peptydy złożone z 21 aminokwasów, które są synte-
zowane przez różne komórki. W komórkach śródbłonka
bierną barierą. Obecnie uznaje się jego kluczową wytwarzana jest endotelina 1. W przypadku odpowied-
rolę w funkcjonowaniu całego organizmu, a nawet niego pobudzenia komórek śródbłonka (np. wskutek nie-
nazywa się go największym narządem ze względu dotlenienia lub niedokrwienia) w ciągu minuty dochodzi
na jego funkcje wydzielnicze, parakrynne i me- do transkrypcji mRNA i syntezy endoteliny 1. Najpierw
taboliczne. Oprócz tego śródbłonek posiada rów- powstaje zbudowana z 203 aminokwasów preproendoteli-
na, która jest przekształcana w proendotelinę 1 (dużą endo-
nież zdolność aktywacji substancji endogennych telinę) składającą się z 39 aminokwasów. Jest ona następnie
(np. przekształcania angiotensyny I do angiotensy- przekształcana do endoteliny 1 za pośrednictwem enzymu
ny II), ich inaktywacji (bradykinina), a także może konwertującego endotelinę (ECE) należącego do metalo-
je samodzielnie syntetyzować (np. NO, prostacy- proteinaz. Endotelina 1 łączy się z dwoma typami recep-
klina, endotelina). Ważniejsze funkcje śródbłonka torów – ETA i ETB. Receptory ETA, które występują prze-
de wszystkim na komórkach mięśni gładkich naczyń oraz
naczyniowego zestawiono na ryc. B 4.2-3. Jasne kardiomiocytach wiążą się z wysokim powinowactwem
Serce i układ krążenia
jest więc, dlaczego utrata integralności śródbłonka z endoteliną 1. Po interakcji endoteliny z receptorem ETA
wywołuje negatywny skutek w przebiegu procesów dochodzi do aktywacji poprzez białko G fosfolipazy C,
chorobowych (np. miażdżycy). w wyniku czego za pośrednictwem IP3 dochodzi do wzro-
Syntezowany przez śródbłonek NO jest szczegól- stu stężenia wewnątrzkomórkowego wapnia, a w rezultacie
do silnego zwężenia naczynia. Receptory ETB występują
nie ważnym czynnikiem rozszerzającym naczynia, głównie na komórkach śródbłonka, w OUN oraz w przewo-
podobnie jak wytwarzana przez cyklooksygenazę dzie pokarmowym. Transdukcja sygnału zachodzi za po-
(COX) prostacyklina. Do rozkurczu naczynia prowa- średnictwem hamującego białka G.
światło
B4
naczynia hamowanie agregacji trombocytów AI BK
AII iaP
BK
PL PLA2 ACE ACE
B2
eNOS G
bigET
AA PGI2
COX
cytrulina
NO
arginina
[Ca2+]i↑ CYP
ECE
CaM
EET
śródbłonek
ET1
warstwa
środkowa rozkurcz skurcz
Ryc. B 4.2-3. Przegląd najważniejszych naczynioruchowych funkcji śródbłonka (zmodyf. według Bussego).
AA – kwasy arachidonowe, AI – Angiotensyna I, ACE – enzym konwertujący angiotensynę, bigET – endotelina duża, BK – bradykinina,
B2 – receptor bradykininowy 2, CaM – kalmodulina, COX – cyklooksygenaza, CYP – cytochrom P-450, eNOS – śródbłonkowa syntaza tlenku
azotu, ECE – enzym konwertujący endotelinę, EET – kwasy epoksyeikozatrienowe, ET1 – endotelina 1, iaP – nieaktywne naczyniowo
peptydy, PGI2 – prostacyklina, PLA2 – fosfolipaza A2, PL – fosfolipidy.
MUTSCHLER-2009.indd 553 2010-01-07 22:13:34
554 Układ naczyniowy i krążenie
Naczynia włosowate. Wymiana substancji pomiędzy ny w pojemności minutowej serca nie mają wpływu
krwią a tkankami, która jest ważnym zadaniem ukła- na wysokość ciśnienia żylnego.
du krążenia, zachodzi w naczyniach włosowatych.
Ich liczbę szacuje się u człowieka na 40 miliardów, Układ naczyń chłonnych. Jak już wspomniano,
a łączną powierzchnię na 600 m2. Erytrocyty prze- ta część płynu, która po przesączeniu przez naczynia
bywają we wnętrzu naczynia włosowatego średnio włosowate nie podlega zwrotnej reabsorbcji, prze-
jedynie 1–2 sekundy, ale ze względu na niewielkie chodzi do układu naczyń chłonnych. Płyn ten dociera
odległości jest to czas wystarczający do dokonania biernie do ślepo zakończonych kapilar układu limfa-
wymiany substancji. tycznego, które leżą w przestrzeni śródmiąższowej.
Miejscowe ukrwienie kapilarne jest regulowa- Z sieci kapilar chłonka przepływa następnie do więk-
ne za pomocą napięcia mięśni gładkich tętniczek, szych naczyń chłonnych, które tworzą między sobą
a także dzięki specjalnym mechanizmom zamyka- liczne połączenia, a w budowie przypominają na-
jącym (zwieraczom prekapilarnym) umieszczonym czynia żylne. Transport chłonki zachodzi wsku-
na początku naczynia włosowatego. Jeżeli pracujący tek rytmicznych skurczów mięśni gładkich naczyń
narząd, np. mięsień, wymaga więcej tlenu, to krew chłonnych, a w przypadku naczyń chłonnych mięś-
przechodzi przez więcej kapilar, czyli narząd jest le- ni szkieletowych transport ten może być dodatkowo
piej zaopatrywany w krew (obszar czynny). W celu wspomagany skurczami tych mięśni.
uzyskania równowagi inne obszary ciała są wówczas Chłonka z dolnej części ciała gromadzi się w prze-
mniej ukrwione (obszar kompensacyjny). Dotyczy wodzie piersiowym (ductus thoracicus), który ucho-
to przede wszystkim naczyń trzewnych i skórnych, dzi do żyły głównej górnej. Chłonka z głowy, szyi
które stanowią swego rodzaju zbiornik. i obszaru kończyn górnych również dociera do żyły
Ściany naczyń włosowatych są przepuszczalne głównej górnej.
dla płynów oraz substancji o małej masie cząstecz- Do układu naczyń chłonnych zalicza się również
kowej i dlatego w obrębie przestrzeni okołokapilar- węzły chłonne, które pełnią funkcję biologicznych
nej dochodzi do ciągłego przesączania i reabsorpcji filtrów i mają za zadanie oczyszczać chłonkę, usuwa-
płynów oraz małych cząsteczek. Ponieważ żylne jąc dzięki fagocytozie ciała obce i czynniki zakaźne,
ciśnienie reabsorpcji jest nieco mniejsze od tętnicze- a oprócz tego wytwarzać limfocyty.
go ciśnienia przesączania, niecała objętość przesą-
czanego płynu jest resorbowana zwrotnie. Transport
około 10% pozostałej objętości zachodzi za pośred-
nictwem układu naczyń limfatycznych (zob. poni- 4.2.2. Metabolizm lipidów
żej). i środki obniżające stężenie
Układ żylny. Żyły krążenia dużego przyjmują prawie
lipidów we krwi
60% całej objętości krwi. Ze względu na dużą roz-
ciągliwość żył ich wypełnienie zmienia się w zależ- 4.2.2.1. Podstawy patofizjologiczne
ności od zmian ciśnienia.
Do powrotnego transportu krwi żylnej oprócz ak- Powstawanie blaszki miażdżycowej jest, jak to wyka-
cji serca i czynności pompy mięśniowej przyczynia zano w licznych badaniach, ściśle związane ze stęże-
się także oddychanie. Podciśnienie w jamie klatki niem lipidów we krwi. Szczególne znaczenie w tym
piersiowej rozciąga żyły leżące w tym obszarze i wy- procesie mają opisane dalej lipoproteiny o małej gę-
wołuje dodatkowo działanie ssące na krew z przylega- stości (low density lipoproteins – LDL). LDL może
jących naczyń. Obniżenie przepony i związany z tym wnikać do ściany naczynia i jest tam w większej czę-
wzrost ciśnienia w jamie brzusznej dodatkowo uła- ści utleniany do oksydowanego LDL (ox-LDL). Ten
twia przepływ krwi w klatce piersiowej. U człowieka ostatni jest następnie fagocytowany przez makrofa-
leżącego ciśnienie krwi spada stopniowo od dużych gi, które przekształcają się w komórki piankowate
naczyń poprzez małe żyły i żyłki, a w naczyniach i w ten sposób formują rdzeń złogu arteriosklerotycz-
włosowatych wynosi około 18 mm Hg. Ciśnienie nego (blaszki miażdżycowej). Ponieważ obniżenie
w końcowym odcinku żyły głównej odpowiedzialne poziomu LDL przez substancje obniżające stężenie
za napełnianie prawego przedsionka określa się mia- lipidów we krwi prowadzi do zmniejszenia tworze-
nem centralnego ciśnienia żylnego. Wynosi ono z re- nia się złogów miażdżycowych w ścianie naczyń,
guły 2–4 mm Hg. leki wpływające na przemiany lipidowe oraz zmniej-
Opory przepływu w układzie żylnym w porówna- szające stężenie lipidów zostaną omówione tutaj,
niu z obszarem tętniczek są niewielkie. Dlatego zmia- a nie w rozdziale B 4.1.
MUTSCHLER-2009.indd 554 2010-01-07 22:13:34
Układ naczyniowy i krążenie 555
4.2.2.2. Lipidy osocza jest cholesterol lub estry cholesterolu, podczas elek-
troforezy zachowują się jak β-globuliny.
Lipidy osocza są heterogenną grupą, do której nale-
Lipoproteina (a) [Lp(a)] zawiera apolipoproteinę
żą:
A, która jest strukturalnie spokrewniona z plazmi-
■ tłuszcze obojętne (triglicerydy), nogenem. W przeciwieństwie do niego Lp(a) dzia-
ła po związaniu z fibryną prozakrzepowo. Stanowi
■ fosfolipidy,
więc ona niezależny czynnik ryzyka arteriosklerozy,
■ cholesterol, na który na razie nie można jeszcze wpływać farma-
kologicznie.
■ estry cholesterolu,
■ wolne kwasy tłuszczowe. Lipoproteiny o dużej gęstości (HDL) zawierają
w sobie szczególnie dużą ilość białek i stosunkowo
Ponieważ lipidy nie rozpuszczają się w wodzie, małą cholesterolu. Są one w stanie pobierać choleste-
są transportowane we krwi nie w postaci wolnej, lecz rol ze złogów w ścianach naczyń i oddać go IDL.
jako tzw. lipoproteiny, czyli w połączeniu z białkami
transportującymi. Białka te określa się mianem apoli- W tab. B 4.2-1 zestawiono lipoproteiny, a na ryc. B
poprotein i dzieli się na kilka typów (np. A, B, C i E). 4.2-4 widnieje schemat ich powstawania, dróg trans-
Lipoproteiny dzieli się przede wszystkim ze względu portu, funkcji oraz rozkładu.
na ich zachowanie podczas wirowania lub elektrofo-
Serce i układ krążenia
rezy. Każdej frakcji obecnie przypisuje się inne funk- W celu lepszego zrozumienia możliwości interwencji
cje. Rozróżnia się następujące frakcje: farmakologicznych w hiperlipidemiach poniżej omó-
wiono przebieg metabolizmu lipoprotein.
■ chylomikrony,
■ lipoproteiny o bardzo małej gęstości (VLDL – very Wchłanianie przez enterocyty. Po spożyciu tri-
low density lipoproteins), glicerydy i cholesterol, a także sterole roślinne
docierają do jelita. Cholesterol i sterole roślinne
■ lipoproteiny o pośredniej gęstości (IDL – interme-
są rozpoznawane przez białko NPC1L1 (białko
diate density lipoproteins),
podobne do białka C1, którego defekt genetyczny
■ lipoproteiny o małej gęstości (LDL – low density powoduje chorobę Niemanna-Picka), które uczest-
B4
lipoproteins), niczy w ich wybiórczym transporcie do wnętrza
enterocytu. Interesujące jest przy tym, że wchła-
■ lipoproteina (a) [Lp(a)],
nianie cholesterolu może zostać kompetycyjnie za-
■ lipoproteiny o dużej gęstości (HDL – high density hamowane przez sterole roślinne. Z drugiej strony
lipoproteins). ich biodostępność w organizmie jest mocno ograni-
czona przez fakt, że po absorpcji do wnętrza ente-
Chylomikrony to widoczne pod mikroskopem ku- rocytu są wypompowywane z powrotem do świat-
leczki lipidowe o średnicy 0,5–1 μm. Zawartość ła jelita przez dwa transportery ABC (ABCG5
białek w chylomikronach wynosi tylko 2%, a część i ABCG8). Defekty genetyczne tego systemu
lipidowa składa się prawie wyłącznie z triglicery- eliminacji prowadzą do wysokiego stężenia roś-
dów. Podczas elektroforezy pozostają one w punk- linnych steroli (sitosterolemii), które jest związane
cie startu. z arteriosklerozą.
Lipoproteiny o bardzo małej gęstości (VLDL) o ma- Metabolizm w enterocytach. Po resorpcji choleste-
sie cząsteczkowej 5 · 106 wędrują podczas elektrofo- rolu we wnętrzu enterocytu dochodzi do jego estryfi-
rezy przed β-globulinami. Transportują one głównie kacji. Odpowiedzialnym za to enzymem jest acylo-
wytwarzane w organizmie tłuszcze obojętne. transferaza cholesterolowa acylokoenzymu A typu 2
(ACAT-2). Estry cholesterolu oraz triglicerydy są na-
Lipoproteiny o pośredniej gęstości (IDL) to czą- stępnie łączone z syntezowaną w jelicie apolipoprote-
steczki VLDL, które oddały zawarte w nich triglice- iną B-48 i triglicerydami za pomocą mikrosomalnego
rydy tkance tłuszczowej lub mięśniowej. białka transportującego triglicerydy (MTP), w wy-
niku czego powstają chylomikrony, które następnie
Lipoproteiny o małej gęstości (LDL) o masie czą- przez przewód piersiowy przedostają się do krążenia
steczkowej 2–3 · 106, których głównym składnikiem ogólnego.
MUTSCHLER-2009.indd 555 2010-01-07 22:13:34
556 Układ naczyniowy i krążenie
Tab. 4.2-1. Zestawienie lipoprotein (zmodyf. według Mahleya i Bersota)
Lipoproteina Skład lipidów Apolipoproteina Szczególne właściwości
chylomikrony triglicerydy i cholesterol A-II, A-IV, B-48, wchłanianie za pośrednictwem
i remnanty (10:1) C-I, C-II,C-III, E Apo-E w wątrobie
VLDL triglicerydy i cholesterol B-100, C-I, C-II, C-III, E lekko podwyższony potencjał
(5:1) aterogenny przy podwyższonym
stężeniu w osoczu
IDL triglicerydy i cholesterol B-100, C-I, C-III, E
(1:1)
LDL estry cholesterolu B-100 wysoki potencjał aterogenny
przy podwyższonym stężeniu w osoczu
Lp(a) estry cholesterolu Apo A, B-100 potencjał aterogenny
przy podwyższonym stężeniu w osoczu
HDL fosfolipidy A-I, A-II, C-I, C-II, C-III, E potencjał antyaterogenny
estry cholesterolu
Metabolizm chylomikronów w osoczu i ich pobie- resorpcji i równocześnie zmniejszenia syntezy de
ranie przez wątrobę. Chylomikrony oddają kwasy novo. Pośrednictwo w tym procesie SREBP wyjaś-
tłuszczowe powstałe po rozkładzie triglicerydów nia również, dlaczego po farmakologicznej zmianie
przez lipazę lipoproteinową (LPL) do tkanki tłusz- stężenia cholesterolu dochodzi do dużej liczby zmian
czowej (w celu magazynowania) oraz do mięśni (jako ekspresji genów.
źródło energii). Do interakcji chylomikronów z lipazą Wewnątrz komórek wątrobowych zresorbowa-
lipoproteinową wymagana jest obecność apolipopro- ny cholesterol jest z jednej strony przekształcany
teiny C-II. Pozostałości chylomikronów (remnanty) do kwasów żółciowych, które następnie za pośred-
z dużą zawartością estrów cholesterolu wiążą się nictwem transportera ABC (ABCB11) wydalane
za pośrednictwem apolipoproteiny E z receptorem są z żółcią do jelita, a z drugiej strony wydzielany
LDL lub z białkiem LRP (LDL-receptor related pro- jest bezpośrednio do żółci. W przypadku tego ostat-
tein) i za ich pośrednictwem przechodzą do wnętrza niego procesu znowu istotną rolę odgrywają opisane
komórek wątroby. wyżej transportery ABC – ABCG5 i ABCG8.
Wydzielany do żółci cholesterol przechodzi
Metabolizm i synteza cholesterolu w wątrobie. z nią następnie do jelita i może tam zostać ponownie
Pula wątrobowego cholesterolu jest uzupełniana wchłonięty. Ta droga transportu cholesterolu pokazu-
dzięki opisywanej wyżej resorpcji egzogennego cho- je, że tylko część wchłanianego z jelita cholesterolu
lesterolu. Z drugiej strony właściwie każda komórka pochodzi z pożywienia. Wyjaśnia to również, dlacze-
organizmu jest w stanie samodzielnie syntetyzować go zmiany w diecie przy hipercholesterolemii często
cholesterol. Jest to wieloetapowy proces, w którym mają ograniczoną skuteczność.
cholesterol syntezowany jest z acetylokoenzymu A, Oprócz wydzielania z żółcią i przekształcaniu
najpierw przekształcanego przez kluczowy enzym do kwasów żółciowych cholesterol z wątroby może
– reduktazę hydroksymetyloglutarylo-CoA do me- ponownie przejść do krążenia ogólnego wraz z apoli-
walonianu, a następnie poprzez związki pośrednie poproteinami (apolipoproteiną B-100), fosfolipidami
– geranyl i farnezyl ostatecznie syntezowany jest cho- i triglicerydami w postaci VLDL.
lesterol. Endogenna synteza cholesterolu oraz jego
resorpcja w wątrobie podlega w większości regula- Metabolizm VLDL, IDL i LDL. Z VLDL, podobnie
cji. Zachodzi ona za pośrednictwem ekspresji wielu jak w przypadku chylomikronów, tkanka tłuszczowa
genów wśród nich m.in. ekspresji genów receptora i mięśniowa pobiera kwasy tłuszczowe odszczepione
LDL, przez tzw. białka wiążące sterolowe elemen- przez lipazę lipoproteinową. W wyniku tego procesu
ty regulacyjne (SREBP – sterol response elements powstają IDL. Część IDL wiąże się następnie z re-
binding proteins) Wysokie wewnątrzkomórkowe ceptorem LDL w wątrobie i jest szybko resorbowa-
stężenie cholesterolu prowadzi do zmniejszenia jego na przez komórki wątrobowe, a część po kolejnym
MUTSCHLER-2009.indd 556 2010-01-07 22:13:34
Układ naczyniowy i krążenie 557
A wątroba tłuszcz i cholesterol
acetylo-CoA z pokarmu
(C2) acetoacetylo-CoA
(C4)
3-hydroksy-3- sole żółciowe,
metyloglutarylo-CoA cholesterol
B48
cholesterol mewalonian E
(C27) C
izopentenylo- przewód
pirofosforan pokarmowy
lanosterol (C5) remnant
(C30)
pirofosforan
geranylu
epoksyd skwalenu (C10)
pirofosforan
skwalen farnezylu
(C30) (C15)
B100 chylo-
C mikron
B48 światło jelita krew
LPL
E ABCG5/G8
Serce i układ krążenia
FS TG
C
ATP
C
VLDL MTP
E
C
NPC1L1 apo
chylomikron C C B48
B100 B100 A
C ACAT 2
E E CE
CETP FS B4
C micele
IDL LDL HDL enterocyt
B
tkanki obwodowe
triglicerydy (TG) C wolny cholesterol SR-BI receptor LDL
estry cholesterolu (CE) FS kwasy tłuszczowe apolipoproteiny LRP
Ryc. B 4.2-4. A Biosynteza cholesterolu, B metabolizm lipidów i lipoprotein, C wchłanianie cholesterolu. LPL lipaza lipoproteinowa
(objaśnienia w tekście).
rozkładzie triglicerydów przekształca się w LDL. Białko prekursorowe – pra-β-HDL jest syntezowa-
Te z kolei wiążą się również za pośrednictwem apo- ne w jelicie i w wątrobie. Oprócz tego pra-β-HDL
lipoproteiny B-100 z receptorem LDL i przechodzą może powstać podczas hydrolizy triglicerydów
dzięki endocytozie do komórek wątroby lub innych z chylomikronów lub VLDL z jednoczesnym uwol-
komórek organizmu, które potrzebują cholesterolu nieniem Apo A-I oraz fosfolipidów. Jest ono w sta-
do budowy swojej błony cytoplazmatycznej oraz in- nie pobierać cholesterol z makrofagów w ścianie
nych funkcji. Oprócz tej fizjologicznej roli LDL ma naczynia. W procesie tym ponownie ważną rolę od-
również, jak opisano wyżej, duże znaczenie w po- grywa białko transportowe makrofagów – ABCA1.
wstawaniu miażdżycy. Dzięki absorpcji cholesterolu z pra-β-HDL powstają
sferyczne, dojrzałe HDL. Występuje w nich acy-
Metabolizm HDL i zwrotny transport cholestero- lotransferaza lecytynowo-cholesterolowa (lecithin
lu. Synteza i funkcje HDL są nadzwyczaj złożone. cholesterol acyltransferase – LCAT), która estryfi-
MUTSCHLER-2009.indd 557 2010-01-07 22:13:34
558 Układ naczyniowy i krążenie
kuje wolny cholesterol. Tak powstające estry cho- Typy IIa, IIb oraz IV stanowią około 95% wszystkich
lesterolu są następnie przenoszone do LDL przez przypadków hiperlipoproteinemii. Określenie typu
białko transportujące estry cholesterolu (choleste- hiperlipoproteinemii jest istotne nie tylko ze względu
ryl ester transfer protein – CETP). Inna droga prze- na odmienny obraz kliniczny i patofizjologię, ale rów-
mian HDL polega na bezpośrednim wchłanianiu nież ze względu na ich odmienne leczenie.
dojrzałych HDL w wątrobie dzięki specyficznym Ze względu na przyczynę rozróżnia się hiperlipo-
receptorom (SR-BI). Za pośrednictwem obu tych proteinemie:
dróg cholesterol pierwotnie odłożony w ścianach
■ pierwotne,
naczyń jest transportowany z powrotem do krążenia
systemowego i wątroby. Ten tzw. zwrotny transport ■ wtórne.
cholesterolu jest interesującym przyczynkiem do le-
czenia uszkodzeń ścian naczyń przez złogi arterio- U podłoża hiperlipoproteinemii pierwotnych leżą
sklerotyczne. Na podstawie badań epidemiologicz- genetycznie uwarunkowane zaburzenia przemiany
nych wykazano, że wysoki poziom HDL w osoczu lipidów, podczas gdy znacznie częściej występujące
związany jest z niskim odsetkiem incydentów ser- postacie wtórne wynikają z nieprawidłowego odży-
cowo-naczyniowych (m.in. zawałów serca). Z dru- wiania (z nadmierną podażą tłuszczów), nadwagi,
giej strony warte rozważenia jest to, że wskutek nadużywania alkoholu, a także chorób metabolicz-
złożonego procesu syntezy HDL nie zawsze jego nych (niedoczynności tarczycy, cukrzycy, dny mo-
podwyższone stężenie może być zaletą. Tak dzieje czanowej).
się na przykład w przypadku wrodzonych defektów
białka CEPT, kiedy to pomimo wysokiego stężenia Leczenie hiperlipoproteinemii. Podstawę leczenia
HDL pacjenci cierpią równocześnie na rozsiany pro- hiperlipoproteinemii stanowi odpowiednia dieta.
ces miażdżycowy. Należy dążyć do:
■ normalizacji masy ciała,
4.2.2.3. Hiperlipoproteinemie ■ odpowiedniego zbilansowania diety (ok. 55% war-
tości kalorycznej powinny stanowić węglowodany,
do 30% tłuszcze, w tym po 10% nienasyconych,
Zaburzenia przemian lipidów polegają przede wszyst-
jednonienasyconych i wielonienasyconych, oraz
kim na hiperlipoproteinemiach, czyli zwiększeniu
10–20% białka),
jednej bądź większej ilości frakcji lipidów. W za-
leżności od rodzaju lipoproteiny, której ilość uległa ■ codziennego dostarczania błonnika (warzywa,
zwiększeniu, rozróżnia się według Fredricksona róż- owoce i in.) w ilości przynajmniej 35 mg oraz
ne typy hiperlipoproteinemii, które zestawiono w tab. zmniejszenia podaży cholesterolu do mniej niż 300
B 4.2-2. mg dziennie.
Tab. 4.2-2. Klasyfikacja hiperlipoproteinemii (według Fredricksona)
Typ Zawartość lipoprotein Nazwa Synonimy
w osoczu
I zwiększone stężenie chylomikronów hiperchylomikronemia egzogenna hipertriglicerydemia
indukowana tłuszczami
IIa zwiększone stężenie LDL hiper-β-lipoproteinemia hipercholesterolemia rodzinna
ksantomatozowa
IIb zwiększone stężenie LDL i VLDL hiper-β- i hiper-pre-β-lipoproteinemia hipercholesterolemia
z hipertriglicerydemią
III zwiększone stężenie IDL broad-β-disease hipercholesterolemia
i chylomikronów z hipertriglicerydemią
IV zwiększone stężenie VLDL hiper-pre-β-lipoproteinemia hipertriglicerydemia endogenna
(indukowana węglowodanami)
V zwiększone stężenie VLDL hiper-pre-β-lipoproteinemia mieszana hipertriglicerydemia
i chylomikronów z hiperchylomikronemią endogenno-egzogenna
MUTSCHLER-2009.indd 558 2010-01-07 22:13:34
Układ naczyniowy i krążenie 559
Jeżeli za pomocą zmian w diecie nie udaje się osiąg- skanemu z grzyba Aspergilllus terreus, można było
nąć wystarczającej normalizacji stężenia lipidów po raz pierwszy w praktyce wykorzystać ten mecha-
we krwi i wciąż istnieje ryzyko wystąpienia miażdży- nizm działania. Do analogów lowastatyny zaliczamy
cy, należy rozważyć zastosowanie dodatkowo leków atorwastatynę, fluwastatynę, simwastatynę i pra-
obniżających stężenie lipidów. wastatynę (tab. B 4.2-3).
Oprócz pożądanego stężenia HDL, które u mężczyzn Lowastatyna i simwastatyna są stosowane w posta-
wynosi > 35 mg/dl a u kobiet 40 mg/dl, wartości ci nieaktywnych laktonów (proleków), które są prze-
stężeń pozostałych lipoprotein w osoczu powinny kształcane głównie w wątrobie do aktywnej postaci
kształtować się następująco: kwasu hydroksylowego z otwartym pierścieniem. Inne
leki z tej grupy stosowane są natomiast wyjściowo
■ u pacjentów bez istniejących innych czynników
w postaci kwasów hydroksylowych.
ryzyka: LDL < 155 mg/dl, cholesterol całkowity
Wszystkie statyny działają podobnie, ale różnią się
< 215 mg/dl, triglicerydy < 200 mg/dl,
między sobą znacząco jeśli chodzi o farmakokinety-
■ u pacjentów z dodatkowymi czynnikami ryzyka (stę- kę. Ważniejsze dane zamieszczono w tab. B 4.2-3.
żenie HDL < 35 mg/dl, płeć męska, palenie tytoniu,
obciążenie rodzinne, nadciśnienie, cukrzyca, choroba W większości dużych badań klinicznych udowodniono
niedokrwienna serca): LDL < 100 mg/dl, choleste- pozytywne efekty inhibitorów reduktazy HMG-CoA oraz
ich korzystny wpływ na chorobowość i śmiertelność.
rol całkowity < 180 mg/dl, triglicerydy < 150 mg/dl.
U pacjentów z bardzo wysokim ryzykiem wskazane Efekty plejotropowe. W przypadku statyn opisano cały
jest osiągnięcie stężenia LDL < 70 mg/dl. szereg działań, które na pierwszy rzut oka nie mają nic
Serce i układ krążenia
wspólnego z działaniem obniżającym poziom lipidów.
W długookresowych badaniach nad lekami obniżają- Do tych tzw. efektów plejotropowych zalicza się np. regula-
cję syntezy tlenku azotu skutkującą rozszerzeniem naczyń,
cymi stężenie lipidów we krwi stwierdzono znaczący efekt antyoksydacyjny a także wpływ na adrenergiczne
spadek zawałów serca oraz zmniejszenie całkowitej mechanizmy transdukcji sygnału, żeby wymienić tylko
śmiertelności. nieliczne. Molekularne podłoże tych efektów plejotro-
powych jest złożone: istotnym mechanizmem wydaje się
jednak fakt, że po zablokowaniu biosyntezy cholesterolu
nie są tworzone również produkty kwasu mewalonowego
4.2.2.4. Leki stosowane (zob ryc. 4.2-4). Wskutek tego nie powstają takie związki
w hiperlipidemiach pośrednie jak geranyl i farnezyl, które są istotnymi czą- B4
steczkami uczestniczącymi w mechanizmach transdukcji
4.2.2.4.1. Statyny (inhibitory reduktazy sygnałów. W ten sposób można wyjaśnić, dlaczego zacha-
mowanie biosyntezy cholesterolu przez statyny ma wpływ
hydroksymetyloglutarylo-CoA) na tak liczne aspekty funkcjonowania komórki.
Szczególnie skutecznie można obniżyć stężenie pod- Farmakokinetyka. Po podaniu doustnym inhibi-
wyższonego poziomu cholesterolu dzięki blokadzie tory reduktazy HMG-CoA są wchłaniane jedynie
reduktazy HMG-CoA, która jest kluczowym enzy- częściowo i podlegają dużemu efektowi pierwszego
mem w uczestniczącym w biosyntezie cholesterolu przejścia, w którym biorą udział różne enzymy CYP.
katalizującym przejście HMG-CoA w kwas mewalo- Wskutek tego metabolizmu oksydacyjnego dochodzi
nowy (zob. ryc. B 4.2-4). do utworzenia ze statyn z wyjątkiem fluwastatyny
Gdy po zablokowaniu enzymu na przykład w ko- i prawastatyny aktywnych metabolitów. Dostępność
mórkach wątroby dochodzi do obniżenia stężenia we- biologiczna poszczególnych związków jest bardzo
wnątrzkomórkowego cholesterolu w hepatocytach, różna. Pożywienie zwiększa wchłanianie lowastatyny
to na zasadzie mechanizmu sprzężenia zwrotnego i simwastatyny, a zmniejsza prawastatyny. Wydalanie
dochodzi do wytworzenia większej ilości recepto- następuje przez nerki oraz z kałem.
rów dla LDL w celu wychwycenia większej ilości
cholesterolu z krwi. Wskutek tego obniża się stę- Dawkowanie. Dawkowanie podano w tab. B 4.2-3.
żenie zarówno LDL jak i cholesterolu całkowitego.
Poza tym inhibitory reduktazy HMG-CoA powodują Działania niepożądane i interakcje. Inhibitory re-
nieznaczne podwyższenie stężenia HDL oraz w za- duktazy HMG-CoA są w większości przypadków
leżności od wartości wyjściowej obniżenie stężenia dobrze tolerowane. Jako działania niepożądane ob-
triglicerydów. serwowano zaburzenia żołądkowo-jelitowe, bóle
Dzięki lowastatynie, silnie działającemu, kompe- głowy, zmęczenie, zaburzenia snu, świąd, suchość
tytywnemu inhibitorowi reduktazy HMG-CoA, uzy- w jamie ustnej, reakcje nadwrażliwości, a poza tym
MUTSCHLER-2009.indd 559 2010-01-07 22:13:34
560 Układ naczyniowy i krążenie
Tab. B 4.2-3. Statyny
Wzór strukturalny Nazwa Preparat handlowy Okres Dawkowanie
międzynarodowa półtrwania (mg)
HO atorwastatyna Sortis 14 10-80
COOH
OH
F
N CH(CH3) 2
O
HN
fluwastatyna Cranoc, 2,3 40-80
HO LOCOL
COOH
OH
CH(CH3) 2
N
HO
prawastatyna Mevalotin protect, 2 10-40
COOH Pravasin protect,
OH
Pravastatin-CT,
O Pravastatin-
ratiopharm u.a.
H5C2
O
H
CH3 CH3
HO
lowastatyna LovaHEXAL, 1,7 10-80
HO O Lovastatin-
ratiopharm,
O Lovastatin STADA,
O
Mevinacor u.a.
H5C2
O
H
CH3 CH3
H3C
simwastatyna Simvabeta, 2 10-80
HO O SimvaHEXAL,
Simvastatin-
O ratiopharm,
O
ZOCOR u.a.
H5C2
O
H3C H
CH3 CH3
H3C
MUTSCHLER-2009.indd 560 2010-01-07 22:13:34
Układ naczyniowy i krążenie 561
wzrost stężenia aminotransferaz w osoczu, a także
bóle mięśniowe, miopatie i rzadko także zmętnienie F
soczewki.
O F
Przedawkowanie i interakcje z innymi środkami N
mogą doprowadzić do miotoksyczności, a w skraj-
nych przypadkach do rabdomiolizy (rozpadu mięśni
poprzecznie prążkowanych), ponieważ wysokie stę- OH
HO
żenia inhibitorów HMG-CoA w tkankach może blo-
kować procesy metaboliczne w komórkach mięśni. nikotynamid
Taki wpływ na metabolizm występuje zwłaszcza wte-
dy, kiedy statyny charakteryzujące się niską biodo-
stępnością i wysokim efektem pierwszego przejścia
nianie cholesterolu. Wiązanie z białkami krwi wy-
zostaną zastosowane łącznie z substancjami hamują-
nosi 99,7%, okres półtrwania substancji wyjściowej
cymi enzymy CYP3A4.
i metabolitów wynosi 22 godziny. Dlatego ezetymib
Równoczesne podanie inhibitorów reduktazy
może być podawany raz dziennie (w standardowej
HMG-CoA z lekami przeciwzakrzepowymi pochod-
dawce 10 mg).
nymi kumaryny może nasilić ich działanie przeciw-
Do działań niepożądanych ezetymibu zalicza się
zakrzepowe (szczególnie w przypadku fluwastatyny,
w pojedynczych przypadkach podwyższenie poziomu
która podobnie jak pochodne kumaryny jest meta-
aminotransferaz w osoczu, które przebiega zwykle
bolizowana przez CYP2C9). Ponieważ antagonizm
Serce i układ krążenia
bezobjawowo i jest odwracalne. Oprócz tego obser-
kompetycyjny dotyczący enzymów metabolizują-
wowano zaburzenia żołądkowo jelitowe.
cych wyjaśnia tylko częściowo interakcje metabo-
liczne prawdopodobnie w grę wchodzą również inne
Terapia skojarzona ezetymibu z innymi lekami
mechanizmy wyjaśniające wpływ statyn na metabo-
obniżającymi stężenie lipidów. Ponieważ hamo-
lizm.
wanie wchłaniania cholesterolu prowadzi do wzrostu
syntezy cholesterolu endogennego, sensowne jest le-
Przeciwwskazania. Stosowanie inhibitorów redukta-
czenie skojarzone wraz z lekami hamującymi syntezę
zy HMG-CoA jest przeciwwskazane w schorzeniach
cholesterolu (statyny). Udowodniony został addycyj-
wątroby, po przebytych miopatiach, a także w ciąży
i okresie karmienia.
ny efekt dotyczący obniżenia stężenia cholesterolu B4
LDL dla kombinacji ezetymibu ze wszsytkimi staty-
nami. W handlu dostępny jest gotowy preparat łączo-
ny ezetymibu z simwastatyną. Ze względu na różne
4.2.2.4.2. Inhibitory wchłaniania cholesterolu
drogi metabolizmu, którym podlegają oba te związku
nie dochodzi między nimi do interakcji farmakokine-
Jak napisano wcześniej, wchłanianie cholesterolu
tycznych. Skojarzenie ezetymibu z fibratami (p. niżej)
w jelicie cienkim, dostarczanego z pożywieniem lub
wydaje się ze względu na różne punkty uchwytu rów-
wydzielanego z żółcią, jest ważnym procesem, któ-
nież stosowne. Ponieważ jednak zgodność tego typu
ry decyduje o ilości znajdującego się w organizmie
kombinacji nie została do tej pory jeszcze wystarcza-
cholesterolu. Podsunęło to pomysł zsyntetyzowania
jąco przebadana, nie poleca się na razie równoczesne-
inhibitorów wchłaniania cholesterolu jako leków ob-
go stosowania ezetymibu i fibratów.
niżających poziom lipidów.
Ezetymib został dopuszczony jako pierwszy lek
w nowej klasie substancji – inhibitorów wchłania- 4.2.2.4.3. Żywice jonowymienne,
nia cholesterolu. Środek ten blokuje wybiórczo biał- sterole roślinne
ko NPC1L1 (p. wyżej) odpowiedzialne za jelietowe oraz estry etylowe kwasów omega-3
wchłanianie cholesterolu oraz podobnych struktural-
nie fitosteroli. Cholestyramina jest zasadową żywicą jonowymien-
Ezetymib jest wskazany (samodzielnie lub w sko- ną, która ma wysokie powinowactwo do kwasów żół-
jarzeniu ze statynami) w leczeniu hipercholestero- ciowych. Po podaniu doustnym kwasy żółciowe wią-
lemii a także sitosterolemii. Po podaniu doustnym zane są częściowo z nierozpuszczalną i nie podlega-
nie jest metabolizowany przez układ enzymów P-450, jącą wchłanianiu żywicą jonowymienną a następnie
tylko przekształcany jest przez glukuronylotransfe- wydalane z kałem. Dzięki temu zwykle niewielkie
razy do glukuronidu, który również blokuje wchła- ze względu na krążenie jelitowo-wątrobowe wydala-
MUTSCHLER-2009.indd 561 2010-01-07 22:13:35
562 Układ naczyniowy i krążenie
nie kwasów żółciowych zwiększa się aż dziesięcio- Jako działania niepożądane występują szczególnie
krotnie. Deficyt ten jest uzupełniany przez syntezę zaburzenia żołądkowo-jelitowe. Przy równoczesnym
kwasów żółciowych z cholesterolu. Wskutek obni- podawaniu leków przeciwzakrzepowych może dojść
żenia się wewnątrzkomórkowego stężenia choleste- do nieznacznego wydłużenia czasu krwawienia.
rolu w wątrobie zwiększa się liczba receptorów dla W tym przypadku wskazane jest uważne monito-
LDL, a co za tym idzie wychwyt LDL przez wątrobę, rowanie współczynników krzepnięcia. Ze względu
a w konsekwencji w przypadku typowego dawkowa- na możliwość znacznego podwyższenia poziomu
nia (p. niżej) stężenie cholesterolu we krwi zmniejsza transaminaz wątrobowych, zalecane jest ostrożne
się w przeciągu dwóch tygodni o około 20–30%. stosowanie preparatu u pacjentów z zaburzeniami
Cholestyramina jest szczególnie wskazana w przy- czynności wątroby.
padku hiperlidemii typu IIa. Ponadto stosuje się
ją w przypadku biegunki spowodowanej żółcią.
Dawkowanie wynosi 12–16 (–24) g dziennie 4.2.2.4.4. Fibraty
w dawkach podzielonych. Stosowanie dużych dawek
żywicy jonowymiennej ze względu na jej działania Prototypem tej grupy leków był klofibrat będący pochod-
niepożądane może prowadzić do niestosowania się ną kwasu arylooksymasłowego, który już nie jest obecnie
stosowany. Obecne w użyciu są natomiast jego pochodne
przez pacjenta do wskazań lekarskich. oraz analogi.
Działania niepożądane to zaparcia lub stolce
tłuszczowe ze względu na zaburzenia wchłaniania
Etofibrat jest preparatem, w którym klofibrat połą-
tłuszczów, a także inne zaburzenia żołądkowo-jelito-
czony jest z kwasem nikotynowym (zob. niżej) za po-
we, oraz w przypadku przewlekłego stosowania – hi-
średnictwem glikolu etylenowego w postaci dwu-
powitaminozy dotyczące witamin rozpuszczalnych
estru. W organizmie jego połączenia estrowe ulegają
w tłuszczach.
szybkiemu rozkładowi w wyniku czego powstają
Podawanie cholestyraminy równocześnie z po-
półestry klofibryny i kwasu nikotynowego z glikolem
chodnymi kumaryny, glikozydami naparstnicy, hor-
etylenowym i dlatego etofibrat nie pojawia się w po-
monami tarczycy lub tetracyklinami może zmniejszać
staci niezmienionej w osoczu.
ich wchłanianie. W celu zapobieżenia tej interakcji
Klofibrat etofiliny jest estrem utworzonym z kwasu
należy zachować odstęp minimum jednej godziny
klofibrynowego i etofiliny. Również ten lek jest szyb-
pomiędzy zażyciem cholestyraminy a któregoś z tych
ko rozkładany w miejscu połączenia estrowego.
leków.
Niektóre sterole blisko spokrewnione z cholesterolem po- Do analogów klofibratu (zob. tab. B 4.2-4) zalicza
chodzenia roślinnego (m.in. sitosterol) zmniejszają wchła- się bezafibrat, fenofibrat i gemfibrozyl.
nianie cholesterolu z przewodu pokarmowego dzięki kom-
petycyjnemu hamowaniu białka transportującego (p. wyżej) Fibraty mają niewielki wpływ na stężenie cholesterolu
a także jego estryfikacji w komórkach jelita. W ten sposób w osoczu. Działają one znacznie silniej na poziom triglice-
zmniejszają one stężenie cholesterolu we krwi. Ze wzglę- rydów, który potrafią zmniejszyć o 30–50%. Terapeutyczne
du na ten sam mechanizm działania wciąż dyskutowany skutki obniżenia poziomu trójglicerydów są polem kontro-
jest kontrowersyjny efekt zmniejszający wchłanianie cho- wersyjnych dyskusji. Pewne jest, że bardzo wysokie stęże-
lesterolu margaryn zawierających sterole. nie triglicerydów prowadzić może do wystąpienia zapalenia
trzustki i powinno być leczone. Należy jednak zauważyć,
Estry etylowe kwasów omega-3 zawierają etylowe że w przypadku wielu pacjentów lepszy efekt na stężenie
triglicerydów może mieć odstawienie alkoholu.
estry syntezowanych półsyntetycznie kwasów tłusz-
czowych omega-3. Istotnym składnikiem jest ester
Mechanizm działania fibratów nie został jeszcze
etylowy kwasu eikozanowego (EPA; 46%) oraz doko-
w pełni wyjaśniony. Od dawna wiadomo, że wzmaga-
zanowego (DHA; 38%), które służąc jako „fałszywe”
ją one aktywność lipazy lipoproteinowej, w wyniku
substraty dla enzymu syntezującego triglicerydy po-
czego zwiększa się liczba cząstek VLDL, które przez
wodują obniżenie stężenia VLDL (zob. ryc. B 4.2-4).
pośrednią postać IDL są przekształcane do LDL.
Preparat stosuje się w leczeniu hipertriglicerydemii,
Oprócz tego dochodzi do zmniejszenia uwalniania
w typie IIb/III hiperlipoproteinemii także w skoja-
VLDL z wątroby i wątrobowej syntezy cholestero-
rzeniu ze statynami, kiedy monoterapia nie odno-
lu. Poziom HDL podwyższa się. Niedawno opisa-
si wystarczającego skutku. Szczególnie wskazany
no, że fibraty wiążą się także z jądrowymi czynni-
jest w przypadku hipertriglicerydemii u pacjentów
kami transkrypcyjnymi PPARα z typu receptorów
po zawale mięśnia sercowego.
aktywowanych przez proliferatory peroksysomów.
Wielostronne działanie fibratów wyjaśnia również
MUTSCHLER-2009.indd 562 2010-01-07 22:13:35
Układ naczyniowy i krążenie 563
fakt, że poprzez PPARα nasilają onę transkrypcję ca- nio przenosić wyników badań nad gemfibrozylem
łego szeregu genów oprócz lipazy lipoproteinowej. na inne leki z tej grupy.
Dane farmakokinetyczne i dawkowanie fibratów ze- Metaboliczne interakcje gemfibrozylu z ceriwastatyną,
stawiono w tab. B4.2-4. z powodu których ta ostatnia musiała niedawno zostać wy-
cofana z rynku, polegały na hamowaniu przez gemfibrozyl
Działania niepożądane tej bardzo heterogennej struk- i jego glukurowane metabolity metabolizmu ceriwastatyny
turalnie grupy leków zostały najlepiej udokumento- przez enzymy CYP2C8. Oprócz tego gemfibrozyl hamo-
wane dla gemfibrozylu. Podobnie jak w przypadku wał wychwyt ceriwastatyny przez wątrobę poprzez blokadę
statyn mogą to być uszkodzenia mięśni, które w przy- białka transportującego (OATP1B1). Wskutek tego docho-
padkach skrajnych mogą skończyć się rabdomiolizą. dziło do osiągania przez ceriwastatynę bardzo wysokich
stężeń w osoczu i w rezultacie do śmiertelnych przypadków
Szczególne znaczenie ma potencjalny wpływ gem- rabdomiolizy.
fibrozylu na metabolizm. On sam i jego glukurowane
metabolity hamują cały szereg enzymów cytochromu
P-450 oraz glukuronylotransferazy, przez co może 4.2.2.4.5. Kwas nikotynowy
dochodzić do istotnych interakcji (np. z przeciwza-
krzepowymi pochodnymi kumaryny lub doustnymi Kwas nikotynowy obniża w sposób zależny od dawki stęże-
lekami przeciwcukrzycowymi). Wpływ na metabo- nie wolnych kwasów tłuszczowych, a także triglicerydów
lizm innych fibratów jest gorzej zbadany. Ze względu i cholesterolu w osoczu. Obniżenie poziomu cholesterolu
jednak na różnice w budowie nie można bezpośred- zachodzi z reguły dopiero po obniżeniu się stężenia trigli-
cerydów. Po odstawieniu leku należy liczyć się z efektem
Serce i układ krążenia
Tab. B 4.2-4. Fibraty
Wzór strukturalny Nazwa Preparat handlowy Okres Dawkowanie
międzynarodowa półtrwania (mg)
CH3 O etofibrat Lipo-Merz retard 16 500-1000
O
O
O
H3C
Cl O
N
B4
O
CH3 O klofibrat etofiliny Duolip n.b. 500
O
N CH3
H3C N
Cl O
N N O
CH3
CH3 bezafibrat Befibrat, 1-2 400
O Bezafibrat AL,
O OH
Bezafibrat-ratiopharm,
H3C Cedur u.a.
NH O
Cl
CH3 fenofibrat CIL, 22 250
Cl O
O CH3 durafenat,
H3C Lipidil,
O CH3 Normalip pro u.a.
O
H3C CH3 gemfibrozyl Gemfi-1 A Pharma, 1,5 900-1200
CH3
Gevilon
O OH
H3C
O
MUTSCHLER-2009.indd 563 2010-01-07 22:13:35
564 Układ naczyniowy i krążenie
Wskazania do stosowania obejmują hiperlipidemie
związane z niskim poziomem cholesterolu HDL.
O W celu uzyskania pożądanego efektu wymagane jest sto-
sowanie wysokich dawek od 1 000 do 2 000 mg dziennie.
NH2 Z powodu możliwości wystąpienia działań niepożądanych
(zob niżej) dawki te należy wprowadzać stopniowo. Nawet
N wtedy jednak w przypadku dawek powyżej 2 g dziennie
wielu pacjentów bardzo źle toleruje ten lek. Z tego powo-
nicotinsäure du uzyskano postać o przedłużonym działaniu, która przy
zachowaniu odpowiedniego dawkowania (stopniowe pod-
wyższanie dawek) w mniejszym stopniu powoduje wystą-
pienie efektów niepożądanych.
Do działań niepożądanych należą w większości efek-
z odbicia. Szczególnie warte uwagi jest wyraźne zwiększe- ty rozszerzenia naczyń krwionośnych. Często obserwuje
nie stężenia cholesterolu HDL. się zespół zaczerwienienia, spadek ciśnienia krwi, świąd
Działanie obniżające stężenie lipidów polega na hamo- oraz dolegliwości żołądkowo-jelitowe. W przypadku za-
waniu mobilizacji tłuszczów (hamowanie lipolizy) poprzez czerwienienia pomaga równoczesne podawanie inhibito-
blokadę lipazy trójglicerydowej oraz (w dużej mierze wyni- rów cyklooksygenazy, np. kwasu acetylosalicylowego.
kającemu z tego) zmniejszaniu syntezy VLDL w wątrobie, W przypadku przewlekłego stosowania może dojść do za-
a co za tym idzie zmniejszeniu tworzenia się LDL. Oprócz burzeń czynności wątroby i zmniejszonej tolerancji wę-
tego wykazano, że kwas nikotynowy powoduje podwyższe- glowodanów.
nie aktywności lipazy lipoproteinowej. Molekularny me- Z powodu interakcji z białkami transportującymi kwas
chanizm działania kwasu nikotynowego polega w skrócie moczowy podczas leczenia kwasem nikotynowym istnieje
na tym, że wiąże się on z dwoma receptorami zależnymi niebezpieczeństwo nasilenia się objawów hiperurykemii.
od białka G (HM74 oraz HM74a). HM74a pośredniczy
w działaniu kwasu nikotynowego w adipocytach poprzez
redukcję wewnątrzkmórkowego stężenia cAMP i zmniej-
szeniu wskutek tego uwalniania wolnych kwasów tłuszczo- 4.2.3. Nadciśnienie
wych. W makrofagach kwas nikotynowy pobudza receptor i leki przeciwnadciśnieniowe
aktywowany przez proliferatory peroksysomów gamma
(PPARγ), który uruchamia geny kodujące białka odpowie-
dzialne za zwrotny transport cholesterolu. Dzięki temu me- 4.2.3.1. Podstawy patofizjologiczne
chanizmowi kwas nikotynowy prowadzi do zwiększonego
wychwytu cholesterolu na obwodzie.
Po podaniu doustnym kwas nikotynowy wchłania się Jako nadciśnienie określa się każdy długotrwa-
szybko i praktycznie w całości. Tylko 12% substancji ły wzrost tętniczego ciśnienia krwi powyżej normy.
wydalane jest w postaci niezmienionej z moczem. W wą- W tab. B 4.2-5 podano definicję i klasyfikację nadciś-
trobie kwas nikotynowy jest m. in. sprzęgany z glicyną nienia według Niemieckiego Towarzystwa do Walki
do kwasu nikotynurowego. Ten szlak metaboliczny zwią-
zany jest z opisanym niżej objawem zaczerwienienia. z Nadciśnieniem. Za ciśnienie optymalne, sprzyjają-
Biodostępność kwasu nikotynowego jest nieznana. U ko- ce zdrowiu uznaje się wartość ciśnienia skurczowego
biet w osoczu obserwuje się wyższe stężenia niż u męż- 120 mmHg a rozkurczowe 80 mmHg.
czyzn. Okres półtrwania wynosi około jednej godziny.
Tab. B 3.2-5. Klasyfikacja zakresów ciśnienia krwi (w mmHg; według wytycznych Niemieckiego Towarzystwa do Walki z Nadciśnie-
niem)
Kategoria Skurczowe Rozkurczowe
Optymalne <120 <80
Prawidłowe <130 <85
Wysokie prawidłowe 130-139 85-89
Nadciśnienie łagodne (stopień 1) 140-159 90-99
podgrupa nadciśnienia granicznego 140-149 90-94
Nadciśnienie średniego stopnia (stopień 2) 160-179 100-109
Nadciśnienie ciężkie (stopień 3) >180 >110
Izolowane nadciśnienie skurczowe >140 <90
podgrupa układowego nadciśnienia granicznego 140-149 <90
MUTSCHLER-2009.indd 564 2010-01-07 22:13:36
Układ naczyniowy i krążenie 565
Jak już wcześniej opisano wartość ciśnienia tętni- 3. sercowo-naczyniowe (np. wskutek zwężenia
czego krwi wynika z pojemności minutowej serca cieśni aorty, w całkowitym bloku serca, w ze-
oraz oporu obwodowego, co oznacza, że może ono spole serca hiperkinetycznego)
wynikać ze zwiększonej pojemności minutowej ser-
4. neurogenne (wskutek organicznych schorzeń
ca, zwiększonego oporu naczyń obwodowych, bądź
układu nerwowego, np. guzów, zapalenia
ze zwiększenia obydwu tych parametrów jednocześ-
mózgu, zapalenia opon mózgowych, zatrucia
nie. U osób młodych nadciśnienie związane jest zwy-
tlenkiem węgla lub talem)
kle ze wzrostem pojemności minutowej serca, podczas
gdy u osób starszych jest ono spowodowane częściej
Z tego przypada na:
zwiększeniem oporu obwodowego. U tych pacjentów
stwierdza się też nierzadko izolowane podwyższenie ■ nadciśnienie pierwotne – ok. 90% przypadków,
ciśnienia skurczowego, w przypadku którego możli-
■ nadciśnienie nerkowe – 6–8%,
wość wystąpienia poważnych powiłkań była w prze-
szłości niedoceniana. ■ nadciśnienie wewnątrzwydzielnicze – ≤ 1%,
■ nadciśnienie sercowo-naczyniowe – ≤ 1%,
Skutki nadciśnienia. Przewlekły podwyższony opór
naczyniowy prowadzi wskutek uwalniania czynni- ■ nadciśnienie neurogenne < 1%.
ków wzrostu m.in. płytkowego czynnika wzrostu
(PDGF – platelet derived growth factor), do pogru- W dalszym ciągu nieznane pozostają przyczyny powsta-
bienia błony wewnętrznej i mięśniówki i w związku wania najczęstszej i najważniejszej postaci nadciśnienia
Serce i układ krążenia
– nadciśnienia pierwotnego. Rozważa się cały szereg moż-
z tym do dalszego zwiększania oporu obwodowego liwych czynników wyzwalających, np. obciążenie rodzin-
(circulus vitiosus). Nadciśnienie jest jedną z najważ- ne, brak ruchu, nadmierny stres, otyłość i in.
niejszych przyczyn występowania miażdżycy, której Mnożą się wskazówki na to, że duże znaczenie w rozwoju
skutkami mogą być: i utrzymywaniu się nadciśnienia pierwotnego mogą mieć
zmiany funkcjonowania ośrodkowego układu nerwowego,
■ w przypadku mózgu udary niedokrwienne, a w szczególności podwyższone napięcie układu współczul-
nego. Oprócz tego w patogenezie podwyższonego ciśnie-
■ w przypadku serca choroba wieńcowa, przerost le- nia tętniczego wykazano związek ze zmniejszeniem wytwa-
wej komory oraz niewydolność serca, rzania endogennych substancji rozszerzających naczynia
(m.in. NO, prostaglandyna E2, bradykinina). W przypad-
■ w przypadku nerek niewydolność. ku osób z nadwagą nadciśnienie rozwija się częściej niż B4
u osób z prawidłową masą ciała, szczególnie jeżeli istnieje
Postacie nadciśnienia. Z klinicznego i patogene- u nich oporność na insulinę i dochodzi w związku z tym
tycznego punktu widzenia rozróżnia się następujące do hiperinsulinemii (zespół metaboliczny). Wszystkie te
obserwacje wskazują na to, że w przypadku nadciśnienia
postacie nadciśnienia: pierwotnego mamy do czynienia ze złożonym i wieloczyn-
nikowym mechanizmem.
A. Nadciśnienie pierwotne (synonim: wrodzone,
idiopatyczne) o ciągle nieznanej przyczynie (zob.
Podział nadciśnienia na stopnie i czynniki pro-
niżej)
gnostyczne. Zalecenia dotyczące podziału nadciś-
B. Nadciśnienie wtórne w następstwie patologicz-
nienia na stopnie według Niemieckiego Towarzystwa
nych zmian w narządach:
do Walki z Nadciśnieniem opisano w tab. B 4.2-5.
1. nerkowe
W celu oceny całkowitego ryzyka sercowo-naczy-
1.1 naczyniowo-nerkowe (wskutek zwężenia niowego opracowano również niekorzystne czynni-
tętnicy nerkowej) ki ryzyka u pacjentów z nadciśnieniem (zob. tab. B
4.2-6).
1.2. miąższowo-nerkowe (np. wskutek prze-
wlekłego kłębuszkowego zapalenia ne-
Ogólna ocena stopnia nadciśnienia i czynników ry-
rek, marskości nerek w przebiegu zapa-
zyka pozwala na wykonanie prognozy rozwoju cho-
lenia odmiedniczkowego nerek, torbielo-
roby (zob. tab. B 4.2-7). I tak na przykład ciężkie
watości nerek, skrobiawicy, guzkowego
nadciśnienie (> 180 mm Hg skurczowe, > 110 mm
zapalenia okołotętniczego, nefropatii
Hg rozkurczowe) przy jednocześnie udokumento-
ciążowej)
wanej niewydolności serca lub chorobie wieńcowej
2. wewnątrzwydzielnicze (w zespole Cushinga, jest związane z bardzo wysokim ryzykiem (praw-
Conna, hipertyreozie, akromegalii, w guzie dopodobieństwo śmierci w przeciągu najbliższych
chromochłonnym nadnercza) 10 lat wynosi > 30%).
MUTSCHLER-2009.indd 565 2010-01-07 22:13:36
566 Układ naczyniowy i krążenie
Tab. B 4.2-6. Czynniki prognostyczne choroby nadciśnieniowej
Sercowo-naczyniowe czynniki ryzyka Choroby lub zmiany narządowe
zmienne: niezmienne: choroba wieńcowa,
nadciśnienie, obciążający wywiad rodzinny, przerost lewej komory serca,
palenie papierosów, płeć męska, niewydolność serca,
cukrzyca, kobiety po menopauzie, ośrodkowe zaburzenia ukrwienia
otyłość, wiek (m.in. przemijające epizody niedokrwienne,
Adipositas, udar, demencja naczyniowa),
brak ruchu, obwodowa niedrożność tętnic,
podwyższone stężenie fibrynogenu przewlekłe choroby nerek
(z mikro- lub makroproteinurią),
retinopatia nadciśnieniowa
Ponieważ prognoza dotycząca pacjentów chorują- liwe do leczenia przyczynowego (np. nieoperacyj-
cych na nadciśnienie zależy również w dużej mierze ne, obustronne nadciśnienie naczyniowo-nerkowe)
od tego w jakim stadium rozpoznano podwyższone to dostępne jest tylko leczenie objawowe. Jest ono
ciśnienie krwi, szczególne znaczenie w tym przypad- niezbędne, ponieważ około 25% wszystkich zgonów
ku mają badania profilaktyczne. wynika bezpośrednio lub pośrednio z nadciśnienia.
Dowiedziono również, że prognozy dotyczące dłu-
Środki stosowane w leczeniu nadciśnienia. Nadciś- gości życia u pacjentów z nadciśnieniem – także
nienie wewnątrzwydzielnicze, sercowo-naczyniowe w przypadku nadciśnienia złośliwego – dzięki lecze-
i neurogenne może być przynajmniej w części przy- niu farmakologicznemu można znacznie wydłużyć,
padków leczone poprzez leczenie choroby je wywo- pod warunkiem, że leczenie to będzie prowadzone
łującej, a więc leczenie przyczynowe. Jeśli chodzi konsekwentnie.
o najczęściej występujące nadciśnienie pierwotne Zarówno przed jak i w trakcie leczenia nadciśnie-
oraz niektóre postaci nadciśnienia wtórnego niemoż- nia należy podjąć następujące kroki:
Tab. B 4.2-7. Klasyfikacja czynników ryzyka (wystąpienia zgonu sercowo naczyniowego, udaru niepowodującego zgonu lub zawału
mięśnia sercowego) w zależności od stopnia ciężkości nadciśnienia oraz innych czynników; por. tab. B 4.2-6
Ciśnienie tętnicze (w mmHg
Inne czynniki ryzyka Prawidłowe Wysokie prawidłowe Nadciśnienie Nadciśnienie Nadciśnienie
lub choroby stopnia 1 stopnia 2 stopnia 3
sk. 120-129 sk. 130-139 sk. 140-159 sk. 160-179 sk. 180
lub lub lub lub lub
rozk. 80-84 rozk. 85-89 rozk. 90-99 rozk. 100-109 rozk. 110
Brak innych średnie średnie lekko znacznie wysokie
czynników ryzyka odwyższone podwyższone
1-2 czynników ryzyka lekko podwyższone lekko podwyższone znacznie znacznie bardzo wysokie
podwyższone podwyższone
3 lub więcej czynników ryzyka znacznie podwyższone wysokie wysokie wysokie bardzo wysokie
lub uszkodzenia narządów
lub cukrzyca
Klinicznie jawna choroba wysokie bardzo wysokie bardzo wysokie bardzo wysokie bardzo wysokie
sercowo-naczyniowa
sk. – ciśnienie skurczowe; roz. – ciśnienie rozkurczowe
MUTSCHLER-2009.indd 566 2010-01-07 22:13:36
Układ naczyniowy i krążenie 567
■ rzucenie palenia, Lekami pierwszego wyboru są obecnie β-blokery,
inhibitory konwertazy angiotensyny, antagoniści re-
■ zmniejszenie konsumpcji alkoholu do < 30 g czy-
ceptora AT1, leki moczopędne i antagoniści kanału
stego alkoholu dziennie,
wapniowego.
■ zwiększenie aktywności fizycznej i unikanie czyn- Należy dążyć do normalizacji ciśnienia krwi,
ników stresujących, tzn. – niezależnie od wieku pacjenta! – do obniże-
nia ciśnienia rozkurczowego krwi poniżej 90 mmHg,
■ ograniczenie podaży soli kuchennej,
a skurczowego < 140 mmHg. Ponieważ zbyt szyb-
■ redukcja masy ciała w przypadku nadwagi, kie obniżenie ciśnienia krwi jest często ze względu
na dolegliwości subiektywne źle tolerowane przez
■ ponowne wnikliwe ustalenie wskazań w przypadku
pacjentów, a szczególnie u osób starszych spowodo-
terapii niesteroidowymi lekami przeciwzapalnymi,
wać może wystąpienie niebezpiecznych komplikacji
glukokortykosteroidami oraz środkami antykon-
(np. niedokrwienia mózgu) należy (za wyjątkiem
cepcyjnymi.
przełomów nadciśnieniowych) obniżać ciśnienie krwi
powoli. Przy wyborze leku przeciwnadciśnieniowego
Zmniejszenie całkowitego ryzyka sercowo-naczynio-
powinno się brać pod uwagę ogólny stan pacjen-
wego można również osiągnąć dzięki konsekwentne-
ta (m.in. wiek, ewentualne choroby towarzyszące)
mu leczeniu cukrzycy.
i stosować w miarę możliwości najmniejszą możliwą
dawkę w celu zmniejszenia ryzyka wystąpienia dzia-
Leczenie farmakologiczne nadciśnienia jest, w związ-
łań niepożądanych.
Serce i układ krążenia
ku ze złożonym mechanizmem regulacji ciśnienia
Monitorowanie leczenia polega na regularnych po-
tętniczego krwi, możliwe z wykorzystaniem wielu
miarach ciśnienia krwi oraz rozpoznawaniu działań
różnie działających substancji. Ich punkty uchwytu
niepożądanych leków.
zestawiono schematycznie na ryc. B 4.2-5.
α-metyldopa,
klonidyna
pień mózgu baroreceptory B4
napięcie układu
współczulnego
β-blokery NA
ciśnienie
objętość krwi CO tętnicze
antagoniści
aldosteronu opór
diuretyki wydalanie obwodowy
sodu aldosteron
antagoniści
β-blokery receptora α,
angiotensyna II antagoniści
kanału
wapniowego,
NA renina inhibitory ACE NA leki rozszerzające
naczynia
angiotensyna I
antagoniści
receptora AT1
Ryc. B 4.2-5. Punkty uchwytu leków przeciwnadciśnieniowych. CO pojemność minutowa serca, NA noradrenalina, ACE konwertaza
angiotensyny.
MUTSCHLER-2009.indd 567 2010-01-07 22:13:37
568 Układ naczyniowy i krążenie
Działania niepożądane wpływające na jakość życia jeśli chodzi o punkty końcowe badań przy porówny-
podczas terapii lekami przeciwnadciśnieniowymi walnej zdolności obniżania ciśnienia krwi.
są dla pacjentów przede wszystkim dlatego trudne Mechanizm obniżania ciśnienia krwi przez β-blokery
do zaakceptowania, że przed rozpoczęciem lecze- pomimo intensywnych i długich badań nie jest jesz-
nia zwykle nie odczuwali oni żadnych dolegliwości. cze w pełni wyjaśniony. Pod uwagę brane są następu-
Dlatego niezmiernie ważne jest poświęcenie uwagi jące punkty uchwytu:
na wyjaśnienie pacjentowi możliwych skutków nie-
■ obniżanie pojemności minutowej serca,
korzystnych długotrwałej terapii.
■ zmniejszenie wyrzutu reniny przez nerki i związanej
z tym syntezy angiotensyny II i uwalniania aldo-
4.2.3.2. Leki przeciwnadciśnieniowe steronu,
■ blokada presynaptycznych receptorów β i zmniej-
4.2.3.2.1. Leki działające szenie wskutek tego uwalniania noradrenaliny,
na układ współczulny
■ zmniejszenie napięcia układu współczulnego przez
działanie na ośrodkowy układ nerwowy.
Spośród leków działających na układ współczulny
omówionych we wcześniejszych rozdziałach, w le-
Osiągane przez poszczególne β blokery obniżenie
czeniu nadciśnienia stosowane są:
ciśnienia krwi jest we wszystkich możliwych sko-
■ blokery receptorów α, jarzeniach z innymi lekami podobne i niezależne
od ich właściwości fizykochemicznych i profilu dzia-
■ blokery receptorów β,
łania.
■ leki sympatykolityczne.
Leki sympatykolityczne, do których należą α2-mi-
Spośród leków blokujących receptory α największe metyki, agoniści receptora imidazolinowego oraz
znaczenie w leczeniu nadciśnienia mają środki bloku- rezerpina, stosowane są w monoterapii lub też w po-
jące wybiórczo receptor α1. Efekt obniżający ciśnie- łączeniu z innymi lekami przeciwnadciśnieniowym
nie krwi wynika z rozszerzenia naczyń krwionośnych wówczas, gdy istnieją przeciwwskazania do stosowa-
wskutek blokady receptorów α. nia innych leków działających na układ współczulny,
Ze względu na powodowanie hipotonii ortosta- np. β-blokerów.
tycznej, a także brak dowodów na wydłużanie przez
nie życia, ta grupa leków nie należy już więcej do le-
ków hipotensyjnych pierwszego wyboru. 4.2.3.2.2. Leki moczopędne
Leki blokujące receptory β są bardzo często sto- Kolejną szczególnie istotną grupą leków stosowa-
sowane w leczeniu nadciśnienia ze względu na jed- nych w leczeniu nadciśnienia są leki moczopędne.
noznacznie pozytywne wyniki badań dotyczących Obniżenie ciśnienia krwi po ich podaniu zachodzi
wydłużania przez nie życia, a także ze względu w dwóch fazach. Początkowe obniżenie ciśnienia
na ich ogólną dobrą tolerancję (przy uwzględnieniu następuje na skutek wzmożonego wydalania jonów
przeciwwskazań). Szczególnie skuteczne są one u pa- sodu. W wyniku zmniejszenia się stężenia jonów
cjentów ze wzmożonym napięciem układu współ- sodu w osoczu dochodzi do spadku objętości osocza
czulnego tzn. z reguły do 55–60 roku życia. Wraz i pojemności minutowej serca, natomiast opór na-
ze starzeniem się, kiedy coraz mniejsze znaczenie ma czyniowy odruchowo nieco wzrasta. Utrzymywanie
nadciśnienie spowodowane nadmiernie wysoką po- się obniżonego ciśnienia krwi podczas drugiej fazy,
jemnością minutową a zwiększa się znaczenie obwo- w której objętość osocza normalizuje się, a wyda-
dowego oporu naczyniowego – skuteczność β-bloke- lanie jonów sodu wraca do wartości podobnych jak
rów maleje. Mogą one jednak w wielu przypadkach przed rozpoczęciem terapii, wynika prawdopodob-
być skutecznie stosowane również u pacjentów w po- nie ze zmniejszonej odpowiedzi mięśniówki gładkiej
deszłym wieku. Należy też zaznaczyć, że z ostatnich naczyń na bodźce powodujące skurcz naczyń wsku-
danych wynika, że istnieje coraz mniej przeciwwska- tek zmniejszonej zawartości jonów sodu w ścianie
zań do stosowania β-blokerów u pacjentów z nadciś- naczynia. Rozważa się także mechanizm regulacji
nieniem. Nowsze badania kliniczne wykazały jednak, w dół (down-regulation) receptorów α, oraz wzmo-
że długodziałające dihydropirydynowe pochodne an- żoną syntezę prostacykliny przez leki moczopędne.
tagonistów wapnia mają przewagę nad β-blokerami Nie stwierdzono jednak bezpośredniego efektu roz-
MUTSCHLER-2009.indd 568 2010-01-07 22:13:37
Układ naczyniowy i krążenie 569
szerzającego naczynia tych leków przy normalnym jaśnia to dlaczego blokery kanału wapniowego mają
dawkowaniu. działanie rozszerzające naczynia i w związku z tym
Leki moczopędne wskazane są w monoterapii przede poprzez zmniejszanie oporu obwodowego obniżają
wszystkim u pacjentów chorujących na nadciśnienie ciśnienie krwi. Działanie obniżające ciśnienie krwi
w podeszłym wieku, a ponadto są one istotnymi le- jest tym większe im wyższy jest wyjściowy poziom
kami stosowanymi w kombinacji z szeregiem leków ciśnienia krwi (u pacjentów z prawidłowymi wartoś-
przeciwnadciśnieniowych, które mają efekt retencyj- ciami ciśnienia krwi stosowane w dawkach terapeu-
ny na jony sodu i wodę (np. leki rozszerzające naczy- tycznych praktycznie nie obniżajągo).
nia). W połączeniach takich preferowane są zwykle
leki długo działające, jak na przykład tiazydy, w prze- Wszystkie blokery kanału wapniowego wiążą się z podjed-
ciwieństwie do diuretyków krótko działających, nostką α1c kanału L, jednak w zależności od budowy w róż-
nych miejscach. Podjednostka ta składa się z czterech po-
np. diuretyków pętlowych. wtarzających się odcinków, które przechodzą przez błonę
W celu zapobieżenia utracie jonów potasu i magnezu, komórkową szcześciokrotnie (S1-S6). Części te są uformo-
u pacjentów z normalną lub nieznacznie upośledzo- wane w kształt pierścienia w ten sposób, że cztery odcinki
ną czynnością nerek można stosować w połączeniu transmembranowe (zawsze S6) tworzą kanał jonowy.
z lekami powodującymi utratę sodu (saluretykami)
Oprócz blokerów kanału wapniowego do antagonistów
leki moczopędne oszczędzające potas np. amiloryd wapnia zalicza się również inne substancje, które w od-
lub triamteren. Obecnie stosuje się niższe niż kie- mienny sposób wpływają na metabolizm wapnia. Należą
dyś dawki saluretyków u pacjentów z nadciśnieniem do nich np. substancje, które wchodzą w reakcję z kalmo-
i w związku z tym istotnie zmniejszyła się częstość duliną – szeroko rozpowszechnionym zależnym od wap-
Serce i układ krążenia
występowania działań niepożądanych – w tym utraty nia białkiem regulacyjnym, bądź też takie, które wpływają
na patologiczny prąd wapniowy występujący w przypadku
potasu i magnezu. Dlatego obecnie takie połączenie niedokrwienia (tzw. blokery przeładowania wapniem – cal-
leków nie jest w zasadzie konieczne. cium overload blockers). Poniżej omówione zostaną tylko
substancje blokujące kanał wapniowy typu L.
4.2.3.2.3. Antagoniści kanału wapniowego Zarówno z chemicznego jak i z farmakologicznego
(antagoniści wapnia) punktu widzenia blokery kanału wapniowego nie two-
rzą jednorodnej grupy substancji. Wśród tzw. czy-
Wprowadzenie do fizjologii. W zwykłej komórce stę- stych blokerów kanału wapniowego, czyli substancji,
żenie wolnych jonów wapnia jest około dziesięć tysięcy które w normalnych dawkach nie wywierają żadnych
B4
razy mniejsze niż w przestrzeni pozkomórkowej. Stężenie
to może być zmieniane lub regulowane poprzez otwieranie dodatkowych działań na inne kanały jonowe, rozróż-
się i zamykanie kanałów wapniowych, uwalnianie i wią- nia się w zależności od punktu uchwytu na kanale
zanie wewnątrzkomórkowego wapnia, a także przez białka wapniowym antagonistów kanału wapniowego:
transportujące jony wapnia znajdujące się w błonach ko-
mórkowych oraz wewnątrzkomórkowych. Wskutek pobu- ■ grupę 1,4-dihydropirydyny (nifedypiny),
dzenia przez różne bodźce zewnętrzne kanały wapniowe
są na krótko otwierane przez co dochodzi do krótkotrwa- ■ grupę werapamilu,
łego dużego wzrostu stężenia jonów wapnia wewnątrz ko- ■ grupę diltiazemu.
mórki, a następnie do wiązania jonów wapnia z białkami
(np. kalmoduliną, zob niżej). Aktywowane białka wywołu-
ją następnie w komórce różnorakie reakcje. Podwyższone Pomimo różnic w budowie między lekami tej gru-
stężenie wewnątrzkomórkowego wapnia może zostać szyb- py istnieją także zwracające uwagę podobieństwa
ko obniżone do poprzedniej wartości dzięki magazynowa- w zakresie farmakokinetyki: wszystkie blokery kana-
niu jonów wapnia w magazynach wewnątrzkomórkowych, łu wapniowego są metabolizowane przez CYP3A4.
a także poprzez transport przez odpowiednie białka.
W związku z tym istnieje możliwość interakcji
ze wszystkimi substancjami, które indukują bądź ha-
Nazwa antagoniści wapnia zostało wprowadzone
mują ten enzym. Przykładowo równoczesne podanie
na określenie substancji, które hamują przezbłono-
z blokerem kanału wapniowego induktora – ryfam-
wy napływ jonów (zmniejszenie prądu wapniowego)
picyny prowadzić może do obniżenia jego stężenia
przez hamowanie tzw. wolnych napięciowozależ-
w osoczu poniżej poziomu terapeutycznego. Z kolei
nych kanałów wapniowych typu L. Dlatego nazy-
równoczesne stosowanie z blokerami kanału wapnio-
wane są one obecnie bardziej poprawnie antagoni-
wego innych substratów dla CYP3A4 (np. leków prze-
stami kanału wapniowego (calcium entry blockers).
ciwhistaminowych, przeciwgrzybiczych, immunosu-
Ponieważ wewnątrzkomórkowe stężenie wolnych
presyjnych, inhibitorów proteazy) prowadzi do pod-
jonów wapniowych zapewnia zdolność do skurczu
wyższenia – w zależności od powinowactwa do en-
komórek gładkich mięśni naczyń krwionośnych, wy-
MUTSCHLER-2009.indd 569 2010-01-07 22:13:37
570 Układ naczyniowy i krążenie
zymu – bądź stężenia blokerów kanału wapniowego Tab. B 4.2-8. Profil działania antagonistów kanału wapniowego
bądź tej drugiej substancji. Również sok z grejpfruta
zawiera flawonoidy, które hamują CYP3A4 w prze- Nife- Wera- Dil-
wodzie pokarmowym oraz zmniejszają ekspresję en- dypina pamil tiazem
zymu. Równoczesne podanie soku z grejpfrutów oraz opór tętnic wieńcowych
pochodnej dihydropirydyny zwiększa w ten sposób
biodostępność blokera kanału wapniowego. opór obwodowy
Za wyjątkiem niechiralnych związków diltiaze- ciśnienie krwi
mu i nifedypiny, wszystkie pozostałe blokery kanału
akcja serca
wapniowego są dostępne w handlu jako mieszaniny
racemiczne, pomimo tego, że oba enancjomery blo- przewodnictwo AV
kują kanał wapniowy z różną siłą.
kurczliwość
Wspólną właściwością farmakodynamiczną wszyst- brak wpływu
odruchowy wzrost akcji serca w przypadku nifedypiny
kich tych trzech grup jest obniżanie stężenia we- o nieprzedłużonym działaniu
wnątrzkomórkowego wapnia, jednakże w sposób
silnie zróżnicowany:
■ w mięśniówce gładkiej naczyń prowadzą do zmniej-
Później opracowano pochodne dihydropirydyny, któ-
szenia napięcia mięśni gładkich i w rezultacie
re dzięki obecności lipofilnych łańcuchów bocznych
do rozszerzenia naczynia,
miały dłuższy okres półtrwania jak amlodypina oraz
■ w sercu zmniejszają aktywność zależnej od wap- takie, które dzięki gromadzeniu się w komórkowych
nia ATPazy miozyny, wskutek czego zmniejsza się błonach lipidowych miały przedłużony czas działania
obrót wysokoenergetycznych fosforanów i równo- – lacydypina, lerkanidypina.
cześnie zapotrzebowanie na tlen. Amlodypina, nikardypina i nisoldypina są stoso-
wane nie tylko w leczeniu nadciśnienia tętniczego,
Poszczególne grupy blokerów kanału wapniowego ale także w chorobie niedokrwiennej serca, felody-
różnią się pomiędzy sobą jeśli chodzi o wpływ na po- pina, isradypina, lacipidyna, lerkanidypina, nilwady-
wstawanie pobudzeń w węźle zatokowym oraz prze- pina i nitrendypina natomiast są dopuszczone jedynie
wodzenie pobudzenia w węźle przedsionkowo-komo- do stosowania w leczeniu nadciśnienia.
rowym. Jedynie pochodne z grupy werapamilu oraz
diltiazemu działają ujemnie chronotropowo na wę- Nimodypina ma wyjątkową pozycję. Jest ona ze wzglę-
zeł zatokowy oraz ujemnie dromotropowo na węzeł du na bardzo wysoką lipofilność a co z tego wynika dużą
zdolność penetracji do ośrodkowego układu nerwowego
przedsionkowo-komorowy. Jeśli chodzi o blokery stosowana w profilaktyce i leczeniu wynikających z niedo-
kanału wapniowego z grupy nifedypiny, to powodują krwienia ubytków neurologicznych w przebiegu skurczów
one w dawkach terapeutycznych przede wszystkim naczyń mózgu po krwawieniach podpajęczynówkowych
rozszerzenie naczyń krwionośnych, a podane w po- (dawkowanie początkowe przez pierwsze 2 godziny 1 mg/
staci szybko uwalnianych, krótkodziałających prepa- godzinę, następnie przy dobrej tolerancji 2 mg/godzinę
we wlewie dożylnym przez około 10 dni; dodatkowo do-
ratów mogą doprowadzić nawet, wskutek wywołania ustnie przez 7 dni sześć razy dziennie po 60 mg). Oprócz
aktywacji układu współczulnego, do odruchowego tego stosowana jest w zaburzeniach wydolności mózgu po-
wzrostu częstości akcji serca. Dzięki pobudzeniu chodzenia organicznego w wieku podeszłym.
układu współczulnego antagonizowany jest wówczas
także ujemny efekt inotropowy. Wzory strukturalne i dawkowanie pochodnych dihy-
W tab. B 4.2-8 przedstawiono profil działania dropirydyny podano w tab. B 4.2-9.
różnych blokerów kanału wapniowego wynikający
z opisanych właściwości. Do działań niepożądanych, które występują przede
wszystkim podczas stosowania szybko- i krótkodzia-
Pochodne 1,4-dihydropirydyny. Pierwszym stoso- łających preparatów, i wynikają z nagłego i masyw-
wanym w leczeniu środkiem zawierającym tę gru- nego rozszerzenia naczyń krwionośnych, należą nie-
pę była nifedypina w postaci o szybkim uwalnia- pożądany spadek ciśnienia krwi, odruchowa tachy-
niu. Następnie wprowadzono do leczenia preparaty kardia, bóle i zawroty głowy, nudności, zaburzenia
o przedłużonym działaniu m.in nitrendypinę i ni- żołądkowo-jelitowe, zaczerwienienie skóry i obrzęki
kardypinę, w przypadku których wyraźnie mniejsza kostek. Szczególnie niepożądana jest tachykardia
jest odruchowa tachykardia po podaniu (zob. wyżej). związana z podwyższonym ryzykiem wystąpienia za-
MUTSCHLER-2009.indd 570 2010-01-07 22:13:37
Układ naczyniowy i krążenie 571
Tab. B 4.2-9. Antagoniści kanału wapniowego
Wzór strukturalny Nazwa Preparat handlowy Okres Dawkowanie
międzynarodowa półtrwania (mg)
I. Leki z grupy nifedypiny
nifedypina Adalat, 1,7-3,4 30-60
NO2 Corinfar,
O O Nifedipin-ratiopharm,
NifeHEXAL u.a.
H3CO OCH3
H3C NH CH3
amlodypina Amlobeta, 35-50 5-10
Amlodipin-
Cl ratiopharm,
O O Amlodipin STADA,
H3CO OC2H5 Norvasc u.a.
O
H3C NH NH2
Cl felodypina Felocor, 18 5-10
Serce i układ krążenia
Felodipin-ratiopharm,
Cl Felodipin STADA,
O O
Modip u.a.
H3CO OC2H5
H3C NH CH3
N isradypina LOMIR, 8 5-10
O vascal
N
O
O
H3CO O
CH(CH3) 2 B4
H3C NH CH3
O lacydypina Motens 13-19 2-4
O C(CH3) 3
O O
H5C2O OC2H5
H3C NH CH3
NO2 lerkanidypina Carmen, 8-10 10-20
Corifeo
O O H3C CH3 CH3
N
H3CO O
H3C NH CH3
NO2 nikardypina Antagonil 8 60-90
O O CH3
N
H3CO O
H3C NH CH3
MUTSCHLER-2009.indd 571 2010-01-07 22:13:38
572 Układ naczyniowy i krążenie
Tab. B 4.2-9. Antagoniści kanału wapniowego (kontynuacja)
Wzór strukturalny Nazwa Preparat handlowy Okres Dawkowanie
międzynarodowa półtrwania (mg)
I. Leki z grupy nifedypiny
NO2 nilwadypina Escor, 15-20 8-16
Nivadil
O O
CH(CH3) 2
H3CO O
NC NH CH3
NO2 nimodypina Nimodipin HEXAL, 1,1-1,7 90
Nimotop
O O
(H3C) 2HC OCH3
O O
H3C NH CH3
nisoldypina Baymycard 10-12 10-20
NO2
O O
H3CO O CH(CH3) 2
H3C NH CH3
NO2 nitrendypina Bayotensin, 8-12 20-40
Nitrendipin beta,
O O Nitrendipin-
ratiopharm,
H3CO OC2H5 Nitrendipin STADA,
u.a.
H3C NH CH3
II. Leki z grupy werapamilu
CH3 NC CH(CH3) 2
werapamil Isoptin, 4 120-360
H3CO N OCH3
VeraHEXAL,
Verapamil AL,
Verapamil-
H3CO OCH3 ratiopharm u.a.
CH3 NC CH(CH3) 2 gallopamil Gallobeta, 3,5-8 100-150
H3CO N OCH3 Procorum
H3CO OCH3
OCH3
III. Leki z grupy diltiazemu diltiazem DiltaHEXAL, 6 180-360
OCH3 Diltiazem-ratiopharm,
Diltiazem STADA,
Dilzem u.a.
S
OCOCH3
N
O
N(CH3) 2
MUTSCHLER-2009.indd 572 2010-01-07 22:13:38
Układ naczyniowy i krążenie 573
wału serca podczas leczenia preparatami nifedypiny Gallopamil różni się chemicznie od werapamilu je-
o nieprzedłużonym działaniu. dynie dodatkową grupą metoksylową i ma podobne
Ponieważ podczas podawania w wysokich daw- do niego właściwości farmakologiczne (dawkowanie
kach doustnych nifedypiny obserwowano u myszy 75–150 mg dziennie).
i szczurów efekty teratogenne, pochodne dihydro-
pirydyny są przeciwwskazane podczas całęgo okre- Diltiazem. Pochodna benzotiazepiny – diltiazem
su trwania ciąży. Kolejnym przeciwwskazaniem – nie jest w żaden sposób spokrewniony chemicznie
jest ciężka hipotonia. z wymienionymi wyżej antagonistami kanału wap-
Inne leki przeciwnadciśnieniowe wzmacniają niowego. Jeśli chodzi o profil działania farmakolo-
efekt obniżający ciśnienie krwi pochodnych dihydro- gicznego jest on podobny do werapamilu.
pirydyny. Po podaniu doustnym diltiazem jest praktycznie
w całości wchłaniany. Jego biodostępność jednak
Leki z grupy werapamilu. Werapamil podobnie jak z powodu wyraźnego efektu pierwszego przejścia
nifedypina po podaniu doustnym jest wchłaniany nie- wynosi około 50%. Związek ten o okresie półtrwania
mal w całości. Podlega on silnemu stereoselektywne- 4–5 godzin jest w organizmie deacetylowany a tak-
mu efektowi pierwszego przejścia przez CYP3A4, że podlega O- i N-demetylacji. Stwierdzono rów-
który metabolizuje silniej działający enancjomer S. nież obecność koniugatów metabolitu fenolowego.
Oprócz tego werapamil metabolizowany jest przez Wydalany jest prawie wyłącznie w postaci metaboli-
CYP1A2. tów przez nerki oraz z żółcią.
Niektóre metabolity wykazują około jednej dzie- Średnia dawka dzienna wynosi 180 mg.
Serce i układ krążenia
siątej działania substancji macierzystej. Wydalanie Działania niepożądane, przeciwwskazania i in-
metabolitów zachodzi głównie przez nerki. terakcje są podobne jak w przypadku werapamilu.
Werapamil wskazany jest w leczeniu nadciśnienia, Z powodu działania teratogennego w badaniach
ale oprócz tego także w chorobie niedokrwiennej ser- na zwierzętach, w przypadku podawania diltiazemu
ca oraz tachykardiach nadkomorowych. u kobiet w wieku rozrodczym należy wykluczyć ist-
Przeciętna dawka dzienna w leczeniu nadciśnienia nienie ciąży.
wynosi 120–240 mg.
Jako działania niepożądane obserwuje się zabu-
rzenia przewodnictwa w mięśniu sercowym (blok 4.2.3.2.4. Inhibitory konwertazy
przedsionkowo-komorowy), bradykardie, niepożą- angiotensyny (IKA)
B4
dane spadki ciśnienia krwi, nasilenie niewydolności
serca, zaczerwienienie skóry, zaparcia (często) oraz Do tej grupy leków zalicza się kaptopril, jedyny in-
skórne reakcje alergiczne. hibitor konwertazy angiotensyny z grupą -SH, oraz
Werapamil jest przeciwwskazany w przypadku jego długo działające pochodne:
bloku przedsionkowo-komorowego II i III stopnia,
■ benazapril,
niewyrównanej niewydolności serca, zespołu węzła
zatokowego, świeżego zawału serca (szczególnie ■ cilazapril,
w przypadku bradykardii) oraz w ciężkiej hipotonii.
■ enalapril,
W przypadku podania werapamilu równocześnie
z innymi lekami antyarytmicznymi zwiększa się nie- ■ fozynopril,
bezpieczeństwo wystąpienia zaburzeń przewodnictwa
■ lizynopril,
lub bradykardii; β-adrenolityki nasilają jego działa-
nie kardiodepresyjne, a leki przeciwnadciśnieniowe ■ meksypril,
– działanie obniżające ciśnienie krwi. W przypadku
■ perindopril,
równoczesnego podania werapamilu i teofiliny do-
chodzi wskutek konkurencji o CYP1A2 do wyraźne- ■ chinapril,
go wzrostu stężenia teofiliny w osoczu. Hamowanie
■ ramipril,
glikoproteiny P przez werapamil wyjaśnia jego inter-
akcje z lekami, które są transportowane przez P-gp. ■ spirapril,
Istotnym klinicznie przykładem interakcji jest inter-
■ trandolapril.
akcja farmakokinetyczna między werapamilem a di-
goksyną, która prowadzi do podwyższenia stężenia
Działanie tej grupy leków przeciwnadciśnieniowych
w osoczu digoksyny.
polega na blokowaniu enzymu konwertującego an-
MUTSCHLER-2009.indd 573 2010-01-07 22:13:38
574 Układ naczyniowy i krążenie
angiotensyna I
angiotensyna II
Asp Arg Val Tyr IIe His Pro Phe His Leu ACE
Ryc. B 4.2-6. Mechanizm działania enzymu konwertującego angiotensynę (ACE).
giotensynę, który katalizuje przekształcenie angioten- natomiast wydalany jest w równej mierze przez nerki
syny I w angiotensynę II, będącej jedną z substancji jak i z żółcią.
najsilniej zwiększających ciśnienie krwi. Dzięki temu
dochodzi do obniżenia obwodowego oporu naczyń Dawkowanie. Zazwyczaj stosowane dawki dobowe
(angiotensyna działa nie tylko wyłącznie bezpośred- inhibitorów konwertazy angiotensyny w leczeniu
nio obkurczająco naczynia, ale także pośrednio po- nadciśnienia zawarte są w tab. B 4.2-10. U pacjentów
przez uwalnianie katecholamin z nadnerczy, ułatwia- z aktywowanym układem renina-angiotensyna-aldo-
nie uwalniania noradrenaliny z zakończeń nerwów steron (np. wskutek leczenia lekami moczopędnymi,
współczulnych oraz podwyższanie napięcia układu dużej utraty wody lub soli bądź ciężkiej niewydolno-
współczulnego poprzez ośrodkowe działanie na area ści serca) może dojść w przypadku podawania inhibi-
postrema). Oprócz tego wskutek zmniejszonego wy- torów konwertazy angiotensyny do znacznego obni-
twarzania angiotensyny II dochodzi do zmniejszenia żenia ciśnienia krwi. W takich przypadkach leczenie
uwalniania aldosteronu i w związku z tym do wy- należy rozpoczynać od niskich dawek i podwyższać
stąpienia słabego efektu diuretycznego. Inhibitory je stopniowo.
konwertazy angiotensyny opóźniają także rozkład
rozszerzających naczynia kinin, ponieważ odpowie- Działania niepożądane. Wśród działań niepożąda-
dzialny za ich biotransformację enzym kininaza II nych opisuje się przede wszystkim suchy kaszel (u
jest identyczny z konwertazą angiotensyny. Wzrost około 5–10% leczonych), a także zaburzenia sma-
stężenia tych peptydów (bradykininy i kallidyny) ku, bóle głowy, nudności, zawroty głowy, niedociś-
wspomaga działanie obniżające ciśnienie krwi. nienie, biegunkę, kurcze mięśniowe, nadwrażliwość
na światło i skórne odczyny alergiczne. Do cięż-
Farmakokinetyka. Za wyjątkiem kaptoprilu i lizyno- szych, choć rzadszych działań niepożądanych zalicza
prilu, które działają bezpośrednio, pozostałe inhibito- się niebezpieczeństwo wystąpienia osterej niewydol-
ry konwertazy angiotensyny są prolekami, z których ności nerek, obrzeków naczynioruchowych, a także
dopiero po hydrolizie powstają właściwe substancje leukopenii. Jeśli chodzi o te działania niepożądane,
czynne (benazaprilat, cilazaprilat, enalaprilat, fozy- to najbardziej narażeni są pacjenci z niewydolnością
noprilat, meksyprilat, perindoprilat, chinaprilat, rami- nerek i dlatego powinni być oni szczególnie dokład-
prilat, spiraprilat, trandolaprilat). Jeżeli niemożliwe nie monitorowani.
jest leczenie preparatami doustnymi, bądź też istnieje
podejrzenie niewystarczającej biodostępności, do- Przeciwwskazania. Inhibitory konwertazy angioten-
stępny jest preparat enalaprilatu do podawania dożyl- syny są przeciwwskazane u pacjentów z obustronnym
nego. Lizynopril wchłania się po podaniu doustnym zwężeniem tętnicy nerkowej, bądź ze zwężeniem
w ok. 25%, natomiast pozostałe związki wchłaniają tętnicy nerkowej jedynej nerki, pacjenci po prze-
się szybko i w dużym odsetku. Z powodu gwałtownej szczepie nerki, chorujący na pierwotny hiperaldo-
dysocjacji z enzymu oraz krótkiego okresu półtrwa- steronizm, a także w trakcie ciąży i karmienia pier-
nia w osoczu kaptopril działa krótko (kilka godzin). sią. Do względnych przeciwwskazań należą ciężkie
Pozostałe inhibitory konwertazy angiotensyny mają choroby autoimmunologiczne i kolagenozy. Leki te
z kolei tak długi okres działania wynikający z powol- należy również ostrożnie stosować u pacjentów z ob-
nej dysocjacji z połączenia z enzymem docelowym, turacyjnymi chorobami płuc.
że mimo krótkich okresów półtrwania w osoczu wy-
starcza ich podanie w jednej dawce dobowej. Interakcje. Z powodu niebezpieczeństwa wystąpie-
Wydalanie zachodzi w przypadku większości nia hiperkaliemii inhibitory konwertazy angiotensy-
związków z tej grupy głównie przez nerki, fozynopril ny nie powinny być – przynajmniej w praktyce ogól-
MUTSCHLER-2009.indd 574 2010-01-07 22:13:39
Układ naczyniowy i krążenie 575
Tab. B 4.2-10. Inhibitory konwertazy angiotensyny
Wzór strukturalny Nazwa Preparat handlowy Okres Dawkowanie
międzynarodowa półtrwania (mg)
CH3 kaptopril ACE-Hemmer- 2 12,5-75
HS N ratiopharm,
Captobeta,
O COOH
Lopirin Cor,
tensobon u.a.
H5C2O O enalapril Benalapril, 11 5-20
CH3 EnaHEXAL,
N Enalapril-ratiopharm,
NH XANEF u.a.
O COOH
NH2 lizynopril Acerbon, 12 10-20
LisiHEXAL,
HO O Lisinopril-ratiopharm,
Lisinopril STADA u.a.
N
NH
O COOH
Serce i układ krążenia
perindopril Coversum 5 2-8
H5C2O O
CH3 H H
N
H7C3 NH
O COOH
trandolapril Udrik 16-24 1-4
H5C2O O
CH3 H H B4
N
NH
O COOH
ramipril Delix, 15 2,5-10
H5C2O O Ramipril-beta,
CH3 H H Ramipril HEXAL,
N Vesdil u.a.
NH
O COOH
spirapril Quadropril 40 3-6
S
H5C2O O
CH3 S
N
NH
O COOH
O CH(CH3) 2 fozynopril dynacil, 11,5 10-40
Fosinorm
H5C2 O O O
P
N
O
COOH
MUTSCHLER-2009.indd 575 2010-01-07 22:13:39
576 Układ naczyniowy i krążenie
Tab. B 4.2-10. Inhibitory konwertazy angiotensyny (kontynuacja)
Wzór strukturalny Nazwa Preparat handlowa Okres Dawkowanie
międzynarodowa półtrwania (mg)
chinapril Accupro, 3 10-40
Quinapril AL,
H5C2O O Quinapril beta,
CH3
Quinapril HEXAL u.a.
N
NH
O COOH
OCH3 meksypril Fempress 30 7,5-30
OCH3
H5C2O O
CH3
N
NH
O COOH
benazepril Benazepril AL, 11 10-40
H5C2O O Benazepril beta,
Benazepril HEXAL,
N COOH Cibacen u.a.
NH
O
H5C2O O cilazapril Dynorm 9 1,25-5
N
N
NH
O COOH
nej – łączone z diuretykami oszczędzającymi potas. przynajmniej po części wynikają z zahamowania rozkładu
Inhibitory syntezy prostaglandyn (niesteroidowe leki bradykininy. Oprócz tego angiotensyna I może być przekształ-
przeciwzapalne) zmniejszają hipotensyjne działanie cana do angiotensyny II przez inne, częściowo miejscowe
układy enzymatyczne (np. chymazy lub chymostatin like an-
inhibitorów konwertazy angiotensyny, chociaż małe giotensin generating enzyme, CAGE). Angiotensyna II może
dawki kwasu acetylosalicylowego nie wpływają też powstać bezpośrednio z angiotensynogenu np. dzięki t-
na ich działanie. Środki znieczulenia ogólnego z ko- PA. Dochodzi wtedy do zjawiska tzw. Angiotensin-Escape-
lei nasilają hipotensyjny efekt inhibitorów konwerta- Phenomen, kiedy to pomimo blokady enzymu konwertują-
zy angiotensyny. cego angiotensynę powstające ilości angiotensyny II mogą
wywierać działanie farmakologiczne. Związki, które działają
na dalszych etapach układu renina-angiotensyna-aldosteron
niż enzym konwertujący angiotensynę, czyli na receptory dla
4.2.3.2.5. Antagoniści receptora angiotensyny II angiotensyny II powinny umożliwić z jednej strony zmniej-
(antagoniści AT1, sartany) szenie działań niepożądanych wynikających z hamowania
rozkładu bradykininy, a z drugiej strony zahamować cały
układ skuteczniej niż inhibitory konwertazy angiotensyny.
Układ renina-angiotensyna-aldosteron może być ha- Dotychczasowe wyniki leczenia za pomocą antagonistów
mowany nie tylko przez inhibitory konwertazy angio- receptora angiotensyny II potwierdziły te założenia.
tensyny dzięki hamowaniu przekształcenia angioten-
syny I w angiotensynę II, ale także przez antagoni-
Receptory angiotensyny II. Jak przedstawiono
stów receptora angiotensyny II.
na ryc. B 4.2-7 angiotensyna II działa za pośredni-
Rozwój tej grupy związków nastąpił na podstawie następują- ctwem receptorów dwóch typów – AT1 i AT2. Podczas
cych założeń: w przypadku terapii inhibitorami konwertazy gdy receptor AT1 odpowiedzialny jest za podwyższa-
angiotensyny, jak napisano powyżej, często dochodzi do wy- nie ciśnienia krwi i proliferację komórek, pobudzenie
stąpienia działań niepożądanych (m.in. suchy kaszel), które receptora AT2 hamuje proliferację.
MUTSCHLER-2009.indd 576 2010-01-07 22:13:39
Układ naczyniowy i krążenie 577
Antagoniści receptora AT1 (sartany). Terapeutycznie równaniu z inhibitorami konwertazy angiotensyny
przydatni są antagoniści, którzy blokują wybiórczo ze względu na fakt, że nie jest blokowany enzym
receptory angiotensyny typu 1. Związki te, zwane rozkładający bradykininę. Większość działań niepo-
sartanami zostały zsyntezowane jako związki naśla- żądanych występuje na poziomie placebo. Rzadko
dujące peptydy będące częściowymi agonistami lub dojść może do wystąpienia senności, zawrotów gło-
antagonistami tego receptora. Pierwszą substancją wy, zaparć, hiperkaliemii, podwyższenia poziomu
wprowadzoną do leczenia w 1995 roku był losartan. transaminaz lub kreatyniny.
Wkrótce potem na rynek weszły kolejne leki, które
mają podobne działanie, a różnią się jedynie farma- Przeciwwskazaniem do stosowania antagonistów AT1
kokinetyką: są ciężka niewydolność nerek i wątroby, kardiomio-
patia przerostowa, a także ciąża i karmienie piersią.
■ irbesartan,
■ kandesartan, Obecnie antagoniści receptora AT1 są stosowani za-
równo u chorych przy rozpoczęciu terapii jak i u pa-
■ walsartan,
cjentów, którzy nie tolerują inhibitorów konwertazy
■ telmisartan, angiotensyny. To czy i jak duża jest przewaga terapeu-
tyczna wybiórczej blokady receptorów angiotensyny
■ olmesartan,
II nad hamowaniem konwertazy angiotensyny pozo-
■ eprosartan. staje wciąż dyskusyjne. W kontekście opisanego wyżej
Angiotensin-Escape-Phenomen przy leczeniu inhibi-
Serce i układ krążenia
Wszyscy antagoniści receptora AT1 są wskazani w le- torami konwertazy angiotensyny wciąż dyskutowane
czeniu nadciśnienia pierwotnego. Losartan i walsar- i badane są połączenia w terapii antagonistów recepto-
tan są również stosowane w leczeniu niewydolności ra AT1 z inhibitorami konwertazy angiotenysyny.
serca.
Do najnowszych odkrytych substancji wpływających na układ
Działania niepożądane takie jak suchy kaszel lub renina-angiotensyna-aldosteron należy bezpośredni inhibitor
reniny – aliskiren. Należy oczekiwać niedługo ostatecznych
obrzęk naczyniowy zgodnie z oczekiwaniami wystę- wyników badań jego przydatności terapeutycznej.
pują rzadziej podczas leczenia tą grupą leków w po-
B4
ciśnienie perfuzji w nerce
angiotensynogen wydzielanie reniny hormony układu współczulnego
produkty rozpadu bradykininy angiotensyna I
ACE inhibitory ACE
antagoniści AT1
bradykinina
angiotensyna II
BK AT2 AT1
Gq Gi Gq
rozszerzenie naczyń obwodowych, hamowanie proliferacji, skurcz naczyń: bezpośredni, pośredni
wzrost przepuszczalności naczyń włosowatych, rozszerzenie naczyń (aktywacja układu współczulnego)
pobudzenie odruchu kaszlowego, retencja jonów sodu
obrzęk naczynioruchowy (rzadko) (pobudzenie wydzielania aldosteronu)
proliferacja
(hiperplazja)
Ryc. B 4.2-7. Punkty uchwytu inhibitorów ACE oraz antagonistów receptora AT1. BK receptor bradykininowy.
MUTSCHLER-2009.indd 577 2010-01-07 22:13:40
578 Układ naczyniowy i krążenie
Tab. B 4.2-11. Antagoniści receptora AT1
Wzór strukturalny Nazwa Preparat handlowy Okres Dawkowanie
międzynarodowa półtrwania (mg)
Cl
N lozartan LORZAAR 2 50-100
HOH2C CH3 Aktiver
N
Metabolit
6-9
N
N
N NH
irbezartan Aprovel, 11-15 150-300
N
Karvea
O CH3
N
N
N
N NH
N kandezartan Atacand, 9 8-16
O CH3
(jako cyleksetyl) Blopress
N
HOOC
N
N
N NH
(H3C) 2HC O
walzartan Cordinate, 9 80-160
CH3
HOOC N DIOVAN,
Provas
N
N
N NH
HO CH3
H3C olmezartan Olmetec, 12-18 10-40
N (jako medoksomil) Votum
O
N CH3
H3C O
O
O
O
N
N
N NH
CH3
telmizartan Kinzalmono, 24 20-80
N
N Micardis
N N CH3
CH3
HOOC
eprozartan Emestar Mono, 5-9 600
S N (jako mesylat) Teveten Mono
CH3
HOOC N
COOH
MUTSCHLER-2009.indd 578 2010-01-07 22:13:40
Układ naczyniowy i krążenie 579
4.2.3.2.6. Inne leki rozszerzające naczynia hydralazyny, która zawiera tylko jedną grupę hydra-
krwionośne przez bezpośredni zynową. Pomimo obniżania ciśnienia krwi zwiększa-
wpływ na mięśnie gładkie ją one poziom ukrwienia nerek, choć stopień filtra-
cji kłębuszkowej pozostaje niezmieniony wskutek
zmniejszonego ciśnienia filtracyjnego.
Do tej grupy substancji należą:
Dihydralazyna wchłania się po podaniu doustnym
■ dihydralazyna i hydralazyna, bardzo szybko, jednak podlega acetylacji w efekcie
pierwszego przejścia, która zachodzi w różnym stop-
■ nitroprusydek sodu,
niu u szybkich i wolnych acetylatorów. W zależności
■ cykletanina, od uwarunkowego genetycznie typu acetylacji okres
półtrwania wynosi między 1 a 8 godzin, czas dzia-
■ lek otwierający kanał potasowy – minoksydyl.
łania około 6–8 godzin, a biodostępność u szybkich
acetylatorów 22–30%. W przypadku wolniej acetylu-
Leki te działają poprzez wpływ na małe tętnice
jących jest ona podwyższona do 38–50%.
i tętniczki obniżając obwodowy opór naczyniowy,
Hydralazyna ze względu na działania niepożą-
a co za tym idzie ciśnienie krwi. Ponadto niektóre
dane (zob. niżej) jest w większości przypadków sto-
z nich zwiększają także objętość żylnych naczyń po-
sowana w preparatach w połączeniu z innymi leka-
jemnościowych.
mi przeciwnadciśnieniowymi (np. z propranololem
Mechanizm działania dihydralazyny i hydralazyny
i bendroflumetiazydem, lub metoprololem i hydro-
wciąż jest niewyjaśniony – rozważany jest hamujący
chlorotiazydem). Umożliwia to obniżenie pojedyn-
Serce i układ krążenia
wpływ na zależne od IP3 uwalnianie wapnia z retiku-
czej dawki z 25 do 10 mg. 90% dawki jest wydalane
lum endoplazmatycznego. Działanie nitroprusydku
przez nerki w postaci metabolitów hydrazynowych.
sodu polega na nagłym uwalnianiu NO (zob. ryc. B
Bez określenia u pacjenta typu acetylacji nie powinno
4.2-3). Minoksydyl zwiększa prawdopodobieństwo
się stosować dawek wyższych niż 50 mg.
otwarcia zależnych od ATP kanałów potasowych,
Działania niepożądane w przypadku obu tych leków
zwiększając w ten sposób potencjał spoczynkowy
mogą wynikać z obniżenia ciśnienia krwi co prowadzi
błony komórkowej (hiperpolaryzacja), co prowadzi
do aktywacji układu współczulnego a także układu re-
do zmniejszenia napływu jonów wapnia poprzez
nina-angiotensyna-aldosteron i wystąpienia wskutek
napięciowozależne kanały wapniowe. Zmniejszone
tego tachykardii, wzrostu pojemności minutowej ser-
w ten sposób stężenie wewnątrzkomórkowych jo-
ca oraz retencji jonów sodu i wody z tworzeniem się
B4
nów wapnia prowadzi przede wszystkim w arterio-
miejscowych obrzęków. Oprócz tego wystąpić mogą
lach do zmniejszenia się napięcia mięśniówki gład-
bóle i zawroty głowy, osłabienie, nudności, zaburzenia
kiej i wskutek tego do obniżenia ciśnienia krwi.
żołądkowo jelitowe i biegunki. Obserwowano również
Dihydralazyna, hydralazyna i minoksydyl stosowane
wystąpienie reakcji alergicznych.
są doustnie w przewlekłym leczeniu podwyższonego
ciśnienia tętniczego. Nitroprusydek sodu natomiast W przypadku długotrwałego stosowania dihydralazyny
stosowany jest jedynie w leczeniu przełomów nad- w dużych dawkach może dojść do rozwinięcia się reumatoi-
ciśnieniowych (zob. niżej). dalnego zapalenia stawów. Jeżeli leczenie nie zostanie wte-
dy przerwane istnieje niebezpieczeństwo, że, szczególnie
Wśród pochodnych hydrazynoftalazyny istotne zna- u osób wolno acetylujących, a także u posiadaczy antygenu
HLA-DR4, dojść może do rozwinięcia się zespołu przy-
czenie hydralazyny uzyskała dihydralazyna obok pominającego toczeń rumieniowaty, który po odstawieniu
leku można leczyć przy użyciu hormonów kory nadnerczy.
Występujące niekiedy parestezje i zapalenia nerwów obwo-
dowych wynikają z antagonistycznego działania dihydrala-
zyny względem witaminy B6 mogą być skutecznie leczone
NH2 przy pomocy tej witaminy.
HN
N N Nitroprusydek sodu (disodu pentacyjanonitrozyl żelaza
N N
(II)) jest kompleksowym związkiem nieorganicznym wraż-
liwym na światło, który dzięki uwalnianiu NO powoduje
HN HN rozszerzenie tętniczek przedwłosowatych oraz zawłosowa-
NH2 NH2 tych żyłek. Substancja ta może być podawana wyłącznie
w postaci ciągłego wlewu dożylnego (najlepiej w postaci
pompy) przy ścisłym monitorowaniu pacjenta, a efekt ob-
hydralazyna dihydralazyna niżający ciśnienie występuje bezpośrednio po rozpoczęciu
wlewu. Efekt jest ściśle zależny od dawki, a ciśnienie krwi
MUTSCHLER-2009.indd 579 2010-01-07 22:13:41
580 Układ naczyniowy i krążenie
można obniżyć do każdej założonej wartości. Ze względu a następnie wydalenie przez nerki, a czas półtrwania
na bardzo krótki czas działania tuż po spowolnieniu bądź wynosi 6–8 godzin.
zakończeniu wlewu ciśnienie krwi ponownie wzrasta, dzię-
ki czemu jego poziom można bardzo łatwo regulować. Lek
ten nie wpływa na układ wegetatywny. Minoksydyl jest to pochodna piperydynodiaminopi-
Nitroprusydek sodu jest stosowany w leczeniu przełomów rymidyny, która działa hipotensyjnie silniej i dłużej
nadciśnieniowych (zob, niżej) oraz podczas określonych niż np. dihydralazyna czy hydralazyna. Ze względu
zabiegów chirurgicznych (np. w operacjach w obrębie gło- jednak na poważne działania niepożądane jest on
wy i gardła, w chirurgii naczyniowej) w celu regulowanego
wskazany w leczeniu nadciśnienia wyłącznie u pa-
obniżania ciśnienia krwi.
Średnie dawkowanie wynosi 3 μg/kg masy ciała na minutę, cjentów, u których inne leki przeciwnadciśnieniowe
maksymalna dawka wynosi 800 μg/min. – także stosowane w kombinacjach – nie były wystar-
W przypadku zbyt szybkiego wlewu może dojść wskutek czająco skuteczne. W takich przypadkach ze względu
nadmiernego spadku ciśnienia krwi do utraty świadomości na wyraźnie występujący efekt przeciwregulacyjny
i utraty tętna. Do objawów przedawkowania podostrego na-
(pobudzenie układu współczulnego, retencja sodu
leżą nudności, wymioty, kurczowe bóle brzucha oraz zanik
odruchów. i wody) minoksydyl musi być stosowany razem z le-
Należy także zaznaczyć, że ostre przedawkowanie nitropru- kiem moczopędnym oraz β-adrenolitykiem lub cen-
sydku sodu prowadzić może do zatrucia cyjankami. W or- tralnie działającym α2-sympatykomimetykiem.
ganizmie z nitroprusydku sodu oprócz NO uwalniają się
bowiem także cyjanki, które pod wpływem odpowiedniej
syntetazy przekształcane są w stukrotnie mniej toksyczne
rodanki (tiocyjanki). W przypadku zbyt wielkiego obcią-
żenia cyjankami możliwości przerobowe tego enzymu
są przekroczone i dochodzi do wzrostu ich stężenia. NH2
W przypadku długotrwałego leczenia nitroprusydkiem O
sodu (więcej niż 2 dni) istnieje również niebezpieczeństwo N N
kumulacji rodanków, wskutek czego dojść może do wy-
stąpienia zaburzeń mowy, osłabienia mięśni oraz reakcji H2N N
psychotycznych. W celu uniknięcia tych działań niepożą-
danych należy w takim przypadku kontrolować stężenie
rodanków w surowicy krwi. minoksydyl
Cykletanina to lek hipotensyjny, którego mecha-
nizm działania nie jest do końca wyjaśniony. Oprócz
pobudzenia śródbłonkowych receptorów muska-
rynowych i uwalniania tlenku azotu powoduje ona
rozszerzenie naczyń krwionośnych prawdopodobnie Po podaniu doustnym minoksydyl jest wchłania-
wskutek hamowania napięciowozależnych kanałów ny prawie w całości. Czas półtrwania w osoczu krwi
wapniowych. W przypadku nadciśnienia pierwotne- wynosi ok. 4 godzin a efekt obniżający ciśnienie krwi
go cykletaninę stosuje się w dawkach od 50 do mak- utrzymuje się powyżej 24 godzin. Najważniejszym
symalnie 200 mg. Co interesujące po podaniu tego ilościowo metabolitem jest glukuronid. Oprócz tego
związku akcja serca raczej się zmniejsza niż zwięk- minoksydyl jest w wątrobie przekształcany do siar-
sza. Eliminacja cykletaniny zachodzi poprzez koniu- czanu minoksydylu, który jest w największej mierze
gację z resztami kwasu glukuronowego i siarkowego, odpowiedzialny za działanie. Wydalanie metabolitów
zachodzi głównie przez nerki.
Dawka dobowa dla dorosłych wynosi zazwyczaj
od 5 do 40 mg. Leczenie rozpoczyna się od daw-
ki 5 mg dziennie i podwyższa następnie powoli aż
O do osiągnięcia wymaganego ciśnienia krwi (dawka
HO maksymalna dobowa wynosi 100 mg).
Szczególnie istotnym działaniem niepożądanym
H3C N Cl jest występujący najczęściej u pacjentów leczonych
minoksydylem nadmierny porost włosów. Zwykle
rozpoczyna się ona na twarzy i ustępuje w ciągu kil-
cykletanina ku miesięcy po odstawieniu leku. Oprócz tego w 3%
przypadków przede wszystkim u pacjentów z upośle-
dzoną czynnością nerek obserwuje się obecność pły-
nu w worku osierdziowym.
MUTSCHLER-2009.indd 580 2010-01-07 22:13:41
Układ naczyniowy i krążenie 581
4.2.3.3. Schemat postępowania nie osiągnie się zadowalającego efektu hipotensyjne-
w leczeniu nadciśnienia go wprowadza się do leczenia drugi lek, przy czym
z reguły jako jeden ze składników leczenia stosowa-
ny jest lek moczopędny lub antagonista kanału wap-
Obecnie przyjmuje się zasadę indywidualnego podej-
niowego. W przypadku kombinacji antagonisty kana-
ścia w leczeniu nadciśnienia, które powinno zgodnie
łu wapniowego z β-blokerem preferowane są długo-
z planem stopniowo zapewnić normalizację ciśnienia
działające pochodne dihydropirydyny.
krwi (< 140/90; u cukrzyków < 130/80). Na ryc. B
Jeżeli leczenie dwuskładnikowe w dalszym ciągu
4.2-8 przedstawiono schemat postępowania opra-
nie daje wystarczającego efektu wskazana jest tera-
cowany przez Niemieckie Towarzystwo do Walki
pia trójskładnikowa:
z Nadciśnieniem.
W przypadku łagodnego lub umiarkowanego nad- ■ lek moczopędny + β-bloker + lek rozszerzający na-
ciśnienia leczenie rozpoczyna się od monoterapii czynia,
przy pomocy β-blokera, inhibitora konwertazy an-
■ lek moczopędny + inhibitor konwertazy angioten-
giotensyny, antagonisty receptora AT1, leku moczo-
syny (lub antagonista AT1) + antagonista kanału
pędnego lub antagonisty kanału wapniowego (leki
wapniowego,
przeciwnadciśnieniowe pierwszego wyboru). β-blo-
kery i inhibitory konwertazy angiotensyny stosuje się ■ lek moczopędny + α2-sympatykomimetyk + lek
zwykle u pacjentów poniżej 55 roku życia, natomiast rozszerzający naczynia.
powyżej tego wieku stosuje się głównie leki moczo-
Serce i układ krążenia
pędne i antagonistów kanału wapniowego. Dzięki leczeniu trójskładnikowemu można osiągnąć
Jeżeli przy zastosowaniu zwykłych dawek dane- satysfakcjonujący poziom ciśnienia krwi w 90–95%
go leku w monoterapii, oraz po zamianie go na inny, przypadków. Pacjenci, u których nie udało się osiąg-
monoterapia
antagonista
B4
β-bloker diuretyk kanału wapniowego inhibitor ACE antagonista AT1
leczenie dwuskładnikowe
diuretyk
plus
antagonista
β-bloker lub kanału wapniowego lub inhibitor ACE lub antagonista AT1
antagonista
kanału wapniowego
plus
β-bloker lub inhibitor ACE lub antagonista AT1
Ryc. B 4.2-8. Schemat stopniowego leczenia nadciśnienia.
MUTSCHLER-2009.indd 581 2010-01-07 22:13:42
582 Układ naczyniowy i krążenie
nąć wystarczającego obniżenia ciśnienia krwi powin- ■ Następnie można podać dożylnie 25 mg urapidy-
ni być kierowani do specjalistycznych klinik, gdzie lu, którego działanie występuje w ciągu 10 minut.
do leczenia zostaną włączone dodatkowe leki, np. mi- Ze względu na znaczne obniżenie ciśnienia krwi
noksydyl. należy liczyć się z możliwością wystąpienia takich
Ponieważ, jak już wspomniano, większość pa- działań niepożądanych jak bóle głowy i palpitacje.
cjentów z nadciśnieniem nie odczuwa związanych
z nim subiektywnych dolegliwości, a po rozpoczęciu Na koniec można podać klonidynę (w dawce 0,075 mg)
leczenia farmakologicznego występują u nich efekty powoli dożylnie, która również działa w ciągu 10 mi-
niepożądane wskutek czego ich samopoczucie polega nut. Jako działanie niepożądane może wystąpić nasi-
pogorszeniu, bardzo ważne są dokładne wyjaśnienia lona sedacja.
i sprawdzenie przestrzegania przez pacjenta zaleceń
lekarskich (compliance). W celu osiągnięcia odpo- Przełom nadciśnieniowy oprócz postępowania wstęp-
wiedniego poziomu przestrzegania przez pacjenta nego wymaga natychmiastowego przekazania pacjen-
zaleceń lekarskich sensowne jest, żeby ograniczyć ta na oddział stacjonarny, gdzie stosuje się również
w miarę możliwości ilość przyjmowanych dziennie wymienione wyżej leki. W przypadku braku zadowa-
przez pacjenta leków dzięki stosowaniu preparatów lającego efektu bądź też ponownego nagłego wzrostu
łączonych. ciśnienia tętniczego podaje się w ciągłym wlewie do-
żylnym nitroglicerynę bądź alternatywnie klonidynę,
urapidyl lub dihydralazynę, w warunkach intensyw-
4.2.3.4. Leczenie nadciśnienia nego nadzoru. Jeżeli nie ma przeciwwskazań można
w sytuacjach nagłych dodatkowo podać dożylnie 20–40 mg furosemidu.
Jeżeli działania te nadal nie przynoszą rezultatu,
a nie można z całą pewnością wykluczyć guza chro-
O sytuacji nagłej w przypadku nadciśnienia mówi-
mochłonnego nadnerczy, poleca się podjąć jeszcze
my wówczas, gdy wskutek podwyższonego ciśnie-
jedną próbę leczenia za pomocą 25 mg urapidylu i.v.
nia krwi dojść może do sytuacji zagrożenia życia
a następnie podać fenoksybenzaminę.
i konieczne jest natychmiastowe jego obniżenie. Taka
sytuacja występuje w przypadku przełomu nadciś-
nieniowego czyli nagłego dużego wzrostu ciśnienia
skurczowego i rozkurczowego przy prawidłowych 4.2.3.5. Leczenie nadciśnienia w ciąży
lub podwyższonych wartościach wyjściowych a tak- i w trakcie karmienia piersią
że w przypadku komplikacji przewlekłego nadciś-
nienia takich jak np. krwotok mózgowy lub ostra Podwyższone ciśnienie krwi u kobiety ciężarnej
niewydolność lewokomorowa z obrzękiem płuc. (stwierdzone w kilku pomiarach po 20. tygodniu
Według zaleceń Niemieckiego Towarzystwa do Walki ciąży ciśnienie krwi przekraczające 140/90) oznacza
z Nadciśnienienim lekarz pogotowia ratunkowego zwiększone zagrożenie dla płodu. Jeżeli wartość ciś-
lub rodzinny powinien w takim przypadku leczyć nienia skurczowego przekracza 170 mmHg a rozkur-
podwyższone ciśnienie za pomocą następujących czowego 110 mmHg oznacza to znacznie podwyż-
leków (aczkolwiek nie ma dostępnych badań porów- szone ryzyko śmiertelności okołoporodowej. Z tego
nawczych dotyczących ich skuteczności): powodu w okresie ciąży wskazane jest leczenie nad-
ciśnienia. Jednak ze względu na niebezpieczeństwo
■ Na początku podać 1,2 mg nitrogliceryny w spray’u
zmniejszenia się perfuzji maciczno-łożyskowej ciś-
lub w kapsułce do rozgryzania. Początek działania
nienie należy zmniejszać ostrożnie (docelowo < 140
występuje w ciągu kilku minut (nitrogliceryna
mmHg ciśnienie skurczowe, < 90 mmHg rozkurczo-
jest oprócz tego lekiem z wyboru w przypadku
we).
obrzęku płuc, niestabilnej dusznicy bolesnej oraz
Do leków przeciwnadciśnieniowych szczególnie
zawału serca).
polecanych w ciąży należy α-metylodopa, selektywni
■ Kolejną opcją jest doustne podanie 5 mg nifedy- antagoniści receptora β1 adrenergicznego (np. ateno-
piny lub nitrendypiny w postaci szybkowchłanial- lol, bisoprolol lub metoprolol). Można również roz-
nej. Również w tym przypadku działanie powinno ważyć zastosowanie dihydralazyny.
rozpocząć się w przeciągu kilku minut (jednakże Przeciwwskazane są leki moczopędne ze wzglę-
w przypadku niestabilnej dusznicy bolesnej i zawa- du na zmniejszanie objętości krwi krążącej i przez
łu serca antagoniści kanału wapniowego są prze- to pogarszanie ukrwienia maciczno-łożyskowego,
ciwwskazani). inhibitory konwertazy angiotensyny oraz antagoniści
MUTSCHLER-2009.indd 582 2010-01-07 22:13:42
Układ naczyniowy i krążenie 583
receptora AT1 również ze względu na zmniejszenie 4.2.4. Leczenie nadciśnienia
ukrwienia maciczno-łożyskowego oraz tworzenia płucnego
wód płodowych oraz antagoniści kanału wapniowe-
go ze względu na działanie embriotoksyczne i terato-
Wszystkie podtypy nadciśnienia płucnego, a szcze-
genne wykazane w próbach na zwierzętach.
gólnie nadciśnienie tętnicy płucnej (pulmonary artery
W przypadku związanych z podwyższonym ciś-
hypertension – PAH), rozpoznaje się wówczas, gdy
nieniem sytuacji nagłych w ciąży można podać dihy-
w spoczynku średnie ciśnienie krwi mierzone w tęt-
dralazynę (5 mg i.v.) lub urapidyl (6,25 mg i.v.).
nicy płucnej wynosi > 25 mmHg, a w trakcie wysiłku
W przypadku gotowości skurczowej można dodatko-
> 30 mmHg.
wo podać siarczan magnezu (4 g i.v.) lub diazepam
Czynniki wyzwalające są w dalszym ciągu niezna-
(5–10 mg i.v.).
ne. Jeśli chodzi o patofizjologię, to w PAH dochodzi
W leczeniu nadciśnienia w okresie karmienia pier-
do proliferacji śródbłonka i mięśniówki gładkiej na-
sią podawane mogą z wyjątkiem leków moczopęd-
czyń (tzw. remodeling naczyniowy), przewagi czyn-
nych wszystkie leki stosowane w ramach zwykłego
ników naczynioskurczowych oraz wzmożonej skłon-
leczenia nadciśnienia. W przypadku leczenia β-blo-
ności do tworzenia zakrzepów przy równocześnie
kerami należy mieć na uwadze fakt, że mogą one
zmniejszonej aktywności fibrynolitycznej w płucnym
osiągnąć wysokie stężenie w organizmie dziecka
łożysku naczyniowym. Dlatego wydaje się słuszne,
i w niektórych przypadkach doprowadzić do spadku
żeby w przeciwieństwie do innych postaci nadciś-
ciśnienia lub zmniejszenia akcji serca.
nienia opisywać nadciśnienie płucne jako waskulo-
patię proliferacyjną. Wynikające z niej zmniejszenie
Serce i układ krążenia
światła naczyń prowadzi do podwyższenia ciśnienia
4.2.3.6. Leczenie nadciśnienia w krążeniu małym, wzrostu obciążenia następczego
u pacjentów z cukrzycą w prawej komorze i w końcu do niewydolności pra-
wego serca.
Cukrzyca i nadciśnienie często występują wspólnie Nadciśnienie płucne jest ciężkim stanem chorobo-
i działają synergistycznie w odniesieniu do wystę- wym, który związany jest ze złymi rokowaniami (pię-
powania incydentów sercowo naczyniowych. Chory cioletnie przeżycie 30–50% chorych). Wyleczenie
na cukrzycę i nadciśnienie ma w porównaniu do oso- możliwe jest tylko dzięki transplantacji.
by z grupy kontrolnej czterokrotnie wyższe ryzyko Leczenie farmakologiczne nadciśnienia płucne- B4
zachorowalności i śmiertelności sercowo-naczynio- go polega na próbie podwyższenia objętości naczyń
wej. Dlatego u chorych na cukrzycę nadciśnienie płucnych poprzez stosowanie leków rozszerzających
musi być leczone farmakologicznie. Wartości doce- naczynia co ma poprawić wydolność serca. W lecze-
lowe wynoszą < 130 mmHg dla ciśnienia skurczo- niu stosuje się:
wego i < 80 mmHg dla rozkurczowego szczególnie
■ iloprost,
jeśli występuje mikroalbuminuria lub nefropatia cuk-
rzycowa. Aby osiągnąć takie wartości ciśnienia pra- ■ antagonistów endoteliny,
wie zawsze wymagane jest stosowanie kombinacji
■ antagonistów PDE5.
leków. Szczególnie wskazane w tej grupie pacjentów
jako leki w kombinacji są inhibitory konwertazy an-
Iloprost jest to pochodna prostacykliny, która jest sto-
giotensyny, w przypadku których wykazano w wielu
sowana w leczeniu pierwotnego nadciśnienia płucne-
badaniach, że mogą spowolnić progresję nefropatii
go. Okres półtrwania wynosi 20 do 30 minut, dawka
cukrzycowej. Istnieją też wskazówki, że również an-
pojedyncza 2,5 do 5 μg podawana w aerozolu
tagoniści receptora AT1 mają efekt nefroprotekcyjny,
a w porównaniu z β-blokerami przy podobnym obni-
W grupie antagonistów endoteliny jako pierwszy
żeniu ciśnienia krwi zmniejszają częstość występo-
w leczeniu nadciśnienia płucnego został wprowa-
wania objawów cukrzycy.
dzony bozentan. Jest to niewybiórczy podwójny
Również u pacjentów z zespołem metabolicznym
antagonista receptorów endotelinowych ETA i ETB.
szczególnie ważne jest staranne ustawienie odpo-
Jego biodostępność wynosi ok. 50%, wiązanie się
wiedniego ciśnienia krwi.
z białkami osocza powyżej 98% a końcowy okres
półtrwania około 5,5 godziny. Bozentan metaboli-
zowany jest w wątrobie za pośrednictwem CYP3A4
oraz CYP2C9. Dzięki zjawisku autoindukcji enzymy
metabolizujące w trakcie przewlekłego stosowania
MUTSCHLER-2009.indd 583 2010-01-07 22:13:42
584 Układ naczyniowy i krążenie
enzymów przebiega zwykle bezobjawowo i jest od-
wracalne po redukcji dawki. U niektórych pacjentów
HOOC
obserwuje się także normalizację poziomu enzymów
mimo kontynuowania terapii. Ze względu na induk-
cję CYP3A4 oraz CYP2C9 podawane równocześ-
H H CH3 CH3 nie leki, które są metabolizowane przez te enzymy
C
C są szybciej przerabiane i możliwe jest, że ich dzia-
OH
łanie ulegnie osłabieniu. Należy wówczas rozważyć
OH
odpowiednie zmodyfikowanie dawkowania. W przy-
padku równoczesnego podania leków przeciwzakrze-
iloprost powych takich jak warfaryna lub jej pochodne, któ-
re są metabolizowane przez CYP2C9, należy ściśle
kontrolować INR (International Normalized Ratio).
W przypadku środków antykoncepcyjnych istnieje
bozentanu zwiększają jego klirens (spadek stężenia możliwość utraty ich antykoncepcyjnego działania
w osoczu do 50–65% wartości pierwotnej). Badania wskutek indukcji enzymatycznej. Należy unikać
in vitro wykazują silne hamowanie wątrobowego biał- równoczesnego podawania inhibitorów CYP3A4
ka transportującego, pompy soli kwasów żółciowych i CYP2C9 (np. flukonazolu), ponieważ prowadzić
(bile salt export pump; BSEP;ABCB11). Leczenie to może do wzrostu stężenia bozentanu. Z tego same-
rozpoczyna się od dawkowania 62,5 mg dziennie go powodu przeciwwskazane jest jednoczesne poda-
przez cztery tygodnie, a następnie dawka podtrzymu- wanie cyklosporyny. Z powodu hamującego wpływu
jąca dwa razy dziennie po 125 mg. W celu uniknięcia glibenklamidu na BSEP również ten lek nie powinien
możliwego, chociaż nieudowodnionego, efektu z od- być podawany wraz z bozentanem.
bicia lek ten powinien być odstawiany powoli.
Kolejnym antagonistą endoteliny stosowanym w lecze-
niu nadciśnienia płucnego jest sitaksentan. Jest to sil-
ny i wysoce wybiórczy antagonista receptora ETA.
C2H4OH Lek ten jest metabolizowany przez CYP2C9 oraz
OCH3 O
O
CYP3A4 z końcowym okresem półtrwania wynoszą-
N cym 10 godzin. Całkowita biodostępność sitaksenta-
N nu wynosi od 70 do 100%. Dawkowany jest w ilości
HN N
O S O N
100 mg raz dziennie.
Kolejny sposób leczenia nadciśnienia płucnego pole-
ga na stosowaniu inhibitorów PDE5, których przed-
C(CH3) 3
stawicielem jest pierwotnie stosowany w leczeniu
zaburzeń wzwodu sildenafil. W nadciśnieniu płuc-
nym lek ten podawany jest w ilości 20 mg trzy razy
bozentan dziennie.
Oprócz zastosowania w leczeniu nadciśnienia płucnego bo-
sentan stosowany jest u pacjentów ze sklerodermią w celu H3C O
zmniejszenia ilości owrzodzeń na palcach rąk i stóp. O
S O
Najczęściej występujące działania niepożądane O O N
bosentanu związane są z jego działaniem blokują- S
NH CH3
cym endotelinę (np. bóle głowy, zaczerwienienie). O
Cl
Szczególnym efektem niepożądanym stosowania
bosentanu jest zależny od dawki wzrost poziomu
sitaksentan
aminotransferaz wątrobowych (AspAt i AlAt) praw-
dopodobnie związany z hamowaniem przez niego
białka BSEP (zob. wyżej). Podwyższenie stężenia
MUTSCHLER-2009.indd 584 2010-01-07 22:13:42
Układ naczyniowy i krążenie 585
Nowe perspektywy. Obecnie w nadciśnieniu płucnym naczyń żylnych na wyrzut katecholamin przy zmianie
eksperymentalnie stosowany jest inhibitor kinazy tyro- pozycji, co skutkuje silnym wzrostem akcji serca oraz
zmniejszeniem amplitudy ciśnienia krwi w wyniku pod-
zynowej imatinib. W kontekście wspomnianej wyżej
wyższenia ciśnienia rozkurczowego i obniżenia skurczo-
patogenezy tego schorzenia jako choroby proliferacyj- wego
nej takie zastosowanie tego leku wydaje się słuszne.
■ postać asympatykotoniczną (hipodiastoliczną) z nie-
wielką zmianą akcji serca i spadkiem zarówno ciśnienia
skurczowego jak i rozkurczowego z powodu niewystar-
4.2.5. Leczenie niedociśnienia czającej odpowiedzi układu sympatycznego na bodźce
ortostatyczne.
i ortostatycznych
zaburzeń ciśnienia krwi Niedociśnienie należy leczyć wyłącznie wówczas,
O niedociśnieniu tętniczym (hipotonii) mówi się wtedy, kiedy u pacjenta występują silnie wyrażone utrudnia-
gdy skurczowe ciśnienie krwi w warunkach spoczynkowych jące funkcjonowanie objawy oraz w grupach ryzyka,
jest mniejsze od 100 (110) mmHg. Podobnie jak w przypad- do których zalicza się pacjentów w wieku podeszłym
ku nadciśnienia rozróżnia się kilka jego postaci. (ryzyko udarów mózgowych, omdleń), cukrzyków
W przypadku niedociśnienia pierwotnego (samoistnego, i alkoholików (niebezpieczeństwo silnych zaburzeń
konstytutywnego) jego wartość ustalana jest w ośrodkach
regulacji krążenia. ortostatycznych, a także ciężarne (zwiększone nie-
Niedociśnienie wtórne występuje jako skutek innych bezpieczeństwo poronień, porodów przedwczesnych,
schorzeń. Najbardziej znane są spadki ciśnienia krwi wsku- zahamowania wzrostu płodu, zwiększona śmiertel-
tek zaburzenia regulacji w trakcie i po zakończeniu chorób ność okołoporodowa). W takich przypadkach przed
Serce i układ krążenia
infekcyjnych. Niedociśnienie wtórne może również wyni- rozpoczęciem leczenia farmakologicznego lub w jego
kać z przyczyn:
trakcie należy zalecić pacjentom unikanie szybkiego
■ sercowo-naczyniowych (np. przy niewydolności serca, wstawania zwłaszcza rano po długim pozostawaniu
zapaleniu mięśnia sercowego, zawale serca, zaburzeniach
rytmu serca, hipowolemii, zatorze płucnym)
w pozycji leżącej. Przydatny jest również wysiłek
fizyczny oraz środki fizyczne jak prysznic naprze-
■ endokrynologicznych (np. przy niedoczynności kory nad- miennie ciepłą i zimną wodą, masaż szczotką a tak-
nerczy, niedoczynności tarczycy, niedoczynności przy-
sadki) że – za wyjątkiem ciężarnych! – dieta bogata w sól
(zwiększanie objętości osocza) oraz napoje zawiera-
■ neurogennych (np. przy neuropatii cukrzycowej, neuro-
jące kofeinę.
patii alkoholowej, udarze mózgu, chorobie Parkinsona) B4
Podstawą leczenia jest wówczas leczenie choroby pierwot- Do założeń farmakologicznej terapii niedociśnienia
nej.
należą:
Ze względu na objawy kliniczne rozróżnia się
■ podwyższenie napięcia żylnego i wskutek tego po-
■ przewlekłe niedociśnienie bezobjawowe
prawa napływu krwi żylnej do serca
■ przewlekłe niedociśnienie objawowe
■ zwiększenie kurczliwości serca i wskutek tego za-
■ niedociśnienie regulacyjne (wynikające z ortostatycz-
nych zaburzeń regulacji ciśnienia) pewnienie wystarczającej pojemności minutowej
nawet przy niskim ciśnieniu napełniania
Przewlekłe niedociśnienie bezobjawowe jest odmianą nor-
malnej regulacji układu krążenia, która nie wywołuje szkód ■ podwyższenie oporu obwodowego i wskutek tego
i w związku z tym oczywiście nie wymaga leczenia. ciśnienia wyjściowego, dzięki czemu podczas
W przypadku drugiej postaci niedociśnienia obecne zmiany położenia zmniejsza się prędkość odpływu
są liczne dolegliwości subiektywne takie jak np. zawroty krwi z odgałęzień tętnic do pojemnościowych na-
głowy, uczucie zimna, meteopatia i in. czyń żylnych przez co wydłuża się czas potrzebny
Niedociśnienie regulacyjne, polega na dysregulacji reak-
cji ortostatycznej podczas zmiany postawy z leżącej na sto- na reakcję organizmu podczas wstawania
jącą. Szczególną postać stanowi reakcja wazowagalna, któ- ■ zmniejszenie wydalania jonów sodu i wskutek tego
ra polega na tym, że po stymulacji nerwu błędnego, często
wskutek sytuacji stresującej związanej ze strachem może zwiększenie objętości krążącego osocza dzięki flu-
dojść do bradykardii i spadku ciśnienia krwi aż do omdle- drokortyzonowi.
nia.
W celu podwyższenia napięcia żylnego najodpowied-
Ze względu na rodzaj zaburzenia ortostatycznego rozróżnia niejsza jest przede wszystkim dihydroergotamina.
się:
Osłabia ona ponadto następstwa zaburzeń ortosta-
■ postać sympatykotoniczną (hiperdiastoliczną) wynika- tycznych związanych z nadmiernym wyrzutem kate-
jącą z niewystarczającej odpowiedzi pojemnościowych
cholamin.
MUTSCHLER-2009.indd 585 2010-01-07 22:13:43
586 Układ naczyniowy i krążenie
W celu podwyższenia oporu obwodowego oraz ciej wskutek spadku ciśnienia krwi. Z powodu zmniejszenia
zwiększenia kurczliwości mięśnia sercowego (wy- zaopatrzenia w tlen dochodzi do kwasicy tkanek i wskutek
tego zaburzeń funkcjonowania dotkniętych narządów.
łącznie w postaci z obniżonym napięciem układu
współczulnego!) stosowane są sympatykomimetyki. Do przyczyn wstrząsu zalicza się:
W przypadku wyboru tego rodzaju leczenia powinno
się stosować wyłącznie leki działające równocześnie ■ niewystarczające wypełnienie naczyń i wynikające z tego
na receptory α i β. Należą do nich zarówno substancje zmniejszenie napływu krwi żylnej do serca wskutek
zmniejszenia objętości krwi w przypadku krwawień,
działające bezpośrednio np. metylosiarczan amezy-
utraty osocza lub płynów np. po wypadkach, zabiegach
nium jak i bezpośrednie sympatykomimetyki np. ety- operacyjnych, poparzeniach, zapaleniu otrzewnej lub zła-
lefryna. maniach (wstrząs hipowolemiczny)
Jeśli chodzi o wybór leku, to dihdroergotamina
jest lekiem pierwszego wyboru w postaci niedociśnie- ■ zaburzenia czynności serca wskutek jego niewydolno-
ści bądź niewystarczającego napełniania lewej komory
nia z podwyższonym napięciem układu współczulne-
spowodowanego zawałem serca, zapaleniem mięśnia
go czyli w sympatykotonicznej postaci zaburzeń or- sercowego, zaburzeniami rytmu serca, tamponadą worka
tostatycznych, hipotonii ortostatycznej ze zwiększo- osierdziowego lub zatorem płuc (wstrząs kardiogenny)
nym spoczynkowym ciśnieniem krwi (częsta u star-
szych pacjentów) oraz w przypadku przewlekłego ■ nieprawidłową funkcję śródbłonka w naczyniach ob-
wodowych spowodowaną endotoksynami podczas sep-
niedociśnienia z tachykardią spoczynkową.
sy (wstrząs septyczny), wskutek reakcji anafilaktycznej
Sympatykomimetyki są natomiast wskazane prze- (wstrząs anafilaktyczny) albo z przyczyn neurogennych
de wszystkim w przypadku asympatykotonicznej po- wskutek niezwykle silnego bólu, urazu głowy lub rdzenia
staci zaburzeń ortostatycznych oraz w przewlekłym kręgowego czy tępych urazów (wstrząs neurogenny)
niedociśnieniu z bradykardią.
Dla wszystkich postaci wstrząsu charakterystyczny jest nie-
prawidłowy stosunek pojemności naczyń do objętości krwi
krążącej.
4.2.6. Leczenie wstrząsu
Na ryc. B 4.2-9 przedstawiono typowy przebieg wstrząsu
Wstrząs sercowo-naczyniowy charakteryzuje się ostrym po- na przykładzie wstrząsu hipowolemicznego.
gorszeniem ukrwienia ważnych życiowo narządów najczęś-
zmniejszenie spadek odruchowa
utrata krwi pojemności aktywacja układu
ciśnienia krwi
minutowej serca współczulnego
zmniejszony uszkodzenie spadek skurcz
powrót mięśnia perfuzji tętniczek
żylny sercowego narządów
rozszerzenie uszkodzenie ścian
skurcz naczyń hipoksja,
żyłek hipowolemia naczyń włosowatych,
pojemnościowych kwasica utrata osocza
podwyższona
martwica lepkość krwi,
tkanek wykrzepianie
wewnątrz-
naczyniowe
Ryc. B 4.2-9. Schemat patogenezy wstrząsu hipowolemicznego.
MUTSCHLER-2009.indd 586 2010-01-07 22:13:43
Układ naczyniowy i krążenie 587
Ośrodek krążenia w rdzeniu przedłużonym reaguje na spa- ■ roztworów elektrolitów
dek ciśnienia wynikający z utraty objętości krwi uogólnio-
ną aktywacją układu współczulnego. Dzięki temu dochodzi ■ płynów zastępczych osocza
z jednej strony do neurogennego skurczu naczyń obwodo-
wych i podwyższenia akcji serca, a z drugiej strony do wy- ■ preparatów osocza
rzutu katecholamin z nadnerczy, które wywołują podobne
■ krwi (w wyjątkowych przypadkach, kiedy koniecz-
efekty.
Dzięki tej reakcji układu współczulnego dochodzi ne jest uzupełnienie nośników tlenu)
do redystrybucji krwi: obwód zostaje prawie całkowi-
cie wyłączony na rzecz ważnych dla życia organów, ta- Leki rozszerzające naczynia. Oprócz wypełnienia
kich jak mózg, serce i płuca (tzw. centralizacja krążenia). objętościowego duże znaczenie mają leki działające
Zaangażowane zostają w ten proces zarówno doprowadza-
na układ krążenia i serce. W fazie centralizacji krą-
jące odgałęzienia tętniczek jak i odprowadzające odgałę-
zienia żył. Jest to bardzo przydatna regulacja krążenia, któ- żenia a także w późniejszych fazach wstrząsu po wy-
ra jednak jeżeli nie zostanie podjęte odpowiednie leczenie starczającym uzupełnieniu objętości konieczne są leki
i wstrząs będzie trwał dłuższy czas ulegnie przełamaniu. rozszerzające naczynia.
Niedotlenienie mózgu prowadzi bowiem do zmniejszenia
aktywności układu współczulnego, aż zaczyna przeważać
Dopamina. Lekiem spełniającym wszelkie wyma-
układ przywspółczulny. W słabo ukrwionych obszarach
również wskutek niedostatecznego zaopatrzenia w tlen do- gania dotyczące leczenia wstrząsu jest dopamina.
chodzi do uwalniania substancji rozszerzających naczynia, Jest to amina biogenna z grupy katecholamin, która
które działają przede wszystkim na tętniczki. Oznacza to, powstaje poprzez dekarboksylację 3,4-dihydroksy-
że ponownie możliwy jest napływ krwi na obwód, a wciąż fenyloalaniny (DOPA). W dawkach 1–2 μg/kg/min
utrudniony odpływ. W związku z tym dochodzi do utra-
Serce i układ krążenia
poprzez pobudzanie receptorów dopaminergicznych
ty płynu do tkanek. Z powodu zmniejszenia się prędko-
ści przepływu krwi dochodzi do agregacji erytrocytów, rozszerza naczynia nerkowe oraz otrzewnej wskutek
co powoduje szybki wzrost lepkości krwi. W końcu krew czego dochodzi do wzrostu przepływu krwi przez ner-
zatrzymuje się całkowicie w obrębie naczyń włosowatych ki i wzmożonej diurezy. W dawkach 2–10 μg/kg/min
i zaczyna krzepnąć. W ten sposób początkowo odwracalny dopamina pobudza dodatkowo receptory β wskutek
wstrząs przechodzi we wstrząs nieodwracalny.
czego dochodzi do wzrostu pojemności minutowej
Oprócz tych zdarzeń zachodzących w obrębie ukła-
du naczyniowego w tkankach dochodzi do następujących serca. W dawkach powyżej 10 μg/kg/min dochodzi
procesów: z powodu niedotlenienia reakcje metaboliczne do pobudzenia receptorów α i przez to do skurczu na-
wymagające tlenu nie są możliwe. Dotyczy to zwłaszcza czyń obwodowych, co w połączeniu ze zwiększoną
tworzenia wysokoenergetycznych fosforanów niezbędnych pojemnością minutową serca prowadzi do wzrostu B4
dla prawidłowego funkcjonowania i utrzymywania struk-
ciśnienia krwi.
tury komórek. Ponieważ produkty powstające podczas
glikolizy nie podlegają dalszej biotransformacji, dochodzi 75% dawki dopaminy jest metabolizowanej do far-
do wzrostu stężenia pirogronianów i mleczanów, a wartość makologicznie nieaktywnego kwasu homowanilino-
pH obniża się (kwasica mleczanowa). wego. Pozostałe 25% przekształcane jest w noradre-
Podczas wstrząsu narządem szczególnie narażonym nalinę i następnie metabolizowane do kwasu wanili-
są nerki, ponieważ są one jednym z najlepiej ukrwionych
nomigdałowego.
narządów i dlatego są szczególnie wrażliwe na zaburzenia
W przypadku długotrwałego podawania dopaminy
przepływu krwi.
jej działanie ulega osłabieniu wskutek tachyfilaksji.
Opisane procesy umożliwiają zrozumienie zasad sku-
Dobutamina. Dobutamina jest dostępna w handlu
tecznego leczenia wstrząsu, którego celem jest:
jako preparat racemiczny. Działa ona silniej inotropo-
■ możliwie szybkie przywrócenie prawidłowej per- wo dodatnio niż dopamina, natomiast wyraźnie sła-
fuzji tkanek biej działa na naczynia obwodowe, ponieważ pobu-
dzanie receptorów α przez enancjomer lewoskrętny
■ usunięcie pierwotnej przyczyny wstrząsu
w znacznym stopniu antagonizowane jest przez dzia-
łanie na receptory β2 enancjomeru prawoskrętnego.
Jeżeli nie dojdzie do usunięcia przyczyny wstrząsu
Oprócz tego dopamina obniża lewokomorowe ciśnie-
to jego leczenie będzie nieskuteczne.
nie napełniania. Jest ona lekiem z wyboru w przypad-
ku wstrząsu kardiogennego (zob. niżej).
Uzupełnienie objętości płynów. Najważniejszym
Dobutamina jest szybko metabolizowana w wątro-
postępowaniem w większości postaci wstrząsu
bie i w tkankach do glukuronidów, a także do farma-
jest uzupełnienie objętości płynów. Należy jak naj-
kologicznie nieaktywnej 3-O-metylodobutaminy.
szybciej doprowadzić objętość płynów krążących
Zazwyczaj stosowana jest w dawkach 2,5–10 μg/
do wartości prawidłowych dzięki zastosowaniu:
kg/min.
MUTSCHLER-2009.indd 587 2010-01-07 22:13:43
588 Układ naczyniowy i krążenie
w fazie początkowej (wstrząs septyczny, anafilak-
tyczny, neurogenny) stosuje się α-sympatykomimety-
ostry spadek ciśnienia ki, głównie adrenalinę i noradrenalinę.
Leki przeciwzakrzepowe, przede wszystkim hepary-
CVP ↓ CVP ↑
↑
na oraz leki fibrynolityczne np. streptokinaza stoso-
wane są w profilaktyce i/lub leczeniu zaburzeń krzep-
nięcia w przebiegu przedłużającego się wstrząsu.
wzrost objętości
Na ryc. B 4.2-10 przedstawiono postępowanie te-
rapeutyczne w przypadku wstrząsu w zależności
od najważniejszych parametrów klinicznych.
normalizacja CPV↑ MAP↓
Leczenie wstrząsu kardiogennego. Ponieważ
w przypadku wstrząsu kardiogennego mamy do czy-
nienia ze szczególnie ciężką, ostrą postacią niewy-
dopamina
dolności serca, jego leczenie polega na:
■ ekonomizacji pracy serca poprzez obniżenie obcią-
żenia wstępnego i następczego (azotany, inhibitory
konwertazy angiotensyny, leki moczopędne, leki
MAP↓ TPR↑ normalizacja MAP↓ TPR↑ rozszerzające naczynia)
■ zwiększeniu siły skurczu przez podawanie leków
dobutamina noradrenalina działających inotropowo dodatnio (agoniści recep-
torów β adrenergicznych, inhibitory fosfodiestera-
MAP↓ zy, lub glikozydy nasercowe)
noradrenalina Szybkie obniżenie obciążenia wstępnego i następcze-
wartości mierzalne go osiągnąć można podając dożylnie diuretyki pętlo-
we, nitroglicerynę lub nitroprusydek sodu. Konieczne
Ryc. B 4.2-10. Leczenie wstrząsu. CVP ośrodkowe ciśnienie jest przy tym monitorowanie mierzonego wewnątrz-
żylne (central venous pressure), MAP średnie ciśnienie tętnicze tętniczo ciśnienia krwi oraz ciśnienia w tętnicy płuc-
(mean arterial pressure), TPR całkowity opór naczyniowy (total nej. Korzystne okazało się również podawanie inhibi-
peripheral resistance). torów konwertazy angiotensyny.
W zależności od konkretnego przypadku skutecz-
ne może okazać się podawanie związków działają-
W przypadku długotrwałego podawania dobuta- cych inotropowo dodatnio, szczególnie dopaminy
miny, podobnie jak w przypadku dopaminy dochodzi i dobutaminy (zob. wyżej).
do osłabienia jej działania wskutek tachyfilaksji. Glikozydy nasercowe stosuje się w przypadku
wstrząsu kardiogennego wyłącznie wtedy, gdy rów-
α-sympatykomimetyki. W przypadku postaci nocześnie występuje migotanie przedsionków, lub
wstrząsu ze zmniejszeniem się oporu obwodowego też w przypadku występującej przed zawałem prze-
OH
HO NH2 HO NH
CH3
HO HO
dopamina dobutamina
MUTSCHLER-2009.indd 588 2010-01-07 22:13:43
Układ naczyniowy i krążenie 589
wlekłej niewydolności mięśnia sercowego. W takim Nie istnieje swoiste leczenie tej choroby. Zalecana
przypadku nie należy obawiać się wynikających z po- jest fizykoterapia w postaci kąpieli naprzemiennie
dawania glikozydów komorowych zaburzeń rytmu w ciepłej i zimnej wodzie.
serca czy też poszerzenia strefy niedokrwienia.
Wszystkie wymienione wyżej środki mogą jednak
okazać się nieskuteczne, jeżeli wskutek zawału doj- 4.2.7.1.2. Organiczne zaburzenia obwodowego
dzie do uszkodzenia znacznej części mięśnia serco- przepływu krwi
wego, co obserwuje się często we wstrząsie kardio-
gennym. Do organicznych zaburzeń obwodowego przepływu krwi
należą:
■ niedrożność tętnic
4.2.7. Leczenie zaburzeń tętniczego ■ zapalenia naczyń
przepływu krwi ■ zator naczyniowy
4.2.7.1. Zaburzenia obwodowego Najważniejszym spośród wymienionych wyżej zabu-
rzeń jest niedrożność tętnic. Jej najczęstszą przyczyną
przepływu krwi jest miażdżyca. W zależności od stopnia ciężkości rozróż-
nia się cztery stadia tej choroby. W stadium I ukrwienie
Ze względu na patogenezę wyróżnia się następujące jest na tyle wystarczające, że dolegliwości pojawiają się do-
zaburzenia tętniczego przepływu krwi: piero po szczególnie dużym wysiłku. W stadium II wskutek
postępującej miażdżycy przepływ, krwi a co za tym idzie
Serce i układ krążenia
■ czynnościowe zaopatrzenie w tlen przede wszystkim naczyń obwodo-
wych jest tak mocno zredukowane, że podczas chodzenia
■ organiczne dochodzi bólu związanego z gromadzeniem się kwaśnych
produktów przemiany, który zmusza do zatrzymania się.
Po krótkim odpoczynku bóle ustępują. Stan taki określa się
mianem chromania przestankowego (claudicatio intermit-
4.2.7.1.1. Czynnościowe zaburzenia tens). Im gorsze jest ukrwienie, tym krótsze są odcinki po-
przepływu krwi konywane bez bólu. W stadium III bóle kończyn pojawiają
się również w spoczynku. W stadium IV występują dodat-
Tego typu zaburzenia przepływu krwi (angioneuro- kowo uszkodzenia skóry z martwicą, co nierzadko oznacza
patie) powstają wskutek nieprawidłowej regulacji konieczność amputacji. ukrwienie jest na tyle wystarcza- B4
jące, że dolegliwości pojawiają się dopiero po szczególnie
naczyniowej wywołanej nieprawidłową odpowiedzią dużym wysiłku. W
na bodźce małych naczyń, której przyczyny znane
są tylko częściowo. Oprócz miażdżycy, kolejną ważną przyczyną obwodowych
Choroba Raynauda charakteryzuje się podwyż- zaburzeń ukrwienia tętniczego jest choroba zakrzepowo-
szoną gotowością skurczową naczyń określonych zarostowa (endangiitis obliterans, choroba Winiwartera-
Buergera). Najistotniejszą jej przyczyną jest palenie pa-
regionów układu krwionośnego (szczególnie w za- pierosów z zaciąganiem się. Proces chorobowy zaczyna się
kresie palców u rąk i stóp). Wskutek skurczu naczyń w tkankach poniżej błony wewnętrznej naczyń, a następnie
dochodzi do długotrwających, przerywanych epizo- obejmuje i ją. Wskutek zgrubienia ścian, światło naczynia
dów niedokrwienia z zaburzeniami czucia i silnymi ulega silnemu zmniejszeniu aż do całkowitego zamknięcia.
bólami. Choroba ta występuje najczęściej u młodych Następnie dochodzi do zakrzepicy małych i średnich na-
czyń. Szczególnie dotknięte procesem chorobowym są koń-
kobiet. Napady tego typu mogą zostać wywołane czyny dolne. Wskutek niedokrwienia dochodzi do powsta-
również przez alkaloidy sporyszu (np. ergotaminę). wania silnych bólów a w końcu martwicy (noga palacza
Leczenie polega na unikaniu czynników wywołu- papierosów)
jących, treningu autogennym oraz podawaniu leków
rozszerzających naczynia. Skuteczni okazali się an- W przypadku zatoru naczyniowego tętnicy dochodzi
do niedoboru krwi w zaopatrywanym przez nią obszarze,
tagoniści kanału wapniowego np. nifedypina w daw- co prowadzi do ostrego i zagrażającego życiu stanu choro-
ce 10–30 mg trzy razy dziennie. Oprócz tego można bowego. Stopień ciężkości oraz wielkość martwicy zależą
stosować antagonistów receptora α1, np. prazosynę nie tylko od umiejscowienia zatoru, ale także od obecno-
(dawkowanie początkowo dwa razy dziennie 0,5 mg, ści krążenia obocznego. Im mniej naczyń obocznych, tym
a następnie stopniowo zwiększając dawkę do 6 mg większe zaburzenia ukrwienia. Szczególne znaczenie kli-
niczne ma zator płucny. Nagłe zamknięcie dużego lub
dwa razy dziennie). jednego z mniejszych odgałęzień tętnicy płucnej prowadzi
do odruchowego pobudzenia nerwu błędnego i wskutek
W akrocyjanozie tętniczki są zwężone, a żyłki roz- tego do zaburzeń oddychania i zaburzeń czynności ser-
szerzone. Ekspozycja na zimno nasila dolegliwości. ca. Zamknięcie jednego z głównych pni tętnicy płucnej
MUTSCHLER-2009.indd 589 2010-01-07 22:13:44
590 Układ naczyniowy i krążenie
jest zwykle śmiertelne. Jeżeli wystąpieniu zatoru płucne- tego wykazać. Wręcz przeciwnie – w przypadku re-
go, który nie był śmiertelny towarzyszy niewydolność le- gularnego podawania ogólnie czy to doustnie czy do-
wokomorowa serca, dochodzi do powstania krwotocznego
żylnie leków rozszerzających naczynia w większości
zawału płuc.
przypadków istnieje niebezpieczeństwo dodatkowego
pogorszenia ukrwienia zagrożonego obszaru bądź
Leczenie zachowawcze niedrożności tętnic polega
to poprzez uogólnione rozszerzenie naczyń prowa-
na eliminacji czynników ryzyka, a także celowanej te-
dzące do spadku ciśnienia, bądź też w przypadku pra-
rapii ruchowej polegającej na progresywnych przery-
widłowego ciśnienia krwi poprzez zjawisko podkra-
wanych ćwiczeniach, aż do granicy bólu. Szczególnie
dania wskutek rozszerzenia naczyń w obszarach nie-
korzystne wyniki osiąga się w ten sposób w stadium
zmienionych chorobowo. Z tego rodzaju zjawiskiem
II: czynnościowe przekrwienie wspomaga tworzenie
należy liczyć się zwłaszcza w przypadku ciężkich
się naczyń obocznych, polepsza upośledzoną aktyw-
zaburzeń ukrwienia.
ność ruchową i hamuje postęp choroby. W stadium III
W stadium III i IV niedrożności tętnic w dalszym
i IV aktywny trening marszowy jest natomiast prze-
ciągu stosuje się jednak dotętnicze (ostatnio również
ciwwskazany.
dożylne) wlewy prostaglandyny E1 (alprostadil).
Postępowanie zabiegowe w celu poszerzenia światła na- Oprócz działania rozszerzającego naczynia oraz
czynia stosowane jest przede wszystkim w przypadku krót- hamującego agregację płytek krwi efekt leczniczy
kiego izolowanego zwężenia lub niedrożności naczynia PGE1 związany jest prawdopodobnie z hamowaniem
i polega na przezskórnej angioplastyce. Za pośrednictwem proliferacji komórek mięśni gładkich naczyń.
cewnika prowadzącego do uszkodzonego naczynia wpro- Dotętnicze dawkowanie wynosi 10–20 μg
wadzany jest cewnik z balonikiem, który po napompowa-
niu rozszerza światło naczynia, bądź też materiał zakrze- za pośrednictwem pompy infuzyjnej po rozpuszcze-
powy blokujący światło naczynia usuwany jest za pomocą niu w roztworze soli fizjologicznej przez 60–120 mi-
promieniowania laserowego (angioplastyka laserowa). nut jeden do dwóch razy dziennie; dożylnie we wle-
wie 40 μg w 50–250 ml soli fizjologicznej dwa razy
Farmakologiczne leczenie obwodowych tętniczych dziennie.
zaburzeń ukrwienia polega na:
Pochodna prostacykliny iloprost stosowana jest w po-
■ hamowaniu agregacji trombocytów
stępującym zakrzepowo-zarostowym zapaleniu na-
■ rozszerzaniu światła naczynia czyń w przypadku niemożności uzyskania rewasku-
laryzacji we wlewie w dawce 0,5–2 ng/kg/min przez
■ poprawianiu płynności krwi
6 godzin. Okres półtrwania wynosi pół godziny. Lek
■ profilaktyce zwężenia naczynia jest metabolizowany poprzez β-oksydację boczne-
go łańcucha karboksylowego. Metabolity usuwane
■ fibrynolizie w przypadku zamknięcia naczynia przez
są głównie przez nerki.
zakrzep lub zator
Poprawianie płynności krwi. Podobnie jak w przy-
Hamowanie agregacji trombocytów. Korzystne
padku leków rozszerzających naczynia wieńcowe
działanie leków hamujących agregację trombocy-
również w przypadku leków stosowanych w leczeniu
tów, np. kwasu acetylosalicylowego czy klopidogrelu
obwodowych zaburzeń ukrwienia (zob. tab. B4.2-12)
w przypadku obwodowych zaburzeń ukrwienia zosta-
niedawno wykazano nowe aspekty ich stosowania.
ło udowodnione w dużych badaniach. Każdy pacjent
Stwierdzono, że oprócz działania rozszerzającego na-
wykazujący objawy niedrożności tętniczej powinien
czynia niektóre z tych związków posiadają właściwo-
być leczony tymi lekami, jeżeli nie ma do tego prze-
ści zmniejszania lepkości krwi, polepszania płynności
ciwwskazań.
krwinek, rozbijania agregatów płytek krwi i zmniej-
szania agregacji trombocytów. Do leków, które były
Stosowanie leków rozszerzających naczynia. Leki
skuteczne w leczeniu pacjentów w drugim stadium
rozszerzające naczynia można stosować w przypad-
w kontrolowanych badaniach klinicznych i wykazały
ku obwodowych zaburzeń ukrwienia jako dodatek
przynajmniej częściowo efekt poprawienia płynności
do leczenia zabiegowego (np. angioplastyki) lub le-
krwi należą:
ków hamujących agregację. Przyjmuje się, że rozsze-
rzenie obocznych naczyń krwionośnych może popra- ■ buflomedil
wić ukrwienie dotkniętego niedokrwieniem narządu.
■ cynaryzyna
Jednak w licznych badaniach podobnie jak w przy-
padku choroby niedokrwiennej serca nie udało się ■ flunaryzyna
MUTSCHLER-2009.indd 590 2010-01-07 22:13:44
Układ naczyniowy i krążenie 591
Tab. B 4.2-12. Leki stosowane w leczeniu obwodowych zaburzeń ukrwienia
Wzór strukturalny Nazwa Preparat handlowy Okres Dawkowanie
międzynarodowa półtrwania (mg)
O buflomedil Bufedil, 2-3 150-300
N
BufloHEXAL,
H3CO OCH3 Buflomedil-CT,
Defluina peri u.a.
OCH3
cynaryzyna in Arlevert 60-70 150-300
N N
F flunarazyna Flunarizin-CT, 430 10
Flunarizin-
Serce i układ krążenia
ratiopharm,
Flunavert,
N N Natil-N
naftidrofuryl Dusodril, 1,2 600
Nafti-CT,
O Naftilong,
O Nafti-ratiopharm u.a. B4
N(C2H5) 2
O
O O CH3 pentoksyfilina Claudicat, 1,6 400
Pento-PUREN,
H3C N N Pentoxifyllin-
ratiopharm,
O N N Trental
CH3
■ naftidrofuryl kające z zaburzeń ukrwienia (niedosłyszenie, nagłe
osłabienie słuchu i in.). Jako mechanizm działania
■ pentoksyfilina
rozważane jest hamowanie fosfodiesterazy związanej
z agregacją trombocytów. Zarówno mechanizm dzia-
Pentoksyfilina wskazana jest w celu przedłużenia
łania pentoksyfiliny jak i jej skuteczność kliniczna
możliwego do pokonania dystansu u pacjentów z prze-
są jednak wciąż dyskutowane.
wlekłą niedrożnością tętnic obwodowych w stadium
drugim kiedy inne środki, takie jak np. trening mar-
Wskazania do stosowania buflomedilu są podobne
szowy, czy zabiegi rozszerzające światło naczynia
jak w przypadku pentoksyfiliny. Mechanizm działa-
lub rekonstrukcyjne są niemożliwe do przeprowadze-
nia jest nieznany, rozważa się blokadę obwodowych
nia, bądź przeciwwskazane. Kolejnym wskazaniem
receptorów α. Dane kliniczne są ograniczone.
są zaburzenia czynności ucha wewnętrznego wyni-
MUTSCHLER-2009.indd 591 2010-01-07 22:13:44
592 Układ naczyniowy i krążenie
Naftidrofuryl stosowany jest w leczeniu przewlekłej
Fibrynoliza zakrzepu lub zatoru zamykającego
niedrożności tętnic obwodowych a także w leczeniu
naczynie. W przypadku trwającego nie dłużej niż
objawów zaburzeń funkcji mózgu u osób w wieku
1–2 miesiące zamknięcia naczynia przez zakrzep, na-
podeszłym w przypadku demencji umiarkowane-
leży rozważyć leczenie fibrynolityczne. Oprócz roz-
go stopnia (nie wynikającej z choroby Alzheimera).
puszczenia zakrzepu leczenie takie prowadzi również
Stosowany jest także w leczeniu objawów uszkodzeń
do obniżenia lepkości krwi poprzez obniżenie pozio-
neurologicznych oraz ubytków czynnościowych
mu fibrynogenu. Skuteczność leczenia zależy przede
po udarach łącznie z kwasem acetylosalicylowym i/
wszystkim od czasu trwania i lokalizacji zakrzepu.
lub dipirydamolem. Naftidrofuryl blokuje receptor 5-
HT2 antagonizując w ten sposób naczynioskurczowe
oraz nasilające agregację płytek działanie serotoni-
ny. Skuteczność naftidrofurylu została potwierdzona 4.2.7.2. Leczenie zaburzeń
przez liczne prospektywne badania kontrolowane ukrwienia mózgowego
z placebo.
Podobnie jak w przypadku naczyń obwodowych,
Cynarazyna ma działanie antyhistaminowe, anty- w mózgu za 90% przypadków zaburzeń ukrwienia
serotoninowe, cholinolityczne oraz blokujące kanał odpowiedzialne są zmiany miażdżycowe naczyń.
wapniowy. Dostępne są jedynie nieliczne dane po- Wśród ostrych zaburzeń niedokrwiennych wyróżnia
twierdzające skuteczność tego leku. się:
■ przemijające ataki niedokrwienne (TIA – transient
Flunaryzyna klasyfikowana jest jako antagonista
ischemic attack) wywoływane są przez wędrujące
kanałów wapniowych, jednak dokładny mechanizm
zlepy trombocytów w miażdżycowo zmienionych
jej działania nie został do tej pory wyjaśniony. Lek
odcinkach naczyń, a występujące wskutek niedo-
ten jest stosowany w objawowym leczeniu zawrotów
krwienia objawy neurologiczne ustępują całkowi-
głowy pochodzenia przedsionkowego powstałych
cie w ciągu 24 godzin
wskutek długotrwałych zaburzeń czynności aparatu
przedsionkowego a także w profilaktyce migreny, ■ przedłużone odwracalne niedokrwienne deficyty
kiedy leczenie β-blokerami jest przeciwwskazane neurologiczne (PRIND – prolonged reversible is-
lub niewystarczająco skuteczne. Również w przypad- chemic neurological deficit) w przypadku których
ku tego leku dane kliniczne dotyczące skuteczności objawy neurologiczne również ustępują, ale naj-
są ograniczone. szybciej w ciągu tygodnia,
■ niedokrwienny udar mózgu (zawał mózgu, apo-
Poprawę mikrokrążenia próbuje się osiągnąć również
pleksja) z jedynie częściowym ustąpieniem obja-
dzięki hemodylucji.
wów neurologicznych,
Hemodylucja hiperwolemiczna polega na dożyl-
nym podawaniu roztworów koloidalnych (nisko- ■ postępujący niedokrwienny udar mózgu z nasi-
cząsteczkowego dekstranu lub hydroksyetylowanej lającymi się objawami bądź brakiem tendencji
skrobi) co prowadzić ma do obniżenia hematokry- do ich ustępowania.
tu, a co za tym idzie zmniejszenia lepkości krwi.
Oprócz tego zmniejszeniu ulega agregacja krwinek Przewlekła niewydolność naczyń mózgowych wyni-
czerwonych. Dzięki podwyższeniu ciśnienia onko- kająca z ugoólnionych zmian miażdżycowych tętnic
tycznego dochodzi zaś do efektu przeciwobrzęko- mózgowych prowadzi od początkowo bezobjawo-
wego. Do wad tego postępowania należą obciąże- wych niewielkich głęboko położonych stref niedo-
nie zwiększoną objętością, szczególnie u pacjentów krwienia do uogólnionego otępienia wielozawałowe-
z niewydolnością serca, stosunkowo krótki czas go.
działania oraz niebezpieczeństwo wystąpienia reak- U podłoża większości chorób otępiennych,
cji anafilaktycznych. np. u pacjentów z chorobą Alzheimera nie występują
Hemodylucja izowolemiczna polega na zmniejsza- jednak zaburzenia przepływu mózgowego tylko inne
niu wartości hematokrytu przez upust krwi i równo- mechanizmy.
czesne podanie roztworów koloidalnych. Unika się Leczenie przemijających ataków niedokrwiennych
w ten sposób obciążenia objętościowego, a oprócz oraz przedłużonych odwracalnych niedokrwiennych
tego efekt hemodylucji utrzymuje się znacząco dłu- deficytów neurologicznych polega na podawaniu in-
żej. hibitorów agregacji trombocytów, a w szczególności
kwasu acetylosalicylowego w dawce 0,1–0,3 g dzien-
MUTSCHLER-2009.indd 592 2010-01-07 22:13:44
Układ naczyniowy i krążenie 593
nie. Kwasu acetylosalicylowego nie powinno się jed- 4.2.8. Leki stosowane
nak stosować podczas leczenia trombolitycznego (p. w chorobach naczyń żylnych
niżej) i do 24 godzin po jego zakończeniu. Ważne
są też inne środki podejmowane w celu zapewnienia
Choroby naczyń żylnych stanowią szczególny prob-
odpowiedniego ciśnienia perfuzji (np. leczenie nie-
lem kliniczny oraz społeczny ze względu na częstość
wydolności serca lub istotnych hemodynamicznie
ich występowania oraz możliwe następstwa. Problem
zaburzeń rytmu serca).
ten dotyczy jednej siódmej części populacji. Oprócz
Także w przypadku niedokrwiennego udaru mózgu
ostrego zapalenia żył powierzchownych (throm-
podstawę leczenia stanowią interwencje internistycz-
bophlebitis), zakrzepicy żył głębokich (phlebothrom-
ne. Leczenie fibryndiczne z użyciem r-tPA stosuje
bose) oraz żylaków duże znaczenie ma przewlekła
się jedynie w sytuacji, kiedy pacjent zostanie przy-
niewydolność żylna (CVI – chronic venous insuffi-
wieziony na specjalistyczny oddział w ciągu trzech
ciency), która jest następstwem zmian w układzie żył
godzin od wystąpienia udaru i zostanie wykluczony
powierzchownych i głębokich. Choroba ta charakte-
krwotok mózgowy. Jednakże należy się wówczas li-
ryzuje się zespołem objawów z uczuciem ciężkości
czyć z możliwością wystąpienia krwawienia. Można
oraz bólu kończyn dolnych po dłuższym pozosta-
dodatkowo stosować wewnątrztętnicze leczenie
waniu w pozycji stojącej lub siedzącej oraz obrzę-
proksymalnego zamknięcia tętnicy środkowej móz-
kami zlokalizowanymi głównie w okolicach kostek.
gu za pośrednictwem aktywatorów plazminogenu,
U jej podłoża leżą zaburzenia odpływu krwi żylnej
ale jest to forma terapii stosowana raczej w pojedyn-
z podwyższonym ciśnieniem żylnym wskutek:
czych przypadkach.
Serce i układ krążenia
U pacjentów z ostrym udarem niedokrwiennym ■ zamknięcia bądź niewydolności zastawkowej żył
mózgu często występuje podwyższone ciśnienie krwi, głębokich wskutek choroby zakrzepowej,
jednak do wartości 180–220/105–120 nie powinno
■ niewydolności układu żył przeszywających,
być obniżane, aby zapewnić utrzymanie ukrwienia
mózgu. Wyższe wartości wymagają natomiast lecze- ■ powierzchownej żylakowatości.
nia za pomocą m.in. kaptoprilu, klonidyny, dihydra-
lazyny oraz metoprololu. Należy unikać dużych wa- Według Widmera rozróżnia się trzy stadia choroby.
hań ciśnienia krwi. W stadium I występują objawy zastoju krwi żylnej
W leczeniu obrzęku mózgu lub podwyższonego w obrębie stopy oraz rozszerzenie żył podskórnych
ciśnienia wewnątrzczaszkowego w przebiegu uda- i śródskórnych (corona phlebectatica paraplantaris).
B4
ru stosuje się diuretyki osmotyczne, np. 500 ml 20% W stadium II pojawiają się dodatkowo zmiany tro-
roztworu mannitolu dziennie w trakcie pierwszych ficzne w obrębie skóry, zmiany pigmentacyjne (hi-
48 godzin po udarze (dłuższe leczenie za pomocą perpigmentacja i depigmentacja), oraz stwardnienie
diuretyków osmotycznych może stwarzać proble- skóry (dermatosclerosis). W stadium III pojawia się
my, ponieważ istnieje wówczas nibezpieczeństwo (czerwone bądź zaleczone) owrzodzenie podudzia
ich przenikania przez uszkodzoną barierę krew-mózg (ulcus cruris).
do tkanek). Obniżenie ciśnienia wewnątrzczaszko-
wego można również osiągnąć za pomocą krótko Zgodnie z patofizjologią zmian leczenie polega nie tylko
działających barbituranów np. tiopentalu. na interwencjach chirurgicznych (wycięcie żylaków, pod-
wiązanie niewydolnych żył przeszywających) lecz także
Skuteczne leczenie przewlekłych zaburzeń na stosowaniu leczenia uciskowego za pomocą opatrunków
ukrwienia mózgowego oraz objawów psychicznych bądź pończoch uciskających, co jest najważniejszym ro-
pochodzenia organicznego jest możliwe jedynie dzajem leczenia.
w ograniczonym stopniu a efekty są zwykle nieza- Przydatne wydaje się również pobudzanie krążenia żyl-
dowalające. nego za pomocą ćwiczeń marszowych, masażu wodnego,
jazdy na rowerze, gimnastyki kończyn oraz wysokiego
Podobnie jak w przypadku leczenia obwodowych ułożenia nóg podczas snu ze względu na znaczenie pompy
zaburzeń ukrwienia istnieje coraz więcej doniesień mięśniowej dla powrotu krwi żylnej.
oraz badań kontrolowanych dotyczących przydat-
ności naftidrofurylu (zob. wyżej), dihydroergotok-
W farmakologicznym leczeniu zaburzeń krążenia żyl-
syny oraz szczególnie często stosowanego wyciągu
nego stosuje się:
Ginko-biloba, jednak do tej pory ich skuteczność
jest kontrowersyjna. Dotyczy to również leków no- ■ środki zwiększające napięcie żył,
otropowych.
■ leki moczopędne,
■ leki przeciwobrzękowe.
MUTSCHLER-2009.indd 593 2010-01-07 22:13:45
594 Układ naczyniowy i krążenie
Środki zwiększające napięcie żył to uwodornio- kająca z czegoś więcej niż efekt miejscowego masażu
ne alkaloidy sporyszu, a zwłaszcza dihydroergota- nie została potwierdzona.
mina. Poprzez zwiększanie napięcia żył poprawia
się powrót krwi żylnej, oraz prędkość przepływu
w żyłach mimo istniejących zmian zakrzepowych. 4.2.9. Leczenie zaburzeń wzwodu
Nie jest możliwa, bądź tylko w ograniczonym stop-
niu możliwa jest wybiórcza poprawa napięcia ścian Wzwód prącia (erekcja) zachodzi wskutek (przemijającego)
żył, bez wpływu na tętnice. Przede wszystkim przy wypełnienia krwią parzystych ciał jamistych prącia (corpo-
wyższych dawkach bądź w przypadku podawania ra cavernosa) podczas pobudzenia seksualnego. Pobudzone
włókna nerwowe układu przywspółczulnego za pośredni-
pozajelitowego dihydroergotaminy istnieje niebez- ctwem NO i jego wtórnego przekaźnika cGMP powodują
pieczeństwo wystąpienia obwodowych zaburzeń rozszerzenie tętnic zaopatrujących prącie, mięśnie gładkie
ukrwienia (ergotismus). Biorąc pod uwagę możli- ciał jamistych ulegają zwiotczeniu i dochodzi do ucisku żył
wość wystąpienia działań niepożądanych, możliwość wewnątrzjamistych.
terapeutycznego zastosowania tych leków stoi pod Zaburzenia wzwodu oznaczają stan, w którym brak lub
niewystarczająca erekcja powoduje niemożność odbycia
znakiem zapytania. satysfakcjonującego stosunku płciowego (impotentia co-
eundi). Z wiekiem zwiększa się częstotliwość występowa-
Leki moczopędne. W początkowym leczeniu obrzę- nia tych zaburzeń. W wieku 65 lat występują one u 25–30%
ków pochodzenia żylnego (np. przed lub równocześ- populacji. Do przyczyn ich występowania należą zmniej-
nie z dopasowaniem pończochy uciskającej) można szony napływ krwi tętniczej do ciał jamistych, bądź zwięk-
szony odpływ krwi żylnej, zbyt niska produkcja testostero-
zastosować diuretyki tiazydowe w niskich dawkach, nu, hiperprolaktynemia, porażenie poprzeczne, stwardnie-
które prowadzą do silnego i długiego wzrostu diu- nie rozsiane, polineuropatia cukrzycowa, także zaburzenia
rezy. Przewlekłe leczenie nie jest wskazane. Należy psychogenne i spowodowane lekami (leki antyandrogenne,
również unikać diuretyków pętlowych ze względu przeciwdepresyjne, przeciwnadciśnieniowe, uspokajające).
na niebezpieczeństwo wzrostu wartości hematokrytu
W przypadku udowodnionego niedoboru androgenów le-
a co za tym idzie podwyższenia lepkości krwi. czenie polega na podawaniu testosteronu. U cukrzyków
Leki przeciwobrzękowe. Należą do nich preparaty, należy uregulować przemianę materii.
które zawierają: W przypadku zaburzeń wynikających z przyczyn tętni-
czych bądź neuropatycznych skuteczne są iniekcje z papa-
■ wyciągi z nasienia Aesculus hippocastanum (kasz- weryny lub rozszerzającej naczynia prostaglandyny – al-
tanowca), lub też uzyskiwane z nich glikozydy tri- prostadilu. Jest to jednak forma leczenia wybitnie uciążli-
terpenowe (escyna) wa dla pacjentów.
■ wyciągi z liści czerwonej winorośli
Wraz z wprowadzeniem na rynek sildenafilu,
■ pochodne flawonoidów, m.in. rutyna lub jej półsyn- wardenafilu i tadalafilu w leczeniu pojawiły się
tetyczne hydroksyetelowane pochodne jak np. tri- trzy podawane doustnie leki o nowym mechanizmie
hydroksyetylorutyna (trokserutyna) lub O-(β-hy- działania. Jako inhibitory fosfodiesterazy 5 (PDE 5),
droksyetylo)-rutozyd która występuje głównie w tętnicach ciał jamistych
prącia, hamują rozpad cGMP i nasilają oraz przedłu-
■ dobesylan wapniowy
żają jego działanie. U większości pacjentów można
za ich pomocą skutecznie leczyć zaburzenia wzwodu.
Środki te zmniejszają przepuszczalność naczyń wło-
Trzy preparaty dostępne w handlu różnią się między
sowatych i zmniejszają w ten sposób przesączanie
sobą głównie właściwościami farmakokinetycznymi,
osocza, wskutek czego zmniejszają, a przynajmniej
a co za tym idzie początkiem oraz czasem działania
ograniczają miejscowe obrzęki i równocześnie po-
(zob. tab. B 4.2-13).
prawiają powrotny prąd żylny. Dokładny mechanizm
działania nie jest znany. Ich skuteczność kliniczna
Leki te są metabolizowane głównie przez cyto-
pomimo kilku pozytywnych wyników kontrolowa-
chrom CYP3A4, a sildenafil również przez CYP2C9
nych badań i częstego stosowania jest dyskusyjna.
do wciąż aktywnego głównego metabolitu, którym
jest N-desmetylosildenafil. Równoczesne podanie
Środki stosowane miejscowo w chorobach żył za-
inhibitorów CYP3A4 może przez to prowadzić do in-
wierają przede wszystkim samą heparynę bądź hepa-
terakcji między lekami.
rynę w połączeniu z innymi związkami lub środka-
Do działań niepożądanych należą częste bóle gło-
mi pochodzenia roślinnego (np. nikotynian benzylu,
wy i zaczerwienienie twarzy, a także niekiedy dole-
Tinctura Arnicae montanae). Ich skuteczność wyni-
gliwości dyspeptyczne, zaburzenia widzenia, nie-
MUTSCHLER-2009.indd 594 2010-01-07 22:13:45
Układ naczyniowy i krążenie 595
Tab. B 4.2-13. Inhibitory fosfodiesterazy 5
Wzór strukturalny Nazwa Preparat handlowy Okres Dawkowanie
międzynarodowa półtrwania (mg)
O CH3 sildenafil Revatio, 3-5 25-100
N VIAGRA
O O HN
N
S
N N
N C3H7
H3C OC2H5
O CH3 wardenafil LEVITRA 4-5 5-20
O O HN
N
S N
N N
N C3H7
H5C2 OC2H5
O tadalafil CIALIS 17,5 10-20
H
CH3
N
Serce i układ krążenia
HN N
O
O
drożność nosa i zawroty głowy. Może dojść również
B4
W porównaniu z podaniem podskórnym biodostępność
do wystąpienia priapizmu. w przypadku tabletki podjęzykowej wynosi mniej niż 20%.
U pacjentów, którzy przyjmują nitraty lub inne do- Głównymi produktami metabolizmu są koniugaty apomor-
finy i N-desmetylowanych pochodnych z kwasem glukuro-
nory tlenku azotu inhibitory PDE 5 są przeciwwska-
nowym i siarkowym. Okres półtrwania w osoczu wynosi
zane ze względu na niebezpieczeństwo wystąpienia około 3 godzin. Wydalanie zachodzi zarówno przez nerki
poważnych, a niekiedy śmiertelnych działań niepożą- jak i z kałem.
danych wskutek zablokowania rozpadu cGMP i wy- Do działań niepożądanych które ulegają osłabieniu
nikającego z tego nasilonego działania NO. Do in- w przypadku regularnego podawania należą przede wszyst-
kim nudności, bóle i zawroty głowy oraz spadek ciśnienia
nych przeciwwskazań należą poważne choroby ser-
tętniczego. Do pozostałych w większości przypadków słabo
cowo-naczyniowe oraz schorzenia wątroby. nasilonych działań niepożądanych zalicza się katar, zapale-
nie gardła, oszołomienie, bóle, nasilony kaszel, uderzenia
Apomorfina jest kolejnym lekiem stosowanym w leczeniu krwi do głowy, poty i in. Apomorfina jest przeciwwskaza-
zaburzeń wzwodu podawanym podjęzykowo. Ten agonista na u pacjentów z niestabilną dusznicą bolesną, niedawno
receptorów dopaminergicznych był wcześniej stosowa- przebytym zawałem serca, ciężką niewydolnością serca lub
ny jako lek przeciwparkinsonowski oraz środek wymiot- niedociśnieniem. Może być ona jednak w przeciwieństwie
ny. Lek ten działa głównie na receptory D2 i stosowany do sildenafilu stosowana łącznie z nitratami, jeżeli nie zo-
jest podjęzykowo (ze względu na silny efekt pierwszego stanie przekroczona dawka 3 mg. Równoczesne podanie
przejścia) w dawce 2–3 mg 20 minut przed planowanym alkoholu zwiększa ryzyko wystąpienia hipotonii. Należy
stosunkiem seksualnym. Podobnie jak sildenafil, apomorfi- również unikać równoczesnego przyjmowania innych le-
na nie jest afrodyzjakiem i w celu osiągnięcia pożądanego ków działających centralnie agonistycznie bądź antagoni-
działania wymagana jest stymulacja seksualna. stycznie na receptory dopaminergiczne.
MUTSCHLER-2009.indd 595 2010-01-07 22:13:45
596 Krew
4.3. Serce
4.3.1. Podstawy anatomiczne Nasierdzie pokrywające powierzchnię serca skła-
i fizjologiczne da się z jednowarstwowego nabłonka oraz leżącej pod
nim cienkiej błony łącznotkankowej. W miejscach
ujść dużych naczyń przechodzą w siebie wewnętrzna
Serce jest położone w przedniej części śródpiersia.
i zewnętrzna blaszka osierdzia.
Jego wielkość odpowiada w przybliżeniu wielkości
zaciśniętej pięści danego człowieka. Średnio u doro-
Zastawki serca. Zastawki serca pełniące funkcje
słej kobiety ciężar serca wynosi 280 g, a u mężczyzny
zaworów, zapobiegają cofaniu się strumienia krwi
320 g. Pod względem czynnościowym można ludzkie
w trakcie rytmicznych skurczów mięśnia sercowego.
serce podzielić na dwie części: prawą i lewą połowę,
Zastawki położone między przedsionkami a komora-
z których każda składa się z kolei z mniejszej części
mi określa się jako zastawki przedsionkowo-komoro-
– przedsionka (atrium) i większej – komory (ventri-
we lub ze względu na ich kształt jako zastawki typu
culus) (zob. ryc. B 4.3-1).
„żaglowego”. Zastawka między prawym przedsion-
kiem a prawą komorą nazywa się zastawką trójdziel-
Przedsionki znajdujące się w górnej części serca
ną, natomiast znajdująca się między lewym przed-
są od siebie oddzielone przegrodą międzyprzedsion-
sionkiem a lewą komorą to zastawka dwudzielna. Aby
kową (septum atriorum). W prawym przedsionku
zapobiec odchyleniu płatków „żaglowych” do przed-
mają swoje ujścia żyły główne, natomiast w lewym
sionków, płatki zastawek są połączone z mięśniem
przedsionku żyły płucne. Z zewnątrz rozgraniczenie
sercowym za pomocą włókien ścięgnistych przez tzw.
między komorami a przedsionkami można rozpoznać
mięśnie brodawkowate. W miejscach ujść obu komór
po bruździe – bruździe wieńcowej (sulcus corona-
znajdują się tzw. zastawki półksiężycowate, z których
rius) (zob. ryc. B 4.3-2).
każda składa się z trzech płatków łącznotkankowych
Płaszczyzna wyznaczona przez bruzdę wieńcową
o wzmocnionym brzegu i kształcie półksiężycowa-
tworzy podstawę serca. W tej płaszczyźnie znajdują
tym. Zastawka w miejscu ujścia pnia płucnego nazy-
się wszystkie zastawki serca (zob. poniżej) i dlate-
wa się zastawką pnia płucnego, a zastawka w miejscu
go określa się ją także jako płaszczyznę zastawkową.
ujścia aorty – zastawką aorty.
Obie komory oddziela od siebie przegroda międzyko-
morowa (septum interventriculare).
Fazy czynności serca. Serce pełni funkcje pompy
Całe serce otacza błona surowicza – osierdzie.
dzięki rytmicznym skurczom i rozluźnianiu. Zgodnie
Składa się ono z dwóch blaszek oddzielonych cienką
warstwą płynu, które są w stosunku do siebie rucho-
me. Wewnętrzna blaszka pokrywająca powierzchnię
serca jest nazywana nasierdziem (zob. poniżej).
pień płucny
aorta
Budowa ściany serca. Ściana serca składa się z trzech
warstw: wewnętrznej – wsierdzia, środkowej – mięś- żyła lewy
nia sercowego oraz zewnętrznej – nasierdzia. główna górna przedsionek
Wsierdzie składa się z warstwy śródbłonka i luź- zastawka aortalna
nej tkanki łącznej. Wyścieła ono całą wewnętrzną prawy zastawka
przestrzeń serca. przedsionek dwudzielna
Warstwa mięśniowa ściany serca ma budowę po- struny
zatoka ścięgniste
dobną do mięśni szkieletowych i tak jak one posiada
wieńcowa mięśnie
poprzeczne prążkowanie oraz siateczkę sarkotubular- brodawkowate
żyła
ną między włókienkami mięśniowymi. W odróżnie- główna dolna przegroda
niu od mięśni szkieletowych w mięśniu sercowym zastawka komorowa
znajduje się siatka rozgałęzionych włókien, gdzie trójdzielna
granice komórek są zaznaczone przez tzw. błyszczące prawa lewa
komora komora
pasma. Ze względu na specjalną architekturę mięśnia
sercowego w dużej mierze możliwe jest koncentrycz- Ryc. B 4.3-1. Przekrój serca w płaszczyźnie czołowej, wg
ne zmniejszenie się komór serca podczas skurczu Leonhardta. Przedsionki i komory są otwarte. Strzałki wskazują
mięśnia sercowego. kierunek krwi.
MUTSCHLER-2009.indd 596 2010-01-07 22:13:45
Krew 597
Objętość czasowa serca, tzn. objętość krwi transpor-
aorta pień towana w określonym czasie wynosi w spoczynku
płucny około 5 l/min (alternatywnie podaje się tzw. indeks
sercowy wynoszący odpowiednio 3 l/min/m2). W wa-
runkach obciążenia, przede wszystkim podczas wy-
lewe uszko siłku fizycznego, może znacznie wzrosnąć objętość
żyła główna serca minutowa serca (do 25 l/min) zarówno w wyniku
górna
lewa tętnica wzrostu objętości wyrzutowej, jak i wzrostu często-
wieńcowa biegnąca ści akcji serca.
w bruździe Wzrost objętości wyrzutowej jest efektem zwięk-
prawe międzykomorowej
uszko serca szonej ilości krwi napływającej żyłami oraz spo-
wodowane tym podwyższone ciśnienie krwi żylnej.
Bardziej rozciągnięte włókna mięśnia sercowego
w następstwie większego napełniania komór są zdol-
ne do silniejszego skrócenia swojej długości i przez
lewa to do wypompowania większej ilości krwi. Ten tryb
komora
przystosowania się nazywany jest mechanizmem
Franka-Starlinga. W odniesieniu do zdrowego serca
żyła główna prawa tętnica wieńcowa prawa mechanizm ten odgrywa rolę tylko przy krótkotrwa-
dolna biegnąca w bruździe wieńcowej komora łym wyrównaniu objętości wyrzutowej i przy dosto-
Serce i układ krążenia
sowaniu do siebie objętości wyrzutowych prawej
Ryc. B 4.3-2. Widok serca od przodu. i lewej połowy serca. W znacznie większym stopniu
praca serca dostosowuje się do obciążenia organi-
zmu przez oddziaływanie układu współczulnego.
Aktywacja układu współczulnego prowadzi do zwięk-
z tym występują fazy skurczowe oraz fazy rozkur- szenia siły skurczu mięśnia sercowego (dodatni efekt
czowe. inotropowy). Silniejsze skurcze mięśnia sercowego
Na początku fazy skurczowej, tzw. okresu skurczu wskutek większego wykorzystania objętości zalega-
izometrycznego, wszystkie układy zastawkowe są za- jącej powodują wypompowanie większej objętości
mknięte. Z tego powodu skurcz mięśnia sercowego wyrzutowej lub przezwyciężają większy obwodowy
B4
przy niezmieniającej się objętości komór wywołuje opór naczyniowy.
szybki wzrost ciśnienia. Następnie, gdy ciśnienie Zmiana częstości akcji serca jest drugą możliwoś-
śródkomorowe przekroczy wartość ciśnienia w du- cią dostosowania pracy serca do potrzeb całego or-
żych tętnicach (ok. 80 mm Hg), otwierają się zastaw- ganizmu. Zmniejszenie napięcia nerwu błędnego lub
ki półksiężycowate i rozpoczyna się faza wyrzutowa. zwiększenie napięcia układu współczulnego wiąże
Początkowo ciśnienie śródkomorowe wzrasta pod- się ze zwiększeniem częstości akcji serca. Nerw błęd-
czas dalszego przebiegu fazy skurczowej i pod jej ko- ny odgrywa większą rolę w dostosowaniu częstości
niec zaczyna się obniżać. akcji serca niż układ współczulny.
Podczas fazy wyrzutowej z komory zostaje wy- Wraz ze wzrostem częstości akcji serca ulega tak-
pompowana objętość skurczowa wynosząca około że zmianie stosunek czasu trwania fazy skurczowej
70 ml. Podobna objętość resztkowa pozostaje w ko- do fazy rozkurczowej. Przy wzroście częstości akcji
morze. serca następuje znacznie większe skrócenie cza-
Wraz ze spadkiem ciśnienia śródkomorowego su trwania fazy rozkurczowej niż fazy skurczowej.
rozpoczyna się pierwsza część fazy rozkurczowej. W wyniku tego czas, w którym krew może napływać
Zastawki półksiężycowate zamykają się i po obniże- do komór oraz przepływać przez naczynia wieńcowe,
niu się ciśnienia poniżej wartości ciśnienia w przed- jest krótszy. Dalsze zwiększenie częstości akcji serca
sionkach otwierają się zastawki przedsionkowo-ko- jest więc nieekonomiczne.
morowe. W ten sposób rozpoczyna się ostatnia faza, W przypadku dłużej trwających obciążeń, poza
tzw. faza wypełniania komór serca, w czasie której czynnościową, regulacją występuje także dostosowa-
krew (w dużej mierze pasywnie) wpływa do komór. nie strukturalne. Włókna mięśniowe stają się grubsze
i dłuższe (przerost mięśnia sercowego), a następnie
Przystosowanie się czynności serca. W okresie dochodzi do rozszerzenia jam serca. Ciężar serca
spoczynku objętość wyrzutowa wynosi około 70 ml, wynoszący prawidłowo 300 g może wzrosnąć nawet
a częstość akcji serca mniej więcej 70 uderzeń/min. do 500 g.
MUTSCHLER-2009.indd 597 2010-01-07 22:13:46
598 Krew
Obciążenie wstępne i następcze serca. Termin ob- rozruszniku serca znajdującym się przy ujściu żyły
ciążenie wstępne (preload) oznacza późnorozkurczo- głównej górnej do prawego przedsionka, i prze-
we wypełnienie, które determinuje pasywnie powsta- wodzone przez mięśniówkę przedsionka do węzła
jące późnorozkurczowe napięcie ściany serca. przedsionkowo-komorowego (węzła AV, czyli węzła
Miarą obciążenia następczego (afterload) jest na- Aschoffa i Tawary).
pięcie ściany serca, które jest potrzebne do przezwy- Zgodnie z nazwą węzeł ten znajduje się w miejscu
ciężenia późnorozkurczowego ciśnienia w aorcie lub przejścia przedsionka w komorę. Dalsze przewodze-
pniu płucnym. Z tego powodu zmniejszenie obciąże- nie bodźca następuje przez pęczek Hisa, jego komo-
nia następczego można uzyskać przez zmniejszenie rowe odnogi (odnogi Tawary) i rozgałęzienia układu
późnorozkurczowego ciśnienia w aorcie lub pniu płuc- bodźcoprzewodzącego (włókna Purkinjego) w komo-
nym, a także przez zmniejszenie średnicy komory. rach.
Ten skomplikowany układ zabezpiecza serce
Układ bodźcotwórczy i bodźcoprzewodzący serca. przed zatrzymaniem jego czynności. Jeżeli zabrak-
Serce należy do narządów pracujących rytmicznie, nie czynności węzła zatokowego jako pierwotnego
które w odpowiednich warunkach kontynuują swoją rozrusznika z około 60–80 bodźcami/min., wówczas
czynność także poza organizmem. Ta zdolność zwią- wytwarzanie bodźców z mniejszą częstością (ok.
zana jest z występowaniem w nim układów bodźco- 50–60 bodźców/min.) następuje w ośrodku drugo-
twórczego i bodźcoprzewodzącego (automatyzm ser- rzędowym – węźle Aschoffa i Tawary. Jeżeli przewo-
ca). Ponadto układ współczulny i przywspółczulny dzenie bodźców zostało przerwane również w węźle
regulują czynność serca, nie mając przy tym wpływu przedsionkowo-komorowym, wytwarzanie bodźców
na jej wywoływanie. przejmują ośrodki trzeciorzędowe z bardzo wolną
W prawidłowych warunkach pobudzenia są wy- częstością wynoszącą mniej więcej 15–25 bodźców/
twarzane w węźle zatokowym (zob. ryc. B 4.3-3), min. (rytm komorowy).
węzeł przedsionkowo-komorowy węzeł zatokowy
+50 +50
(mV) (mV)
0 0
-50 -50
-100 -100
pęczek Hisa
+50 mięsień przedsionkowy
+50
(mV)
(mV)
0
0
-50
-50
-100
300 ms -100
włókna Purkinjego mięsień komorowy
+50 +50
(mV) (mV)
0 0
-50 -50
-100 -100
Ryc. B 4.3-3. Układ bodźcotwórczy i bodźcoprzewodzący serca (ciemnoniebieski) z postaciami potencjałów czynnościowych, które
sa charakterystyczne dla odpowiednich włókien.
MUTSCHLER-2009.indd 598 2010-01-07 22:13:46
Krew 599
Potencjał spoczynkowy. Włókna mięśniowe serca, razy dłużej aniżeli odpowiedni proces pobudzenia
tak jak wszystkie dające się pobudzać włókna nerwo- we włóknie nerwowym.
we i mięśniowe, mają potencjał spoczynkowy pomię- Potencjał czynnościowy jest osiągany poprzez licz-
dzy wnętrzem komórki (ujemny ładunek) a przestrze- ne ukierunkowane przepływy prądu wywołane akty-
nią pozakomórkową (dodatni ładunek). Ten potencjał wacją określonych kanałów jonowych. Uczestniczące
błonowy wynosi około –90 mV. Wytwarza go głów- w tym procesie kanały jonowe, ich zależna od czasu
nie potencjał dyfuzyjny jonów potasu. aktywacja oraz kierunek transportu przedstawiono
na ryc. B 4.3-4.
Potencjały czynnościowe i czas refrakcji. Poten-
cjały czynnościowe potrzebne dla czynności serca We włóknach mięśnia sercowego prawie przez cały
są wytwarzane w tzw. komórkach rozrusznikowych, czas trwania potencjału czynnościowego występuje
które mają zdolność spontanicznej depolaryzacji bezwzględna refrakcja tzn. w tym czasie nie jest moż-
(zob. ryc. B 4.3-3). liwe powstanie nowego pobudzenia. Dopiero pod
W niektórych komórkach po wystąpieniu poten- koniec potencjału czynnościowego we włóknach
cjału czynnościowego pojawia się natychmiast spon- mięśnia sercowego następuje względna refrakcja
taniczna powolna depolaryzacja (potencjał generato- (okres zmniejszonej pobudliwości), zanim ponownie
rowy) z powodu zmniejszenia przepuszczalności dla osiągną swoją pełną pobudliwość. Okresy refrakcji
jonów potasowych. Po osiągnięciu potencjału progo- chronią serce przed długotrwałym skurczem przez
wego powstaje następny potencjał czynnościowy. następujące szybko po sobie bodźce lub pobudzenia.
W komórkach rozrusznikowych węzła zatokowego Naprzemienne występowanie skurczów i rozkurczów
Serce i układ krążenia
depolaryzacja zachodzi szybciej niż w pozostałych jest więc konieczne dla ochrony serca.
komórkach układu bodźcoprzewodzącego. Dlatego
te komórki (aktualny rozrusznik) prawidłowo deter-
minują rytm czynności serca. Dopiero gdy zabraknie napływ do środka ITo IKr
ich czynności, wolniejsza spontaniczna depolaryza- wypływ na zewnątrz IKs
cja w innych grupach komórek może osiągnąć próg IK1
i przez to determinować rytm czynności serca (po-
INa
tencjalne rozruszniki). Bodźce pochodzące z układu If
współczulnego przyspieszają spontaniczną depola- ICa-T
ryzację komórek rozrusznikowych, natomiast bodźce ICa-L B4
z układu przywspółczulnego ją spowalniają.
W przeciwieństwie do komórek rozrusznikowych
napływ
włókna mięśniówki roboczej serca prawidłowo Na+
(tzn. w warunkach niepatologicznych) nie posia- T-Typ
dają zdolności spontanicznej depolaryzacji. Można napływ
Ca2+
w nich wywołać potencjał czynnościowy jedynie L-Typ
przez depolaryzację przyległego miejsca. Podobnie
do mięśni szkieletowych, zaczynając od potencja- przejściowy IT01
wypływ K+
łu spoczynkowego wynoszącego około –90 mV IT02
w wyniku wzrostu przepuszczalności dla jonów
sodu, dochodzi do szybkiego wzrostu potencjału IKs
prostownik
do mniej więcej +30 mV. Po tej fazie szybkiego opóźniony
wzrostu potencjału występuje dłużej trwająca faza IKr
plateau (szczególnie charakterystyczna dla mięś- prostownik
niówki serca), która jest głównie spowodowana napływowy IK1
zwiększoną przepuszczalnością błony komórkowej prąd If
dla jonów wapnia, natomiast zmniejszoną dla jonów rozrusznika
potasu. W wyniku tego efekty wywierane przez po- wymiennik
wolny dokomórkowy napływ Ca2+ oraz odpowiedni Na+, Ca2+
wypływ K+ z komórki są takie same. Dopiero gdy Na+, K+-
przepuszczalność dla Ca2+ zmniejszy się, a dla K+ ATPaza
znowu wzrośnie, dochodzi do pełnej repolaryzacji.
Potencjał czynnościowy w komórkach mięśnia ser- Ryc. B 4.3-4. Potencjał czynnościowy serca z odpowiedzialnymi
cowego trwa w całości ok. 300 ms, tzn. prawie sto kanałami jonowymi (zmodyfikowano wg Rodena).
MUTSCHLER-2009.indd 599 2010-01-07 22:13:47
600 Krew
Sprzężenie elektromechaniczne. Podobnie jak kurczliwych, oddziałują z troponiną C i w ten sposób
w przypadku innych mięśni, proces pobudzenia roz- umożliwiają wiązanie miozyny do aktyny, co prowa-
przestrzeniający się w sercu wywołuje skurcz włó- dzi do skrócenia włókien i skurczu. Pobranie jonów
kien mięśniowych. Aktywację aparatu kurczliwego wapnia przez retikulum sarkoplazmatyczne pro-
wywołaną przez potencjał czynnościowy określa się wadzi do wygaszenia sygnału, w czym ważną rolę
w tym przypadku jako sprzężenie elektromechanicz- odgrywa ATPaza wapniowa retikulum endoplazma-
ne. Kluczową rolę w tym procesie odgrywają jony tycznego (SERCA). Na aktywność SERCA wpływa
wapnia. fosfolamban, który poprzez fosforylację powoduje
W trakcie depolaryzacji jony Na+ i Ca2+ przecho- wzrost jej aktywności. Uwolniona część wapnia, któ-
dzą ze śródmiąższu do miocytów przez kanały sodo- ra nie zostanie pobrana przez SERCA, jest ponownie
we lub kanały wapniowe typu L. Dodatkowo, dzięki transportowana na zewnątrz komórki za pomocą wy-
napływowi jonów wapnia, z retikulum sarkoplazma- miennika Na+/Ca2+ (białka NCX). NCX wymienia
tycznego uwalniane są jeszcze większe ich ilości, przy trzy jony sodu na jeden jon wapnia.
czym duże znaczenie w tym procesie odgrywa recep-
tor rianodynowy. Po uwolnieniu z retikulum sarko- Procesy zachodzące podczas sprzężenia elektrome-
plazmatycznego jony Ca2+ dyfundują do elementów chanicznego zestawiono na ryc. 4.3-5.
+
AP [Ca2 ]i
przestrzeń
zewnątrzkomórkowa
skurcz
sarkolemma 200 ms cewka
poprzeczna
receptor receptor
rianodynowy rianodynowy
retikulum
Ca2+ sarkoplazmatyczne Ca2+
Ca2+ Ca2+
ATP
ATP
Ca2+ SERCA SERCA Ca2+
Ca2+
P
aktyna
PKA
miozyna
cAMP sarkomer
AC
ATP 3Na+ 2K+
Gs
β1
ATP ATP
skurcz
rozkurcz
Ca2+ 3Na+ Ca2+
Ryc. B 4.3-5. Schemat sprzęgania elektromechanicznego w mięśniu sercowym. Wzrost wewnątrzkomórkowego stężenia Ca2+
(niebieskie strzałki) dzięki napływowi Ca2+ przez niezależne od napięcia kanały wapniowe typu L i następujące uwalnianie Ca2+
z retikulum sarkoplazmatycznego z udziałem receptora rianodynowego prowadzi do skurczu. Uwolnienie Ca2+ z cytoplazy (czer-
wone strzałki) odbywa się poprzez SERCA w retikulum sarkoplazmatycznym albo przez wymiennik Na+/Ca2+ oraz ATPazę Ca2+
w błonie plazmatycznej i skutkuje relaksacją. Wykres nad ryciną pokazuje czasowy przebieg potencjału czynnościowego, następu-
jące uwalnianie jonów Ca2+ oraz skurcz. Podane stężenie cytozolowe Ca2+ wynoszące 10–7 M odnosi się do rozluźnionej komórki
mięśnia sercowego. AC – cyklaza adenylanowa, AP – potencjał czynnościowy, PKA – kinaza białkowa A, SERCA – ATPaza wapniowa
retikulum endoplazmatycznego.
MUTSCHLER-2009.indd 600 2010-01-07 22:13:47
Krew 601
W ten sposób potencjał czynnościowy odgrywa po- staje dostarczona większa ilość krwi, a tym samym
dwójną rolę: więcej tlenu.
W spoczynku zapotrzebowanie 100 g tkanki serca
■ powoduje uwalnianie jonów wapnia (efekt wyzwa-
na tlen wynosi około 10 ml/min, natomiast w wyni-
lający),
ku zwiększenia przepływu krwi w tętnicach wieńco-
■ wywołuje napływ jonów wapnia w celu napełnienia wych maksymalna ilość dostarczanego tlenu może
magazynów (efekt napełnienia). wzrosnąć do mniej więcej 65 ml/min. Zatem rezerwa
wieńcowa jest około pięć razy większa od zapotrze-
Krążenie wieńcowe. Krew jest doprowadzana bowania spoczynkowego.
do serca przez dwie tętnice wieńcowe, które mają
swój początek w aorcie tuż za zastawkami półksię- W autoregulacyjnym zwiększeniu przepływu krwi
życowatymi. przez tętnice wieńcowe pośredniczy przede wszyst-
Przez lewą tętnicę wieńcową (arteria coronaria kim adenozyna. Niedobór tlenu w mięśniu sercowym
sinistra), zaopatrującą w krew większość mięśniów- w wyniku niedostatecznej resyntezy ATP prowadzi
ki lewej komory, przepływa 4/5 krwi doprowadzanej do uwalniania adenozyny, która wywołuje rozszerze-
do naczyń serca. Początkowo lewa tętnica wieńcowa nie naczyń wieńcowych. Również aktywacja układu
biegnie między lewym uszkiem sercowym a pniem współczulnego wywołuje rozszerzenie tętnic wień-
płucnym, a następnie rozgałęzia się na gałąź okala- cowych.
jącą (ramus circumflexus) i gałąź międzykomorową
przednią (ramus interventricularis anterior). Gałąź Termin mięsień sercowy jako element oporu tęt-
Serce i układ krążenia
okalająca biegnie w bruździe wieńcowej do tylnej nic wieńcowych oznacza ciśnienie wywierane przez
powierzchni serca, natomiast gałąź międzykomoro- mięsień sercowy na tętnice wieńcowe. Wskutek ryt-
wa przednia w bruździe podłużnej w dół. micznego przebiegu czynności serca przepływ przez
Prawa tętnica wieńcowa (arteria coronaria dex- tętnice wieńcowe nie zachodzi w sposób ciągły, lecz
tra) rozpoczyna się pod prawym uszkiem sercowym wykazuje rytmiczne wahania zależne od faz skurczo-
i dalej biegnie w bruździe wieńcowej. Na tylnej po- wych. W tętnicach podwsierdziowych lewej komory
wierzchni serca rozgałęzia się ona na gałąź między- podczas skurczu mięśnia sercowego w następstwie
komorową tylną, biegnącą w tylnej bruździe podłuż- ściśnięcia tętnic przez kurczącą się mięśniówkę serca
nej do koniuszka serca. nie może płynąć krew. Zatem część mięśnia sercowe-
Układ żył serca jest podobny do układu jego tęt- go jest zaopatrywana w krew głównie w czasie fazy
B4
nic. Krew z żył zbiera się w zatoce wieńcowej (sinus rozkurczowej. Z tego powodu rozkurcz mięśniówki
coronarius), która uchodzi do prawego przedsionka. serca oraz czas trwania fazy rozkurczowej mają de-
Tylko niewielka część krwi żylnej napływa poprzez cydujący wpływ na przepływ krwi przez mięśniówkę
małe żyły bezpośrednio do jam serca. serca.
Ilość krwi przepływającej w danym przedziale
czasu przez tętnice wieńcowe zależy przede wszyst-
kim od: 4.3.2. Niewydolność serca
■ ciśnienia perfuzyjnego, tzn. od ciśnienia krwi w tęt-
nicach wieńcowych, 4.3.2.1. Podstawy patofizjologiczne
■ oporu tętnic wieńcowych.
Według definicji Światowej Organizacji Zdrowia
termin niewydolność serca oznacza ograniczoną
Drugi czynnik składa się z dwóch elementów, naczyń
zdolność wytrzymania obciążeń fizycznych z powodu
krwionośnych i mięśnia sercowego (element pozana-
wykrywalnych zaburzeń czynności serca. Przy napły-
czyniowy).
wie dostatecznej ilości krwi żyłami serce jako pompa
nie jest w stanie wykonać pracy potrzebnej do zaopa-
Opór naczyniowy wynika z napięcia mięśniówki
trzenia organizmu w krew, czyli pojemność minuto-
gładkiej tętnic wieńcowych: zmniejszone napięcie
wa serca jest za mała.
wiąże się ze zwiększeniem przepływu krwi. Serce
z prawidłowymi tętnicami wieńcowymi dysponuje
dużą rezerwą wieńcową (zdefiniowaną jako maksy- Według nowszej definicji (Packer, 1998), ułatwiającej lep-
sze zrozumienie nowych form leczenia niewydolności ser-
malny przepływ krwi przez tętnice wieńcowe w po- ca, stwierdzono, że przewlekła niewydolność serca jest zło-
równaniu z przepływem podstawowym), tzn. w razie żonym zespołem klinicznym, uwarunkowanym zaburzenia-
potrzeby całemu sercu lub obszarowi z hipoksją zo- mi czynności serca, który charakteryzuje się występowa-
MUTSCHLER-2009.indd 601 2010-01-07 22:13:48
602 Krew
niem hemodynamicznych, nerkowych i neurohumoralnych ność serca. Pozasercowe powikłania w przebiegu
mechanizmów kompensacyjnych. przewlekłej choroby serca (np. wysoka gorączka,
tyreotoksykoza) są innymi możliwymi przyczynami
W zależności od tego, których części serca dotyczą ostrej niewydolności serca.
zmiany patologiczne, rozróżnia się niewydolność Możliwym podłożem przewlekłej niewydolności
prawej lub lewej połowy serca albo prawej i lewej serca są przede wszystkim choroba wieńcowa, nad-
polowy serca (uogólnioną). Według czasu, w którym ciśnienie, kardiomiopatie i zaburzenia rytmu serca.
powstaje lub istnieje niewydolność serca, rozróżnia Rzadziej przyczynami są nabyte wady serca, toksycz-
się ostrą i przewlekłą niewydolność serca. ne uszkodzenia serca (np. przez alkohol, rtęć, arsen),
Odpowiednio do stopnia ciężkości stanu można czy upośledzenie napełniania jam serca albo funkcji
rozróżniać niewydolność obciążeniową i niewydol- pompowania (np. przy wysięku osierdziowym lub za-
ność spoczynkową albo (wg NYHA) niewydolność ciskającym zapaleniu osierdzia) (zob. ryc. B 4.3-6).
od I do IV stopnia (zob. tabela B 4.3-1). W następstwie niedostatecznej sprawności pom-
powania zostają uruchomione różne mechanizmy
Jedynie w przypadku, kiedy niewydolność serca jest uwa- kompensacyjne, których zrozumienie wyjaśnia
runkowana zmniejszeniem kurczliwości mięśnia sercowe- właściwość niewydolności serca polegającą na tym,
go, występuje niewydolność mięśnia sercowego. Termin
niewydolność serca nie jest równoznaczny z „niewydolnoś- że jest to postępujący proces chorobowy. Tak więc
cią mięśnia sercowego”; niewydolność mięśnia sercowego przewlekłej niewydolności serca z powodu zmniej-
jest jedynie jedną z postaci niewydolności serca, jednak szonej objętości wyrzutowej serca, która prowadzi
często występującą i szczególnie ważną. do zmniejszenia ilości krwi przepływającej przez
układ tętniczy oraz przez nerki, towarzyszy długo
Epidemiologia i rokowanie. Rocznie w USA poja- utrzymująca się aktywacja układu renina-angioten-
wia się 400 000 nowych zachorowań na niewydol- syna-aldosteron (RAAS). Aktywacja RAAS wywo-
ność serca i jest to jedno z najczęstszych rozpoznań łuje zwężenie naczyń oraz zatrzymanie NaCl i wody,
w zakresie chorób wewnętrznych. 2–5% ludności co ułatwia powstawanie obrzęków. Aktywność reni-
w wieku między 65. i 75. r.ż. oraz prawie 10% w wie- ny w osoczu stanowi parametr prognostyczny niewy-
ku powyżej 80. r.ż. choruje na niewydolność serca. dolności serca: im wyższa jest aktywność reniny, tym
Przy ciężkiej niewydolności serca (NYHA IV, tabela gorsze jest rokowanie.
B 4.3-1) śmiertelność w okresie 12 miesięcy może Jednocześnie w wyniku stymulacji tylnego pła-
wynosić nawet do 50% i odpowiada pod tym wzglę- ta przysadki dochodzi do uwalniania większej ilości
dem zaawansowanym chorobom nowotworowym. adiuretyny (wazopresyny), która poprzez aktywację
receptorów V2 w kanalikach zbiorczych nefronów
Przyczyny. Możliwą przyczyną ostrej niewydolności powoduje zatrzymanie wody. Istnieje również hipote-
serca jest przede wszystkim zanik czynności większej za, że receptory V1 przyczyniają się do uogólnionego
części tkanki mięśnia sercowego z powodu zawału zwężenia naczyń krwionośnych.
mięśnia sercowego lub, rzadziej, rozsiane zapalenie Ponadto u pacjentów z niewydolnością serca na-
mięśnia sercowego. Poza tym ostre przeciążenie ser- stępuje znaczne zwiększenie stężenia aldosteronu
ca, np. przy nagłym wzroście ciśnienia w krążeniu w osoczu. Poza aktywacją układu RAAS przyczyną
płucnym z powodu zatoru płucnego lub rozerwania tego jest także zmniejszenie wątrobowego klirensu
się zastawki serca, może wywołać ostrą niewydol- aldosteronu w następstwie zmniejszonego przepływu
Tabela B. 4.3.1. Podział przewlekłej niewydolności sercowej wg New York Heart Association (NYHA)
I. Choroba serca bez ograniczenia sprawności fizycznej. Codzienne obciążenie fizyczne nie wywołuje nadmiernego wyczerpania,
zaburzeń rytmu serca, duszności czy dusznicy bolesnej.
II. Choroba serca z niewielkim ograniczeniem sprawności fizycznej. Brak dolegliwości w spoczynku.
Większe obciążenie fizyczne powoduje wyczerpanie, zaburzenia rytmu serca, duszność lub dusznicę bolesną.
III. Choroba serca z większym ograniczeniem sprawności fizycznej przy codziennych czynnościach.
Brak dolegliwości w spoczynku. Niewielkie obciążenie fizyczne wywołuje wyczerpanie,
zaburzenia rytmu serca, duszność lub dusznicę bolesną.
IV. Choroba serca z dolegliwościami podczas wszystkich czynności fizycznych oraz w spoczynku. Konieczność leżenia w łóżku.
MUTSCHLER-2009.indd 602 2010-01-07 22:13:48
Krew 603
IHD, nadciśnienie,
kardiomiopatia,
wada zastawek i. in.
systoliczne i/lub
diastoliczne zaburzenie
glikozydy nasercowe funkcji przedsionka
zmniejszona zdolność zwiększone obciążenie
wyrzutowa serca wstępne, zwiększone
tachykardia ciśnienie żylne
skurcz
naczyń
glikozydy zmniejszona powstawanie odmy
nasercowe perfuzja narządów,
dysfunkcja baroreceptorów
Serce i układ krążenia
diuretyki
bloker AT1
aktywacja układu aktywacja zatrzymanie sodu
współczulnego RAAS i wody
β-bloker
inhibitory ACE
blokowanie receptorów β, angiotensyna II ↑
aktywacja kinazy B4
β-adrenoceptorów, blokery AT1
aktywacja inhibitorowego
białka G aldosteron ↑ antagoniści
aldosteronu
przebudowa mięśnia
sercowa, włóknienie
Ryc. B 4.3-6. Patomechanizmy przewlekłej niewydoności serca z możliwościami interwencji farmakologicznej. RAAS – układ renina-
angiotensyna-aldosteron.
krwi przez wątrobę. U pacjentów z niewydolnością także pośrednio, przez dodatkową aktywację układu
serca zmieniony jest czas działania aldosteronu. RAAS, do zwiększenia obciążenia następczego, a po-
U zdrowych ludzi efekt wywołany przez aldoste- nadto stanowi istotny czynnik w procesie powstawa-
ron w postaci zatrzymania sodu w organizmie zanika nia zaburzeń rytmu serca. Wielu pacjentów z zaawan-
w ciągu krótkiego czasu (aldosterone escape), nato- sowaną niewydolnością serca umiera nagle z powodu
miast u chorych na niewydolność serca utrzymuje się komorowych zaburzeń rytmu. Z tego względu znacz-
długo. ny wzrost stężenia katecholamin, a także wzrost
Inną istotną cechą przewlekłej niewydolności ser- aktywności reniny w osoczu stanowią niekorzystne
ca jest zwiększenie aktywności układu współczulnego czynniki prognostyczne.
za pośrednictwem baroreceptorów. Wysokie stężenia Prawdopodobnie inne mediatory również wpływa-
katecholamin prowadzi nie tylko bezpośrednio, lecz ją na przebieg choroby: np. w przebiegu niewydol-
MUTSCHLER-2009.indd 603 2010-01-07 22:13:48
604 Krew
ności serca synteza NO – substancji rozszerzającej wydolności serca dochodzi do zaburzenia skurczów
naczynia – może być ograniczona. Przede wszystkim mięśnia sercowego.
u pacjentów z ciężką niewydolnością serca wzrasta Natomiast przy rozkurczowym zaburzeniu czyn-
wytwarzanie endoteliny – endogennej substancji ności następuje zmniejszenie podatności lewej komo-
zwężającej naczynia. ry, tzn. wzrost sztywności mięśniówki. Najczęstszą
Wiele badań wykazało, że u pacjentów z niewy- przyczyną rozkurczowego zaburzenia czynności
dolnością serca liczba sprawnych czynnościowo ko- jest zwiększenie ilości tkanki łącznej w mięśniu ser-
mórek mięśnia sercowego ulega dalszemu zmniej- cowym pojawiające się po obumarciu miocytów, za-
szeniu w wyniku zaprogramowanej śmierci komórek zwyczaj wskutek nadciśnienia.
(apoptozy). Wymiar i znaczenie apoptozy w prze-
biegu niewydolności serca stanowią jednak kwestię Objawy niewydolności serca. Niewydolność pra-
kontrowersyjną. wej połowy serca prowadzi do wzrostu ciśnienia
w prawym przedsionku i dużych żyłach w krąże-
Ogólnie można stwierdzić, że serce ograniczone pod wzglę- niu dużym, tzn. zastoju krwi w dużym krwiobiegu.
dem swojej czynności ulega dalszemu uszkadzaniu przez Następstwem są obrzęki określonych części ciała.
różne czynniki humoralne:
Obrzęki pojawiają się głównie wokół kostek, może
■ Zwiększona aktywność układu RAAS i uwarunkowana też dojść do uwarunkowanego zastojem nagroma-
tym stymulacja receptora AT1 prowadzi do aktywacji ki-
naz wewnątrzkomórkowych (ras, p38, c-jun). Wskutek
dzenia się płynu w jamie brzusznej (wodobrzusza).
wzmożonej ekspresji embrionalnych genów dochodzi na- Typowym zjawiskiem jest również powiększenie się
stępnie do postępującego przerostu serca. Angiotensyna wątroby (wątroba zastoinowa) oraz wydalanie białka
II może także indukować apoptozę. Inhibitory acetylo- z moczem (proteinuria zastoinowa).
cholinoesterazy lub blokery receptora AT1 działają anta- W pozycji leżącej następuje mobilizacja płynu
gonistycznie w stosunku do złożonych działań wywiera-
nych przez angiotensynę II.
obrzękowego. Prowadzi to do nasilonej produkcji
i częstego oddawania moczu w nocy (nykturii).
■ Silnie podwyższone stężenia aldosteronu (aż do 20-
krotnie przewyższających normę) wywołują prolifera- Znaczne zmniejszenie zdolności pompowania prawej po-
cję fibroblastów, która może prowadzić do zwłóknienia łowy serca z powodu jej niewydolności prowadzi również
okołonaczyniowego. Ze względu na to, że aldosteron do obniżenia się ilości krwi dostarczanej do lewej połowy
jest również wytwarzany miejscowo i z tego powodu, serca. Bezpośrednim efektem jest spadek pojemności mi-
mimo podania inhibitorów acetylocholinoesterazy, miej- nutowej serca. Może to prowadzić do spadku ciśnienia tęt-
scowo mogą występować jego duże stężenia co daje pod- niczego w krążeniu dużym, co dodatkowo powoduje niedo-
stawę do leczenia niewydolności serca z użyciem jego stateczne ukrwienie tkanek w tej części układu krążenia.
antagonistów.
■ Zwiększona impulsacja układu współczulnego wraz
z podwyższonymi stężeniami katecholamin powodują
Przy ostrej niewydolności lewej połowy serca wzra-
obniżenie progu wrażliwości receptorów β1. Wzrost ak- sta ciśnienie w lewym przedsionku i w żyłach płuc-
tywności kinazy receptora adrenergicznego β powoduje nych. Zastój krwi w naczyniach krwionośnych płuc
rozprzęganie receptorów od rozpoczętego procesu prze- prowadzi do wzrostu ciśnienia w krążeniu małym,
twarzania sygnału. Dochodzi do intensywnego wytwa- wskutek czego następuje wzmożone przechodzenie
rzania inhibitorowych białek G, a aktywność cyklazy
adenylanowej ulega zmniejszeniu. Katecholaminy in-
płynu z naczyń włosowatych płucnych do tkanki śród-
dukują przerost, przebudowę i stres oksydacyjny w ko- miąższowej i pęcherzyków płucnych.
mórkach mięśnia sercowego. Są one uważane za czyn- Pojawia się obrzęk płuc, powodujący ciężkie zabu-
niki wyzwalające zaprogramowaną śmierć komórek, rzenia wentylacji i wymiany gazów oddechowych,
prowadzącą do obumarcia dalszych kardiomiocytów. a także uczucie duszności (asthma cardiale).
Tym zjawiskom można przeciwdziałać podając leki β-
adrenolityczne.
Przewlekła niewydolność lewej połowy serca
objawia się dusznością (trudnościami w oddychaniu,
■ Poza tym, w odpowiedzi na rozciąganie, kardiomiocyty którym towarzyszy subiektywne odczuwanie duszno-
syntetyzują tzw. mózgowy peptyd natriuretyczny (BNP).
Wraz z przedsionkowym peptydem natriuretycznym ści), która może nasilić się aż do orthopnoe (duszno-
(ANP) i peptydem natriuretycznym C (CNP) należy on ści występującej przede wszystkim w pozycji leżącej,
do rodziny endogennych neuropeptydów, które posiadają zmuszającej do przybrania pozycji wyprostowanej),
właściwości wazodylatacyjne, natriuretyczne i diuretycz- a także prowadzić do sinicy (sine zabarwienie skóry
ne. Rekombinowane BNP jest dostępne w USA pod na- i błon śluzowych wskutek niedostatecznego nasy-
zwą nezyrytyd.
cenia krwi tlenem) oraz uwarunkowanego zastojem
krwi zapalenia oskrzeli. Przewlekła niewydolność le-
Jednostki chorobowe (skurczowa i rozkurczowa wej połowy serca prowadzi, podobnie jak niewydol-
niewydolność serca). W przypadku skurczowej nie- ność prawej połowy serca, do przerostu mięśnia ser-
MUTSCHLER-2009.indd 604 2010-01-07 22:13:49
Krew 605
cowego i zwiększenia objętości krążącej krwi (zob. 4.3.2.2.1. Diuretyki
wyżej). Dodatkowo przy niedostatecznym zaopa-
trzeniu narządów w tlen może powstać czerwienica,
Diuretyki są często stosowane w leczeniu niewydol-
która z powodu zwiększenia lepkości krwi powoduje
ności serca, ponieważ stymulują wydalanie chlor-
dalszy wzrost obciążenia serca.
ku sodowego i płynów z organizmu i dzięki temu
Najczęstsze końcowe stadium chorób, które pier-
zmniejszenie obciążenia wstępnego i następczego.
wotnie doprowadzają do uszkodzenia lub przecią-
Diuretyki pętlowe wykazują dobrą skuteczność
żenia lewej połowy serca, stanowi przejście procesu
przy ostrej niewydolności serca. Rozszerzenie żył
chorobowego w niewydolność lewej i prawej połowy
pojawiające się przed efektem diuretycznym po-
serca (uogólnioną niewydolność serca), ponieważ
woduje zmniejszenie obciążenia wstępnego serca.
długotrwałe znaczne podwyższenie ciśnienia krwi
Nie jest ono, jak w przypadku nitratów, wywoła-
w krążeniu małym przy zastoju krwi w płucach wtór-
ne bezpośrednim oddziaływaniem na mięśniówkę
nie obciąża także prawą połowę serca.
gładką naczyń, lecz dochodzi do niego dzięki pros-
taglandynom uwalnianym do krwiobiegu z nerek.
Dlatego u pacjentów z ciężką niewydolnością nerek
4.3.2.2. Leki stosowane lub po nefrektomii diuretyki pętlowe stosowane w le-
w leczeniu niewydolności serca czeniu niewydolności serca nie działają natychmiast.
W późniejszym czasie zmniejszenie objętości osocza
Dawniej w farmakologicznym leczeniu niewydolno- w następstwie intensywnej diurezy przyczynia się
Serce i układ krążenia
ści serca zazwyczaj próbowano uzyskać normalizację do działania zmniejszającego obciążenie wstępne.
zdolności pompowania za pomocą substancji działa- Przykładowo w leczeniu niewydolności serca daw-
jących inotropowo dodatnio. W tym celu stosowano kowanie furosemidu wynosi 20–40 (–100) mg i.v.
przede wszystkim glikozydy nasercowe. Dzięki jed- Wszystkie grupy diuretyków są również bardzo
noznacznym wynikom badań klinicznych w ostatnim skuteczne w leczeniu przewlekłej niewydolności ser-
dziesięcioleciu nastąpiła jednak zmiana paradygmatu ca.
leczenia niewydolności serca: coraz częściej stosuje Poprawa stanu chorego pod względem zastoju
się substancje, które nie działają pierwotnie inotropo- krwi, zmniejszenia ciśnienia w jamach serca oraz
wo dodatnio, lecz których działanie polega bardziej w krążeniu małym, a także spadku naczyniowego
na ekonomizacji pracy serca i jednocześnie powo- oporu obwodowego, prowadzi do zwiększonej tole- B4
duje zniesienie szkodliwych humoralnych reakcji rancji na obciążenia.
kompensacyjnych (zwiększona aktywność układu Dobre wyniki uzyskuje się dzięki stosowaniu
współczulnego i układu RAAS). Do tej grupy należą diuretyków w połączeniu z inhibitorami enzymu
substancje czynne, które: konwertującego angiotensynę i glikozydami naser-
cowymi. Należy jednak brać pod uwagę fakt, że u pa-
■ przeciwdziałają zatrzymaniu chlorku sodowego
cjentów leczonych wcześniej diuretykami z powodu
i płynów w organizmie (diuretyki),
aktywacji układu RAAS inhibitory enzymu konwer-
■ oddziałują na układ RAAS (inhibitory enzymu tującego angiotensynę można podawać jedynie za-
konwertującego angiotensynę, blokery receptora czynając od małych dawek, a następnie stopniowo
AT1, antagoniści aldosteronu) lub je zwiększając i prowadząc odpowiednią kontrolę
(ryzyko dużego spadku ciśnienia krwi). Należy także
■ zmniejszają wzmożoną impulsację w układzie
brać pod uwagę to, że hipokalemia uwarunkowana
współczulnym (leki β-adrenolityczne).
stosowaniem diuretyków zwiększa wrażliwość serca
na działanie glikozydów nasercowych.
Poza stosowaniem leków, w leczeniu niewydolności
serca należy wykorzystać również wszystkie możli-
we środki niefarmakologiczne, takie jak: 4.2.2.2. Inhibitory enzymu
■ zmniejszenie nadwagi, konwertującego angiotensynę
(inhibitory ACE)
■ kontrola ilości dostarczanych płynów,
■ normalizacja poziomu cholesterolu, Liczne duże badania kliniczne wykazały, że stosowa-
nie inhibitorów ACE u pacjentów z niewydolnością
■ unikanie konsumpcji alkoholu i palenia tytoniu,
serca (NYHA II-IV) znacznie zmniejsza śmiertel-
■ regularne wykonywanie ćwiczeń fizycznych. ność, a także nasilenie objawów i liczbę koniecznych
MUTSCHLER-2009.indd 605 2010-01-07 22:13:49
606 Krew
hospitalizacji. Stosowanie inhibitorów ACE okazało receptory AT1, przede wszystkim w przypadku, kiedy
się korzystne również u pacjentów, u których objawy występują działania niepożądane inhibitorów ACE
nie występują lub w grupach pacjentów narażonych wywołane blokadą rozkładu bradykininy (np. odru-
wprawdzie na duże ryzyko z powodu innych chorób chowy kaszel spowodowany podrażnieniem).
układu sercowo-naczyniowego, ale u których (jesz- Obecnie coraz częściej dyskutuje się nad tym,
cze) nie występuje niewydolność serca. czy jednoczesne podawanie inhibitora ACE i blokera
Inhibitory ACE są więc lekiem z wyboru w lecze- receptorów AT1 niesie dodatkowe korzyści w porów-
niu pacjentów z ograniczoną funkcją lewej komory naniu z monoterapią (Dawki początkowe i docelowe,
(frakcja wyrzutowa < 35%), niezależnie od towarzy- zob. tabela B 4.3-3).
szącego zespołu objawów.
Ze względu na możliwość wystąpienia bardzo sil- Tabela B 4.3-3. Dawkowanie antagonistów AT1 przy przewle-
nego efektu przy pierwszym podaniu inhibitora ACE, kłej niewydolności serca (wg Hoppe)
początkowa dawka powinna być mała, a pacjenta
należy monitorować w czasie pierwszych godzin Antagonista AT1 Dawka początko- Dawka docelo-
po przyjęciu leku. Dawki początkowe i docelowe in- wa/dzień (mg) wa/dzień (mg)
hibitorów ACE przy przewlekłej niewydolności serca kandesartan 4 32
podano w tabela B 4.3-2.
eprosartan 300 400-800
Tabela B 4.3-2. Dawkowanie inhibitorów ACE przy przewlekłej
niewydolności serca (wg Hoppe) irbesartan 75 150-300
losartan 12,5 50-100
Inhibitor ACE Dawka początko- Dawka docelo-
wa/dzień (mg) wa/dzień (mg) olmesartan 10 40
benazepryl 2,5 2 × 5-10 telmisartan 20 40-80
kaptopryl 3 × 6,25 3 × 25-50 walsartan 2 × 40 2 × 160
enalapryl 2,5 2 x 10
fozynopryl 10 20
lizynopryl 2,5 5-20 4.3.2.2.4. Antagoniści aldosteronu
perindopryl 2 4 (spironolakton, eplerenon)
kwinapryl 2,5-5 5-10
Jak wspomniano wyżej, u osób z niewydolnością
ramipryl 1,25-2,5 10 serca stężenie aldosteronu jest podwyższone, a jego
działanie nasilone. Nawet po podaniu inhibitora ACE
trandolapryl 1 4
nie dochodzi do pełnej blokady produkcji aldosteronu
w nadnerczach. Być może jest on również uwalniany
za pośrednictwem innych mechanizmów (np. wy-
U wielu pacjentów początkowo wzrasta stęże- dzielanie aldosteronu zależne od potasu), dlatego
nie kreatyniny w osoczu, jednak później najczęściej z patofizjologicznego punktu widzenia w leczeniu
utrzymuje się ono na stałym poziomie. Z tego wzglę- niewydolności serca uzasadnione jest stosowanie an-
du w pierwszej fazie leczenia powinno się kontrolo- tagonistów aldosteronu (np. spironolaktonu lub eple-
wać czynność nerek. Z powodu zagrożenia hiperkale- renonu), przede wszystkim w ciężkich przypadkach.
mią, przynajmniej w ogólnej praktyce lekarskiej, na-
leży unikać stosowania diuretyków oszczędzających W badaniu RALES (Randomized Aldactone Evaluation
potas w połączeniu z inhibitorami ACE. Study – randomizowane badanie dotyczące stosowania
preparatu Aldactone) poza standardową terapią (inhibitor
ACE, diuretyk pętlowy, glikozyd nasercowy) dodatkowo
stosowano spironolakton w małych dawkach (25 mg/dz.)
4.3.2.2.3. Leki blokujące lub placebo u pacjentów z niewydolnością serca III lub IV
receptory angiotensynowe stopnia wg NYHA. Badanie wykazało 30-procentowy spa-
(blokery receptorów AT1, sartany) dek liczby zgonów przy znacznie mniejszej liczbie hospita-
lizacji i jednocześnie zmniejszenie objawów niewydolności
serca w grupie pacjentów leczonych spironolaktonem.
Alternatywą dla stosowania inhibitorów ACE w le- Pierwotnie spironolakton powstał i został zarejestrowa-
czeniu niewydolności serca stanowią leki blokujące ny jako diuretyk oszczędzający potas, natomiast wskazanie
MUTSCHLER-2009.indd 606 2010-01-07 22:13:49
Krew 607
do stosowania jego analogu – eplerenonu –ogranicza się Tabela B 4.3-4. Dawkowanie β-blokerów przy przewlekłej nie-
dotychczas do niewydolności serca. Eplerenon jest dodat- wydolności serca (wg Hoppe)
kowo wskazany do stosowania w standardowym leczeniu
u pacjentów z dysfunkcją lewej komory serca (LVEF ≤
40%) i klinicznymi objawami niewydolności serca po nie- β-bloker
β-bloker Dawka początko- Dawka docelo-
dawnym przebyciu zawału mięśnia sercowego (50 mg raz wa*/dzień (mg) wa/dzień (mg)
dziennie). metoprolol 12,5-25 200
bisoprolol 1,25 10
4.2.2.5. Blokery receptorów karwedilol 2 × 3,125 2 × 25
β-adrenergicznych (β-blokery) nebiwolol 1,25 10
* podwojenie dawki nie częściej niż co 14 dni,
Długo uważano, że stosowanie leków β-adrenolitycz- jeśli poprzednia dawka jest tolerowana
nych jest przeciwwskazane przy niewydolności ser-
ca ze względu na ich działanie inotropowo ujemne.
Jednak w wielu klinicznych badaniach wykazano,
średnio reguluje się układ RAAS. Pozytywne efekty
że w wyniku dodatkowego podawania β-blokerów
wywierane przez tę grupę leków polegają więc głów-
bez częściowej aktywności agonistycznej znacznie
nie na normalizacji homeostazy humoralnej i dlatego
obniża się śmiertelność. Z tego względu leki β-adre-
pojawiają się one z pewnym opóźnieniem. Po około
nolityczne należą obecnie do niezbędnych środków
Serce i układ krążenia
trzech miesiącach leczenia wzrasta frakcja wyrzuto-
w leczeniu niewydolności serca.
wa serca przy jednoczesnym spadku późnorozkur-
W pierwszych badaniach klinicznych dotyczących stosowa- czowego ciśnienia w komorach (ekonomizacja pracy
nia metoprololu brały udział jedynie małe grupy pacjentów, serca).
natomiast w opublikowanym w 1999 r. badaniu MERIT-HF
(interwencyjne badanie randomizowane dotyczące stoso- Dotychczas nie zostało wyjaśnione, czy w leczeniu
wania metoprololu CR/Zok przy zastoinowej niewydolno- pacjentów z niewydolnością serca powinno się sto-
ści serca) obejmującym 3991 pacjentów z niewydolnością
serca stopnia III lub IV wg NYHA wykazano zmniejszenie sować β-bloker hamujący dodatkowo receptory α
ogólnej umieralności o 35%. i w wyniku tego rozszerzający naczynia, jak np. kar-
wedilol, czy β-bloker wywołujący rozszerzenie na- B4
czyń krwionośnych na skutek uwalniania NO.
To zjawisko, które początkowo uważano za paradoks,
polegające na poprawie sprawności pompowania ser-
ca, a także rokowania przy długotrwałym podawaniu
leku wywierającego pierwotnie działanie inotropowo 4.2.2.6. Glikozydy nasereowe
ujemne, można wyjaśnić następująco: bezpośrednio
po rozpoczęciu terapii, która powinna być przepro- Czynnikiem wyzwalającym niewydolność serca jest
wadzana w sposób stopniowy pod ścisłą kontrolą, jego ograniczona czynność. Z tego względu należy
począwszy od bardzo małych dawek (ok. 10% dawki zwiększyć sprawność pompowania za pomocą sub-
końcowej), początkowo dochodzi – zgodnie z oczeki- stancji działających inotropowo dodatnio, aby w ten
waniami – do zmniejszenia frakcji wyrzutowej i ciś- sposób leczyć chorobę. Zgodnie z tym założeniem
nienia krwi. glikozydy nasercowe były przez długi czas lekiem
z wyboru w leczeniu niewydolności serca. Wyniki
W efekcie, pod wpływem β-blokera, liczba recepto- dużych badań klinicznych zmieniły jednak opinie
rów β, która uprzednio była zmniejszona z powodu dotyczące takiego sposobu leczenia. W badaniu DIG
nadmiernej stymulacji ze strony układu współczulne- (Digitalis Investigation Group, czyli Grupa Badawcza
go (odwrócenie regulacji w dół), ulega normalizacji, ds. Glikozydów Naparstnicy, 1997; 7788 pacjentów,
zmniejsza się rozprzężenie receptorów β od ich drogi kontrola placebo) nie wykazano wpływu podawania
przetwarzania sygnału oraz ekspresja genu kodują- digoksyny na umieralność, nastąpiła natomiast po-
cego białka G, wywierającego działanie hamujące. prawa pod względem wytrzymałości obciążeniowej
Zapobiega to również apoptozie komórek mięśnia ser- i liczby hospitalizacji. Tak jak dawniej, także obecnie
cowego (albo przynajmniej ją zmniejsza) indukowa- glikozydy nasercowe zaliczają się do standardowych
nej przez wysokie stężenia katecholamin. Dochodzi substancji wykorzystywanych w leczeniu niewydol-
poza tym do zmniejszenia wydzielania reniny wywo- ności serca, przy czym nadal nie są lekiem z wyboru
łanego przez układ adrenergiczny i w ten sposób po- oraz nie stosuje się ich jako monoterapii.
MUTSCHLER-2009.indd 607 2010-01-07 22:13:49
608 Krew
Działania. Glikozydy nasercowe: współczulnym, towarzyszącej niewydolności serca,
a także ze zmniejszeniem częstotliwości akcji serca.
■ zwiększają siłę skurczu mięśnia sercowego (działa-
Zmniejszenie częstości akcji serca, będące efektem
nie inotropowo dodatnie),
działania glikozydów nasercowych, oznacza ekonomi-
■ spowalniają częstość skurczów (działanie chrono- zację pracy serca. Jest to szczególnie korzystny efekt
tropowe ujemne), w przypadku występowania tachykardii nadkomoro-
wej lub arytmii. Utrudnienie przewodzenia pobudzenia
■ utrudniają przewodzenie bodźców (działanie dro-
często bywa korzystne w przypadku migotania/trzepo-
motropowe ujemne) oraz
tania przedsionków (umożliwia powrót rytmu zatoko-
■ ułatwiają heterotopowe wytwarzanie pobudzeń wego), jednak może zaburzać przewodzenie bodźców
w wyniku obniżenia progu pobudliwości (działa- z przedsionków do komór. Heterotopowe wytwarzanie
nie batmotropowe dodatnie), co początkowo może pobudzeń jest w każdym wypadku niepożądane, po-
prowadzić do skurczów dodatkowych, a po podaniu nieważ może prowadzić do komorowych skurczów
dawek toksycznych nawet do migotania komór. dodatkowych i tachykardii komorowej (zob. poniżej).
Działania pożądane i niepożądane są więc ze sobą ści-
Zwiększenie siły skurczów mięśnia sercowego, za- śle powiązane, a zakres stężeń terapeutycznych tej gru-
równo w przypadku zdrowego, jak i niewydolnego py substancji jest niewielki.
serca, prowadzi do zwiększenia maksymalnej szyb-
kości wzrostu ciśnienia w lewej komorze. W niewy- Założenia strukturalne. Glikozydy nasercowe za-
dolnym sercu dochodzi do zwiększenia pojemności wierają steroidy jako składnik aglikonowy (geniny,
minutowej, skrócenia nadmiernie rozciągniętych włó- np. digitoksygeniny). Jest on połączony wiązaniem
kien mięśniowych i zmniejszenia się patologicznie glikozydowym z dezoksycukrami (np. digitoksozą),
powiększonego serca. Opróżnianie komór jest spraw- których polarność w znacznym stopniu określa właś-
niejsze, tzn. zmniejsza się objętość krwi zalegająca ciwości farmakokinetyczne (zob. tabela B 4.3-5).
w komorach w czasie rozkurczu. Jednocześnie ule-
ga zwiększeniu rozkurczowe napełnianie jam serca,
Do glikozydów nasercowych, które mają znaczenie
co prowadzi do zmniejszenia się ciśnienia w żyłach.
terapeutyczne, należą:
Poprawa sprawności pompowania serca wiąże się też
ze zmniejszeniem nasilonej impulsacji w układzie ■ digoksyna,
Tabela B 4.3-5. Zestawienie glikozydów nasercowych
Glikozyd Genina Cukier Preparat handlowy
digitoksoza acetylodigi- glukoza
toksoza
digitoksyna digitoksygenina 3 – – Digimed,
Digimerck,
Digitoxin
β-acetylodigoksyna digoksygenina 2 1 – β-acetylodigoxin,
Digostada,
Novodial,
Stillacor
digoksyna digoksygenina 3 – – Digacin,
Digoxin,
Lanicor,
Lenoxin
metylodigoksyna digoksygenina 3*) – – Lanitop
gitoksygenina = 16β-hydroksy-digitoksygenina
digoksygenina = 12β-hydroksy-digitoksygenina
*) .....................................................................................
MUTSCHLER-2009.indd 608 2010-01-07 22:13:50
Krew 609
wewnątrzkomórkowego stężenia jonów sodowych,
stanowiący stały element błony komórkowej wymie-
O O niacz Na+/Ca2+, który w warunkach fizjologicznych
wymienia 3 zewnątrzkomórkowe jony Na+ na l we-
CH3 wnątrzkomórkowy jon Ca2+, transportuje mniej jo-
CH3 H
nów Ca2+ z komórki do przestrzeni zewnątrzkomór-
kowej. W efekcie w fazie rozkurczowej więcej jonów
H OH wapniowych gromadzi się w siateczce sarkoplazma-
HO tycznej, a w następującej potem fazie skurczowej
H
większa ilość jonów wapniowych uwalnia się z ma-
digitoksygenina gazynów. Skutkuje to nasileniem sprzężenia elektro-
mechanicznego oraz większą siłą skurczu (efekt ino-
tropowo dodatni).
Spadek wewnątrzkomórkowego stężenia jonów
H3C O OH
potasowych i obniżenie potencjału spoczynkowego
błony komórkowej prowadzą do zmniejszenia pręd-
HO
kości przewodzenia bodźców. Objawy toksyczne rów-
OH
nież można wytłumaczyć wpływem transportu jonów
przez błonę komórkową: wskutek jeszcze silniejsze-
D-digitoksoza
go hamowania ATP-azy Na+/K+ dochodzi do dalsze-
Serce i układ krążenia
go zmniejszenia wewnątrzkomórkowego stężenia jo-
nów potasowych, natomiast zawartość jonów wapnia
■ acetylodigoksyna, ulega takiemu zwiększeniu, że zostaje przekroczona
zdolność magazynowania siateczki śródcytoplazma-
■ metylodigoksyna,
tycznej i pojawiają się potencjały następcze, które
■ digitoksyna. mogą doprowadzić do dodatkowych skurczów.
Należą one do grupy glikozydów naparstnicy i są izo- Kinetyka. Glikozydy nasercowe o zastosowaniu te-
lowane z naparstnicy purpurowej (Digitalis purpu- rapeutycznym różnią się między sobą przede wszyst-
rea) i wełnistej (Digitalis lanata). Pierwotne główne kim pod względem:
B4
glikozydy z naparstnicy purpurowej to purpurea-gli-
■ możliwości resorpcyjnych,
kozydy A i B, które przy C3 aglikonu zawsze posia-
dają podstawnik tetrasacharydowy, składający się ■ okresu półtrwania w osoczu (i przez to tzw. wskaź-
z trzech cząsteczek digitoksozy i jednej cząsteczki nikiem zaniku działania w ciągu doby),
glukozy. W przypadku pierwotnych lanata-glikozy-
■ czasu działania i niebezpieczeństwa kumulacji,
dów, lanatozydów A, B i C, digitoksoza znajdująca
się obok glukozy jest acylowana. ■ głównej drogi wydalania (przez nerki, przez drogi
żółciowe).
Profil działania. Wszystkie glikozydy nasercowe
działają farmakodynamicznie tak samo, a różnią się W tabeli B 4.3-6 zebrano dane na temat charaktery-
między sobą jedynie pod względem charakterystyki styki farmakokinetycznej niektórych glikozydów na-
farmakokinetycznej! parstnicy.
Mechanizm działania. Wiązanie się glikozydów Najważniejsze reakcje biotransformacji digitoksyny
nasercowych z zależną od magnezu ATP-azą Na+/ to:
K+ prowadzi – zależnie od stężenia glikozydu –
■ hydroksylacja na C12 i przemiana w digoksynę,
do jej częściowego zablokowania, a w efekcie rów-
nież do hamowania transportu jonów sodu z wnętrza ■ odszczepienie cząsteczek digitoksozy,
komórek do przestrzeni zewnątrzkomórkowej oraz
■ uwodornienie podwójnego wiązania w pierście-
transportu jonów potasu z przestrzeni zewnątrzko-
niu laktonowym z wytworzeniem mniej aktywnej
mórkowej do wnętrza komórek.
dihydrodigitoksyny,
Prowadzi to do wzrostu stężenia jonów Na+ we-
wnątrz komórki oraz obniżenia wewnątrzkomór- ■ sprzęganie z wytworzeniem glukuronidów lub
kowego stężenia jonów K+. Ze względu na wzrost półestrów kwasu siarkowego.
MUTSCHLER-2009.indd 609 2010-01-07 22:13:50
610 Krew
Tabela B 4.3-6. Parametry glikozydów naparstnicy
Glikozyd Dawka podtrzy- Okres Biodostępność Droga
mująca (mg/dzień) półtrwania (h) (%) eliminacji
digoksyna 0,25 40 60-80 przez nerki (80%),
z kałem (20%)
β-acetylodigoksyna 0,3 40 90 przez nerki (80%)
z kałem (20%)
metylodigoksyna 0,2 48 79 przez nerki (60%)
metabolizm
do digoksyny
digitoksyna 0,05-0,07 168-192 >90 przez nerki (60%)
z kałem (40%)
Metabolity hydroksylowane oraz uboższe w cukier dolności serca jedynie glikozydami nasercowymi
wykazują jeszcze aktywność biologiczną, a wydzie- byłoby niewystarczające).
lane z żółcią koniugaty są częściowo ponownie roz-
■ tachykardiach i tachyarytmiach nadkomorowych,
szczepiane przez bakterie jelitowe. Uwolniony gliko-
a także trzepotaniu i migotaniu przedsionków (por.
zyd może ulegać ponownemu wchłanianiu (krążenie
rozdz. 4.3.3.1).
jelitowo-wątrobowe). Wyjaśnia to długi okres działa-
nia digitoksyny.
Ze względu na wąski zakres terapeutyczny, stosowa-
W cząsteczce digoksyny, podobnie jak w przypad-
nie glikozydów wymaga dużej ostrożności i szczegó-
ku digitoksyny, możliwe jest uwodornienie wiązania
łowej wiedzy na temat ich właściwości. Na podsta-
podwójnego w pierścieniu laktonowym, odszczepie-
wie częstości zaleceń lekarskich można wnioskować,
nie cukru, a także sprzęganie.
że glikozydy nasercowe bywają jeszcze ciągle stoso-
β-acetylodigoksyna ulega deacylacji w ścianie je-
wane bez wyraźnego wskazania.
lita i/lub w wątrobie, a następnie podlega dalszej dea-
cetylacji, jak digoksyna.
Dawkowanie. Stosuje się dawkowanie indywidual-
Metylodigoksyna (β-metylodigoksyna), która jest
ne, kontrolując stężenia osoczowe. W tabeli B 4.3-6
tylko częściowo demetylowana, charakteryzuje się nie-
podano parametry dotyczące dawkowania niektórych
co inną kinetyką niż digoksyna. Ze względu na dużą
glikozydów nasercowych.
lipofilność łatwo dociera do OUN, przez co istnieje
U pacjentów ze zwiększoną wrażliwością na gli-
większe zagrożenie ośrodkowymi działaniami niepo-
kozydy nasercowe należy je dawkować szczególnie
żądanymi.
ostrożnie.
W odniesieniu do wydalania glikozydów naparst-
Dotyczy to pacjentów, u których występują:
nicy należy zwrócić uwagę, że około 60% digoksy-
ny, ale tylko około 35% digitoksyny ulega wydala- ■ hipokalemia (silnie zwiększone[!] powinowactwo
niu nerkowemu. Przy niewydolności nerek należy glikozydów nasercowych do ATP-azy Na+/K+),
zmniejszyć dawkowanie digoksyny odpowiednio
■ hiperkalcemia,
do klirensu kreatyniny, co pozwoli uniknąć toksycz-
nych działań niepożądanych. W przypadku digitok- ■ zaburzenia wydalania glikozydów nasercowych
syny nie jest konieczne dostosowanie dawki. (przede wszystkim preparatów digoksyny) przy
niewydolności nerek,
Wskazania. Stosowanie glikozydów nasercowych
■ zapalenie mięśnia sercowego,
jest wskazane przy:
■ choroba wieńcowa (zob. poniżej) wskutek niedo-
■ niewydolności mięśnia sercowego, przede wszyst-
boru ATP spowodowanego niedostatecznym za-
kim przy umiarkowanie ciężkiej i ciężkiej prze-
opatrzeniem w tlen,
wlekłej niewydolności mięśnia sercowego. Można
je stosować jedynie jako dodatek do inhibitorów ■ podeszły wiek (powyżej 70 r.ż.),
ACE i β-blokerów (leczenie przewlekłej niewy-
■ niedowaga.
MUTSCHLER-2009.indd 610 2010-01-07 22:13:50
Krew 611
Oznaczanie stężenia w osoczu jest wprawdzie odpo- kardii komorowej, a nawet do stanów majaczenio-
wiednią metodą do sprawdzenia dawkowania, jednak wych oraz drgawek. Migotanie przedsionków może
nie zastępuje dokładnej obserwacji klinicznej. doprowadzić do śmierci.
W lżejszych zatruciach leczenie polega na natych-
Działania niepożądane. Jak już wspomniano, gliko- miastowym odstawieniu glikozydu nasercowego
zydy nasercowe cechują się wąskim zakresem tera- i monitorowaniu pacjenta.
peutycznym. Ryzyko działań niepożądanych w posta- Przy ciężkich przypadkach stosuje się środki zapo-
ci arytmii, stanu oszołomienia, bólów głowy, zabu- biegające wchłanianiu i powodujące przerwanie krą-
rzeń widzenia (zwłaszcza zaburzeń widzenia barw), żenia jelitowo-wątrobowego (monitorowane płukanie
a także – głównie pochodzenia ośrodkowego – nud- żołądka, podanie węgla aktywnego lub cholestyrami-
ności i wymiotów istnieje nawet jeżeli nie osiągnie ny). Przyspieszona eliminacja za pomocą hemoper-
się jeszcze poziomu pełnego działania. U pacjentów fuzji jest możliwa jedynie w przypadku digitoksyny,
w podeszłym wieku mogą wystąpić stany splątania ale nie digoksyny.
i halucynacje. Częstość występowania działań niepo- W razie ciężkich zatruć glikozydami naparstnicy
żądanych wynosi około 20%. skutecznym środkiem jest podanie surowicy owczej
Co ciekawe, w badaniach in vitro dla digitoksyny przeciw glikozydom naparstnicy (fragmenty przeciw-
wykazano występowanie efektu hamującego wzrost ciał Fab).
nowotworu. Badania epidemiologiczne wydają się Przy zaburzeniach rytmu typu bradykardii wskaza-
potwierdzać ten efekt. ne jest podanie atropiny, a w razie braku efektu lecze-
nia farmakologicznego, zastosowanie stymulatora.
Serce i układ krążenia
Przeciwwskazania. Stosowanie glikozydów naserco- Jeżeli występują zaburzenia rytmu typu tachykar-
wych jest przeciwwskazane przy ciężkich bradykar- dii, podaje się jony potasu we wlewie dożylnym (10
diach, komorowych zaburzeniach rytmu (szczególnie mmol KC1 przez l godzinę) pod stałą kontrolą EKG
w tachykardiach komorowych), przerostowo-zapo- i stężenia elektrolitów (przeciwwskazane przy hiper-
rowej kardiomiopatii oraz w przypadku podejrzenia kaliemii i bloku przedsionkowo-komorowym!).
zatrucia glikozydami nasercowymi.
Interakcje. Saluretyki, środki przeczyszczające, hor- 4.3.2.2.7. Katecholaminy
mony kory nadnerczy, insulina i amfoterycyna B na- przy ostrej niewydolności serca
silają działanie glikozydów nasercowych, ponieważ
B4
ich działanie prowadzi do utraty potasu. Sole wapnia Inotropowo dodatnie działanie katecholamin, pole-
podane pozajelitowo również zwiększają siłę dzia- gające na pobudzeniu receptorów β-adrenergicznych
łania glikozydów. Natomiast triamteren i amiloryd, w układzie współczulnym oraz stymulacji cyklazy
poprzez zatrzymywanie potasu, oraz cholestyramina, adenylanowej za pośrednictwem białka G, zostało
poprzez zmniejszenie wchłaniania glikozydów, osła- omówione wyżej. Katecholaminy są odpowiednim
biają ich działanie. Chinidyna podwyższa stężenie di- lekiem w leczeniu szczególnych przypadków niewy-
goksyny i jej pochodnych w osoczu (prawdopodobnie dolności serca przy intensywnej terapii, ponieważ wy-
na skutek współzawodnictwa o glikoproteinę P w jeli- wierają one nie tylko efekt inotropowo dodatni, lecz
cie). Werapamil i nifedypina, również inhibitory gliko- także chronotropowy dodatni, ułatwiają ektopowe
proteiny P, podwyższają stężenia digoksyny w osoczu tworzenie pobudzeń i wzmagają zużycie tlenu przez
za pośrednictwem takiego samego mechanizmu. mięsień sercowy. Cechują się również szybką utratą
Induktory enzymatyczne, jak np. rifampicyna, działania na skutek desensybilizacji. Przy ostrej nie-
poprzez indukcję glikoproteiny P mogą natomiast wydolności serca, przede wszystkim przy wstrząsie
powodować duże zmniejszenie stężenia digoksyny kardiogennym, pod ścisłą kontrolą parametrów ser-
w osoczu. cowych i układu krążenia stosuje się:
■ dopaminę,
Zatrucie glikozydami nasercowymi. Już po prze-
kroczeniu dawki potrzebnej do uzyskania pełnego ■ dobutaminę.
efektu leczniczego o 1,5–3 razy należy liczyć się
z wystąpieniem objawów zatrucia. Przy zatruciu gli-
kozydami nasercowymi wymienione działania niepo- 4.3.2.2.8. Inhibitory fosfodiesterazy
żądane pojawiają się w nasilonej postaci. W ciężkich
przypadkach może dochodzić do całkowitego bloku Do krótkotrwałej terapii u pacjentów z ciężką niewy-
przedsionkowo-komorowego, bradykardii lub tachy- dolnością serca, których nie udaje się leczyć za po-
MUTSCHLER-2009.indd 611 2010-01-07 22:13:51
612 Krew
mocą innych leków, pod ścisłą kontrolą (monitoro- zostać odpowiednio dopasowana. Enoksymon z okre-
wanie EKG) można stosować następujące inhibitory sem półtrwania w osoczu wynoszącym 4–6 godzin
fosfodiesterazy III: głównie ulega wydalaniu nerkowemu w postaci sul-
fonowanego metabolitu.
■ milrynon,
Jako dawki początkowe podaje się 50 μg/kg
■ enoksymon. milrynonu przez 10 minut, po tym następuje wlew
podtrzymujący – 0,375 – 0,75 μg/kg przez minutę.
Blokada fosfodiesterazy prowadzi do zwiększenia Dawka początkowa enoksymonu podawana powo-
stężenia cAMP, w wyniku czego na serce wywiera- li dożylnie wynosi 0,5 – 1 mg/kg, a wlew podtrzy-
ne są działania inotropowo dodatnie i chronotropowo mujący – 0,5 mg/kg z odstępem 30-minutowym.
dodatnie, a także dochodzi do rozszerzenia naczyń. Alternatywnie można podawać dawkę początkową
Wzrasta objętość wyrzutowa i pojemność minutowa we wlewie (90μg/kg/min).
serca, natomiast zmniejsza się późnorozkurczowa ob- Podobnie jak przy stosowaniu innych substancji
jętość lewej komory oraz opór naczyń obwodowych. czynnych, mogą pojawić się działania niepożądane,
Ze względu na profil działania tę grupę substancji zwiększające stężenie cAMP oraz uwarunkowane
określa się jako inodylatatory (substancje zwiększa- tym ektopowe tworzenie pobudzeń, czyli arytmia.
jące siłę skurczu mięśnia sercowego i jednocześnie Najczęściej występują arytmie komorowe, rzadziej
rozszerzające naczynia krwionośne – przyp. tłum.). nadkomorowe. Rozszerzenie naczyń powoduje ryzy-
Wbrew początkowym oczekiwaniom – działanie ino- ko wystąpienia hipotonii. Inne działania niepożądane
tropowo dodatnie i jednocześnie zmniejszanie obcią- to trombocytopenia, gorączka, zaburzenia żołądko-
żenia następczego uważano za szczególnie obiecu- wo-jelitowe, bóle głowy, bóle mięśniowe i wzrost
jące – nie nadają się one do terapii długookresowej, stężenia transaminaz. Przy podawaniu enoksymonu
ponieważ wywołują znaczne działania niepożądane obserwuje się oligurię i zakrzepowe zapalenie żyły
(zob. poniżej). w miejscu wstrzyknięcia.
Przeciwwskazania to ciężka zaporowa kardiomio-
O NH CH3 patia, wyraźna hipowolemia, tachykardia, tętniak ko-
mory serca oraz okres ciąży i karmienia piersią.
NC Dotychczas nie są znane żadne istotne interakcje.
N
4.3.2.2.9. Azotany
milrinon
Przy ostrej niewydolności serca z zastojem krwi
w płucach z grupy organicznych azotanów stosuje
We wszystkich dotychczas przeprowadzonych badaniach się nitroglicerynę w dawce 0,8–1,6 (–2,4) mg pod-
klinicznych stosowanie inhibitorów fosfodiesterazy pro- językowo co 5–10 min, a przy niedostatecznym
wadziło do zwiększenia liczby zgonów. Tym samym gli- działaniu, pod ścisłą kontrolą stanu pacjenta, 0,5–3
kozydy nasercowe są jedynymi substancjami działającymi
pozytywnie inotropowo, których długotrwałe stosowanie
(–6) mg/godz. dożylnie za pomocą pompy infuzyjnej.
u chorych z niewydolnością serca nie wpływa na skróce- Korzystny efekt jest uwarunkowany przede wszyst-
nie przeżywalności. Z tego względu stosowanie inhibito- kim zmniejszonym napływem krwi żylnej i obniże-
rów fosfodiesteraz jest ograniczone czasowo. Nie można niem obciążenia wstępnego, a także zmniejszeniem
jednak zapominać o tym, że zaawansowana niewydolność (mniej nasilonego) obciążenia następczego.
serca (NYHA III lub IV) wiąże się z występowaniem bar-
dzo męczących objawów. Znaczna poprawa jakości życia
Wspomagająco, przy przewlekłej niewydolności
w wyniku stosowania inodylatatorów coraz częściej wywo- serca, przede wszystkim przy współistniejącej nie-
łuje dyskusję na temat tego, czy mimo zwiększonego ryzy- wydolności wieńcowej, można stosować dwuazotan
ka wystąpienia zgonu nie jest jednak uzasadnione czasowo izosorbidu, monoazotan izosorbidu i czteroazotan
ograniczone podawanie tej grupy substancji przy niewydol- pentaerytrytylu.
ności serca.
Dla milrynonu okres półtrwania w osoczu wynosi 4.3.2.2.10. Leczenie zaburzeń
2–3 godziny. Substancja jest w większości (83%) rytmu serca w przebiegu
wydalana drogą nerkową w postaci niezmienionej. niewydolności serca
Niewielka część dawki ulega glukuronidacji. W przy-
padku pacjentów z niewydolnością nerek dawka musi
MUTSCHLER-2009.indd 612 2010-01-07 22:13:51
Krew 613
Przy postępującej niewydolności serca dużym prob- Duża częstość występowania zaawansowanej niewydolno-
lemem są komorowe arytmie (zob. poniżej) powo- ści serca i związane z nią dotychczas złe rokowanie spraw-
dzano również możliwości zastosowania metod niefarma-
dujące ryzyko nagłej śmierci z przyczyn sercowych.
kologicznych. Jedną z nich jest operacyjne zmniejszenie
Dodatkowo u pacjentów w podeszłym wieku leczenia przerośniętej lewej komory (częściowe wycięcie lewej ko-
może także wymagać migotanie przedsionków zwią- mory, operacja Battisty), prowadzące do zwiększenia frak-
zane z niewydolnością serca. Z tego względu należy cji wyrzutowej. Ponadto testuje się układy stymulatorów,
szczególnie dobrze kontrolować przeciwarytmiczną które mają poprawiać hemodynamikę.
terapię niewydolności serca oraz uwzględnić fakt, że,
wbrew oczekiwaniom, wiele leków przeciwarytmicz-
nych nie poprawia rokowania dotyczącego pacjentów 4.3.2.4. Leczenie ostrej
z niewydolnością serca, ale je pogarsza. W przypadku niewydolności serca
jednego leku przeciwarytmicznego klasy III (amioda-
ron) wykazano jednak, że zmniejsza zaburzenia ryt- W leczeniu ostrej niewydolności serca, w zależności
mu serca nie zwiększając przy tym śmiertelności. od ciśnienia skurczowego, stosuje się środki moczo-
pędne, substancje rozszerzające naczynia oraz leki
działające inotropowo dodatnio (zob. tabela B 4.3-
4.3.2.3. Plan etapowego 8).
leczenia przewlekłej
niewydolności serca
4.3.3. Zaburzenia rytmu serca
Serce i układ krążenia
Na podstawie dotychczasowych doniesień można i leki przeciwarytmiczne
stwierdzić, że skuteczne leczenie farmakologicz-
ne przewlekłej niewydolności serca stanowi złożo- 4.3.3.1. Podstawy patofizjologiczne
ny problem i podlegało ono znaczącym zmianom
w ostatnich latach. Aktualny plan etapowego leczenia Zmiany rytmu serca powstają na skutek oddziaływa-
przedstawiono w tabeli B 4.3-7. nia na tworzenie i/lub przewodzenie bodźców.
B4
Tabela B 4.3-7. Plan etapowego leczenia przewlekłej niewydolności serca (wg Hoppe i wsp.)
Lek Bezobjawowa dysfunkcja NYHA II NYHA III /
LV/NYHA I NYHA IV
inhibitor ACE wskazany wskazany wskazany
β-bloker (bez ISA) po zawale serca, wskazany* wskazany*
przy nadciśnieniu
tiazyd przy nadciśnieniu przy zatrzymaniu płynów wskazany,
albo zatrzymaniu płynów do potencjonowania działania
w wywiadzie diuretyków pętlowych
diuretyk petlowy – przy zatrzymaniu płynów wskazany
albo zatrzymaniu płynów
w wywiadzie
antagonista aldosteronu po zawale mięśnia sercowego po zawale mięśnia sercowego wskazany
bloker receptorów AT1 przy nietolerancji inhibitorów ACE przy nietolerancji inhibitorów ACE przy nietolerancji inhibitorów ACE
glikozydy nasercowe przy migotaniu przedsionków przy migotaniu przedsionków
z częstoskurczem z częstoskurczem,
w rytmie zatokowym
po poprawie
ciężkich objawów**
ISA – wewnętrzna aktywność sympatomimetyczna
* tylko u pacjentów ze stabilnym stanem hemodynamicznym, podawać powoli i pod ścisłą kontrolą
** z niskimi poziomami docelowymi we krwi
MUTSCHLER-2009.indd 613 2010-01-07 22:13:51
614 Krew
Tabela B 4.3-8. Farmakoterapia przy ostrej niewydolności serca (wg wytycznych European Society of Cardiology).
ostra niewydolność serca
podanie tlenu,
furosemidu,
triazotanu glicerolu
skurczowe ciśnienie krwi skurczowe ciśnienie krwi skurczowe ciśnienie krwi
> 100 mg Hg 85–100 mm Hg < 85 mm Hg
rozszerzenie naczyń substancje inotropowe
rozszerzenie naczyń oraz substancje inotropowe oraz dopamina i adrenalina
(dobutamina, inhibitory PDE)
przy efektywnym leczeniu:
doustne podawanie furosemidu/
inihibitora AC
Jeżeli u osoby dorosłej w spoczynku częstość akcji Tachykardie zatokowe występują przy wysiłku fizycznym
serca przekracza 100 uderzeń/min, wówczas mówi się i w stanach pobudzenia. Niewytrenowane serce reaguje
o tachykardii. Częstość akcji serca < 60 uderzeń/min na zwiększone wymagania nieekonomiczną tachykardią.
W czasie gorączki wzrost temperatury ciała o 1° C powodu-
określa się jako bradykardię. Termin arytmia ozna-
je zwiększenie częstości akcji serca o około 10 skurczów/
cza nieregularne skurcze mięśnia sercowego. Skurcz min. Tachykardię kompensacyjną, zazwyczaj wywołaną
dodatkowy pojawia się w wyniku działania bodź- przez układ współczulny, stwierdza się we wstrząsie, po za-
ca wyzwolonego poza prawidłowym rytmem serca, wale, przy niewydolności lub zapaleniu mięśnia sercowego
co powoduje przejściową zmianę prawidłowego ryt- oraz niedokrwistości. Ponadto tachykardia stanowi ważny
objaw nadczynności tarczycy.
mu podstawowego.
Następstwem tachykardii zatokowej jest początkowy
Zaburzenia tworzenia fal przewodzenia wywodzą- wzrost minutowej pojemności serca. Jeżeli jednak częstość
ce się z węzła zatokowego określa się jako nomoto- akcji serca przekracza wartość graniczną (210 minus wiek
powe, natomiast pochodzące z ośrodków wtórnych pacjenta w latach), dochodzi do pogorszenia hemodynami-
lub trzeciorzędowych lub z kurczącej się mięśniówki ki (zmniejszonego wypełniania komór) i przepływu krwi
przez tętnice wieńcowe (skrócony czas trwania rozkur-
jako ektopowe (heterotopowe). Zaburzenia ektopowe
czu).
mogą powstwać w przedsionkach (nadkomorowe) lub
w komorach (komorowe) (zob. tabela B 4.3-9). Bradykardie zatokowe występują u wyczynowych spor-
W zaburzeniach przewodzenia bodźców rozróżnia towców trenujących wytrzymałość, u których doszło
się zaburzenia w okolicy węzła zatokowego (przewo- do wzmożenia impulsacji nerwu błędnego. Są wyrazem
fizjologicznego procesu adaptacji. Patologiczne zmniej-
dzenie zatokowo-przedsionkowe), granicy między
szenie częstości akcji serca stwierdza się u pacjentów
przedsionkami i komorami (przewodzenie przedsion- ze wzmożoną impulsacją nerwu błędnego spowodowaną
kowo-komorowe) lub zaburzenia układu bodźcoprze- innymi przyczynami, np. wzrostem ciśnienia śródczaszko-
wodzącego komór. wego z powodu guza mózgu lub urazu mózgu. (W wyni-
Do nomotopowych zaburzeń tworzenia fal prze- ku podwyższonego ciśnienia śródczaszkowego dochodzi
do niedoboru tlenu w mózgu, na co układ współczulny re-
wodzenia należy tachykardia zatokowa, bradykardia
aguje z większą wrażliwością niż układ przywspółczulny.)
i arytmia, natomiast do zaburzeń ektopowych zalicza Poza tym zmniejszenie częstości akcji serca występuje przy
się różne formy skurczów dodatkowych, nadkomorową niedoczynności tarczycy i po przebyciu niektórych zakażeń
tachykardię napadową, tachykardie komorowe oraz (np. po durze plamistym lub zakażeniu wirusowym). Jeżeli
trzepotanie czy migotanie przedsionków i komór. nie nastąpi odpowiedni wzrost pojemności wyrzutowej,
MUTSCHLER-2009.indd 614 2010-01-07 22:13:52
Krew 615
Tabela B 4.3-9. Schemat podziału zaburzeń rytmu serca
zaburzenia powstawania pobudzeń zaburzenia przewodzenia pobudzeń
zatokowo- przedsionkowo-
nomotopowe ektopowe /przedsionkowe -komorowe wewnątrzkomorowe
nadkomorowe komorowe
tachykardia zatokowa, skurcze dodatkowe dodatkowe skurcze
baradykardia zatokowa, przedsionkowe, nadkomorowa komorowe, tachykardia
arytmia zatokowa tachykardia napadowa, komorowa, trzepotanie
trzepotanie przedsionków, komór, migotanie komór
migotanie przedsionków
Serce i układ krążenia
bradykardia prowadzi do zmniejszenia pojemności minu- jednak tylko część fal pobudzenia podlega przewodzeniu
towej serca. do komór (np. przewodzenie 2: l lub 3:l). Częściowy blok
przedsionkowo-komorowy (zob. poniżej) jest przy tym
Fizjologiczne arytmie zatokowe występujące – częściej uwarunkowany czasem refrakcji w komorowym układzie
u młodzieży niż ludzi w podeszłym wieku – w związku bodźcoprzewodzącym. Trzepotanie przedsionków powsta-
z oddychaniem (np. wydechowe obniżenie częstości, wde- je prawie zawsze na podłożu chorób serca ze zmianami
chowy wzrost częstości). Patologiczne arytmie zatokowe organicznymi, przede wszystkim miażdżycy tętnic wień-
obserwuje się w chorobie wieńcowej, ostrym zapaleniu cowych oraz po zawale mięśnia sercowego. Ponadto trze- B4
mięśnia sercowego i ekstremalnie wzmożonej czynności potanie przedsionków pojawia się przy wadach zastawek
tarczycy (tyreotoksykozy). Szczególną postać patologicz- powstałych w wyniku nadmiernego rozciągnięcia ściany
nej arytmii zatokowej stanowi zespół chorego węzła zato- przedsionka.
kowego (sick sinus syndrome), w przebiegu którego wystę- W przypadku migotania przedsionków (częstość 350–
pują różne zaburzenia (m.in. utrzymująca się bradykardia 600 uderzeń/min) liczne fale pobudzenia krążące po przed-
zatokowa, naprzemienna tachykardia i bradykardia, niedo- sionkach prowadzą do zniesienia skutecznych skurczów
stateczny wzrost częstości akcji serca przy obciążeniu). przedsionków. Zmieniający się blok przewodzenia bodź-
ców powoduje, że skurcze komór następują w nieregular-
Nadkomorowe napadowe tachykardie z częstością akcji nych odstępach (niemiarowość zupełna). Migotanie przed-
serca 150–220 uderzeń/min często sprowadza się do tzw. sionków obserwuje się przede wszystkim przy wadach
pobudzeń krążących. Dodatkowo, np. w zespole Wolffa- zastawek w lewej połowie serca, głównie w przypadku ste-
Parkinsona-White’a, poprzez dodatkową drogę przewodze- nozy mitralnej. (W wyniku zwężenia zastawki dwudzielnej
nia stanowiącą bezpośrednie połączenie przedsionków z ko- zwiększa się ciśnienie w lewym przedsionku, a nadmierne
morami, dochodzi do ponownej depolaryzacji przedsion- rozciągnięcie przedsionka wyzwala zaburzenie tworzenia
ków. Nadkomorowe napadowe tachykardie są albo dziedzi- bodźców.)
czone, albo występują po zapaleniach mięśnia sercowego,
przy niedoborze tlenu, hipokalemii oraz w niewydolności Komorowe skurcze dodatkowe występują u pacjentów
serca z towarzyszącym zastojem krwi w przedsionkach. z labilnością psychowegetatywną, a także u osób z chorobą
Duża częstość akcji serca skraca czas trwania rozkurczów serca na podłożu organicznym (np. miażdżyca tętnic wień-
i skurczów do tego stopnia, że obniża się sprawność pom- cowych, uszkodzenie mięśnia sercowego). Objawy kliniczne
powania serca. Dlatego często dochodzi do zmniejszenia zależą od tego, czy skurcze dodatkowe występują pojedyn-
ukrwienia ważnych dla życia narządów, zwłaszcza mózgu czo (maks. 10/min), z większą częstością (ponad 10/min)
i nerek. lub w salwach oraz czy są one monotopowe, tzn. wywodzą
się z jednego ośrodka lub politopowe, tzn. wywodzą się
Trzepotanie i migotanie przedsionków to stosunkowo z wielu ośrodków. Monotopowe skurcze dodatkowe wska-
częste zaburzenia tworzenia bodźców w przedsionkach zują na występowanie miejscowego uszkodzenia mięśnia
ze szczególnie wysoką częstotliwością tworzenia bodź- sercowego, a politopowe na rozległe uszkodzenie mięśnia
ców. sercowego. Skurcze dodatkowe komorowe są szczególnie
Podczas trzepotania przedsionków z częstością 200–300/ niebezpieczne, jeżeli występują one w tzw. fazie wrażli-
min ich czynność jest wprawdzie jeszcze skoordynowana, wej, czyli w pierwszej części załamka T w EKG, ponieważ
MUTSCHLER-2009.indd 615 2010-01-07 22:13:52
616 Krew
w ten sposób może zostać łatwo wywołane migotanie ko- (Cardiac Arrhythmias Suppresion Trial, badanie do-
mór (zob. poniżej). tyczące zwalczania arytmii), w którym stwierdzono,
że liczba nagłych zgonów z przyczyn sercowych oraz
Komorowe tachykardie występujące przy ciężkich orga-
nicznych uszkodzeniach serca, np. zawale mięśnia serco- umieralność ogólna w grupie pacjentów z zawałem
wego, stanowią ciągłe zagrożenie życia, ponieważ mogą mięśnia sercowego leczonej flekainidem i enkainidem
łatwo przechodzić w trzepotanie lub migotanie komór. była znacznie większa niż w grupie, której podawano
Wówczas nie jest już możliwe skoordynowane kurczenie placebo. Duże badania również potwierdziły zwięk-
się komór. W przypadku trzepotania komór jest wprawdzie
szoną umieralność w wyniku stosowania innych le-
jeszcze pompowana niewielka ilość krwi, mimo to docho-
dzi do utraty przytomności i wstrząsu. Migotanie komór ków przeciwarytmicznych klasy I i III. Dlatego leki
w wyniku braku pompowania krwi (czynnościowe zatrzy- przeciwarytmiczne należy stosować z zachowaniem
manie akcji serca) wywołuje w krótkim czasie zgon, o ile jak największej ostrożności, a wskazanie do ich po-
nie utrzyma się czynności pompowania za pomocą natych- dawania istnieje tylko wtedy, gdy występują wyraźne
miastowego zewnętrznego masażu serca do czasu zastoso-
subiektywne dolegliwości wskutek zaburzeń hemo-
wania innych środków leczniczych (defibrylacji).
Poza wymienionymi uszkodzeniami mięśnia sercowe- dynamiki lub arytmia jest oceniana jako szczególnie
go przyczyną trzepotania lub migotania komór może być niebezpieczna. Jednak nawet w takich przypadkach
niedobór tlenu, wychłodzenie organizmu, przedawkowanie leczenie środkami przeciwarytmicznymi przynosi
środków stosowanych do narkozy i in. Migotanie komór rzeczywiste korzyści pod względem rokowania jedy-
jest także najczęstszą przyczyną zgonu przy porażeniu prą-
nie u części pacjentów. Należy także uwzględnić fakt,
dem elektrycznym.
że u wielu pacjentów z zaburzeniami rytmu serca
Przy zaburzeniach przewodzenia bodźców upośledzone – zwłaszcza z izolowanymi skurczami dodatkowymi
jest przewodzenie lub rozprzestrzenianie się depolaryzacji – nie występuje żadna patologia serca i nie wymagają
w przedsionkach i/lub komorach. Przewodzenie bodźców oni leczenia środkami przeciwarytmicznymi. Z tego
może być przy tym opóźnione, częściowo lub całkowicie
względu przed zastosowaniem leku przeciwaryt-
zablokowane. Zgodnie z tym rozróżnia, się trzy stopnie
ciężkości zaburzeń przewodzenia bodźców: micznego niezbędne jest ustalenie szczegółowego
I stopień – opóźnienie przewodzenia bodźców, rozpoznania oraz dokładne monitorowanie pacjenta
II stopień – sporadyczne występowanie nieprzewodze- podczas prowadzenia leczenia.
nia bodźców z przedsionków do komór (częściowy blok),
III stopień – całkowite przerwanie przewodzenia (cał- Do leczenia ciężkich arytmii serca w coraz większej mie-
kowity blok). rze stosuje się niefarmakologiczne formy terapii. Należy
do nich przykładowo wszczepienie defibrylatora, który po-
Poszczególne stopnie nie są od siebie jednoznacznie odgra- przez układ czujników rozpoznaje zaburzenia rytmu serca
niczone. Przyczynami zaburzeń przewodzenia bodźców są: i w razie potrzeby generuje bodziec defibrylujący. Używanie
miażdżyca tętnic wieńcowych, zawał serca, hiperkalemia, tego rodzaju urządzeń poprawia rokowanie. Ponadto w no-
zapalenie mięśnia sercowego, a także przedawkowanie gli- wych badaniach wykazano, że stosowanie defibrylatora
kozydów naparstnicy czy chinidyny (zob. poniżej). z lekiem przeciwarytmicznym (np. sotalolem) zmniejsza
częstość generowania bodźca defibrylującego przez defi-
brylator (poprawa jakości życia) bez zwiększania śmiertel-
4.3.3.2. Leki przeciwarytmiczne ności. Poza tym elektrokonwersja przewlekłego migotania
przedsionków udaje się łatwiej przy jednoczesnym podaniu
leku przeciwarytmicznego (np. klasy III). W przyszłości
Leki przeciwarytmiczne są substancjami stosowany- być może częściej będą występowały wskazania do stoso-
wania farmakologicznej terapii przeciwarytmicznej w połą-
mi w celu normalizacji nieprawidłowej akcji serca. czeniu z metodami fizycznymi.
W zależności od rodzaju zaburzeń rytmu, stosowany
środek powinien:
■ zwiększać lub zmniejszać częstość akcji serca, 4.3.3.2.1. Leki stosowane
w leczeniu zaburzeń
■ hamować ektopowe tworzenie bodźców i/lub rytmu serca typu bradykardii
■ zwiększać lub zmniejszać prędkość przewodzenia
bodźców. W krótkotrwałym farmakologicznym leczeniu aryt-
mii typu bradykardii (włącznie z blokiem przedsion-
Należy jednak koniecznie zwrócić uwagę na to, kowo-komorowym) stosuje się:
że leki przeciwarytmiczne, szczególnie klasy I (zob.
■ agonistów receptorów β-adrenergicznych,
poniżej), działają także proarytmogennie, tzn. mogą
wywołać arytmie, u 20% leczonych pacjentów. ■ środki parasympatykolityczne.
Zostało to potwierdzone w 1989 r. w badaniu CAST
MUTSCHLER-2009.indd 616 2010-01-07 22:13:52
Krew 617
Jeśli terapia farmakologiczna z wykorzystaniem tych ■ leki przeciwarytmiczne klasy IV (antagoniści wap-
substancji nie jest skuteczna lub występują poważ- nia, blokery kanałów wapniowych).
niejsze zaburzenia przez dłuższy czas, to musi zostać
wszczepiony elektryczny stymulator. Poza substancjami należącymi do wymienionych klas,
dla wielu antyarytmików zidentyfikowano odpowied-
Agoniści receptorów β-adrenergicznych. Substan- nie kanały jonowe, przez blokadę których substancje
cje pobudzające receptory β, takie jak adrenalina, te mogą rozwijać swoje działanie. Związek między
izoprenalina i orcyprenalina, powodują szybsze nara- potencjałem czynnościowym a kanałami jowowymi
stanie potencjału czynnościowego oraz skracają czas odpowiedzialnymi za jego powstawanie pokazu-
trwania potencjału czynnościowego i czas refrakcji. je ryc. B 4.3-4. Rycina B 4.3-7 uwidacznia punkty
Te efekty są głównie wynikiem podwyższenia napły- uchwytu poszczególnych klas antyarytmików na po-
wu jonów wapnia do komórki w fazie plateau oraz tencjale czynnościowym oraz EKG.
przyspieszenia wypływu jonów potasu podczas fazy
repolaryzacji potencjału czynnościowego.
Już przy stosowaniu zalecanych dawek, a szcze- A C
gólnie w przypadku przedawkowania agonistów re-
1
ceptorów β-adrenergicznych, istnieje ryzyko arytmii 2 potencjał
wskutek zwiększonej pobudliwości mięśniówki ko- rozrusznika klasa II
0 (faza 4
mór (wzmaganie aktywności rozruszników ektopo- 3
wych). Inne działania niepożądane to drżenia mięś- faza 4
Serce i układ krążenia
niowe, stany lękowe i wzmożona potliwość. szybka
depolaryzacja klasa I
Parasympatykolityki. Zwiększenie częstości ak- (faza 0)
B QRS
cji serca można wywołać poprzez stymulację układu
współczulnego, a także przez blokadę nerwu błędnego. T
P faza plateau klasa IV
W tym celu leczniczo stosuje się atropinę (pojedyn- (faza 2)
cza dawka 0,5 mg), a także czwartorzędowy związek
aminowy bromek ipratropium (dawkowanie początko-
repolaryzacja
we 0,5 mg i.v. lub 5–15 mg p.o., przy terapii ciągłej klasa III
20–45 mg/dz. p.o.). Bromek ipratropium dopuszczono
PQ QT (faza 3) B4
do leczenia bradykardii zatokowych. Biodostępność
Ryc. B 4.3-7. A – sercowy potencjał czynnosciowy, B – EKG
leku podawanego drogą doustną wynosi 1,6 godziny,
z powierzchni ciała, C – punkty uchwytu działania leków prze-
wydalanie zachodzi głównie z żółcią. ciwarytmicznych.
4.3.3.2.2. Leki stosowane w leczeniu zaburzeń
rytmu serca typu tachykardii
4.3.3.2.2.1. Leki przeciwarytmiczne klasy I
i skurczów dodatkowych
Wspólną cechą leków przeciwarytmicznych kla-
Ze względu na dużą liczbę substancji stosowanych
sy I (zob. tabela B 4.3-10), które są również określane
w leczeniu arytmii typu tachykardii oraz ich zróż-
jako antagoniści sodu (blokery kanałów sodowych),
nicowanych mechanizmów działania, podzielono
leki przeciwarytmiczne stabilizujące błonę komórko-
je na różne klasy (wg Vaughana Williamsa). Zgodnie
wą lub przeciwmigotaniowe, jest to, że dzięki blo-
z tą klasyfikacją rozróżnia się:
kadzie szybkich kanałów sodowych (INa) prowadzą
■ leki przeciwarytmiczne klasy I (blokery kanałów one do zmniejszenia prędkości narastania potencjału
sodowych), czynnościowego, a tym samym do zmniejszenia szyb-
kości przewodzenia.
■ leki przeciwarytmiczne klasy II (blokery recepto-
Dodatkowo leki przeciwarytmiczne klasy I powo-
rów β),
dują wolniejsze narastanie potencjału w stymulatorze,
■ leki przeciwarytmiczne klasy III, substancje, prze- podwyższenie progu depolaryzacji oraz wydłużenie
dłużające czas trwania potencjału czynnościowego całkowitego czasu trwania repolaryzacji. Prowadzą
bez wpływania na aktywność rozruszników (leki także do zmniejszenia siły skurczu serca (działanie
blokujące kanał potasowy) oraz inotropowo ujemne).
MUTSCHLER-2009.indd 617 2010-01-07 22:13:52
618 Krew
Tabela B 4.3-10. Leki przeciwarytmiczne klasy I
Wzór strukturalny Nazwa Preparaty handlowe Okres Dawkowanie
międzynarodowa półtrwania (mg)
H2C H chinidyna Chinidin-Duriles 6 400-800 (VES)
800-1600 (VHF)
H3CO HO
N
H
HO ajmalina Gilurytmal 1,5 10-50 i.v.
OH
N
C2H5
N H H
CH3
OH detajmium Tachmalcor 13 75-300
HO
N(C2H5) 2
+ OH
N
C2H5
N HH
CH3
HO
CH3 prajmalina Neo-Gilurytmal 4-7 60-100
+
OH
N
C2H5
N HH
CH3
CH3 lidokaina Xylocain (Kardiologie), 0,8 50-100 i.v.,
NH C2H5 Xylocitin-cor potem 200 mg/h
N jako długotrwały
O C2H5 wlew
CH3
CH3 CH3 meksyletyna Mexitil 5-12 400-900
O
NH2
CH3
O flekainid Flecaidine, 20 100-200
F3C O NH Tambocor
NH
O CF3
O propafenon Propafenon, 3-17 450-900
Rytmofenon
O
C3H7
NH
OH
MUTSCHLER-2009.indd 618 2010-01-07 22:13:53
Krew 619
Poszczególne substancje cechują się natomiast ostrożnie. Aby uniknąć zwiększenia częstości skur-
różnymi właściwościami pod względem: czów komorowych, przede wszystkim na początku
leczenia, przy tachykardiach nadkomorowych zaleca
■ wpływu na czas trwania potencjału czynnościo-
się podawanie preparatów naparstnicy przed poda-
wego przez hamowanie repolaryzujących kanałów
niem leku przeciwarytmicznego klasy IA. U pacjen-
K+ (przedłużony czas trwania odcinka QT w EKG
tów bez uszkodzenia mięśnia sercowego efekt ino-
z powierzchni ciała),
tropowy ujemny ma niewielkie znaczenie, natomiast
■ sily i czasu trwania hamowania szybkiego doko- u pacjentów z niewydolnością serca może zagrażać
mórkowego napływu jonów sodu w fazie 0 poten- pracy serca. Z tego powodu przed podaniem leków
cjału czynnościowego (przedłużony interwał QRS przeciwarytmicznych działających inotropowo ujem-
w EKG z powierzchni ciała) oraz nie, które w grupie pacjentów z niewydolnością serca
należy stosować ze szczególną ostrożnością, niezbęd-
■ zależności działania od częstości akcji serca (use
ne jest leczenie niewydolności serca.
dependence).
Działaniem niepożądanym wszystkich substan-
cji działających podobnie do chinidyny, związanym
Tak samo, jak inne kanały jonowe, kanał sodowy może ze zmniejszeniem kurczliwości mięśnia sercowego,
znajdować się w trzech stanach czynnościowych (otwarty, jest obniżenie ciśnienia krwi. Do innych działań nie-
nieczynny, zamknięty), które występują kolejno po sobie pożądanych należą uwarunkowane antycholinergicz-
w ramach jednego potencjału czynnościowego. Leki prze- ną składową działania zaburzenia żołądkowo-jelito-
ciwarytmiczne klasy l wiążą się z kanałem sodowym, kiedy we, suchość w jamie ustnej, utrudnienie mikcji i za-
Serce i układ krążenia
znajduje się on w stanie otwartym. Jeżeli czas trwania wią-
zania się danej substancji z kanałem jest krótki (np. lidoka- burzenia akomodacji. U wielu pacjentów chinidyna
iny), efekt jej działania jest zauważalny tylko w przypadku wywołuje biegunkę. Przedawkowanie może skutko-
występowania dużej częstości akcji serca. Leki wiążące się wać zaburzeniami przewodzenia bodźców (ewentual-
na długi czas (np. propafenon) są również skuteczne przy nie blokiem przedsionkowo-komorowym), politopo-
spoczynkowych częstościach akcji serca. wymi skurczami dodatkowymi i asystolią.
Stosowanie leków przeciwarytmicznych klasy IA
Zgodnie z tymi właściwościami leki przeciwaryt- jest przeciwwskazane przy zdekompensowanej nie-
miczne klasy I dzieli się na klasę IA, IB i IC. wydolności serca, bradykardii, zaburzeniach przewo-
dzenia bodźców oraz zatruciu glikozydami naparst-
Leki przeciwarytmiczne klasy IA. Do tej klasy IA, nicy.
B4
czyli substancji działających chinidyno-podobnie, Trójpierścieniowe leki przeciwdepresyjne i neuro-
poza chinidyną należą także ajmalina, detajmium leptyki nasilają ich działanie przeciwarytmiczne.
i prajmalina (zob. tab B 4.3-10).
Antyarytmiki klasy IA blokują szybki dokomórko- Chinidyna jest stereoizomerem chininy i, podobnie
wy napływ jonów sodu oraz przedłużają czas trwania do niej, również wywiera działanie przeciw zarodź-
potencjału czynnościowego (przedłużony czas trwa- com zimnicy oraz wywołuje skurcze macicy.
nia zespołu QRS i odstępu QT). Oprócz silnego hamowania szybkiego napływu jo-
Chinidyna wykazuje działanie antycholinergicz- nów sodu, małe dawki chinidyny blokują też kanał IKr
ne, które wywiera częściowo antagonistyczny efekt (wewnętrzny korygujący kanał potasowy), a większe
w stosunku do ich bezpośredniego działania na serce. dawki hamują również inne kanały potasowe.
Dlatego dominacja działania antycholinergicznego Poza wymienionymi efektami wywieranymi
przy podawaniu małych dawek chinidyny może do- na serce, przedawkowanie objawia się zaburzenia-
prowadzić do wzrostu częstości akcji serca i poprawy mi widzenia i słuchu oraz nudnościami, wymiotami,
przewodnictwa (tzw. paradoksalne działanie chinidy- zawrotami głowy i stanami majaczeniowymi (tzw.
ny). cinchonismus). Podobnie jak chinina, chinidyna pro-
W przeciwieństwie do tego, większe dawki chini- wadzi często do reakcji alergicznych.
dyny blokują przewodnictwo przedsionkowo-komo- Wskutek farmakokinetycznej interakcji chinidyna
rowe (zob. poniżej). wzmaga działanie digoksyny (ale nie digitoksyny).
Przy uwzględnieniu ww. ograniczeń, leki prze- Ze względu na to, że digoksyna nie ulega metabolizo-
ciwarytmiczne o działaniu podobnym do chinidy- waniu, przyczyną tej interakcji nie jest kompetycyjne
ny mogą być stosowane przy trzepotaniu i migota- hamowanie enzymu metabolizującego, lecz hamowa-
niu przedsionków, tachykardiach nadkomorowych nie glikoproteiny P.
i komorowych, a także skurczach dodatkowych, przy Ponadto chinidyna jest silnym inhibitorem
czym wskazanie do stosowania powinno się określać CYP2D6. Dlatego jednoczesne podanie innych sub-
MUTSCHLER-2009.indd 619 2010-01-07 22:13:53
620 Krew
stratów CYP2D6 (np. metoprololu, propafenonu) nie wpływa na czas trwania potencjału czynnościo-
prowadzi do poważnych interakcji lekowych. wego, ale bardzo silnie blokuje szybki dokomórkowy
napływ jonów sodu w fazie 0 potencjału czynnościo-
Ajmalina, uboczny alkaloid z Rauwolfia serpentina, wego prowadząc do poszerzenia zespołów QRS przy
oraz detajmium i prajmalina, produkty częściowo spoczynkowej częstości akcji serca.
syntetycznej modyfikacji ajmaliny, wywierają na ser- Wskazania, tak jak napisano wyżej, zostały bardzo
ce podobne działanie jak chinidyna. Przy stosowaniu ograniczone, przede wszystkim przy komorowych
dawek terapeutycznych efekt inotropowy ujemny arytmiach z tachykardią. Substancje te stosuje się
jest jednak mniejszy. Co ciekawe, mimo posiadania przede wszystkim do osiągania rytmu zatokowego
struktury aminy czwartorzędowej prajmalina po po- u pacjentów z nadkomorowymi arytmiami typu ta-
daniu doustnym wchłania się dobrze z przewodu po- chykardii, szczególnie takich z migotaniem przed-
karmowego. sionków.
Leki przeciwarytmiczne klasy IB. Do tej klasy anty- Propafenon jest substratem dla CYP2D6 o wyraźnie
arytmików zalicza się lidokainę i meksyletynę (zob. nieliniowej farmakokinetyce, dlatego przy powolnym
tabela B 4.3-10). metabolizowaniu należy się liczyć z wystąpieniem
Substancje te oddziałują głównie na komory podwyższonych stężeń w osoczu i zwiększoną liczbą
i w mniejszym stopniu na przedsionki serca, przy działań niepożądanych.
czym w przeciwieństwie do leków działających po-
dobnie do chinidyny, tylko w niewielkim stopniu Dla flekainidu również wykazano biotransforma-
wpływają na czas trwania potencjału czynnościowe- cję katalizowaną przez CYP2D6. Ze względu na to,
go. że większość substancji jest usuwana drogą nerkową
Leki przeciwarytmiczne klasy IB zmniejszają rów- w postaci niezmienionej, osoby o powolnym meta-
nież szybkość depolaryzacji: przy niższym (mniej bolizmie z normalną funkcją nerek wykazują jednak
ujemnym) spoczynkowym potencjale błony komór- podwyższone stężenie osoczowe.
kowej bardziej niż przy prawidłowym potencjale spo-
czynkowym, oraz przedłużają czas regeneracji kana-
łów sodowych przy dużych częstościach akcji serca. 4.3.3.2.2.2. Leki przeciwarytmiczne klasy II
Działania niepożądane odpowiadają częścio-
wo wywoływanym przez leki działające podobnie Blokery receptorów β-adrenergicznych, które opisano
do chinidyny. Po podaniu dużych dawek może dojść w rozdz. B 1.13.5.2, nadają się do leczenia tachykar-
do stanu pobudzenia OUN oraz drgawek. dii zatokowych, napadowych tachykardii nadkomo-
rowych oraz komorowych skurczów dodatkowych
Prototypem tej grupy leków przeciwarytmicznych z powodu ich działania antyadrenergicznego. Obok
jest środek miejscowo znieczulający – lidokaina. amiodaronu (zob. poniżej) jest to jedyna klasa leków
Blokuje ona kanały sodowe zależne od napięcia przeciwarytmicznych, dla której wykazano zmniej-
znajdujące się w stanie otwartym i nieaktywnym, szenie umieralności. Ze względu na ich dobrą toleran-
ale nie w stanie spoczynku. Jej działanie jest więc cję, przy stosowaniu się do przeciwwskazań, są uwa-
tym lepsze, im częściej kanały sodowe zmieniają żane za grupę podstawowych leków przeciwarytmicz-
swój stan w jednostce czasu, co oznacza, że lidokaina nych. Należy zwrócić uwagę na fakt, że obniżają one
działa przede wszystkim przy dużych częstotliwoś- przewodnictwo przedsionkowo-komorowe.
ciach pracy serca. Dlatego stosuje się ją w arytmiach
komorowych typu tachykardii. Ze względu na duży
efekt pierwszego przejścia nie podaje się jej drogą 4.3.3.2.2.3. Leki przeciwarytmiczne klasy III
doustną.
Substancje z tej grupy, do której należą sotalol,
Meksyletyna jest chemicznie spokrewniona z lido- amiodaron i ibutylid, cechują się tym, że powodują
kainą, posiada podobny profil działania, ale, w prze- przedłużenie czasu trwania potencjału czynnościo-
ciwieństwie do tej pierwszej, może być podawana wego, głównie przez blokadę kanałów wapniowych.
doustnie. Do ważnych działań niepożądanych wywoływanych
przez tę klasę leków są charakterystyczne zaburze-
nia rytmu serca (arytmii torsade-de-pointes) wskutek
Leki przeciwarytmiczne klasy IC. Ta grupa, do której przedłużenia potencjału czynnościowego (przedłuże-
należą flekainid i propafenon (zob. tabela B 4.3-10), nie odstępu QTC), które stanowią zagrożenie życia.
MUTSCHLER-2009.indd 620 2010-01-07 22:13:53
Krew 621
Sotalol jest dotychczas jedynym β-blokerem, który cjalnym inhibitorem CYP1A2, CYP2C9, CYP2D6
poza blokowaniem β-adrenoreceptorów wykazuje i CYP3A4. Głównym metabolitem jest N-dezetylo-
typowe właściwości leku przeciwarytmicznego klasy amiodaron, poza tym powstają także metabolity de-
III. Oba enancjomery sotalolu podawanego w postaci jodowane.
racematu wykazują zbliżoną skuteczność pod wzglę- Amiodaron wykazuje także poważne działania
dem działania blokującego kanał potasowy. Wbrew niepożądane. Szczególnie często pojawiają się zło-
oczekiwaniom, w badaniach czystego D-enancjo- gi na powierzchni rogówki. Poza tym może dojść
meru, niepowodującego blokowania β-adrenorecep- do uczulenia na światło, zaburzeń czynności tarczycy
torów, nie uzyskano jednak żadnych pozytywnych (zarówno niedoczynności, jak i nadczynności), a tak-
wyników. że do zmian w tkance śródmiąższowej płuc z zaburze-
Po podaniu doustnym sotalol jest prawie całkowi- niami oddychania oraz zaburzeń czynności wątroby.
cie wchłaniany z przewodu pokarmowego (biodostęp- Podobnie jak chinidyna, amiodaron podawany jed-
ność ok. 100%). Okres półtrwania w osoczu wynosi nocześnie z digoksyną podwyższa jej stężenie w oso-
7–18 godz., a wydalanie zachodzi przez nerki. czu, co jest prawdopodobnie wywołane inhibicją P-
Dawkowanie wynosi początkowo 160 mg/dz. glikoproteiny. Amiodaron może także nasilać działa-
z kontrolą częstości akcji serca, która nie powinna nie przeciwzakrzepowe pochodnych dikumarolu.
spadać poniżej 55 uderzeń/min. W razie potrzeby
można je zwiększyć do 320–480 mg/dz. Ze względu na kliniczne znaczenie amiodaronu po-
wstała jego pochodna – dronedaron. Oczekuje się,
Amiodaron jest pochodną benzofuranu, która działa że przy takiej samej skuteczności dronedaron będzie
Serce i układ krążenia
zarówno przy nadkomorowych, jak i komorowych wykazywał znacznie korzystniejszy profil działań
zaburzeniach rytmu serca. Blokuje wiele repolaryzu- niepożądanych.
jących i depolaryzujących kanałów jonowych i dla-
tego dotychczas nie udało się przyporządkować jego Ibutylid jest IKr-blokerem, który jest wskazany
działania tylko jednemu mechanizmowi molekular- do konwersji trzepotania przedsionków u pacjentów
nemu. Należy podkreślić przede wszystkim jego sku- ze sprawnie działającą lewą komorą. Szybkość kon-
teczność w przypadku arytmii, które nie poddają się wersji maleje wraz z wydłużaniem się okresu wystę-
leczeniu za pomocą innych leków przeciwarytmicz- powania zaburzeń rytmu. Substancja wykazuje wy-
nych (dawka nasycająca 600 mg/dz. przez 8–10 dni, raźny efekt pierwszego przejścia i dlatego podaje się
dawka podtrzymująca 200 mg/dz. z przerwą week- ją tylko pozajelitowo (1 mg jako krótkotrwały wlew
B4
endową). Poza blokerami β-adrenoreceptorów amio- przez 10 minut, powtarzany maksymalnie 1 raz).
daron jest dotychczas jedynym lekiem przeciwaryt- Okres połowicznego zaniku wynosi 6 godzin.
micznym, za pomocą którego udało się zmniejszyć
umieralność w porównaniu z podawaniem placebo.
Poza tymi korzystnymi właściwościami amiodaron
charakteryzuje się jednak również znaczącymi wada- OH C2H5
mi. Okres półtrwania wynoszący 20–100 dni jest bar- N
O (CH2) 6CH3
dzo długi, ponadto duże ilości amiodaronu gromadzą H3C
się w tkankach. Z tego powodu trudno jest regulować S
NH
dawki. Jest on metabolizowany przez CYP3A4 (bio- O
dostępność 46%), ale dodatkowo jest także poten- ibutylid
O
(CH2) 3CH3
4.3.3.2.2.4. Leki przeciwarytmiczne klasy IV
I
O C2H5
N Leki przeciwarytmiczne klasy IV obejmują antagoni-
O C2H5
I
stów wapnia (blokery kanałów wapniowych) o właś-
ciwościach przeciwarytmicznych, takie jak: werapa-
amiodaron mil, jego analog gallopamil, oraz diltiazem.
Te substancje hamują dokomórkowy napływ jo-
nów wapnia przez powolne, zależne od napięcia ka-
MUTSCHLER-2009.indd 621 2010-01-07 22:13:53
622 Krew
nały wapniowe i wskutek tego zmniejszają szybkość
depolaryzacji powolnych potencjałów czynnościo- NH2
wych w węźle zatokowym i przedsionkowo-komoro-
N N
wym oraz przedłużają przewodzenie między przed-
sionkami a komorami. Poza tym wydłużają one czas HOH2C O N N
efektywnej refrakcji i hamują potencjały następcze,
które mogą doprowadzić do zaburzeń rytmu serca. HO OH
Z powodu tych właściwości farmakodynamicz-
nych stosowanie leków przeciwarytmicznych klasy adenozyna
IV jest wskazane przy nadkomorowych zaburzeniach
rytmu typu tachykardii.
Dawkowanie werapamilu przy stosowaniu poza-
jelitowym wynosi 5 mg powoli i.v., natomiast przy Częstymi działaniami niepożądanymi są: nagłe
podawaniu doustnym 3 razy dziennie po 40–80 mg. zaczerwienienie, duszność, skurcz oskrzeli, nudności
Gallopamil podaje się 3–4 razy dziennie po 50 mg, i zawroty głowy, natomiast sporadycznie pojawiają
a diltiazem 3 razy dziennie po 60–120 mg. się nadmierna potliwość, palpitacje serca, uczucie go-
rąca i stan oszołomienia. Bardzo rzadko może dojść
do zagrażających życiu asystolii lub arytmii komo-
4.3.3.2.2.5. Glikozydy nasercowe rowych.
Stosowanie adenozyny jest przeciwwskazane
Glikozydy nasercowe mogą być stosowane w le- w przypadku bloku przedsionkowo-komorowego II
czeniu tachykardii nadkomorowych oraz migotania i III stopnia, trzepotania i migotania przedsionków,
i trzepotania przedsionków z szybkim przewodze- a także obturacyjnej choroby płuc.
niem przedsionkowo-komorowym, w celu zmniej- Dipirydamol wzmaga działanie adenozyny poprzez
szenia częstości skurczów komór. hamowanie pobierania dokomórkowego, ale należy
Stosowanie glikozydów nasercowych jest prze- unikać łączenia tych leków. Pochodne ksantyny osła-
ciwwskazane przy arytmiach komorowych, ze wzglę- biają działanie adenozyny.
du na ryzyko migotania komór.
4.3.3.2.2.7. Magnez
4.3.3.2.2.6. Adenozyna
Stosowanie jonów magnezu jako leku przeciwaryt-
Adenozyna nadaje się do leczenia wymagających micznego omówiono w rozdz. B 7.2.2.4.3.
leczenia napadowych tachykardii nadkomorowych.
Jej stosowanie jest wskazane przede wszystkim
w przypadku, kiedy nie można podać innych leków
4.3.4. Choroba wieńcowa serca
przeciwarytmicznych (np. werapamilu). W wyniku
oddziaływania na receptory A1 w sercu i wywołanego
przez to hamowania cyklazy adenylanowej, dochodzi 4.3.4.1. Podstawy patofizjologiczne
do otwierania kanałów potasowych w węźle zatoko-
wym. Adenozyna blokuje także kanały wapniowe W ten sam sposób, jak w innych tętnicach, rów-
w węźle przedsionkowo-komorowym, wywołując nież w tętnicach wieńcowych przede wszystkim
w ten sposób efekt dromotropowy ujemny. Efektem w dużych pniach, mogą powstać zmiany miażdży-
tego jest wzrost spoczynkowego potencjału błony cowe. Miażdżyca tętnic wieńcowych, prowadząca
komórkowej, hamowanie przewodnictwa przedsion- do ich zwężenia albo, przez dodatkowe powstawa-
kowo-komorowego oraz zmniejszenie częstości akcji nie zakrzepów, do częściowego lub całkowitego za-
serca. Adenozyna nie wywiera działania na komory mknięcia ich światła, stanowi najważniejszą przy-
serca. czynę choroby wieńcowej, która może przyjmować
Ze względu na bardzo szybką dezaminację do inozy- postać od bezobjawowej choroby wieńcowej, poprzez
ny oraz wychwyt przez erytrocyty, okres połowicznego dusznicę bolesną aż do zawału mięśnia sercowego,
półtrwania wynosi tylko kilka sekund. Dlatego adeno- a nawet wtórnego zgonu z przyczyn sercowych (udar
zyna musi być podawana dożylnie w bolusie. Dawka serca). Około 1/3 wszystkich zgonów jest spowodo-
początkowa wynosi 3 mg, ale przy słabym działaniu wana zawałem mięśnia sercowego, dlatego wydaje
można ją zwiększyć maksymalnie do 12 mg. się, że konieczne jest zastosowanie we właściwym
MUTSCHLER-2009.indd 622 2010-01-07 22:13:54
Krew 623
czasie środków zapobiegawczych, mających na celu getatywna, arytmia, niewydolność serca oraz nad-
eliminację czynników ryzyka powstania miażdżycy mierne zapotrzebowanie na tlen z powodu wzmo-
tętnic wieńcowych. żonej pracy serca (np. przy nadciśnieniu, wadach
Pewnymi czynnikami ryzyka są: zastawek serca) lub za niska zawartość tlenu we krwi
(np. przy niedokrwistościach, methemoglobinemii,
■ palenie tytoniu,
zatruciu tlenkiem węgla).
■ nadwaga, Napad dusznicy bolesnej dotyczy przede wszyst-
kim warstw mięśnia sercowego położonych w pobliżu
■ nadciśnienie,
wsierdzia, ponieważ powoduje wzrost późnorozkur-
■ hiperlipoproteinemia, czowego ciśnienia w lewej komorze. Jest to związane
ze zwiększeniem elementu oporu dla tętnic wieńco-
■ cukrzyca.
wych, który stanowi mięśniówka serca. Wskutek tego
zmniejsza się przepływ krwi w warstwach znajdują-
Częsta jest także kombinacja tych czynników, zwłasz-
cych się w pobliżu wsierdzia.
cza przy zespole metabolicznym. Niedostateczny wy-
Ze względu na występowanie lub możliwość
siłek fizyczny, hektyczny tryb życia i utrzymujący się
wywołania napadu rozróżnia się następujące formy
stan psychicznej frustracji również mogą sprzyjać po-
dusznicy bolesnej:
jawieniu się choroby wieńcowej.
W profilaktyce należy uwzględnić wymienione wy- ■ stabilną dusznicę bolesną (dusznicę obciążenio-
żej czynniki ryzyka. Osobom palącym tytoń powinno wą),
Serce i układ krążenia
się zwracać uwagę na zagrożenia związane z tym nało-
■ niestabilną dusznicę bolesną.
giem. U pacjentów z nadciśnieniem należy przeprowa-
dzić skuteczne leczenie tej nieprawidłowości. Osoby
Dla stabilnej dusznicy bolesnej charakterystycz-
z nadwagą powinny zmniejszyć swoją masę ciała dzię-
ne jest to, że objawy zależą od obciążenia i szyb-
ki stosowaniu diety niskotłuszczowej i niskowęglo-
ko ustępują po przerwaniu obciążenia. Oznacza to,
wodanowej. Podwyższone stężenie lipidów we krwi
że w spoczynku objawy jeszcze nie występują. Jeśli
należy dodatkowo obniżać za pomocą środków die-
obciążenie wzrasta albo wskutek pobudzenia psy-
tetycznych, natomiast leki obniżające stężenie tłusz-
chicznego dochodzi do aktywacji układu współczul-
czów stosować tylko w przypadku, kiedy zastosowane
nego, spowodowany tym wzrost zużycia tlenu może
środki nie są wystarczające. Szczególne znaczenie ma
wywołać napad dusznicy bolesnej. Ze względu na to,
B4
fakt, że u osób uprawiających regularnie sport znacz-
że koreluje on z częstością akcji serca, przynajmniej
nie rzadziej dochodzi do zawału mięśnia sercowego,
przez pewien okres czasu, pojawia się on przy indywi-
a w przypadku jego wystąpienia mają trzykrotnie
dualnej, zawsze podobnej częstości akcji serca i stąd
większą szansę przeżycia niż osoby niewytrenowane.
pochodzi nazwa stabilna dusznica bolesna. Stabilna
dusznica bolesna podlega z kolei podziałowi we-
Dusznica bolesna (Angina pectoris). W przypadku dług systemu CCS (Canadian Cardiovascular Society
tej choroby pojawia się dysproporcja między dostar- – Kanadyjskiego Towarzystwa Kardiologicznego).
czaną ilością tlenu a jego zużyciem (niewydolność Podobnie jak w przypadku klasyfikacji niewydolności
wieńcowa) przy zmniejszonej, a w zaawansowanych serca według schematu NYHA, dokonano podziału
przypadkach w dużym stopniu zniesionej rezerwie na cztery podgrupy. CCS I obejmuje pacjentów, u któ-
wieńcowej. Mięsień sercowy jako narząd pracujący rzych występują dolegliwości jedynie przy dużym wy-
bez przerwy, w którym nie może wystąpić dług tle- siłku fizycznym. Przy CCS II obserwuje się niewielkie
nowy, jest szczególnie wrażliwy na niedostateczne upośledzenie prawidłowej aktywności fizycznej spo-
zaopatrzenie w tlen. wodowane dusznicą bolesną, natomiast w przypadku
W napadzie dusznicy bolesnej u pacjenta wystę- CCS III występuje znaczne upośledzenie. CCS IV
puje charakterystyczne uczucie ucisku za mostkiem, charakteryzuje się występowaniem dusznicy bolesnej
jakby klatka piersiowa była ograniczona przez obręcz przy najmniejszym obciążeniu fizycznym oraz bólami
beczki lub ściśnięta w imadle (stąd określenie angina w spoczynku. W wyniku tego postać CCS IV prze-
pectoris = ciasnota klatki piersiowej). Ból promie- kształca się w niestabilną dusznicę bolesną.
niuje aż do lewego barku i ramienia, sporadycznie pa- Według Braunwalda niestabilną dusznicę boles-
cjenci odczuwają także dolegliwości w okolicy karku ną dzieli się na trzy klasy: I – u pacjentów bez dole-
i obojczyka albo mają rozstrój żołądka. gliwości w spoczynku w ostatnich 2 miesiącach oraz
Poza miażdżycą, do najważniejszych przyczyn z trzema lub więcej epizodami dusznicy dziennie
dusznicy bolesnej należą: niewłaściwa regulacja we- przy niewielkim obciążeniu; II – do tej klasy zalicza-
MUTSCHLER-2009.indd 623 2010-01-07 22:13:54
624 Krew
ją się pacjenci, u których w okresie ostatniego mie- wzrostu stężenia cukru we krwi oraz stężenia enzy-
siąca, ale nie podczas ostatnich 48 godzin, wystąpiły mów mięśniowych w surowicy, a także do typowych
podostre dolegliwości w spoczynku; III – pacjenci zmian w EKG. (Niektóre zawały mięśnia sercowe-
z ostrymi dolegliwościami w spoczynku (jeden lub go mają jednak przebieg bezobjawowy, co oznacza,
więcej epizodów) także podczas ostatnich 48 godzin. że nie zostają one zauważone przez pacjentów.)
Przy niestabilnej dusznicy bolesnej zmienia się Najczęstszą przyczyną zawału serca jest zamknięcie
więc częstość i nasilenie objawów. Jednego dnia światła pnia tętnicy wieńcowej najczęściej w wyniku
mogą się nagle pojawić bardzo ciężkie napady dusz- pęknięcia blaszki miażdżycowej. Co ciekawe, często
nicy bolesnej, podczas gdy następnego dnia napad zamykane są te naczynia, których światło zostało zwę-
wywołuje dopiero znaczny wysiłek fizyczny. Zakłada żone przez blaszki tylko w niewielkim stopniu. Można
się, że przyczyną dużego zróżnicowania nasilenia ob- z tego wnioskować, że blaszki są tam jeszcze niesta-
jawów są zakrzepy o zmiennej wielkości, znajdujące bilne i dlatego łatwiej ulegają odrywaniu. W efekcie
się na płytkach miażdżycowych, rozerwanie płytki następuje nagłe zamknięcie naczynia. Leczenie zapo-
miażdżycowej z utworzeniem zakrzepu oraz skurcze biegawcze polega zatem na stabilizacji płytek.
tętnic wieńcowych. Spastyczne zwężenie tętnic tłu-
maczy się tym, że w przypadku stenozy ekscentrycz- Zawał serca jest szczególnie groźny w przypadku, gdy
nej, niezmieniona część ściany tętnicy może jeszcze dodatkowo pojawią się powikłania. Najważniejszymi
reagować spastycznym skurczem. powikłaniami są:
Szczególną postać niestabilnej dusznicy boles-
nej stanowi dusznica przedzawałowa (dusznica ■ zaburzenia rytmu serca, a szczególnie arytmie ko-
crescendo), w przypadku której, wskutek zwiększa- morowe (zob. wyżej),
jącego się zwężenia tętnicy wieńcowej, dochodzi
do silnego zmniejszenia się obciążenia koniecznego ■ mechaniczna niewydolność mięśnia sercowego
do wywołania napadu. z zagrażającym obrzękiem płuc lub wstrząsem kar-
U wszystkich pacjentów z niestabilną dusznicą bo- diogennym,
lesną istnieje duże ryzyko wystąpienia zawału mięś-
nia sercowego i dlatego wskazane jest prowadzenie ■ wytworzenie się tętniaka ściany serca,
leczenia stacjonarnego.
Szczególną formą spoczynkowej dusznicy bolesnej ■ powstanie skrzeplin na ścianie jam serca w pobliżu
z pojawiającymi się jedynie okresowo zaburzeniami tętniaka, które mogą spowodować zatory w narzą-
ukrwienia w następstwie skurczów naczyń jest dusz- dach zaopatrywanych przez układ dużego krwio-
nica bolesna Printzmetala. biegu (np. w mózgu lub tkankach obwodowych),
Co ciekawe, przy niedostatecznym ukrwieniu serca ■ prawie zawsze śmiertelne pęknięcie serca, czyli ro-
nie zawsze dochodzi do dolegliwości typu dusznicy zerwanie tkanki w obszarze wystąpienia zawału
bolesnej. Jeżeli brak jest tego rodzaju dolegliwości, z krwawieniem do jamy osierdzia (tamponada serca)
mimo zaburzeń przepływu krwi przez tętnice wieńco- albo oderwanie się mięśnia brodawkowatego i przez
we (wykrywanych np. w EKG obciążeniowym), wów- to zniszczenie aparatu mocującego zastawkę mitralną.
czas mamy do czynienia z bezobjawowym niedo-
krwieniem mięśnia sercowego. Klinicznie rozróżnia O ile nie wynikną tego rodzaju powikłania lub pa-
się pacjentów, u których zawsze brakuje objawów, oraz cjent przeżyje po ich wystąpieniu, rejon objęty zawa-
takich z epizodami niedokrwienia objawowego i bez- łem zabliźnia się, czyli tkanka mięśniowa zostaje za-
objawowego. Przyczyną braku objawów w pierwszej stąpiona tkanką łączną. Mięsień sercowy może w ten
grupie pacjentów jest (ogólne) zmniejszenie odczuwa- sposób ponownie podjąć swoją prawidłową funkcję
nia bólu, natomiast w drugiej grupie przebieg objawów albo powstaje przewlekła niewydolność serca.
wynika z różnego czasu trwania fazy ischemicznej.
Zawał serca (zawał mięśnia sercowego). Jeżeli Ostry zespół wieńcowy (OZW). Dawniej najczęściej
w którejś części serca dojdzie do nagłego przerwa- rozpatrywano wyżej wymienione różne formy choro-
nia przepływu krwi, to w tym obszarze następuje ob- by wieńcowej serca oddzielnie. Obecnie postacie za-
umarcie tkanki mięśnia sercowego (martwica), czyli grażające życiu określa się wspólną nazwą ostry ze-
zawał serca, któremu najczęściej towarzyszy silny spół wieńcowy (acute coronary syndrom, ACS). Tym
ból i odczucie ucisku w klatce piersiowej. Prowadzi samym OZW obejmuje niestabilną dusznicę bolesną,
to do spadku ciśnienia krwi, gorączki, leukocytozy, transmuralny i nietransmuralny zawał serca. Dzięki
MUTSCHLER-2009.indd 624 2010-01-07 22:13:54
Krew 625
badaniom EKG można dokonać dokładniejszego roz- ■ zmniejszając zużycie tlenu przez zmniejszenie kur-
różnienia. U pacjentów, których EKG nie wykazało czliwości, częstości akcji serca i/lub napięcia ścian
obecności uniesienia odcinka ST wystąpił nietrans- mięśnia sercowego,
muralny zawał serca (non ST-elevation myocardial
■ zwiększenie ilości dostępnego tlenu przede wszyst-
infarction, NSTEMI), natomiast u pacjentów z unie-
kim w wewnętrznych warstwach ścian serca przez
sionym ST – zawał transmuralny (ST-elevation myo-
przedłużenie czasu trwania rozkurczu i zmniejsze-
cardial infarction, STEMI).
nie pozanaczyniowego elementu oporu tętnic wień-
cowych,
4.3.4.2. Leki stosowane w chorobie ■ zlikwidowanie skurczów tętnic wieńcowych.
wieńcowej
(leki przeciw dusznicy bolesnej) W ten sposób likwiduje się lub przynajmniej zmniej-
sza dysproporcję między ilością dostarczanego tlenu
Po tym, jak w doświadczeniach na zwierzętach udało się a zapotrzebowaniem na niego.
zwiększyć przepływ krwi przez tętnice wieńcowe za po-
mocą środków rozszerzających tętnice wieńcowe, a prze- Celem leczenia jest:
de wszystkim tętniczki, przyjęto, że za ich pomocą można
także leczyć dolegliwości stenokardialne u ludzi. Obecnie
dowiedziono, że nie jest to właściwa metoda terapeutyczna, ■ w przypadku napadu dusznicy bolesnej spowodo-
ponieważ tętniczki znajdujące się za miejscem zwężenia wanie szybkiego jego ustąpienia,
zostały już maksymalnie rozszerzone w wyniku niedoboru
Serce i układ krążenia
tlenu. Substancje rozszerzające wyłącznie tętniczki straciły ■ profilaktyka lub przynajmniej zmniejszenie kolej-
więc znaczeniu w leczeniu dusznicy bolesnej. nych napadów dusznicy bolesnej oraz
Skuteczne leczenie dusznicy bolesnej można osiąg- ■ poprawa rokowania, przede wszystkim zmniejsze-
nąć w następujący sposób (zob. ryc. B 4.3-8): nie ryzyka wystąpienia zawału serca.
antagoniści wapnia, β-bloker B4
(β-bloker)
zmniejszenie zmniejszenie częstości
kurczliwości serca akcji serca, wydłużenie
fazy rozkurczu
zmniejszenie zwiększenie dostępności
zapotrzebowania serca tlenu (przede wszystkim
na tlen w wewnętrznych warstwach
ścian serca)
zmniejszenie napięcia
ścian serca
zmniejszenie objętości zmniejszenie oporu
napełniania (zmniejszenie obwodowego (zmniejszenie
obciążenia wstępnego) obciążenia następczego)
azotany antagoniści wapnia,
(azotany)
Ryc. B 4.3-8. Zmniejszenie zużycia tlenu przez serce oraz zwiększenie dostępności tlenu przez leki stosowane w chorobie wieńcowej.
MUTSCHLER-2009.indd 625 2010-01-07 22:13:54
626 Krew
Dostępne grupy substancji to: czyli zmniejszenie obciążenia następczego). Poza
tym nitraty powodują rozszerzenie tętnic wieńco-
■ azotany („związki nitrowe”),
wych przebiegających w nasierdziu oraz ustąpienie
■ blokery β-receptorów oraz skurczów tętnic wieńcowych.
W wyniku zmniejszenia obciążenia wstępnego
■ antagoniści wapnia.
i następczego oraz uwarunkowanego tym spowol-
nienia pracy mięśnia sercowego, obniża się zapotrze-
bowanie serca na tlen, a w następstwie zmniejszenia
4.3.4.2.1. Azotany („związki nitrowe”) się pozanaczyniowego elementu oporu tętnic wień-
cowych uzyskuje się poprawę ilości dostarczanego
Określenie azotany oznacza w medycynie estry kwa- tlenu.
su azotowego powodujące poprawę stanu w duszni- Działanie związków nitrowych jest szczególnie
cy bolesnej. Różne substancje, należące do tej grupy korzystne u pacjentów z chorobą wieńcową podczas
i wymienione w tabela B 4-3.11, wykazują bardzo obciążenia. Oznaką poprawy sprawności mięśnia ser-
podobne działanie pod względem farmakodynamicz- cowego jest wzrost zdolności wytrzymania obciążeń
nym, a różnią się jedynie właściwościami farmako- przez pacjenta.
kinetycznymi, czyli przede wszystkim początkiem Ze względu na opisane działania azotany są waż-
i czasem trwania działania oraz tolerancją nitratów nymi lekami w objawowym leczeniu choroby wień-
(zob. poniżej). cowej. Dotychczas nie udało się jednak udowodnić
Poprzez oddziaływanie na mięśniówkę żył, nitra- istnienia pozytywnego efektu w postaci zmniejszenia
ty powodują ich rozszerzenie i w ten sposób zwięk- umieralności.
szają wypełnienie układu żylnego krwią (venous
pooling). Wskutek tego zmniejsza się napływ krwi Mechanizm działania. W wyniku redukującej bio-
żylnej do serca, objętość wypełnienia, a także rozkur- transformacji azotany zostają w organizmie prze-
czowe napięcie ścian serca (preload reduction, czyli kształcone we właściwą substancję czynną – mo-
zmniejszenie obciążenia wstępnego). Równocześnie, notlenek azotu (NO). (Są zatem typowymi proleka-
ale w sposób wtórny i tylko przez krótki czas, w wy- mi.) NO stymuluje cytozolową cyklazę guanylanową,
niku rozszerzenia dużych pni tętniczych zmniejsza która katalizuje tworzenie cyklicznego guanozyno-
się ciśnienie w aorcie, obniża się opór obwodowy monofosforanu (cGMP) z guanozynotrifosforanu
i skurczowe napięcie ścian serca (afterload reduction, (GTP). cGMP powoduje z kolei zmniejszenie we-
Tabela B 4.3-11. Azotany
Wzór strukturalny Nazwa Preparaty handlowe Okres Pojedyncza
międzynarodowa półtrwania dawka
(mg)
ONO2 triazotan glicerolu Corangin nitrospray, 2-4 0,2-2,4
Nitroglicerinum,
O2NO ONO2 Trinitrosan
O2NO H diazotan izosorbitolu Aerosonit, 30-60 (ISDN) 20-60
O Cardiosorbid, 300 (IS-5-N)
Sorbonit, 120 (IS-2-N)
O ISDN-ratiopharm
H ONO2
HO H monoazotan izosorbitolu Corangin, 300 20-60
O Effox,
somonit,
O Izonit
H ONO2
O2NO ONO2 tetraazotan pentaerytrylu Nitrason N, 240-600 50-80
Pentalong, (aktywny metabolit)
O2NO ONO2
Pentaerythritol
MUTSCHLER-2009.indd 626 2010-01-07 22:13:55
Krew 627
wnątrzkomórkowego stężenia jonów wapnia i przez
to zmniejszenie napięcia naczyń (zob. ryc. B 4.3-9). stres mechaniczny
Podanie nitratów przyczynia się do doprowadze- stres mechaniczny
receptory aktywuje receptor
nia z zewnątrz ważnej cząsteczki sygnałowej, która (acetylocholina, bradykinina,
jest również produkowana endogennie. substancja P i.in.)
Akt
+
↑ [Ca2 ]i
Znaczenie i powstawanie NO. NO jest centralnym Gq
mediatorem w układzie sercowo-naczyniowym,
kalmodulina Ca2+-kalmodulina
w ośrodkowym i obwodowym układzie nerwowym
oraz w procesach immunologicznych. Od dawna eNOS
znany śródbłonkowy czynnik rozluźniający (EDRF (nieaktywny)
eNOS
– endthelium derived relaxing factor) okazał się właś- (aktywny)
nie monotlenkiem azotu. Poza wazodylatacją, NO cytrulina + NO arginina
wykazuje także działanie hamujące agregację trom-
bocytów oraz adhezję monocytów.
NO jest biosyntetyzowany z L-argininy w reakcji komórka śródbłonka
katalizowanej przez syntazę NO (NOS). NOS jest he- NO
moproteiną wykazuąjcą strukturalne podobieństwo sGC
do rodziny P-450.
Serce i układ krążenia
cGMP GTP
Istnieją trzy izoformy syntazy NO: PKG
rozkurcz
– forma indukowalna (iNOS), która występuje w obwodo- komórka mięśnia gładkiego
wych komórkach krwi, w śródbłonku i mięśniach gład-
kich naczyń krwionośnych może być silnie aktywowana Ryc. B 4.3-9. Biosynteza i punkty uchwytu NO.
w odpowiedzi na patologiczne stymulatory (np. endotok-
syny powstające w szoku).
jonów wapnia. Wyjaśnia to, dlaczego sepsa związana z LPS
– konstytutywna forma śródbłonkowa (eNOS), której obec- wiąże się z silną wazodylatacją. Z drugiej strony, ze wzglę-
B4
ność dowiedziono w śródbłonku, w kardiomiocytach, du na efekt cytotoksyczny, uwalnialnianie dużych ilości NO
w komórkach mezangium w nerkach oraz w trombo- stanowi niespecyficzny mechanizm obronny przeciw wni-
cytach. Tej formie przypisuje się szczególne znaczenie kającym obcym organizmom albo komórkom rakowym.
w podtrzymywaniu napięcia ścian naczyń krwionośnych,
a tym samym w regulacji ciśnienia krwi. Opisany mechanizm wyjaśnia, dlaczego w rejonie
naczyń wieńcowych ze zmianami miażdżycowy-
– konstytutywna forma neuronalna (nNOS), znajdowana mi z częściowo wydolnym śródbłonkiem dochodzi
w tzw. neuronach produkujących NO. Powstający tam do niewystarczającej produkcji NO, który jednak
monotlenek azotu posiada funkcje atypowego neurotrans- można uzupełnić przez podanie nitratów.
mitera (np. modulacja transmisji synaptycznej).
Tolerancja na azotany. Zależnie od wysokości daw-
Głównym mechanizmem regulacyjnym dla form konsty- ki i długości okresu stosowania, po pewnym – zazwy-
tutywnych (iNOS oraz eNOS) jest wewnątrzkomórkowe czaj już w ciągu 24 godzin – obserwuje się znaczne
stężenie jonów wapnia. Jeśli dojdzie do podwyższenia osłabienie hemodynamicznego działania azotanów
wewnątrzkomórkowego stężenia jonów wapnia spowodo- (tolerancję na działanie azotanów). Dotychczas
wanego acetylocholiną lub bradykininą, NOS może zostać nie zostało do końca wyjaśnione, w jaki sposób po-
przekształcony z formy nieaktywnej w aktywną za pomocą wstaje tolerancja na działanie azotanów.
kompleksu wapń-kalmodulina. Najważniejszym aktywato-
ram fizjologicznym dla eNOS jest jednak naprężenie sta- Przez długi czas promowano hipotezę zakładającą, że przy
tyczne (shear stress) wywierane przez strumień krwi w na- powtarzanym podawaniu nitratów grupy SH potrzebne
czyniach, które aktywuje specyficzne kaskady sygnałowe. do ich biotransformacji nie są dostępne w wystarczającym
stopniu i dlatego nie dochodzi do powstawania NO. W efek-
Te ostatnie mogą przeprowadzać eNOS w formę aktywną. cie powstaje tolerancja na działanie azotanów. Obecnie ten
iNOS jest aktywowany przez cytokiny oraz elementy bak- pogląd został obalony. Badania wykazały, że przyczyną
terii (np. lipopolisacharydy, LPS) niezależnie od stężenia tolerancji na działanie azotanów jest najprawdopodobniej
MUTSCHLER-2009.indd 627 2010-01-07 22:13:55
628 Krew
wzmożone uwalnianie reaktywnych form tlenu następujące Nitrogliceryna podana dożylnie osłabia działanie
po podaniu azotanów. Reaktywne formy tlenu powodują heparyny. Zaleca się szczególną ostrożność przy jed-
wzmożone przekształcanie NO w azotyn nadtlenku, wsku-
noczesnym podawaniu nitratów i inhibitorów PDE-5.
tek czego zachodzi słabsza aktywacja cyklazy guanylano-
wej. Dyskutuje się również nad możliwością wzmożonego W tym przypadku dochodzi do znacznego nasilenia
wytwarzania angiotensyny II i zwiększonego uwalniania wazodylatacji z wyraźnym spadkiem ciśnienia krwi.
endoteliny ze śródbłonka wskutek rozszerzenia naczyń
wywołanego przez NO. Jeżeli te teorie zostaną potwier- Triazotan glicerolu, wcześniej niewłaściwie pod
dzone, to podanie przeciwutleniaczy (np. witaminy C),
względem chemicznym, nazywany nitrogliceryną,
inhibitorów ACE i/lub antagonistów endotelin umożliwia-
łoby uniknięcie tolerancji na działanie azotanów. Poza tym ulega biotransformacji w wątrobie, erytrocytach
ostatnio doniesiono, że za tolernację na nitraty może rów- i śródbłonku z przekształceniem w prawie lub w ogó-
nież odpowiadać hamowanie mitochondrialnej dehydroge- le niedziałające metabolity – diazotan i monoazotan
nazy aldehydowej (ALDH 2), która odgrywa ważną rolę glicerolu oraz glicerynę. Biodostępność po podaniu
w ich bioaktywacji.
podjęzykowym ok. 40%, a po zastosowaniu przez-
skórnego systemu terapeutycznego – ok. 70%.
Stosowanie odpowiednich schematów dawkowania Najszybciej, ale i najkrócej działa triazotan glice-
pozwala na uniknięcie powstania hemodynamicznej rolu – nie w postaci o przedłużonym działaniu, lecz
tolerancji na działanie azotanów, jednak nie zapobie- jako kapsułka do rozgryzienia lub w aerozolu, wchła-
ga powstawaniu uszkadzających śródbłonek reaktyw- niający się w jamie ustnej albo gardłowej. Obecnie,
nych form tlenu. Do takich schematów należy tzw. podobnie jak wcześniej, triazotan glicerolu jest naj-
„podawanie ekscentryczne” (dwie dawki dziennie ważniejszym środkiem w leczeniu ostrego napadu
w odstępie siedmiu godzin) wolno działających azo- dusznicy bolesnej. Po podaniu podjęzykowym działa
tanów. Podczas nocnej przerwy w podawaniu azota- w ciągu kilku sekund lub minut.
nów może jednak dojść do częstszego występowania W przeciwieństwie do tego nie jest uzasadnione
napadów dusznicy bolesnej. doustne stosowanie triazotanu glicerolu w profilak-
tyce dusznicy bolesnej w postaci preparatów o prze-
Farmakokinetyka. Azotany po podaniu doustnym, dłużonym działaniu, ponieważ ich działanie jest nie-
a nitrogliceryna i diazotan izosorbitolu (zob. poniżej) pewne z powodu dużego efektu pierwszego przejścia.
także po podaniu podjęzykowym, wchłaniają się szyb- Należy również unikać przezskórnego profilaktycz-
ko i dobrze. Przede wszystkim w wątrobie następuje nego podawania triazotanu glicerolu w postaci pla-
redukcyjne rozszczepianie grup estrowych kwasu azo- strów, ponieważ szybko dochodzi do rozwoju tole-
towego i sprzęganie z wytworzeniem nieaktywnych rancji na działanie azotanów.
metabolitów, wydalanych głównie droga nerkową.
Diazotan izosorbitolu (ISDN) może być stosowany
Dawkowanie. Średnie dawki pojedyncze podano zarówno w ostrym napadzie dusznicy bolesnej, gdzie
w tabela B 4-3-11. jednak działa nieco wolniej niż triazotan glicerolu, jak
też w profilaktyce dusznicy bolesnej, jako tzw. azotan
Działania niepożądane. Działania niepożądane o długim czasie działania. Mimo że po podaniu do-
są z reguły następstwem działania rozszerzającego ustnym już przy pierwszym przejściu przez wątrobę
naczynia. W efekcie, przede wszystkim na początku większość diazotanu izosorbidu ulega biotransfor-
leczenia, często mogą występować bóle głowy („azo- macji, powstające przy tym monoazotan izosorbidu
tanowe bóle głowy”), a także zawroty głowy, nudno- (ISMN), 2-monoazotan izosorbitolu oraz 5-mono-
ści, osłabienie i zaczerwienienie skóry. Ponadto przy azotan izosorbitolu (zob. tabela B 4.3-11) wykazują
podawaniu dużych dawek istnieje ryzyko silniejsze- jeszcze biologiczną aktywność i mają dłuższy czas
go spadku ciśnienia krwi z odruchową tachykardią. działania (dla 5-ISMN okres półtrwania w osoczu
Donoszono również, jak wspomniano wyżej, o uszko- wynosi ok. 4 godziny). ISDN jest stosowany głównie
dzeniu śródbłonka (dysfunkcja śródbłonkowa). w postaci preparatów o opóźnionym działaniu.
Przeciwwskazania. Stosowanie nitratów jest prze- Monoazotan izosorbitolu (ISMN) został wprowa-
ciwwskazane w ciężkich stanach z hipotonią, zwłasz- dzony do handlu jako oddzielna substancja ze wzglę-
cza we wstrząsie, a także przy przerostowej zaporo- du na długi okres półtrwania i dobrą biodostepność.
wej kardiomiopatii. Cechuje się słabą lipofilnością i opóźnionym począt-
kiem działania, dzięki czemu jest to odpowiedni lek
Interakcje. Azotany wzmagają działanie przeciw- do stosowania w profilaktyce dusznicy bolesnej, na-
nadciśnieniowe leków obniżających ciśnienie krwi. tomiast nie w celu przerywania napadu.
MUTSCHLER-2009.indd 628 2010-01-07 22:13:55
Krew 629
Tetraazotan pentaerytrytylu (PETN) jest również
stosowany wyłącznie w postaci preparatów o prze- –
O O N
dłużonym działaniu. Substancja wyjściowa wyka-
H5C2 +
N
zująca wysoką potencję w badaniach in vitro, przy- O N N
molsydomina
puszczalnie nie wchłania się, lecz już w przewodzie O
pokarmowym ulega przekształceniu w triazotan
z udziałem nieswoistych esteraz. Po wchłonięciu się
–
triazotan jest metabolizowany z wytworzeniem ak- O N
+
tywnych metabolitów – di- i monoazotanu pentaery- HN N
N
trylu (okres półtrwania wynosi odpowiednio 4 lub 10 SIN-1
O
godzin). Metabolity są sprzęgane z kwasem glukuro-
nowym i podlegają krążeniu jelitowo-wątrobowemu.
Co ciekawe, tolerancja na działanie azotanów
O N
oraz dysfunkcja śródbłonkowa PETN jest znacz-
NC N
nie mniejsza niż w przypadku innych azotanów. N
SIN-1A
Prawdopodobną przyczyną jest, że poprzez indukcję O
hemoksygenazy i wzmożoną ekspresję ferrytyny dzia-
ła ona przeciwutleniająco oraz nie inaktywuje mito-
chondrialnej dehydrogenazy aldehydowej. Częstość
SIN-1C
bólów głowy po zastosowaniu nitratów jest również
Serce i układ krążenia
NO + NC N
mniejsza w przypadku PETN. N
O
4.2.4.1.2. Molsydomina
Ryc. B 4.3-10. Odszczepianie NO z cząsteczki molsydominy.
Pochodna sydnoniminy, molsydomina, wykazuje
podobny profil działania w jak inne azotany, jednak
silniej zmniejsza obciążenie wstępne. Z powodu sto-
sunkowo późnego początku działania (ok. 20 min ne wyniki odnoszą się także do ludzi. Molsidominę
po podaniu) nadaje się do stosowania w profilaktyce należy stosować tylko u pacjentów w podeszłym wie-
B4
dusznicy bolesnej, natomiast nie do przerywania na- ku lub u pacjentów, u których nie są tolerowane inne
padu. leki, albo wykazują one niedostateczną skuteczność.
Molsydomina jest prolekiem, z którego w wyniku Pozytywne działania leku podawanego wieczorem
enzymatycznego odszczepienia reszty etoksykarbo- dla uniknięcia rozwoju nietolernacji na działanie azo-
nylowej w organizmie powstaje 3-morfolinosydnoni- tanów nie zostały dotychczas jednoznacznie dowie-
mina (SIN-1). Wskutek nieenzymatycznego otwarcia dzione.
pierścienia SIN-1 ulega przekształceniu w postać Działania niepożądane i przeciwwskazania są ta-
z otwartym pierścieniem (SIN-lA), od której zostaje kie same, jak w przypadku azotanów.
odszczepiona właściwa substancja czynna NO (zob.
ryc. B 4.3-10).
W przeciwieństwie do powstwania NO przy poda- 4.3.4.2.3. Blokery β-adrenoreceptorów
waniu azotanów, w przypadku molsidominy nie są ko-
nieczne reduktazy ani kofaktory. Leki β-adrenolityczne są odpowiednie do stosowania
Po doustnym podaniu wchłania się ona szybko w profilaktyce napadów dusznicy bolesnej. Blokując
i całkowicie. Okres półtrwania wynosi 1–1,5 go- receptory β, osłaniają serce przed nadmierną impulsa-
dziny. Metabolity molsidominy są wydalane prawie cją adrenergiczną układu współczulnego, spowalniają
w całości przez nerki. częstość akcji serca oraz lekko osłabiają kurczliwość
Standardowe dawkowanie wynosi 2 razy dziennie mięśnia sercowego. W następstwie tego zmniejsza się
po 8 mg (w przypadku form o przedłużonym działa- zapotrzebowanie serca na tlen. Działania te są szcze-
niu). gólnie nasilone w stanach obciążenia fizycznego
Ze względu na to, że u szczurów po podaniu bar- i emocjonalnego. Niekorzystnym zjawiskiem jest po-
dzo wysokich dawek powstały guzy nosa, ograniczo- czątkowy wzrost późnorozkurczowego ciśnienia i ob-
no wskazania do stosowania molsidominy, chociaż jętości, a przez to zwiększenie elementu oporu tętnic
w dużej mierze wykluczono możliwość, iż otrzyma- wieńcowych. U pacjentów z utajoną niewydolnością
MUTSCHLER-2009.indd 629 2010-01-07 22:13:56
630 Krew
serca ten element może się ujawnić po podaniu leku w komórkach węzła zatokowego. Działanie iwabra-
β-adrenolitycznego. dyny jest zależne od częstości akcji serca, ponieważ
W przypadku potwierdzonego rozpoznania dusz- może ona rozwijać swoje działanie wewnątrz kanału
nicy bolesnej uwarunkowanej skurczem tętnic, leki tylko przy otwartym kanale.
β-adrenolityczne powinno się podawać jedynie z sub- Mimo dobrego wchłaniania, ze względu na silny
stancjami rozszerzającymi naczynia (azotanami, an- efekt pierwszego przejścia, biodostępność wynosi
tagonistami wapnia). tylko 40%. CYP3A4 metabolizuje iwabradynę do N-
Znaczenie blokerów β-adrenoreceptorów w lecze- metylowanego, aktywnego metabolitu. (Dlatego prze-
niu choroby wieńcowej wynika z tego, że skutecz- ciwwskazane jest jednoczesne podawanie iwabrady-
ność wielu z tych substancji (np, metoprolol, pro- ny oraz inhibitorów CYP3A4.) Induktory CYP3A4
pranolol, tymolol) potwierdzono w kontrolowanych mogą osłabiać to działanie. Okres półtrwania wynosi
badaniach, dotyczących wtórnej profilaktyki zawału 11 godzin.
serca, (tzw. kardioprotekcyjne działanie β-blokerów). Iwabradyna jest wskazana do objawowego lecze-
Stosowanie leków β-adrenolitycznych w leczeniu nia przewlekłej stabilnej postaci dusznicy bolesnej
choroby wieńcowej jest wskazane zwłaszcza u pa- u pacjentów z normalnym rytmem zatokowym, u któ-
cjentów po przebytym zawale serca lub przy współ- rych stosowanie β-blokerów jest przeciwwskazane
istniejącym nadciśnieniu. albo źle tolerowane.
Dawkowanie wynosi 5 mg dwa razy dziennie.
Jako działanie niepożądane mogą występować
4.3.4.2.4. Blokery kanałów If bradykardie. Specyficznym działaniem niepożąda-
nym wywoływanym przez iwabradynę są tzw. fosfe-
Jak pokazano na ryc. B 4.3-4, kanał wolnego prze- ny, czyli wrażenia wzrokowe powstające w szczegól-
pływu przepuszczalny dla jonów Na+, K+ i Ca2+ nych warunkach, niezależnie od zewnętrznego źródła
(pacemaker current, If) wywołuje potencjały czyn- światła, które występują u 15% pacjentów w ciągu
nościowe, a przez to akcję serca w komórkach samo- pierwszych 2 miesięcy po rozpoczęciu leczenia.
depolaryzujących. Z tego względu hamowanie kana- Fosfeny są efektem hamowania przez iwabradynę
łu If prowadzi do redukcji częstości akcji serca bez również kanałów Ih w siatkówce oka.
wpływu układu adrenergicznego, a tym samym bez
działania inotropowego ujemnego. Niecodzienny
fakt aktywacji tych kanałów przez hiperpolaryzację 4.3.4.2.5. Blokery kanałów wapniowych
oraz wewnątrzkomórkowy cAMP przyczyniło się (antagoniści wapnia)
do powstania ich nazwy – If (f – funny). Do tej pory
sklonowano cztery izoformy kanałów If. Należą one Blokery kanałów wapniowych, opisane już w rozdz.
do rodziny HCN (hyperpolarisation-activated, cyclic B 4.4.2.4.2.3, powodują bezpośrednie spowolnienie
nucleotide-gated channels, HCN–HCN4). Kanały pracy serca poprzez hamowanie sprzęgania elektro-
HCN występują także poza mięśniem sercowym. mechanicznego oraz wywołanego tym zmniejszeniem
kurczliwości (działanie inotropowe ujemne), a także
Iwabradyna jest pierwszą dostępną substancją dzia- niebezpośrednie odciążenie serca dzięki zmniejsze-
łającą na tę strukturę docelową. W stanie aktywnym niu obciążenia następczego i – mniej wyraźnie – ob-
regulowanym hiperpolaryzacją hamuje ona kanały If ciążenia wstępnego (zob. ryc. B 4.3-8). W obrębie
tętnic wieńcowych blokery kanałó4.3.5. wapniowych
działają przede wszystkim na większe pnie tętnicze,
gdzie są w stanie spowodować ustąpienie skurczu
tętnic wieńcowych. Ze względu na te właściwości
mogą być stosowane do profilaktyki napadów dusz-
O CH3 OCH3
nicy bolesnej oraz leczenia dławicy Prinzmetala (zob.
H3CO
N wyżej).
N OCH3
H3CO
4.3.4.2.6. Inne leki stosowane
iwabradyna w leczeniu choroby wieńcowej
Dipirydamol blokuje transporter adenozyny, który przeno-
si adenozynę w osoczu, m.in. w erytrocytach lub trombocy-
MUTSCHLER-2009.indd 630 2010-01-07 22:13:56
Krew 631
batyd), mają zastosowanie w leczeniu ostrego zespołu
wieńcowego. Poza tym w OZW wskazane jest także
C2H4OH
podawanie heparyny (niefrakcjonowanej lub mało-
N
cząsteczkowej). Przy stosowaniu kombinacji tych
N N
N C2H4OH leków (kwasu acetylosalicylowego i inhibitorów gli-
HOH4C2 N koproteiny Ilb/IIIa oraz heparyny) należy zwracać
N N
uwagę na zwiększone ryzyko wystąpienia krwawień.
C2H4OH N
4.3.4.2.8. Stenty uwalniające leki
dipirydamol (drug-eluting stents)
Typowe leczenie w przypadku ostrego zespołu
tach, i w ten sposób zwiększa jej stężenie przy receptorach wieńcowego polega na przezskórnej angioplasty-
w naczyniach krwionośnych. W efekcie rozszerzeniu ule- ce balonowej (PTCA). Po wprowadzeniu cewnika
gają przede wszystkim śródścienne tętniczki układu tętnic do odpowiedniego naczynia krwionośnego próbuje
wieńcowych, dzięki czemu wzrasta przepływ krwi przez
mięsień sercowy. Jednak jak opisano wcześniej, naczynia te
się poszerzyć zwężone miejsce za pomocą balona
są już maksymalnie rozszerzone w niedotlenionych obsza- i w ten sposób ponownie umożliwić przepływ krwi.
rach. Jeżeli przy napadzie dusznicy bolesnej poda się dipiry- W trakcie zabiegu mogą wystąpić dwa problemy.
damol, wskutek nadmiernego przepływu krwi w obszarach Z jednej strony istnieje ryzyko przerwania naczynia
Serce i układ krążenia
nieobjętych niedotlenieniem obniża się ciśnienie perfuzyj- krwionośnego, z drugiej strony interwencja może
ne w obszarach źle zaopatrywanych w tlen, które w wyniku
tego są jeszcze gorzej ukrwione (tzw. zjawisko podkrada-
prowadzić do proliferacji komórek mięśni gładkich,
nia wieńcowego). Wyniki uzyskane w badaniach dotyczą- które ponownie zamykają naczynie. Aby zapobiec tej
cych profilaktycznego stosowania dipirydamolu w leczeniu restenozie, w latach 90. XX w. rozpoczęto wkładanie
choroby wieńcowej także nie były przekonujące. Natomiast do naczyń małych metalowych rurek (stentów), które
dipirydamol można podawać jako inhibitor agregacji trom- rozszerzają się za pomocą cewnika z balonem i w ten
bocytów, przede wszystkim w połączeniu z kwasem acety-
losalicylowym. Dane z większych badań wskazują na to,
sposób naczynie pozostaje otwarte. W związku z tym
że takie połączenie jest odpowiednie do wtórnego zapo- zaczęto również produkować stenty, które, po wpro-
biegania udarom mózgu. Farmakokinetyka dipirydamolu wadzeniu do naczyń krwionośnych, uwalniają leki
nie została dotychczas wystarczająco scharakteryzowana. mające zapobiegać proliferacji komórek (drug-elu-
B4
ting stents, DES). Obecnie używa się dwóch takich
Nikorandyl jest przykładem substancji aktywującej kanały
potasowe (KATP) oraz powodujacej uwalnianie NO o dzia-
systemów, jeden z nich uwalnia sirolimus, a drugi –
łaniu przeciwdusznicowym. paklitaksel. Oba systemy prowadzą do długotrwałego
zapobiegania restenozie. Niejednokrotnie dochodziło
Trimetazydyna jest lekiem o nowym mechanizmie dzia- natomiast do trombozy, mogącej stanowić zagrożenie
łania, który dotychczas nie został dopuszczony do leczenia dla życia. Z tego powodu szczególnie ważne jest dłu-
wieńcowej choroby serca. Jego działanie antyischemiczne
nie polega na poprawie hemodynamiki, lecz na zwiększa-
gookresowe (przynajmniej przez 1 rok) hamowanie
niu tolernacji ischemii poprzez zapobieganie kwasicy we- agregacji trombocytów za pomoca kwasu acetylosa-
wnątrzkomórkowej. Substancja posiada także właściwości licylowego i klopidogrelu.
przeciwutleniające.
Obecnie opracowuje się szereg podobnych produktów,
przy czym szczególnie interesujące są stenty resorbujące
(np. z magnezu), które powleczono lekiem (tzw. projekt
4.3.4.2.7. Inhibitory agregacji trombocytów
DREAM: drug eluting absorbable metal stents).
Inhibitory agregacji trombocytów, odpowiednio
do opisanej patogenezy, mają szczególne znaczenie 4.3.4.3. Zgodne z wytycznymi
w leczeniu ostrego zespołu wieńcowego. Dlatego po-
leczenie ostrych zespołów
dawanie kwasu acetylosalicylowego (100 mg/dz.)
powoduje znaczne zmniejszenie umieralności. U pa-
wieńcowych
cjentów z nietolerancją kwasu acetylosalicylowego
można podawać inhibitory ADP takie, jak tiklopidynę Zgodnie z dotychczas obowiązującymi wytycznymi,
lub klopidogrel, przy czym ze względu na lepszą to- w przypadku ostrego zespołu wieńcowego bez unie-
lernacją powinno się stosować przede wszystkim klo- sienia odcinka ST (NSTEMI) postępuje się w nastę-
pidogrel. Inhibitory glikoproteiny IIb/IIIa (np. eptifi- pujący sposób: niezależnie od objawów należy jak
MUTSCHLER-2009.indd 631 2010-01-07 22:13:56
632 Krew
najszybciej (najpóźniej w ciągu 48 godzin) zaprze- cową lub bezpośrednio po jej zakończeniu dostają
stać stosowania inwazyjnej diagnostyki. Sposób po- klopidogrel. Leczenie klopidogrelem kontynuuje się
stępowania przedstawiono na ryc. B 4.3-11. co najmniej przez 9 miesięcy. Do zwalczania bólu
Wszyscy pacjenci natychmiast otrzymują kwas stosuje się silne analgetyki (np. siarczanu morfiny, 10
acetylosalicylowy (250–500 mg i.v.). Leczenie nale- mg powoli i.v. lub s.c.), a do uspokojenia psychicz-
ży kontynuować dożywotnio, dawkując 100 mg/dz. nego – trankwilizery (np. diazepam, 5–10 mg powoli
W przypadku niestabilnej dusznicy bolesnej, dodatko- i.v. lub s.c.).
wo do ASS podaje się dożylnie heparynę niefrakcjo- Ostry zespół wieńcowy z uniesieniem odcinka ST
nowaną lub niskocząsteczkową. Do hemodynamicz- (STEMI) leczy się zgodnie z wytycznymi w nastę-
nego odciążania serca często stosuje się także triazo- pujący sposób: w ciągu pierwszych 12 godzin wska-
tan glicerolu (0,8–2,4 mg podjęzykowo, nie stosować zane jest leczenie reperfuzyjne. Dodatkowo, podsta-
przy silnym niedociśnieniu!). Dotychczas brakuje wową metodą leczenia jest interwencja cewnikowa.
jednak badań wskazujących na wzrost przeżywalno- Fibrynolizę wywołaną lekami stosuje się jedynie
ści. Metoprolol lub inny β-bloker podaje się dożylnie w przypadku, gdy podawanie interwencyjne może
przy wysokim ciśnieniu krwi lub częstotliwości ser- rozpocząć sie z opóźnieniem ponad 90 minut w sto-
ca. Przy konieczności przeprowadzenia interwencji sunku do rozpoczęcia lizy. Badania kliniczne wyka-
kardiologicznej (cewnikowanie serca, dylatacja balo- zały, że przedszpitalne rozpoczęcie fibrynolizy przed
nowa) podaje się dożylnie antagonistów glikoprotei- rozpoczęciem właściwego leczenia stacjonarnego
ny IIb/IIIa (przy nieznanym stanie wieńcowym tirofi- może okazać się korzystne dla poprawy rokowania.
ban lub eptifibatyd, w przeciwnym razie abciksimab). Jako terapię towarzyszącą powinno się podawać ASS
Poza tym wszyscy pacjenci przed angiografią wień- (250–500 mg i.v.) i heparynę (najczęściej 60 I.E./kg
badanie kliniczne, 12-kanałowe EKG,
monitoring, pobieranie próbek krwi
brak uniesienia odcinka ST (NSTEMI)
ASS/klopidogrel, heparyna, β-bloker,
triazotan gliceryny, morfina, ew. diazepam
wysokie ryzyko niskie ryzyko
początkowa strategia zachowawcza
początkowa strategia inwazyjna kontrola troponiny co 6-12 godz.
inhibitor Gp IIb/IIIa,
angiografia wieńcowa pozytywna 2 × negatywna
dylatacja balonowa, implantacja stentu nie nadaje się pozostałe leki stosowane
albo operacja by-pass do rewaskularyzacji w chorobie wieńcowej
Ryc. B 4.3-11. Postępowanie terapeutyczne przy ostrym zespole wieńcowym bez uniesienia odcinka ST (NSTEMI).
MUTSCHLER-2009.indd 632 2010-01-07 22:13:56
Krew 633
jako wlew dożylny, max. 5000 I.E.). Przy braku prze- wiednio poniżej 130 lub 85 mm Hg, to samo dotyczy
ciwwskazań wskazane jest również wczesne podawa- pacjentów wysokiego ryzyka, np. cukrzyków i osób
nie β-blokerów oraz inhibitorów ACE lub antagoni- z przewlekłą niewydolnością nerek. Jeśli nie ma
stów AT1. Nie jest natomiast wskazane profilaktyczne przeciwwskazań, blokery β-adrenoceptorów stosuje
podawanie leków przeciwarytmicznych. się u wszystkich pacjentów z chorobą niedokrwienną
Leczenie analgetykami i trankwilizerami odbywa serca. Inhibitory ACE albo alternatywnie blokery AT1
się tak, jak opisano powyżej. są wskazane razem z diuretykami, przede wszystkim
przy wartościach ciśnienia krwi ponad 130/85 mm Hg
lub objawach niewydolności serca. Poza tym niektó-
rym pacjentom zaleca się podawanie statyn. Stężenie
4.3.4.4. Wtórna profilaktyka choroby
glukozy we krwi powinno wynosić na czczo poniżej
niedokrwiennej serca (IHD) 6 mmol/l. Do hamowania agregacji trombocytów po-
daje się kwas acetylosalicylowy, a przy przeciwwska-
Zarówno w pierwotnej jak i wtórnej prewencji obo- zaniach do stosowania ASS albo przy działaniach
wiązują te same kryteria dotyczące unikania lub niepożądanych – klopidogrel. U pacjentów z trzepo-
wpływu czynników ryzyka. Skurczowe i rozkurczo- taniem przedsionków konieczne jest podawanie anty-
we ciśnienie krwi powinno zostać ustalone odpo- koagulantów.
Serce i układ krążenia
B4
MUTSCHLER-2009.indd 633 2010-01-07 22:13:57
MUTSCHLER-2009.indd 634 2010-01-07 22:13:57
Układ oddechowy 635
5. Układ oddechowy
5.1. Podstawy anatomiczne i fizjologiczne
Układ oddechowy służy do pobierania niezbędnego elementami tego chrzęstnego rusztowania oraz pod
do życia tlenu, a wydalania ditlenku węgla. nimi leżą mięśnie gładkie, które dzięki swoim właś-
ciwościom mają wpływ na średnicę oskrzeli.
Płuca i drogi oddechowe. Płuca składają się z płuca Ściana wewnętrzna tchawicy i oskrzeli wyścielona
prawego i płuca lewego, wypełniających odpowied- jest tzw. nabłonkiem oddechowym, którego rzęski,
nio oba boczne obszary klatki piersiowej. Każde płu- poruszając się w kierunku jamy ustnej, uniemożli-
co pokryte jest opłucną trzewną (pleura visceralis), wiają przemieszczanie się w głąb dróg oddechowych
która niewielką ilością płynu surowiczego oddzielo- zanieczyszczeń wdychanych wraz z powietrzem oraz
na jest od opłucnej ściennej (pleura parietalis), sty- wspomagają usuwanie wydzieliny oskrzelowej. Pod
kającej się z wewnętrzną powierzchnią ściany klatki nabłonkiem rozmieszczone są gruczoły mieszane,
piersiowej, przeponą i śródpiersiem. Obecność płynu produkujące wydzielinę surowiczą i śluzową.
między obiema warstwami – określanymi wspólnie
jako opłucna – umożliwia ich ślizganie się względem Oskrzela i pęcherzyki płucne. Drobne oskrzela
siebie. We wnęce płuca (hilus pulmonis), w której dzielą się na oskrzeliki, które po kolejnym rozgałę-
oskrzela główne i naczynia wnikają do płuca, opłuc- zieniu się uchodzą do przewodów pęcherzykowych
na trzewna przechodzi w opłucną ścienną. Głębokie (ductus alveolares), a te do pęcherzyków płucnych.
szczeliny dzielą płuca na płaty, przy czym płuco pra- Począwszy od oskrzelików, następuje zanik chrzą-
Układ oddechowy
we na trzy, a lewe na dwa. stek wzmacniających, rzęsek, nabłonek staje się co-
Powietrze wdychane w sposób fizjologiczny raz cieńszy i w efekcie przechodzi w płaski nabłonek
przechodzi przez nos, gdzie jest ogrzewane, nawil- pęcherzykowy.
żane i oczyszczane. W przypadku oddychania wy- Pęcherzyki płucne mają kształt półkuli o średnicy
muszonego pobierane jest dodatkowo przez usta, 0,2–0,3 mm. Ich liczba wynosi około 300 milionów,
skąd przedostaje się do gardła (pharynx) będącego a łączna powierzchnia waha się w granicach 80–
aż do krtani (larynx) odcinkiem wspólnym układu 90 m2. Otoczone są gęstą siecią naczyń włosowatych,
oddechowego i pokarmowego. Rozdzielenie drogi przez które przepływa krew żylna z tętnicy płucnej. B5
pokonywanej przez powietrze i pokarm następuje Dzięki kontaktowi krwi w naczyniach włosowatych
w okolicy krtani. Powietrze płynie bowiem dalej sa- z powietrzem w pęcherzykach płucnych możliwa
modzielnie tchawicą (trachea) i oskrzelami główny- jest wymiana gazów oddechowych.
mi do wielokrotnie rozgałęzionego drzewa oskrze-
lowego płuc. Wentylacja. Wentylacja polega na krążeniu powie-
Tchawica jest szeroką, łącznotkankową rurą trza w drogach oddechowych. Wymiana gazowa
o ścianach zbudowanych z podkowiastych chrząstek w pęcherzykach płucnych (wentylacja pęcherzyko-
tchawiczych połączonych włóknami mięśni gładkich. wa) możliwa jest dzięki rytmicznym, naprzemiennym
Na wysokości 5 kręgu piersiowego tchawica rozga- wdechom i wydechom powietrza. Podczas wdechu
łęzia się na 2 oskrzela główne skierowane ukośnie do pęcherzyków dostarczane jest powietrze boga-
w dół, w kierunku wnęk płuc. Każde z oskrzeli dzieli te w tlen, podczas wydechu usuwane jest natomiast
się na kanaliki o coraz mniejszym świetle, tworząc powietrze ubogie w tlen, ale nasycone ditlenkiem
w ten sposób drzewo oskrzelowe. węgla. Ruch powietrza w drogach oddechowych
Ściany oskrzeli wzmocnione są chrząstkami, możliwy jest dzięki rytmicznemu, naprzemiennemu
w przypadku większych oskrzeli, lub płytkami, rozszerzaniu się i zwężaniu klatki piersiowej. Po-
w przypadku mniejszych oskrzeli, dzięki czemu przy większenie pojemności klatki piersiowej następuje
zmianie ciśnienia rurki oskrzelowe zachowują swój w wyniku uniesienia łuków żebrowych i spłaszczenia
kształt, a ich światło pozostaje otwarte. Pomiędzy przepony.
MUTSCHLER-2009.indd 635 2010-01-07 22:13:57
636 Układ oddechowy
Wdech jest procesem aktywnym, polegającym symalnym wydechu może być wydmuchnięta z płuc
na zwiększeniu pojemności klatki piersiowej na sku- w ciągu 1 sekundy (FEV1).
tek skurczu mięśni oddechowych. Wraz z rozsze-
rzaniem się płuc ciśnienie wewnątrzpłucne staje się Wymiana gazów oddechowych. Ciśnienie par-
niższe niż ciśnienie atmosferyczne, w wyniku czego cjalne O2 w pęcherzykach płucnych wynosi około
strumień nowo pobranego powietrza może wpłynąć 100 mm Hg. W krwi żylnej w naczyniach włosowa-
do pęcherzyków płucnych. tych płuc jest znacznie niższe i osiąga przeważnie 40
Wydech w czasie spokojnego oddychania jest pro- mm Hg. Różnica ciśnień parcjalnych O2 może być
cesem w znacznym stopniu pasywnym. Płuca bo- więc równa 60 mm Hg. Odwrotnie jest w przypadku
wiem, połączone z klatką piersiową poprzez war- ciśnienia parcjalnego CO2, które w początkowym od-
stewkę płynu opłucnowego, kurczą się i powracają cinku naczyń włosowatych kształtuje się na poziomie
do pozycji wyjściowej dzięki swojej elastyczności. 46 mm Hg i jest o 6 mm Hg wyższe niż w pęcherzy-
Objętość pojedynczego wdechu w trakcie spokoj- kach płucnych. Taka różnica ciśnień parcjalnych obu
nego oddychania jest mała w porównaniu z całkowitą gazów jest korzystna, ponieważ umożliwia ich wy-
objętością powietrza zawartego w płucach. W razie mianę przez błony pęcherzyków i naczyń (zob. ryc.
potrzeby możliwe jest jednak pogłębienie normal- B 5.1-2). Wystarczająco szybka dyfuzja CO2, mimo
nego wdechu lub wydechu poprzez nabranie lub niewielkiej różnicy ciśnień parcjalnych tego gazu,
wypuszczenie dodatkowej objętości wdechowej lub wynika natomiast z tego, że współczynnik dyfuzji
wydechowej. Całkowite usunięcie powietrza z płuc w tkankach płucnych jest w przypadku CO2 prawie
nie następuje jednak nigdy, nawet przy bardzo głę- 23 razy wyższy niż w przypadku O2.
bokim wydechu. Pewna objętość resztkowa pozostaje Wymiana na drodze dyfuzji jest efektywna tylko
zawsze w pęcherzykach płucnych oraz doprowadza- wtedy, gdy powierzchnia wymiany jest duża, a droga
jących drogach oddechowych. (zob. ryc. B 5.5-1). dyfuzji krótka. Warunki panujące w płucach w peł-
Istotnymi parametrami funkcji płuc, szczególnie ni spełniają te wymogi. Łącznie pęcherzyki płucne
ważnymi przy diagnozowaniu i kontroli przebiegu tworzą bowiem powierzchnię wynoszącą 80–90 m2,
astmy oskrzelowej, są maksymalny przepływ wyde- a droga dyfuzji rzadko przekracza 1 μm. W stanach
chowy (peak flow) oraz objętość wydechowa pierw- patologicznych w wyniku zmniejszenia powierzch-
szosekundowa, tj. objętość powietrza, która po mak- ni wymiany oraz wydłużenia drogi dyfuzji może
20-30 lat 50-60 lat 20-30 lat
dodatkowa
objętość
wdechowa
pojemność
wdechu
pojemność
całkowita
pojemność 5,1 4,4 4,4 (l)
życiowa objętość wdechu
(oddechu)
dodatkowa
objętość
wydechowa
funkcjonalna
pojemność
resztkowa
objętość 1,6 2,2 1,4 (l)
resztkowa
Ryc. B 5.1-1. Objętość i pojemność płuc. Podane wartości pojemności życiowej oraz objętości resztkowej (po prawej) powinny
uwzględniać ich zależność od wieku oraz płci.
MUTSCHLER-2009.indd 636 2010-01-07 22:13:57
Układ oddechowy 637
Perfuzja płucna. Przepływ krwi przez płuca wynosi
pęcherzyk w spoczynku 5–6 l/min. Jego ciągłość utrzymywana
CO2 O2 jest dzięki różnicy ciśnień między tętnicą płucną i le-
nabłonek wym przedsionkiem, wynoszącej średnio 8 mm Hg.
pęcherzykowy
W porównaniu z dużym układem krążenia naczynia
śródmiąższ
płucne charakteryzuje więc bardzo niewielki opór
O2 Hb nabłonek naczyń przepływu, który wraz ze wzrostem przepływu krwi
CO2 H2O włosowatych
spowodowanym rozszerzeniem naczyń płucnych
H+ HCO-3 HbO2 erytrocyt
i otwarciem rezerwowych naczyń włosowatych może
być dalej redukowany.
plazma Regulacja oddychania. Celem regulacji oddycha-
nia jest optymalne dopasowanie stopnia wentylacji
do aktywności metabolicznej organizmu w spoczynku
i w czasie wysiłku, a także utrzymania w granicach
normy ciśnienia parcjalnego O2 i CO2 oraz wartości
wymiana gazowa pH krwi tętniczej. Głębokość oddechów i ich często-
w płucach
tliwość muszą być przy tym tak do siebie dopasowane,
aby czynności oddechowe odbywały się przy możli-
żyła płucna wie najmniejszym nakładzie energii. Osiągnięcie tego
jest możliwe dzięki złożonemu systemowi regulacyj-
tętnica płucna
aorta nemu, w jaki wyposażony jest organizm.
Regularne następstwo wdechów i wydechów
jest wynikiem współdziałania grup komórek nerwo-
wych w tyłomózgowiu, w tzw. ośrodkach oddecho-
wych. Naprzemienne impulsy neuronalne, wywołu-
jące reakcję w postaci wdechu i wydechu umożli-
Układ oddechowy
wiają powstanie określonego rytmu oddechowego,
który jednak może być dostosowywany do aktual-
żylna tętnicza nych potrzeb organizmu. Wzmożenie czynności od-
prężność O2 40 mm Hg 100 mm Hg dechowych następuje w wyniku wzrostu ciśnienia
prężność CO2 46 mm Hg 40 mm Hg parcjalnego CO2, spadku ciśnienia parcjalnego O2
kwasowość pH 7,37 7,40 oraz obniżenia wartości pH krwi tętniczej. Wrażliwe
wysycenie O2 75% >97% na zmiany ciśnienia parcjalnego gazów oraz warto-
zawartość O2 140-180 ml/l 180-230 ml/l* ści pH są m.in. chemoreceptory obwodowe leżące
zawartość CO2 po obu stronach szyi w miejscu rozgałęzienia tęt-
B5
530 ml/l 490 ml/l
nicy szyjnej i tworzące tzw. kłębek szyjny (glomus
* zależne od stężenia hemoglobiny
caroticum) oraz w bezpośrednim sąsiedztwie łuku
Ryc. B 5.1-2. Wymiana gazowa w płucach. Parametry krwi żyl- aorty (arcus aorticum). Przyspieszenie oddechu
nej (prawy przedsionek, żyły płucne; niebieska) i tętniczej (żyły towarzyszące spadkowi ciśnienia parcjalnego O2
płucne, aorta; czerwona) w stanie spoczynku. Rysunek powięk- następuje wyłącznie na skutek działania bodźców
szonego fragmentu ilustruje wymianę gazową w płucach. z chemoreceptorów, wzrost ciśnienia parcjalnego
CO2 i stężenia jonów wodorowych wywołuje tę re-
akcję tylko w bardzo niewielkim stopniu. Wpływ
CO2 i jonów H na czynności oddechowe odbywa się
nastąpić ograniczenie ilości wymienianych gazów. więc za pośrednictwem chemowrażliwych struktur
Do ograniczenia powierzchni wymiany dochodzi w pniu mózgu.
np. w rozedmie płuc na skutek zaniku przegród pę- Oprócz bodźców swoistych proces oddychania
cherzykowych oraz w niedodmie płuc (brak przepły- może ulegać zmianom na skutek działania bodźców
wu powietrza w w niektórych częściach płuc). Wy- nieswoistych, do których należą m.in. bodźce bólowe
dłużenie drogi dyfuzji następuje przede wszystkim i termiczne. Wzmożenie wentylacji mogą stymulo-
w obrzęku płuc jako efekt gromadzenia się płynu wać także hormony (np. adrenalina i progesteron).
śródkomórkowego oraz w zwłóknieniu płuc (zob. ryc. Wysokość ciśnienia parcjalnego O2 we krwi jest re-
B 5.1-1) w wyniku odkładania się tkanki łącznej. gulowana również dzięki receptorom w śródbłonku
MUTSCHLER-2009.indd 637 2010-01-07 22:13:58
638 Układ oddechowy
naczyń krwionośnych, w komórkach mięśni gładkich HIF-1 alfa), przyczyniających się do większej eks-
naczyń krwionośnych i erytrocytach. W śródbłonku presji erytrocytów.
w sytuacji obniżenia się ciśnienia parcjalnego O2 do-
chodzi do podwyższenia stężenia jonów Ca2+, co po- Formy oddychania. Określenie eupnoe oznacza od-
woduje uwolnienie działających wazodylatacyjnie dech prawidłowy, odbywający się bez udziału świa-
tlenku azotu (NO) oraz prostacykliny (PGI2). Ko- domości, wykonywany z częstotliwością 16 odde-
mórki mięśni gładkich zawierają wrażliwe na ATP chów/min. Terminu hypnoe używa się dla nazwania
kanały potasowe, które są aktywowane w wyniku oddechu pogłębionego, wykonywanego z normalną
spowodowanego hipoksją zmniejszenia się ilości lub zwiększoną częstotliwością, a tachypnoe dla
ATP/ADP. Następująca wówczas hiperpolaryzacja oddechu przyspieszonego. W przypadku oddechu
błon komórkowych oraz osłabienie komórek mięśni utrudnionego i odczuwania w związku z nim nieprzy-
gładkich prowadzą do wazodylatacji. Odpowiedzią jemnych doznań subiektywnych (np. duszność, nie-
erytrocytów na spadek ciśnienia parcjalnego O2 możność głębszego oddychania) mówi się o dyspnoe.
jest wzmożone uwalnianie ATP, co stymuluje pro- Nasilenie się tych dolegliwości w stopniu utrudnia-
dukcję NO i PGI2. Przy hipoksji utrzymującej się jącym oddychanie także w pozycji leżącej określa
przez dłuższy czas dochodzi ponadto do indukcji się mianem orthopnoe. Asfikcja oznacza brak odde-
czynników transkrypcyjnych (np. transkrypcyjne- chu lub jego znaczne osłabienie w wyniku porażenia
go czynnika indukowanego niedotlenieniem 1 alfa, ośrodków oddechowych.
5.2. Zaburzenia oddychania
Zmiany chorobowe w układzie oddechowym prowa- chodzącego z wdychanego pyłu kwarcowego. Szcze-
dzą w wielu przypadkach do zaburzeń wentylacji, gólnie zagrożeni tą chorobą są górnicy i kamieniarze,
które ze względu na przyczyny ich rozwoju dzieli ponieważ przez długi czas są narażeni na działanie
się na: pyłu. Zwłóknienia płuc rozwijają się również w za-
awansowanym stadium sarkoidozy (choroby Boecka)
■ restrykcyjne,
– zapalnej choroby tkanki mezenchymalnej. Wiele
■ obturacyjne. jest natomiast czynników mogących wywołać roz-
siane śródmiąższowe zwłóknienia płuc. Przykładem
tego typu zwłóknień jest zwłóknienie popromienne,
5.2.1. Restrykcyjne zaburzenia które może być skutkiem zbyt intensywnego działa-
wentylacji nia promieni rentgenowskich.
W przypadku zmniejszenia sprawnie funkcjonujące-
go miąższu płucnego (np. po resekcji płuca, w niedo-
dmie) lub ograniczenia rozszerzalności układu two- 5.2.2. Obturacyjne zaburzenia
rzonego przez płuca, tchawicę i przeponę może dojść wentylacji
do powstania restrykcyjnych zaburzeń wentylacji.
Przyczyną tego rodzaju ograniczeń mogą być rozle- Do obturacyjnych zaburzeń wentylacji zalicza się te
głe zrosty warstwy opłucnej, pozostałe po rozległym choroby płuc, w których przebiegu następuje zwęże-
zapaleniu płuc, lub zwłóknienia płuc. nie dróg oddechowych, a tym samym zwiększenie
oporu przepływającego przez nie strumienia powie-
Zwłóknienie płuc jest chorobą polegającą na odkła- trza (zob. ryc. B 5.2-1).
daniu się w płucach, w sposób ogniskowy lub rozsia-
ny, dodatkowej tkanki łącznej. Zniszczeniu ulegają Najważniejszymi tego typu chorobami są:
przy tym zwykle również włókna sprężyste, płuca
■ astma oskrzelowa,
tracą częściowo swoją rozciągliwość i elastyczność.
■ przewlekła obturacyjna choroba płuc (POChP),
Do zwłóknień płuc zalicza się silikozę powstającą
na skutek działania wolnego kwasu krzemowego po- ■ rozedma płuc.
MUTSCHLER-2009.indd 638 2010-01-07 22:13:58
Układ oddechowy 639
przewlekłe za- rozedma
palenie oskrzeli płuc
brak obturacji dróg
oddechowych (pacjenci
niekwalifikowani jako
pacjenci z POChP)
blue POChP pink nieodwracalne
bloater puffe
Ryc. B 5.2-1. Podział obturacyjnych chorób dróg oddechowych. obturacja dróg
oddechowych
Obturacja dróg oddechowych w przebiegu astmy oskrzelowej
jest odwracalna. Pacjenci astmatyczni z wykazywaną w badaniu
funkcjonalnym długotrwałą obturacją resztkową zaliczani są do
grupy mieszanej astmatyczno-POChP. Przewlekłe choroby płuc odwracalne
(POChP) są charakterystyczne dla pacjentów, u których nieod-
wracalna obturacja dróg oddechowych utrzymuje się nawet
po zastosowaniu terapii optymalnej. Blue bloater (typ zapalny)
i pink puffer (typ rozedmowy) są ekstremalnymi odmianami kli-
nicznymi. Istnieją jednak pacjenci z z przewlekłym zapaleniem grupa mieszana astma
oskrzeli i/lub z rozedmą płuc, u których nie dochodzi do obtura- astma-POChP oskrzelowa
cji i w związku z tym nie są klasyfikowani jako pacjenci z POChP
(według Grimmingera, Suttorpa, Seegera).
W przypadku astmy oskrzelowej – przynajmniej leczenia optymalnego. Przewlekłe zapalenie oskrze-
Układ oddechowy
w jej początkowym stadium – obturacja dróg odde- li oraz rozedma płuc z obstrukcją występują u tych
chowych jest odwracalna. Pacjenci z ostrą obturacją pacjentów jednocześnie. Ekstremalnymi odmianami
w znacznym stopniu odwracalną, ale z przewlekłą klinicznymi schorzenia są blue bloater (typ zapalny)
obstrukcją resztkową wykazywaną w płucnym bada- oraz pink puffer (typ rozedmowy). Pacjentów z prze-
niu funkcjonalnym zaliczani są do grupy mieszanej wlekłym zapaleniem oskrzeli i/lub z rozedmą bez ob-
astmatyczno-POChP. W przewlekłej obturacyjnej jawów obstrukcji nie klasyfikuje się jako pacjentów
chorobie płuc (POChP) nieodwracalna obturacja dróg z POChP aż do momentu wystąpienia wyraźnej ob-
oddechowych utrzymuje się nawet przy zastosowaniu strukcji dróg oddechowych (ryc. B 5.2-1).
B5
5.3. Astma oskrzelowa
5.3.1. Patofizjologia
astmy oskrzelowej
Ze względu na częstość występowania astma oskrze- mięśniówki oskrzeli. Obecnie traktowana jest jako
lowa zaliczana jest w krajach uprzemysłowionych zapalna choroba dróg oddechowych z nadreaktyw-
do chorób społecznych. W krajach zachodnich na ast- nością drzewa oskrzelowego i zmienną obstrukcją
mę oskrzelową zapada około 5% dorosłych i około dróg oddechowych. Towarzyszącymi jej objawami
10% dzieci. Rozwojowi tej choroby sprzyjają czyn- są napadowa duszność (szczególnie w nocy i nad
niki genetyczne oraz czynniki zewnętrzne, np. częsta ranem) z towarzyszącymi świstami wydechowymi,
antybiotykoterapia, zachodni styl życia, a także kon- świsty i furczenia stwierdzane w badaniu osłucho-
takt z substancjami alergizującymi. Długo funkcjono- wym, a także kaszel i odkrztuszanie lepkiej prze-
wało przekonanie, że astma oskrzelowa jest chorobą zroczystej wydzieliny. Zwężenie dróg oddechowych
MUTSCHLER-2009.indd 639 2010-01-07 22:13:58
640 Układ oddechowy
A B
alergen
IgE
przeciwciała
alergen IgG
degranulacja blokada alergenu
alergen
Ryc. B 5.3-1. Degranulacja komórki tucznej (mastocytu) spowodowana pomostowaniem za pomocą alergenu dwóch przeciwciał
IgE (A) i blokada alergenu przez przeciwciała IgG w ramach tzw. odczulania (B).
spowodowane jest działaniem mediatorów, szczegól- (uwrażliwienie), które są wiązane na powierzchni
nie mediatorów zapalanych, wywołujących: komórek tucznych, a także granulocytów zasado-
i kwasochłonnych (zob. ryc. B 5.3-1). Jeżeli przy
■ skurcz mięśniówki oskrzeli,
ponownym kontakcie z alergenem nastąpi przy jego
■ obrzęk ściany oskrzeli, udziale pomostowanie dwóch sąsiadujących ze sobą
przeciwciał IgE, to w wyniku degranulacji tych ko-
■ wzmożone wydzielania gęstego, ciągnącego się
mórek uwalniane są mediatory zapalne. Reakcja
śluzu.
ta obejmuje początkowo komórki tuczne znajdujące
się na powierzchni błony śluzowej oskrzeli. Uwol-
W początkowym stadium choroby zdarzają się okresy
niane mediatory uszkadzają nabłonek oskrzelowy
bezobjawowe, w miarę jej rozwoju śluz wydzielany
i prowokują w ten sposób liczne komórki tuczne oraz
jest intensywniej, również między napadami, i tylko
komórki zapalne w głębiej położonych warstwach
częściowo usuwany. W ten sposób tworzy się dodat-
tkanek również do wydzielania mediatorów. Przez
kowe ryzyko wtórnych infekcji bakteryjnych.
uszkodzony nabłonek alergeny wnikają do obszaru
W zależności od czynników wyzwalających rozróż-
podnabłonkowego, gdzie są pochłaniane przez ma-
nia się:
krofagi. Te, oddzielając część białkową od alergenu
■ astmę alergiczną egzogenną, i prezentując ją specyficznemu receptorowi komórko-
wemu T na limfocytach T CD4+, stają się komórkami
■ astmę endogenną, dzieloną na np. astmę infekcyjną,
prezentującymi antygen (APC, antigen presenting
astmę rozwijającą się na skutek działania drażnią-
cells). Aktywowane w ten sposób limfocyty T odłą-
cych bodźców fizycznych lub chemicznych, astmę
czają m.in. interleukinę-2 (IL-2), która za pośredni-
polekową.
ctwem receptorów IL-2 prowadzi autokrynnie do na-
silonej proliferacji komórek T (ekspansja klonalna,
Wymienione formy alergii rzadko występują w czy- zob. ryc. B 5.3-2). Pod wpływem IL-4 limfocyty T
stej postaci, częściej natomiast w formie mieszanej. różnicują się m.in. na komórki pomocnicze Th2, któ-
Astma oskrzelowa alergiczna egzogenna jest ty- re za pośrednictwem komórek osoczowych stymulują
powa dla dzieci, tylko 10% ogółu pacjentów z tym produkcję przeciwciał IgE, a za pośrednictwem IL-5
schorzeniem stanowią dorośli. Polega ona na powsta- rekrutują i aktywują granulocyty eozynofilne i bazo-
niu nadwrażliwości zależnej od IgE. W ostatnich la- filne. Taka kolejność reakcji powoduje uwalnianie
tach występuje coraz częściej – niejednokrotnie ra- już istniejących i syntezę kolejnych mediatorów od-
zem z alergicznym nieżytem nosa oraz wypryskiem powiedzialnych za powstanie wyżej opisanych typo-
atopowym – u osób z atopią. wych objawów astmy, tj. skurczu oskrzeli, wzmo-
Różnego rodzaju alergeny (pyłek roślin, roztocze żonego wydzielania śluzu o zwiększonej gęstości,
kurzu domowego, naskórek zwierząt itp.) induku- a także obrzęku ściany oskrzeli w przypadku reakcji
ją u osób z atopią powstanie przeciwciał typu IgE utrzymującej się przez długi czas. Z biegiem czasu
MUTSCHLER-2009.indd 640 2010-01-07 22:13:59
Układ oddechowy 641
alergen
IgE
komórki tuczne
(mastocyty)
warstwa
nabłonek śluzowa
otwór połączenia ścisłego mediatory
(tight junctions) (np. histamina)
alergen
proliferacja
CD4
IL-2
MHC-II TCR
obszar podnabłonkowy
limfocyt T (CD4+)
makrofagi komórka prezentująca IL-4
antygen (różnicowanie)
IgE IL-4
komórka plazmowa komórki TH2
(produkcja IgE)
cytokiny
Układ oddechowy
(IL-5 i in.)
komórki tuczne eozynofile bazofile
(wiązanie IgE)
mediatory
B5
mediatory
reakcja natychmiastowa reakcja opóźniona reakcja późna
histamina, leukotrieny, cytokiny
serotonina, prostaglandyna,
proteazy czynnik aktywujący płytki
i in.
zwężenie
mięśnie gładkie
konsekwencje patofizjologiczne
oskrzelików
błona śluzowa obrzęk błony
i terapeutyczne
zapalny śluzowej
światło stan wzmożone
wydzielanie śluzu
o zwiększonej gęstości
terapia
w granicach normy obturacja
np. β2 sympatykomimetyki,
glukokortykosteroidy inhalacyjne
Ryc. B 5.3-2. Schemat patogenezy alergicznej astmy oskrzelowej (według Grimmingera, Suttorpa, Seegera).
MUTSCHLER-2009.indd 641 2010-01-07 22:13:59
642 Układ oddechowy
diatory zapalne niszczą barierę nabłonkową i przez
IgE to wdychane substancje drażniące mogą podrażnić
alergen
wrażliwe włókna C nieadrenergicznego i niecholi-
nergicznego układu nerwowego (NANC). Wywoła-
ne podrażnienie przenoszone jest w wyniku odruchu
aksonowego na mięśnie oskrzelowe, komórki śród-
błonkowe w naczyniach włosowatych i gruczoły pod-
rybosom śluzówkowe. Uwolnienie neuropeptydów, takich jak
substancja P, powoduje utrzymywanie się opisanych
wyżej patomorfologicznych mechanizmów zwężenia
oskrzeli, zwiększenie przepuszczalności naczyń wło-
sowatych (obrzęk) oraz wydzielanie większej ilości
śluzu.
komórki tuczne Oturacja oskrzelowa może być wywołana również
przez leki. Skurcz oskrzeli mogą wywołać przede
wszystkim blokery receptorów β-andrenergicznych
oraz leki przeciwbólowe i przeciwzapalne hamujące
biosyntezę prostaglandyn.
spontaniczne mediatory lipidowe synteza mRNA Jak już wspomniano, poszczególne formy astmy
uwolnienie uwolnione z błony i białek
cząsteczki w ciągu w ciągu są ze sobą powiązane, a granice pomiędzy nimi mogą
preformowane minut godzin być płynne. Szczególnie w późniejszych stadiach,
histamina prostaglandyna interleukiny w których oprócz podłoża alergicznego często poja-
serotonina leukotrieny α TNF i in. wia się nadreaktywność oraz reakcje odruchowe.
substancja P czynnik
proteazy i in. aktywujący Stan zapalny prowadzi do wzmożonej reaktywno-
płytki i in. ści (nadreaktywności) drzewa oskrzelowego. Nad-
wrażliwość zwiększa się w miarę nasilania się proce-
Ryc. B 5.3-3. Spontaniczne uwolnienie lub utworzenie nowych su chorobowego, w zaawansowanych stadiach nawet
mediatorów zapalnych po aktywacji komórek tucznych. bardzo niewielkie ilości substancji o działaniu bron-
chokonstrykcyjnym mogą zaburzyć funkcję płuc.
Ważna rolę w przypadku astmy oskrzelowej od-
napady astmy mogą być powodowane nie tylko przez grywają leukotrieny. W moczu i we krwi pacjentów
alergeny swoiste, ale również bodźce nieswoiste. z tą chorobą stwierdzono podwyższone stężenie tzw.
Ważne w patogenezie mediatory zapalne mogą leukotrienów cysteinowych (CystLT) LTC4, LTD4
być zmagazynowane w ziarnistościach komórek i LTE4, powstających z uwolnionego kwasu arachi-
(np. histamina) lub tworzone de novo pod wpływem donowego w wyniku jego połączenia się z białkiem
bodźców (np. leukotrieny, prostaglandyny, czynnik membranowym (five lipooxygenase activing prote-
aktywujący płytki – PAF), ewentualnie w ramach in – FLAP), a następnie utlenienia przez 5-lipook-
syntezy białkowej (np. cytokiny) (zob. ryc. B 5.3-3). sygenazę (5-LOX) do leukotrienu A4 (LTA4). Ten
Głównym źródłem mediatorów w początkowej fazie niestabilny produkt przejściowy albo przy udzia-
reakcji astmatycznej (tzw. reakcji natychmiastowej) le hydrolazy LTA4 ulega przemianie w LTB4, albo
są komórki tuczne obecne stale w tkance płucnej, alternatywnie, w wyniku przeniesienia reszty glu-
natomiast w reakcji późnej i przewlekłym stanie za- tationowej przez syntetazę LTC4 na LTA4 tworzy
palnym duże znaczenie mają granulocyty eozynofilne leukotrien cysteinowy LTC4. W dwóch kolejnych
i bazofilne przyciągane chemotaktycznie przez inter- przemianach enzymatycznych powstają pozostałe
leukinę i czynnik stymulujący tworzenie granulocy- leukotrieny cysteinowe LTD4 i LTE4. Leukotrieny
tów i makrofagów (GM-CSF). cysteinowe syntezowane są głównie przez granu-
Astma endogenna nie rozwija się na skutek działa- locyty eozynofilne i komórki tuczne. Za pośredni-
nia alergenów specyficznych, które mogłyby wywoły- ctwem rozmieszczonych w drzewie oskrzelowym
wać ostre ataki, lecz nadreaktywności drzewa oskrze- receptorów CysLTl stymulują wytwarzanie śluzu,
lowego. U chorych na ten rodzaj astmy nawet pier- powodują ucieczkę płynu z naczyń, a tym samym
wotnie nieswoiste czynniki drażniące, np. zakażenia, obrzęk, skurcz oskrzeli oraz uszkodzenie nabłonka
wdychanie dymu tytoniowego, zimnego powietrza, oskrzelowego.
mgły lub powietrza zanieczyszczonego, powodują W zależności od ciężkości objawów astmę oskrze-
nadmierną reakcję oskrzeli. Przyjmuje się, że me- lową można podzielić na 4 stopnie:
MUTSCHLER-2009.indd 642 2010-01-07 22:14:00
Układ oddechowy 643
β2-sympatyko-
mimetyki,
komórki tkanek glukokortykosteroidy, ksantyna,
oskrzelowych kwas kromoglikanowy, parasympa-
nedokromil tyktolityki
komórki tuczne
reakcja
natychmiastowa,
mediatory zwężenie
makrofagi zapalne oskrzeli
komórki
nabłonkowe
glukokortykosteroidy,
kwas kromoglikanowy,
nedokromil reakcja późna:
wtargnięcie zwężenie oskrzeli
mediatory zwiększona przepusz-
eozynofile czalność naczyń odma
zapalne
uszkodzenie nabłonka
oskrzelowego
neutrofile
wzmożone wydzielanie
śluzu o podwyższonej
limfocyty gęstości
nadreaktywność oskrzeli
Ryc. B 5.3-4. Pochodzenie i działanie mediatorów zapalnych w drogach oddechowych i miejsca działania leków przeciwastmatycz-
nych.
Układ oddechowy
czynniki szkodliwe
(np. dym papierosowy, zimne powietrze,
zanieczyszczenia z powietrza)
wzmożone
wydzielanie śluzu śluz bariera nabłonkowa
B5
uszkodzenie
nabłonka
gruczoł śluzowy
naczynie krwionośne doprowadzające
hipersekrecja włókna C nieadrenergicznego
śluzu i niecholinergicznego układu
nerwowego (NANC)
odruch
ekstrawazacja cholinergiczny
plazmy
komórki mięśni gładkich
zwężenie oskrzeli,
hipertrofia, hiperplazja
Ryc. B 5.3-5. Patogeneza astmy oskrzelowej o podłożu niealergicznym (według Barnesa).
MUTSCHLER-2009.indd 643 2010-01-07 22:14:00
644 Układ oddechowy
stan zapalny w oskrzelach
komórki zapalne fibroblasty,
(eozynofile, makrofagi) miofibroblasty
tworzenie składników
MMPs macierzy pozakomórkowej
(nierównowaga MMP/TIMP) (kolagen, elastyna, fibronektyna)
rozkład składników odkładanie składników
macierzy pozakomórkowej macierzy pozakomórkowej
(kolagen, glikozaminy i in.) w drogach oddechowych
cytokiny ułatwienie utrata włóknienie zgrubienie
i peptydy migracji integralności okołooskrzelowe blaszki
chemotaktyczne komórek tkanek siateczkowej
przewlekłe zapalenie remodeling dróg oddechowych
Ryc. B 5.3-6. Długotrwałe skutki stanu zapalnego w oskrzelach w przebiegu astmy oskrzelowej (według Kroegla i in.). MMPs – meta-
loproteinazy macierzy, TIMP – tkankowe inhibitory metaloproteinaz.
■ stopień 1: astma epizodyczna, w której najwyżej dowie i funkcjonowaniu (remodeling dróg oddecho-
dwa razy w tygodniu dochodzi do krótkotrwałych wych, zob. ryc. B 5.3-6). Według najnowszych badań
napadów przy zachowaniu prawidłowych wartości istotna jest w takich sytuacjach równowaga pomiędzy
PEF; metaloproteinazami macierzy (matrix metalloprotei-
nases – MMP) i ich endogennymi inhibitorami (tissue
■ stopień 2: astma przewlekła lekka, w której dolegli-
inhibitors of metalloproteinases – TIMP). MMP są za-
wości występują najwyżej raz dziennie, a wartości
wierającymi cynk enzymami, które rozkładają prote-
PEF wynoszą co najmniej 80% wartości prawidło-
oglikany i białka macierzy pozakomórkowej tkanki
wych;
łącznej (kolagen, glikozaminy i in). Na skutek procesu
■ Stopień 3: astma przewlekła średnio ciężka, dla któ- zapalnego zaburzenie proporcji między metaloprotei-
rej charakterystyczne są częstsze, przede wszystkim nazami macierzy i ich inhibitorami może spowodować
nocne napady i wartości PEF w zakresie 60–80% przesunięcie równowagi między rozkładem i regenera-
wartości prawidłowych; cją macierzy zewnątrzkomórkowej.
■ stopień 4: astma przewlekła ciężka, w której chory
prawie nigdy nie jest całkowicie wolny od objawów,
a wartości PEF stanowią mniej niż 60% wartości 5.3.2. Leczenie astmy oskrzelowej
prawidłowych; wydolność fizyczna jest znacznie (leki przeciwhistaminowe)
ograniczona, zdarzają się również ostre pogorsze-
nia, niekiedy zagrażające życiu. Celem leczenia astmy jest poprawa jakości życia oraz
rokowań poprzez:
W przypadku niedostatecznego leczenia przewlekłe-
■ kontrolę objawów,
go stanu zapalnego dróg oddechowych po dłuższym
czasie dochodzi do nieodwracalnych zmian w ich bu- ■ unikanie ostrych pogorszeń,
MUTSCHLER-2009.indd 644 2010-01-07 22:14:01
Układ oddechowy 645
■ osiągnięcie możliwie prawidłowej czynności płuc, wiążą alergeny, nie dopuszczając do ich reakcji z IgE
na powierzchni mastocytów (zob. ryc. B 5.3-1).
■ utrzymanie normalnej aktywności fizycznej,
Warunkiem skuteczności odczulania jest zastoso-
■ (jak najdalej idące) unikanie działań niepożąda- wanie szczepionki z właściwym antygenem, wczes-
nych. ne rozpoczęcie oraz konsekwentne prowadzenie
leczenia przez kilka lat. Wielu chorych uczulonych
Leczenie astmy można zacząć już na poziomie reakcji jest na liczne, częściowo nieustalone alergeny i w ta-
alergen-przeciwciało i wtedy określa się je jako przy- kich przypadkach nie jest możliwe całkowite odczu-
czynowe. Może ono jednak polegać na usuwaniu lub lenie. Prawdopodobieństwo sukcesu terapeutycznego
przynajmniej ograniczaniu skutków reakcji alergen- zmniejsza się ponadto w przypadku dłuższego trwania
-przeciwciało, ewentualnie na uwalnianiu mediatorów choroby, w starszym wieku oraz w uczuleniu na wie-
albo na zapobieganiu lub przynajmniej ograniczaniu le alergenów. W związku z tym odczulanie na jeden
zakresu odruchowego skurczu oskrzeli. Takie lecze- znany alergen należy rozpoczynać jak najwcześniej.
nie nazywane jest objawowym. Wobec dużej liczby Odczulanie jest możliwe w przypadku uczulenia
różnych mediatorów uczestniczących w patogenezie np. na pyłki roślin oraz roztocze kurzu domowego,
astmy jest oczywiste, że leki znoszące działanie tylko ponieważ unikanie tego typu alergenów oraz farma-
jednego z nich są mniej korzystne od leków wpływa- koterapia nie są wystarczające do kontrolowania ob-
jących na więcej procesów w przebiegu tej choroby. jawów. W związku z ograniczoną skutecznością oraz
możliwymi powikłaniami (zob. poniżej) stosowanie
tej formy leczenia jest kwestią sporną.
5.3.2.1. Leczenie przyczynowe Ze względu na ryzyko wystąpienia ciężkich, nie-
kiedy zagrażających życiu, reakcji anafilaktycznych
jako odpowiedzi na wstrzyknięcie alergenu odczu-
Leczenie przyczynowe jest możliwe pod warunkiem:
lanie może być wykonywane tylko pod warunkiem
■ utrzymania karencji, zachowania odpowiednich środków ostrożności
(konieczna jest możliwość podania leków przeciw-
■ przeprowadzeniu odczulania,
wstrząsowych). Ryzyko wystąpienia takich reakcji
Układ oddechowy
■ wiązania IgE przez monoklonalne przeciwciała zwiększa się wraz ze stężeniem alergenu w szcze-
anty-IgE. pionce.
Karencja jest unikaniem kontaktu z alergenem. Za- Monoklonalne przeciwciała. Ze względu na rolę,
pewnia to pobyt w wysokich górach lub nad morzem jaką monoklonalne przeciwciała anty-IgE odgrywa-
i choć zazwyczaj nie trwa zbyt długo, jego dobro- ją we wczesnej i późnej reakcji alergicznej, mogą
czynne działanie nie powinno być podawane w wąt- być rozpatrywane jako najważniejszy element tera-
pliwość. U wielu chorych dochodzi w tym czasie pii. Humanizowane przeciwciało anty-IgE omalizu-
do znacznej poprawy, a nierzadko również do zmniej- mab, zawierające około 5% białka mysiego, wiąże
B5
szenia nadreaktywności drzewa oskrzelowego. się precyzyjnie z epitopem w obrębie fragmentu Fc
W miarę możliwości należy także unikać innych w IgE. Tworząc kompleksy z krążącymi cząsteczka-
czynników prowokujących napady astmy, np. zimne- mi IgE, zapobiega ich łączeniu się z receptorami IgE
go powietrza oraz dymu. na powierzchni komórek tucznych (zob. ryc. B 5.3-7).
Odczulanie jest zasadne tylko w astmie alergicz- Długotrwałe leczenie przeciwciałami anty-IgE
nej, a jego celem jest spowodowanie niewrażliwości prawdopodobnie prowadzi również do zmniejszenia
pacjenta na alergen. Osiąga się to przez ustalenie aler- gęstości receptorów IgE na powierzchni komórek
genu powodującego objawy i podawani go podskór- zapalnych.
nie w formie szczepionki w rosnących dawkach. Omalizumab jest wskazany w ramach leczenia
Mechanizm działania szczepionek odczulają- uzupełniającego w przypadku pacjentów (od 12 roku
cych nie został całkowicie wyjaśniony. Zakłada się, życia) z ciężką przewlekłą astmą alergiczną, wykazu-
że wstrzykiwanie alergenu stymuluje powstawanie jącą odczyn dodatni w testach skórnych oraz pogłę-
dużej ilości limfocytów T supresorowych hamujących biającą się pomimo terapii inhalacyjnej glukokorty-
reakcję alergiczną. Rozważa się również możliwość, kosteroidami i β2-sympatykomimetykami.
że w warunkach stałej stymulacji antygenowej oprócz Omalizumab wstrzykiwany jest podskórnie. Jego
przeciwciał IgE tworzone są również, w coraz więk- metabolizm i eliminacja następują w wątrobie bądź
szej ilości, przeciwciała IgG. Immunoglobuliny te w układzie retikularno-histiocytarnym, a okres poło-
nie wiążą się z mastocytami, lecz krążą we krwi i tam wiczego rozpadu wynosi około 26 dni.
MUTSCHLER-2009.indd 645 2010-01-07 22:14:01
646 Układ oddechowy
tworzenie kompleksu z wolnym IgE
IgE
blokada
wiązania IgE
omalizumab
P
receptor IgE
zahamowanie
P
produkcji IgE
P
P
komórka plazmowa
komórka tuczna
Ryc. B 5.3-7. Hamowanie przez przeciwciała anty-IgE (omalizumab) reakcji immunologicznej powodującej uwalnianie IgE.
Omalizumab kieruje się w stronę epitopu fragmentu Fc w IgE, wiążącego się z receptorem IgE.
Dawkowanie i częstość podawania zależą od ilości inhalatory proszkowe nie nadają się dla pacjentów, którzy
IgE i masy ciała. W przypadku pacjentów, u których mają ograniczone przepływy oddechowe (np. dzieci, oso-
by starsze). Ponieważ ilości substancji docierającej do płuc
IgE wynosi mniej niż 76 IU/ml, korzystne działa- i miejsca działania mogą się bardzo różnić w zależności
nie terapeutyczne jest bardzo mało prawdopodobne. od sposobu inhalacji, zmiana typu inhalatora może wyma-
Ponieważ organizm cały czas produkuje IgE, oma- gać dopasowania dawki.
lizumab powinien być podawany przez 4 tygodnie,
ponieważ umożliwi to utrzymanie niskiego poziomu
IgE.
Działaniami niepożądanymi towarzyszącymi po- 5.3.2.2.1. Leki o działaniu przeciwzapalnym
dawaniu leku mogą być odczyn i ból w miejscu
iniekcji, obrzmienia, rumień, świąd i ból głowy. Ograniczenie reakcji zapalnej w błonie śluzowej
oskrzeli w przebiegu astmy można osiągnąć za po-
mocą:
5.3.2.2. Leczenie objawowe ■ substancji hamujących uwalnianie mediatorów,
■ glukokortykosteroidów,
Leczenie objawowe astmy oskrzelowej może polegać
na: ■ antagonistów receptorów leukotrienów CysLT1.
■ hamowaniu stanu zapalnego i obniżaniu nadreak-
Dzięki ograniczeniu wydzielania mediatorów leki
tywności oskrzeli,
o działaniu przeciwzapalnym, stosowane przez dłuż-
■ znoszeniu skurczu oskrzeli. szy czas, powodują ograniczenie nacieku z komó-
rek zapalnych w tkankach, zmniejszenie obstrukcji
Wiele leków przeciwastmatycznych stosuje się w formie dróg oddechowych oraz nadreaktywności na bodźce
inhalacji. Dzięki temu osiąga się stężenia terapeutyczne kurczące oskrzela (zob. ryc. B 5.3-8). W przypadku
leków w oskrzelach przy znacznym ograniczeniu (niepo-
żądanych) działań systemowych. Do niedawna dostępne wszystkich tzw. kontrolerów, z wyjątkiem glukokor-
były tylko zawiesiny freonowe w postaci aerozoli do- tykosteroidów przyjmowanych w dużych dawkach,
zowanych. Podanie dawki leku było prawie niezależne efekt terapeutyczny pojawia się bardzo powoli. Leki
od oddechu. W najnowszych preparatach (np. z bezchlo- te nadają się więc tylko do zapobiegania napadom,
rowym hydrofluoroalkanem jako nośnikiem) substancja a w celu ograniczenia stanu zapalnego w drogach
czynna ma postać roztworu. Rozpuszczony lek dociera
nawet do drobnych odgałęzień oskrzeli. W stosowanych oddechowych muszą być stosowane regularnie i dłu-
inhalatorach proszkowych substancja czynna ulega roz- gofalowo, niezależnie od aktualnego samopoczucia
pyleniu pod wpływem silnego wdechu. W związku z tym chorego.
MUTSCHLER-2009.indd 646 2010-01-07 22:14:01
Układ oddechowy 647
objawami astmy alergicznej, w przypadku pacjentów
przed
rozpoczęciem dorosłych przeważnie nie przynoszą zadowalających
leczenia efektów.
0,35
0,3 Mechanizm działania nie jest jeszcze szczegółowo
wyjaśniony. W pobudzonych komórkach tucznych
0,25
metacholina (mg)
wykazano jedynie blokadę kanałów chlorkowych
0,2 i związane z tym ograniczenie uwalniania mediato-
0,15
rów. Przypuszcza się ponadto, że substancje te hamu-
ją wytwarzanie określonych cytokin w makrofagach.
0,1 Ze względu na dużą polarność i wynikającą z niej
0,05 niewielką lipofilność kwas kromoglikanowy i ne-
dokromil wchłaniają się bardzo słabo i w związku
0 z tym działają tylko miejscowo. W przypadku astmy
4 8 12
czas trwania leczenia alergicznej muszą być inhalowane w postaci zmi-
kronizowanego (w specjalnym rozpylaczu) proszku,
placebo nedokromil roztworu lub aerozolu z dozownika ciśnieniowego.
kwas kromoglikanowy glukokortykosteroid Połknięte nie ulegają resorpcji i są prawie całkowicie
wydalane z kałem.
Ryc. B 5.3-8. Wzrost poziomu metacholiny niezbędnej do ob- Dawkowanie kromoglikanu sodowego w posta-
turacji oskrzeli przy inhalacji stabilizatorami komórek tucznych. ci proszku lub roztworu wynosi 20 mg cztery razy
dziennie, w przypadku stosowania aerozolu ciśnie-
niowego tylko 2 mg cztery razy dziennie. Nedokro-
mil inhaluje się 2–4 razy dziennie po 4 mg.
5.3.2.2.1.1. Leki hamujące uwalnianie Działaniami niepożądanymi występującymi w przy-
mediatorów padku wdychania proszku są miejscowe podrażnie-
nia w drogach oddechowych, przechodzące niekiedy
Układ oddechowy
Do stosowanych leków hamujących uwalnianie me- w skurcz oskrzeli. Często również smak nedokromilu
diatorów należą: odczuwany jest jako nieprzyjemny.
Przy jednoczesnym podawaniu innych leków
■ kwas kromoglikanowy,
nie ma ryzyka niepożądanych interakcji.
■ nedokromil. Kromony stosuje się również w alergicznym za-
paleniu spojówek oraz alergicznym nieżycie nosa,
Kwas kromoglikanowy i nedokromil (kromony). a także w chorobach alergicznych przewodu pokar-
Stosowane profilaktycznie leki te zapobiegają w jed- mowego, np. w alergiach pokarmowych.
nakowym stopniu reakcji natychmiastowej i późnej.
B5
Kromony rzadko podawane są dzieciom z typowymi
5.3.2.2.1.2. Glukokortykosteroidy
HOOC O O COOH Kortyzol i jego pochodne należą do najsilniejszych
leków przeciwzapalnych. W astmie oskrzelowej
OH ograniczają reakcje zapalne w tkankach, zmniejsza-
O O O O ją wydzielanie śluzu, poprawiają oczyszczanie rzęs-
kowe, zmniejszają obrzęk błony śluzowej oskrzeli,
częściowo przeciwdziałają uszkodzeniu nabłonka
kwas kromoglikanowy
i wzmacniają (m.in. przez zwiększoną ekspresję
receptorów β-adrenergicznych) działanie leków
C3H7 C2H5
β2-sympatykomimetycznych (tzw. efekt beta-tole-
HOOC O N COOH
rancyjny). Dopiero w dużych lub bardzo dużych daw-
kach wykazują bezpośrednio w oskrzelach działanie
spazmolityczne.
O O
nedokromil Glukokortykosteroidy stosowane w leczeniu
wziewnym. Wziewne miejscowo działające gluko-
MUTSCHLER-2009.indd 647 2010-01-07 22:14:01
648 Układ oddechowy
kortykosteroidy są lekami z wyboru w długotermino- przyjmowaniu dużych dawek (stopień 4, zob. tab. B
wym leczeniu astmy. Najczęściej stosowane są: 5.3-1) należy liczyć się z możliwością wystąpienia
ogólnoustrojowych działań niepożądanych w postaci
■ dipropionian beklometazonu,
zahamowania czynności kory nadnerczy, osteoporozy
■ budezonid, i zaćmy. W takich przypadkach konieczne jest stoso-
■ cyklezonid,
■ 17-propionian flutikazonu, O
■ mometasonfuroat. O O C2H5
CH3 O C2H5
HO
Dipropionian beklometazonu i cyklezonid są proleka- CH3 H O
mi, a ich hydroliza do postaci czynnej odbywa się pod CH3
Cl H
wpływem esteraz płucnych w pozycji C-21. Dzięki O
temu udaje się uniknąć zarówno ogólnoustrojowego,
dipropionian beklometazonu
jak i miejscowego niepożądanego działania leku.
Typowe dawkowanie wynosi dla dipropionianu
beklometazonu 0,5–1 mg, dla budezonidu 0,4–0,8 mg, O CH2OH
dla cyklezonidu 0,08–0,16 mg, dla 17-propionianu CH3 O C3H7
HO
flutikazonu 0,25–0,5 mg/dzień a dla mometasonfu- O
CH3 H
roatu 0,2–0,4 mg/dzień (zob. tab. B 5.3-1). H
H H
Skuteczność leczenia jest uzależniona od techniki O
(ilość połykanej substancji czynnej powinna być jak budezonid
najmniejsza) oraz regularności przeprowadzania in-
halacji. W leczeniu zapobiegawczym, podobnie jak O
kromony, stosowane są inhalatory proszkowe zawie- O CH(CH3) 2
rające glukokortykosteroidy i aerozole dozowane.
O
Ich działanie terapeutyczne ujawnia się jednak dopie- CH3 O
ro po kilku dniach. W przypadku ostrych napadów HO
CH3 H O
astmy są nieskuteczne, ponieważ nie wykazują dzia-
H
łania rozszerzającego oskrzela. H H
Podanie miejscowe stosunkowo niewielkich da- O
wek leku pozwala uniknąć ogólnoustrojowych dzia- cyklezonid
łań niepożądanych. Nawet w sytuacji przypadkowego
połknięcia choćby niewielkiej dawki substancji czyn-
nej jej biologiczna dostępność z powodu wysokiego O S CH2F
efektu pierwszego przejścia jest niewielka (zob. tab. CH3 O C2H5
HO
B 5.3-1), a ryzyko pojawienia się działań niepożąda-
CH3 H O
nych znikome. CH3
Działaniami niepożądanymi objawiającymi się F H
miejscowo są chrypka (odwracalna miopatia mięś- O
niówki krtani) i suchość w jamie ustnej wynikające F
z dużej koncentracji leku. W przypadku pojawie-
17-propionian flutikazonu
nia się chrypki wskazane jest zredukowanie dawki,
zmiana techniki inhalowania lub czasowe odstawie-
nie leku. U 5% pacjentów obserwuje się również O CH2Cl
drożdżakowe zapalenie jamy ustnej. Zakażeniom CH3 O
HO O
tego typu zapobiegają w większości przypadków leki CH3 H O
przeciwgrzybicze, które można przyjmować bez ko- CH3
nieczności przerywania leczenia. Wykonywanie in- Cl H
halacji glukokortykosteroidami przed posiłkami po- O
zwala nie tylko uniknąć infekcji grzybiczych, ale tak- mometasonfuroat
że znacząco zmniejsza chrypkę. Przy długotrwałym
MUTSCHLER-2009.indd 648 2010-01-07 22:14:02
Układ oddechowy 649
Tabela B 5.3-1. Dawkowanie (dawki dzienne) wziewnych glukokortykosteroidów u dorosłych i dzieci według schematu długotermi-
nowego leczenia astmy oskrzelowej. DB – dostępność biologiczna
Sto- Dipropionian Budezonid Cyklezonid 17-propionian Mometasonfuroat
pień beklometazonu* (doustnie DB ok. 11%) (doustnie DB < 1%) flutikazonu (doustnie DB < 1%)
(doustnie DB ok. 20%) (doustnie DB < 1%)
dorośli dzieci dorośli dzieci dorośli dzieci dorośli dzieci dorośli dzieci
(od 5 r.ż.) (od 6 r.ż.) (< 12 r.ż.) (od 4 r.ż.) (< 12 r.ż.)
1 - - - - - - - - - -
2 500 μg < 400 μg 400 μg < 400 μg 80 μg - 250 μg < 200 μg 200 μg -
3 1000 μg = 400 μg 800 μg = 400 μg 160 μg - 500 μg = 200 μg 400 μg -
4 2000 μg > 400 μg 1600 μg > 400 μg > 160 μg - 1000 μg > 200 μg 800 μg -
* dawka ze składników drobnocząsteczkowych powinna być zredukowana o połowę (zob. informacje producentów)
wanie profilaktyki osteoporozy poprzez przyjmowa- jący leczenie przeciwastmatyczne. Wskazaniem
nie soli wapnia i witaminy D. do jego zastosowania jest leczenie uzupełniające
u chorych na astmę lekką lub średnio ciężką (stopnie
Glukokortykosteroidy stosowane systemowo. 2–3), której kontrola za pomocą glukokortykostero-
W związku z poważnymi efektami niepożądanymi idów wziewnych oraz doraźnie stosowanych krótko
długotrwale systemowe stosowanie glukokortykoste- działających β2-sympatykomimetyków jest niewy-
roidów jest uzasadnione tylko wtedy, gdy inne me- starczająca (zob. poniżej). Montelukast nie nadaje się
tody lecznicze nie przynoszą skutku. Należy jednak do leczenia ostrych napadów astmy.
utrzymywać wziewne podawanie glukokortykostero-
idów, aby w leczeniu ogólnym można było stosować Po doustnym podaniu montelukast jest szybko wchła-
ich możliwie najmniejsze dawki. niany. Jego dostępność biologiczna przy podaniu
Układ oddechowy
W typowych przypadkach ogólna glukokortyko- doustnym wynosi średnio 64%. Ponieważ montelu-
terapia astmy polega na doustnym podawaniu pred- kast jest metabolizowany głównie przez CYP3A4,
nizolonu. Dawka (podtrzymująca) wynosi 5–10 mg szczególnie u dzieci wskazana jest ostrożność przy
dziennie. podawaniu go jednocześnie z innymi lekami induku-
Duża dawka glukokortykosteroidów, podana do- jącymi lub hamującymi aktywność CYP3A4. Okres
żylnie, jest konieczna i ratująca życie w ciężkich na- półtrwania określa się na 2,7–5,5 godz.
padach astmy, a szczególnie w stanie astmatycznym Dawkowanie montelukastu wynosi u dorosłych
(status asthmaticus). 10 mg jednorazowo przed snem, a u dzieci między 6
i 14 rokiem życia 5 mg na dzień.
B5
W trakcie przyjmowania leku mogą pojawić się
5.3.2.2.1.3. Antagoniści receptorów działania niepożądane w postaci bólu głowy, bólu
leukotrienów CysLT1 brzucha, kaszlu, biegunki, niestrawności i gorączki.
Opisano pojedyncze przypadki alergicznego ziarni-
Antagoniści receptorów leukotrienów1 cysteinowych
(lukasty) łączą się selektywnie z receptorem CysLT1
i tym samym hamują prowokowane przez leukotrie-
ny cysteinowe wytwarzanie śluzu, obrzęk, skurcz O
–
Na
+
oskrzeli oraz uszkodzenie nabłonka oskrzelowego.
W badaniach klinicznych leki te wykazały skutecz- O
S
ność w astmie wysiłkowej, astmie prowokowanej
przez zimne powietrze lub alergeny oraz w astmie Cl N
indukowanej przez niesteroidowe leki przeciwzapal- HO
ne. Ze względu na niewystarczające działanie leki te H3C CH3
nie mogą jednak zastąpić wziewnych glukokortyko-
steroidów. Pierwszym i dotychczas jedynym na rynku montelukast sodowy
niemieckim (i polskim) antagonistą receptora CysLT1
jest montelukast, wprowadzony jako lek uzupełnia-
MUTSCHLER-2009.indd 649 2010-01-07 22:14:02
650 Układ oddechowy
niakowego zapalenia naczyń (zespół Churga-Straus- ■ teofilina,
sa) oraz nadwrażliwości (reakcje anafilaktyczne,
■ antagoniści receptorów muskarynowych (parasym-
obrzęk naczynioruchowy, pokrzywka).
patykolityki).
Lekami z tej samej grupy są zafirlukast, pranlu-
kast, pobilukast i iralukast.
β2-sympatykomimetyki. W mięśniówce oskrze-
li β2-sympatykomimetyki pobudzają receptory β2
związane z białkiem G do stymulacji cyklazy adeny-
5.3.2.2.1.4. Inne leki o działaniu hamującym
lanowej, w związku z czym następuje wzmożona pro-
stan zapalny
dukcja cAMP aktywującego kinazę proteinową typu
A. W wyniku szeregu reakcji dochodzi do przerwania
Leki przeciwhistaminowe blokujące wyłącznie recep-
skurczu oskrzeli. Ponadto β2-sympatykomimetyki
tor H1 wykazują niewystarczającą skuteczność w lecze-
pobudzają aktywność ruchową rzęsek, dzięki czemu
niu astmy, mimo że histamina należy do mediatorów
usprawniają usuwanie śluzu oraz hamują wydzielanie
uwalnianych w czasie degranulacji komórek tucznych.
mediatorów zapalnych, co umożliwia ich ograniczo-
Odnosi się to właściwie również do ketotifenu wyka-
ne działanie profilaktyczne. Substancje z tej grupy
zującego dodatkowo działanie stabilizujące mastocyty.
nie wykazują jednak działania przeciwzapalnego,
Badania kliniczne wskazują jednak, że leki z tej grupy,
w związku z czym w długotrwałym leczeniu przy 3
które oprócz blokady receptorów H1 działają na gra-
i 4 stopniu nie mogą zastąpić wziewnych glukokor-
nulocyty kwasochłonne (np. cetyryzyna i loratadyna),
tykosteroidów. Podawane są najczęściej wziewnie,
mogą być skuteczne w leczeniu astmy.
ponieważ w ten sposób unika się w dużym stopniu
ich działania niepożądanego (drżenia, tachykardia,
komorowe zaburzenia rytmu, wzrost ciśnienia krwi)
5.3.2.2.2. Leki rozszerzające oskrzela
(zob. tab. B 5.3-2).
Krótko działające wziewne β2-sympatykomimety-
Lekami stosowanymi w stanach skurczowych oskrze-
ki są lekami pierwszego rzutu w doraźnym leczeniu
li są:
ostrego skurczu oskrzeli. Rozszerzenie oskrzeli na-
■ β2-sympatykomimetyki, stępuje w ciągu kilku minut po aplikacji i utrzymuje
β2 sympatykomimetyki
K+
β2
cyklaza
Gs adenylanowa P
Ca2+– aktywowany kanał K+
PDE
↑ + ↑
IP3 / [Ca2 ] rozszerzenie oskrzeli
AMP cAMP ATP
↑
MLCK
teofilina
PKA
Na+/
K+-ATP-azy ↑
Ryc. B 5.3-9. Punkty uchwytu i działania β2-sympatykomimetyków i teofiliny (według Bernasa). MLCK – kinaza lekkiego łańcucha
miozyny, PDE – fosfodiestrazy, PKA – kinazy proteinowe typu A, IP3 – inozytolotrifosforan.
MUTSCHLER-2009.indd 650 2010-01-07 22:14:03
Układ oddechowy 651
Tabela B 5.3-2. Dane o β2-sympatykomimetykach istotne w leczeniu astmy (według Lemmera)
Substancja Nazwa handlowa Sposób Dostępność biologiczna Okres półtrwania Czas działania
czynna (przykłady) podawania (doustnie) [%] (godz.) (godz.)
Działania krótkotrwałe (3-6 godz.)
fenoterol Berotec wziewnie, doustnie 1,5 3 3-5
reproterol Bronchospasmin wziewnie, doustnie 1-2 4-6
salbutamol SalbuHEXAL, wziewnie, doustnie 25 2-7 3-6
Sultanol
terbutalina
Bricanyl, doustnie 10-15 3-4 3-6
Terbutalin STADA
tulobuterol doustnie
Atenos, Brelomax 2-4 3-6
Działanie długotrwałe (12-24 godz.)
bambuterol Bambec doustnie 10-12 10 24
a
clenbuterol Spiropent doustnie 100 1 (34) 14
a
formoterol Foradil, wziewnie, doustnie 65 2-3 (5-8) 12
Formoterol-ratiopharm
salmeterol Serevent wziewnie 3,5 12
a
eliminacja dwufazowa
Układ oddechowy
się przez 3–6 godzin. Substancjami o takim działaniu do wytworzenia się aktywnego β2-sympatykomime-
są fenoterol, reproterol, salbutamol, terbutalina i tulo- tyku terbutaliny (zob. ryc. B 5.3-10).
buterol (zob. tab. B 5.3-2). Dawkowanie powinno być możliwie jak najmniej-
sze. Ograniczenie dawki można osiągnąć przez połą-
Wziewne β2-sympatykomimetyki o długim czasie czenie z innymi lekami przeciwastmatycznymi.
działania (12–24 godz.), jak clenbuterol, formote-
rol i selmaterol, są wskazane w profilaktyce ataków Teofllina. Działanie rozszerzające oskrzela, ale nieco
astmy średniej (stopień 3) i ciężkiej (stopień 4). słabsze niż β2-sympatykomimetyki wykazuje pochod-
Ze względu jednak na swój późny początek dzia- na ksantyny – teofilina i jej rozpuszczalne w wodzie
B5
łania przy długim czasie działania nie nadają się sole z organicznymi zasadami, np. etylenodiaminą.
do ukierunkowanego leczenia obturacji oskrzelo- Mechanizm działania teofiliny nie jest jeszcze w peł-
wej. Stosowane powinny być zawsze z wziewnymi ni poznany. W wysokim stężeniu hamuje ona fosfo-
glukokortykosteroidami. diesterazę rozkładającą cAMP do AMP, co prowadzi
Długoterminowe leczenie ogólnoustrojowe β2- do zwiększonego stężenia cAMP w komórkach (zob.
-sympatykomimetykami (zob. tab. B 5.3-2) jest wska- ryc. B 5.3-9), a to z kolei do rozszerzenia oskrzeli.
zane tylko w wyjątkowych przypadkach, gdy za- Teofllina blokuje ponadto receptory adenozyny, któ-
stosowanie preparatów wziewnych do opanowania rych stymulacja powoduje skurcz oskrzeli i uwalnia-
objawów astmy okazuje się niewystarczające. Do- nie histaminy. Dyskutowane jest również bezpośred-
bre efekty przynosi natomiast stosowanie substancji nie działanie przeciwzapalne tego leku.
o przedłużonym działaniu. Osiąga się je albo za po- Uwagę należy zwrócić na zawężony zakres lecze-
mocą substancji, które dzięki budowie cząsteczkowej nia z towarzyszącymi działaniami niepożądanymi
mają długi czas działania, albo przez podanie proleku, w przypadku wyższych stężeń w osoczu (zob. poni-
który dopiero we krwi lub w tkankach rozwija swo- żej), duże różnice międzyosobnicze okresu półtrwa-
je właściwości. W przypadku bambuterolu fenolowe nia w osoczu (zob. tab. B 5.3-3) oraz okołodobową
grupy OH istotne przy wiązaniu receptorów są ma- zmienność kinetyki teofiliny przy podawaniu doust-
skowane w wyniku estryfikacji kwasem karbami- nym. Leczniczo korzystne stężenie w surowicy wy-
nowym. Reakcja hydrolizy estrowej prowadzi więc nosi 5–15 μg/ml.
MUTSCHLER-2009.indd 651 2010-01-07 22:14:03
652 Układ oddechowy
Tabela B 5.3-3. Okres półtrwania trwania teofiliny w surowicy daczkę, nadczynność tarczycy, zaburzenia rytmu ser-
ca, obturacyjną kardiomiopatię przerostową oraz
Pacjenci Okres półtrwania choroby wątroby.
(godz.) Jako (zależne od dawki) działania niepożąda-
Dorośli ne, szczególnie w przypadku stężenia we krwi > 20
niepalący 7- 9 μg/ml, mogą wystąpić zaburzenia w obrębie ośrod-
palący 4- 5
kowego układu nerwowego (niepokój, bezsenność,
Dzieci (> 1 rok) 3- 5 nudności, bóle głowy), a ponadto tachykardia i tachy-
wcześniaki > 24 arytmia oraz dolegliwości żołądkowo-jelitowe. Opi-
niewydolnością nerek lub serca > 24 sywano przypadki zgonów po zbyt szybkim podaniu
dożylnym.
Stężenie teofiliny we krwi zwiększają antybiotyki
makrolidowe, cymetydyna, cyprofloksacyna i enok-
Teofilina podawana dożylnie lub doustnie w posta- sacyna, obniżają natomiast induktory enzymatyczne
ci roztworu sprawdziła się w leczeniu ciężkich napa- (np. barbiturany i karbamazepina, składniki dymu
dów astmy oraz stanu astmatycznego (zob. poniżej). tytoniowego). β2-sympatykomimetyki działają syner-
Teofilina o przedłużonym działaniu powinna być gistycznie.
przyjmowana doustnie, ponieważ w ten sposób moż-
na nie tylko wydłużyć czas działania, ale również Antagoniści receptora muskarynowego (parasym-
w znacznym stopniu uniknąć niepożądanych wzro- patykolityki). Antagoniści receptora muskarynowe-
stów stężenia we krwi. go mają działanie rozszerzające oskrzela. Pochodne
Dawki dobiera się indywidualnie. Średnia daw- trzeciorzędowe, podawane doustnie w formie resor-
ka podtrzymująca wynosi 200–800 mg. Szczególna bowalnej, powodują zagęszczenie śluzu i utrudnia-
ostrożność zalecana jest w przypadku chorych na pa- ją odkrztuszanie na skutek hamowania wydzielania
CH3 OH
N O NH
H3C C(CH3) 3
O CH3
O N
CH3
O
bambuterol
CH3 OH CH3 OH
N O NH N O NH
H3C C(CH3) 3 H3C C(CH3) 3
O CH3 O
O N OH O
H
O
hydroksybambuterol
OH
O NH
H C(CH3) 3
O
H
terbutalina
Ryc. B 5.3-10. Metabolizm bambuterolu.
MUTSCHLER-2009.indd 652 2010-01-07 22:14:04
Układ oddechowy 653
oskrzelowego oraz porażenia nabłonka rzęskowego. 5.3.2.2.3. Preparaty złożone
Znacznie lepsze są pochodne czwartorzędowe, które
wchodzą w skład aerozoli. W astmie oskrzelowej je- Krajowe i międzynarodowe sposoby leczenia astmy
dynym obecnie stosowanym antagonistą receptorów zalecają u pacjentów z przewlekłą astmą w stopniu
muskarynowych jest bromek ipratropium. Działanie 3 i 4 jednoczesne podawanie β2-sympatykomimety-
tego leku rozpoczyna się w ciągu 3–5 minut po inha- ków o przedłużonym działaniu i wziewnych gluko-
lacji i utrzymuje się przez 4–6 godzin. Powodowane kortykosteroidów (zob. tab. B 5.3-4). Skutkiem tego
przez niego rozszerzenie oskrzeli jest jednak nieco było wprowadzonie inhalatora zawierającego ksy-
słabsze niż w przypadku β2-sympatykomimetyków, nofonian salmeterolu i 17-propionian flutikazonu
dlatego jego rola w leczeniu astmy oskrzelowej lub fumaran formoterolu i budezonid (zob. poniżej).
jest bardzo niewielka. Takie połączenia bardziej spełniają oczekiwania
Dawka bromku ipratropium jednorazowo wynosi chorych. Jak dotąd nie opisano nowych działań nie-
250–500 μg. pożądanych. Ze względu na brak możliwości zmia-
Suchość wywołana zahamowaniem wydzielania ny proporcji poszczególnych składników preparaty
śliny jest rzadko występującym działaniem niepożą- zawierające ich połączenia nie nadają się w przy-
danym. W jaskrze i zaburzeniach w oddawaniu mo- padku chorych rozpoczynających leczenie, wyma-
czu lek jest przeciwwskazany. gających ustalenia pierwszych dawek oraz chorych
Bromek tiotropium, antagonista receptorów mu- z astmą niestabilną.
skarynowych, nie jest dopuszczony w leczeniu astmy Połączenie czwartorzędowego parasympatykoli-
oskrzelowej. Substancja ta stosowana jest w przewle- tyku z β2-sympatykomimetykiem umożliwia reduk-
kłej obturacyjnej chorobie płuc (POChP, zob. poni- cję dawki sympatykomimetyka, więc zastosowanie
żej). takiej kombinacji jest w niektórych przypadkach
Tabela B 5.3-4. Podział według stopnia ciężkości i schematu długoterminowego leczenia astmy oskrzelowej u dorosłych (zalece-
nia Niemieckiej Ligi ds. Zwalczania Chorób Dróg Oddechowych oraz Niemieckiego Towarzystwa Pneumologicznego i Medycyny
Oddechowej)
Układ oddechowy
Astma przerywana Astma stała
1 2 lekka 3 średnia 4 ciężka
symptomy FEV1/PEF symptomy FEV1/PEF symptomy FEV1/PEF symptomy FEV1/PEF
dzień: 1 × tydz. dzień: 1 × tydz. dzień: codziennie > 60% do dzień: stale
noc: 2 × mies. ≥ 80% noc: > 2 × mies. 80% noc: > 1 × mies. noc: często 60%
< 80%
Leczenie przewlekłe (controller) B5
brak wziewne glukokortykosteroidy: wziewny glukokortykosteroid: wziewny glukokortykosteroid:
małe dawki małe lub średnie dawki duże dawki
oraz oraz
(substancje alternatywne β2-sympatykomimetyk β2-sympatykomimetyk o długim
u dzieci: kromony o długim działaniu działaniu (LABA)
lub montelukast (LABA)
ponadto jedna
zamiast LABA, lub kilka
ew. dodatkowo: dodatkowych możliwości:
zwiększanie teofilina
dawki przy astmie alergicznej
montelukast omalizumab
teofilina doustny glukokortykosteroid
doustny β2-sympatyko- w jak najmniej
mimetyk skutecznych dawkach
Potrzeby leczenia (reliever)
wziewny szybko wziewny szybko wziewny szybko wziewny szybko
i krótko działający i krótko działający i krótko działający i krótko działający
β2-sympatykomimetyk β2-sympatykomimetyk β2-sympatykomimetyk β2-sympatykomimetyk
MUTSCHLER-2009.indd 653 2010-01-07 22:14:04
654 Układ oddechowy
korzystne. Brak jest natomiast jednoznacznych ocen Do optymalizacji leczenia farmakologicznego
połączenia β2-sympatykomimetyku z kwasem kro- dąży się poprzez łączenie dwóch lub trzech leków,
moglikanowym. a nie przez zwiększanie dawki jednego leku. Pogor-
szenia w przebiegu choroby wymagają zwiększenia
dawki (eskalacji) bądź podania kolejnej substancji
5.3.2.2.4. Schemat leczenia astmy czynnej. Jeżeli stan ogólny pacjenta jest stabilny
przez kilka miesięcy, można spróbować zmniejszyć
Celem leczenia astmy jest usunięcie jej objawów, dawkę leku (deeskalacja).
unikanie pogorszeń, prawidłowe funkcjonowanie Oprócz stosowania leków niezmiernie ważne
płuc, a w przypadku dzieci – ich normalny rozwój. jest również pouczanie pacjentów i wykonywanie
Przy astmie na podłożu alergicznym wskazane jest, przez nich samodzielnych pomiarów za pomocą
w miarę możliwości, unikanie kontaktu z alergenami miernika maksymalnego przepływu peak-flow.
(np. sierścią zwierząt, roztoczy, pyłków). Odczulanie
daje korzystne efekty tylko w przypadku niektórych
pyłków i jadu osy. O wyborze odpowiednich leków 5.3.2.2.5. Leczenie ciężkich napadów astmy
decyduje stopień zaawansowania astmy (zob. poni- i stanu astmatycznego
żej) i leczenie wstępne.
Schemat długotrwałego leczenia został przedsta- Przez pojęcie stanu astmatycznego rozumie się prze-
wiony w tab. B 5.3-4. Elementem wszystkich 4 stop- wlekły silny skurcz oskrzeli, który może się rozwijać
ni astmy oskrzelowej jest terapia według aktualnych stopniowo w ciągu wielu godzin albo bardzo gwał-
potrzeb, reliever, której zadaniem jest łagodzenie townie. W każdym przypadku jest to stan zagroże-
nagłych napadów duszności. Spośród leków najle- nia życia i wymaga oprócz zabiegów fizykalnych
piej sprawdzają się w takich sytuacjach β2-sympa- (np. mechanicznego odsysania wydzieliny) odpo-
tykomimetyki o szybkim działaniu i krótkim okresie wiedniego i intensywnego leczenia z podaniem:
połowiczego rozpadu (zob. tab. B 5.3-2), ponieważ
■ tlenu (2–4 l/min) przez sondę nosową (należy uwa-
nie gromadzą się w organizmie i tym samym pozwa-
żać na hiperkapnię),
lają uniknąć ogólnoustrojowych działań niepożąda-
nych. Reliever działają jednak objawowo i nie przy- ■ wziewnego β2-sympatykomimetyku o krótkim
noszą korzyści profilaktycznych, dlatego począwszy działaniu (np. salbutamolu),
od stopnia 2 astmy oskrzelowej jako leczenie dłu-
■ glukokortykosteroidu dożylnego (w zależności od
goterminowe, controller, wprowadza się dodatko-
potrzeb, np. 100–250 mg prednizolonu),
wo wziewne glukokortykosteroidy. Od stopnia 3
wziewny glukokortykosteroid łączony jest z β2-sym- ■ teofiliny (pacjentom, którzy wcześniej nie przyjmo-
patykomimetykiem o długim działaniu w celu unik- wali leku, do 5 mg/kg) w formie wlewki (w przy-
nięcia napadów astmy. W przypadku utrzymywania padku wcześniejszego leczenia teofiliną konieczne
się objawów zasadne staje się włączenie do leczenia jest wykonanie badania krwi).
montelukastu lub teofiliny. Warto jednak pamiętać,
że ze względu na ograniczony zakres działania zna- Pacjentom niespokojnym lub ze stanami lękowymi
czenie teofiliny w leczeniu długotrwałym w ostatnich można podać środki uspokajające lub przeciwlękowe
latach się zmniejszyło. W ciężkiej przewlekłej astmie ze względu na ryzyko depresji ośrodka oddechowe-
w stopniu 4 muszą być zastosowane także doustne go, ale tylko po wykluczeniu zwiększonego stężenia
glukokortykosteroidy. ditlenku węgla we krwi (hiperkapni).
MUTSCHLER-2009.indd 654 2010-01-07 22:14:04
Układ oddechowy 655
5.4. Przewlekła obturacyjna choroba płuc (POChP,
przewlekłe obturacyjne zapalenie oskrzeli)
5.4.1. Patofizjologia przewlekłej strzenie oskrzeli, nowotwór oskrzeli, gruźlica, astma
obturacyjnej choroby płuc oskrzelowa, niewydolność lewokomorowa).
Charakterystyczny dla POChP jest szybki spa-
dek parametru funkcji płuc. Wśród jego przyczyn
Objawami charakterystycznymi dla przewlekłego
na pierwszym miejscu wymienia się palenie papiero-
obturacyjnego zapalenia oskrzeli są wzmożona pro-
sów, znacząco przyspieszające i tak postępujący wraz
dukcja śluzu w drogach oddechowych oraz kaszel.
z wiekiem spadek funkcji płuc (zob. ryc. B 5. 4-1).
Według definicji Światowej Organizacji Zdrowia
Powtarzające się infekcje wirusowe mogą powodo-
(WHO) o przewlekłym zapaleniu oskrzeli można mó-
wać uszkodzenie pokrytego rzęskami nabłonka od-
wić wtedy, gdy u danego pacjenta przez przynajmniej
dechowego, a tym samym gromadzenie się śluzu.
trzy kolejne miesiące w ciągu dwóch lat utrzymywał
Na wzrost zachorowalności wpływa także stopień za-
się kaszel niespowodowany żadną wyraźną przyczy-
nieczyszczenia powietrza w miejscu zamieszkania.
ną. W dalszym przebiegu, kiedy dodatkowo następu-
Niektóre podstawowe patomechanizmy POChP
je zwężenie dróg oddechowych, przewlekłe zapalenie
nie są jeszcze wyjaśnione. Przypuszcza się, że czą-
oskrzeli przechodzi w przewlekłe obturacyjne zapa-
steczkowe mechanizmy reakcji zapalnej są inne niż
lenie oskrzeli, kwalifikowane następnie jako POChP
w przypadku astmy oskrzelowej. Stany zapalne
(chronic obstructive pulmonary disease). W przeci-
dróg oddechowych oraz miąższu płuc przenoszone
wieństwie do astmy oskrzelowej, przewlekła obtura-
są przede wszystkim przez granulocyty obojętno-
cja w POChP nigdy nie jest całkowicie odwracalna
chłonne, makrofagi i limfocyty T CD8+, które po-
(zob. por. ryc. B 5.2-1).
przez aktywację różnego rodzaje proteaz (np. elasta-
POChP jest najczęstszą przewlekłą chorobą płuc.
zy, metaloproteinazy macierzowe – MMP) prowadzą
Mężczyźni zapadają na nią trzykrotnie częściej niż
Układ oddechowy
do zwłóknienia dróg oddechowych, a w efekcie do
kobiety. W praktyce ogólnomedycznej stwierdzana
zniszczenia miąższu płuc. Rozwijająca się w następ-
jest u co szóstej dorosłej osoby niepalącej i prawie
stwie tego nadreaktywność drzewa oskrzelowego
u co drugiej palącej. Prawidłowe zdiagnozowanie
powoduje odruchowe uwalnianie acetylocholiny wa-
POChP wymaga wykluczenia wielu chorób sercowo-
runkującej degranulację komórek tucznych ściany
-płucnych, mogących dawać podobne objawy (np. roz-
oskrzeli i uwalnianie mediatorów stanu zapalnego.
Acetylocholina i mediatory wspólnie przyczyniają
się do skurczu oskrzeli, wzmożonego wydzielania
gęstego, ciągnącego się śluzu oraz rozwoju odmy.
B5
Czynniki wziewne zaburzają natomiast funkcję
4 rzęsek, co uniemożliwia transport śluzu i prowadzi
osoby niepalące do jego zalegania. Powstające w ten sposób warun-
3 ki sprzyjają rozwojowi wtórnych infekcji bakteryj-
osoby palące nych, wywoływanych głównie przez Gram-dodatnie
FEV1 (litry)
(30 papierosów
w ciągu dnia) szczepy Haemophilus influenze, Pneumococcus,
2 Staphylococcus. Mediatory uczulające i toksyny
osoby, które
przestały produkowane przez bakterie zaostrzają, w opisany
palić wyżej sposób, proces chorobowy. Ogniska zapalne
1
mogą być rozproszone w drzewie oskrzelowym lub
duszność przy normalnej aktywności obejmować krtań (tracheitis), duże oskrzela (bron-
0 chitis) bądź oskrzela obwodowe (bronchiolitis).
25 55 75
wiek (lata) Przyjmuje się również, że silny przewlekły kaszel
jest dodatkowym czynnikiem powodującym zmia-
Ryc. B 5.4-1. Spadek natężonej objętości wydechowej pierw- ny w strukturze chorobowo zmienionych oskrzeli
szosekundowej (FEV1) w zależności od wieku. Wykres uwzględ- i oskrzeli obwodowych. Hipoteza ta poparta jest ob-
nia osoby palące, niepalące i osoby, które przestały palić (we- serwacjami, z których wynika, że rozwój choroby ule-
dług Grimmingera, Suttorpa, Seegera). ga spowolnieniu po zahamowaniu odruchu kaszlu.
MUTSCHLER-2009.indd 655 2010-01-07 22:14:05
656 Układ oddechowy
5.4.2. Leczenie przewlekłej Według nowszych badań klinicznych w przypadku
obturacyjnej choroby płuc astmy oskrzelowej wziewni antagoniści receptorów
muskarynowych wykazują lepsze działanie rozkur-
(POChP) czające oskrzela niż krótko działające β2-sympaty-
komimetyki. Pogląd ten jest zgodny z główną rolą
Podobnie jak w przypadku astmy oskrzelowej, opra- acetylocholiny w patofizjologii POChP i doprowadził
cowano schemat stopniowego leczenia POChP w za- do opracowania związku o nazwie bromek tiotro-
leżności od nasilenia się choroby (zob. tab. B 5.4-1 pium, który powinien być podawany wziewnie tylko
i B 5.4-2). Nadrzędnym zaleceniem jest odstawienie raz dziennie. Bromek tiotropium działa selektywnie
palenia oraz unikanie drażniącego wpływu czynni- na receptory płucne M3, czyli dysocjuje z nich o wie-
ków pochodzących z otoczenia. Leczenie objawowe le wolniej niż z innych receptorów M, dzięki czemu
POChP (zob. tab. B 5.4-2) przypomina, poza kilkoma możliwe jest jego długotrwałe rozkurczające działa-
różnicami, leczenie astmy oskrzelowej. nie na oskrzela.
Tabela B 5.4-1. Podział stopniowego nasilenia POChP. Stopień nasilenia POChP oceniany jest w zależności od natężonej objęto-
ści wydechowej pierwszosekundowej (FEV1), stosunku FEV1/VC (VC – pojemność życiowa) oraz objawów (według Niemieckiej Ligi
ds. Zwalczania Chorób Dróg Oddechowych)
Stopień nasilenia Kryteria oceny
0 (grupa ryzyka) spirometria w granicach normy
przewlekłe objawy (kaszel, odkrztuszanie plwociny)
I (lekkie) FEV1 > 80%, VC < 70%
z/bez objawów (kaszel, odkrztuszanie plwociny)
II (umiarkowane) 30% < FEV1 < 80%, FEV1 / VC < 70%
z/bez objawów (kaszel, odkrztuszanie plwociny)
III (ciężkie) FEV1 < 30%, FEV1 / VC < 70%
FEV1 < 50% oraz niewydolność oddechowa lub oznaki prawokomorowej niewydolności serca
Tabela B 5.4-2. Schemat stopniowego długoterminowego leczenia POChP. Sposób leczenia uzależniony jest od stopnia nasilenia
choroby (według Niemieckiej Ligi ds. Zwalczania Chorób Dróg Oddechowych)
Stopień nasilenia Leczenie farmakologiczne Leczenie niefarmakologiczne
0 brak leczenia unikanie czynników zagrożenia (kuracja antynikotynowa)
I w razie potrzeby β2-sympatykomimetyki szczepienia ochronne
i/lub środki antycholinergiczne
przy braku poprawy β2-sympatykomimetyki dodatkowa rehabilitacja:
i/lub środki antycholinergiczne ćwiczenia fizyczne, psychoterapia,
dodatkowo teofilina odpowiednia dieta, szkolenie pacjenta
II jw.; ponadto leczenie wziewnymi
glukokortykosteroidami dłużej niż 3 miesiące,
dalsze zalecenia adekwatnie
do uzyskanego efektu leczenia
III dodatkowo sprawdzanie, czy jest wskazanie inne możliwości: oddychanie w domu,
do długoterminowej terapii za pomocą O2 chirurgia klatki piersiowej, transplantacja płuca
MUTSCHLER-2009.indd 656 2010-01-07 22:14:05
Układ oddechowy 657
Zalecana dawka dzienna bromku tiotropium wy- Słuszność wprowadzenia wziewnych glukokor-
nosi 18 μg. Nie jest on prawie w ogóle metabolizo- tykosteroidów od II stopnia POChP budzi kontro-
wany. Po podaniu wziewnym jest w 14% wydalany wersje. Według nowszych badań tylko u 10–15%
z moczem. pacjentów z POChP wyraźna poprawa funkcji płuc
Najczęstszym działaniem niepożądanym (> 10%) jest efektem leczenia glukokortykosteroidami. Zgod-
jest suchość w ustach. Inne przypominające atropinowe nie z tym wziewne glukokortykosteroidy powinny
działania niepożądane występują znacznie rzadziej. być podawane indywidualnie, wyłącznie w przy-
padku wyraźnie efektywnego leczenia. Ze względu
Leki antycholinergiczne mogą być zarówno poda- na częste u pacjentów z POChP infekcje zaleca się
wane samodzielnie, jak i łączone z β2-sympatykomi- również szczepienia przeciw grypie i pneumokokom,
metykami o przedłużonym działaniu, co w pewnym a w przypadku każdej rozwijającej się bakteryjnej su-
stopniu wzmacnia wywoływany przez nie efekt roz- perinfekcji – antybiotykoterapię. Przy dużym zafleg-
kurczowy. mieniu wskazane są leki wykrztuśne.
5.5. Leczenie alergicznego nieżytu nosa
Alergiczny nieżyt nosa, podobnie jak astma alergicz- geny, na które pacjent narażony jest przez cały rok.
na, należy do grupy chorób atopowych, a jej współ- Ponieważ w przebiegu przewlekłego alergicznego
czynnik chorobowości wynosi 10–20%. W zasadzie nieżytu nosa mogą wystąpić powikłania i trudno od-
jest ona IgE-zależną reakcją zapalną błony śluzowej wracalne skutki (zmiany w obrębie błony śluzowej
nosa, którą poprzedza faza uczulenia na określony nosa, przewlekłe zapalenie zatok, zapalenie ucha
alergen. Alergiczny nieżyt nosa może być warunko- środkowego, zaburzenia węchu), schorzenie to po-
wany genetycznie lub przez czynniki zewnętrzne. winno być leczone bardzo konsekwentnie. Pacjen-
Jego mechanizmy patologiczne przypominają me- tom podaje się przede wszystkim leki przeciwhista-
chanizmy alergicznej astmy oskrzelowej. minowe H1, kwas kromoglikanowy oraz glukokor-
Układ oddechowy
Sezonowy alergiczny nieżyt nosa powodowany tykosteroidy w postaci aerozoli donosowych (zob.
jest przez pyłki, a nieżyt całoroczny – przez aler- tab. B 5.5-1).
Tabela B 5.5-1. Leczenie alergicznego nieżytu nosa
Sezonowy
Dolegliwości Lekkie Umiarkowane
lekkie głównie objawy głównie objawy
B5
Objawy: lub przemijające nosowe oczne
Leczenie: doraźnie doustne leki przeciw- glukokortykosteroidy glukokortykosteroidy donosowe oraz
histaminowe donosowe DNCG* miejscowo do oczu
lub (od początku okresu pylenia) lub
miejscowe leki przeciwhistaminowe oraz codziennie doustne leki
lub miejscowe leki przeciwhistaminowe przeciwhistaminowe
DNCG* do nosa lub
i/lub oczu DNCG*
Przewlekły
Dorośli Dzieci
stały kontakt z alergenem: objawy przemijające: DNCG* donosowo
glukokortykosteroidy donosowe doustne leki przeciwhistaminowe, codziennie doustne
doraźnie leki obkurczające leki przeciwhistaminowe
naczynia
przy braku skuteczności lub stałym kontakcie z alergenem:
glukokortykosteroidy donosowe
* DCNG – kromoglikan disodowy
MUTSCHLER-2009.indd 657 2010-01-07 22:14:05
658 Układ oddechowy
5.6. Leki przeciwkaszlowe
Leki przeciwkaszlowe omówiono już w rozdz. B 1.5.4.
5.7. Leki o działaniu wykrztuśnym
Substancje ułatwiające lub przyspieszające usuwanie nego śluzu powoduje obniżenie jego lepkości. Po-
wydzieliny śluzowej z oskrzeli i tchawicy określane są przez stymulację tworzenia dużej ilości lizosomów
jako leki o działaniu wykrztuśnym (zob. tab. B 5.7-1). i aktywację enzymów hydrolitycznych bromheksyna
W grupie tej wyróżniono leki: powoduje rozkład kwaśnych mukopolisacharydów
śluzu oskrzelowego.
■ sekretolityczne,
■ mukolityczne, Ambroksol jest biologicznie aktywnym głównym
metabolitem bromheksyny. Dodatkowym efektem
■ stymulujące transport śluzu.
działania ambroksolu jest zmniejszenie napięcia
powierzchniowego poprzez stymulację tworzenia
Ponieważ wyraźne odgraniczenie od siebie poszcze-
aktywnego powierzchniowo surfaktantu (zob. poni-
gólnych grup i uniknięcie płynnego przechodzenia
żej), co uniemożliwia przyleganie śluzu do nabłonka
jednej w drugą nie jest możliwe, podział ten nie bę-
oskrzelowego.
dzie przytaczany w dalszej części podręcznika.
Po podaniu doustnym ambroksol jest wprawdzie
Stosowanie leków wykrztuśnych w znacznej mie-
całkowicie resorbowany, ale około 30% substancji
rze oparte jest na doświadczeniu. Mimo niezmiernie
czynnej ulega zmetabolizowaniu jeszcze przed osiąg-
częstego uwzględniania w procesie leczenia podważa
nięciem krążenia dużego. Łącznie 90% substancji
się wartość terapeutyczną wielu z tych substancji.
pojawia się w moczu w formie metabolitów (kwas
W trakcie leczenia ważne jest dostarczanie do or-
dibromoantranilowy, glukuronidy). Okres półtrwania
ganizmu odpowiedniej ilości płynów.
w surowicy wynosi około 10 godzin.
Rolę substancji wykrztuśnych pełnią następujące
Działania niepożądane w formie dolegliwości żo-
substancje, mieszaniny lub grupy substancji:
łądkowo-jelitowych, objawów alergicznych na skórze
■ leki zawierające saponiny oraz środki wymiotne, i błonach śluzowych, duszności i gorączki z dreszcza-
mi występują stosunkowo rzadko.
■ olejki eteryczne,
■ bromheksyna i jej metabolit ambroksol, Acetylocysteina, ewentualnie jej aktywny metabolit
cysteina, obniża lepkość śluzu oskrzelowego poprzez
■ acetylocysteina,
rozbicie mostków disiarczkowych w części białkowej
■ karbocysteina. cząsteczki śluzu.
Na skutek nasilonego efektu pierwszego przejścia
Preparaty saponinowe (np. Primulae radix, Polyga- tylko 10% niezmienionego leku dociera do krwiobie-
lae radix) oraz wymiotne (np. Ipecacuanhae radix, gu. Oprócz cysteiny, w wątrobie powstają dicetylocy-
emetyna) działają prawdopodobnie wyłącznie na dro- steina i cystyna, a także różnorodne disiarczki. Okres
dze odruchowej przez stymulację aferentnych włó- półtrwania w surowicy wynosi dla cysteiny około l
kien przywspółczulnych. Olejki eteryczne (np. olejek godziny.
anyżkowy, eukaliptusowy, mentolowy, tymiankowy, W czasie przyjmowaniu leku rzadko obserwowa-
terpentynowy) bezpośrednio stymulują wydzielanie no działania niepożądane w postaci zaburzeń żo-
śluzu. łądkowo-jelitowych, bólów głowy, szumu w uszach
Stosowanie olejków eterycznych w przypadku oraz reakcji alergicznych. Podawanie z acetylocyste-
niemowląt i dzieci budzi zastrzeżenia ze względu iną doustnych leków przeciwbakteryjnych (penicy-
na ich niepewne działanie i ryzyko skurczu krtani lub liny, tetracykliny, cefalosporyny, aminoglikozydów)
pobudzenia ośrodkowego układu nerwowego. wymaga zachowania przynajmniej dwugodzinnego
odstępu między jednym a drugim środkiem, ponie-
Bromheksyna stymuluje komórki gruczołowe waż acetylocysteina może spowodować ich inakty-
do wytwarzania śluzu. Wzrost objętości produkowa- wację.
MUTSCHLER-2009.indd 658 2010-01-07 22:14:06
Układ oddechowy 659
Tabela B 5.7-1. Leki wykrztuśne
Wzór strukturalny Nazwa Preparaty handlowe Okres Pojedyncza
międzynarodowa półtrwania dawka
(godz.) (mg)
Br bromheksyna Bisolvon, 16 8
Bromhexin Krewel
CH3 Meuselbach,
N Bromhexin-
Br -ratiopharm
NH2
Br ambroksol AmbroHEXAL, 10 30
Ambroxol AL,
H Ambroxol-ratiopharm,
N Mucosolvan
Br
NH2
OH
O acetylocysteina ACC, 1 200
Acemuc,
HN CH3 Fluimicil,
HS NAC-ratiopharm
COOH
NH2 karbocysteina Transbronchin 1-3 750
HOOC S
COOH
Układ oddechowy
Acetylocysteina stosowana jest nie tylko jako lek sływanie, można osiągnąć poprzez stymulację czyn-
wykrztuśny, zalecana jest również w leczeniu zatruć ności rzęsek. Taki efekt może przynieść podanie
paracetamolem. β2-sympatykomimetyków, których korzystne działa-
nie w obturacyjnych chorobach płuc polega na znie-
Karbocysteina, która w przeciwieństwie do acety- sieniu skurczu oskrzeli oraz w pewnym stopniu rów-
locysteiny nie posiada reaktywnych grup tiolowych, nież na pobudzeniu ruchliwości rzęsek. Funkcję
nie może reagować bezpośrednio z cząsteczkami ślu- wspomagającą transport wydzieliny pełnią oprócz
zu. Przyjmuje się, że substancja ta stymuluje wytwa- β2-sympatykomimetyków także wymienione wcześ-
rzanie śluzu o niskiej lepkości, hamując jednocześnie niej olejki eteryczne, wykazujące działanie antysep-
B5
syntezę wysokiej lepkości. Efektem takich reakcji tyczne i słabe spazmolityczne. Kliniczne znaczenie
jest zmniejszenie się ilości wytwarzanego śluzu. ich właściwości jest jednak wątpliwe.
Działanie sekretomotoryczne, czyli wzmożony
ruch wydzieliny oskrzelowej i skuteczniejsze odka-
MUTSCHLER-2009.indd 659 2010-01-07 22:14:06
660 Układ oddechowy
5.8. Surfaktant
Napięcie powierzchniowe pęcherzyków płucnych Podanie leku w dawkach 50–100 mg fosfolipidu
jest znacznie mniejsze niż teoretycznie oczekiwane na kg masy ciała powinno nastąpić na drodze do-
w przypadku zawierającej wodę warstwy granicznej. tchawiczego wkroplenia bezpośrednio po urodzeniu.
Płynna warstwa pokrywająca powierzchnię pęche- W razie konieczności surfaktant można zaaplikować
rzyków musi więc zawierać substancje obniżające ponownie.
napięcie powierzchniowe. Substancje te określa się Działania niepożądane mogą wystąpić w postaci
mianem surfaktantu. przejściowej niedrożności dróg oddechowych i krwa-
Surfaktant składa się w 90% z mieszaniny fosfoli- wienia płucnego. Dzieci leczone surfaktantem sto-
pidów (głównie poddanej estryfikowanej fosfatydylo- sunkowo częściej niż nieleczone zapadają na infekcje
choliny oraz fosfatydyloglicerolu) oraz z obojętnych oraz posocznicę.
lipidów i białek. Przeciwwskazania nie są znane.
Rolą surfaktantu jest ochrona pęcherzyków płuc-
nych przed zapadaniem się w końcowej fazie wyde-
chu, umożliwia on także równomierną wentylację,
ułatwia wymianę tlenu i ditlenku węgla oraz popra-
wia transport śluzu.
(CH2) 14CH3
Niedobór surfaktantu jest przyczyną zespołu nie- O
O
wydolności oddechowej u wcześniaków, który cechu- O
H3C(CH2) 14 O H
je się ciężkimi poporodowymi zaburzeniami oddechu
O
i jest przyczyną wysokiej śmiertelności. Szczególnie H3C –
+ O P O
zagrożone są dzieci o masie urodzeniowej mniejszej N
H3C O
niż 1500 g (poród przed 30 tygodniem ciąży). CH3
Znaczne zmniejszenie liczby zgonów noworod-
ków, skrócenie czasu stosowania respiratora oraz dipalmitoilofosfatydylocholina
zmniejszenie częstości dysplazji oskrzelowo-płuc-
nych występujących po dłuższym stosowaniu odde-
chu wspomaganego można osiągnąć dzięki dotcha-
wiczemu wkraplaniu surfaktantu bydlęcego.
5.9. Leczenie infekcji wirusem oddechowym
Wirus oddechowy (respiratory syncytial virus miesięcy były leczone z powodu dysplazji oskrze-
– RSV) jest najczęstszą przyczyną infekcji dróg lowo-płucnej, mogą otrzymać zapobiegawczo pali-
oddechowych u noworodków, szczególnie wcześ- wizumab. Paliwizumab jest humanizowanym prze-
niaków, oraz małych dzieci. Pomiędzy wrześniem ciwciałem monoklonalnym IgG1K, wiążącym się
i kwietniem (sezon RSV) w Europie ponad połowa z epitopem A wirusa RSV.
wszystkich hospitalizacji dzieci poniżej 2 roku życia Paliwizumab aplikuje się raz w miesiącu domięś-
spowodowana jest schorzeniami dróg oddechowych niowo w dawce 15 mg/kg masy ciała.
wywołanych RSV. Noworodki urodzone w 35 lub Działanie niepożądane leku objawia się gorączką,
wcześniejszym tygodniu ciąży, które na początku odczynem w miejscu wstrzyknięcia, nerwowością,
sezonu RSV mają mniej niż 6 miesięcy, oraz dzie- zakażeniem górnych dróg oddechowych oraz kasz-
ci poniżej 2 roku życia, które w ciągu ostatnich 6 lem.
MUTSCHLER-2009.indd 660 2010-01-07 22:14:06
Układ oddechowy 661
5.10. Leczenie mukowiscydozy
Mukowiscydoza (cystic fibrosis) jest najczęstszą zę istnieje ryzyko niedożywienia. Dieta pacjentów
dziedziczną autosomalno-recesywną chorobą, która z tą chorobą musi więc być bogata w tłuszcze, należy
prowadzi do śmierci. Polega na defekcie białka okre- jednak przestrzegać odpowiedniej substytucji enzy-
ślanego jako CFTR (cystic fibrosis transmembrane mów trzustkowych oraz witamin rozpuszczalnych
conductance regulator), które w błonie podstawnej w tłuszczach (A, D, E, K).
komórek nabłonka pełni funkcję kanału jonów Cl–.
Skutki defektu CFTR widoczne są przede wszystkim Leki znoszące skurcz oskrzeli. Rozszerzenie drze-
w narządach z gruczołami egzokrynnymi, szczegól- wa oskrzelowego i związana z tym poprawa klirensu
nie w płucach i trzustce. rzęskowego uzyskiwane są poprzez podanie β2-sym-
Ze względu na zmniejszone wydzielanie jonów patykomimetyków oraz leków antycholinergicznych.
Cl– i związaną z tym wzmożoną resorpcję zwrotną Rzeczywiste skutki takiej terapii wywołują jednak
jonów Na+ ślina staje się mniej uwodniona i zwięk- duże kontrowersje. Teofilina stosowana jest stosun-
sza się jej lepkość. Zawiera ona ponadto więcej DNA kowo rzadko.
i aktyny F z autolizowanych granulocytów obojęt-
nochłonnych, co również przyczynia się do wzrostu Sekretolityki. Podwyższona lepkość wydzieliny
jej lepkości. Ponieważ śluz o dużej lepkości nie może oskrzelowej u chorych na mukowiscydozę jest wska-
być wydalony i przez to zmniejsza klirens rzęskowy, zaniem do podania leków o działaniu wykrztuśnym.
dochodzi do wytworzenia się mechanizmu tzw. błęd- Skuteczność acetylocysteiny i innych leków powodu-
nego koła: zastój wydzieliny, zatkanie dróg oddecho- jących rozrzedzenie śluzu jest podważana. Z powodu
wych, zakażenie ze stanem zapalnym, zastój wydzie- dużej zawartości DNA w wydzielinie oskrzelowej
liny. Szczególne znaczenie ma również niewydolność można podjąć próbę podania rekombinowanej ludz-
egzokrynna trzustki, wynikająca z zaburzeń funkcji kiej dezoksyrybonukleinazy (rhDN-azy, dornazy; co-
wydzielniczych tego gruczołu i prowadząca do zabu- dziennie 2500 jednostek u chorych powyżej 5 roku
rzeń odżywiania. życia). Efektami niepożądanymi takiego leczenia
Układ oddechowy
Obecnie około 40% pacjentów z mukowiscydo- są: zapalenie gardła, zapalenie krtani, chrypka, bóle
zą stanowią dorośli. Dzieci rodzące się z tą chorobą w klatce piersiowej, wysypki skórne, wytwarzanie
mają w dzisiejszych czasach zdecydowanie większą przeciwciał IgG lub IgM.
szansę dożycia wieku dorosłego niż jeszcze przed 10
laty. Przyczynowe leczenie mukowiscydozy jest jed- Leki przeciwbakteryjne. Zmniejszenie klirensu rzęs-
nak w dalszym ciągu niemożliwe. Obecnie dostępne kowego sprzyja rozwojowi drobnoustrojów w drze-
metody leczenia obejmują: wie oskrzelowym. Często stwierdzanymi bakteriami
są gronkowiec złocisty, Pseudomonas aeruginosa,
■ zabiegi fizjoterapeutyczne,
Haemophilus influenzae i in. Leczenie przeciwbak-
B5
■ optymalizację odżywiania, teryjne, które zawsze powinno następować dopiero
po zbadaniu plwociny i wykonaniu antybiogramu,
■ podawanie leków rozkurczających oskrzela, leków
jest trudne. Rozwój oporności bakterii może bowiem
sekretolitycznych i leków przeciwbakteryjnych,
mieć niekiedy tragiczne skutki. Dlatego najczęściej
■ regulację przezmembranowego transportu jonów stosuje się kombinacje różnych leków antybakteryj-
Cl– i Na+, nych.
■ zwalczanie miejscowych stanów zapalnych po-
Regulacja transportu jonów Cl– i Na+. Zmniejsze-
przez podawanie miejscowych środków przeciw-
nie lepkości wydzieliny oskrzelowej można pró-
zapalnych.
bować osiągnąć za pomocą amiloridu. Na skutek
zablokowania kanałów potasowych w wydzielinie
Fizjoterapia. Zabiegi fizykoterapeutyczne i rehabilita-
oskrzelowej pozostaje bowiem wówczas więcej
cja mają wspomóc odkrztuszanie wydzieliny oskrze-
wody. Słuszność podania w takiej sytuacji trifo-
lowej, odciążyć mięśnie oddechowe oraz utrzymać
sforanu urydyny (UTP), który miałby aktywować
ruchomość klatki piersiowej.
specjalne kanały Cl–, nie została jak dotąd potwier-
dzona.
Odżywianie. Na skutek niewydolności egzokryn-
nej trzustki u większości chorych na mukowiscydo-
MUTSCHLER-2009.indd 661 2010-01-07 22:14:07
MUTSCHLER-2009.indd 662 2010-01-07 22:14:07
Układ trawienny 663
6. Układ trawienny
6.1. Podstawy anatomiczne i fizjologiczne
Rolą układu trawiennego jest wchłanianie substan- gruczołach: śliniance przyusznej (glandula paro-
cji odżywczych, witamin, soli mineralnych i wody tis), śliniance podżuchwowej (glandula subman-
oraz wydalanie części produktów przemiany mate- dibularis) oraz śliniance podjęzykowej (glandula
rii. Do postaci wchłanianej, w której pokarm może sublingualis), z których poprzez przewody wypro-
być resorbowany przez organizm, jest doprowadzany wadzające uchodzi do jamy ustnej. W ciągu doby
na drodze trawienia, tj. na skutek enzymatycznego produkowane jest ok. 1,5 l śliny, której skład zależy
rozpadu węglowodanów, białek i tłuszczów. od właściwości spożytego pokarmu (wydzielanie
Na ryc. B 6.1-1 przedstawiono narządy układu tra- płynnej, spłukującej śliny po spożyciu pokarmów
wiennego. suchych, wydzielanie lepkiej śliny trawiennej po po-
karmach płynnych). W trakcie aktu połykania, który
rozpoczyna się dowolnie, a kontynuowany jest od-
6.1.1. Jama ustna i gardło ruchowo, wymieszany ze śliną pokarm przechodzi
przez gardło (pharynx) do przełyku (oesophagus).
W jamie ustnej, stanowiącej początkowy odcinek Ponieważ w gardle krzyżują się drogi oddechowe
układu trawiennego, pokarm (stały) ulega rozdrob- i pokarmowe, w celu uniknięcia przedostania się
nieniu, wymieszaniu ze śliną i uformowaniu w kęsy. pokarmu do leżącej przed przełykiem tchawicy przy
Ślina wytwarzana jest w trzech dużych, parzystych połykaniu zamyka się wejście do krtani.
Układ trawienny
gardło
przełyk B6
wpust
dwunastnica odźwiernik
jelito czcze
okrężnica poprzeczna
okrężnica wstępująca
jelito zstępujące
jelito kręte
jelito ślepe okrężnica esowata
wyrostek robaczkowy
odbyt
Ryc. B 6.1-1. Schemat układu trawiennego (według Benninghoffa-Groerttlera).
MUTSCHLER-2009.indd 663 2010-01-07 22:14:07
664 Układ trawienny
6.1.2. Przełyk sów), której główne działanie polega na zwiększaniu
wydzielania HCl przez komórki okładzinowe (zob.
poniżej).
Przełyk jest rurą o długości 22–25 cm, leżącą między
tchawicą i kręgosłupem, której ściana zbudowana
jest, patrząc od góry, w jednej trzeciej części wysoko-
ści z mięśni poprzecznie prążkowanych, a w dwóch 6.1.3.2. Motoryka i opróżnianie żołądka
trzecich z mięśni gładkich. Rolą tego narządu jest wy-
łącznie transport miazgi pokarmowej. Pusty żołądek wygląda jak spłaszczona rura mięś-
W górnym i dolnym odcinku przełyku znajdują się niowa, której ściany wewnętrzne silnie przylegają
struktury zamykające jego światło, określane jako gór- do siebie. Przyjęcie pokarmu – warunkowane prze-
ny i dolny zwieracz przełyku. Podczas aktu połykania de wszystkim przez przekaźniki NANC (nieadrener-
górny zwieracz otwiera się, umożliwiając przesunięcie giczne i niecholinergiczne przekaźniki autonomicz-
kęsa do przełyku. Przesuwanie pokarmu w kierunku nego układu nerwowego) – prowadzi do rozluźnienia
żołądka następuje wskutek skurczów mięśni okrężnych mięśniówki gładkiej, tj. rozciągnięcia ściany żołądka
(perystaltyki). W momencie dotarcia fali perystaltycz- bez zwiększenia ciśnienia wewnątrzżołądkowego.
nej do dolnego odcinka przełyku następuje otwarcie Wymieszanie i dalsze rozdrobnienie spożytego po-
zwieracza dolnego i przesunięcie pokarmu do żołąd- karmu, który jednocześnie przekształca się w miazgę
ka. Skoordynowany proces przesuwania pokarmu pokarmową (chymus), następuje przy zamkniętym
jest kontrolowany przez ośrodkowy układ nerwowy. odźwierniku na skutek silnych skurczów mięśniówki
Bodźce nerwowe kierujące tą czynnością przebiega- żołądka.
ją za pośrednictwem nerwu błędnego do mięśniówki W momencie opróżniania żołądka następuje krótko-
przełyku. trwałe otwarcie odźwiernika i jednoczesne, spowodo-
wane ruchami perystaltycznymi części odźwierniko-
wej, przesunięcie miazgi pokarmowej do dwunastnicy.
Proces ten regulowany jest neuronalnie i hormonalnie.
6.1.3. Żołądek Impulsy przewodzone przez parasympatyczny nerw
błędny pobudzają motorykę. Również odruchowe
6.1.3.1. Budowa żołądka opróżnianie żołądka warunkowane jest impulsami do-
prowadzanymi przez ten nerw. W przypadku zbyt du-
W żołądku człowieka rozróżnia się wpust (cardia), żego przepełnienia żołądka, dużej koncentracji tłusz-
dno (fundus), trzon żołądka (corpus ventriculi), część czów oraz reakcji kwasowej w początkowym odcinku
odźwiernikową (antrum) oraz odźwiernik (pylorus). dwunastnicy opróżnianie żołądka hamują hormony
Powierzchnia przednia i tylna żołądka są oddzielone żołądkowo-jelitowe, a szczególnie sekretyna i chole-
krzywizną małą i dużą. cystokinina, wytwarzane w błonie śluzowej jelita cien-
Błona śluzowa żołądka wyścielona jest jednowar- kiego i transportowane do żołądka wraz z krwią. Prze-
stwowym nabłonkiem walcowatym z dołkami żołąd- mieszczenie się treści żołądka do dalszych odcinków
kowymi (foveolae gastricae), do których uchodzą przewodu pokarmowego podlega więc hormonalnemu
swoiste gruczoły żołądkowe. Gruczoły znajdujące się procesowi sprzężenia zwrotnego. Nie są to jednak je-
w okolicy wpustu i odźwiernika wytwarzają, podob- dyne procesy hormonalne biorące udział w kierowa-
nie jak komórki nabłonka, wyłącznie śluz, natomiast niu motoryką żołądka. Wciąż jeszcze dyskutowany
gruczoły trzonu i dna – śluz, kwas solny i enzymy jest wpływ dopaminy i serotoniny.
proteolityczne. W związku z tym w błonie śluzowej
trzonu i dna znajdują się trzy rodzaje komórek:
■ oboczne wytwarzające śluz (komórki śluzowe uj- 6.1.3.3. Wydzielanie żołądkowe
ścia i szyjki),
Gruczoły żołądka wydzielają w ciągu doby 2–3 l soku
■ okładzinowe wydzielające kwas solny i czynnik
żołądkowego, który jest prawie izotonicznym z krwią
wewnątrzpochodny,
roztworem kwasu solnego o pH 0,8–1,5, zawierają-
■ główne uwalniające enzymy proteolityczne. cym enzymy trawienne, śluz i czynnik wewnątrzpo-
chodny niezbędny do resorpcji witaminy B12. Kwas
W komórkach G błony śluzowej części odźwierni- solny powoduje denaturację białek pokarmowych,
kowej żołądka odbywa się synteza gastryny (dużej, przez co ułatwia ich rozpad enzymatyczny oraz za-
małej i minigastryny o różnej zawartości aminokwa- pewnia wartości pH korzystne dla działania enzymów
MUTSCHLER-2009.indd 664 2010-01-07 22:14:07
Układ trawienny 665
żołądkowych, dzięki czemu nieczynny pepsynogen są za pomocą enzymu H+/K+-ATP-azy ponownie ak-
może ulec przekształceniu w różne pepsyny. Ponadto tywnie transportowane do komórki.
działa zabójczo na bakterie wprowadzane do żołądka Mediatorami powodującymi wydzielanie HCl są:
z pokarmem.
■ acetylocholina,
Wydzielanie kwasu solnego. Kwas solny wytwa- ■ histamina,
rzają w żołądku komórki okładzinowe, których kana-
■ gastryna.
liki wewnętrzne są połączone ze światłem gruczołu.
Istotnym etapem procesu wydzielniczego jest czynny
W żołądku wchodzą one w reakcje ze swoimi spe-
transport protonów przez błonę kanalika wydzielni-
cyficznymi receptorami z błony komórkowej: ace-
czego do światła kanalika (zob. ryc. B 6.1-2). Trans-
tylocholina z receptorem muskarynowym (receptor
port ten następuje w kierunku przeciwnym do dużego
M3), histamina z receptorem H2 i gastryna z recep-
gradientu stężeń jonów H+ (około 1 × 106, tj. pH = 6),
torami cholecystokininy typu 2 (zob. ryc. B 6.1-3).
ponieważ wartość pH w komórce wynosi 7,0–7,2, na-
Zachodząca przy tym silna interakcja między hista-
tomiast w kanaliku wydzielniczym ok. l. Energia nie-
miną i gastryną oraz nieco słabsza między histaminą
zbędna do czynnego transportu pochodzi z rozpadu
i acetylocholiną wynika ze stymulowania przez ace-
ATP. Enzymem katalizującym ten proces jest enzym
tylocholinę i gastrynę procesu uwalniania histaminy
ATP-aza protonowo-potasowa (H+/K+-ATP-aza),
oraz współdziałania między przekaźnikiem wtórnym
obecny w błonie mikrokosmków wydzielniczych.
cAMP i Ca2+ przy pobudzeniu pompy protonowej.
Enzym ten składa się z dwóch podjednostek (α i β)
Procesy te wyjaśniają, dlaczego blokada receptora
i jest aktywowany przez cAMP i jony Ca2+. Powodu-
H2 przez jego antagonistów zmniejsza nie tylko wy-
je wymianę protonów na jony potasowe w stosunku
dzielanie stymulowane przez histaminę, ale również
równoważnym. Protony pochodzą przede wszystkim
wpływ gastryny i acetylocholiny na wydzielanie
z reakcji dysocjacyjnych kwasu węglowego, prowa-
kwasu. Prostaglandyny E2, oddziałując na komórki
dzących do powstania równoważnych ilości HCO3–.
okładzinowe, również hamują wydzielanie kwasu.
Jon HCO3– przenika zgodnie z gradientem stężeń
do krwi, gdzie wymienia jon Cl–.
Śluz żołądkowy. Śluz żołądkowy produkowany
W żołądku oprócz protonów są uwalniane tak-
jest w komórkach powierzchniowych błony śluzowej
że jony chlorkowe i potasowe, przy czym jony K+
i w komórkach okładzinowych gruczołów. Jest lepki
i przyczepny dzięki zawartym w nim wysokocząstecz-
kowym glikoproteinom. Śluz pokrywający ściany
żołądka zawiera wodorowęglany i dzięki temu może
Układ trawienny
błona śluzowa światło chronić je przed samotrawieniem przez HCl i pepsynę
+ (bariera błony śluzowej). Zasadniczym czynnikiem
K+ K
ochronnym jest jednak brak jakichkolwiek uszkodzeń
we wszystkich komórkach powierzchniowych błony
K+
śluzowej, który wynikałby z dobrego ukrwienia błony
3 Na+ śluzowej lub szybkiej regeneracji powierzchniowych
ATP
2 K+
K+ ATP H+ kanaliki uszkodzeń. W mechanizmie ochronnym uczestniczy
CO2+H2O
CA
H + prostaglandyna E2. B6
CO2
HCO3- Cl- Wytwarzanie pepsyny. W komórkach głównych gru-
Cl- czołów żołądkowych wytwarzany jest pepsynogen,
stanowiący mieszaninę przynajmniej 7 proenzymów
/-Cl -
pH = 7,0 - 7,2 proteolitycznych. Po jego egzocytotycznym wydzie-
3
HCO
leniu ulega aktywacji do czynnej proteazy – pepsyny,
HCO3- komórki przez odszczepienie części inhibicyjnej. Reakcje te
okładzinowe są zapoczątkowywane przez kwas solny soku żołąd-
kowego i kontynuowane autokatalitycznie.
Ryc. B 6.1-2. Wydzielanie HCl przez komórki okładzinowe. Jony
H+ są wpompowywane do kanalików wewnątrzkomórkowych
w wyniku aktywności enzymu H+/K+-ATP-azy. Razem z proto- Regulacja wydzielania soku żołądkowego. Po-
nami przez specjalne kanaliki przechodzą do światła także jony dobnie jak motoryka i opróżnianie żołądka, również
Cl– i K+. CA – anhydrataza węglanowa. wydzielanie soku żołądkowego jest regulowane ner-
MUTSCHLER-2009.indd 665 2010-01-07 22:14:07
666 Układ trawienny
błona śluzowa światło
neurony aferentne
receptory bodźce
kwas arachidonowy (chemiczne,
mechaniczne)
COX-1 NLPZ
nerw
błędny
PGE2
neurony EP3
MZ/ histam
eferentne ECL ina Gi
Gs
H2
AC Cl-
Gq
cAMP kinazy
GZ
CCK2
gastryna proteinowe
ATP H+ HCI
krew K +
Gq Ca2+
M3
lina K+
acetylocho
pozazwojowe komórka
włókna okładzinowa
parasympatyczne
pompa
proteinowa
(H+/K+-ATP-azy)
Ryc. B 6. 1-3. Stymulacja komórki okładzinowej przez 3 typy receptorów (M3, CCK2 i H2) i hamowanie komórki okładzinowej przez
jeden typ receptorów (EP3). Pompa protonowa jest pobudzana przez Ca2+ i cAMP. Hamowanie syntezy cAMP i zmniejszenie koncen-
tracji jonów Ca2+ powoduje blokadę aktywności pompy protonowej. MZ – komórka tuczna, ECL – komórka enterochromafinopo-
dobna, GZ– komórka G, COX-1 – cyklooksygenaza-1, PGE2 – prostaglandyna E2, AC – cyklaza adenylanowa, EP3 – receptor prosta-
glandyny E3, H2 – receptor histaminowy typu 2, CCK2 – receptor cholecystokininy typu 2, NLPZ – niesteroidowe leki przeciwzapalne.
Neurony eferentne (kolor zielony), neurony aferentne (kolor czerwony).
wowo i hormonalnie. Wydzielanie soku żołądkowego ponadto histamina, uwalniana w wyniku pobudzenia
dzieli się na następujące po sobie fazy: komórek tucznych lub innych komórek ją wytwarza-
jących (np. komórek enterochromafinopodobnych –
■ głowową,
enterochromaffin-like cells, ECL). W sposób pośredni
■ gastryczną, gastryna zwiększa ponadto wytwarzanie i uwalnianie
histaminy.
■ jelitową (zob. ryc. B 6.1-4).
W fazie żołądkowej pokarm przesuwający się przez
Głowowa faza wydzielania jest regulowana nerwowo. żołądek wyzwala wydzielanie soku żołądkowego
Wrażenia zapachowe i smakowe wyzwalają dośrod- przez przesuwający się pokarm. Rozciąganie ścian
kowe impulsy nerwowe, które w ośrodkowym ukła- żołądka oraz bodźce chemiczne, np. produkty roz-
dzie nerwowym pobudzają włókna nerwu błędnego. padu białek, jony wapnia, kofeina lub alkohol, przy-
Pobudzenie nerwu błędnego powoduje uwolnienie czyniają się do powstania miejscowych odruchów
w ścianie żołądka acetylocholiny. Z jednej strony ace- cholinergicznych i uwolnienia gastryny. Obniżenie
tylocholina pobudza bezpośrednio komórki okładzi- wartości pH poniżej 3 hamuje uwalnianie gastryny.
nowe i główne (przez receptor M3), z drugiej strony
pobudzenie nerwu prowadzi do uwolnienia gastryny W fazie jelitowej następuje najpierw zwiększenie,
z komórek G błony śluzowej części odźwiernikowej a następnie zmniejszenie ilości wydzielanego soku
(antrum). Poprzez układ krwionośny gastryna docie- żołądkowego. W momencie znalezienia się pokarmu
ra do komórek okładzinowych i uruchamia wydziela- niekwaśnego w dwunastnicy następuje uwolnienie
nie HCl. W wydzielaniu kwasu solnego uczestniczy gastryny z komórek G tego narządu. Przesunięcie
MUTSCHLER-2009.indd 666 2010-01-07 22:14:08
Układ trawienny 667
stryny i pepsyny w żołądku, a także sekretyny w jeli-
cie cienkim. Spowalnia ponadto endokrynną i egzok-
obraz faza rynną funkcję trzustki (sekrecję insuliny i glukagonu
głowowa oraz wodorowęglanu i enzymów trawiennych) i obni-
ża – przy niezmienionym ogólnoustrojowym ciśnie-
niu krwi – ukrwienie okolicy unerwionej przez nerw
trzewny o 20–30%.
zapach Pobudzenie emocjonalne spowodowane stresem,
podenerwowaniem lub gniewem przyspiesza, a stra-
smak
pepsynogen chem lub smutkiem spowalnia wydzielanie soku żo-
łądkowego oraz motorykę żołądka.
nerw błędny
HCI
gastryna 6.1.4. Jelito cienkie
faza
żołądkowa Jelito cienkie, w którym wchłaniana jest większość
rozdrobnionych cząstek pokarmowych o niskim cię-
gastryna żarze molekularnym, dzieli się na trzy odcinki:
sekretyna ■ dwunastnicę (duodenum),
faza
jelitowa ■ jelito czcze (jejunum),
Ryc. B 6.1-4. Schemat przedstawiający fazy wydzielania soku ■ jelito kręte (ileum).
żołądkowego (według Thewsa, Mutschlera, Vaupela).
Dwunastnica ma około 25 cm długości. Kształtem
przypomina podkowę, obejmującą częścią wklęsłą
głowę trzustki. Do części wstępującej uchodzi prze-
wód trzustkowy wyprowadzający (ductus pancreati-
do dwunastnicy kwaśnej miazgi pokarmowej uwal- cus) i przewód żółciowy (ductus choledochus), łączą-
nia sekretynę, która zmniejsza wydzielanie kwasu ce się ze sobą na odcinku końcowym.
solnego, zwiększa natomiast uwalnianie pepsynoge- Dwunastnica łączy się z mającym około 1,2 m dłu-
nu. Hamowanie wydzielania soku żołądkowego od- gości jelitem czczym, a to przechodzi w jelito kręte
bywa się również pod wpływem cholecystokininy, mające około l,8 m długości.
Układ trawienny
szczególnie wtedy, gdy do górnej części dwunastni- Szczególną cechą jelita cienkiego jest znaczne
cy przedostanie się miazga pokarmowa zawierająca zwiększenie powierzchni chłonnej na skutek obec-
tłuszcze. ności fałdów błony śluzowej, kosmków i mikro-
kosmków. Fałdy są najgęstsze i najdłuższe (do 8 mm)
Związki te rozprowadzane są w organizmie wraz z krwią, w dwunastnicy i jelicie czczym, a powstają na skutek
dlatego zaliczane są do hormonów. Ponieważ częściowo od- uwypuklenia błony podstawowej. Na nich tworzą się
działują również na sąsiednie komórki bądź służą do prze-
kazywania sygnałów między komórkami, nie jest możliwe palcowate kosmki o wysokości l mm. Ich nabłonek B6
wyraźne oddzielenie hormonów tkankowych od neuroprze- zbudowany jest z tzw. enterocytów, posiadających
kaźników. mikrokosmki, ściśle do siebie przylegające wyrostki
protoplazmatycznne, skierowane do światła do jelita.
Oprócz omówionych wyżej związków istnieją inne W ten sposób powierzchnia chłonna jelita jest po-
hormony żołądkowo-jelitowe uczestniczące w regu- większona 600-krotne, co w jelicie cienkim tworzy
lacji czynności wydzielniczej i motorycznej. w sumie powierzchnię 200 m2.
Enteroglukagon oraz polipeptyd insulinotropo- Oprócz błony śluzowej ścianę jelita cienkiego bu-
wy zależny od glukozy (GIP) hamują wydzielanie duje również warstwa mięśni okrężnych i podłużnych
HCl w żołądku, a wspomagają uwalnianie insuliny oraz błona surowicza tworzona przez brzuszną war-
z trzustki. stwę otrzewnej. W ścianie jelita cienkiego znajdują
Somatostatyna, powstająca nie tylko w podwzgó- się wegetatywne sploty nerwowe, tj. zwój podślu-
rzu, ale również w wielu innych organach, np. w ko- zówkowy (plexus submucosus), unerwiający błonę
mórkach D błony śluzowej żołądka i jelita cienkiego śluzową, oraz zwój krezkowy, unerwiający mięś-
oraz trzustce, hamuje wydzielanie kwasu solnego, ga- niówkę (plexus myentericus). Błona mięśniowa jelita
MUTSCHLER-2009.indd 667 2010-01-07 22:14:08
668 Układ trawienny
cienkiego wykonuje dwa rodzaje skurczów: odcinko- Określenie jelito ślepe wynika z tępego zakończe-
we oraz robaczkowe (perystaltyczne). nia tego odcinka jelita. W jego górnym zakończeniu
Dla motoryki jelita cienkiego charakterystyczne uchodzi bocznie jelito kręte. Przez znajdującą się
są ruchy skurczowe, które umożliwiają dokładne wy- tu zastawkę krętniczo-kątniczą (valva ileocaecalis)
mieszanie masy pokarmowej z sokiem trzustkowym, treść pokarmowa stopniowo przesuwana jest do jelita
żółcią oraz wydzieliną gruczołów jelitowych, a także grubego.
ruchy robaczkowe, które przesuwają treść pokarmo- Wyrostek robaczkowy jelita ślepego (appendix
wą do dalszych odcinków przewodu pokarmowego. vermiformis) odchodzi od jelita ślepego. Jego dłu-
Ruchy te są wywoływane rozciąganiem się ściany gość wynosi 2–20 cm, przekrój 0,5–1 cm.
jelita i podlegają kontroli zwoju krezkowego. Ruchy Okrężnica (colon) łączy się z jelitem ślepym i dzieli
kosmków są natomiast stymulowane przez bodźce się na okrężnicę wstępującą, poprzeczną, zstępującą
ze zwoju podśluzówkowego, wzmacnia je natomiast i esowatą (colon ascendens, transversum, descendes,
działanie peptydu powstającego w wyniku kontaktu sigmoideum). Szerokość światła tego odcinka jelita
kwaśnej miazgi pokarmowej z błoną śluzową dwu- grubego wynosi 6–8 cm, długość około l,3 m. Typo-
nastnicy. we dla okrężnicy są trzy taśmy (taeniae), tj. podłużnie
Niezależnie od względnie niewielkiego wpływu przebiegające prążkowane pasma błony mięśniowej,
nerwów przywspółczulnych i współczulnych czyn- oraz uwypuklenia ściany jelita (haustra coli) oddzie-
ności motoryczne i wydzielnicze przewodu żołądko- lone od siebie przewężeniami powstałymi na skutek
wo-jelitowego znajdują się pod niewielkim wpływem miejscowych skurczów mięśni okrężnych.
układu przywspółczulnego i współczulnego oraz pod Końcowym odcinkiem jelita grubego jest odbyt-
kontrolą układu nerwowego. Aktywacja następuje nica (rectum), o długości 15–20 cm, zakończona
w wyniku pobudzenia receptorów rozciągających, w odbycie (anus) zwieraczem wewnętrznym zbudo-
które poprzez synapsy znajdujące się w ścianie jelita wanym z mięśni gładkich oraz zwieraczem zewnętrz-
wywołują miejscowe skurcze odruchowe. nym utworzonym z mięśni poprzecznie prążkowa-
nych. Zewnętrzne warstwy mięśniówki podłużnej
Sok jelitowy. W ciągu doby wytwarzane jest 2,5 1 nie występują w postaci taśm, ale ponownie tworzą
soku jelitowego, wydzielającego się pod wpływem zamkniętą warstwę okrężną.
bodźców mechanicznych i chemicznych. Gruczoły W przeciwieństwie do jelita cienkiego błona śluzo-
jelita cienkiego (tzw. gruczoły Lieberkühna) uwal- wa jelita grubego nie zawiera kosmków, lecz szczegól-
niają płyn izotoniczny z krwią, zawierający bardzo nie głębokie i gęsto umiejscowione krypty jelitowe.
niewielkie ilości enzymów. W wyniku oddzielenia się Nabłonek krypt oraz nabłonek powierzchniowy za-
komórek śluzowych do dwunastnicy mogą się jednak wierają komórki kubkowe wytwarzające śluz. Część
dostać enzymy znajdujące się w rąbku szczoteczko- komórek nabłonkowych zaopatrzona jest w nabłonek
wym tych komórek. Gruczoły dwunastnicy (Brunne- szczoteczkowaty odpowiedzialny za resorpcję.
ra) wytwarzają ciągnącą, śluzowatą wydzielinę, która W odbytnicy pod błoną śluzową znajduje się tzw.
ze względu na wysokie stężenie wodorowęglanów strefa hemoroidalna, nagromadzenie naczyń tętni-
ma pH o wartości 8–9. Na czynność wydzielniczą czych, które współuczestniczą razem z mięśniówką
dwunastnicy mogą wywierać wpływ także gastryna, w zamknięciu odbytu. Ruchy jelita grubego umoż-
sekretyna i cholecystokinina. liwiają ugniatanie zawartości jelita i przesuwanie
jej w kierunku odbytu. W wyniku resorpcji wody oraz
elektrolitów jest ona zagęszczana.
6.1.5. Jelito grube Oprócz powolnych ruchów robaczkowych mięśni
okrężnych w krótkich odcinkach jelita jeden do trzech
razy w ciągu dnia tworzą się silne fale perystaltycz-
Jelito grube, ostatni odcinek przewodu pokarmowe-
ne, rozpoczynające się w jelicie ślepym i dochodzące
go, dzielone jest na następujące odcinki:
do esicy, które przesuwają kał do odbytnicy i w ten
■ jelito ślepe (caecum) z wyrostkiem robaczkowym sposób rozpoczynają defekację.
(appendix vermiformis),
■ okrężnicę (colon),
■ odbytnicę (rectum). 6.1.6. Wątroba i drogi żółciowe
W jelicie grubym w wyniku zagęszczania treści jelita Wątroba leży pod prawym uwypukleniem przepo-
formowany jest kał (stolec). ny i jest narządem pełniącym najważniejszą funkcję
MUTSCHLER-2009.indd 668 2010-01-07 22:14:09
Układ trawienny 669
Płytka komórki wątrobowej może znaleźć się
przewód przewód wątrobowy w dwóch położeniach. Między przeplatającymi się
pęcherzy- beleczkami komórek wątrobowych znajdują się za-
kowy toki wątrobowe (kapilary wątrobowe), które łączą
przewód żółciowy wspólny
się między sobą i tworzą promienistą sieć kapilar.
pęcherzyk trzustka
W ich ścianie znajdują się oprócz komórek śródbłon-
żółciowy przewód
wyprowadzający ka także komórki gwiaździste należące do układu
mononuklearno-fagocytarnego (dawniej układu sia-
dwunastnica teczkowo-śródbłonkowego) i wykazujące zdolność
do fagocytozy. Między zatokami wątrobowymi i ko-
mórkami wątrobowymi istnieje szczelina (przestrzeń
okołozatokowa Dissego), do której wnikają mikro-
Ryc. B 6.1-5. Pozawątrobowe drogi żółciowe, dwunastnica kosmki komórki wątrobowej. W ten sposób tworzą
i trzustka w swoim prawidłowym położeniu w nadbrzuszu.
się optymalne warunki do resorpcji związków, które
przez liczne rozstępy ściany kapilary przedostały się
do przestrzeni okołozatokowej Dissego.
w procesie przemiany materii. Ze względu na wytwa- Między komórkami wątrobowymi, ale obok za-
rzanie i wydzielanie żółci jest również największym tok wątrobowych, od których są zawsze oddzielo-
gruczołem wewnątrzwydzielniczym. Podzielona ne, przebiegają włosowate przewodziki żółciowe.
jest na dwa płaty, większy prawy i mniejszy lewy. Ich ściana zbudowana jest z błony komórek wątro-
Masa wątroby wynosi około 1500 g. bowych. Włosowate przewodziki żółciowe rozpo-
Na powierzchni wklęsłej, przez znajdujące się czynają się w części środkowej zrazika i biegną od-
tu tzw. wrota wątroby wchodzą do tego narządu dwa środkowo w kierunku obwodu zrazika wątrobowego,
naczynia doprowadzające krew: tętnica wątrobowa gdzie w okolicy tzw. pola okołowrotnego, tj. miejsca
(arteria hepatica) oraz żyła wrotna (vena portae), łączenia się zrazików, uchodzą do międzyzrazikowe-
a wychodzą przewody żółciowe (ductus hepatici). go przewodu żółciowego.
Tętnica wątrobowa zaopatruje wątrobę w krew Komórki wątrobowe charakteryzuje obecność
tętniczą. Żyłą wrotną (vena portae) doprowadza- licznych mitochondriów oraz szczególnie wyraźnie
na jest krew żylna z nieparzystych narządów jamy zaznaczone retikulum endoplazmatyczne.
brzusznej, a wraz z nią składniki pokarmowe wchło-
nięte z żołądka, jelit i wątroby. Po przejściu przez ka- Wydzielanie żółci. W ciągu doby organizm produku-
pilary wątrobowe, tj. sinusoidy, krew przepływa żyłą je 600–800 ml żółci. Skład i szybkość powstawania
wątrobową (vena hepatica) do żyły głównej dolnej żółci zależą od rodzaju i ilości spożytego pożywienia.
Układ trawienny
(vena cava inferior). Wartość pH wynosi od 7,4 do 8,5.
Obydwa przewody żółciowe łączą się tuż po opusz- Oprócz jonów nieorganicznych prawie izotonicz-
czeniu wątroby w przewód wątrobowy wspólny (duc- na z krwią żółć zawiera przede wszystkim kwasy
tus hepaticus communis) (zob. ryc. B 6.1-5). Miejsce żółciowe (np. kwas dezoksycholowy, kwas cholo-
jego zakończenie podkreśla odgałęzienie przewodu wy; przeważnie w postaci sprzężonej z tauryną albo
pęcherzykowego (ductus cysticus) prowadzącego glicyną), barwniki żółciowe, cholesterol, fosfolipidy
do pęcherzyka żółciowego. Część przewodu żółcio- i niektóre enzymy (np. fosfatazę zasadową). W żółci B6
wego łącząca się z przewodem wątrobowym wspól- mogą znajdować się również leki oraz ich metabolity,
nym określana jest jako przewód żółciowy wspólny które tą drogą są częściowo wydalane.
(ductus choledochus). Razem z przewodem trzustko- W drogach żółciowych, a szczególnie w pęcherzy-
wym (zob. poniżej) uchodzi on przeważnie do wstę- ku żółciowym, którego pojemność wynosi 10–15 ml,
pującego ramienia dwunastnicy. skład żółci się zmienia. Na skutek utraty wody stę-
żenie barwników żółciowych i cholesterolu wzrasta
Budowa zrazików wątrobowych. Elementami bu- 5–10-krotnie, natomiast w wyniku resorpcji zwrotnej
dującymi wątrobę są zraziki o średnicy 1–2 mm, jonów sodowych, chlorkowych i wodorowęglano-
których liczba u człowieka wynosi 50 000–100 000. wych do naczyń krwionośnych obniża się stężenie
Poszczególne zraziki oddzielone są od siebie słabą elektrolitów.
powięzią łącznotkankową. W poprzecznym przekroju Wydzielanie żółci regulują hormony żołądkowo-
histologicznym przypominają kształtem sześciokąt. -jelitowe oraz układ autonomiczny. W trakcie trawie-
Każdy zrazik wątrobowy zbudowany jest z licznych nia jej ilość wzrasta dwukrotnie przy zwiększaniu
komórek tworzących tzw. beleczki. stężenia wodorowęglanów. Wydzielanie żółci zwięk-
MUTSCHLER-2009.indd 669 2010-01-07 22:14:09
670 Układ trawienny
szają sekretyna, nasilony przepływ wątrobowy krwi Odpowiedzialne za rozkład białek trypsyna, chy-
oraz pobudzenie nerwu błędnego. motrypsyna, karboksypeptydaza są enzymami wy-
W trakcie spożywania pokarmu żółć spływa bez- dzielanymi przez komórki groniaste (pęcherzyko-
pośrednio do dwunastnicy. W okresach braku trawie- we) trzustki w postaci nieczynnej i aktywowanymi
nia przedostaje się do pęcherzyka żółciowego, gdzie w dwunastnicy pod wpływem enteropeptydazy oraz
jest zagęszczana i gromadzona i skąd po uwolnieniu w sposób autokatalityczny pod wpływem trypsyny.
cholecystokininy wywołanym skurczami pęcherzyka Obecność lipazy, fosfolipazy A i enterazy umoż-
żółciowego przelewa się do dwunastnicy. liwia trawienie tłuszczów. Lipaza trzustkowa, en-
zym najważniejszy w procesie trawienia tłuszczów,
wydzielana jest wprawdzie w postaci czynnej,
ale do jej działania konieczna jest obecność kwasów
6.1.7. Trzustka żółciowych. W przypadku fosfolipazy A aktywacja
następuje w dwunastnicy.
Trzustka (pancreas) jest narządem wydzielniczym, Wymienione enzymy czy nieczynne proenzymy
w którym nieregularnie rozmieszczone są wewnątrz- są wytwarzane w komórkach groniastych i gromadzo-
wydzielnicze skupiska komórek, tzw. wysp Langer- ne w postaci ziaren zymogenu. Pobudzenie czynności
hansa. Część zewnątrzwydzielnicza trzustki wydziela wydzielniczej powoduje opróżnienie pęcherzyków
enzymy trawienne. Ten ważący około 100 g narząd i przesunięcie ich zawartości do światła końcowego
położony jest w górnej części jamy brzusznej, poniżej odcinka przewodów gruczołowych. Jednocześnie
żołądka. Można podzielić go na trzy części: głowę rozpoczyna się nowa synteza.
trzustki przylegającą do wklęsłej powierzchni dwu- Regulacja wydzielania soku trzustkowego może
nastnicy, trzon trzustki oraz ogon wyprowadzający odbywać się drogą nerwową i hormonalną. Fazą
trzustki. głowową wydzielania trzustkowego określa się etap
Przewód trzustkowy (ductus pancreaticus), prze- wydzielania odbywający się pod wpływem bodźców
wód wyprowadzający trzustki, przechodzi wzdłuż ca- nerwowych. Bodźce smakowe i zapachowe, wystę-
łej długości trzustki i, jak wyżej wspomniano, razem pujące przed i w trakcie spożywania pokarmu powo-
z przewodem żółciowym wspólnym uchodzi do dwu- dują wydzielanie odruchowe. Identyczne działanie
nastnicy. ma sam widok pokarmu lub jego wyobrażenie.
Na przekroju histologicznym widoczna jest struk- Następująca w drugiej kolejności faza żołądkowa
tura zrazikowa. Zraziki są utworzone z końcowych rozpoczyna się po przedostaniu się pokarmu do żołąd-
odcinków, tzw. gron (acini). Odcinki łączące zagięte ka. W wyniku mechanicznego rozciągania ściany tego
do poszczególnych gron tworzą połączenia pomiędzy narządu następuje wzmożenie wydzielania na skutek
odcinkiem końcowym gruczołu i przewodem wypro- bodźców miejscowych i uwolnienie hormonów żo-
wadzającym. łądkowo-jelitowych, tj. sekretyny, cholecystokininy
i gastryny. Działanie sekretyny powoduje uwolnienie
Wydzielanie trzustkowe. Izotoniczna z krwią wy- znacznej ilości silnie zasadowej, ale ubogiej w enzy-
dzielina trzustkowa jest wytwarzana w ilości około my wydzieliny trzustkowej. Cholecystokinina i ga-
1–1,5 1 na dobę i transportowana do dwunastnicy stryna zwiększają ilość enzymów wydzielanych przez
przez przewód trzustkowy. Ze względu na znaczną gruczoły groniaste (komórek pęcherzykowych).
zawartość węglowodorów jej wartość pH wyno- Trzecia faza, faza jelitowa, rozpoczyna się po przej-
si 8,0–8,4. Wydzielina trzustki razem z zasadową ściu do dwunastnicy kwaśnej treści żołądkowej bądź
żółcią i sokiem jelitowym zobojętnia kwaśny sok produktów przemiany tłuszczów i białek. Pobudzenie
żołądkowy, skutkiem czego miazga pokarmowa jelitowe wspomaga wydzielanie soku trzustkowego
w dwunastnicy ma odczyn obojętny lub słabo zasa- poprzez uwolnienie sekretyny i cholecystokininy.
dowy. Powstające środowisko sprzyja działaniu en- Sole kwasów żółciowych również pobudzają czyn-
zymów z soku trzustkowego, dla których optymalne ność zewnątrzwydzielniczą trzustki.
pH wynosi 7,8.
Sok trzustkowy zawiera enzymy rozkładające
węglowodany, białka i tłuszcze, co umożliwia im sa-
modzielne trawienie pokarmu, tj. w przypadku braku 6.1.8. Trawienie
innych enzymów trawiennych.
Enzymem obecnym w soku trzustkowym w posta- Enzymy niezbędne do rozdrabniania pokarmu na części
ci aktywnej jest rozkładająca węglowodany α-amy- o rozmiarach umożliwiających wchłanianie są obecne
laza. w soku żołądkowym, jelitowym i trzustkowym.
MUTSCHLER-2009.indd 670 2010-01-07 22:14:09
Układ trawienny 671
Trawienie węglowodanów. Przeważająca część wystarcza do rozbicia 80% tej ilości tłuszczów, która
(około 60%) węglowodanów spożywanych w ciągu dotrze do środkowego odcinka dwunastnicy.
doby pochodzi ze skrobi. Tylko 30% uzyskiwanych W procesie trawienia tłuszczów aktywna jest nie tyl-
jest z sacharozy i około 10% z laktozy. Oprócz wy- ko lipaza, ale również inne enzymy trzustkowe. Fosfo-
mienionych dwucukrów spożywane są także niewiel- lipaza A oddziela od lecytyny jeden kwas tłuszczowy,
kie ilości cukrów prostych w postaci glukozy i frukto- co powoduje powstanie lizolecytyny. Obecne w po-
zy. Innym węglowodanowym składnikiem pożywie- żywieniu estry cholesterolu są hydrolizowane przez
nia jest glikogen pochodzenia zwierzęcego. odpowiednie esterazy do cholesterolu i kwasów żół-
Skrobia jest mieszaniną składającą się w 20% ciowych.
z liniowej amylozy i w 80% z rozgałęzionej amylo-
pektyny. Amylopektyna przypomina pod względem
budowy glikogen. Część tworzona przez amylozę
oddzielana jest przez enzym 1,4-α-amylazę zawarty 6.1.9. Resorpcja
w ślinie i w soku trzustkowym. Produktami końco-
wymi tego rozpadu są maltoza i maltotrioza. Amy- Produkty powstałe w wyniku procesu trawienia mu-
lopektyna i glikogen rozkładane są przez enzymy szą przeniknąć przez ścianę jelita najpierw do krwi
obecne w nabłonku szczoteczkowym enterocytów. lub limfy, a następnie wraz z nimi dotrzeć do wszyst-
W nabłonku tym znajdują się również disacharyda- kich komórek ciała. Narządem odpowiedzialnym
zy, które przekształcają maltozę, sacharozę i laktozę za resorpcję składników pokarmowych jest jelito
w cukry proste. cienkie. W zależności od wartości stężenia resorpcja
odbywa się na drodze dyfuzji lub transportu czyn-
Trawienie białek. Hydroliza białek odbywa się pod nego.
wpływem endopeptydaz, które rozszczepiają wiąza- Przejście monosacharydów przez warstwę lipido-
nia peptydowe cząsteczek. Białka rozkładane są naj- wą błony komórkowej utrudnia ich dobra rozpusz-
pierw na polipeptydy, a następnie na oligopeptydy. czalność w wodzie. Z kolei dobra rozpuszczalność
Zadaniem egzopeptydaz jest odcinanie aminokwasów cukrów prostych w wodzie utrudnia ich przecho-
na końcu łańcucha C- lub N- i w ten sposób stopnio- dzenie przez warstwę lipidową błony komórkowej.
wy rozkład poli- lub oligopeptydów. Do endopepty- Ze względu na tę właściwość glukoza musi być trans-
daz należą pepsyna soku żołądkowego, oraz trypsyna portowana czynnie. Prawdopodobnie w podobny
i chymotrypsyna soku trzustkowego. Egzopeptydazy sposób odbywa się również przenikanie galaktozy.
występują w soku trzustkowym i w rąbku szczotecz- W przypadku fruktozy możliwa jest dyfuzja wspoma-
kowym enterocytów. gana za pomocą nośników, odbywająca się zgodnie
Trawienie białek zaczyna się w żołądku. Ma z gradientem stężeń.
Układ trawienny
to jednak znaczenie drugorzędne, ponieważ tylko Resorpcja naturalnych L-aminokwasów odbywa
10–15% białek pokarmowych jest hydrolizowanych się według czterech swoistych systemów transportu
przez pepsynę. Najważniejszy etap w przemianie bia- aminokwasów obojętnych, kwaśnych, zasadowych
łek przypada na dwunastnicę. Produkcja aktywnych oraz iminokwasów. Polipeptydy resorbowane są tyl-
tutaj peptydaz trzustkowych zaczyna się 10–20 min ko w bardzo niewielkim zakresie.
po spożyciu pokarmu i trwa tak długo, jak długo biał- Tłuszcze zmienione pod wpływem lipazy w wol-
ko znajduje się w jelicie. ne kwasy tłuszczowe, glicerynę i monoglicerydy B6
są przyjmowane przez enterocyty. Te przekształca-
Trawienie tłuszczów. W żołądku tłuszcze są rozbi- ją obecne w naturalnych tłuszczach kwasy tłusz-
jane na kropelki o średnicy około 10 nm. Następnie czowe z 16–18 atomami węgla ponownie w tri-
w zasadowym środowisku dwunastnicy, w obecności glicerydy i otoczone osłonką białkową przekazują
białek, lecytyny i kwasów żółciowych w wyniku pro- do limfy jako tzw. chylomikrony. Przy braku kwa-
cesu emulsyfikacji ulegają rozbiciu na jeszcze mniej- sów żółciowych i lipazy trzustkowej krótko- i śred-
sze kropelki o średnicy 100 nm. niołańcuchowe triglicerydy mogą być wchłonięte
W dwunastnicy lipaza trzustkowa wspomagana również w swojej niezmienionej formie przez ko-
przez kolipazę gromadzi się na granicy faz emulgo- mórki śluzowe, z których przede wszystkim przez
wanej cząsteczki tłuszczu. Zapoczątkowana w ten żyłę wrotną dostają się do wątroby. Z tego powo-
sposób hydroliza prowadzi do oddzielenia od acy- du w przypadku chorób jelit, wątroby lub trzustki,
loglicerolu kwasów tłuszczowych związanych z C-1 którym towarzyszą stolce tłuszczowe, podaje się
i C-3, w wyniku czego 2-monoacyloglicerol pozosta- średniołańcuchowe triglicerydy zamiast innych
je niezajęty. Ilość lipazy uwolnionej przez trzustkę tłuszczów.
MUTSCHLER-2009.indd 671 2010-01-07 22:14:09
672 Układ trawienny
6.2. Choroba wrzodowa
6.2.1. Podstawy patofizjologiczne szym opiniom podawanie glukokortykosteroidów
(bez NLPZ) nie zwiększa ryzyka rozwoju choroby
wrzodowej. Patogenetyczny udział może mieć na-
Zaburzenia trawienia, tzn. schorzenia, do których
tomiast zarzucanie do żołądka treści dwunastnicy
przyczyniły się kwas solny i enzymy, dotyczą górnej
z zawartymi w niej kwasami żółciowymi, które jako
części przewodu żołądkowo-jelitowego, tj. przełyku,
naturalne detergenty rozpuszczają fosfolipidy błony
żołądka i dwunastnicy. W zależności od umiejsco-
komórkowej i ułatwiają uszkadzanie błony śluzowej
wienia i patogenezy rozróżnia się chorobę refluksową
przez protony.
przełyku (gastro-esophageal reflux disease – GERD),
chorobę wrzodową żołądka (ulcus ventriculi) oraz
Przy wrzodzie dwunastnicy (Ulcus duodeni) przewa-
chorobę wrzodową dwunastnicy (ulcus duodeni).
żają czynniki agresywne. Prawie zawsze całkowita
Wrzód trawienny jest ostro ograniczonym ubytkiem
ilość wydzielanego kwasu solnego jest podwyższona
obejmującym śluzówki i głębiej położone warstwy
(hyperchlorhydria), a sekrecja HCO3– w trzustce nie-
tkanek. W przypadku nadżerki uszkodzeniu ulega
wystarczająca.
wyłącznie błona śluzowa.
Morfologiczne zmiany błony śluzowej przełyku
Brak równowagi między czynnikami agresywnymi
określane są mianem choroby refluksowej przełyku.
i obronnymi pojawia się również w chorobie refluk-
Makroskopowe nadżerki lub owrzodzenia świadczą
syjnej przełyku. Dodatkowo towarzyszy mu zwiot-
o refluksowym zapaleniu przełyku.
czenie zwieracza przełyku, zwłaszcza w pozycji le-
U osoby zdrowej mechanizmy obronne błony ślu-
żącej, co sprzyja zarzucaniu treści pokarmowej z żo-
zowej żołądka (bariera błony śluzowej z nieuszkodzo-
łądka do przełyku.
nym nabłonkiem powierzchniowym, wystarczającą
ilością śluzu i prawidłowym ukrwieniem) przeciwsta-
Dolegliwości związane z wrzodem występują prze-
wiają się mechanizmom agresywnym (bezpośrednio
ważnie okresowo w formie kurczowego i piekącego
zwiększonemu wydzielaniu HCl oraz pepsynogenu,
bólu w nadbrzuszu. Mają różny czas utrzymywania się,
pośrednio nasilonym bodźcom cholinergicznym oraz
a ich pojawienie się jest związane ze spożyciem pokar-
wzmożonemu wydzielaniu gastryny i zasiedlaniu żo-
mu. Stopień nasilenia dolegliwości klinicznych nie od-
łądka bakteriami Helicobacter pylori).
zwierciedla zakresu zmian w obrębie błony śluzowej,
W chorobie wrzodowej dużą rolę odgrywa –
w związku z czym nie mogą one stanowić podstawy
oprócz skłonności warunkowanych genetycznie
do postawienia diagnozy. Wrzody, szczególnie te indu-
– napięcie nerwu błędnego, które wzmaga się pod
kowane NLPZ, mogą przebiegać bezobjawowo.
wpływem stresu oraz emocji. O wiele istotniejsze
w jej patogenezie jest jednak zasiedlenie błony ślu-
Istotnymi powikłaniami choroby wrzodowej są:
zowej żołądka przez bakterie Helicobacter pylori.
Szczep ten wykryto u 95% pacjentów z wrzodem ■ krwawienie (15% w ciągu 10 lat),
dwunastnicy i u 60–70% pacjentów z wrzodem żo-
■ perforacja wrzodu (przedziurawienia w obrębie
łądka. Nawrotowe owrzodzenie dwunastnicy prawie
brzucha) z silnymi dolegliwościami bólowymi,
zawsze jest skutkiem zakażenia Helicobacter pylori.
wstrząsem i obronnym napięciem mięśni brzucha
Jednakże tylko u niewielkiej części nosicieli Helico-
(5–10%),
bacter pylori rozwija się wrzód trawienny.
■ zwężenie odźwiernika.
Ponieważ często dodatkowymi objawami towarzy-
szącymi chorobie wrzodowej żołądka (Ulcus ventri-
culi) są przewlekłe zanikowe zapalenie żołądka (zob.
poniżej) oraz zmiany składu śluzu żołądkowego, 6.2.2. Leczenie choroby wrzodowej
przyjmuje się, że oprócz zasiedlenia przez Helico-
bacter pylori przyczyną tej choroby jest zaburzenie Cele terapeutyczne. W leczeniu wrzodu trawienne-
mechanizmów obronnych błony śluzowej. Istotnym go celem terapeutycznym jest:
czynnikiem patogenetycznym są również blokery
■ zlikwidowanie bólu,
syntezy prostaglandyny z grupy niesteroidowych le-
ków przeciwzapalnych (NLPZ). Wbrew wcześniej- ■ przyspieszenie gojenia się wrzodu,
MUTSCHLER-2009.indd 672 2010-01-07 22:14:09
Układ trawienny 673
■ eradykacja Helicobacter pylori, bą wrzodową. Zalecenie specjalnej diety o działaniu
leczniczym jest niemożliwe, ponieważ nie ma sposobu
■ zapobieganie powikłaniom,
odżywiania skutecznego przy tego typu schorzeniach.
■ zapobieganie nawrotom.
Gojenie się wrzodu powinno być potwierdzane
6.2.2.1. Blokery H+/K+-ATP-azy
badaniem endoskopowym (we wrzodzie żołądka
należy wykluczyć raka), ponieważ brak objawów (inhibitory pompy protonowej)
nie musi koniecznie oznaczać wygojenia owrzo-
dzenia. W ocenie wyników terapeutycznych należy Zastosowanie blokady H+/K+-ATP-azy umożliwia
uwzględnić liczne przypadki samoistnego wylecze- prawie całkowite zahamowanie wydzielania kwasu
nia wrzodu żołądka lub dwunastnicy. Jeszcze nie- solnego. Zgodnie z uproszczoną zasadą: nie ma kwa-
dawno nawet stosowanie środków farmakologicz- su – nie ma wrzodu, inhibitory pompy protonowej
nych nie pozwalało wywierać wpływu na przebieg są lekami pierwszego wyboru w leczeniu wrzodu
choroby wrzodowej. Obecnie jednak poprzez era- trawiennego oraz refluksowego zapalenia przełyku.
dykację Helicobacter pylori (zob. poniżej) możliwe Wszystkie dotychczas dostępne związki są pochod-
jest całkowite wyleczenie. nymi benzimidazolu (zob. tab. B 6.2-1):
Przed rozpoczęciem lub na początku terapii zabu-
■ omeprazol,
rzeń trawienia należy pouczyć pacjenta o szkodliwym
oddziaływaniu palenia, nadmiernego spożycia kawy ■ lansoprazol,
i skoncentrowanych alkoholi, nieregularnego odży-
■ pantoprazol,
wiania oraz przyjmowania niesteroidowych leków
przeciwzapalnych na dolegliwości związane z choro- ■ rabeprazol.
Tabela B 6.2-1. Inhibitory pompy protonowej
Wzór strukturalny Nazwa Nazwa handlowa Okres Dawka
międzynarodowa półtrwania dzienna
(godz.) (mg)
NH O omeprazol Antra, 0,6-1,0 20(-40)
S N Omebeta,
H3CO N CH3 OMEP,
Omeprazol STADA
Układ trawienny
H3C OCH3
NH O esomeprazol Nexium 0,8-1,2 20-40
S N
H3CO N CH3
H3C OCH3
NH O lansoprazol Agopton, 0,9-1,6 15-30 B6
S N Lansoprazol-CT,
N Lansoprazol-ratipharm,
CF3 Lansoprazol STADA
H3C O
NH O pantoprazol Pantozol, 0,9-1,9 20-40
S N Rifun
F2HC
O N
H3CO OCH3
NH O rabeprazol Pariet 0,9-1,5 20
S N
N
H3C O OCH3
MUTSCHLER-2009.indd 673 2010-01-07 22:14:10
674 Układ trawienny
A B pochodna spiryny kwas sulfenowy
H+ OCH3 OCH3
H3C CH3 H3C CH3
N+ N+
HN S S
O N NH
omeprazol OH
NH
H3CO
omeprazol Ð H2O OCH3
K+ H+/K+- + H2O
OCH3
-ATP-azy
H3C CH3
a ATP
zanie
okładzinórka
ow
nieodwracalne wią N+
kom
-azy
an H+ /K -ATP
+ S
N N
H+ sulfenamid
jelito cienkie
OCH3
tętniczka komórka światło
okładzinowa
Ryc. B 6.2-1. Bioaktywacja omeprazolu i nieodwracalne hamowanie aktywności H+/K+-ATP-azy. (A) Omerpazol jest resorbowany
w jelicie cienkim, a następnie przenoszony za pośrednictwem układu krwionośnego do kanalików komórek okładzinowych. (B)
W środowisku kwaśnym omeprazol przechodzi w spirozwiązek. Poprzez odłączenie cząsteczki wody powstaje czynny metabolit,
sulfenamid, hamujący nieodwracalnie H+/K+-ATP-azy.
Esomeprazol pojawił się na rynku przed kilkoma Inhibitory pompy protonowej nie mają wprawdzie
laty razem z S-enancjomerem omeprazolu (racemat). działania przeciwbakteryjnego, jednak poprzez stałe
Nie jest on jednak skuteczniejszy niż R-omeprazol, zahamowanie zwiększania się wartości pH ułatwiają
ponieważ oba enancjomery przekształcane są w po- eradykację Helicobacter pylori (zob. poniżej).
stać czynną, nie chiralną (zob. poniżej). Inhibitory pompy protonowej są wskazane w le-
Pochodne benzimidazolu są prolekami, które dopie- czeniu choroby wrzodowej dwunastnicy (Ulcus duo-
ro w środowisku kwaśnym przekształcają się w odpo- deni), choroby wrzodowej żołądka (Ulcus ventriculi),
wiadające im czynne sulfenamidy. Po przejściu przez zespołu Zollingera-Ellisona, a także symptomatycz-
żołądek (tabletki powinny posiadać powłokę odporną nej nadżerkowej lub wrzodziejącej choroby reflukso-
na działanie kwasu żołądkowego, zob. poniżej) są re- wej przełyku. Ponadto mogą być stosowane w profi-
sorbowane w jelicie cienkim i za pośrednictwem ukła- laktyce nawracających wrzodów żołądka oraz w ra-
du krwionośnego przenoszone do kanalików komórek mach terapii długoterminowej mimo potencjalnych
okładzinowych. W panującym tutaj środowisku kwaś- zagrożeń wynikających ze stałego obniżania ilości
nym tworzą przejściowy spirozwiązek, który następ- produkowanego kwasu solnego (np. długotrwała hi-
nie ulega przekształceniu w odpowiedni kwas sulfe- pergastrynemia, zasiedlenie żołądka przez bakterie).
nowy. Po odłączeniu cząsteczki wody kwas sulfenowy Inhibitory pompy protonowej zaleca się również jako
tworzy aktywny metabolit, cykliczny sulfinyloamid jeden ze środków w przypadku krwawiących wrzo-
(zob. ryc. B 6.2-1), który reaguje z podjednostką α dów. Zdolność trombocytów do agregacji jest bo-
H+/K+-ATP-azy i tworzy mostek disulfidowy. W efek- wiem o wiele większa w środowisku lekko kwaśnym
cie następuje zmniejszenie ilości ATP przyłączanego i neutralnym niż w kwaśnym.
do enzymu oraz zahamowanie defosforylacji zależnej Wiedząc, że jednoczesne stosowanie kilku leków
od jonów K+, a tym samym zablokowanie całego en- może również przyczynić się do powstania wrzodu
zymu. Regeneracja enzymów jest możliwa wyłącznie żołądka, nie należy wprowadzać inhibitorów pompy
przez ich tworzenie na nowo, dlatego działanie po- protonowej bezkrytycznie, bez wyraźnego wskazania
chodnych benzimidazolu, mimo ich krótkiego okresu do leczenia nimi.
półtrwania, obejmuje 1–3 dni (zob. tab. B 6.2-1).
MUTSCHLER-2009.indd 674 2010-01-07 22:14:10
Układ trawienny 675
Farmakokinetyka. Ze względu na swoją niestabil- Ponadto najczęściej po pozajelitowym podaniu in-
ność w środowisku kwaśnym inhibitory pompy pro- hibitorów pompy protonowej występowały znaczne
tonowej muszą być podawane w formie preparatów zaburzenia wzroku i słuchu. Dlatego leki z tej grupy
opornych na działanie soku żołądkowego. Dostępność ze wskazaniem do podania pozajelitowego powinny
biologiczna omeprazolu wynosi na początku leczenia być aplikowane nie w formie pigułek, lecz krótkiego
około 40% i wzrasta do 65% po wielokrotnym poda- wlewu.
niu oraz dużych dawkach; w przypadku lansoprazolu, Przeciwwskazaniem do podania inhibitorów pom-
pantoprazolu i rabeprazolu pozostaje cały czas na tym py protonowej są zaburzenia funkcji wątroby.
samym poziomie. Zwiększenie dostępności biolo-
gicznej omeprazolu w stanie stacjonarnym nie wy-
nika więc ze wzrostu wartości pH żołądka, ale naj-
6.2.2.2. Antagoniści receptora
prawdopodobniej z nasycenia metabolizmu pierw-
szego przejścia. Substancja czynna niewykorzystana histaminowego H2
w komórkach okładzinowych jest biotransformowa-
na w wątrobie do odpowiednich pochodnych hydrok- Rola antagonistów receptora histaminowego H2 po-
sylowych i sulfonów, wydalanych przede wszystkim lega na blokowaniu konkurencyjnym receptorów
przez nerki. Wytwarzanie spirozwiązków w środowi- histaminowych komórek okładzinowych błony ślu-
sku kwaśnym przez dołączenie protonu powoduje, zowej żołądka, hamowaniu zarówno podstawowego,
że inhibitory pompy protonowej są wysycane w za- jak i stymulowanego histaminą wydzielania kwasu
leżności od wartości pH w miejscu działania: im niż- solnego oraz zmniejszaniu niekonkurencyjnym wy-
sza jest wartość pH związku, a wyższa wartość pKa, dzielania kwasu uwalnianego w wyniku pobudzenia
tym silniej lub szybciej związek zostanie wysycony za pośrednictwem nerwu błędnego i przez gastrynę.
i zadziała. W związku ze stosunkowo wysoką war- Dostępne są następujące leki o działaniu antago-
tością pKa (4,9) rabeprazol in vitro o wiele szybciej nistycznym w stosunku do receptora H2 (zob. tab.
ulega przekształceniu w aktywny sulfenamid niż inne B 6.2-2):
substancje z tej grupy (pKa omeprazolu wynosi 4,13,
■ cymetydyna,
lansoprazolu – 4,01, pantoprazolu – 3,96), a swoje
działanie rozpoczyna już pierwszego dnia terapii. ■ ranitydyna,
Kwestią dyskusyjną jest jednak to, czy zastosowanie
■ famotydyna,
rabeprazolu jest zawsze klinicznie ważną zaletą.
Inhibitory pompy protonowej metabolizowane ■ nizatydyna.
są przede wszystkim za pomocą CYP2C19 i w mniej-
szym stopniu przez CYP3A, dzięki czemu możliwa U pacjentów z chorobą wrzodową antagoniści re-
Układ trawienny
jest ich interkacja z warfaryną, diazepamem i feny- ceptora H2 łagodzą objawy bólowe i przyspieszają
toiną oraz z innymi lekami biotransformowanymi proces gojenia się. Kliniczna skuteczność tych le-
w wyniku utleniania (opóźniony metabolizm). Sub- ków jest taka sama. Różne jest natomiast nasilenie
stancje z tej grupy hamują wchłanianie witaminy B12 ich działania, związane z tym dawkowanie i możliwe
oraz ketokonazolu, a zwiększają resorpcję digoksyny. objawy niepożądane (zob. poniżej). Przyjęcie wie-
Makrolidy, takie jak klarytromycyna, erytromycyna czorem przeciętnej dawki leku utrzymuje wartość
i roksytromycyna, zwiększają zdolność omeprazolu pH żołądka na poziomie większym niż 5 przez 4–6 B6
do biotransformacji. Omeprazol natomiast wpływa godz. (dla famotydyny 12 godz.). Uzyskanie szyb-
na ich zawartość w osoczu. kiego i skutecznego wyleczenia choroby wrzodowej,
a zwłaszcza choroby refluksowej przełyku, możliwe
Dawkowanie. W przypadku choroby wrzodowej żo- będzie natomiast wtedy, gdy wartość pH soku żołąd-
łądka i dwunastnicy oraz choroby refluksowej przeły- kowego będzie większa niż 4 przez 15 godz./dzień.
ku dawkowanie jak w tab. B 6.2-1. Odsetek wyleczeń po zastosowaniu antagonistów
receptora H2 jest mniejszy niż po inhibitorach pom-
Działania niepożądane. Inhibitory pompy protono- py protonowej, dlatego w leczeniu wrzodu trawien-
wej wywołują prawie identyczne działania niepożą- nego są one lekami drugiego wyboru. W przypadku
dane, przy czym omeprazol był najdłużej badanym pacjentów poddawanych intensywnemu leczeniu
lekiem. Do najczęściej zgłaszanych działań niepo- ze wskazaniem do „inhibitorów kwasowych” częś-
żądanych należą: dolegliwości żołądkowo-jelitowe, ciej stosowane są leki z grupy antagonistów receptora
zawroty głowy, nudności, bóle głowy, zwiększone histaminowego H2 niż z grupy inhibitorów pompy
stężenie enzymów wątrobowych i wykwity skórne. protonowej. Nie prowadzą one bowiem do całkowi-
MUTSCHLER-2009.indd 675 2010-01-07 22:14:11
676 Układ trawienny
Tabela B 6.2-2. Antagoniści receptora histaminowego H2
Wzór strukturalny Nazwa Nazwa handlowa Okres Dawka
międzynarodowa półtrwania dzienna
(godz.) (mg)
NH NH
CH3
cymetydyna Cimebeta, 1,5-2 800
N S CimeHEXAL,
N Cimetidin AL,
NH CH3 CN Cimetidin-CT
O NH NH ranitydyna Ranibeta, 2-3 300
(H3C) 2N S CH3 Ranitidin-ratiopharm,
Sostril,
NO2 Zantic
N NH NH nizatydyna Nizax Lill 1,5-2 300
(H3C) 2N S CH3
S
NO2
NH2 famotydyna Fadul, 2,5-4 40
H2N N SO 2NH2 Famobeta,
N
S N Famotidin-ratiopharm,
NH2 S PEPDUL
tego zahamowania wydzielania kwasu żołądkowego, długo utrzymującą się podwyższoną wartość pH,
dzięki czemu zachowana jest kwasowa bariera chro- sprzyjają bakteryjnemu zasiedleniu żołądka, a tym
niąca żołądek przed zasiedleniem przez bakterie. samym powstawaniu infekcji dróg oddechowych.
Poza leczeniem choroby wrzodowej leki z grupy Na skutek, uzależnionego od dawki, antyandro-
antagonistów receptora histaminowego H2 mogą być gennego działania cymetydyna może wywołać zabu-
stosowane w zapobieganiu odnawiania się wrzodu. rzenia potencji, oligospremię i ginekomastię. Z tego
Jednak również w tym przypadku lepsze działanie względu oraz faktu, że w wyniku podawania innych
wykazują inhibitory pompy protonowej. blokerów receptorów H2 zaburzenia te występują
o wiele rzadziej niż w przypadku cymetydyny, nie na-
Farmakokinetyka. Po podaniu doustnym leki będą- leży stosować jej w procesie leczenia. U pacjentów
ce antagonistami receptora histaminowego H2 wchła- w podeszłym wieku oraz u pacjentów z zaburzeniami
niają się szybko z przewodu żołądkowo-jelitowego. czynnościowymi wątroby i nerek obserwowano rów-
Ich okres półtrwania w osoczu wynosi 1,5–4 godz., nież zamroczenia i halucynacje.
w przypadku podawania w jednej dawce dobowej
różnice są nieistotne. Substancje z tej grupy antago- Przeciwwskazania. Przeciwwskazania bezwzględne
nistów są metabolizowane w różnym stopniu, a część są nieznane. Szczególnie istotne jest ustalenie wska-
niezmetabolizowana wydalana jest przez nerki. zań u kobiet w ciąży i w okresie laktacji.
Dawkowanie. W procesie leczenia wrzodów tra- Interakcje. Cymetydyna hamuje reakcje zależne
wiennych blokery H2 podaje się przeważnie wieczo- od cytochromu P-450. Klinicznie istotne jest nasi-
rem. Zalecana dawki cymetydyny wynosi 800 mg, lenie i wydłużenie działania biotransformowanych
ranitydyny i nizatydyny – 300 mg, famotydyny w wyniku utlenienia benzodiazepin i pośrednich le-
– 40 mg. ków przeciwzakrzepowych (z wyjątkiem fenproku-
monu), lidokainy, fenytoiny i teofiliny oraz nasilone
Działania niepożądane. Bóle głowy (zwłaszcza działania niepożądane trójcyklicznych leków prze-
po przyjęciu famotydyny i nizatydyny), nudności, za- ciwdepresyjnych.
wroty głowy, biegunki lub zaparcia, zaburzenia sma- Hamowanie enzymów mikrosomalnych przez inne
ku (cymetydyna), bóle stawów i mięśni, przemijające blokery H2 jest o wiele słabsze i mniej istotne.
zwiększenie stężenia transaminaz w osoczu.
Wlewy dużych dawek leku stosowane w leczeniu
lub w profilaktyce wrzodu stresowego, powodujące
MUTSCHLER-2009.indd 676 2010-01-07 22:14:11
Układ trawienny 677
6.2.2.3. Leki neutralizujące Tabela B 6.2-3. Leki neutralizujące (wybór)
Preparat zawiera:
Leki neutralizujące są substancjami, które neutra- (nazwa handlowa)
lizują lub wiążą żołądkowy kwas solny. Wskazane
są w nadkwaśności i jej następstwach, np. zgadze Gaviscon kwas alginowy,
czy warunkowanych obecnością kwasu dolegliwoś- wodorotlenek glinu
ciach żołądkowych, oraz w szybkim leczeniu symp- Gelusil krzemian magnezowo-
tomatycznym (np. aż do momentu zadziałania inhi- -glinowy
bitorów pompy protonowej) w przypadku choroby Maalox, Maaloxan wodorotlenek glinu,
wrzodowej żołądka i dwunastnicy. wodorotlenek magnezu
Pojedyncza dawka zalecana do skutecznej zmiany
Phosphalugel fosforan glinu
wartości pH w przypadku wrzodu żołądka i dwunast-
nicy odpowiada ilości leku neutralizującego, która Rennie węglan wapnia,
może zobojętnić 20–50 mmol kwasu solnego. W przy- węglan magnezu
padku zgagi i dolegliwości żołądkowych spowodowa- Riopan uwodniony zasadowy siarczan
nych obecnością kwasu zalecana pojedyncza dawka glinowo-magnezowy
(= magaldrat)
neutralizująca wynosi 20–25 mmol. Kwestią dysku-
Solugastril tlenek glinu,
syjną jest zdolność leków neutralizujących nie tylko węglan wapnia
do działania buforującego, ale także wiązania kwasów
Talcid uwodniony zasadowy węglan
żółciowych, stymulowania syntezy prostaglandyn oraz glinowo-magnezowy
pełnienia funkcji miejscowej substancji ochronnej. (= hydrotalyid)
W procesie leczenia choroby wrzodowej żołądka Trigastril tlenek glinu,
i dwunastnicy, a także choroby refluksowej przełyku wodorowęglan magnezu,
węglan wapnia
leki neutralizujące mają mniejsze znaczenie niż inhi-
bitory pompy protonowej lub antagoniści receptora
H2 (zob. poniżej). Z tego powodu stosowane są wy-
łącznie w celu natychmiastowego złagodzenia dole-
gliwości bólowych, aż do momentu, jak już wspo- Jony magnezu działają osmotycznie, z czego wy-
mniano, zadziałania inhibitorów pompy protonowej. nikają ich właściwości przeczyszczające.
Skutecznymi i dobrze znoszonymi lekami neutralizu- Wodorotlenek glinu neutralizuje kwas solny (tworzy
jącymi są: chlorek glinu) oraz częściowo go adsorbuje. Z ogól-
Układ trawienny
nej ilości tworzących się jonów glinu resorbowany
■ związki magnezu,
jest 1%, jej zwiększenie powodują kwaśne napoje.
■ związki glinu W związku z tworzeniem się w dwunastnicy nie-
rozpuszczalnych fosforanów glinu pacjentom z nie-
(lub ich mieszanina), np. tlenek magnezu, trójkrze- wydolnością nerek podaje się wodorotlenek glinu
mian magnezowy, wodzian magnezowo-glinowy w celu obniżenia podwyższonego stężenia fosfora-
(zob. tab. B 6.2-3). nów we krwi. U pacjentów dializowanych długo-
Leki neutralizujące należy przyjmować 1–3 godz. trwałe podawanie może doprowadzić do encefalo- B6
po posiłku oraz przed udaniem się na spoczynek lub patii wynikającej z nagromadzenia jonów magnezu
w razie potrzeby. Ze względu na właściwości absorp- w mózgu. W celu zmniejszenia jelitowego wchłania-
cyjne leki z tej grupy nie powinny być stosowane nia fosforanów zaleca się włączenie soli wapnia lub
z innymi lekami. seweramelu.
U pacjentów z prawidłową czynnością nerek dłu-
Tlenek i wodorotlenek magnezu są trudno rozpusz- gotrwałe podawanie wodorotlenku glinu stwarza
czalne w wodzie i reagują powoli z kwasem solnym niebezpieczeństwo powstania niedoboru fosforanów.
w żołądku, tworząc chlorek magnezu. Z tego związku Brak tych związków prowadzi do nadczynności gru-
w jelicie cienkim tworzą się fosforany i węglany, któ- czołów przytarczycznych, a ta do osteoporozy.
re są resorbowane w 10% i wydalane przez nerki. Ze względu na wydłużony czas przechodzenia
W przypadku zaburzeń funkcji nerek długotrwałe przez jelita wodorotlenek glinu może powodować
podawanie tych związków magnezu może doprowa- zaparcia. Przyczyną tego jest najprawdopodobniej
dzić do hipermagnezemii. wiązanie kwasów żółciowych.
MUTSCHLER-2009.indd 677 2010-01-07 22:14:11
678 Układ trawienny
Działanie uwodnionego zasadowego siarczanu gli-
nowo-magnezowego w pewnym stopniu odpowiada OR
działaniu mieszaniny wodorowęglanu magnezu i wo- RO O
dorowęglanu glinu. Związki te, wzajemnie wydłu- RO O OR
żając swój czas działania, wydłużają czas przejścia
OR
przez jelita. Przy długotrwałym podawaniu istnieje O
RO OR
możliwość tworzenia się krzemianowych kamieni
nerkowych. OR
W funkcji leku neutralizującego często stosowany jest nadal R = SO 3[A l2(OH) 5] n H2O
węglan wapnia. Podobnie jak wodorowęglan sodu (zob.
poniżej), reaguje on wprawdzie w żołądku z kwasem sol-
nym i tworzy ditlenek węgla, ale ilość uwalnianego CO2
jest stosunkowo niewielka i w związku z tym nieszkodli- sukralfat
wa. W jelicie cienkim powstają węglany i fosforany, ilość
resorbowanego tutaj wapnia warunkowana jest wieloma
czynnikami i przeważnie wynosi 10–35%.
Działaniami niepożądanymi towarzyszącymi długotrwa-
łemu stosowaniu węglanu wapnia mogą być hiperkalcemia funkcja nerek. Długotrwała resorpcja jonów glinu
i odkładanie soli wapniowych w różnych tkankach, przede Al3+ może bowiem spowodować encefalopatię, a jed-
wszystkim w nerkach (nefrokalcynoza). Spory budzi wciąż noczesne stosowanie tetracyklin może spowodować
wpływ jonów wapnia (acid rebound) na wzmożone wy- zmniejszenie ich resorpcji.
dzielanie soku żołądkowego. Węglan wapnia ma również
lekkie działanie obstrukcyjne.
Związkiem stosowanym coraz rzadziej jest wodorowę-
glan sodu. W trakcie neutralizacji kwasu solnego wodo- 6.2.2.5. Pochodne prostaglandyny E
rowęglanem w bardzo krótkim czasie uwalnia się bowiem
duża ilość ditlenku węgla, co powoduje niepożądane wzdę- Mizoprostol jest jedyną pochodną prostaglandyny E do-
cia. Ponadto przy doustnym przyjmowaniu wodorowęgla- stępną na rynku farmaceutycznym i stosowaną w leczeniu
nu sodu następuje prawie całkowita resorpcja jonów sodu, choroby wrzodowej. Działanie tego związku polega na ak-
w związku z czym w organizmie zaczyna przeważać od- tywizowaniu czynników osłonowych oraz oddziaływaniu
czyn zasadowy. poprzez receptory typu EP3 na komórki okładzinowe i ha-
mowanie w ten sposób sekrecji kwasu solnego (zob. ryc.
B 6.1-3).
Leki neutralizujące z wielowartościowymi kationami
mogą zmniejszać resorpcję soli kuchennej, tetracy-
Po podaniu doustnym mizoprostol jest szybko resor-
klin i inhibitorów gyrazy poprzez adsorpcję i tworze-
bowany i biotransformowany do aktywnego kwasu
nie kompleksów.
mizoprostolanowego. Okres półtrwania w osoczu
wynosi 20–40 minut. Wydalany jest głównie w for-
mie metabolitów za pośrednictwem nerek.
6.2.2.4. Sukralfat Ze względu na poważne działanie niepożądane
mizoprostol może być podawany tylko w leczeniu
Sukralfat, zasadowa sól glinowa sulfonowanej sacha- towarzyszącym choroby wrzodowej, jednocześnie
rozy, tworzy na powierzchni wrzodu kompleksowe z niesteroidowymi lekami przeciwzapalnymi. Roz-
połączenia z zasadowymi białkami i w ten sposób wiązaniem jest wprowadzenie leku będącego połą-
uniemożliwia wnikanie do błony śluzowej czynników czeniem mizoprostolu i diklofenaku.
agresywnych, takich jak kwas solny, pepsyna i żółć. Dawka zalecana w terapii choroby wrzodowej wy-
Ponadto sukralfat i uwolnione jony glinu wzmagają nosi 800 μm, a w profilaktyce choroby wrzodowej
syntezę prostaglandyn. Po podaniu sukralfatu szyb- 400–600 μm w ciągu dnia.
ciej zostaje wyleczona zarówno choroba wrzodowa Oprócz biegunek (20–40%) i bólu brzucha (do
żołądka, jak i choroba wrzodowa dwunastnicy. Sku- 20%) pacjenci przyjmujący mizoprostol zgłaszają
teczność sukralfatu jest jednak niższa niż skuteczność działania niepożądane w formie mdłości, bólu głowy
inhibitorów pompy protonowej czy antagonistów re- i odrętwienia. Rzadko dochodzi do zaburzenia men-
ceptora histaminowego H2. struacji.
Sukralfat dawkuje się 4 razy dziennie 1 g. Mizoprostol jest przeciwwskazany u kobiet w ciąży
Działaniem niepożądanym towarzyszącym przyj- ze względu na możliwość wystąpienia skurczu maci-
mowaniu sukralfatu są zaparcia. Przeciwwskazaniem cy i związane z tym niebezpieczeństwo poronienia.
do leczenia sukralfatem jest mocno ograniczona Nie może być podawany także w zapaleniu jelit.
MUTSCHLER-2009.indd 678 2010-01-07 22:14:12
Układ trawienny 679
6.2.2.6. Leki cholinolityczne u 10–20% pacjentów. Dlatego związki bizmutu są stosowa-
ne wyłącznie w połączeniu z innymi substancjami czynny-
(parasympatykolityki) mi jako schemat rezerwowy leczenia.
Ostateczna wartość związków bizmutu w leczeniu cho-
Leki cholinolityczne poprzez kompetycyjne blokowanie roby wrzodowej jest niewyjaśniona, szczególnie ze wzglę-
receptora muskarynowego hamują sekrecję kwasu solnego du na brak danych farmakokinetycznych i danych dotyczą-
i pepsynogenu. Identyczny efekt można wprawdzie osiąg- cych potencjalnego zagrożenia. Wydalanie bizmutu z orga-
nąć za pomocą klasycznych cholinolityków, np. atropiny, nizmu odbywa się jeszcze przez kilka miesięcy po zakoń-
ale dopiero po dawkach wywołujących silne działania nie- czeniu leczenia. Ponadto dawniejsze badania wykazywały
pożądane (suchość w ustach, zaburzenia akomodacji, czę- występowanie encefalopatii i osteodystrofii jako skutków
stoskurcz i in.). leczenia długotrwałego. Najskuteczniejsze okazuje się le-
czenie opierające się na ścisłym przestrzeganiu dawek leku
Pirenzepina ma większe powinowactwo do receptorów i trwające 4–8 tygodni, z następującą bezpośrednio po nim
muskarynowych zwojowych (receptorów M1) niż do za- przynajmniej 12-tygodniową przerwą.
zwojowych (receptory M2 i M3), dlatego zmniejsza wydzie- Azotan bizmutu stosuje się 2 razy dziennie po 400 mg.
lanie soku żołądkowego bardziej selektywnie niż atropina. Działaniem niepożądanym są nudności i wymioty. Po-
Podana doustnie pirenzepina wchłania się w 30%, nadto w czasie pasażu żołądkowo-jelitowego ulega prze-
wydalana jest natomiast przede wszystkim w postaci nie- kształceniu w siarczan bizmutu barwiący kał na kolor czar-
zmienionej z moczem i kałem (okres półtrwania w osoczu ny.
wynosi około 10 godz.). Przyjmowana powinna być 2 razy Niewydolność nerek, ciąża i okres laktacji są przeciw-
dziennie po 50 mg. wskazaniami do przyjmowania związków bizmutu.
Działania niepożądane pirenzepiny, choć słabsze, przy- Związki bizmutu hamują wchłanianie tetracykliny.
pominają zasadniczo te, które ujawniają inne leki cholinoli-
tyczne. Tolerancja i skuteczność tej substancji jest jednak o
wiele mniejsza niż innych inhibitorów pompy protonowej
i antagonistów receptora histaminowego H2, w związku 6.2.2.8. Zapobieganie nawrotom.
z czym jest rzadko włączana w proces leczenia farmakolo- Leczenie powodujące eradykację
gicznego choroby wrzodowej.
Przeważająca część pacjentów po wyleczeniu wrzodu
dwunastnicy szybko odczuwa dolegliwości wynika-
jące z nawrotu choroby – u niektórych wznowa nastę-
puje w ciągu 12 miesięcy. Do niedawna zmniejszenie
ilości przypadków odnawiania się wrzodu osiągano
O poprzez profilaktyczne podawanie antagonistów re-
HN
ceptora histaminowego H2 (zob. powyżej) w połą-
czeniu z leczeniem czynnego wrzodu. Profilaktycz-
N ne działanie blokerów H2 nie trwa jednak dłużej niż
Układ trawienny
N
O N N CH3 okres jego stosowania.
Istotny spadek liczby nawrotów ( o < 20%) uzy-
skuje się poprzez eradykację, czyli całkowitą elimi-
pirenzepina nację Helicobacter pylori (HP). U pacjentów z tej
grupy stwierdzono także normalizację zwiększonego
wydzielania gastryny i pepsynogenu.
O eradykacji można mówić wtedy, gdy w wyniku biopsji
B6
błony śluzowej żołądka wykonanej po 4 tygodniach (lub
później) od momentu zakończenia leczenia nie stwierdzi się
obecności bakterii. W przypadku supresji brak jest wpraw-
6.2.2.7. Związki bizmutu dzie Helicobacter pylori zarówno w trakcie leczenia, jak
i bezpośrednio po jego zakończeniu, jednak w ciągu kilku
tygodni szczep ponownie zasiedla błonę śluzową.
Związki bizmutu były do niedawna powszechnie stosowa-
ne w tzw. lekach na żołądek, jednak ze względu na nie-
bezpieczeństwo zatrucia bizmutem znacznie ograniczono Pomimo wrażliwości Helicobacter pylori in vitro
ich stosowanie. Związki bizmutu leczą wrzody trawienne na działanie wielu leków przeciwzakażeniowych erady-
równie szybko jak antagoniści receptora histaminowego kacja tego szczepu z błony śluzowej żołądka jest trud-
H2. Sole bizmutu, podobnie jak sukralfat, działają osło- na. Terapia trójskładnikowa, trwająca 7 dni i składająca
nowo na błonę śluzową żołądka i wiążą kwasy żółciowe.
Ponadto niszczą wprawdzie Helicobacter pylori, jednak się z jednego inhibitora pompy protonowej oraz dwóch
supresja bakterii osiągana w leczeniu jednoskładnikowym leków przeciwzakażeniowych, uważana jest za terapię
jest bardzo krótkotrwała, a eradykacja udawała się jedynie z wyboru (zob. tab. B 6.2-4). Uzyskana dzięki niej era-
MUTSCHLER-2009.indd 679 2010-01-07 22:14:12
680 Układ trawienny
Tabela B 6.2-4. Plan leczenia eradykacyjnego Helicobacter pylori
Dawka dzienna (mg) Czas trwania leczenia
Francuska terapia trójskładnikowa
inhibitory pompy protonowej 2 x 1 dawka standardowa* 7 dni
+
klarytromycyna 2 x 500 7 dni
+
amoksycylina 2 x 1000 7 dni
Włoska terapia trójskładnikowa
inhibitory pompy protonowej 2 x 1 dawka standardowa* 7 dni
+
klarytromycyna 2 x 250-500 7 dni
+
metronidazol 2 x 400 7 dni
Rezerwowy plan leczenia, terapia czteroskładnikowa
inhibitory pompy protonowej 2 x 1 dawka standardowa* 1-10 dni
+
sole bizmutu 4 x tgl. 4-10 dni
+
tetracyklina 4 x 500 4-10 dni
+
metronidazol 3 x 400 4-10 dni
* Dawki standardowe: omeprazol 20 mg, lansoprazol 30 mg, pantoprazol 40 mg
dykacja jest większa niż 90%. Połączenie inhibitora terapię czteroskładnikową (zob. tab. B 6.2-4). Wyniki
pompy protonowej z klarytromycyną i amoksycyliną leczenia eradykacyjnego ocenia się po 6 tygodniach
(tzw. francuska terapia trójskładnikowa) może jednak od momentu zakończeniu postępowania leczniczego
wywołać reakcję alergiczną. Natomiast w przypadku za pomocą testu 13C-mocznikowo-oddechowego albo
zestawienia inhibitora pompy protonowej z klarytro- gastroskopowo oraz na podstawie badań na obecność
mycyną i metronidazolem (tzw. włoska terapia trój- Helicobacter pylori.
składnikowa) może wystąpić oporność na metronida- Szczepionka przeciw Helicobacter pylori jest
zol. Jako terapię rezerwową można zastosować tzw. w trakcie badań.
MUTSCHLER-2009.indd 680 2010-01-07 22:14:12
Układ trawienny 681
6.3. Zapalenie żołądka i jego leczenie
Zapaleniem żołądka określa się proces zapalny błony pełności i brakiem apetytu. U części pacjentów do-
śluzowej żołądka przebiegający bez wrzodów, prze- legliwości te ustępują po podaniu leków zobojętnia-
wlekłe lub ostro. jących.
Wyleczenie ostrego zapalenia żołądka, wywołane-
go stresem, spożywaniem zbyt dużej ilości alkoholu, Zapalenie żołądka typu B (stosunkowo częste) wy-
przyjmowaniem niesteroidowych leków przeciwza- woływane jest przez infekcje bakteryjne, najczęściej
palnych lub infekcjami następuje w ciągu kilku dni przez Helicobacter pylori. W przypadku silnych
od momentu ustania wpływu szkodliwych czynni- objawów i potwierdzonej badaniami obecności He-
ków. licobacter pylori konieczne jest przeprowadzenie
Przewlekłe zapalenia żołądka dzieli się w zależno- eradykcji szczepu. Zapaleniu żołądka typu B często
ści od etiologii na trzy rodzaje; dobór metody lecze- towarzyszy hipochlorhydria.
nia uzależniony jest od rodzaju zapalenia żołądka.
Zapalenie żołądka typu C (stosunkowo rzadkie)
Zapalenie żołądka typu A (stosunkowo rzadkie) ma podłoże chemiczno-toksyczne, jego przyczyną
współwystępuje często z chorobami autoimmu- jest najczęściej długotrwałe przyjmowanie nieste-
nologicznymi. Dochodzi bowiem do wytworzenia roidowych leków przeciwzapalnych lub zarzucanie
przeciwciał przeciw komórkom okładzinowym, żółci. W przypadku gdy zmiana leku powodującego
H+/K+-ATP-azie lub czynnikowi wewnątrzpochod- zapalenie nie jest możliwa, należy podać lek z grupy
nemu (niezbędny w procesie wchłaniania witaminy inhibitorów pompy protonowej lub antagonistów re-
B12). Na skutek atrofii komórek okładzinowych mogą ceptora histaminowego H2. Prokinetyki oraz substan-
wystąpić bezkwaśność (achlorhydria) oraz zaburze- cje wiążące żółć, np. leki neutralizujące, są wskazane
nia żołądkowo-jelitowe z towarzyszącym uczuciem w leczeniu zapaleń wywołanych zarzucaniem żółci.
Układ trawienny
B6
MUTSCHLER-2009.indd 681 2010-01-07 22:14:12
682 Układ trawienny
6.4. Przewlekłe zapalne choroby jelit
6.4.1 Podstawy patofizjologiczne społeczeństwa. Etiologia i patogeneza obu chorób
są nieznane. Przyjmuje się, że czynniki genetyczne
Pojęcie przewlekłe zapalne choroby jelit obejmuje dwie i środowiskowe mogą przyczynić się do patologicz-
choroby: colitis ulcerosa i chorobę Crohna. W ostat- nej aktywności systemu immunologicznego (zob. ryc.
nich latach znacznie wzrosła częstość występowania B 6.4-1). Rolę dyspozycji genetycznych udowodnio-
obu chorób. Szacuje się, że cierpi na nie 0,01–0,1% no jednak tylko w przypadku choroby Crohna. U bli-
choroba Crohna (MC) colitis ulcerosa (CU)
- predyspozycje genetyczne - czynniki środowiskowe
(np. NOD2) (np. dbałość o higienę)
- autoodporność - antygen bakteryjny
nabłonek światło
antygen
makrofag
CD 4
CD4+-limfocyt T
IL-1 MHC-II TCR
aktywowany komórka prezentująca
IL-8 makrofag TNF-α antygen
aktywowany limfocyt T
IL-12
blaszka właściwa
komórka Th1 komórka Th2
(choroba Crohna) (MC) (colitis ulcerosa) (CU)
IFN-γ, IL-2, IL-10,
TNF-α IL-4,
IL-5
naczynie krwionośne
monocyt monocyt limfocyt
Ryc. B 6.4-1. Schemat patofizjologicznej aktywacji systemu immunologicznego błony śluzowej przy przewlekłym zapaleniu jelita
grubego. Zob. tekst. TCRT – receptor komórkowy, IL – interleukina, TNF-α – czynnik martwicy nowotworu, INF-γ – interferon γ.
MUTSCHLER-2009.indd 682 2010-01-07 22:14:12
Układ trawienny 683
sko 50% pacjentów stwierdzono bowiem mutacje w formie nadżerek, martwicy i owrzodzeń (zob. ryc.
w obrębie genu NOD2 (nucleotide-binding oligome- B 6.4-1).
rization domain-2-)/CARD15- (caspase recruitment Colitis ulcerosa jest z klinicznego punktu widzenia
domain family, member 15-), który warunkuje po- przewlekłym nawracającym zapaleniem powierzch-
wstanie wewnątrzkomórkowego sensora, w normal- niowych warstw błony śluzowej okrężnicy. Zaczyna
nych warunkach odpowiedzialnego za unieszkodli- się z reguły w okolicy odbytnicy, skąd stopniowo
wianie bakterii. Zarówno wspomniane mutacje, jak rozprzestrzenia się na okoliczne odcinki okrężnicy.
i inne jeszcze nieznane czynniki mogą prowadzić W obrazie endoskopowym widoczne są owrzodzenia
do częściowej utraty tolerancji na jelitową florę bak- o nieostrych brzegach i widocznych zwłóknieniach
teryjną. Antygeny bakteryjne przenikają wówczas ba- oraz zanik prawidłowych naczyń krwionośnych. Hi-
rierę jelitową i są prezentowane w blaszce właściwej stologicznie można natomiast stwierdzić obecność
przez makrofagi prezentujące antygen komórkom tzw. zasklepek (nagromadzeń granulocytów w za-
CD4+ limfocytów T. W przypadku colitis ulcerosa głębieniach). Pacjenci skarżą się przede wszystkim
skutkuje to przede wszystkim proliferacją komórek na krwawo-śluzowe biegunki z towarzyszącymi im
pomocniczych Th2 oraz następującym po niej tworze- kurczowymi bólami brzucha. Dolegliwości postępują
niem interleukin (IL-4, IL-5, IL-10 i in.). Dla choroby skokowo lub stopniowo. Możliwe są zarówno gwał-
Crohna charakterystyczna jest natomiast aktywacja towne pogorszenia, jak i okresy remisji.
komórek pomocniczych Th1, które produkują INF-γ, Choroba Crohna jest również zapaleniem przewle-
IL-2 oraz TNF-α. Makrofagi uaktywnione przez te kłym, obejmującym jednak cały przewód pokarmo-
cytokiny uwalniają kolejne cytokiny, takie jak IL-1, wy (od jamy ustnej do otworu odbytowego). Główne
IL-8, IL-12 i TNF-α, powodujące migrację komórek zmiany chorobowe umiejscowione są przede wszyst-
zapalnych do naczyń krwionośnych. Ta złożona re- kim w dolnym odcinku jelita cienkiego i/lub w jelicie
akcja zapalna niszczy jelitowe komórki nabłonkowe, grubym. Zmiany patologiczne obejmują wszystkie
co pociąga za sobą miejscowe uszkodzenia tkanek warstwy ścian jelita (zapalenie przyścienne), rów-
rozległe
colitis ulcerosa
aktywność od nieznacznej do średniej duża aktywność bądź szybki przebieg
Układ trawienny
standardowo: aminosalicylan glukokortykosteroidy (doustnie lub pozajelitowo),
(doustnie) 3-4 g/dzień ekwiwalent prednisolonu 1 mg/kg masy ciała
lub dodatkowo aminosalicylan 3-4 g/dzień
w przypadku braku wrażliwości na steroidy B6
cyklosporyna 4 mg/kg masy ciała dożylnie
(ponad 24 godz.) dziennie
lub
takrolimus 0,1 mg/kg masy ciała doustnie
lub 0,01 mg/kg masy ciała dożylnie
w przypadku braku postępów
w leczeniu wskazanie
do zabiegu operacyjnego
azatiopryna 2-2,5 mg/kg masy ciała dziennie
przewlekły aktywny przebieg lub
6-merkaptopuryna 1-1,5 mg/kg masy ciała dziennie
Ryc. B 6.4-2. Indukcja remisji w colitis ulcerosa (według Siegmunda i Zeitza).
MUTSCHLER-2009.indd 683 2010-01-07 22:14:13
684 Układ trawienny
nież te głębokie. Pojawiają się m.in. nacieki limfo-
cytów i komórek plazmatycznych oraz ziarniniaki
żołądek
nabłonkowe i owrzodzenia. Często dochodzi również
do wytworzenia przetok. jelito czcze
jelito kręte
mesalazy
na powle okrężnica
kana ety
oporna locelulozą
6.4.2. Leczenie colitis ulcerosa na działa
nie soku
żołądko
wego po
wlekana
mesalaz
Wybór sposobu leczenia colitis ulcerosa zależy yna
od stopnia zaawansowania i rozległości choroby. Gdy olsala
zyna
stan zapalny obejmuje tylko odbytnicę, wskazane sulfas
jest leczenie miejscowe za pomocą czopków, wlewek alazy
na
lub preparatów piankowych. Jeżeli zajęte są także
dalsze odcinki jelita grubego, konieczne jest syste-
matyczne leczenie.
W ostrych atakach choroby, czyli wykazujących
tendencję do remisji, lekiem pierwszego wyboru
Modyfikacja Miejsce Rodzaj
są aminosalicylany zawierające kwas 5-aminosa- uwalniania uwalniania
licylowy jako składnik czynny (zob. ryc. B 6.4-2).
Pozytywnych efektów leczenia można jednak ocze- 5-ASA (mesalazyna) początkowy pH niezależne
powlekana odcinek jelita
kiwać dopiero po 2–4 tygodniach. Kwas 5-amino- etylocelulozą cienkiego
salicylowy dostępny jest w postaci formuł galeno-
5-ASA (mesalazyna) dystalny odcinek PH niezależne
wych, co pozwala na terapię dostosowaną do loka- oporna na działanie soku jelita cienkiego w jelicie cienkim
lizacji choroby (zob. ryc. B 6.4-3). Aby zapobiec żołądkowego powlekana
wchłanianiu już w początkowych odcinkach jelita 5-ASA-5-ASA początkowy bakteryjne oddziele-
cienkiego, 5-ASA podawany jest w formie powleka- (olsalazyna) odcinek okrężnicy nie w okrężnicy
nych tabletek odpornych na działanie kwasu solnego
5-ASA-sulfadryna początkowy bakteryjne oddziele-
w żołądku, w formie czopków, wlewek doodbytni- (sulfalazyna) odcinek okrężnicy nie w okrężnicy
czych lub pianek, a także kowalencyjnie powiązany
z innym związkiem (np. olsalazyna, sulfasalazyna, Ryc. B 6.4-3. Miejsca uwalniania różnych formuł galenowych
zob. poniżej). przez aminosalicylany.
Mechanizm działania preparatów zawierających
5-ASA jest złożony. Za istotną przyjmuje się stymu-
lację podtypu γ receptora aktywowanego prolifera- Dawkowanie w leczeniu ostrych rzutów dla mesa-
tora peroksysomu (PPARγ), przyczyniającego się lazyny i olsalazyny wynosi 3–4 g, a dla sulfasalazyny
również do integralności bariery jelitowej. Ponadto 4–6 g dziennie.
przez hamowanie czynnika transkrypcyjnego NF-kB Działaniami niepożądanymi mesalazyny i olsala-
powinna zostać zablokowana produkcja cytokin, zyny są biegunka, bóle i zawroty głowy, zwiększone
a przez inhibowanie 5-lipooksygenazy – synteza stężenie transaminaz, methemoglobinemia, reakcje
leukotrienów. Także opisane zahamowanie produk- nadwrażliwości. Sulfasalazyna wywołuje dodatkowo
cji prostaglandyn wydaje się mieć znaczenie drugo- działania niepożądane, które można wyjaśnić obec-
rzędne, ponieważ potencjalne blokery syntezy pro- nością składnika sulfamidowego. Powoduje ponadto
staglandyn, np. niesteroidowe leki przeciwzapalne, oligospermię i agranulocytozę (rzadko). Mesalazyna
oddziałują negatywnie na przewlekłe choroby za- jest lekiem pierwszego wyboru.
palne jelit. Przeciwwskazania oraz interakcje odpowiadają
Sulfasalazyna i olsalazyna wchłaniają się powoli, tym wywołanym przez salicylany bądź sulfasalacznę
w związku z czym podane doustnie docierają do jelita oraz tym charakterystycznym dla sulfonamidów.
grubego (zob. ryc. B 6.4-3). W jelicie grubym nastę-
puje redukcja grupy azowej przez bakterie E. coli. W przypadku bardzo zaawansowanego bądź przebie-
Z sulfasalazyny powstają w związku z tym mesala- gającego piorunująco colitis ulcerosa stosuje się do-
zyna i sulfapirydyna, a z olsalazyny dwie cząsteczki datkowo oprócz 5-ASA także glukokortykosteroidy
mesalazyny (zob. ryc. B 6.4-4). Produkty rozpadu – ekwiwalenty prednizolonu. Podawanie leku (doust-
są (częściowo) wchłaniane w jelicie grubym. nie lub pozajelitowo) należy rozpocząć od dawki nie-
MUTSCHLER-2009.indd 684 2010-01-07 22:14:13
Układ trawienny 685
przekraczającej w ciągu doby 1mg/kg masy ciała (do-
ustnie lub doodbytniczo) i kontynuować je aż do uzy- nieskomplikowany remisja po skomplikowanym
skania remisji (zob. ryc. B 6.4-2). Wycofywanie leku przebieg przebiegu, pełnym rzucie
colitis ulcreosa i indukowana cyklosporyną
należy przeprowadzać stopniowo przez 2 miesiące. lub
Jeżeli proces zapalny zlokalizowany jest w dystalnym takrolimusem
odcinku okrężnicy, pojawiające się ogólnoustrojowe
działania niepożądane mogą być leczone miejsco-
wo. Dobre efekty można osiągnąć dzięki preparatom
piankowym, ponieważ wchodzące w ich skład sub- aminosalicylan (doustnie) azatiopryna 2-2,5 mg/kg
stancje pomocnicze utrudniają wchłanianie (np. pian- 1-2 g/dzień; dziennie
przy zajęciu lub
ka doodbytnicza Colifoam zawiera octan hydrokor- 6-merkaptopuryna
dystalnego odcinka jelita
tyzonu). Skuteczne są również glukokortykosteroidy 0,5-1,5 g/dzień 1-1,5 mg/kg dziennie
domiejscowe z podwyższonym efektem pierwszego w postaci czopków
przejścia, np. budezonid, który w 90% ulega mecha- lub
1-4 g/dzień w formie wlewów
nizmowi efektu pierwszego przejścia i jest podawany
albo miejscowo w postaci wlewek doodbytniczych,
albo doustnie w kapsułkach powlekanych opornych w przypadku braku tolerancji
na działanie soku żołądkowego. Metabolity cechuje
niewielkie powinowactwo do receptorów steroido-
wych. E. coli Nissle
(doustnie 200 mg/dzień
W przypadku chorych niewrażliwych na leki ste-
roidowe możliwe jest zastosowanie cyklosporyny
Ryc. B 6.4-5. Utrzymanie remisji w przypadku colitis ulcreosa
(dziennie 4 mg/kg masy ciała podane dożylnie na 24
(według Siegmunda i Zeitza).
godz.) alternatywnie z takrolimusem (0,1 mg/kg masy
ciała doustnie lub 0,01 mg/kg masy ciała dożylnie).
Ze względu na ograniczony zasięg terapeutyczny choroba jest przewlekle aktywna, azatiopryna (2–2,5
cyklosporyny wskazana jest kontrola jej działania mg/kg masy ciała dziennie) lub 6-merkaptopuryna
i w razie potrzeby zmiana dawki. Po uzyskaniu re- (1–1,5 mg/kg masy ciała dziennie) są lekami z wy-
akcji należy rozpocząć leczenie immunosupresyjne boru. Podawanie azatiopryny lub 6-merkaptopuryny
azatiopryną (2–2,5 mg/kg masy ciała dziennie) lub 6- wymaga sprawdzania morfologii krwi, γ-GT oraz po-
merkaptopuryną (1–1,5 mg/kg masy ciała dziennie) ziomu enzymów trzustki.
i kontynuować je aż do momentu remisji (zob. ryc. B Jeżeli w wyniku leczenia farmakologicznego
6.4-5). Również w przypadku pacjentów, u których nie uzyska się wyraźnej poprawy stanu pacjenta, na-
Układ trawienny
O O OH
S
NH N
HOOC N
HOOC N N COOH
N
HO B6
HO salazosulfapirydyna osalazyna
HOOC NH2 O O HOOC NH2 OH
S
NH N
HO HO H2N COOH
H2N
kwas 5-aminosalicylowy sulfapirydyna kwas 5-aminosalicylowy kwas 6-aminosalicylowy
(kwas 5-amino-2-hydroksybenzoesowy) = mesalazyna
Ryc. B 6.4-4. Biotransformacja salazosulfapirydyny (sulfasalazyny) do kwasu 5-aminosalicylowego i sulfapirydyny oraz olsalazyny do
dwóch cząsteczek kwasu 5-aminosalicylowego pod wpływem flory bakteryjnej.
MUTSCHLER-2009.indd 685 2010-01-07 22:14:13
686 Układ trawienny
leży rozważyć przeprowadzenie zabiegu operacyjne- Po zakończeniu leczenia colitis ulcerosa o cięż-
go (zob. ryc. B 6.4-2). kim przebiegu konieczne jest ponaddwuletnie lecze-
nie podtrzymujące remisję. Zalecane jest podawanie
Przebiegające bez powikłań i zakończone remisją w tym czasie azatiopryny w dawce 2–2,5 mg/kg masy
colitis ulcerosa wymaga długoterminowgo (ponad ciała dziennie (zob. ryc. B 6.4-5).
2 lata) leczenia podtrzymującego remisję za pomocą
preparatów 5-ASA (np. mesalazyna 1–2 g/dzień do-
ustnie; przy zajęciu odcinka dystalnego także miej- 6.4.3. Leczenie choroby Crohna
scowo). W ten sposób redukuje się również ryzyko
rozwoju chorób nowotworowych okrężnicy. W przy-
Proces leczenia choroby Crohna oraz colitis ulcero-
padku nietolerancji 5-ASA sprawdzają się w tej roli
sa ma bardzo podobny charakter (zob. ryc. B 6.4-6).
probiotyki, np. preparaty zawierające probiotyczny
W przypadku ostrych, ale lekkich rzutów stosuje się
szczep E. coli Nissle 1917.
5-ASA (mesalazynę, 3–4 g/dzień); jej skuteczność
lekki rzut ciężki rzut
miejsce rzutu ew. operacja
(np. w przypadku ropni,
przetok, zwężeń)
odcinek krętniczo-
jelito kręte okrężnica -kątniczy
preparaty 5-ASA budesonid glukokortykosteroid (1 mg/kg masy ciała
(np. mesalazyna (9 mg/dzień) ekwiwalentu prednizolonu)
3-4 g/dzień
odpowiedź na leczenie odpowiedź na terapię
pozytywna negatywna pozytywna negatywna
plus
azatiopryna
terapia (2,5 mg/kg masy ciała dziennie)
podtrzymująca remisję
lub
6-merkaptopuryna
(1,5 mg/kg masy ciała dziennie)
lub
metotreksat
przewlekły (15-25 mg domięśniowo tygodniowo)
aktywny przebieg (lek drugiego wyboru)
lub
infliksimab (5 mg/kg co 8 tygodni)
przy braku tolerancji
Ryc. B 6.4-6. Terapia ostrych rzutów i uzyskanie remisji w chorobie Crohna.
MUTSCHLER-2009.indd 686 2010-01-07 22:14:14
Układ trawienny 687
jest jednak mniejsza niż w colitis ulcerosa. Przy lo- kliniczną, jak i wyleczenie uszkodzeń potwierdzo-
kalizacji choroby Crohna na odcinku krętniczo-kąt- nych endoskopowo. Podstawą do opracowania za-
niczym dobre efekty przynosi podawanie budezoni- sad tej terapii była obserwacja, na której podstawie
du (9 mg/dzień). Pacjenci z ciężkimi rzutami mogą stwierdzono u pacjentów z chorobą Crohna uwal-
otrzymywać początkowo glukokortykosteroidy nianie przez chorobowo zmieniony odcinek jelita
(ekwiwalent prednizolonu w dawce 1 mg/kg masy dużych ilości prozapalnych cytokin TNF-α – głów-
ciała dziennie). Przy występujących już wcześniej nej cytokiny odpowiedzi immunologicznej typu Th1
częstych rzutach zaleca się łączenie glukokortykoste- (zob. ryc. B 6.4-1). Białko wiążące etanercept, które
roidu z azatiopryną (2–2,5 mg/kg masy ciała dziennie) jako rozpuszczalny receptor TNF-α jest równocześ-
lub 6-merkaptopuryną (1–1,5 mg/kg masy ciała dzien- nie antagonistą receptora TNF-α, okazało się niesku-
nie). Bardzo ciężkie rzuty są wskazaniem do podania teczne w leczeniu choroby Crohna. Przypuszcza się,
glukokortykosteroidu w połączeniu z antybiotykiem że infliksimab, wiążąc przyłączony do błony TNF-α,
(mezlocylina/metronidazol pozajelitowo lub cypro- w przeciwieństwie do etanerceptu prowadzi do roz-
floksacyna/metronidazol doustnie). Ich skuteczności padu podnabłonkowych komórek zapalnych.
nie potwierdzono jednak w wystarczającym stopniu. Utrzymanie remisji u pacjentów z chorobą Croh-
W przewlekłej aktywnej chorobie Crohna wska- na jest dosyć trudne. Niekiedy udaje się to osiągnąć
zane jest długotrwałe podawanie azatiopryny (2,5 w sposób farmakologiczny, podając azatioprynę
mg/kg masy ciała dziennie) lub 6-merkaptopuryny (2–2,5 mg/kg masy ciała dziennie) lub 6-merkapto-
(1,5 mg/kg masy ciała dziennie). Lekiem drugiego purynę (1–1,5 mg/kg masy ciała dziennie). Dokładne
wyboru jest metotreksat (2,5 mg domięśniowo tygo- wskazówki dotyczące pomagającego utrzymać remi-
dniowo). sję dawkowania podaje się również dla metotreksatu
U pacjentów z chorobą Crohna, których organi- (15 mg/tydz. domięśniowo) lub infliksimabu (5–10
zmy nie reagują na działanie 5-ASA, glukokortyko- mg/kg masy ciała co 8 tygodni). Efekt wywoływa-
steroidów i azatiopryny, terapia immunomodulująca ny przez 5-ASA jest bardzo słabo widoczny, dlatego
infliksimabem będącym przeciwciałem chimerycz- związek ten jest zalecany przez niektórych autorów
nym TNF-α może spowodować zarówno poprawę tylko w remisji uzyskanej w sposób chirurgiczny.
Układ trawienny
B6
MUTSCHLER-2009.indd 687 2010-01-07 22:14:14
688 Układ trawienny
6.5. Czynnościowe zespoły jelitowe i ich leczenie
6.5.1. Leczenie zespołu jelita Tegaserodu nie należy stosować w czasie ciąży
drażliwego i karmienia oraz u dzieci.
Zespół jelita drażliwego (colon irritabile, okrężnica
spastyczna) należy do najczęstszych chorób żołądko- 6.5.2. Zaparcie
wo-jelitowych. Kobiety dotyka 2–3 razy częściej niż Zaparciem określa się opóźnione wydalanie suchego i twar-
mężczyzn. Obejmuje zaburzenia czynnościowe i bóle dego stolca. Wynika ono albo z opóźnionego przesuwania
jelita grubego, które nie są spowodowane rozpozna- treści jelitowej (przy zachowaniu prawidłowego mechani-
walną choroba organiczną tej części przewodu pokar- zmu defekacji), albo z zaburzeń odruchu wydalania.
Przyczyną opóźnionego pasażu jelitowego mogą być
mowego. Objawami najczęściej zgłaszanymi przez czynniki dietetyczne (wypełnienie jelita przez składniki po-
chorych są nieregularne stolce (zaparcia lub biegunki), żywienia ubogiego w substancje balastowe), zmiany w ob-
uczucie sytości, wzdęcia i bóle brzucha. Ponieważ rębie ściany jelita (np. guzy, przewlekłe stany zapalne),
ich przyczyny są nieznane, leczenie ukierunkowa- zaburzenia endokrynne (np. nadczynność tarczycy) oraz
ne jest na zwalczanie najważniejszych dolegliwości. czynnościowe i organiczne zaburzenia układu nerwowego
(np. stres, urazy kręgosłupa). Zaparcia mogą powodować
W przypadku gdy są nimi zaparcia, można podać leki także niektóre leki psychotropowe oraz opioidy.
balastowe (zob. poniżej). Kontrowersje budzi sto- Zaburzenia mechanizmu opróżniania towarzyszą scho-
sowanie leków rozkurczających, cholinolitycznych, rzeniom odbytu (np. żylakom odbytu, przetoce odbytu), utra-
przeciwdepresyjnych, antagonistów dopaminy, leków cie odruchu odbytniczego lub osłabieniu tłoczni brzusznej.
wiatropędnych, opioidów czy leków kojących.
Wychodząc z założenia, że serotonina odgrywa Leki przeczyszczające przyspieszają wydalanie
ważną rolę w procesie nocycepcji w trzewiach oraz stolca. Pod wpływem reklam są one jednak stoso-
w regulacji funkcji jelit, postanowiono wprowadzić wane zbyt często i bez wyraźnych wskazań. Tym-
do leczenia zaparciowej postaci zespołu jelita drażli- czasem tylko nieliczni pacjenci z przewlekłymi za-
wego tegaserod, będący częściowym antagonistą re- parciami zmieniają tryb życia i sposób odżywiania
ceptora 5-HT4. Działający prokinetycznie tegaserod zgodnie z zaleceniami lekarza (np. wzbogacają dietę
jest jednak dopuszczony w leczeniu tylko w niektó- w pokarmy bogate w składniki balastowe). Więk-
rych krajach. Może być stosowany u kobiet, ponie- szość woli sięgnąć po leki przeczyszczające. Dla-
waż wyłącznie u nich, jak pokazały wyniki przepro- tego do obrotu należy wprowadzać wyłącznie leki
wadzonych na dużą skalę badań klinicznych, jest zna- cechujące się najmniejszym działaniem niepożąda-
cząco bardziej skuteczny niż placebo (45–55% kobiet nym.
reagowało na placebo, a 60% na tegaserod). Zasadniczo należy odradzać stosowanie leków
Dostępność biologiczna tegaserodu kształtuje się przeczyszczających w celu „oczyszczenia krwi” lub
na poziomie 10%, przy przyjmowaniu go w trakcie odchudzania. Leki przeczyszczające nie działają od-
posiłków spada do 5%. Okres półtrwania w osoczu truwająco ani nie obniżają trwale masy ciała.
wynosi 11 godz.
Dawkowany powinien być 2 razy dziennie po 6 mg Mechanizm działania leków przeczyszczających.
(przed posiłkami) przez maksymalnie 12 tygodni. Działanie większości leków przeczyszczających
Działaniem niepożądanym towarzyszącym przyj- powoduje powiększenie objętości zawartości jelita
mowaniu tegaserodu jest przede wszystkim biegun- i wyzwolenie fali perystaltycznej w wyniku zwięk-
ka. Odnotowano również pojedyncze przypadki cyst szenia ciśnienia wewnętrznego w jelicie uzyskanego
na jajnikach oraz omdleń. przez:
■ pęcznienie w wyniku wchłaniania wody (środki
pęczniejące),
HN NH
■ osmotyczną retencję wodny (środki przeczyszcza-
N (CH2) 4CH3
NH NH jące),
H3CO ■ zahamowanie resorpcji Na+ i wody z jelita i/lub
zwiększenie wydzielania wody do wnętrza jelita
tegaserod (środki przeczyszczające antyresorpcyjne i odwad-
niające).
MUTSCHLER-2009.indd 688 2010-01-07 22:14:14
Układ trawienny 689
w nielicznych przypadkach poważne w skutkach za-
osłabianie burzenia (np. odwodnienie przy niewystarczającej
odruchu opróżniania podaży wody lub niedrożność jelit wywołaną przy-
jęciem środków pęczniejących). Przy długotrwa-
łym stosowaniu leków przeczyszczających bardzo
prz rzeczy
yjm szcza
p
często dochodzi do zaburzeń równowagi elektroli-
owa jących
nka
tycznej, szczególnie do utraty potasu, co również
nie śr
biegu
przyczynia się do zaparć (circulus vitiosus, zob.
odków
ryc. B 6.5-1). Działania takiego nie wykazują środ-
ki pęczniejące.
utrat
ata du
y
6.5.2.1. Środki pęczniejące
utr rata so
wod
pota
ut
a su
Łagodnie działającymi środkami przeczyszczającymi
wzmo
żone wydzielanie są występujące naturalnie lub otrzymywane w sposób
aldosteronu półsyntetyczny pęczniejące, nieulegające strawieniu
polisacharydy. Należą do nich np. substancje zawarte
w siemieniu lnianym, nasionach indyjskiej babki ja-
Ryc. B 6.5-1. Mechanizm błędnego koła tworzący się przy dłu- jowatej, otrębach pszennych oraz bazorynie, będącej
gotrwałym przyjmowaniu środków przeczyszczających. nierozpuszczalną w wodzie mieszaniną polisachary-
dów z naturalnej żywicy tragant.
Zwyczajowo stosuje się kilkanaście gramów (1 ły-
żeczkę do herbaty) 1–3 razy dziennie.
Do tej grupy należą także środki ułatwiające pasaż
Przyjmując preparaty pęczniejące, należy wypi-
mas kałowych:
jać odpowiednią ilość wody w celu niedopuszczenia
■ tzw. środki wygładzające, które umożliwiają uzy- do sklejenia się treści jelitowej i związanej z tym
skanie na ścianach jelita efektu przypominającego niedrożności jelit. Aby zmniejszyć ilość środka pęcz-
naoliwienie, niejącego potrzebną do uzyskania efektu przeczysz-
czającego, łączy się go z innymi środkami przeczysz-
■ leki wywołujące odruch opróżniania.
czającymi.
W skład wielu leków przeczyszczających, np. pochod-
Układ trawienny
Rozpuszczalne w wodzie środki pęczniejące (np. otręby
nych antrachinonu i difenolu (zob. poniżej), wchodzą pszenne, nasiona indyjskiej babki jajowatej) mogą przy-
również substancje zwiększające perystaltykę, wpły- czynić się do obniżenia poziomu cholesterolu w osoczu.
wające na pracę mięśni gładkich. Ich skuteczność jest jednak niższa niż w przypadku in-
nych środków zmniejszających stężenie lipidów. Niejasne
jest również, czy jest to swoisty efekt leczniczy, czy wynik
Wskazania. Jednorazowe lub krótkotrwałe przyjmo- zmiany składu pożywienia.
wanie środków przeczyszczających jest wskazane
w celu opróżnienia jelit przed badaniem radiologicz- B6
nym lub endoskopowym, a także przed zabiegami chi-
rurgicznymi. Leki te stosuje się ponadto w przypadku 6.5.2.2. Środki osmotyczne
bolesnych wypróżnień (np. wskutek przetok odbytu) o działaniu przeczyszczającym
oraz w celach zapobiegawczych lub leczniczych przy
zaparciach wywołanych przewlekłym podawaniem Woda, ponieważ łatwo ulega resorpcji, nie ma dzia-
opioidów (np. w terapii nowotworowej). łania przeczyszczającego. Po dodaniu do niej sub-
W przewlekłych zaparciach niewarunkowanych stancji trudno wchłanialnych, odpowiednio do ciś-
stosowaniem opioidów konieczna jest najpierw zmia- nienia osmotycznego tej substancji w podawanym
na trybu życia i nawyków żywieniowych, a dopiero roztworze, gdy jest on normotoniczny, dochodzi
w przypadku braku pozytywnych rezultatów można do zmniejszenia resorpcji wody z jelita, natomiast
rozważyć podanie środków przeczyszczających. gdy jest hipertoniczny, następuje oddanie wody z roz-
tworu do światła jelita i dzięki temu wydalenie dużej
Działania niepożądane. Krótkotrwałe przyjmo- ilości kału. Czas rozpoczęcia defekacji zależy od ilo-
wanie środków przeczyszczających powoduje tylko ści i stężenia roztworu, wskazane jest jednak unikanie
MUTSCHLER-2009.indd 689 2010-01-07 22:14:15
690 Układ trawienny
OH
OH
O OH OH OH OH
CH2OH CH2OH
HOH2C OH HOH2C HOH2C
HOH2C O O HOH2C O O O HOH2C O O OH
HO OH HO OH HO OH
OH OH OH
laktoza laktuloza laktitol
podawania roztworów hipertonicznych ze względu tyki. Makrogol nie jest ani resorbowany, ani metabo-
na niebezpieczeństwo odwodnienia. lizowany.
Solne środki przeczyszczające. Jest to grupa związ-
ków, do których należą siarczan magnezu i siarczan 6.5.2.3. Leki przeczyszczające hamujące
sodu (sól gorzka i sól glauberska), fosforan sodu oraz resorpcję i nawadniające
cytrynian sodu. Najczęściej stosuje się sól gorzką
i sól glauberską, które przyjmowane są w dawce 10–
Leki z tej grupy hamują wchłanianie jonów sodowych
20 g w roztworze możliwie najbardziej izotonicznym
i wody wskutek blokady ATP-azy zależnej od jonów
w stosunku do płynu tkankowego (MgSO4 · 7 H2O
Na+ i K+ (działanie antyresorpcyjne). Jednocześnie
o stężeniu 3,3%, Na2SO4 · 10 H2O o stężeniu 4,2%).
wspomagają one w różnym stopniu napływ elektro-
Przy dłuższym podawaniu środków przeczysz-
litów i wody do jelita (działanie odwadniające), prze-
czających zawierających jony sodowe może dojść
de wszystkim przez zwiększenie przepuszczalności
do retencji płynów i nadciśnienia. Po podaniu środ-
w obrębie obwódki zamykającej (tight junctions).
ków przeczyszczających zawierających jony magne-
zu w przypadku pacjentów z niewydolnością nerek
Olej rycynowy. Olej rycynowy składa się najczęściej
należy liczyć się z możliwością wydzielania zbyt ma-
z triglicerydów kwasu rycynolowego (kwas 12-hy-
łej ilości magnezu, co może powodować osłabienie
droksylowy). W jelicie cienkim z nieczynnego trigli-
siły skurczów mięśni, osłabienie odruchów i obniże-
cerydu uwalniany jest przez lipazę kwas rycynolowy
nie ciśnienia krwi.
działający w tej części przewodu pokarmowego.
Stosowanie oleju rycynowego jako środka prze-
Alkohole cukrowe i cukry. Osmotycznymi środka-
czyszczającego jest skuteczne i pozbawione działań
mi przeczyszczającymi są również alkohole cukrowe
niepożądanych. Jednak ze względu na niechęć pa-
mannitol, sorbitol, laktitol oraz cukry laktoza i lak-
cjentów do jego zażywania nadaje się bardziej do le-
tuloza.
czenia zaparć ostrych niż przewlekłych.
W jelicie laktuloza, laktoza i laktitol ulegają pro-
Najczęściej zalecana dawka wynosi 1,5–6 g. Okres
cesowi fermentacji wywołanemu przez szczepy bak-
leczenia nie powinien przekroczyć 14 dni ze względu
terii jelitowych, w wyniku czego ulegają rozkładowi
na możliwość utraty wody, potasu i sodu.
do kwasów (kwas octowy, propionowy, masłowy
i mlekowy) pobudzających perystaltykę.
Polietylenoglikol. Środkiem przeczyszczającym COOH
o działaniu osmotycznym jest polietylenoglikol 4000 (CH2) 5CH3
(makrogol 4000). Makrogol w formie proszku wią-
że wodę, z którą jest przyjmowany, i transportuje OH
ją do jelita grubego. Dzięki dostarczonej w ten spo-
kwas rycynolowy
sób wodzie stolec ulega rozmiękczeniu, powiększa
swoją objętość, co powoduje przyspieszenie perystal-
MUTSCHLER-2009.indd 690 2010-01-07 22:14:15
Układ trawienny 691
Glikozydy antrachinowe. Chemicznie spokrewnione Glikozydy antrachinowe uaktywniają się w jelicie
glikozydy hydroksyantrachinowe (oprócz glikozydów cienkim dopiero po rozerwaniu wiązania glikozydo-
hydroksyantronowych i hydroksydiantronowych) za- wego i zredukowaniu ich przez bakterie E. coli do an-
warte w aloesie, korze kruszyny pospolitej, owocach tronów lub antranoli. Z tego też powodu podane do-
szakłaku pospolitego, liściach strączyńca wąskolist- ustnie zaczynają działać dopiero po 8–10 godzinach.
nego, kłączu rzewienia dłoniastego i innych roślinach Większość antrachinonów wydalana jest z ka-
leczniczych znajdują zastosowanie jako środki prze- łem. Tylko niewielka część tych związków usuwana
czyszczające. Wolne od glukozy pochodne antrachi- jest z organizmu z moczem, który pod ich wpływem
nonów nazywane są emodynami. W emodynie aloesu przybiera ciemny kolor.
Dawkowanie zależy od zawartości związku czyn-
nego w ekstrakcie. Sennozydy stosuje się w dawce
OH
20–25 mg.
HO OH
Ostre działania niepożądane występują rzadko.
Kwestią wciąż dyskutowaną dotyczącą glikozydów
HOH2C O O O OH
antrachinowych są właściwości rakotwórcze lub
ułatwiające wzrost guza. Związków tych nie stosuje
się w ciąży ze względu na ich właściwości genotok-
COOH
syczne.
H H
COOH
Syntetyczne leki przeczyszczające – difenole. Sub-
stancjami czynnymi należącymi do tej grupy są:
HOH2C O O O OH ■ bisakodyl,
OH
■ pikosiarczan sodu.
HO
OH
Najstarszą substancją czynną oraz związkiem macie-
sennozyd A rzystym leków z tej grupy jest fenoloftaleina, niesto-
sowana ze względu na poważne działania niepożą-
dane (reakcje nadwrażliwości, krwotoczne zapalenie
jelit i in.).
grupa metylowa przy C-3 ulega utlenieniu do grupy
hydroksymetylowej, a w rzewieniu do grupy karbok-
Bisakodyl. Lek zawierający bisakodyl podany do-
sylowej. Emodyną kruszyny jest 6-hydroksychryzo-
Układ trawienny
ustnie jest częściowo resorbowany w jelicie cienkim
fanol. Naturalne, występujące w liściach strączyńca
po deacetylacji hydrolitycznej w świetle i w błonie
glikozydy hydroksydiantronowe są nazywane senno-
śluzowej jelita. Następnie w wątrobie ulega glu-
zydami. W celach leczniczych stosuje się wyłącznie
kuronizacji, a produkty tej reakcji są wraz z żółcią
wyciągi.
wydzielane do jelita (krążenie wątrobowo-jelitowe).
Ponieważ hydrofilne glukuronidy nie są resorbowane
w jelicie cienkim, przedostają się do jelita grubego,
OH O OH
w którym w wyniku działania drobnoustrojów dzielą B6
się, tworząc postać aktywną – wolny difenol. Dostęp-
ne w aptekach doustne formy bisakodylu posiadają
R
otoczkę odporną na działanie soku żołądkowego,
O
dzięki czemu susbstancja czynna może być uwalnia-
na dopiero w jelicie cienkim i grubym. Według now-
szych badań czynny difenol tworzony jest w sposób
R
enzymatyczny nie tylko w jelicie grubym, ale rów-
CH3 chryzofanol nież na całej długości układu wydalniczego.
Zażycie tabletek przeczyszczających opornych
CH2OH emodyna aloesu na działanie soku żołądkowego przed snem przynosi
COOH rzewień efekt dopiero po upływie około 10 godzin, zażycie
ich rano na pusty żołądek skutkuje już po upływie
około 6 godzin. Po aplikacji doodbytniczej bisakodyl
MUTSCHLER-2009.indd 691 2010-01-07 22:14:16
692 Układ trawienny
działa już po 30–60 minutach, ponieważ omija układ
krążenia jelitowo-wątrobowego.
Pikosiarczan sodu jest analogiem bisakodylu. W pi- C2H5 O
kosiarczanie sodu wspólna dla obydwu związków po- O (CH2) 3CH3
H3C(CH2) 3 O
chodna dihydrodifenylopirydylu ulega jednak estryfi- OO S O C2H5
kacji kwasem siarkowym zamiast kwasem octowym. –
O Na
+
Ponadto w przeciwieństwie do bisakodylu jest roz-
kładany przez bakterie do wolnego difenolu wyłącz- dokuzan sodu
nie w jelicie grubym, stosowany per se jest prawie
nieresorbowany.
Początek działania następuje przeciętnie 10–12
godz. po zażyciu doustnym.
Dawkowanie obydwu substancji jest jednakowe
i wynosi 5–10 mg. czynny rozmiękcza kał i czyni jego powierzchnię
bardziej śliską. Wciąż jednak nie rozstrzygnięto,
czy w niewielkich dawkach zawartych w tych połą-
HO czeniach rzeczywiście powoduje on addycyjny efekt
OH przeczyszczający. Związek jest dobrze tolerowany.
Do tego samego celu służy paraffinum liquidum. Jako olej
mineralny nie jest trawiony i nie ulega resorpcji. Powinien
O być stosowany krótkotrwale. Przy dłuższym stosowaniu
istnieje ryzyko:
O
■ hipowitaminozy witamin rozpuszczalnych w tłuszczach,
fenoloftaleina ponieważ nie są powtórnie wchłaniane,
■ zaburzeń trawienia,
■ reakcji spowodowanej obecnością ciała obcego, tj. kro-
H3C O O CH3
pelki tłuszczu wchłoniętej w obrębie brzucha.
O O
N 6.5.2.5. Substancje wpływające
na odruch defekacji
bisakodyl
Wielowartościowe alkohole, szczególnie glicerol
(gliceryna) i sorbit, oraz osmotyczne środki prze-
O O czyszczające w formie czopków lub mikrolewatyw
O O
S S – +
mogą być stosowane w celu pobudzenia odruchu de-
+ – O Na
Na O O O fekacji. Zaleca się szczególnie niemowlętom i małym
dzieciom.
N
pikosiarczan sodu
6.5.2.6. Antagoniści receptorów
opioidowych
o działaniu peryferyjnym
Alwimopan jest selektywnym antagonistą receptora opio-
idowego μ, który badany jest obecnie pod kątem zastoso-
6.5.2.4. Środki ułatwiające pasaż mas wania w leczeniu zaparć indukowanych opioidami i poope-
kałowych racyjnej niedrożności jelit. W warunkach fizjologicznych
alwimopan jest jonem amfoterycznym, w związku z czym
nie powinien przekraczać bariery krew-mózg. Antagoni-
Do niektórych leków przeczyszczających dodawany zowane są więc tylko peryferyjne, odpowiedzialne za za-
jest dokuzan sodu (sól sodowa estru kwasu sulfobur- parcie działania opioidów, a nie te w obrębie ośrodkowego
sztynowego), który jako związek powierzchniowo układu nerwowego (szczególnie znieczulenia).
MUTSCHLER-2009.indd 692 2010-01-07 22:14:16
Układ trawienny 693
6.5.3. Leczenie biegunek Wzmożoną motorykę jelitową jako przyczynę biegunek
podaje się również przy nadczynności tarczycy.
Biegunką są częste (częściej niż 3 razy dziennie) wypróż-
nienia stolca wodnistego lub papkowatego. W chorobach Biegunki powinny być leczone w zależności
jelita cienkiego lub górnych odcinków jelita grubego wy- od ich przyczyny. Podstawowymi środkami zarad-
dalane są duże ilości kału ze znaczną zawartością wody,
choroby dystalnego odcinka okrężnicy powodują częste czymi powinno być podawanie:
oddawanie małych ilości kału. ■ płynów i elektrolitów,
W patogenezie biegunek rozróżnia się następujące mecha- ■ opioidów.
nizmy wyzwalające:
■ niedostateczne wchłanianie substancji osmotycznie czyn- Wspomagająco można podawać środki adsorbujące
nych z jelita (biegunka osmotyczna), oraz ściągające lub liofilizowane drożdżaki.
■ zwiększone wydzielanie elektrolitów i wody do jelita W przypadku biegunek spowodowanych zarazka-
(biegunka wydzielnicza), mi inwazyjnymi wskazane jest zastosowanie leków
■ zwiększoną przepuszczalność błony śluzowej jelita, przeciwinfekcyjnych.
■ zaburzenia motoryki jelita.
Podaż płynów i elektrolitów. Ostre biegunki po-
Często kilka mechanizmów działa jednocześnie. dróżników oraz biegunki letnie mają samorzutny
przebieg i nie wymagają podawania leków. W czasie
Biegunka osmotyczna może być wywoływana przez zespół leczenia najważniejsze jest przyjmowanie wystarcza-
złego trawienia lub złego wchłaniania, a także przyjmowa-
nie trudno wchłanialnych substancji (por. środki osmotycz- jącej ilości płynów i elektrolitów. Szczególnie nie-
ne o działaniu przeczyszczającym, np. zawarte w „bezcu- mowlęta i małe dzieci oraz osoby w podeszłym wie-
krowych” cukierkach, w dżemie dla chorych na cukrzycę ku są bardzo szybko zagrożone niebezpieczeństwem
i in.). Niespożywanie tego typu produktów powoduje ustą- odwodnienia. Uzupełnienie płynów i elektrolitów
pienie biegunki. jest możliwe, jeśli mechanizmy resorpcji nie uległy
Biegunka wydzielnicza jest bardzo często wywołana przez uszkodzeniu. W większości przypadków wystarcza
toksyny bakteryjne, które w wyniku interakcji z białkiem doustne podawanie roztworów glukozowo-elektro-
G uaktywniają cyklazę adenylanową komórek śluzowych, litowych. Korzystny efekt łącznego podawania jo-
zwiększając tym samym wytwarzanie cAMP. Przyczyną nów sodowych i glukozy polega na ich aktywnym
biegunki wydzielniczej mogą być toksyny przecinkowca transporcie z jelita do enterocytów i następującym
cholery, toksyny Salmonelli i Shigelli oraz toksyny gron-
kowców i patogennych szczepów Escherichia coli. Znaczna w wyniku tego zwiększeniu resorpcji tej symultanicz-
część biegunek letnich i biegunek podróżników jest powo- nej aplikacji oraz równoczesnym – warunkowanym
dowana przez toksyny E. coli. Przy zakażeniu wywołanym osmotycznie – wzmożonym przyjmowaniu wody.
Układ trawienny
przez gronkowce i Eschericha coli dochodzi zazwyczaj W przypadku utraty dużej ilości wody i elektrolitów
do ujawnienia symptomów warunkowanych obecnością konieczne jest nawadnianie pozajelitowe.
toksyn, natomiast zarazki inwazyjne, np. Shigella i Yersini,
wnikają do ściany jelita i wywołują zapalenia miejscowe Do uzupełnienia elektrolitów utraconych przy
i układowe. cholerze najlepiej nadaje się tzw. roztwór WHO, któ-
Innymi przyczynami biegunki wydzielniczej mogą być ry zawiera 20 g glukozy, 3,5 g NaCl, 3 g cytrynia-
substancje wewnątrzpochodne, np. wazoaktywny peptyd nu sodu i l,5 g KC1 w 1 l wody. W leczeniu innych
jelitowy (VIP) lub niewchłonięte kwasy żółciowe. Biegun- zapaleń bakteryjnych i wirusowych jelit stosuje się
ki wywołane zastojem żółci stwierdza się po resekcji jelita B6
krętego, głównego miejsca resorpcji wtórnej kwasów żół- mieszankę, w której zawartość Na+ jest zmniejszona
ciowych. Kwasy żółciowe, które przedostały się do okręż- o jedną trzecią. W bardzo ciężkich biegunkach ilość
nicy, zwiększają ilość wody i elektrolitów wpływających płynu, którą należy uzupełnić w ciągu 6 godz. o utra-
do tego odcinka jelita i wywołują biegunkę. Jeżeli utrata cone elektrolity i wodę, wynosi 5–10% masy ciała.
kwasów żółciowych jest większa niż ich synteza w wątro-
bie, to w wyniku niedostatecznej resorpcji tłuszczu poja-
wiają się stolce tłuszczowe (steatorroe). Opioidy. Loperamid, blokując obwodowe recepto-
Biegunka wydzielnicza w przeciwieństwie do biegunki ry opioidowe jelita cienkiego, hamuje perystaltykę
osmotycznej może pojawiać się i utrzymywać u pacjentów i z dobrym skutkiem stosowany jest w biegunce spo-
głodujących. wodowanej zaburzeniami motoryki oraz biegunce po-
dróżników. Brak ośrodkowego działania loperamidu
Zwiększona przepuszczalność błony śluzowej jelita powo-
duje ciężkie biegunki w przebiegu chorób zapalnych jelita wyjaśnia jego niewielka dostępność do ośrodkowego
cienkiego i grubego, np. colitis ulcerosa (zob. powyżej) lub układu nerwowego. Nowsze badania wykazują, że lo-
raka okrężnicy. peramid dociera wprawdzie do OUN, ale jest natych-
miast eliminowany przez białko MDR-I.
MUTSCHLER-2009.indd 693 2010-01-07 22:14:16
694 Układ trawienny
Szigeloza i jersinioza o ciężkim przebiegu ze
znaczną biegunką i krwawieniem z przewodu pokar-
mowego wymagają – poza dostarczaniem płynów
N
OH i elektrolitów – stosowania leków przeciwzapalnych.
W szigelozie podaje się chinolony albo ampicylinę,
Cl
w jersiniozie klotrimoksazol lub tetracykliny i cefa-
O N(CH3) 2 losporyny III generacji. Ponieważ pałeczki Shigella
charakteryzuje się niekiedy wielokierunkową opor-
loperamid nością R-plazmidową, przed rozpoczęciem leczenia
należy wykonać antybiogram.
Choroby przewlekle. Biegunki trwające dłużej niż
dwa tygodnie wymagają szczegółowego rozpoznania
Loperamid jest niewskazany w biegunkach wy-
i leczenia dostosowanego do aktualnego stanu pa-
wołanych przez bakterie enteroinwazyjne, ponieważ
cjenta (biegunka jest objawem, a nie chorobą).
unieruchomienie jelita grozi zwiększonym wytwa-
W biegunce wywołanej zastojem żółci mogą być
rzaniem toksyn i zmniejszonym ich wydalaniem.
stosowane wymienniki jonowe (np. cholestyramina),
Dawkowanie loperamidu ustalane jest indywidu-
w stolcach tłuszczowych średniołańcuchowe triglice-
alnie. Dawka początkowa u dorosłych wynosi 4 mg.
rydy.
Sposób leczenia biegunki w colitis ulcerosa lub
Środki adsorpcyjne, ściągające, liofilizowane
chorobie Crohna został opisany w rozdziale B 6.4.
drożdże. Wspomagająco lub w lżejszych przypad-
kach biegunki można stosować środki adsorpcyjne, W przypadku biegunek opornych na leczenie (np. zakaże-
zwłaszcza węgiel leczniczy (carbo medicinalis), nie HIV, guzy hormonozależne) dobre efekty może przy-
nieść leczenie somatostatyną.
środki ściągające (preparaty zawierające garbniki)
oraz liofilizaty Saccharomyces boulardii. Ponadto
wskazane są leki pęczniejące (np. pektyny). Me- regulacja stolca, spożywanie dużej
chanizm (słabego) działania przeciwbiegunkowego uchyłkowatość ilości płynów, dieta bogata w substancje
(faza I) balastowe, ew. środki pęczniejące
środków adsorpcyjnych, ściągających i Saccharomy-
ces boulardii polega na hamowaniu wiązania toksyn
do błony śluzowej jelita. Środki ściągające powodują
uszczelnienie i marskość najwyższych warstw komó- uchyłkowatość karencja żywieniowa, żywienie
1. rzut pozajelitowe, leki przeciwzakaźne
rek oraz hamują wydzielanie z tkanek zmienionych ( faza II) (chinolony, metronidazol),
zapalnie. leki rozkurczające
Środki te można podawać małym dzieciom, które
nie mogą być leczone opioidami.
Leki przeciwzakaźne. Antybiotyki oraz środki de- poprawa brak poprawy
zynfekujące jelita (pochodne hydroksychinoliny)
nie powinny być stosowane w leczeniu zwykłych dieta bogata resekcja
biegunek letnich oraz w leczeniu biegunek podróż- w błonnik, z zespoleniem
ników, ponieważ nie potwierdzono ich skuteczności środki pęczniejące pierwotnym
i możliwe jest wystąpienia działań niepożądanych.
Długotrwałe podawanie dużych dawek preparatów zawie- zapalenie uchyłków, resekcja z zespoleniem pierwotnym, ew.
zapalenie okrężnicy okresowa przetoka zewnętrzna na okrężnicy
rających pochodne hydroksychinoliny spowodowało roz- z utajonymi perforacjami
wój przede wszystkim w Japonii choroby określanej jako (fazy III i IVa)
SMON (subacute myelo-optico-neurophathy). Towarzyszą
jej polineuropatia, zanik szlaków piramidowych, zaburze- profilaktyka z zachowaniem
nia czynności pęcherza, odbytnicy i wzroku. diety bogatej w błonnik
Salmonelozy (np. tyfusu) nie należy leczyć rutynowo zapalenie okrężnicy resekcja
przemieszczenie
antybiotykami, ponieważ antybiotykoterapia przedłu- z wolną perforacją i wytworzenie
(faza IVb) sztucznego odbytu zwrotne
ża wydalanie drobnoustrojów chorobotwórczych.
Ryc. B 6.5-2. Podział i leczenie uchyłkowatości.
MUTSCHLER-2009.indd 694 2010-01-07 22:14:16
Układ trawienny 695
6.5.4. Uchyłkowatość i jej leczenie dzi do rozwoju właściwej choroby uchyłkowej (faza
II – diverticulitis), (faza III – peridiverticulitis, faza
Uchyłkami są nazywane uwypuklenia ściany jelita. IV – pericolitis). Częstość występowania zwiększa
Powstają przeważnie w wyniku jej osłabienia w miej- się z wiekiem (około 10% u 40-letnich osób, 80%
scu skrzyżowania naczyń. Dolegliwości towarzy- u 80-letnich).
szące uchyłkowatości symptomatycznej (faza I) Leczenie lżejszych przypadków ma charakter za-
nie są bezpośrednio związane z uchyłkiem, lecz wy- chowawczy (zob. ryc. B 6.5-2), natomiast ciężki prze-
nikają z zaburzeń motoryki jelita. Dopiero w wyniku bieg choroby jest wystarczającą kwalifikacją do zabie-
powstania stanów zapalnych wielu uchyłków docho- gu chirurgicznego.
6.6. Leczenie uzupełniające enzymami trawiennymi
i kwasami; pobudzanie wytwarzania kwasów
Enzymy trawienne. Podawanie enzymów trawien- Skrótowy opis działania enzymów został przedsta-
nych, przede wszystkim enzymów trzustkowych (pan- wiony w tab. B 6.6-1.
kreatyny), jest wskazane przy zewnątrzwydzielniczej Enzymy trawienne przyjmowane są ponadto
niewydolności trzustki (np. jako skutek przewlekłego w zaburzeniach trawienia bez niedoboru pankreaty-
zapaleniu trzustki, usunięcia trzustki lub przy muko- ny (szczególnie po obfitych posiłkach). Osiągnięte
wiscydozie). Pankreatyna jest proszkiem otrzymanym dzięki temu dobre wyniki polegają mniej lub bardziej
z trzustki ssaków, najczęściej świni, w której oprócz na efekcie placebo.
lipazy, amylazy i proteazy (trypsyna, chymotrypsyna, Enzymy trawienne jako substancje białkowe
karboksypeptydazy) znajdują się również inne enzy- nie są resorbowane w przewodzie pokarmowym
my. Dawki enzymów muszą być duże. W przypadku w postaci niezmienionej, lecz w przeważającej części
całkowitej utraty czynności trzustki zapotrzebowanie rozkładane lub denaturyzowane przez soki trawienne
enzymu na jeden posiłek wynosi 40 000–80 000 jed- lub bakterie. Aktywność enzymów w kale jest bardzo
nostek FIP lipazy, 30 000–60 000 jednostek FIP amy- niewielka.
lazy i 2500–5000 jednostek FIP proteazy.
Kwasy. W przewlekle zanikowym zapaleniu błony
Np. jednostka FIP (Fédération Internationale Pharmaceuti- śluzowej żołądka obniżona jest liczba gruczołów dna,
Układ trawienny
que) lipazy jest ilością enzymu, która przy pH równym 9,0 a tym samym zmniejszone wydzielanie kwasu solnego
i w temperaturze 37°C jest niezbędna do uwolnienia jed-
nego mikroekwiwalentu kwasu tłuszczowego z glicerydu i pepsynogenu. Skutkiem tego mogą być zaburzenia
na minutę. żołądkowo-jelitowe z towarzyszącym im uczuciem
sytości i utratą łaknienia. U części pacjentów dolegli-
wości te ustępują po podaniu kwasu solnego (acidum
hydrochloricum dilutum, 30–60 kropli na szklankę
wody) albo kwasu cytrynowego (pojedyncza dawka B6
Tabela B 6.6-1. Przegląd farmakologicznego działania najważ-
niejszych enzymów pankreatyny wieprzowej 0,25–1,0 g). Ponieważ zastosowane ilości są zbyt
małe, aby mogły zastąpić wewnątrzpochodny kwas
solny, ich działanie należy uznać, jak w przypadku
Enzymy Reakcja katalizowana wielu środków terapeutycznych w przewodzie żołąd-
lipaza redukcja trójglicerydów do wolnych kowo-jelitowym, za efekt placebo.
kwasów tłuszczowych i monoglicerydów Pewne pobudzenie produkcji soku żołądkowego
amylaza redukcja zawierających glukozę lub kwasu solnego można uzyskać, podając kofeinę
polisacharydów do disacharydów bądź alkoholowe preparaty zawierające w swoim
proteazy składzie związki goryczkowe (Vinum Condurango,
trypsyna, redukcja białek nalewka z gencjany).
chymotrypsyna do oligopeptydów Uzupełnianie enzymów żołądkowych w przypad-
karboksypeptydazy redukcja białek do peptydów ku bezkwaśności nie jest konieczne, ponieważ pep-
i aminokwasów syna ma drugorzędne znaczenie w procesie trawienia
białka.
MUTSCHLER-2009.indd 695 2010-01-07 22:14:17
696 Układ trawienny
6.7. Związki wpływające na motorykę żołądka i jelit
6.7.1. Związki wspomagające jest stosowany jako antymimetyk. Dodatkowo wpły-
motorykę żołądka wa także na receptory serotoninowe. Przyjmowany
doustnie wchłania się bardzo szybko. W przeciwień-
i jelit (prokinetyki)
stwie do domperidonu przekracza barierę krew-mózg.
Jego okres półtrwania w osoczu wynosi około 4 go-
W celu przyspieszenia opróżniania żołądka i jedno-
dzin. Wydalany jest przez nerki, częściowo niezmie-
cześnie przyspieszenia przejścia pokarmu przez jelito
niony i w formie sprzężonej.
cienkie w dolegliwościach czynnościowych ze strony
Dawka dobowa wynosi 30 mg.
obu narządów (np. autonomicznej neuropatii cukrzy-
Z powodu blokady receptorów dopaminowych
cowej oraz napadach bólów migrenowych) stosowa-
mogą wystąpić działania niepożądane przejawiające
ne są następujące substancje (zob. tab. B 6.7-1):
się zmęczeniem, zawrotami głowy, obniżonym na-
■ metoklopramid, strojem oraz pozapiramidowymi objawami ruchowy-
mi przypominającymi objawy towarzyszące przyj-
■ domperidon,
mowaniu leków neuroleptycznych. Ponieważ ustaje
■ cizaprid (w trakcie rejestracji). hamowanie wydzielania prolaktyny przez dopaminę,
może również dojść do hiperprolaktynemii, na którą
Ze względu na poważne działania niepożądane rzad- składają się mlekotok, zaburzenia miesiączkowania,
ko zaleca się leki cholinergiczne. nadmierny rozrost sutków u osobników płci męskiej
Klinicznie względny prokinetyczny efekt działania i spadek libido.
obu pierwszych leków ograniczony jest do górnego Przeciwwskazaniami są guz chromochłonny nad-
odcinka przewodu żołądkowo-jelitowego, cizaprid nerczy oraz rak zależny od prolaktyny. Z powodu
działa również na okrężnicę. motorycznych zaburzeń pozapiramidowych, które
u dzieci mogą się szczególnie nasilać, metoklopramid
Metoklopramid działa antagonistycznie na recep- nie powinien być podawany niemowlętom i małym
tory dopaminowe (receptory D2), w związku z czym dzieciom.
Tabela B 6.7-1. Prokinetyki
Wzór strukturalny Nazwa Nazwa handlowa Okres Dawka
międzynarodowa półtrwania dzienna
(godz.) (mg)
O C2H5 metoklopramid Gastronerton, 3-4 30
MCP HEXAL,
Cl N MCP-ratiopharm,
NH C2H5
Paspertin
H2N OCH3
F cizaprid w trakcie rejestracji 8-10
O N O
Cl
NH
OCH3
H2N OCH3
O domperidon DOMPERIDON AL, 7 30
NH DOMPERIDON HEXAL,
N DOMPERIDON-ratiopharm,
Motlium
N
Cl
N
O
NH
MUTSCHLER-2009.indd 696 2010-01-07 22:14:17
Układ trawienny 697
Równoczesne podawanie neuroleptyków wzmac- preparatów zawierających cizaprid przez Federalny Insty-
nia działania niepożądane. tut Leków i Produktów Medycznych (BfArM). Szczegól-
nie zagrożeni są pacjenci, którzy w ramach współleczenia
otrzymują leki hamujące działanie enzymu CYP3A4 istot-
Domperidon blokuje peryferyjne receptory dopa- nego w biotransformacji cizapridu (np. makrolidy, syste-
minowe. Podawany doustnie jest bardzo szybko mowe azolowe preparaty przeciwgrzybicze, inhibitory
wchłaniany. Ze względu na wyraźny efekt pierwsze- proteazy HIV). Może to bowiem prowadzić do trzykrot-
go przejścia dostępność biologiczną domperidonu nego wzrostu stężenie cizapridu w osoczu. Doświadczenia
przeprowadzone na zwierzętach dowodzą teratogennego
nie przekracza 20%. Okres jego półtrwania w osoczu
działania cizapridu.
wynosi 7 godz. Wydalany jest natomiast przez nerki
lub z żółcią.
Agoniści motyliny. Nawet subantybiotyczne dawki
Dawka dobowa wynosi 30 mg.
erytromycyny powodują pobudzenie receptorów mo-
Działania niepożądane domperidonu są podobne
tyliny oraz silne skurcze odźwiernika i dwunastnicy.
do objawów wywoływanych przez metoklopramid.
Analogi erytromycyny niedziałające antybiotycznie
Wyjątek stanowią jedynie zaburzenia ośrodkowe,
są badane jako potencjalne leki prokinetyczne.
które w tym przypadku mają niewielkie nasilenie.
Cizaprid jest częściowym antagonistą receptorów se-
rotoninowych (receptorów 5-HT4), dlatego nie działa 6.7.2. Leki hamujące motorykę
antymimetycznie i nie powoduje pozapiramidowych żołądka i jelit
ruchowych działań niepożądanych.
W celu zmniejszenia wzmożonej motoryki żołądka
Częstymi działaniami niepożądanymi są zaburzenia żołąd- i jelit stosuje się substancje, które nie stymulują re-
kowo-jelitowe, uczucie zmęczenia i zawroty głowy. Po- ceptorów opioidowych w jelicie cienkim (zob. lope-
nadto zgłaszane są wprawdzie rzadko, ale ciężkie zaburze-
nia rytmu serca (wielokształtny częstoskurcz komorowy, ramid) oraz nie przyspieszają działania acetylocho-
częstoskurcz komorowy, migotanie komór, nagła śmierć liny. Do tych ostatnich należą leki spazmolityczne
sercowa). Stały się one powodem wstrzymania rejestracji neuromuskulotropowe i muskulotropowe.
6.8. Leki stosowane w chorobach wątroby
Układ trawienny
Duża liczba znajdujących się na rynku preparatów
zapalenia wątroby typu A, B, C, D i E; zob. tab. B 6.8-
nie powinna sprawiać wrażenia, że leczenie farmako-
1) i niepierwotne wirusy hepatotropowe (np. wirusy
logiczne chorób wątroby, z wyjątkiem zapalenia wą-
Epsteina-Barr, wirus cytomegalii, wirus Coxsackie,
troby (zob. poniżej), jest możliwe w nieograniczonym
odry, adenowirus). Zakażenia wirusami przenoszony-
zakresie. Dokładne badania wykazały, że ani duże
mi drogą jelitową (wirusowe zapalenie wątroby typu
dawki witamin, ani tzw. środki lipotropowe, np. cho-
A i E) można uniknąć dzięki konsekwentnemu prze-
lina lub metionina, nie wpływają korzystnie na cho-
strzeganiu zasad higieny lub dzięki szczepieniom, jak B6
roby wątroby. Zdarza się bowiem, że z pożywieniem
w przypadku wirusowego zapalenia wątroby typu A.
są one dostarczane w niewystarczających ilościach.
Infekcje wywołane wirusem zapalenia wątroby typu
Roślinne preparaty skojarzone, np. sylimarol (suchy
B, C i D są przenoszone przede wszystkim pozajelito-
ekstrakt z mleczka ostu), stosowane są w toksycznych
wo, mają charakter przewlekły i podwyższają ryzyko
uszkodzeniach wątroby oraz jako adjuwant w przewle-
rozwoju raka wątrobokomórkowego. Infekcja będąca
kle zapalnych chorobach wątroby i marskości wątroby.
skutkiem zakażenia wirusem zapalenia wątroby typu
Skuteczność tych preparatów jest jednak wątpliwa.
G ma wprawdzie charakter przewlekły, ale niepostę-
pujący. Zgodnie z ustawą o ochronie przed infekcja-
mi wirusowe zapalenie wątroby jest chorobą wyma-
6.8.1. Wirusowe zapalenia wątroby gająca zgłoszenia.
– profilaktyka i leczenie
Wirusowe zapalenie wątroby typu A. Wywoływane
Zapalenie wątroby mogą wywołać liczne wirusy. Roz- jest przez wirus zapalenia wątroby typu A (HAV), wi-
różnia się pierwotne wirusy hepatotropowe (wirusy rus RNA, należący do pikornawirusów, który jest pa-
MUTSCHLER-2009.indd 697 2010-01-07 22:14:17
698 Układ trawienny
Tabela B 6.8-1. Cechy pierwotnych wirusów hepatotropowych
typu A typu B typu C typu D typu E
(HAV) (HBV) (HCV) (HDV) (HEV)
Rodzina wirusów pikornawirusy hepadnawirusy flawiwirusy ? hepatowirusy
Genom pojedyncza spirala RNA podwójna spirala DNA pojedyncza spirala RNA pojedyncza spirala RNA pojedyncza spirala RNA
Główna droga jelitowo lub przez jelitowo lub przez
przenoszenia pozajelitowo pozajelitowo pozajelitowo
układ pokarmowy układ pokarmowy
Czas wylęgania wirusa 2-6 tygodni 1-6 miesięcy 2-8 tygodni 1-6 miesięcy 2-9 tygodni
Infekcja
przewlekła nie tak tak tak nie
Profilaktyka
aktywna tak tak nie tak nie
bierna tak tak nie tak nie
togenny wyłącznie dla naczelnych. HAV jest prze- tropikalnych), wskazane jest zastosowanie immuni-
noszony przede wszystkim przez skażoną żywność zacji biernej za pomocą humanizowanej immuno-
i wodę pitną. Na obszarach o bardzo niskim pozio- globuliny.
mie higieny zakażone są całe populacje, w krajach
uprzemysłowionych liczba chorych z HAV ciągle się Wirusowe zapalenie wątroby typu B. W przeci-
zmniejsza (odsetek infekcji wynosi około 3%). HAV wieństwie do pozostałych wirusów zapalenia wątroby
powoduje wytwarzanie swoistych przeciwciał IgM wirus hepatitis B (HBV, tzw. cząsteczka Dane) nale-
oraz IgG (IgM oraz IgG anty-HAV), które mają duże ży do wirusów DNA (wirusy hepadna). Składa się on
znaczenie w diagnostyce. W wyniku przebytego za- z osłonki (surface), jądra (core), częściowo podwójnie
każenia HAV nabywa się na nie odporności do końca skręconej spirali DNA i polimerazy DNA. W diagno-
życia. Wirusowe zapalenie wątroby typu A nie ma styce konieczne jest uwzględnienie HBV-DNA, HBc-
przewlekłego przebiegu, zazwyczaj ustępuje samo- Ag (antygen jądra), HBs-Ag (antygen osłonki) oraz
rzutnie i nie wymaga leczenia. Rzadkim powikłaniem Hbe-Ag (postać wydzielnicza HBc-Ag). Odpowiada-
HAV jest ostra niewydolność wątroby, wymagająca
transplantacji tego narządu.
Profilaktyka zakażeń HAV może polegać na ak-
tywnej immunizacji za pomocą inaktywowanych wi-
rusów hepatitis A, zarówno u dorosłych, jak i u dzie- IgG-anty-HBc
ci. Preparaty VAQTA bądź VAQTA K pro infantibus
stężenie
zawierają wysokooczyszczone proteiny wirusa zapa- HBV-DNA
lenia wątroby typu A, a ich podanie powoduje uak- HBe-Ag anty-HBe
tywnienie odporności organizmu. IgM-anty-HBc
HBs-Ag
Szczepienie profilaktyczne przeciw hepatitis A
jest wskazane m.in. w przypadku osób zagrożonych infekcja anty-HBs
ze względów zawodowych (personel medyczny, pra- HBV hepatitis
cownicy kanalizacji miejskiej itp.), podróżujących
do krajów z wysokim odsetkiem zachorowań na HAV, 0 2 4 6 8 10
homoseksualistów oraz osób chorych na hemofilię miesiące
wymagającą przetaczania krwi.
Ryc. B 6.8-1. Serologiczny przebieg wyleczonego ostrego za-
Działaniami niepożądanymi szczepionki są bie-
każenia wirusem zapalenia wątroby typu B. Hbe-Ag – antygen
gunki, wymioty, nieznaczne zwiększenie stężenia Hbe, Hbs-Ag – antygen osłonki, anty-HBc – przeciwciała przeciw
enzymów wątrobowych, a po niektórych preparatach jądru wirusa zapalenia wątroby typu B, anty-HBe – przeciwciała
również zapalenie dróg oddechowych i kaszel. przeciw rozpuszczalnemu antygenowi, anty-HBs – przeciwcia-
Jeżeli szczepienie nie może odbyć się we właś- ła przeciw otoczce wirusa zapalenia wątroby typu B. Anty-Hbs
ciwym czasie (np. po bliskim kontakcie z osobą za- staje się pozytywny, gdy zanika Hbs-Ag, co oznacza wyleczenie
każoną HAV lub krótkotrwałym pobycie w krajach hepatitis B (według Herolda).
MUTSCHLER-2009.indd 698 2010-01-07 22:14:18
Układ trawienny 699
jące im przeciwciała to anty-HBc, anty-HBs oraz anty- rapia interferonem kończy się całkowitym wylecze-
-HBe. Kolejność wytwarzania antygenów i przeciw- niem choroby.
ciał przedstawiona jest na ryc. B 6.8-1. Dodatni wynik Częstymi działaniami niepożądanymi pojawiają-
testu hybrydyzacyjnego wskazuje na namnażanie wi- cymi się w trakcie leczenia interferonem są gorączka,
rusa HBV-DNA, a tym samym na zakażenie. zmęczenie, bóle głowy, bóle mięśni, dreszcze oraz
Hepatitis B zaliczane jest na całym świecie do naj- spadek masy ciała, wypadanie włosów, trombopenia,
częstszych chorób zakaźnych. Przenoszone jest głów- depresja i spadek odporności na działanie bakterii.
nie drogą pozajelitową, płciową lub w warunkach Przeciwwskazaniem do leczenia interferonem α
okołoporodowych. Prawdopodobieństwo zakażenia jest postępująca marskość wątroby, przebyte ciężkie
się HBV jest prawie stukrotnie większe niż praw- choroby serca, równocześnie przeprowadzane lecze-
dopodobieństwo zakażenia się HIV. U prawie 25% nie immunosupresyjne, choroby psychiczne lub auto-
zakażonych osób dorosłych dochodzi do rozwoju immunizacyjne, choroba alkoholowa lub uzależnienie
ostrego wirusowego zapalenia wątroby kończącego od leków, znaczna leukopenia lub trombocytopenia.
się wyleczeniem i eliminacją wirusa, u prawie 2%
zakażonych wirusowe zapalenie wątroby staje się Kolejną uznaną możliwością leczenia jest podawanie
chorobą przewlekłą (jeżeli trwa dłużej niż 6 miesię- nukleozydowego analogu lamiwudyny. Związek ten
cy, zob. tab. B 6.8-2), której odległym powikłaniem hamuje polimerazę DNA, stanowiącą podstawę nie-
może być marskość wątroby lub zrakowacenie komó- zbędną do replikacji HBV. Odsetek odpowiedzi orga-
rek wątrobowych. nizmu na lamiwudinę jest porównywalny z wynikami
Standardowym leczeniem pacjentów z przewlekle- uzyskanymi w przypadku monoterapii interferonem.
nawrotowym HBV (potwierdzonym testem hybrydy- U 20% pacjentów leczonych lamiwudyną wystąpiła
zacyjnym na obecność HBV-DNA) jest monoterapia oporność.
interferonem α w dawce 3 razy od 5 do 10 mln IU. Najczęściej zgłaszanymi działaniami niepożąda-
podskórnie na tydzień (zob. tab. B 6.8-2). U 40% pa- nymi są ogólne złe samopoczucie, zmęczenie, zaka-
cjentów pojawia się tym samym tzw. serokonwersja żenia dróg oddechowych, bóle głowy, nudności, wy-
(od Hbe-Ag do anty-Hbe), w związku z czym do zba- mioty i biegunka.
dania obecności HBV-DNA nie można już użyć testu
hybrydyzacyjnego, lecz czułych metod polimeryzacji W leczeniu przewlekłego hepatitis B u dorosłych
łańcuchowej (PCR). U 5–10% pacjentów z HBV te- pacjentów dopuszczony jest dipiwoksyl adefowiru.
Układ trawienny
25% 65%
infekcja HBV
5-10%
ostre wirusowe zapalenie wątroby trwanie wirusa infekcja subkliniczna
0,1-1% 99% 100% B6
śmiertelne wyleczone 30% 70% wyleczenie
przewlekłe hepatitis bezobjawowe nosicielstwo Hbs-Ag
marskość wątroby pierwotny rak wątroby
Ryc. B 6.8-2. Prawdopodobny przebieg infekcji HBV u dorosłego pacjenta, który nie jest immunosupresyjny (według Herolda).
MUTSCHLER-2009.indd 699 2010-01-07 22:14:18
700 Układ trawienny
Tabela B 6.8-2. Postępowanie lecznicze w przypadku przewlekłego wirusowego zapalenia wątroby typu B
HBe-Ag-pozytywny Hbe-Ag-negatywny Hbe-Ag-negatywny Oporność na lamiwudynę
i trwanie wirusa i trwanie wirusa i wysokie trwanie wirusa
> 100 000 kopii/ml < 100 000 kopii/ml (wariant HBe-minus)
* interferon-αα: leczenie niewskazane, lamiwudyna: zalecane wprowadzenie adefowiru
4-6 miesięcy podskórnie w przypadku wysokich wartości 100 mg dziennie doustnie
3×9-10 mln IU na tydzień lub enzymów wątrobowych zalecana przez min. 52 tygodnie
5-6 mln IU dziennie biopsja w celu dalszej diagnostyki
i ew. leczenie specjalistyczne lub
alternatywnie:
adefowir:
** lamiwudyna: 10 mg dziennie doustnie
100 mg dziennie doustnie przez min. 48 tygodnie
przez 52 tygodnie ew.
do 6 miesięcy po serokonwersji
Hbe-Ag do anty-HBe
** adefowir:
100 mg dziennie doustnie
przez 48 tygodni ew.
do 6 miesięcy po serokonwersji
HBe-Ag
* Przy > pięciokrotnie podwyższonym poziomie transaminaz zalecane jest podanie jako pierwszego interferonu
** Przy < dwukrotnie podwyższonym poziomie transaminaz zalecane jest podanie jako pierwszego analogu nukleozydowego/nukleotydowego
Po podaniu doustnym przekształca się w organizmie Kolejnym związkiem możliwym do zastosowania
w adefowir, monofosforan adenozyny i tym samym w terapii przewlekłego wirusowego zapalenia wątro-
nukleozydowy analog, który w komórce po przejściu by typu B jest entekawir. Analog guanozyny prze-
w difosforan hamuje wirusową polimerazę DNA. kształca się w organizmie w trójfosforan, hamujący
Ponieważ humanizowana polimeraza DNA hamo- selektywnie polimerazę HBV. Dotychczas nie zaob-
wana jest dopiero przy porównywalnie wyższych serwowano oporności.
stężeniach, adefowir był stosunkowo dobrze znoszo- Dawkowanie entecawiru wynosi 0,5 mg, w przy-
ny w badaniach klinicznych. W dotychczasowych padku oporności na lamiwudyna doustnie 1 mg dzien-
badaniach klinicznych nie stwierdzono oporności, nie.
w związku z czym adefowir może być podawany tak- Działaniami niepożądanymi, zgłaszanymi naj-
że w przypadku oporności na lamiwudynę. częściej w czasie badań klinicznych, były ból głowy,
Dawkowanie wynosi doustnie 10 mg dziennie mdłości, zawroty głowy i uczucie zmęczenia.
przez minimum 48 tygodni po serokonwersji Hbe-Ag
do anty-HBe. Rekombinowana szczepionka (Engerix-B dorośli,
Działania niepożądane są zależne od przyjmowa- Engerix-B dzieci) jest standardowym szczepieniem
nej dawki, a objawiają się bólem głowy, mdłościami, dzieci. Powinna być również podana dorosłym prze-
biegunką i zaburzeniem funkcjonowania nerek. bywającym w warunkach o podwyższonym ryzy-
ku zarażenia się HBV. Dostępna jest kombinowana
szczepionka przeciw hepatitis A i B (Twinrix dorośli,
O Twinrix dzieci). W przypadku już stwierdzonej eks-
N pozycji we krwi dodatniego antygenu Hbs-Ag prze-
CH2 NH
prowadzana jest pasywna immunizacja hiperimmu-
HOH2C N N NH2 noglobuliną.
HO Wirusowe zapalenie wątroby typu C. Hepatitis C
rozwija się na skutek zarażenia wirusem HCV (6 roz-
poznanych genotypów), należącym do grupy wirusów
entekawir RNA. Wirus ten atakuje oprócz komórek wątroby także
komórki macierzyste szpiku kostnego i limfocyty krwi.
MUTSCHLER-2009.indd 700 2010-01-07 22:14:18
Układ trawienny 701
Tabela B 6.8-3. Postępowanie terapeutyczne w przypadku W przypadku pegylacji dochodzi do wytworzenia się wią-
ostrego zapalenia wątroby typu C zania kowalencyjnego między polyetylenoglikolem a od-
powiadającym mu interferonem. Celem tej reakcji jest zna-
czące spowolnienie zarówno enzymatycznej odbudowy
Wszystkie genotypy interferonu, jak i wydzielania nerkowego.
24-tygodniowa monoterapia interferonem α
(4 tygodnie 5 mln UI interferonu α-2b codziennie podskórnie, W przeciwieństwie do peginterferonu α-2a peginter-
20 tygodni 5 mln UI interferonu α-2b 3 × tygodniowo podskórnie) feron α-2b zawiera rozgałęziony łańcuch polietyleno-
glikolu, w związku z czym okres półtrwania pegin-
* Ważne: leczenie należy rozpocząć w ciągu pierwszych 4 tygodni terferonu α-2b mógł być wydłużony z 30 do 80 godz.
(okres półtrwania standardowych interferonów wy-
nosi 3–4 godz.). Pegylowane interferony muszą być
dlatego wstrzykiwane tylko jeden raz w tygodniu.
Przed wprowadzeniem badania krwi konserwowanej Peginterferon α-2a powinien być podawany
na obecność przeciwciał HCV transfuzja krwi była w dawce 180 μg/tydzień a peginterferon α-2b w daw-
najczęstszym źródłem infekcji. W Niemczech około ce 1,0–1,5 μg/kg masy ciała tygodniowo. Dawka ry-
1% społeczeństwa choruje na HCV (głównie osoby bawiryny zależy od masy ciała pacjenta, od genotypu
z hemofilią, uzależnione od leków oraz dializowane). HCV oraz zastosowanego peginterferonu. Najczęś-
Przenoszenie wirusa następuje przede wszystkim dro- ciej zalecane dawki dobowe mieszczą się w granicach
gą pozajelitową, a także płciową oraz w warunkach od 800 do 1200 mg i podawane są w dwóch równych
okołoporodowych. Ponad 50% zakażeń HCV ma cha- częściach rano i wieczorem (zob. tab. B 6.8-4). Czas
rakter przewlekły. U 20–30% pacjentów po 20 latach trwania leczenia skojarzonego wynosi 48 tygodni
rozwija się marskość wątroby, a spośród nich u 30% w przypadku pacjentów zakażonych HCV o genoty-
rak wątrobokomórkowy. Ponieważ duży odsetek przy- pie 1 lub 4 oraz 24 tygodnie w przypadku pacjentów
padków ostrego wirusowego zapalenia wątroby typu C zainfekowanych HCV o genotypie 2 lub 3. Odsetek
nabiera cech przewlekłych, wskazane jest stosowanie uzyskanych w ten sposób odpowiedzi wirusologicz-
przez 24 tygodnie monoterapii interferonem α-2a lub nych (brak dowodu obecności w surowicy RNA
interferonem α-2b (5 mln IU dziennie przez 4 tygo- HCV) waha się od 55 do 90%. Terapia może jednak
dnie, a następnie trzykrotnie 5 Mio. j.m. tygodniowo okazać się korzystna również w przypadku pacjentów
przez kolejnych 20 tygodni). Dzięki temu w ponad bez trwałej odpowiedzi wirusologicznej.
20% przypadków można zapobiec przejściu HCV Działania niepożądane leczenia skojarzonego róż-
w stan przewlekły (zob. tab. B 6.8-3). nią się bardzo nieznacznie od tych towarzyszących
Stosowane obecnie standardowe leczenie prze- monoterapii interferonem (zob. poniżej). Należy zwró-
Układ trawienny
wlekłego HCV polega na skojarzonym podawaniu cić uwagę na teratogenne działanie rybawiryny, możli-
pegylowanego interferonu i rybawiryny (zob. tab. wość spowodowania przez nią hemolizy osmotycznej
B 6.8-4). W przypadku oporności na ribawirynę na- (spadek poziomu hemoglobiny z jednoczesnym wzro-
leży zastosować monoterapię pegylowanym interfe- stem poziomu retikulocytów, bilirubiny i kwasu mo-
ronem. czowego) oraz zmniejszenie się liczby limfocytów.
B6
Tabela B 6.8-4. Postępowanie lecznicze w przypadku przewlekłego wirusowego zapalenia wątroby typu C
HCV o genotypie 1 lub 4 HCV o genotypie 2 lub 3
pozytywny efekt leczenia u 55% pozytywny efekt leczenia u 80-90%
48 tygodni: 1,5 μg/kg masy ciała peginterferonu α-2b 1 × tyg. 24 tygodnie: 1,5 μg/kg masy ciała peginterferonu α-2b
podskórnie + rybawiryna (> 10,6 mg/kg masy ciała dz. w postaci tabletek) 1 × tyg. podskórnie + rybawiryna (800-1200 mg dz.
→ 800-1200 mg. dz. odpowiednio do masy ciała odpowiednio do masy ciała w postaci tabletek)
lub lub
48 tygodni: 180 μg peginterferonu α-2a 1 × tyg. podskórnie + rybawiryna 24 tygodnie: 180 μg peginterferonu α-2a
(1000 mg dz. przy masie ciała < 75 kg, 1200 mg. dz. przy masie ciała 1 × tyg. podskórnie + rybawiryna
> 75 kg w postaci tabletek) (800 mg dz. w postaci tabletek)
MUTSCHLER-2009.indd 701 2010-01-07 22:14:19
702 Układ trawienny
Rekombinowany interferon alfacon-1 (jest kotyków. Ponieważ prawdopodobnie nie wywołuje
wprawdzie dopuszczony w leczeniu HCV, jednak typowego zapalenia wątroby, nie wymaga leczenia.
ze względu na konieczność trzykrotnego podawania Około 80% zakażonych HGV jest zarażonych tak-
go w ciągu tygodnia nie jest stosowany standardowo. że HCV. Niekiedy dochodzi również do koinfekcji
Zbudowany jest ze 166 aminokwasów. z HAV i HBV. Leczenie przeprowadzane jest w ta-
kich przypadkach odpowiednio do koinfekcji.
Jak dotychczas nie wynaleziono jeszcze szczepion-
ki przeciw HCV. Obecność w surowicy przeciwciał
anty-HCV nie oznacza jeszcze ochrony przed prze- 6.8.2. Leczenie innych chorób
wlekłym wirusowym zapaleniem wątroby. wątroby
Wirusowe zapalenie wątroby typu D. Wirus hepati-
Stłuszczenle wątroby. Stłuszczenie wątroby najczęś-
tis D (delta) jest niekompletnym wirusem RNA, któ-
ciej wynikające ze spożywania alkoholu w nadmier-
ry do replikacji wykorzystuje osłonkę HBV. Warun-
nych ilościach nie wymaga leczenia farmakologicz-
kiem rozwoju wirusowego zapalenia wątroby typu
nego. Konieczna jest jednak abstynencja alkoholowa,
D jest więc przebiegające równocześnie ostre lub
a w razie potrzeby także redukcja masy ciała.
przewlekłe wirusowe zapalenie typu B i przewaga
wywołujących je wirusów. Rokowanie jest gorsze niż
Niewydolność wątroby, marskość wątroby. Przy
dla hepatitis B. Interferon α-2b jest wprawdzie sku-
niewydolności wątroby lub (postępującej) marskości
teczny, niezmiernie rzadko jednak pozwala na osiąg-
wątroby odpowiednim postępowaniem leczniczym
nięcie trwałego wyleczenia, w związku z czym jego
jest obniżenie stężenia amoniaku we krwi. NH3 po-
stosowanie wciąż budzi kontrowersje. Niewielkie
wstający w jelicie grubym w wyniku procesu trawie-
jest dotychczasowe doświadczenie w zakresie bada-
nia jest metabolizowany w wątrobie. Jeżeli jednak
nia analogów nukleozydów. Wprawdzie niektórzy
na skutek zaburzenia pracy wątroby lub nadciśnienia
pacjenci leczeni np. lamiwudyną stali się HBV-DNA
w żyle wrotnej NH3 z pominięciem wątroby dostaje
negatywni, prawie nigdy jednak nie doszło do zaniku
się za pośrednictwem dużego krwiobiegu bezpośred-
Hbs-Ag czy zmiany w przebiegu choroby. Podobnie
nio do mózgu, dochodzi do jego uszkodzenia (tzw.
leczenie skojarzone składające się z interferonu α-2b
encefalopatia wątrobowa). Obniżenie stężenia amo-
i rybawiryny nie przynosi lepszych efektów niż mo-
niaku można osiągnąć:
noterapia interferonem.
Szczepienie przeciw wirusowemu zapaleniu wą- ■ hamując resorpcję amoniaku poprzez obniżenie
troby typu B (zob. powyżej) zapobiega również wiru- wartości pH w okrężnicy za pomocą laktulozy lub
sowemu zapaleniu wątroby typu D, pod warunkiem, laktitolu,
że wcześniej nie doszło do zakażenia HBV.
■ zmniejszając ilość amoniaku wytwarzanego
w okrężnicy poprzez podanie trudno wchłaniają-
Wirusowe zapalenie wątroby typu E. U podstaw
cych się leków przeciwbakteryjnych (np. neomy-
wirusowego zapalenia wątroby typu E leży infekcja
cyny lub paromomycyny),
wirusem HEV z grupy wirusów RNA. Podobnie jak
w przypadku hepatitis A, jego przeniesienie następuje ■ usprawniając przemianę wstępną amoniaku po-
wraz z kałem lub z zakażoną wodą pitną. HEV wystę- przez poprawienie patologicznego wzoru składu
puje przede wszystkim w Azji Południowo-Wschod- aminokwasów organizmu za pomocą dostarcza-
niej, na niektórych obszarach Afryki, w Ameryce nych pozajelitowo aminokwasów esencjonalnych
Środkowej i Południowej oraz w Turcji. Chociaż ni- lub asparaginianu ornityny.
gdy nie nabiera charakteru przewlekłego, może mieć
przebieg śmiertelny, zwłaszcza u kobiet ciężarnych, W pierwotnej marskości żółciowej kwas ursode-
wśród których liczba zgonów przekracza 20%. Z po- oksycholowy (zob. poniżej) usprawnia przynajmniej
wodu braku leczenia specyficznego HEV może być
leczone wyłącznie symptomatycznie. Dotychczas
brak jest szczepionki przeciw tej chorobie.
H3C(CH2) 11 O C2H4OH
O
Wirusowe zapalenie wątroby typu G. Wirus hepa- 8-9
titis G (HGV) należy do zawierającej jedną nić RNA
polidokanol
rodziny wirusów Flaviviridae. Do zakażenia docho-
dzi najczęściej wśród osób uzależnionych od nar-
MUTSCHLER-2009.indd 702 2010-01-07 22:14:19
Układ trawienny 703
krótkotrwale zastój żółci i związane z tym dolegli- lub okołonaczyniowe zastrzyki z 1-procentowego
wości. Ważna jest terapia objawów towarzyszących, roztworu eteru laurylowego makrogolu (polidokano-
takich jak świąd, syndrom Sicca i osteoporoza. lu). Możliwymi komplikacjami po zabiegu są m.in.
krwawienie lub perforacja przełyku.
Wodobrzusze w marskości wątroby można leczyć diu- We wskazówkach dotyczących zabiegu zaleca się
retykami. obniżenie ciśnienia wrotnego poprzez zmniejszenie
ukrwienia obszaru unerwionego przez nerw trzewny.
Krwawienia z żylaków przełyku. Do endoskopo- Odpowiednimi do tego celu substancjami są octan
wego zamknięcia krwawiących żylaków przełyku terlipresyny, somatostatyna oraz nieselektywne blo-
powstałych wskutek nadciśnienia wrotnego w prze- kery receptorów β.
biegu marskości wątroby nadają się donaczyniowe
6.9. Leki żółciotwórcze i żółciopędne;
środki rozpuszczające kamienie żółciowe
Leki żółciotwórcze zwiększają ilość wytwarzanej żółci w wą- Przeciwwskazaniem do stosowania leków żółciotwórczych
trobie, leki żółciopędne wspomagają opróżnianie pęcherzyka jest ostre zapalenie wątroby, ropień pęcherzyka żółciowego
żółciowego. Pojęcia te zastąpiły niedokładne określenie cho- oraz mechaniczna niedrożność dróg żółciowych wywołana
lagoga. Terapeutyczne znaczenie tych substancji jest jednak np. przez kamienie żółciowe lub guz.
niewielkie pomimo ich dosyć częstego stosowania.
Substancją o działaniu spazmolitycznym i żółciopędnym
Leki żółciotwórcze. Działanie żółciotwórcze wykazują jest hymekromon.
kwasy żółciowe, które po resorpcji z jelita cienkiego jako
substancje odpowiedzialne za wytwarzanie żółci powodują Leki żółciopędne stosuje się w zaburzeniach przepływu
jej wydzielanie. Są to żółć wołowa (fel tauri) oraz kwas żółci (dyskineza) z pęcherzyka żółciowego i dróg żółcio-
dehydrocholowy. Syntetycznym lekiem żółciotwórczym wych oraz w celu wykonania badania radiologicznego
jest febuprol. oceniającego zdolność opróżniania pęcherzyka żółciowe-
go. Roztwór siarczanu magnezu (10–30-procentowy) lub
Leki żółciotwórcze są wskazane tylko wówczas, gdy ko- roztwór sorbitolu (80-procentowy) powodują odruchowe
nieczne jest wzmożenie przepływu żółci przez odprowa- opróżnianie pęcherzyka żółciowego. W przypadku zabu-
dzające drogi żółciowe. Dotyczy to np. zalegania złogów rzenia motoryki dróg żółciowych wspomagająco działają
piasku żółciowego. także leki rozkurczające.
Układ trawienny
OH CH3
O O
(CH2) 3CH3
HO O O
febuprol B6
hymekromon
H3C COOH H3C COOH
CH3 CH3
CH3 H CH3 H
H H H H
HO OH HO OH
H H
kwas chenodeoksycholowy kwas ursodeoksycholowy
MUTSCHLER-2009.indd 703 2010-01-07 22:14:19
704 Układ trawienny
Kamica żółciowa (cholelithiasis). Kamica żółciowa ■ wydzielania cholesterolu z żółcią (kwas ursode-
jest najczęstszą chorobą dróg żółciowych i pęcherzyka oksycholowy, kwas chenodeoksycholowy),
żółciowego. Zapadalność na tę chorobę wśród osób doro-
słych wynosi 10%. Kobiety dotyka dwukrotnie częściej niż ■ syntezy cholesterolu przez blokowanie reduktazy
mężczyzn. Kamienie tworzą się najczęściej w pęcherzyku HMG-CoA (kwas chenodeoksycholowy),
żółciowym, a nieco rzadziej w całym układzie dróg żół-
ciowych. W zależności od związku stanowiącego główny ■ resorpcji jelitowej cholesterolu (kwas ursodeoksy-
składnik kamienie żółciowe dzieli się na kamienie choleste- cholowy).
rolowe, które zawierają cholesterol (> 70%), sole wapnia
oraz białko matriksowe (częstość występowania 75–80%), U 60% pacjentów poddanych leczeniu w ciągu 6–24
oraz kamienie pigmentowe, które zawierają bilirubinę (czę- miesięcy następuje rozpuszczenie kamieni żółcio-
stość występowania 20–25%). Czynnikami sprzyjającymi
wych. Zagrożenie nawrotem choroby po zakończeniu
tworzeniu się kamieni żółciowych są hipercholesterolemia
oraz zaburzenia opróżniania pęcherzyka żółciowego. Tylko terapii dotyczy 25% pacjentów.
u 25% wszystkich nosicieli kamieni występuje kolka ka- Dawkowanie kwasu chenodeoksycholowego wy-
micowa. nosi 15 mg/kg masy ciała, a kwasu ursodeoksycholo-
wego 10 mg/kg masy ciała.
Farmakologiczne rozpuszczanie kamieni żółcio- Opisywane działania niepożądane kwasu chenode-
wych. Próba farmakologicznego rozpuszczenia ka- oksycholowego to przede wszystkim zaburzenia żo-
mieni żółciowych może zakończyć się sukcesem tyl- łądkowo-jelitowe, biegunki oraz przemijające zwięk-
ko w przypadku kamieni cholesterolowych. Są one szenie stężenie transaminaz w osoczu. Kwas ursode-
wykrywalne jedynie w badaniu sonograficznym, po- oksycholowy jest lepiej znoszony.
nieważ w badaniu radiologicznym się nie ujawniają. Obydwie wymienione substancje są przeciwwska-
Warunkiem przeprowadzenia zabiegu jest prawidło- zane w chorobach zapalnych dróg żółciowych, nie-
wa czynność pęcherzyka żółciowego oraz brak cho- drożności przewodu żółciowego wspólnego lub pę-
rób wątroby. cherzyka, w ciężkich zaburzeniach czynnościowych
Rozpuszczanie kamieni żółciowych następuje pod wątroby lub nerek, w często występujących kolkach
wpływem kwasu chenodeoksycholowego (cheno- oraz w okresie ciąży.
diol) lub kwasu ursodeoksycholowego (ursodiol). Cholestyramina i leki neutralizujące zawierające
Ich działanie polega na hamowaniu: glin ograniczają ich wchłanianie.
MUTSCHLER-2009.indd 704 2010-01-07 22:14:20
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 705
7. Nerki i drogi moczowe,
gospodarka wodno-elektrolitowa
i kwasowo-zasadowa
7.1. Podstawy anatomiczne i fizjologiczne
Nerki pełnią podstawową rolę w utrzymaniu homeo- węglanów (węglanów), a tym samym utrzymanie
stazy całego organizmu. Biorą bowiem udział w ta- fizjologicznej wartości pH (izohydria),
kich procesach, jak:
■ (długotrwała) regulacja ciśnienia krwi poprzez
■ wydzielanie produktów przemiany materii układ renina-angiotensyna-aldosteron,
(np. związków znajdujących się prawidłowo mo-
■ odtwarzanie białka osoczowego (np. albumin, lizo-
czu, np. mocznika, kwasu moczowego i kreatyny,
zymu, białka wiążącego retinol) i hormonów pep-
a także substancji obcych (ksenobiotyki) i ich me-
tydowych (np. insuliny, parathormonu).
tabolitów. Jednocześnie odpowiadają za zatrzy-
mywanie komórek krwi i białek osoczowych oraz
Nerki syntetyzują ponadto ważne hormony (kalcy-
odzyskiwanie z moczu w wyniku resorpcji kana-
triol, erytropoetynę) oraz są narządem wykonaw-
likowej ważnych substratów (np. glukozy, amino-
czym hormonów pozanerkowych (np. wazopresyny,
kwasów),
aldosteronu, kalcytoniny, peptydów natriuretycz-
■ kontrola gospodarki wodno-elektrolitowej po- nych, parathormonu). Może w nich także zachodzić
przez regulowane wydzielanie wody i elektrolitów glukoneogeneza.
(np. jonów Na+, K+, Ca2+, Mg2+, Cl– i fosforanów)
i tym samym utrzymywaniu stałej objętości pły-
nu zewnątrzkomórkowego (izowolemia), stęże- 7.1.1. Anatomia makroskopowa
nia osmotycznego (izotonii) i równowagi jonowej
(izojonii), nerki
■ regulowanie gospodarki kwasowo-zasadowej po-
Nerki są narządem parzystym, przypominającym
przez wydzielanie protonów ew. retencję wodoro-
kształtem fasolę, położonym w grzbietowej części
jamy brzusznej poniżej przepony i po obu stronach
kręgosłupa. Brzeg wypukły skierowany jest bocznie,
warstwa na wklęsłym brzegu dośrodkowym znajduje się wnę-
warstwa rdzeniowa
gosodarka wodno-elektr.
korowa nerki (kora) nerki (rdzeń) ka nerki (hilus), z której odchodzą naczynia, nerwy
torebka brodawka
i moczowód. Nerki i drogi moczowe,
włóknista nerkowa U osoby dorosłej długość przekroju podłużnego
miedniczka nerki wynosi 10–12 cm, długość przekroju poprzecz-
nerkowa nego 5–7 cm, a masa około 100–200 g.
tętnica
nerkowa Powierzchnię nerki chroni mocna torebka włókni-
kielich
nerkowy żyła sta. W obrazie mikroskopowym przekroju podłużne-
nerkowa go (ryc. B 7.1-1) wyróżnia się:
tkanka moczowód
tłuszczowa ■ korę nerki (cortex renalis),
■ rdzeń nerki (medulla renalis), B7
■ miedniczkę nerkową (pelvis renalis) z moczowo-
dem.
słupy nerkowe Zewnętrzna warstwa korowa nerki jest jasna i drobno-
ziarnista, wewnętrzna warstwa rdzeniowa jest ciem-
Ryc. B 7.1-1. Przekrój podłużny nerki. niejsza i podłużnie prążkowana. Wnikając w rdzeń,
MUTSCHLER-2009.indd 705 2010-01-07 22:14:20
706 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
kora tworzy słupy nerkowe dzielące rdzeń na 8–17
piramid, które podstawą przylegają do kory a wierz- część kręta kłębuszek nerkowy tętniczka doprowadzająca
chołkami zbiegają się w centrum nerki. Wierzcho- tętniczka odprowadzająca
łek każdej piramidy, nazywany brodawką nerkową,
uchodzi do woreczkowatego kielicha nerkowego.
część
Wychwytują one gotowy mocz (mocz ostateczny) kręta
i odprowadzają go do zbiornika w miedniczce ner- kanalik
kowej. proksymalny
(kręty)
Zaopatrywanie nerki w krew następuje przez odcho- część prosta
dzące od aorty tętnice nerkowe (arteria renali), które
po dotarciu do wnęki rozgałęziają się na tętnice mię- kanalik
dzypłatowe (arteriae interlobares). Te między pira- dystalny
midami biegną ku korze nerki i u podstawy piramid pętla Henlego
tworzą tętnice łukowate (arteriae arcuatae), od któ-
rych odchodzą tętnice międzypłacikowe (arteriae kanalik zbiorczy
interlobulares). Od tych ostatnich, w regularnych
odstępach odgałęziają się zaopatrujące poszczegól-
ne ciałka nerkowe (zob. niżej) tętniczki zaopatrują- odcinek
przejściowy
ce (vasa afferentia), które najpierw tworzą (pierw-
szą) sieć kapilarną, a następnie łączą się ponownie
w naczynia odprowadzające (vasa efferentia). W tym
miejscu powstaje drugi, okołokanalikowy system ka- Ryc. B 7.1-2. Schemat budowy nefronu.
pilarny, którego zadaniem jest zaopatrywanie w krew
kanaliki i kanalik zbiorczy (zob. niżej).
Układ żylny zbudowany analogicznie do układu Ściany kapilar kłębuszków nerkowych zbudowane
tętniczego odprowadza krew żylną do żyły nerkowej są z dwóch warstw: śródbłonka składającego się z komó-
(vena renalis). rek posiadających liczne „okienka” oraz błony podstawnej,
w której zgromadzone są ujemnie naładowane glikozami-
noglikany (tzw. bariera anionowa). Na zewnątrz od błony
podstawnej odchodzą natomiast wypustki stopowate tzw.
7.1.2. Anatomia mikroskopowa nerki podocytów, tj. komórek nabłonkowych torebki Bowmana.
Wypustki te tworzą wokół kapilarów warstwę z licznymi
Podstawowym elementem strukturalnym nerek
jest nefron (B 7.1-2). Każda nerka ludzka zawie-
ra około 1 miliona tych jednostek morfologicznych
i funkcjonalnych. Nefron składa się z: kanalik dystalny
komórki plamki
■ ciałek nerkowych, gęstej
komórki tętniczka
■ aparatu kanalikowego. nabłonkowe odprowa-
dzająca
Ciałko nerkowe. Ciałko nerkowe (ryc. B 7.1-3) skła- tętniczka
da się z kłębka naczyń włosowatych i otaczającej doprowa-
go torebki Bowmana. Blaszka wewnętrzna torebki dzająca
Bowmana okrywa kłębuszek nerkowy, blaszka ze-
wnętrzna okrywa torebkę od zewnątrz i przechodzi
w ścianę kanalika proksymalnego. Jak wspomniano,
krew tętnicza dostaje się do kłębuszka nerkowego torebka
naczyniami doprowadzającymi (vasa afferentia) naczynia włosowate Bowmana
a opuszcza go przez naczynia odprowadzające (vasa kłębka nerkowego
kanalik proksymalny
efferentia). Obydwa położone blisko siebie naczynia (kręty)
tworzą biegun naczyniowy ciałka nerkowego, na-
przeciw którego, na początku aparatu kanalikowego,
Ryc. B 7.1-3. Ciałko nerkowe z komórkami plamki gęstej (macu-
znajduje się biegun moczowy.
la densa) (wg Kristić). Tętniczka doprowadzająca jest otwarta.
MUTSCHLER-2009.indd 706 2010-01-07 22:14:20
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 707
szparami filtracyjnymi i stanowią ważny element błony łęzienia naczyń doprowadzających do drugiej sieci
filtracyjnej naczynia włosowatego kłębuszka nerkowego. kapilarnej, która oplata aparat kanalikowy znajdujący
Cienka silnie ujemnie naładowana błonka rozciągnięta
się w korze i zewnętrznej strefie rdzenia nerki. We-
między sąsiadującymi ze sobą wypustkami stopowaty-
mi zamyka „okienka” w śródbłonku. Torebkę Bowmana wnętrzna warstwa rdzenia jest natomiast zaopatrywa-
od światła kapilara oddzielają więc trzy warstwy, tworzące na przez długie odcinki naczyń prostych (vasa recti),
wspólnie tzw. barierę filtracyjną o łącznej powierzchni oko- które odchodzą od tętnic łukowatych (arteriae arcua-
ło 1 m2. Między poszczególnymi kapilarami, a w większej tae) i naczyń doprowadzających najniżej położonych
ilości przy biegunie naczyniowym, zgromadzone są komór-
kłębuszków nerkowych.
ki mezangialne tworzące dla kłębuszka rodzaj rusztowania.
Dzięki zdolności do fagocytozy mogą one usuwać złogi
gromadzące się w obszarze filtracyjnym, a zawartość akty-
ny i miozyny decyduje o ich kurczliwości. 7.1.3. Ukrwienie nerek
Wszystkie ciałka nerkowe położone są w obszarze
Łączna ilość krwi przepływającej przez obie nerki do-
warstwy korowej, przy czym te leżące bliżej po-
rosłego człowieka wynosi 1,2 l/min lub 1700 1/dzień,
wierzchni mają kłębuszki korowe, a te powyżej gra-
co odpowiada 20–25% pojemności minutowej ser-
nicy korowo-rdzeniowej kłębuszki przyrdzeniowe.
ca w pozycji spoczynkowej. Na korę nerki przypada
około 90% tej całości, na rdzeń zaledwie 10%. Nerki
Aparat kanalikowy. W przypadku aparatu kanaliko-
należą tym samym do najlepiej ukrwionych narzą-
wego istotny jest stosunek niezwykle długiego ukła-
dów, co jest konieczne do uzyskania wystarczającej
du kanalikowego do jego przekroju. W układzie ka-
ilości przesączu kłębuszkowego oraz warunkiem
nalikowym rozróżnia się następujące odcinki:
efektywnie pełnionej funkcji oczyszczającej.
■ kanaliki proksymalne z częścią krętą (pars convo- Pomiary przepływu naczyniowego u ludzi prze-
luta s. contorta) i częścią prostą (pars recta), prowadza się przez pośrednie oznaczenie tzw. kwasu
paraaminohipurowego (klirens PAH). PAH jest sub-
■ odcinek przejściowy (część cienka pętli Henlego),
stancją, która po jednorazowym przejściu przez nerkę
■ kanaliki dystalne z częścią krętą (pars convoluta s. jest prawie całkowicie eliminowana z krwi. Dlatego
contorta) i częścią prostą (pars recta), wartość klirensu (CLPAH) jest identyczna z przepły-
wem nerkowym osocza (RPF):
■ kanalik zbiorczy.
CLPAH = RPF
Część prosta kanalika proksymalnego i dystalnego
oraz odcinek przejściowy określane są wspólnie jako
pętlą Henlego. Kręte odcinki kanalików leżą w części
korowej nerki, odcinki proste przeważnie bliżej czę- 7.1.4. Przesączanie kłębuszkowe,
ści rdzeniowej. powstawanie moczu
Na niewielkim odcinku kanalik dystalny styka się pierwotnego
z tętniczką doprowadzającą (vas efferens) oraz z połą-
gosodarka wodno-elektr.
czonym z nim ciałkiem nerkowym. Obszar nabłonka Z osocza przepływającej krwi odsącza się w kłębusz-
w miejscu zetknięcia się zawiera wiele jąder i nazy- kach nerkowych prawie bezbiałkowy ultrafiltrat, Nerki i drogi moczowe,
wany jest plamką gęstą (macula densa) (zob. B 7-3). mocz pierwotny. Możliwość przejścia cząsteczek
Komórki leżące w obrębie plamki wraz z komórkami z naczyń włosowatych do wnętrza torebki Bowmana
sąsiadującymi z naczyniem doprowadzającym two- zmniejsza się stopniowo, począwszy od cząsteczek
rzą aparat przykłębuszkowy. wielkości 1,5 nm. Przy cząsteczkach większych niż
Po kontakcie z ciałkiem nerkowym część krę- 4 nm przepuszczalność zanika całkowicie (tzw. filtr
ta (pars convoluta) kanalika dystalnego przechodzi kłębuszkowy). Ograniczona filtracja dotyczy rów-
w kanalik zbiorczy, który zbiera i odprowadza mocz nież cząsteczek o masie od 5000 Da do 50 000 Da.
z brodawek nerkowych. Każdy kanalik zbiorczy Przez nieuszkodzony filtr zatrzymywane są prawie B7
ma liczne dopływy z sąsiednich nefronów. Kanaliki całkowicie także najmniejsze białka osocza, albu-
zbiorcze łączą się pod ostrym kątem w większe ka- miny, ponieważ ich masa cząsteczkowa przekracza
naliki, które w wierzchołku piramidy, brodawce ner- 70 000 Da.
kowej, uchodzą do kielicha nerkowego. Przeciętna Filtracja kłębuszkowa zależy jednak nie tyl-
długość kanalika zbiorczego wynosi 22 mm. ko od szerokości szczelin filtracyjnych, ale przede
Zarówno kanaliki proksymalne i dystalne, jak i ka- wszystkim od efektywnego ciśnienia filtracyjnego
nalik zbiorczy są zaopatrywane w krew przez odga- (Peff) panującego w kłębuszkach nerkowych oraz
MUTSCHLER-2009.indd 707 2010-01-07 22:14:21
708 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
od liczby czynnych kłębuszków. Na efektywne ciś- metabolicznych nerek i nie jest toksyczny. Dlatego
nienie filtracyjne składa się ciśnienie krwi w kapi- obowiązuje równanie:
larach (PKap) pomniejszone o koloidalne ciśnienie
osmotyczne osocza krwi (Pkod) i ciśnienia hydrosta- CL inulina = GFR.
tyczne w torebce Bowmana (PBow = wartość ciśnienia
hydrostatycznego pomniejszona o wartość koloidal- Rolę czynnika endogennego dla określenia GFR
nego ciśnienia osmotycznego). Ponieważ ciśnienie może pełnić również kreatynina, posiadająca podob-
krwi w początkowym odcinku naczyń włosowatych ne właściwości do inuliny, a przy podwyższonym
kłębuszka wynosi 48 mm Hg, koloidalne ciśnienie stężeniu w osoczu krwi powodująca również ograni-
osmotyczne 25 mm Hg a ciśnienie w torebce Bow- czone przesączanie kanalikowe. Klirens kreatyniny
mana 12 mm Hg, efektywne ciśnienie filtracyjne wy- dostarcza więc wprawdzie, szczególnie w przypadku
nosi 11 mm Hg: niewydolności nerek, niedokładne (zawyżone) war-
tości GFR, jest jednak, ze względu na możliwość
Peff = PKap – PKod – PBow = 48 – 25 – 12 = 11 mm Hg pominięcia infuzji, o wiele łatwiejszy do oznaczenia.
Kreatynina powstaje – we względnie stałych ilościach
Do końca naczyń włosowatych kłębuszków ner- – z fosfokreatyny w wyniku metabolizmu mięśnio-
kowych efektywne ciśnienie filtracyjne zmniejsza wego. W przypadku zmniejszenia się wartości GFR
się w bardzo niewielkim stopniu, natomiast znacz- konieczny jest w związku z tym wzrost stężenia kre-
nie zwiększa się koloidalne ciśnienie osmotycz- atyniny w osoczu krwi.
ne, co spowodowane jest wzrostem stężenia białek
osocza na skutek odsączania ultrafiltratu. W efekcie W przeciwieństwie do oznaczenia klirensu pomiar
na końcu naczyń włosowatych efektywne ciśnienie jej stężenia kreatyniny w osoczu krwi jest niewystarczający
do dokładnej oceny funkcjonowania nerek, ponieważ uza-
filtracyjne spada do zera. leżniony jest nie tylko od wydalania kreatyny przez ner-
ki, ale również od produkowanej w mięśniach ilości tego
Przesączanie kłębuszkowe. Objętość przesączu od- związku. To wyjaśnia, dlaczego nawet przy prawidłowym
filtrowanego w jednostce czasu we wszystkich kłę- poziomie kreatyny możliwe jest znaczne ograniczenie
buszkach określa się jako wskaźnik przesączania klę- funkcjonowania nerek, jak np. u osób starszych, z powodu
znacznego zmniejszenia masy mięśniowej i spadku wydaj-
buszkowego (GFR). U młodych mężczyzn wynosi on ności pracy mięśni.
125 ml/min, u młodych kobiet 110 ml/min. W ciągu
doby wytwarzanych jest 180 1 moczu pierwotnego,
Za pomocą mikropunkcji naczyń kapilarnych kłę-
co oznacza, że całkowita objętość osocza wynosząca
buszków nerkowych można także ustalić poziom fil-
3 l jest w ciągu doby poddawana procesowi oczysz-
tracji w poszczególnych kłębuszkach.
czania nerkowego około 60 razy.
U pacjentów w starszym wieku GFR jest często znacznie Regulacja współczynnika przesączania klębusz-
zmniejszony. Spadek zdolności filtracyjnej nerki w wieku kowego. Warunkiem równomiernego przesączania
podeszłym nie wynika jednak wyłącznie z procesu starzenia klębuszkowego jest stałe ukrwienie ew. ciśnienie
się organizmu, ale raczej ze zmian chorobowych w obrębie w kapilarach kłębuszków. Zapewnia je autoregulacja
nerki (np. nadciśnienia, miażdżycy, zapalenia kłębuszko- przepływu, w której biorą udział zarówno naczynia
wego nerek). Wyjaśnia to duże różnice międzyosobnicze
czynności nerek u starszych ludzi. doprowadzające, jak i odprowadzające.
Działanie komponentu mięśniowego polega
na tym, że przy spowodowanym otwarciem kanałów
Do oznaczenia GFR używa się klirensu substancji
wapniowych wzroście ciśnienia w tętniczkach nerko-
egzogennych lub endogennych, które mogą być bez
wych i wynikającym z tego zwiększeniem wewnątrz-
przeszkód filtrowane w kłębuszkach nerkowych,
komórkowego stężenia jonów wapniowych podnosi
a w aparacie kanalikowym ani nie podlegają wchła-
się napięcie mięśniówki a przepływ krwi zaczyna
nianiu zwrotnemu, ani nie ulegają przesączeniu.
przebiegać sprawniej (efekt Baylissa).
Czynnikiem egzogennym do oznaczenia klirensu,
Innym mechanizmem składającym się na auto-
który musi być wprowadzony na drodze dożylnej in-
regulację jest kanalikowo-kłębuszkowe sprzężenie
fuzji długotrwałej, aby w trakcie wykonywania po-
zwrotne (TGF – tubuloglomerular feedback), tzn.
miaru możliwe było utrzymanie jego stałego stężenia
przepływ zwrotny od kanalika do bieguna naczynio-
w osoczu, może być należąca do polisacharydów inu-
wego ciałka nerkowego. Przy rosnącym ciśnieniu
lina. Związek ten nie tylko spełnia podane wymaga-
w kapilarach kłębuszka zwiększa się wielkość fil-
nia, ale ponadto nie bierze udziału w czynnościach
tracji i przepływ płynu w kanalikach. Skutkiem tego
MUTSCHLER-2009.indd 708 2010-01-07 22:14:21
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 709
może być niewystarczająca ilość NaCl resorbowana 7.1.5. Transport kanalikowy.
zwrotnie z płynu kanalikowego a tym samym wzrost Wytwarzanie moczu
stężenia NaCl w początkowych odcinkach kanalików
dystalnych. Mechanizm ten oddziałuje na komórki
pierwotnego
plamki gęstej, powodując zwiększenie aktywności
zawartych w nich symporterów Na+/K+/2Cl¯ i wzrost Przesączony mocz pierwotny przechodzi przez apa-
stężenia wymienionych trzech jonów w komórkach rat kanalikowy i kanalik zbiorczy, po czym w wy-
nerkowych. W wyniku rozpadu ATP i ADP rozpo- niku procesu wchłaniania i sekrecji przekształcany
czyna się wzmożona produkcja adenozyny, która jest w mocz (końcowy).
stymuluje receptory A1 i podwyższa przez to stęże-
nie jonów Ca2+ w mięśniówce naczyń krwionośnych.
Wzrost stężenia jonów wapniowych prowadzi zaś 7.1.5.1. Kanalikowe wchłanianie zwrotne
do skurczu tętniczek i redukcji ilości krwi dostarcza-
nej do kapilar kłębuszków oraz obniżenia ciśnienia
Podczas przejścia przez różne odcinki kanalików
wewnątrzkapilarnego.
skład moczu pierwotnego ulega dużej zmianie. Więk-
Obecnie dyskutuje się również m.in. o roli mecha-
sza część rozpuszczonych składników oraz 99% wody
nizmów, w których bierze udział prostaglandyna E2
jest wchłaniania zwrotnie i przechodzi do krwiobie-
i inne mediatory (np. peptydy natriuretyczne) oraz
gu. Tym samym należy rozróżnić transport czynny,
miejscowy układ renina-angiotensyna-aldosteron
wymagający nakładu energii, oraz transport bierny
stymulowany za pośrednictwem baroreceptorów
(dyfuzję, transport za pośrednictwem nośników) (tab.
w naczyniach doprowadzających.
B 7.1-1, ryc. B 7. 1-4).
Opisane mechanizmy gwarantują z jednej strony,
że w przypadku zmiany średniego ciśnienia tętnicze-
Kanalik proksymalny. W kanaliku proksymalnym
go mieszczącego się w przedziale od 90 do 190 mm
ulegają resorpcji nie tylko elektrolity (zob. niżej),
Hg będzie utrzymane stałe ciśnienie w kapilarach
ale również glukoza i aminokwasy (kotransport z jo-
kłębuszkowych, a z drugiej strony, że w przypadku
nami sodu) oraz minimalne ilości białek obecnych
wzrostu ilości filtrowanego NaCl nastąpi przeciw-
w przesączu. Z reguły związki te ulegają prawie cał-
działanie utracie soli i wody poprzez przejściowe
kowicie resorpcji wtórnej. Jedynie w przypadku, gdy
zmniejszenie objętości przesączu.
A. Kanalik proksymalny B. Gruby przewód wznoszący C. Kanalik zbiorczy
światło komórka obszar światło komórka obszar światło komórka obszar
ukrwiony ukrwiony ukrwiony
+ 78 mV – 76 mV + 78 mV – 72 mV + 20 mV – 72 mV
gosodarka wodno-elektr.
A Nerki i drogi moczowe,
HCO3- Na+ 3Na + AR
H+ 2 Cl- Cl- ATP
Na+ 3Na+ K+ 2K+
2K+
ATP
Na+ 3Na+
K+
ATP
+ 2K+
H K+ K+
CA CA Cl-
K+ K+
-
H2CO3 CO2 H2CO3 HCO3
Cl-
Na+ Akwa-
Na + HCO3-
poryna H2O B7
+
Na
ADH
V2
- PKA
Gs
Cl
cAMP↑
Ryc. B 7.1-4. Mechanizm resorpcji NaCl i wody w nefronach (wg Gregera).
MUTSCHLER-2009.indd 709 2010-01-07 22:14:21
710 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
Tabela B 7.1-1. Mechanizmy transportu w kanalikach (wybór; wg Thewsa, Mutschlera, Vaupela)
Transportowana Kanalik proksymalny Pętla Henlego Kłębuszek dystalny Kanalik zbiorczy
substancja (TAL)
resorpcja Na+ a symporter Na+/substrat symporter Na+/2Cl-/K+ symporter Na+/Cl¯ kanał Na+ (ENaC)
antyporter Na+/H+
pozakomórkowo
(FR ≈ 65%) (FR ≈ 25%) (FR 5%) (FR ≈ 4%)
b 3 Na+/2K+
resorpcja Cl- a pozakomórkowo symporter Na+/2Cl¯/H+ symporter Na+/Cl¯ antyporter Na+/HCO3¯
antyporter Cl¯/zasada pozakomórkowo
(FR ≈ 60%) (FR ≈ 30%) (FR ≈ 5%) (FR ≈ 4%)
b kanał Cl- kanał Cl- kanał Cl- kanał Cl-
symporter K+/Cl-
resorpcja wody a,b kanały wodne pozakomórkowo (tylko DH zależne
(akwaporyna-1) w ramieniu zstępującym) ø akwaporyna-2
pozakomórkowo
(FR ≈ 67%) (FR ≈ 13%) (FR ≈ 10-18%)
resorpcja K+ a pozakomórkowo symporter Na+/2Cl¯/H+ K+/H+-ATPazy (międzykomórkowo)
pozakomórkowo
(FR ≈ 65%) (FR ≈ 20%) (FR ≈ 0-10%)
b kanał K+ kanał K+
sekrecja K+ a kanał K+ (komórki główne)
b 3 Na+/2 K+-ATPazy
resorpcja Ca2+ a pozakomórkowo pozakomórkowo kanał Ca2+
(FR ≈ 60%) (FR ≈ 30%) (FR ≈ 8%)
b Ca2+-ATPazy
antyporter 3 Na+/Ca2+
resorpcja Mg2+ a pozakomórkowo pozakomórkowo kanał Mg2+
(FR ≈ 15-20%) (FR≈70%) (FR ≈ 5-10%)
b antyporter 3 Na+/Mg2+
resorpcja a symporter Na+/Pi nieznane
fosforanów (FR ≈ 70-80%) (FR ≈ 10%)
b
resorpcja a symporter Na+/glukozy
glukozy (SGLT 2, początkowy odcinek proksymalny)
symporter 2Na+/glukozy
(SGLT1, końcowy odcinek proksymalny)
(FR ≈ 100%)
b GLUT 2
resorpcja a symporter Na+/AK
aminokwasów (AK) antyporter (dwuzasadowe
AK+cystyna)
b (FR ≈ 100%)
nośniki AK
resorpcja moczu a dyfuzja, nośnik moczu
pozakomórkowo (UT 1, dolny odcinek
(FR≈50%) kanalika zbiorczego)
(FR ≈ 70%)
b nośnik moczu (UT4)
MUTSCHLER-2009.indd 710 2010-01-07 22:14:21
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 711
Tabela B 7.1-1. Mechanizmy transportu w kanalikach (wybór; wg Thewsa, Mutschlera, Vaupela) (kontynuacja)
Transportowana Kanalik proksymalny Pętla Henlego Kłębuszek dystalny Kanalik zbiorczy
substancja (TAL)
sekrecja moczu a,b nośnik moczu
(UT 2, cieńsza część
pętli Henlego
(FE ≈ 50%)
resorpcja a,b antyporter moczan/aniony
kwasu moczowego (FR ≈ 90%)
TAL – odcinek wstępujący, a – szczytowy, b – boczno-podstawny, FR – resorpcja frakcyjna, FE – ekskrecja frakcyjna
ich stężenie w przesączu kłębuszkowym przekroczy W wyniku opisanych procesów transportowych
wartość progową, są wydalane z moczem. Wartości płyn kanalikowy staje się słabo hipotoniczny. Prowa-
progowe są szczególnie istotne przy wchłanianiu dzi to do pozakomórkowej resorpcji wody, przy czym
zwrotnym glukozy. Prawidłowe stężenie glukozy – w odniesieniu do leków rozpuszczalnych – chlorek
w osoczu, a tym samym w przesączu kłębkowym, sodu jest transportowany razem z nią. Pozostała część
wynosi 0,6–1,0 g/1. W tych stężeniach nośnik w kana- jonów sodowych i chlorkowych – również przezko-
likach proksymalnych jest w stanie umożliwić całko- mórkowo – dyfunduje ponownie do krwi. Oznacza
wite wchłonięcie przesączanej glukozy. Obowiązuje to więc, że tylko część jonów sodowych podlega
to również wtedy, gdy po posiłku bogatym w węglo- przezkomórkowemu wchłanianiu zwrotnemu. Ko-
wodany stężenie glukozy wzrośnie do wartości 1,4 rzystne dla organizmu jest to, że tylko jedna trzecia
g/1. Przy wartościach 1,6–1,8 g/1 osiągany jest próg, jonów sodowych musi być transportowana czynnie,
przy którym wszystkie miejsca uchwytu przenośni- tzn. przy nakładzie energii, natomiast dwie trzecie
ków glukozy są wysycone (osiągnięcie maksymalnej przedostaje się ponownie do krwiobiegu biernie, tzn.
możliwości transportu). Dalsze zwiększanie stężenia bez wykorzystania ATP.
powoduje wydalanie glukozy z moczem (glikozu-
rię). Pętla Henlego. W obrębie pętli Henlego, a zwłaszcza
Około 60–70% chlorku sodu i wody przesączo- w jej grubym ramieniu wstępującym (ryc. B 7.1-2),
nych w kłębuszkach jest resorbowane w kanaliku wchłanianych jest około 20–30% chlorku sodu,
proksymalnym. Na tym odcinku kanalika również ale tylko 10% wody. Zasadnicza różnica między
znaczna część jonów potasu przedostaje się z powro- pętlą Henlego a kanalikiem proksymalnym polega
tem z moczu pierwotnego do krwiobiegu. na tym, że o ile aktywny transport jonowy na tym
Konieczny do resorpcji spadek stężenia jonów odcinku kanalika odbywa się wprawdzie (podob-
sodu postępujący od światła kanalika w kierunku na- nie jak w całym aparacie kanalikowym) również
gosodarka wodno-elektr.
błonka kanalika jest utrzymywany dzięki znajdującej za pośrednictwem pompy Na+/K+ w błonie około-
się w tzw. błonie okołokanalikowej (boczno-pod- kanalikowej, o tyle jeden jon sodowy, dwa jony po- Nerki i drogi moczowe,
stawnej) pompie Na+/K+, właściwemu „motorowi” tasu i jeden jon chlorkowy mogą być przenoszone
odzyskiwania Na+ i powiązanych z nim substancji. wspólnie ze światła kanalika do komórek kanalików
Równowagę dla przyjęcia do komórek jonów sodo- za pomocą symporterów w błonie luminalnej (sym-
wych stanowi oddanie protonów do światła kanalika portery Na+/K+/2Cl¯). Recyrkulacja jonów potasu
(antyport błonowy sodu i potasu) i zachodząca tam odbywa się przez kanały potasowe w błonie lumi-
reakcja z węglowodorami zakończona powstaniem nalnej.
CO2 i wody. CO2 dyfunduje do wnętrza komórki Wchłanianie chlorków w kanalikach proksymal-
i w obecności anhydrazy węglanowej powtórnie wy- nych odbywa się przede wszystkim pozakomórko- B7
twarza dwuwęglany, które mogą opuścić komórkę wo, w ramieniu wstępującym pętli Henlego, które
kanalika przez błonę okołokanalikową, oraz protony. w znacznym stopniu jest nieprzepuszczalne dla ma-
Jony sodowe mogą być transportowane ze światła łych anionów. Wszystkie jony chlorkowe są trans-
kanalika do komórek nabłonka na drodze antyportu portowane przezkomórkowo. W grubym wstępują-
z jonami wodorowymi, jak już wspomniano, oraz cym ramieniu pętli Henlego odbywa się wchłanianie
w symporcie razem z glukozą, aminokwasami, siar- magnezu. Około dwóch trzecich filtrowanych jonów
czanami, mleczanami i in. magnezowych jest tu (biernie i przeważnie pozako-
MUTSCHLER-2009.indd 711 2010-01-07 22:14:22
712 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
mórkowo) przeprowadzonych ze światła kanalików nów sodowych w kanaliku dalszym, stężenie aldoste-
do krwiobiegu. ronu i wartość pH (hipokaliemia alkaliczna).
Kanalik dystalny i kanalik zbiorczy. W początkowym Objętość wody resorbowanej w tych odcinkach kana-
odcinku kanalika dystalnego wchłanianiu zwrotne- lików zależy od bilansu płynów. Jeżeli w organizmie
mu podlega około 5% przesączanych chlorków sodu pojawi się konieczność oszczędzania wody i tym sa-
i 10–20% wody. Resorpcja jonów sodowych i chlor- mym tworzenia niewielkiej ilości moczu końcowe-
kowych odbywa się na zasadzie kotransportu. go, następuje zwiększenie resorpcji zwrotnej w ka-
W końcowych odcinkach kanalików dystalnych nalikach dystalnych. W takim przypadku 99–99,5%
oraz w kanaliku zbiorczym jony sodowe przechodzą objętości przesączu powraca do krwiobiegu. Kiedy
przez kanały sodowe do komórek kanalika. Częstość jednak w wyniku dostarczenia do organizmu znacz-
i czas otwarcia kanałów sodowych, a tym samym re- nej ilości płynów musi być usunięta duża ilość wody,
sorpcja jonów sodowych uzależniona jest od stęże- jej ponowne przyjmowanie do krwiobiegu w kanali-
nia aldosteronu (zob. niżej) ewentualnie od stężenia kach dystalnych ulega ograniczeniu (por. B 7.1.5.3.).
białka indukującego syntezę aldosteronu: im więcej Ze względu na tę możliwość regulacji proces wchła-
aldosteronu jest uwalnianego z kory nadnerczy, tym niania wody w kanalikach dystalnych i w kanaliku
więcej jest otwartych kanałów sodowych. zbiorczym określa się jako warunkowy transport
W przypadku jonów potasu kierunek transportu wodny.
w kanalikach dystalnych jest uzależniony od meta-
bolizmu potasu. Jeżeli podaż potasu w pożywieniu Wzmocnienie przeciwprądowe w mechanizmie
jest za mała, następuje jego wchłanianie zwrotne, je- zagęszczania moczu. W dotychczasowych wy-
żeli dieta zawiera w produkty bogate w potas, zwięk- jaśnieniach dotyczących transportu kanalikowego
sza się jego sekrecja do światła kanalika. Wydzielanie na różnych odcinkach aparatu kanalikowego pomi-
potasu jest tym większe, im wyższe jest stężenie jo- nięto istniejące między nimi zależności przestrzenne.
ramię ramię transport aktywny
zstępujące wstępujące
kora
wniaknie
adiuretyny
0,3 0,3 0,1 0,3 0,3
Na+ Na+
H2O H2O
zewnętrzna H2O Na+
część rdzeniowa 0,6 0,6 0,4 Na+ 0,6 Na+ 0,6
Na+ Na+ H2O
gradient podłużny
H2O H2O
0,9 0,9 H2O 0,7 0,9
Na+ Na+ 0,9
Na+ Na+
H2O H2O H2O
1,2 1,2 1,0 Na+ 1,2 1,2
wewnętrzna H2O
część rdzeniowa
Harnstoff Harnstoff
1,2
pętla Henlego naczynie proste kanalik zbiorczy
Ryc. B 7.1-5. Wzmocnienie przeciwprądowe w części rdzennej nerki przy adiurezie. Wzrost stężenia osmotycznego w kierunku
wierzchołka brodawki zaznaczono kolorem o zmieniającym się stopniu natężenia (wartości liczbowe podano w osmol/l)(wg Thewsa,
Mutschlera, Vaupela).
MUTSCHLER-2009.indd 712 2010-01-07 22:14:22
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 713
Wydłużony kształt pętli Henlego wskazuje, że układ różnymi kwasami lub różnymi zasadami, ale nigdy
przestrzenny warunkuje znaczenie czynnościowe. między kwasami i zasadami. W głębszych odcin-
Wychodząc z takiego założenia, zbadano, że za- kach kanalików część uwolnionych związków ulega
gęszczenie moczu odbywa się zgodnie z zasadą tzw. wchłanianiu zwrotnemu. Ponieważ zarówno wydzie-
wzmocnienia przeciwprądowego. Zasadę tę wyjaśnia lanie jak i wchłanianie zwrotne mogą być regulowa-
ryc. B 7.1-5. ne farmakologicznie, oba mechanizmy mają bardzo
Aktywny transport jonów sodowych ze światła duże znaczenie.
kanalika do tkanki śródmiąższowej odbywa się prze-
de wszystkim w ramieniu wstępującym. Ponieważ
ta część pętli Henlego jest właściwie nieprzepusz- 7.1.5.3. Regulacja ilości i składu moczu
czalna dla wody, która w związku z tym nie może
być tutaj resorbowana, w świetle każdego odcinka
Ilość moczu wydalanego w ciągu doby mieści się
ramienia wstępującego stężenie jonów sodowych
w przedziale 500–3900 ml. Średnio wynosi 1500 ml.
nieco się zmniejsza, a w połączonym z nim odcinku
Prawidłowy ciężar właściwy moczu wynosi 1,015–
tkanki śródmiąższowej wzrasta. W tej sytuacji woda
1,025, wartość pH nie powinna być mniejsza niż 4,8
z ramienia zstępującego (przepuszczalnego dla wody)
i większa niż 7,5. Tab. 7.1-2. zawiera dane dotyczą-
przenika, zgodnie z gradientem stężeń, do tkanki
ce ilości związków wydalanych z moczem w ciągu
śródmiąższowej a jony sodowe dyfundują z tkanki
doby.
śródmiąższowej do ramienia zstępującego pętli. Wy-
W celu długotrwałego zachowania równowagi
nikiem tych procesów jest stopniowy wzrost ciśnie-
w organizmie praca nerek musi zmieniać się ade-
nia osmotycznego w kierunku końcowej części pętli,
kwatnie do rodzaju związków, które mają być wyda-
gdzie osiąga najwyższą wartość 1,2 osmol/1. Powsta-
lone. Owo dostosowanie następuje przede wszystkim
ły w ten sposób gradient podłużny istnieje nie tylko
dzięki sprzężeniu zwrotnemu, które podlega kontroli
w pętli Henlego, lecz również w tkance śródmiąż-
ośrodkowej. Nerka jest więc ważnym elementem sy-
szowej: we wszystkich przestrzeniach wypełnionych
stemu służącego regulacji gospodarki wodnej, elek-
płynem, leżących na tej samej wysokości, utrzymuje
trolitycznej oraz równowagi kwasowo-zasadowej.
się taka sama osmolarność. Odpowiednio do tego
przy przejściu moczu przez kanalik zbiorczy, o ile
Regulacja wydalania wody. Przez zwiększenie
pod wpływem wazopresyny jego błony w wyniku ot-
lub zmniejszenie ilości moczu nerka przyczynia się
warcia kanałów wodnych stają się przepuszczalne dla
do utrzymania równowagi metabolizmu wody w orga-
wody, następuje wzrost osmolarności, aż do osiągnię-
nizmie. W czasie niedoboru wody, np. w wyniku zbyt
cia w moczu końcowym – przy antydiurezie (zob. ni-
żej) – wspomnianej wartość l,2 osmmol/1.
W przypadku diurezy wodnej wzmocnienie prze-
Tabela B 7.1-2. Prawidłowe ilości substancji stałych obecnych
ciwprądowe działa w taki sam sposób. Ponieważ jed-
w moczu w ciągu 24 godzin
nak ze względu na niedobór wazopresyny zmniejsza
przepuszczalność wody w kanaliku zbiorczym, wy-
gosodarka wodno-elektr.
równanie stężeń z otaczającą tkanką śródmiąższową Związek Ilość wydzielana
staje się niemożliwe. Stężenie elektrolitów w moczu
(g) Nerki i drogi moczowe,
końcowym opuszczającym nerki jest wówczas mniej- Azot amoniowy 0,4-1,0
sze niż tkance śródmiąższowej. Jony wapnia 0,05-0,4
Jony potasu 1,0-5,0
Jony magnezu 0,05-0,15
7.1.5.2. Wydzielanie kanalikowe Jony sodu 3,0-6,0
Chlorki 6,0-9,0
Wydzielanie w kanalikach i kanaliku zbiorczym, które Fosforany (w przeliczeniu na P)Sulfat 0,7-1,5
zależy od komórkowej przemiany materii i jest proce- Siarczany 1,8-3,5 B7
sem czynnym, obejmuje organiczne kwasy i zasady. Kreatynina 0,5-1,8
Najbardziej znanymi przykładami związków łatwo Mocznik 20-30
eliminowanych przez kanaliki są kwas p-aminohi- Kwas moczowy 0,25-0,75
purowy i penicylina. Wydzielanie kwasów przebiega Kwas hipurowy 0,1-1,0
niezależnie od wydzielania zasad, w związku z czym
Kwas szczawiowy do 0,03
hamowanie kompetycyjne istnieje tylko między
MUTSCHLER-2009.indd 713 2010-01-07 22:14:23
714 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
sekwencji tego wzmożenie wchłaniania zwrotnego
wartość wody (anty diureza). Zwiększenie osmolarności krwi
należna
wyzwala jednocześnie uczucie pragnienia.
neurony
wydzielające W przypadku nadmiernego nawodnienia zmniejszo-
regulator płat
tylny ne stężenie osmotyczne hamuje uwalnianie wazopre-
podwzgórza
przysadki syny. Przy niskim stężeniu ADH ograniczane jest prze-
nikanie wody, a na skutek tego wydalane są duże ilości
hipotonicznego moczu (diureza wodna).
receptory Ośrodek w podwzgórzu odpowiedzialny za go-
receptory ciśnienia
objętościowe osmotycznego adiuretyna spodarkę wodną organizmu z jednej strony decydu-
je o zmianach osmolarności, z drugiej pozostaje pod
wielkości
regulowane wpływem pojemnościowych receptorów przedsionka
objętość krwi osmolarność serca. Zmniejszenie objętości kompartmentu cen-
tralnego wzmaga więc pragnienie (pragnienie hipo-
płyny zewnątrz- wolemiczne), co zmusza do przyjmowania większej
komórkowe nerki ilości płynów a jednocześnie, poprzez uwalnianie
adiuretyny prowadzi do zmniejszonego wydalania
układ uporządkowany moczu. Wzrost objętości krwi skutkuje działaniem
odwrotnym, polegającym na hamowaniu uwalniania
wielkości zaburzone ADH i wzmaganiu wydalania moczu. Wpływ objęto-
(np. odwodnienie)
ści krwi na diurezę określa się jako odruch Gauera-
Henry’ego.
Ryc. B 7.1-6. Schemat utrzymania stałej objętości krwi pod
wpływem wazopresyna (wg Thewsa, Mutschlera, Vaupela).
Regulacja wydzielania Na+. Podobnie jak wydziela-
nie wody wydzielanie jonów sodowych podlega hor-
małej podaży płynów lub utraty znacznej ilości krwi, monalnemu sprzężeniu zwrotnemu.
wydalanie moczu ulega zmniejszeniu. Dostarczenie
płynów, np. poprzez wypicie większej ilości wody
lub na drodze infuzji pozajelitowej (por. Terapia in- stężenie jonów
spadek ciśnienia ↑
fuzyjna), skutkuje zwiększonym wydalaniem moczu. krwiobiegu Na+ w osoczu
Funkcjonowanie nerek regulowane jest w takich sy-
tuacjach hormonalnie (ryc. B 7.1-6): wzrost stężenia
osmotycznego we krwi lub w przestrzeni pozakomór- enzym
ANP renina ↑ konwertujący
kowej – rejestrowany przez osmoreceptory podwzgó- angiotensynę
rza – powoduje uwolnienie wazopresyny, a w kon-
angiotensyna angiotensyna I ↑ angiotensyna II ↑
nabłonek
kanalikach skurcz
naczyń
komórki
przykłębuszków katecholamina ↑
nerkowy
przepływ krwi ↓
komórki plamki tętniczka
gęstej doprowa- aldosteron ↑
dzająca pragnienie ↑
tętniczka
odprowa- błona podstawna
dzająca ADH ↑
komórki mięśni gładkich
Ryc. B 7.1-8. Schemat układu renina-angiotensyna-aldosteron
(zmieniony wg Gregera). ANP – przedsionkowy peptyd sodo-
Ryc. B 7.1-7. Aparat przykłębuszkowy (wg Guytona). pędny.
MUTSCHLER-2009.indd 714 2010-01-07 22:14:23
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 715
pragnienie receptory ciśnienia osmotycznego
NS podwzgórze receptory objętościowe
kora
nadnerczy serce
przysadka
hiperkalemia
wazopresyna
aldosteron atriopeptyna
objętość osmolarność
angiotensyna II
krew EZF
enzym konwertujący
angiotensynę (ACE)
iponatremia,
angiotensyna I hipowolemia
spadek ciśnienia krwi
renina
angiotensynogen
nerka
Ryc. B 7.1-9. Zestawienie kierunków kontroli osmolarnosć oraz objętości krwi oraz objętości płynów pozakomorkowych. EZF – płyny
pozakomórkowe. W przypadku braku Mg2+ nie dohodzi do uwolnienia atriopeptyny (wg Thewsa, Mutschlera, Vaupela).
Hormonem najważniejszym w metabolizmie sodu w tym zakresie). W samych nerkach angiotensyna
jest aldosteron. Uwalniany jest on przez nerkę w wy- II przede wszystkim zmniejsza przepływ krwi przez
niku wielostopniowego procesu, w którym decy- naczynia doprowadzające, co skutkuje osłabieniem
dującą rolę odgrywa aparat przykłębuszkowy nerki przesączania kłębuszkowego.
(complexus iuxtaglomerularis – ryc. B 7.1-7) skła- Bodźcem wpływającym w sposób istotny na uwal-
dający się z krótkiego segmentu kanalika dystalnego nianie reniny a tym samym na uruchomienie układu
i przedkłębuszkowego odcinka naczynia doprowa- renina-angiotensyna-aldosteron jest niedobór w oso-
dzającego. czu jonów Na+, hipowolemia oraz znaczne obniżenie
W miejscu zetknięcia, w okolicy kanalika zloka- ciśnienia tętniczego.
gosodarka wodno-elektr.
lizowane są wysokie, wąskie komórki (tzw. plamka Substancjami endogennymi, które w przeciwień-
gęsta), przy naczyniu znajdują się natomiast ziarniste stwie do aldosteronu nie zatrzymują sodu, lecz działa- Nerki i drogi moczowe,
komórki nabłonkowe (tzw. komórki przykłębuszko- ją sodopędnie i jednocześnie rozszerzająco na naczy-
we). Te ostatnie wytwarzają reninę, która powoduje nia doprowadzające, są peptydy sodopędne. Należą
przemianę angiotensynogenu, będącego α2-globu- do nich przedsionkowy peptyd sodopędny (atriopep-
liną, w dekapeptyd, angiotensynę I. Po oddzieleniu tyna, ANP), mózgowy peptyd sodopędny (BNP) oraz
dwóch aminokwasów angiotensyna I przekształca peptyd natriuretyczny typu C (CNP). Ich działanie do-
się w angiotensynę II (ryc. B 7.1-8). Oddzielenie chodzi do skutku poprzez stymulację rozpuszczalnej
kolejnego aminokwasu prowadzi do powstania hep- cyklazy guanylanowej (GC-A, -B i -C) i utworzenie
tapeptydu, angiotensyny III. Angiotensyna II i sła- cyklicznego guanozynomonofosforanu. Do uwolnie-
biej działająca angiotensyna III uwalniają aldosteron nia ANP dochodzi w przypadku poszerzenia przed- B7
z kory nadnerczy. sionka, BNP przy obciążeniu ciśnieniowym i obję-
tościowym serca. Podwyższony poziom ANP i BNP
Angiotensyna II jest ponadto jedną z substancji naj- w osoczu świadczy więc o niewydolności serca.
silniej obkurczających naczynia, w wyniku czego Na ryc. B 7.1-9 przedstawiono mechanizmy kon-
zwiększa opór naczyń obwodowych i podwyższa ciś- troli osmolarności, objętości krwi i objętości płynów
nienie krwi. (Angiotensyna III działa słabiej również pozakomórkowych.
MUTSCHLER-2009.indd 715 2010-01-07 22:14:24
716 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
Regulacja równowagi kwasowo-zasadowej. Po- nami, anionami słabych kwasów organicznych oraz
nieważ ilość protonów powstających w organizmie z wytwarzanym przez nerkę NH3. Wraz ze wzrostem
w wyniku procesów metabolicznych jest nieustannie stopnia zakwaszenia moczu następuje intensyfikacja
zbyt duża, są one usuwane albo podczas oddychania procesu tworzenia się w komórkach kanalików NH3
– poprzez reakcję z węglowodorami i wzmożone wy- z glutaminy a następnie łączenia się go w świetle ka-
dychanie z dwutlenkiem węgla – albo przez nerki. nalika z jonem wodoru w NH4+, dla którego błony
Wydzielanie nerkowe wolnych jonów wodorowych są prawie nieprzepuszczalne.
jest niewielkie, najniższa osiągalna wartość pH mo- Niektóre komórki w kanaliku zbiorczym są zdolne
czu człowieka wynosi 4,5. Proces ten odbywa się do odbywającej się przy udziale ATP aktywnej sekre-
zazwyczaj po reakcji protonów z substancjami bufo- cji protonów do moczu.
rowymi, szczególnie z pierwszorzędowymi fosfora-
MUTSCHLER-2009.indd 716 2010-01-07 22:14:25
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 717
7.2. Gospodarka wodna,
elektrolitowa i kwasowo-zasadowa
7.2.1. Gospodarka wodna osmozy rozróżnia się izotoniczne (izoosmolarne),
hipertoniczne (hiperosmolarne) i hipotoniczne (hipo-
osmolarne) zaburzenia nawodnienia.
Ciało człowieka w przeważającej części składa się
W tab. B 7. 2-1. zestawiono różne zaburzenia go-
z wody. W przypadku noworodka stanowi ok. 75%
spodarki wodnej oraz ich przyczyny i następstwa.
masy ciała, w przypadku dorosłego mężczyzny ok.
50–70%. Zawartość wody w ciele kobiety jest znacz-
nie mniejsza, ponieważ u kobiet ważniejszym ele-
mentem budulcowym jest tkanka tłuszczowa, w któ- 7.2.2 Gospodarka elektrolitowa
rej skład wchodzi zaledwie 10–30% wody. Odniesie-
nie zawartości wody do beztłuszczowej masy ciała 7.2.2.1. Podział elektrolitów
pokazuje, że u obu płci jest ona względnie stała i wy-
nosi 73%. Przestrzeń pozakomórkowa, na którą składa się prze-
strzeń osoczowa i przestrzeń śródmiąższowa, łącznie
Minimalne zapotrzebowanie na wodę dorosłego z przestrzenią wewnątrzkomórkową wykazują cha-
wynosi 1,5 l/dobę, ponieważ 0,9 l/dobę jest tracone rakterystyczne rozmieszczenie kationów i anionów.
w wyniku parowania, a nerka w celu wydzielenia W osoczu w grupie kationów przeważają jony
związków występujących w prawidłowym moczu sodowe, których stężenie wynosi 142 mmol/l. Stęże-
musi wytworzyć przynajmniej 0,5 l/dobę moczu koń- nia jonów K+, Ca2+ i Mg2+ są niewielkie. W grupie
cowego. Zapotrzebowanie na wodę w przypadku nie- anionów dominujące pod względem stężenia są jony
mowlęcia wynosi minimum 0,3 l/dobę. Cl–. Znaczący wpływ na wartość stężenia ogólnego
wywiera również obecność węglowodorów i białka.
Bilans wodny. Ze względu na warunki klimatyczne Fosforany, siarczany i kwasy organiczne występują
i nawyki żywieniowe panujące w Europie Środkowej tylko w niewielkich ilościach.
dobowy obrót wody na tym obszarze wynosi 2,5 1. Bardzo podobny do osocza pod względem skła-
Jedną połowę tej ilości stanowią pobierane płyny, du jest płyn śródmiąższowy. Jedyna różnica między
drogą woda zawarta w pokarmach stałych oraz woda nimi wynika z tego, że na skutek bardzo ograniczo-
powstała w wyniku utleniania. Przeważająca część nej przepuszczalności ścian naczyń kapilarnych dla
wody wydalana jest w postaci moczu, pozostała ilość białek ich stężenie w płynie śródmiąższowym jest ni-
usuwana jest na drodze niezauważalnego parowania skie.
przez skórę (parowanie transepidermalne) oraz przez Zupełnie inne jest stężenie jonów w płynie we-
płuca. Niewielkie ilości wydalane są także z kałem. wnątrzkomórkowym. Największą część kationów
stanowią jony K+, wśród anionów przeważają jony
gosodarka wodno-elektr.
i Cl –, a także białka. Nerki i drogi moczowe,
U niemowlęcia względny obrót wody jest znacznie większy
niż u dziecka czy u dorosłego (ok. 10% masy ciała dzie-
cka porównaniu z 3–4% masy ciała dorosłego), w związku
z czym łatwo może dojść do zaburzeń bilansu wodnego
(zob. niżej). 7.2.2.2. Kontrola izojonii
Na utratę znacznych ilości wody (aż do l,5 1/godz.)
narażone są osoby pracujące w wysokich temperaturach
(np. hutnicy). W takich przypadkach wraz z obficie wy- Funkcjonowanie narządów i tkanek jest w znacznym
dzielanym potem organizm pozbawiany jest jednocześnie stopniu uzależnione od równowagi stężeń poszcze-
NaCl, co musi być wyrównywane podażą soli. gólnych jonów wewnątrz i na zewnątrz komórki (izo-
jonia). B7
Zaburzenia gospodarki wodnej. Ponieważ woda
stanowi największy ilościowo składnik ciała, zabu- Jony sodowe i chlorki. W zależności od nawyków
rzenia jej gospodarki mogą zakłócić przebieg różnych żywieniowych dorosły wchłania i wydziela dziennie
funkcji organizmu, a niekiedy nawet stać się przyczy- przeciętnie 2–8 g (90–350 mmol) jonów sodowych
ną śmierci. Ujemny bilans wodny powoduje dehydra- oraz 2,5–10 g (70–280 mmol) chlorków (zaleca się
cję (odwodnienie), dodatni bilans wodny hiperhydra- maksymalnie 0,1 g/kg masy ciała NaCl dziennie).
cję (przewodnienie) organizmu. Zgodnie z prawami Pomimo zmieniającej się podaży pozakomórko-
MUTSCHLER-2009.indd 717 2010-01-07 22:14:25
718 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
Tabela B 7.2-1. Zaburzenia gospodarki wodnej
Zaburzenie Charakterystyka Przyczyny Skutki ew. objawy
zaburzenia
Odwodnienie
izotoniczne zmniejszenie ilości wody biegunka, wymioty, krwotok, oparzenia zmniejszenie objętości osocza,
i substancji o działaniu osmotycznym, częstoskurcz, spadek ciśnienia
zwłaszcza NaCl krwi lub wstrząs
hipertoniczne zmniejszone przyjmowanie niedostateczna podaż wody, przewlekłe niepokój, majaczenie, śpiączka,
lub zwiększona utrata wody choroby nerek, moczówka prosta, cukrzyca wstrząs, śmierć po utracie więcej niż 40%
wody wchodzącej w skład organizmu
hipotoniczne pozakomórkowy niedobór sodu, niedostateczna podaż sodu lub podaż zmniejszenie objętości osocza,
przemieszczenie płynów czystej wody po wymiotach, spadek ciśnienia krwi, zniechęcenie
do przestrzeni wewnątrzkomórkowej biegunkach oraz obfitym poceniu, lub śpiączka
niewydolność kory nadnercza
Nadmierne nawodnienie
izotoniczne nadmiar płynu pozakomórkowego uogólniony obrzęk, infuzja nadmiernej ilości w zależności od choroby podstawowej,
roztworu izotonicznego np. niewydolności serca
hipertoniczne pozakomórkowy nadmiar sodu nadczynność kory nadnerczy, obrzęk lub ostra niewydolność nerek
podaż mineralokortykosteroidów
hipotoniczne nadmierna podaż wody, wprowadzenie nadmiernej ilości nudności, wymioty, apatia,
zbyt duża retencja wody płynów infuzyjnych bezelektrolitowych, niewydolność serca, obrzęk płuc
wzmożone wydzielanie wazopresyny
we stężenia jonów Na+ i Cl– pozostają w zasadzie Jony wapnia i fosforany. Jony wapnia – wspólnie
niezmienione, co dla organizmu ma szczególne z fosforanami – budują kości i zęby. Zewnątrzko-
znaczenie, ponieważ wszystkie jony wspólnie okre- mórkowe jony Ca2+ ograniczają przepuszczalność
ślają objętość płynów pozakomórkowych. Regula- błon i wskutek tego zmniejszają pobudliwość ner-
cja stężenia Na+ następuje w wyniku wydzielania wów i komórek mięśniowych. Poza tym hamują
nerkowego, wspomaganego przez układ renina- wydzielanie paratohormonu (zob. niżej), wzmaga-
angiotensyna-aldosteron oraz sodopędny peptyd ją uwalnianie w żołądku jonów H+ i redukują ilość
przedsionkowy. Ściśle jest z tym związana regulacja produkowanych przez trzustkę jonów HCO3–. Pod-
wydzielania jonów Cl–. Dodatkowo jest ona jednak wyższenie wewnątrzkomórkowego stężenia Ca2+ sty-
warunkowana stanem równowagi kwasowo-zasa- muluje skurcz mięśni, pracę gruczołów, wydzielanie
dowej, ponieważ w resorpcji chlorków w cewce hormonów i transmiterów oraz przemiany metabo-
zbiorczej czynny udział bierze antyporter dwuwę- liczne (np. glikolizę). Ponadto jony wapnia regulują
glan/chlorki. aktywność niektórych kanałów jonowych (kanałów
K+, Cl–) oraz enzymów (np. cyklazy adenylanowej).
Jony potasowe. Jony potasowe określają w znacz- Biorą również udział w kompleksowych procesach
nym stopniu elektroneutralność i osmolarność komórkowych jak migracja komórek, namnażanie się
wewnątrz komórek, wpływają na aktywność en- komórek, apoptoza i decydują o krzepliwości krwi.
zymatyczną oraz umożliwiają przepływ bodźców Dobowe zapotrzebowanie osoby dorosłej na Ca2+
w komórkach nerwowych i mięśniowych. Dzienne wynosi 0,8–1 g (20–25 mmol), a nieorganiczny fosfo-
zapotrzebowanie na jony potasu wynosi u dorosłego ran ok. l g (33 mmol). W okresie ciąży ilości te powin-
2–5 g (50–125 mmol). Kontrola zewnątrzkomórko- ny być o 50% większe. Wystarczające ilości wapnia
wego stężenia jonów K+ następuje poprzez wydzie- dostarczane są z reguły w pożywieniu – szczególnie
lanie nerkowe odbywające się przy współudziale wartościowe są pod względem zawartości tego pier-
aldosteronu, przy czym ilość wydzielana musi od- wiastka mleko i produkty mleczne oraz wiele warzyw.
powiadać podaży. Za wewnątrzkomórkowe stężenie (W przypadku osób w podeszłym wieku podaż Ca2+
jonów K+, które jest 30-krotnie wyższe niż w prze- w pożywieniu jest najczęściej niewystarczająca).
strzeni zewnątrzkomórkowej, odpowiada Na+/K+- Homeostaza wapniowo-fosforanowa podlega kon-
ATP-aza. troli paratohormonu i 1,25 dihydroksy-cholekalcyfe-
MUTSCHLER-2009.indd 718 2010-01-07 22:14:25
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 719
rolu (aktywna postać witaminy D3, kalcytriol) oraz W hiponatremii spowodowanej niedoborem sodu
w niewielkim stopniu kalcytoninie. bilans sodu jest ujemny na skutek utraty zbyt dużej
ilości tego pierwiastka albo jego niewystarczającej
Jony magnezowe. Jony magnezowe wpływają podaży. Dochodzi wówczas do izotonicznego lub hi-
na przebieg wielu reakcji enzymatycznych zacho- potonicznego odwodnienia.
dzących w komórce. Poprzez blokowanie kanałów O hiponatremii wynikającej z nadmiernego roz-
jonowych (kanałów K+, Ca2+) zmniejszają pobudli- cieńczenia płynów ustrojowych lub nieprawidło-
wość neuromięśniową, a w dużym stężeniu blokują wego rozdziału jonów sodu mówi się wówczas,
uwalnianie hormonów i neurtransmiterów (np. acety- gdy przy prawidłowym lub nawet zwiększonym
locholiny w płytce nerwowo-mięśniowej). W wyniku stężeniu sodu w organizmie stężenie jonów sodo-
interakcji z Na+/K+-ATPazą i kanałami K+ wspomaga wych w przestrzeni pozakomórkowej jest obniżo-
transport K+ do wnętrza komórki. ne. Objaw ten towarzyszy zmniejszonemu wydala-
Dostarczając organizmowi dziennie 0,3 g (12 mmol) niu wody (np. przy skąpomoczu ew. bezmoczu lub
magnezu zapewnia się jego zrównoważony bilans. przy zwiększonym uwalnianiu diuretyny) oraz przy
Można to uzyskać przez odpowiednie odżywianie. zaburzeniu transportu jonów sodowych do i na ze-
Szczególnie bogate w magnez są warzywa strączko- wnątrz komórki (np. na skutek poważnych zaburzeń
we, zboża, orzechy, migdały, banany i piwo. Z całości przemiany materii albo niedoboru potasu). Hipo-
magnezu pochodzącego z pożywienia w przewodzie natremia spowodowana niewłaściwym rozcieńcze-
pokarmowym wchłaniane jest około 30%, przy czym niem płynów ustrojowych występuje w uogólnio-
przyswajanie regulowane jest hormonalnie adekwatnie nym obrzęku, a zwłaszcza obrzęku wywołanym
do aktualnych potrzeb organizmu. Kalcytriol i soma- niewydolnością serca, zespołem nerczycowym albo
tropina stymulują jelitową resorpcję Mg2+, natomiast w marskości wątroby (zob. nadmierne nawodnienie
aldosteron i kalcytonina ją hamują. izotoniczne).
Objawem klinicznym towarzyszącym hiponatre-
mii spowodowanej niedoborem sodu są najczęściej
hipotoniczne zaburzenia krążenia, natomiast w przy-
7.2.2.3. Zaburzenia elektrolityczne padku hiponatremii wynikającej z rozcieńczenia
płynów ustrojowych lub nieprawidłowego rozdziału
Zaburzenia elektrolitowe mogą być zarówno następ- jonów sodowych najczęściej obserwuje się dolegli-
stwem chorób nerek, jak również czynników poza- wości neurologiczne (np. mdłości, bóle głowy, apatię,
nerkowych, np. zaburzeń hormonalnych (m.in. nie- brak koncentracji).
wydolności kory nadnercza). Rozpoznawane są prze-
ważnie na podstawie badań stężenia jonów w oso- Hipernatremia. W hipernatremii stężenie jonów
czu krwi ewentualnie w surowicy. Należy przy tym w osoczu przekracza 150 mmol/l. W porównaniu
uwzględnić to, że, szczególnie w przypadku jonów, z hiponatremią stan ten występuje o wiele rzadziej,
których stężenie wewnątrzkomórkowe jest większe a jego przyczyną może być zwiększona podaż lub re-
niż pozakomórkowe (np. jony potasu), ich stężenie tencja sodu, a także ujemny bilans wody. Zwiększona
gosodarka wodno-elektr.
w surowicy może być jeszcze prawidłowe, pomi- podaż sodu albo retencja sodu prowadzą do nadmier-
mo że stężenia w tkankach może już być zmienio- nego hipertonicznego nawodnienia. Przyjmowanie Nerki i drogi moczowe,
ne. Chcąc przeprowadzić dokładne badania należy zbyt małej ilości płynów lub utrata wody bez istotnej
więc określić całkowitą ilość elektrolitów w ciele utraty sodu powodują odwodnienie hipertoniczne.
(np. oznaczanie całkowitej ilości potasu za pomocą
techniki izotopowej). Hipokaliemia. W hipokaliemii stężenie jonów pota-
su w osoczu nie przekracza 3,5 mmol/l. Przyczyną
Hiponatremia. Hiponatremię rozpoznaje się wów- może być oprócz niewystarczającej podaży potasu,
czas (tab. B 7.2-2), gdy stężenie jonów sodowych także jego nerkowa lub żołądkowo-jelitowa utrata
w osoczu wynosi mniej niż 135 mmol/l. Klinicznie oraz zaburzenia transportu dokomórkowego i poza- B7
dopuszczalne są stężenia < 130 mmol/1, za stanowią- komórkowego.
ce zagrożenie stężenia < 125 mmol/1. W zależności Niewystarczająca podaż potasu występuje niekie-
od przyczyny rozróżnia się hiponatremię dy w wieloletnim alkoholiźmie lub u pacjentów z jad-
łowstrętem (anorexia nervosa).
■ spowodowaną niedoborem sodu,
Nerkowa utrata potasu występuje przy zwiększo-
■ wynikającą z rozcieńczenia płynów ustrojowych nym stężeniu aldosteromu lub kortyzolu we krwi
lub nieprawidłowego rozdziału jonów sodowych. (pierwotny i wtórny hiperaldosteronizm; zespół Cus-
MUTSCHLER-2009.indd 719 2010-01-07 22:14:25
720 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
Tabela B 7.2-2. Zaburzenia przemiany elektrolitowej
Zaburzenia Charakterystyka Przyczyny Skutki i objawy
elektrolityczne zaburzenia
Hiponatremia stężenie sodu duża utrata sodu, zaburzenia odwodnienie izotoniczne
w surowicy dystrybucji sodu w niewydolności lub hipotoniczne na skutek
< 135 mmol/l serca, marskości wątroby i in. utraty sodu, nadmierne nawodnienie
izotoniczne lub hipotoniczne
w zaburzeniach dystrybucji sodu i in.
Hipernatremia stężenie sodu zwiększona podaż sodu, nadmierne nawodnienie hipertoniczne
w surowicy retencja sodu, utrata wody przy zwiększonej podaży lub retencji Na+,
> 150 mmol/l odwodnienie hipertoniczne
w wyniku utraty wody
Hipokalemia stężenie potasu niewystarczająca podaż potasu osłabienie mięśniowe,
w surowicy (np. w alkoholizmie), utrata potasu zaburzenia żołądkowo-jelitowe,
< 3,5 mmol/l w hiperaldosteronizmie, zespół Cushinga, apatia, zaburzenia czynności nerek,
leczenie saluretykami, przewlekłe zmiany w EKG
nadużywanie leków przeczyszczających,
zaburzenia dystrybucji potasu
w ostrych zasadowicach
Hiperkalemia stężenie potasu zmniejszone wydzielanie potasu podobnie jak w hipokalemii
w surowicy w przewlekłej niewydolności nerek, ostrej
> 5,5 mmol/l niewydolności nerek, niedoborze
mineralokortykoidów, w leczeniu
diuretykami oszczędzającymi potas
Hipokalcemia stężenie wapnia niewystarczająca podaż wapnia, wzmożona reaktwyność układu
w surowicy niewydolność gruczołów przytarczycznych, nerwowego, tężyczka,
< 2 mmol/l upośledzona resorpcja w zespole zaburzenia czucia
złego wchłaniania
Hiperkalcemia stężenie wapnia nadczynność gruczołów utrata masy ciała, utrata łaknienia,
w surowicy przytarczycznych, zatrucie zaparcia, zaburzenia rytmu serca,
> 2,7 mmol/l witaminą D, złośliwe guzy hiperkalcynuria,
w ciężkich przypadkach śpiączka
Hipomagnezemia stężenie magnezu przewlekłe choroby jelit, podobne jak w hipokalcemii,
w surowicy nadczynność gruczołu tarczowego, ponadto częstoskurcz,
< 0,7 mmol/l hiperaldosteronizm zaburzenia rytmu serca
Hipermagnezemia stężenie magnezu niewydolność nerek, niedoczynność zahamowanie czynności ośrodkowych,
w surowicy gruczołu tarczowego, choroba Addisona, zaparcia, działanie podobne do działania
> 1,25 mmol/l przyczyny jatrogenne spowodowane środków kuraropodobnych
podaniem dużych dawek magnezu
hinga) w terapii tiazydami i diuretykami pętlowymi, Hiperkaliemia. W hiperkaliemii stężenie pota-
a także w niektórych przewlekłych chorobach nerek. su w osoczu przekracza 5,5 mmol/l. Przyczyną
Utrata potasu z przewodu pokarmowego jest spowo- jest zwiększona podaż i retencja nerkowa tego pier-
dowana biegunkami lub przewlekłym nadużywaniem wiastka lub jego przejście z komórek do tkanki śród-
leków przeczyszczających. miąższowej.
Zaburzenia dystrybucji potasu są m.in. następ- Hiperkaliemia spowodowana nadmierną podażą
stwem ostrej zasadowicy lub przedawkowania in- zewnątrzustrojowej wynika zazwyczaj z błędnego
suliny (wejście K+ do komórki w wyniku wymiany postępowania terapeutycznego, np. zbyt szybkiego
z H+). dożylnego wlewu roztworów zawierających potas.
Objawami hipokaliemii są osłabienie mięśniowe, Do zmniejszonego wydzielania potasu dochodzi
dolegliwości żołądkowo-jelitowe (wiotkość żołądka, w przypadku przewlekłej niewydolności nerek, ostrej
zaparcia), apatia, zaburzenia funkcji nerek (np. zmniej- niewydolność nerek, niedoboru mineralokortykoidów
szona zdolność koncentracji) oraz zaburzenia rytmu oraz terapii tzw. diuretykami oszczędzającymi potas
serca ze zmianami widocznymi w EKG. (np. spironolaktonem). Wzmożone przechodzenie K+
MUTSCHLER-2009.indd 720 2010-01-07 22:14:25
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 721
z przestrzeni wewnątrzkomórkowej do przestrzeni Hipomagnezemia. Hipomagnezemia (stężenie
pozakomórkowej jest spowodowane m.in. kwasicą magnezu w surowicy poniżej 0,7 mmol/l) często
lub zatruciem glikozydami nasercowymi. występuje w chorobie alkoholowej, w przewlekłych
Objawy wywołane hiperkaliemią przypominają chorobach jelit (np. zapalenie jelita grubego), w nad-
objawy hipokalemii. czynności tarczyc, nadczynności przytarczyc oraz
w hiperaldosteroniźmie. Również towarzyszące kwa-
Hipokalcemia. O hipokalcemii mówi się wówczas, sicy nadmierne nerkowe wydzielanie magnezu może
gdy stężenie wapnia w surowicy spada poniżej 2,2 doprowadzić do niedoboru tego pierwiastka w orga-
mmol/l. Przyczyną tego może być : nizmie.
Objawy kliniczne hipomagnezemii przypominają
■ niewystarczająca podaż z pokarmem, zwłaszcza
te występujące w hipokalcemii (objawy tężyczkowe),
w okresie ciąży i karmienia,
a oprócz nich pojawia się jeszcze częstoskurcz i inne
■ niedobór witaminy D, zaburzenia rytmu serca. Ponieważ jednak niedobór
magnezu zawsze łączy się z innymi zaburzeniami
■ niedoczynność gruczołów przytarczycznych (pier-
przemiany materii, trudno jest ustalić dokładną przy-
wotna i wtórna niedoczynność przytarczyc),
czynę tych objawów.
■ tworzenie się trudno wchłaniających się kamieni
wapniowych w ostrym zapaleniu trzustki, Hipermagnezemia. Dla hipermagnezemii typowe
jest stężenie magnezu w surowicy przekraczające l
■ zmniejszone wchłanianie w zespole złego wchła-
mmol/l. Jako przypadek kliniczny traktowana jest jed-
niania oraz w niewydolności nerek. W następ-
nak dopiero wówczas, gdy stężenie magnezu w suro-
stwie ograniczonego przesączania kłębuszkowego
wicy wynosi 2 mmol/1. Wśród przyczyn tego zabu-
wzrasta stężenie fosforanów w osoczu; zwiększa
rzenia elektrolitowego wymienia się oprócz czynni-
się również stężenie fosforanów w świetle jelita,
ków jatrogennych (podanie zbyt dużej ilości magnezu
co pociąga za sobą obniżenie resorpcji wapnia;
w płynach infuzyjnych, podanie zbyt dużych dawek
ponadto tworzone są niewystarczające ilości 1,25-
siarczanu magnezu jako środka przeczyszczającego),
dihydroksy-witaminy D3, a przez to następuje
także niewydolność nerek, niedoczynność tarczycy
zmniejszenie wydzielania fosforanów i resorpcji
czy choroba Addisona (zob. wyżej).
wapnia.
Objawami hipermagnezemii są obniżona pobud-
liwość mięśni poprzecznie prążkowanych (działanie
Najważniejszym objawem hipokalcemii jest zwięk-
podobne do działania związków kurarynowych), za-
szona pobudliwość całego układu nerwowego, która
parcia i zahamowanie czynności ośrodkowego ukła-
klinicznie przejawia się tężyczką (tetania), tzn. to-
du nerwowego. Przy stężeniu w surowicy powyżej 5
nicznymi kurczami mięśni, przejściowy paraliż (pa-
mmol/1 następuje porażenie czynności oddechowej
restezje) oraz skurczami mięśniówki gładkiej.
i zatrzymanie czynności serca w fazie rozkurczu.
Tab. B 7.2-2.zawiera zestawienie ważniejsze zabu-
Hiperkalcemia. Przyczyną hiperkalcemii (stężenie
rzeń przemiany elektrolitowej.
gosodarka wodno-elektr.
wapnia w surowicy powyżej 2,7 mmol/l) może być
m.in. pierwotna i wtórna nadczynnością przytarczyc, Nerki i drogi moczowe,
unieruchomienie, zatrucie witaminą D, sarkoidoza
(rzadko), choroba Addisona lub (rzadko) nadczynność 7.2.2.4. Zastosowanie terapeutyczne
gruczołu tarczowego. Częstym powodem jej rozwoju soli potasu, wapnia i magnezu
są również złośliwe guzy, zwłaszcza oskrzeli, pier-
si i kobiecych narządów płciowych, które albo dają 7.2.2.4.1. Preparaty potasu
przerzuty osteoblastyczne prowadzące do zwiększo-
nego uwalniania wapnia z kości, albo wytwarzają Preparaty potasu wskazane są w przypadku objawów
substancję podobną do parathormonu (hiperkalcemia niedoboru potasu (stężenie potasu w surowicy < 3,5
nowotworowa). mmol/1). O ile jest to możliwe, potas powinien być B7
Objawem hiperkalcemii jest utrata masy ciała, podawany doustnie lub w wyjątkowych przypadkach,
utrata łaknienia, zaparcia, bębnica, wielomocz i zabu- np. w śpiączce cukrzycowej, dożylnie. Jego średnia
rzenia rytmu serca. Ponadto hiperkalcemia może wy- pojedyncza dawka wynosi 20–40 mmol (maksymal-
wołać psychozę, a w ciężkich przypadkach śpiączkę. na dawka dobowa 150 mmol). Ze względu na miej-
Wzmożone nerkowe wydzielanie wapnia zwiększa scowo drażniące działanie zaleca się przyjmowanie
niebezpieczeństwo kamicy nerkowej. potasu w formie preparatów o przedłużonym działa-
MUTSCHLER-2009.indd 721 2010-01-07 22:14:26
722 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
niu lub rozcieńczonych roztworów (z zastosowaniem brakiem magnezu – wciąż niejednoznaczne. Istnieją
tabletek musujących). wprawdzie opinie, że podawane doustnie jony mag-
W profilaktyce niedoboru potasu spowodowanego nezu nadają się do leczenia różnych typów arytmii
lekami diuretycznymi (saluretycznymi) lepsze efekty oraz działają (słabo) przeciwnadciśnieniowo, nie ma
przynosi stosowanie diety bogatej w potas oraz diu- jednak jednoznacznych dowodów takiego ich działa-
retyków oszczędzających ten pierwiastek niż substy- nia. Profilaktyczne przyjmowanie magnezu w okresie
tucji farmakologicznej preparatami potasu, ponieważ ciąży zmniejsza niebezpieczeństwo wystąpienia rzu-
egzogenne jony potasowe wprowadzone podczas sa- cawki ciążowej.
lurezy są szybko wydalane przez nerki. Ważne jest, aby preparaty magnezowe były przyj-
mowane w wystarczająco wysokich dawkach, tzn.
nie niższych niż 10 mmol Mg2+ /dziennie.
7.2.2.4.2. Preparaty wapnia
Preparaty wapnia (oprócz leków neutralizujących) 7.2.3. Gospodarka
stosuje się w niedoborze wywołanym niewłaściwym kwasowo-zasadowa
odżywianiem lub złym wchłanianiem, a także przy
osteoporozie (i niedoczynności przytarczyc. Zaleca-
Wartość pH krwi tętniczej waha się od 7,37 do 7,43,
nie ich w chorobach alergicznych jest oceniane ne-
a średnia wartość wynosi 7,40. Krew człowieka ma
gatywnie.
więc odczyn lekko zasadowy. Pomimo zmieniają-
Pozajelitowo (np. w ostrej hipokalcemii przebie-
cej się ilości napływających do krwi kwaśnych pro-
gającej z objawami tężyczki) podaje się sole wapnia
duktów przemiany materii jej pH ma stałą wartość.
w połączeniu z organicznymi kwasami węglowymi,
Utrzymanie tej wartości jest konieczne do prawid-
a zwłaszcza z kwasem glukuronowym (calcium glu-
łowego przebiegu przemiany materii w komórkach,
conicum) w dawce 2,25–4,5 mmol. Wstrzykiwanie
ponieważ aktywność wszystkich enzymów metabo-
dożylne powinno przebiegać bardzo powoli i pod
licznych jest uzależniona od wartości pH.
stałym nadzorem pracy serca, ponieważ zbyt szyb-
Utrzymanie zrównoważonej wartości pH krwi za-
ka infuzja do żyły Ca2+ stwarza niebezpieczeństwo
leży od systemu buforowego krwi, wymiany gazowej
znacznego obniżenia ciśnienia krwi (czas wstrzyki-
w płucach oraz procesów wydzielniczych nerki.
wania 2,25 mmola wynosi 3 minuty).
Do stosowania doustnego najlepiej nadaje się wę-
System buforowy krwi. System buforowy krwi
glan wapnia, przede wszystkim ze względu na wyso-
(i przestrzeni śródkomórkowej) jest tworzony przede
ką, około 40-procentową zawartość wapnia, a także
wszystkim przez system wodorowęglanów z CO2 jako
mleczan wapnia z 13-procentową zawartością wap-
bezwodnik kwasu oraz HCO3– jako zasadę korespon-
nia. (Glukonian wapnia oraz laktobionian wapnia
dującą. System ten jest bardzo efektywny, ponieważ
zawierają tylko 9 ew. 6,6% wapnia). Dawka dobowa
obydwa elementy występują we krwi we względnie
wynosi około 1000 mgCa2+/dzień.
wysokich stężeniach.
Wśród układów buforujących krwi istotną rolę
pełnią białka (bufor białkowy). Niezwykle ważna
7.2.2.4.3. Preparaty magnezu
jest również hemoglobina ze względu na jej wysokie
stężenie oraz wchodzącą w jej skład histydynę, której
Zastosowanie soli magnezu jako leków neutralizują-
pierścień imidazolowy może przyłączać protony.
cych i przeczyszczających omówiono w B 6.2.2.3.
Poza usunięciem objawów niedoboru magnezu,
Układ buforowy stanowią również nieorganiczne
będącym najważniejszym wskazaniem do podania
fosforany (bufor fosforanowy), przy czym fosforan
zawierających go preparatów, Mg2+ (przeważnie
pierwszorzędowy (H2PO4–) działa jako kwas, a dru-
w formie siarczanu wapnia) stosowany jest dożylnie
gorzędowy fosforan (HPO4 2–) jako korespondująca
w określonych typach zaburzeń rytmu serca, szcze-
zasada. Ze względu na stosunkowo niewielkie stęże-
gólnie w wielokształtnym częstoskurczu komorowym
nie fosforanów we krwi ich efekt buforujący jest nie-
(torsade de pointes) oraz zaburzeniach rytmu serca
wielki.
po terapii glikozydami nasercowymi, a także w sta-
nach przedrzucawkowych i rzucawkach (eclampsid).
Regulacja pH na drodze oddechowej, nerkowej
Wskazania do podania doustnych preparatów magne-
i wątrobowej przemiany materii. Dwutlenek węgla
zowych są – pomijając objawów niedoboru magne-
powstający jako produkt końcowy tlenowej przemiany
zu i ich następstwa, np. skurcze łydek spowodowane
MUTSCHLER-2009.indd 722 2010-01-07 22:14:26
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 723
materii (w spoczynku 10 mmol/min lub 14 mol/dzień ca, zaburzeń rytmu serca, hipotonii (aż do wstrząsu),
w pozycji spoczynkowej) jest eliminowany z krwi utraty świadomości, a nawet śpiączki.
w sposób ciągły w wyniku oddychania. Czynność
oddechowa zapobiega więc zakwaszeniu organizmu
przez wydalanie lotnego bezwodnika kwasu węglo- 7.2.4. Wlew kroplowy w zaburzeniach
wego. W przypadku nagromadzenia kwasów we krwi gospodarki wodnej,
następuje natychmiast przyspieszenie oddechu, dzię-
ki czemu możliwe staje się wzmożone usuwanie CO2
elektrolitowej i równowagi
i powrót pH do normy. Przy zwiększeniu stężenia za- kwasowo-zasadowej
sad czynność oddechowa ulega spowolnieniu (hipo-
wentylacja), wzrasta natomiast ciśnienie cząstkowe Celem terapii infuzyjnej w zaburzeniach wodno-
CO2 i wskutek tego stężenie jonów H+, a wzrost pH elektrolitowych oraz równowagi kwasowo-zasado-
przynajmniej częściowo jest redukowany. wej jest przywrócenie homeostazy. Rozróżnia się
Oprócz kwasu węglowego produktami procesu przy tym:
przemiany materii są także nielotne kwasy, wśród
■ zapotrzebowanie podstawowe,
których przeważa ilościowo kwas siarkowy. Dzięki
nim możliwe jest nerkowe wydalanie wytworzonych ■ zapotrzebowanie korygujące.
jonów H+.
W procesie regulacji gospodarki kwasowo-zasa- Zaistnienie zapotrzebowania podstawoego wynika
dowej uczestniczy również wątroba. Przy rozpadzie ze zwykłej utraty wody i elektrolitów (wydalanie
tlenowym aminokwasów powstają CO2 lub HCO3– moczu, kału, potu, przeznabłonkowa utratę wody).
i NH3 lub NH4+, które zazwyczaj są całkowicie Z organizmu dorosłego pacjenta ubywa dziennie
wykorzystywane w wątrobie do syntezy mocznika. około 2,5 l wody, 85–120 mmol sodu i 60–90 mmoli
Jeżeli jednak zużycie nie jest całkowite, wówczas potasu.
w wyniku sprzęgania z glutaminą następuje elimi-
nacja NH4+. Glutamina przedostaje się do krążenia Zapotrzebowanie korygujące warunkowane jest przez
nerkowego i współuczestniczy w kanalikowym wy- aktualne nawodnienie i stężenie elektrolitów, oraz
dzielaniu H+. Eliminację protonów zapewnia także straty patologiczne, np. wymioty, biegunkę, wypły-
rozpad tlenowy kwasu mlekowego, kwasu octowego wanie płynów z przetoki.
i kwasu cytrynowego.
W celu uniknięcia zbyt małej lub nadmiernej podaży
Zaburzenia gospodarki kwasowo-zasdowej. Je- płynów (hipo- i hiperperfuzji) konieczne jest dokład-
żeli w wyniku choroby dochodzi do nagromadzenia ne określenie bilansu doprowadzanych i odprowadza-
kwasów lub zasad, wówczas żaden z systemów bu- nych płynów oraz oznaczenie stężenia składników
forowych nie jest w stanie utrzymać wartość pH krwi osocza. Na podstawie dokonanych obliczeń ustala się
na prawidłowym poziomie wynoszącym 7,40. W za- plan wlewu dożylnego. Hiperinfuzja (bilans dodatni)
leżności od przesunięć pH rozróżnia się: stwarza niebezpieczeństwo nadmiernego nawodnie-
gosodarka wodno-elektr.
nia, hipoinfuzja (bilans ujemny) grozi odwodnie-
■ kwasicę (pH < 7,37),
niem. Nerki i drogi moczowe,
■ zasadowicę (pH > 7,43).
Rodzaje płynów infuzyjnych. Roztwory podstawo-
Jeżeli nieprawidłowe wartości pH są wynikiem zabu- we są pozbawionymi elektrolitów roztworami o za-
rzenia funkcji płuc, kwasica lub zasadowica ma cha- wartości węglowodanów (5–10%).
rakter oddechowy, jeżeli są spowodowane zaburze-
niami przemiany materii (np. cukrzycy lub marskość Skład roztworów zasadowych (roztwory zbilanso-
wątroby), określa się je jako metaboliczne. Ponieważ wane) jest tak ustalany, by 2–3 l płynu infuzyjnego
zaburzenia czynności nerek (np. bezmocz) również pokrywało dobowe zapotrzebowanie na wodę i elek- B7
mogą skutkować zmianami pH, zaburzenia nerkowe trolity.
i metaboliczne określa się wspólną nazwą kwasicy
lub zasadowicy nieoddechowej. Ciśnienie osmotyczne izotonicznych roztworów elek-
Znaczniejsze zmiany pH wywołują poważne na- trolitów odpowiada ciśnieniu osocza.
stępstwa, mogące niekiedy nawet zagrażać życiu. Koncentraty elektrolitów zawierają niezbędne jony
Obniżenie wartość pH poniżej 7,20 może więc pro- w stężonym roztworze. Dodawane są do roztworów
wadzić do zmniejszenia pojemności minutowej ser- podstawowych.
MUTSCHLER-2009.indd 723 2010-01-07 22:14:26
724 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
Tabela B 7.2-3. Skład roztworów infuzyjnych
Rodzaj roztworu
infuzyjnego Na+ K+ Mg2+ Ca2+ Cl- Mleczan- HCO3-
koncentrat elektrolitowy 3000 3000 500 3000 2000 2000
całkowity roztwór elektrolitowy 140 4 1 2,5 106 45
roztwór zastępujący sok żołądkowy+ 60 15 120
roztwór zastępujący wydzielinę jelit 140 15 1 2,5 117 45
roztwór podstawowy++ 54 24 2,5 51 25
pooperacyjny roztwór podstawowy+++ 100 20 2,5 2,5 100
+ z dodatkiem 37 mmol/l lizyny, 8 mmol/l glicyny
++ z dodatkiem 7 mmol/l fosforanu
+++ z dodatkiem 50 g/l węglowodanów, 20 mmol/l octanu, 10 mmol/l jabłczanu
Płyny elektrolitowe zawierają elektrolity w stężeniu W kwasicy metabolicznej należy leczyć, o ile
równym lub zbliżonym do osocza. jest to możliwe, chorobę podstawową a tym samym
przyczynę kwasicy (np. podać insulinę w cukrzyco-
W płynach półelektrolitowych znajduje się połowa wej kwasicy ketonowej, pro. S 411). W przypadku,
zawartości elektrolitów osocza. Po dodaniu do nich gdy w procesie leczenia groźnej postaci kwasicy nie-
węglowodanów nabierają właściwości izotonicz- zbędne jest zastosowanie substancji buforowej (za-
nych. Wykorzystywane są przede wszystkim w celu zwyczaj przy wartości pH < 7,15 < 7,2), szczególnie
uzupełnienia utraconej wody. wskazane są roztwory wodorowęglanu sodu. Ich po-
dawanie powinno być kontrolowane poprzez ozna-
Roztwory zastępcze są roztworami infuzyjnymi słu- czanie poziomu kwasów i zasad, przy czym w celu
żącymi do uzupełnienia (kwaśnego) soku żołądkowe- uniknięcia zasadowicy wywołanej hiperinfuzją nale-
go lub (zasadowej) wydzieliny jelita cienkiego. ży dążyć jedynie do osiągnięcie pH > 7,2, a nie całko-
witego wyrównania kwasicy. Działaniem niepożąda-
Roztwory korekcyjne o odczynie zasadowym lub nym wodorowęglanów sodu może być hipernatremia
kwaśnym przeznaczone są do korygowania niepra- i hiperosmolarność.
widłowego pH organizmu. Wystąpienie hipernatremii jest przciwwskazaniem
do podania NaHCO3. Alternartywnie można wów-
Roztwory zastępcze i korekcyjne stosowane są łącz- czas wprowadzić trometamol (2-amino-2-metylopro-
nie i określane jako roztwory korygujące. pan-1,3-diol, bufor Tris). Wskazanie to nie dotyczy
jednak pacjentów z niewydolnością oddechową, po-
W tab. B 7.2-3. podano skład niektórych roztworów infu- nieważ trometamol, najprawdopodobniej w wyniku
zyjnych. zmniejszenia stężenia CO2 krwi, powoduje depresję
oddychania.
Terapia zaburzeń gospodarki wodnej i elektrolito-
wej. Leczenie zaburzeń gospodarki wodnej i elektro-
litowej powinno być dobierane indywidualnie, w za-
leżności od rodzaju zaburzeń. Możliwe do wyboru CH2OH
środki są przedstawione w tab. B 7.2-4. H2N CH2OH
CH2OH
Terapia zaburzeń gospodarki kwasowo-zasado-
wej. W kwasicy oddechowej konieczne jest zwięk- trometamol
szenie wentylacji, w razie potrzeby przez wprowa-
dzenie oddechu zastępczego.
MUTSCHLER-2009.indd 724 2010-01-07 22:14:26
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 725
Tabela 7.2-4. Sposoby leczenia zaburzeń gospodarki wodnej i elektrolitowej
Rodzaj Leczenie
zaburzenia
Odwodnienie
izotoniczne infuzja płynu elektrolitów
hipotoniczne infuzja roztworów glukozy lub roztworów półelektrolitowych z dodatkiem węglowodanów;
po zmetabolizowaniu części węglowodanów podana woda może być wykorzystana
jako tzw. wolna woda osmotyczna
hipertoniczne powolne podawanie hipertonicznego roztworu NaCl przy stałym nadzorze pacjenta
lub doustne podawanie NaCl
Nadmierne nawodnienie
izotoniczne podawanie diuretyków
hipertoniczne przy niezaburzonej czynności nerek podawanie diuretyków, przy niewydolności nerek hemodializa
w celu usunięcia jonów sodowych, infuzja roztworów półelektrolitowych w celu doprowadzenia wolnej wody osmotycznej
hipotoniczne powolne podawanie hipertonicznego roztworu NaCl jak w odwodnieniu hipotonicznym
Hipokalemia
podawanie chlorku potasu doustnie lub pozajelitowo (maksymalnie 20–30 mmol/godz.)
przy stałej kontroli EKG i stężenia potasu w osoczu
Hiperkalemia
wstrzyknięcia kompleksowych soli wapnia, infuzja glukozy z insuliną
w celu spowodowania wewnątrzkomórkowego wiązania potasu,
lub hemodializa; eliminacja jonów potasu przez doustne podawanie wymienników kationowych (np. Resonium® A)
Hipokalcemia
podanie doustnie soli wapnia, a w cięższych przypadkach (tężyczka) dożylnie glukonianu wapnia
Hiperkalcemia
infuzja 0,9% roztworu NaCl i diuretyku pętlowego typu furosemidu, w hiperkalcemii zagrażającej życiu infuzja
sterylnego izotonicznego roztworu kwasu ortoborowego wersenianu lub hemodializa
Hipomagnezemia
w lżejszych przypadkach Mg2+ w dawce 10 mmol/dz., w ciężkiej hipomagnezemii
z napadami drgawkowymi powolne dożylne podawanie siarczanu magnezu
Hipermagnezemia
w przypadku porażenia czynności oddechowej lub zaburzeniach rytmu serca
gosodarka wodno-elektr.
podawanie dożylnie Ca2+ lub hemodializa
Nerki i drogi moczowe,
Nagłe przypadki zasadowicy oddechowej można kaliemii, którą można wyrównać, podając chlorek
opanować przez zastosowanie oddychania wstecz- potasu. Zastosowanie roztworów zakwaszających,
nego do butelki plastykowej (wzrost stężenia CO2 zawierających wymienialne kationy (np. argininę),
we krwi), w przypadku zaburzeń o charakterze prze- lub wykonanie wlewu z mocno rozcieńczonego roz-
wlekłym, np. w chorobach płuc, konieczne jest wyle- tworu HCl jest konieczne tylko w wyjątkowych sy-
czenie choroby podstawowej. tuacjach. B7
Zasadowica metaboliczna prowadzi do wydziela-
nia nadmiernej ilości K+ przez nerki, a to do hipo-
MUTSCHLER-2009.indd 725 2010-01-07 22:14:26
726 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
7.3. Patogenetyczne mechanizmy obrzęku
Obrzękiem nazywane jest patologiczne gromadzenie Tabela B. 7. 3-1. Przyczyny tworzenia się obrzęków
się płynu w przestrzeni śródkomórkowej lub w ko-
mórkach tkanek.
Zwiększenie ciśnienia kapilarnego krwi na skutek:
Puchlina wodna jest nagromadzeniem się płynu
w jamach ciała, wodobrzuszem określa się natomiast znacznego nadciśnienia tętniczego
obecność puchliny w jamie brzusznej. rozszerzenia naczyń oporowych
U podłoża obrzęku mogą leżeć następujące pato- zwężenia żyłek
mechanizmy (tab. B 7.3-1): zastoju krwi żylnej w chorobach serca
(niewydolność prawokomorowa, zwężenie zastawki tętnicy płucnej,
■ wzrost ciśnienia kapilarnego, zwężenie zastawki trójdzielnej)
■ zmniejszenie koloidowego ciśnienia osmotycznego utrudnienia odpływu krwi żylnej
(zakrzepica żylna, żylaki)
(ciśnienia onkotycznego),
długotrwałego stania lub siedzenia
■ zaburzenia gospodarki elektrolitycznej, hiperwolemii
■ zwiększenie przepuszczalności kapilar, Spadek ciśnienia koloidosmotycznego
w niedoborze białek na skutek:
■ zaburzenia odpływu chłonki,
niedożywienia
■ zwiększona produkcja albo zmniejszony rozpad al- żołądkowo-jelitowej utraty białek
dosteronu. (przewlekłe wymioty, biegunki, upośledzone wchłanianie),
zmniejszonej syntezy białek w wątrobie
W warunkach fizjologicznych w początkowym odcin- (ciężka przewlekła choroba wątroby,
izolowanego zaburzenia syntezy białek)
ku kapilarów płyn z naczyń przedostaje się do prze-
strzeni śródmiąższowej. Panujące tutaj ciśnienie białkomoczu nerkowego
(objaw nerczycy, kłębuszkowe zapalenie nerek)
kapilarne krwi przewyższa bowiem koloidowe ciś-
przezskórna utrata białek: oparzenia,
nienie osmotyczne (onkotyczne) w kapilarach, co po- wysiękowe zapalenia skóry
woduje przeciśnięcie płynu do przestrzeni śródmiąż-
szowej. W środkowym odcinku kapilar wartości oby- Zaburzenia elektrolitowe na skutek:
dwu ciśnień są bardzo zbliżone, w związku z czym wzmożonej retencji sodu m.in. w kłębuszkowym
nie występują tu żadne przesunięcia objętościowe zapaleniu nerek lub niewydolności serca
płynu pomiędzy przestrzenią wewnątrz- i zewnątrz- Zwiększona przepuszczalność kapilar na skutek:
komórkową. Przy końcu kapilarów, gdzie ciśnienie niedokrwienia, niedotlenienia
onkotyczne przewyższa ciśnienie krwi w kapilarach, rozwoju stanu zapalengo
następuje powrót płynu z przestrzeni śródmiąższowej nadwrażliwości
do naczynia. Łączna ilość płynu powracająca do ka-
działania toksyn bakteryjnych
pilar jest jednak mniejsza niż łączna ilość płynu wy-
działania szkodliwych czynników chemicznych i mechanicznych
pływającego. Ta część, która pozostała na zewnątrz,
jest zbierana przez systemu naczyń limfatycznych Zaburzenia odpływu limfy na skutek:
i za pośrednictwem przewodu piersiowego (duc- wrodzonego niewłaściwego przebiegu naczyń limfatycznych
tus thoracicus) wprowadzana ponownie do układu bakteryjnego zapalenia naczyń chłonnych
krwionośnego. zaczopowania przez robaki (filarioza) lub komórki guza
Łatwo przewidzieć, że ilość płynu w przestrzeni bliznowacenie (po napromienowaniu)
śródmiąższowej zwiększa się wówczas, gdy prąd operacyjnego usunięcia naczyń limfatycznych
płynu wypływającego zwiększa się na skutek wzro-
stu ciśnienia kapilarnego, gdy ciśnienie onkotyczne Wzmożone wydzielanie aldosteronu
lub zmniejszony jego wydalanie
i wraz z nim zasysanie z przestrzeni śródmiąższowej
w obszarze zakończeń kapilarów zmniejsza się lub zmniejszenie objętości krążącego osocza
lub zaburzenia funkcji nerek
gdy dochodzi do zaburzeń odpływu chłonki.
Wzrost ciśnienia kapilarnego może być również Przyczyny idiopatyczne (nieznane)
skutkiem długotrwałego przebywania w pozycji sto-
MUTSCHLER-2009.indd 726 2010-01-07 22:14:27
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 727
jącej lub siedzącej, rozszerzenia naczyń oporowych
i/lub zwężenia żyłek (np. we wstrząsie), miejscowo
zaburzenia
przy upośledzonym odpływie żylnym (np. wskutek pompy
żylaków lub zakrzepu żylnego) albo ogólnego w wy- ssąco-tłoczącej
niku chorób serca z towarzyszącym im zastojem żyl-
nym krwi lub marskości wątroby. zmniejszenie
pojemności
W marskości wątroby część miąższu wątroby jest zastępo- minutowej serca
wana przez tkankę łączną, a tym samym przepływ wątrobo-
wy krwi przez żyłę wrotną staje się utrudniony i zwiększa zwiększone
wydzielanie aktywacja zmniejszenie
się ciśnienie krwi w żyle wrotnej (nadciśnienie wrotne). Za- hormonu układu przepływu
stój krwi w żyle wrotnej prowadzi do nagromadzenia płynu antydiuretycz- współczulnego osocza w nerkach
w jamie brzusznej (wodobrzusze). nego
Obrzęk spowodowany obniżeniem koloidowego ograniczenie
ciśnienia osmotycznego powstaje z powodu spożywa- uwalnianie reniny ilości
przesączu
nia zbyt małej ilości produktów białkowych (obrzęki kłębuszkowego
głodowe), ograniczonej syntezy białek (np. w cięż-
kich chorobach wątroby) lub utraty znacznej ilości zwiększone
białek (np. w zespole nerczycowym, oparzeniach, wydzielanie
wysiękowych zapaleniach skóry). angiotensynyn II
Przyczyną obrzęku mogą być zaburzenia przemia-
ny elektrolitów ze zwiększoną retencją sodu. Wystę- zwiększone
pują one w zapaleniu kłębuszkowym nerek (przebie- wydzielanie
aldosteronu
gającym jednocześnie z uszkodzeniem kapilarów),
w niewydolności serca lub na skutek przedawkowa-
nia hormonów kory nadnerczy. retencja chlorku
sodu i wody
Do obrzęku z uszkodzeniem ściany kapilarów
(np. przy niedokrwieniu, hipoksji, reakcji zapal-
nej lub alergicznej, zatruciu toksynami bakteryjny- wzrost ciśnienia
mi) dochodzi w wyniku przenikania białek osocza żylnego
do przestrzeni śródmiąższowej, co powoduje obniże-
nie wewnątrzkomórkowego zasysania i przechodze-
obrzęk
nie płynu do tkanek.
Zaburzenia odpływu chłonki mogą być następ-
stwem m.in. zapalenia węzłów chłonnych, zaczopo-
wania naczyń chłonnych przez nicienie (filaria) lub Ryc. B 7.3-1. Schemat patogenezy obrzęków w niewydolności
serca.
komórki nowotworowe, blizn popromiennych lub
gosodarka wodno-elektr.
powstałych w wyniku usunięcia naczyń lub węzłów
chłonnych, np. w ramach operacji usunięcia raka sut- Nerki i drogi moczowe,
ka. we czy wątrobowe. Na ryc. B 7.3-1. przedstawiono
Pojawienie się obrzęków często ma miejsce do- w sposób schematyczny patogenezę obrzęków w nie-
piero wówczas, gdy zmniejszenie objętości krążą- wydolności serca.
cego osocza wywołanego nagromadzeniem płynów
w tkankach spowoduje zwiększenie wydzielania al- Wspólną cechą wszystkich obrzęków jest to, że wzmo-
dosteronu lub ograniczenie jego rozkładu w przypad- żone wydzielanie sodu prowadzi do ich wypłukania.
ku zaburzenia funkcji wątroby, a tym samym wzrost Postępowanie terapeutyczne powinno być ukierun-
wchłaniania zwrotnego sodu i wody w kanalikach kowane przede wszystkim na usunięcie pierwotnej B7
dystalnych. przyczyny tworzenia się obrzęku: w obrzęku serco-
wym należy więc leczyć niewydolność mięśnia ser-
Niezależnie od wymienionych patomechanizmów cowego, w obrzęku wątrobowym zatrzymać postęp
obrzęki można podzielić w zależności od narządów marskości wątroby, w nerczycy natomiast doprowa-
biorących w niej udział, np. obrzęki sercowe, nerko- dzić do uszczelnienia błony kłębuszka nerkowego.
MUTSCHLER-2009.indd 727 2010-01-07 22:14:27
728 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
7.4. Leki moczopędne
Środki powodujące wzmożone wydzielanie moczu jonów sodu i chloru, a jednocześnie zwiększają wyda-
nazywane są lekami moczopędnymi (diuretykami). lanie wody. Optymalna będzie taka substancja czynna,
Jeżeli wraz ze zwiększeniem wydzielania wody na- która spowodowałaby wydalenie elektrolitów w sto-
stępuje zwiększenie wydalanie soli, wówczas mówi sunku stężeń równym ich stosunkowi stężeń w płynie
się o saluretykach lub natriuretykach (diuretykach śródmiąższowym. Nie ma jednak takiego diuretyku
w ścisłym znaczeniu). – przynajmniej w postaci jednej substancji. Tiazydy
i diuretyki pętlowe powodują nie tylko wzmożone
Akwaretykami są związki, których zadaniem jest spowo- usuwanie z organizmu NaCl, ale również utratę jonów
dowanie zwiększonego wydalania wody. Nie ma jednak potasu i magnezu, diuretyki oszczędzające potas pro-
akwaretyków, których działanie po doustnym podaniu by-
łoby skuteczne. wadzą natomiast do retencji potasu, a wtórnie do reten-
cji magnezu. Próbowano więc za pomocą preparatów
łączonych (tab. B 7.4-3), tj. poprzez jednoczesne po-
Diuretyki, pomimo działania w obrębie nerek,
dawanie tiazydu lub diuretyku pętlowego z substancją
nie są „lekami nerkowymi”, tzn. nie mogą ani złago-
czynną oszczędzającą potas, osiągnąć neutralny bilans
dzić przebiegu czy wyleczyć chorób nerek, ani opóź-
potasu i magnezu pomimo zwiększonego wydzielania
nić rozpoczęcia dializy w przypadku niewydolności
NaCl. Tego typu działanie jest jednak możliwe tylko
nerek. Liczne diuretyki (tiazydy, diuretyki oszczę-
pod warunkiem ścisłego nadzoru poziomu elektroli-
dzające potas, ale nie diuretyki pętlowe, por. niżej)
tów we krwi, a szczególnie poziomu potasu.
prowadzą – przynajmniej krótkotrwale w przypadku
przyjmowania większych dawek – w wyniku osła- Oprócz bezpośredniego działania nerkowego diuretyki wy-
bienia przesączania kłębuszkowego do zmniejszenia kazują również – w zależności od grupy, z której pochodzą
wydzielania substancji, które powinny wejść w skład – pośrednie działanie pozanerkowe, objawiające się poza-
moczu, i dlatego przejściowo mogą pogorszyć nie- nerkowo. Pozytywny efekt działania diuretyków pętlowych
wydolność nerek. Terapeutyczną korzyścią stosowa- w ostrej niewydolności serca, pojawiający się w bardzo
krótkim czasie po ich podaniu dożylnym, polega na rozsze-
nia diuretyków może być więc wyłącznie ich zdol- rzeniu naczyń żylnych spowodowanym uwolnieniem pro-
ność do podwyższania ekstrakcji elektrolitów i wody. staglandyn. Działanie przeciwnadciśnieniowe diuretyków
Konieczny do tego wpływ na procesy transportu warunkowane jest częściowo, jak już wyżej wspomniano
jest tylko pozornie swoisty dla nerek: ponieważ stę- (tab. B 7.4-3), osłabieniem reaktywności naczyń wynika-
żenie diuretyków w kanaliku w czasie pasażu przez jącym ze zmniejszenia zawartości sodu w mięśniówce na-
czyń. Oszczędzający potas triamteren zawiera natomiast
nefrony bardzo wzrasta, działanie nerkowe (diure- działający pozanerkowo antyarytmiczny składnik czynny
tyczne) jest ważniejsze niż działanie w innych narzą- o działaniu pozanerkowym, które nie wynika z retencji po-
dach. W żadnej inna grupie leków nie ma tak dużej tasu, ponieważ należący do tej samej grupy amilorid nie ma
zależności między działaniem farmakodynamicznym takiej właściwości.
i farmakokinetycznym jak w przypadku diuretyków.
Punkty uchwytu. Punkty uchwytu różnych grup diu-
Do obecnie stosowanych grupy diuretyków należą: retyków zaznaczono na ryc. B 7.4-1. Inhibitory anhy-
drazy węglanowej uaktywniają się przede wszystkim
■ benzotiazydy (tiazydy) i ich pochodne,
w kanaliku proksymalnym, diuretyki pętlowe w obrę-
■ diuretyki pętlowe, bie części grubościennej ramienia wstępującego pętli
Henlego, tiazydy w części początkowej kanalika dy-
■ diuretyki oszczędzające potas.
stalnego, diuretyki oszczędzające potas w końcowym
Rozwój tych skutecznych i dobrze tolerowanych leków odcinku kanalika dystalnego i w kanaliku zbiorczym.
spowodował prawie całkowite wycofanie starszych prepa- Punkt uchwytu określa profil diuretyku i jego działa-
ratów. Dotyczy to zwłaszcza leków zawierających olejki nie niepożądane.
eteryczne lub pochodne ksantyny. Także diuretyki osmo-
tyczne oraz inhibitory anhydrazy węglanowej, za wyjąt- Siła działania diuretyków. Diuretyki, które wyka-
kiem niewielu wyjątkowych wskazań, właściwie nie są sto-
sowane. zują zbliżoną do liniowej zależność między dawką
a działaniem (ryc. B 7.4-2) i w których przypadku
wraz ze zwiększaniem dawki następuje nasilenie diu-
Działania. Jak wspomniano na początku, saluretyki
rezy, tworzą grupę diuretyków o wysokiej efektyw-
zwiększają wydzielanie niektórych jonów, szczególnie
ności. Należą do nich diuretyki pętlowe.
MUTSCHLER-2009.indd 728 2010-01-07 22:14:27
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 729
kami o rożnach miejscach działania jest właściwym
diuretyki postępowaniem. Jego trafność z klinicznego punktu
oszczędzające potas widzenia potwierdza leczenie niewydolności nerek
inhibitory anhydrazy
węglanowej za pomocą diuretyku pętlowego połączonego z tiazy-
dem: nawet niewielkie dawki obu substancji są sku-
teczniejsze i jednocześnie wywołujące mniej działań
tiazydy kora niepożądanych niż jeszcze niedawno powszechnie
rdzeń stosowane w ramach leczenia jednoskładnikowego
duże dawki diuretyku pętlowego. Ponadto jednoczes-
ne krótkotrwałe podawanie działającego proksymal-
diuretyki pętlowe nie inhibitora anhydrazy węglanowej oraz diuretyku
pętlowego powoduje w wielu przypadkach przeła-
manie wyżej opisanej oporności warunkowanej nad-
Ryc. B 7.4-1. Miejsca działania diuretyków w obrębie nefronów. mierną resorpcją proksymalną.
Wskazania. Głównymi wskazaniami do podania diu-
retyku są:
Diuretyki, w których przypadku krzywa przedsta-
■ ostry obrzęk (np. obrzęk płuc),
wiająca zależność między dawką a działaniem szybko
spłaszcza się, co oznacza, że od pewnego momentu ■ przewlekły obrzęk,
zwiększanie dawki nie pociąga za sobą zwiększenia
■ choroba nadciśnieniowa,
siły działania, określane są jako diuretyki o umiarko-
wanej efektywności. Do tej grupy należą tiazydy oraz ■ ostra, jak również przewlekła niewydolność serca.
diuretyki oszczędzające potas.
Wskazaniami dodatkowymi są:
Należy jednak podkreślić, że siła działania i maksy-
■ wymuszona diureza przy zatruciach,
malny efekt diuretyku zależą nie tylko do substancji
czynnej, ale również, w takim samym lub większym ■ moczówka prosta wywołana cukrzycą,
stopniu, od czynności nerek i stanu chorobowego
■ (w przypadku inhibitorów anhydrazy węglanowej)
pacjenta. W przypadku chorób ze zmniejszoną efek-
jaskra oraz zapobieganie chorobie wysokościowej.
tywną objętością krwi krążącej, np. w niewydolno-
ści krążenia z obrzękami albo w marskości wątroby
Słuszność stosowania diuretyków w obrzękach
z wodobrzuszem, resorpcja proksymalna sodu i wody
nie wymaga uzasadnienia, ponieważ substancja czyn-
zwiększa się – przeważnie w wyniku stymulacji ukła-
na spełnia wówczas rolę diuretyku lub saluretyku,
du renina-angiotensyna-aldosteron – o 70–80%. Podaż
zwiększającego wydalanie NaCl i wody niezbędne
potasu i wody w dalszych odcinkach kanalika dystal-
do usunięcia obrzęku. Diuretyki mogą jednak, jak
nego ulega w związku z tym redukcji i jednocześnie
gosodarka wodno-elektr.
następuje zmniejszenie podatności, a niekiedy nawet
całkowity zanik podatności tej części nerki na dzia- Nerki i drogi moczowe,
łanie wszystkich diuretyków (tzw. oporność diurety-
działanie >
ków). W przypadku chorób nerek zwiększone wyda-
lanie NaCl i wody po podaniu diuretyku zmniejsza się
adekwatnie do stopnia upośledzenia funkcji nerek.
diuretyki o wysokiej
Mechanizmy działania. Mechanizmy działania diu- efektywności
retyków przedstawiono w tab. B 7.4-3. oraz przy opi- (diuretyki pętlowe)
sie poszczególnych grup. B7
Sekwencyjna blokada nefronu za pomocą diure- diuretyki o umiarkowanej
efektywności (tiazydy,
tyku. Ponieważ diuretyki wiążą się tylko z częścią diuretyki oszczędzające potas)
układu kanalikowego i w związku z tym w położo-
nych dystalnie odcinkach może wystąpić wzmocnio- dawka >
ne (kompensacyjne) wchłanianie zwrotne, sekwen-
cyjna blokada nefronu dwoma (lub więcej) diurety- Ryc. B 7.4-2. Zależność między dawką a działaniem diuretyku.
MUTSCHLER-2009.indd 729 2010-01-07 22:14:28
730 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
anhydraza
inhibitory anhydrazy węglanowa wydalanie Na+ ↑
węglanowej
wydalanie dwuwęglanu K+ ↑,
wodobrzusze
2 Cl-
+
Na K+
wydalanie Na+, Cl- ↑
diuretyki pętlowe
wydalanie K+, Mg2+, Ca2+ ↑
Na+ Cl-
tiazydy, wydalanie Na+, Cl- ↑
analogi tiazydów wydalanie K+, Mg2+ ↑
wydalanie Ca2+ ↓
amilorid, wydalanie Na+, Cl- ↑
triamteren
wydalanie K+, Mg2+ ↓
Na+
eplerenon, A aldosteron – białko indukowane ↓
spironolakton
AR wydalanie Na+, Cl- ↑
wydalanie K+, Mg2+ ↓
Ryc. B 7. 4-3. Schemat działania diuretyków. A – aldosteron, AR – receptor aldosteronu; ––< efekt bezpośredni, ----< efekt pośredni.
już wspomniano, uzupełnić swoistą terapię choroby Ważnym działaniem niepożądanym diuretyków
podstawowej, ale nie mogą jej zastąpić. pętlowych i tiazydów jest utrata jonów potasowych,
oraz zwiększone wydzielanie jonów magnezu. Z jed-
W obrzękach nerkowych diuretyki mogą wprawdzie usunąć nej strony, zaburzenia te polegają na nasileniu wymia-
obrzęki, ale ich główna przyczyna, utrata białek wynikająca ny jonów sodowych na jony potasowe w końcowym
z nieszczelnego filtru nerkowego, pozostanie. W przypadku
wodobrzusza warunkowanego chorobami wątroby diurety- odcinku kanalika dystalnego – na skutek ograniczo-
ki ani nie wzmagają produkcji albumin, ani nie obniżają nej resorpcji zwrotnej jonów sodowych w początko-
nadciśnienia wrotnego. Jednak również w tym przypadku wych odcinkach kanalika wzrasta podaż jonów sodo-
leczenie wodobrzusza jest tylko objawowe. wych w końcowym odcinku dystalnym, a z drugiej,
na spowodowanej działaniem NaCl aktywacji układu
Ponadto nie każdy obrzęk należy ewentualnie można renina-angiotensyna-aldosteron.
leczyć diuretykami. Ograniczenie to dotyczy obrzę- W przypadku szybkiego usunięcia obrzęku za po-
ków u kobiet ciężarnych, u których diuretyki stwa- mocą silnie działającego diuretyku dochodzi do utraty
rzają niebezpieczeństwo zwiększenia lepkości krwi płynu wewnątrznaczyniowego, który nie może również
i wynikającego z tego pogorszenia zaopatrzenia pło- szybko być uzupełniony z przestrzeni zewnatrznaczy-
du w tlen. niowej. Skutkiem tego jest odwodnienie ze wzrostem
Diuretyki mogą być krótkotrwale stosowane hematokrytu (wzrost stężenia hemu), a tym samym
w leczeniu przewlekłych, stwardniałych obrzęków zwiększenie lepkości krwi i ryzyko zakrzepicy.
lub wyraźnych zaburzeń odpływu wynikających Diuretyki pętlowe i tiazydy pogarszają tolerancję
z niewydolności żylnej. Nie mogą one jednak zastąpić glukozy. Może to prowadzić do ujawnienia się utajonej
leczenia uciskowego, ale jedynie wspomóc je w fazie cukrzycy lub w rozpoznanej cukrzycy do konieczności
początkowej. przestawienia na insulinę pacjenta przyjmującego do-
Stosowanie diuretyków w nadciśnieniu krwi oraz tychczas doustny lek przeciwcukrzycowy. W przypad-
w niewydolności serca omówiono wyżej. ku pacjentów leczonych stale insuliną może nastąpić
konieczność zwiększenia dawki tego leku.
Działania niepożądane. Wszystkie diuretyki mogą Kolejnym działaniem niepożądanym obydwu grup
spowodować zaburzenia równowagi elektrolitowej diuretyków jest ich wpływ na metabolizm lipidów:
i wodnej. stężenie triglicerydów osocza wzrasta podobnie jak
MUTSCHLER-2009.indd 730 2010-01-07 22:14:28
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 731
stężenie lipoprotein o małej gęstości oraz lipoprote- grup sulfamidowych w cząsteczce ma układ pierście-
in o bardzo małej gęstości. U pacjentów z tenden- niowy. Tiazydy wymienione w tab. B 7.4-1 mają iden-
cją do wzrostu stężenia kwasu moczowego we krwi tyczny profil działania i właściwie nie różnią się mię-
zwiększa się niebezpieczeństwo wystąpienia dny. dzy sobą możliwościami klinicznego zastosowania.
W przypadku diuretyków oszczędzających potas Najczęściej stosowanym diuretykiem jest hydro-
najważniejszym działaniem niepożądanym jest reten- chlorotiazyd.
cja potasu zagrażająca hiperkaliemią.
Wystąpienie i nasilenie wymienionych działań Ta grupa substancji jest dobrym przykładem tego, że pozna-
niepożądanych jest zawsze zależne od przyjmowanej nie jednej nowej substancji czynnej powoduje pojawienie
się na rynku farmaceutycznym licznych tylko w niewielkim
dawki leku. Jeżeli, zgodnie z obecnym stanem wiedzy stopniu zmodyfikowanych substancji o identycznym profi-
(np. w leczeniu choroby nadciśnieniowej), podawane lu działania.
dawki będą mniejsze niż te zalecane w przeszłości,
działania niepożądane będą nieznaczne lub nie poja- Oprócz pochodnych dihydro-benzotiadiazyny za-
wią się wcale. Diuretyki prawidłowo dobrane i pra- częto stosować w procesie leczenia wiele kolejnych
widłowo dawkowane należą do substancji czynnych pochodnych sulfonamidów (tab. B 7.4-1) charaktery-
zarówno skutecznych, jak i dobrze tolerowanych. zujących się podobnym działaniem leczniczym i wy-
wołujących zbliżone działania niepożądane (analogi
Przeciwwskazania. Przeciwwskazaniem do stoso- tiazydów). Różnice pomiędzy tymi związkami doty-
wania diuretyków pętlowych i tiazydów jest niewy- czą przede wszystkim kinetyki eliminacji. Np. chlor-
dolność nerek z bezmoczem, a ze względu na po- talidon jest związkiem o istotnie dłuższym okresie
wodowaną przez nie eliminację potasu również stan półtrwania niż hydrochlorotiazyd.
przedśpiączkowy i śpiączka wątrobowa oraz ciężka
hipokaliemia; podając diuretyki oszczędzające potas Działania. Tiazydy (i analogi tiazydów) zwiększają
należy uważać na hiperkaliemię i poważne zaburze- wydzielanie jonów sodowych i chlorkowych, wzmożo-
nia czynności nerek. Diuretyków nie zaleca się rów- ne jest również wydzielanie jonów potasowych i mag-
nież w hiponatremii. nezowych. Obniżone jest natomiast wydalanie jonów
wapniowych i fosforanowych! Tym tiazydy różnią się
Interakcje. Połączenie diuretyków pętlowych z anty- jakościowo od innych grup diuretyków.
biotykami aminoglikozydowymi i pochodnymi pla- W przeciwieństwie do inhibitorów anhydrazy
tyny zwiększa ich nefrotoksyczność – przede wszyst- węglanowej tiazydy działają skutecznie w kwasicy
kim przy niewystarczającej objętości podawanych metabolicznej i w jej terapii długoterminowej, jed-
płynów. Ryzyko działania ototoksycznego zwiększa nak ich przedłużone podawanie osłabia saluretyczne
się przy jednoczesnym podawaniu dużych dawek działanie na skutek uruchamiania w organizmie me-
szybko podawanych diuretyków pętlowych z an- chanizmu regulacji odwrotnej (zwiększone wydziela-
tybiotykami aminoglikozydowymi. Niesteroidowe nie reniny, wzrost wytwarzania angiotensyny II oraz
leki przeciwzapalne lub przeciwreumatyczne, które zwiększone wydzielanie aldosteronu).
działają poprzez hamowanie syntezy prostaglandyn,
gosodarka wodno-elektr.
osłabiają działanie większości diuretyków. Wskutek Mechanizm działania. Mechanizmem działania tia-
zwiększonego wydzielania potasu diuretyki pędowe zydów określono zmniejszenie przyswajania NaCl Nerki i drogi moczowe,
i tiazydy nasilają działanie glikozydów nasercowych ze światła początkowego odcinka kanalika dystal-
i stabilizujących leków zwiotczających mięśnie. Jed- nego do komórek nabłonkowych kanalika na skutek
noczesne podawanie glukokortykosteroidów i leków zahamowania kotransportu Na+/Cl–.
przeczyszczających z diuretykami pętlowymi i tiazy-
dami zwiększa niebezpieczeństwo hipokaliemii. Farmakokinetyka. Tiazydy są szybko i na ogół
w znacznym stopniu resorbowane z jelita i na drodze
filtracji kłębuszkowej oraz transportu aktywnego wy-
7.4.1. Tiazydy (dwutlenki dzielane w kanaliku proksymalnym. Ich biotransfor- B7
macja nie przebiega jednakowo. O ile np. hydrochloro-
dihydro-benzotiadiazyny) tiazyd właściwie nie podlega przemianie metabolicz-
i ich pochodne nej, o tyle bemetizyd w formie niezmienionej obecny
jest w moczu końcowym w niewielkiej ilości.
Tiazydy (dwutlenki dihydro-benzotiadiazyny) i ich
prototyp, hydrochlorotiazyd są dwucyklicznymi po- Wskazania. Tiazydy ewentualnie ich analogi mogą
chodnymi sulfonamidów, w których jedna z dwóch być stosowane we wszystkich podanych wyżej wska-
MUTSCHLER-2009.indd 731 2010-01-07 22:14:28
732 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
Tabela B 7.4-1. Pochodne dihydro-benzotiodiazyny (tiazydy) i analogi tiazydów sulfonamidowych
Wzór strukturalny Nazwa Nazwa handlowa Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dobowa (mg)
a) tiazydy
R2 NH R1
NH
H2NO2S S
O O
R1 R2
hydrochlorotiazyd Esidrix, 6-12 25
H Cl HCT-beta,
HCT-CT,
HCT HEXAL i in.
F bendroflumetiazyd składnik Docireticu, 3 5
F
pertenso N,
Tensoflux
F
bemetyzyd składnik dehydro 3-6 25
Cl
sanol tri,
diucomb
CH3
b) analogi tiazydu
Cl R2
H2NO2S R1
R1 R2
H3C O mefruzyd Składnik Sail-Adalat, 7 25
O Sali-Prent
H
S N
O CH3
O chlortalidon Hygroton 40-60 25
HN H
OH
CH3 ksypamid Aquaphor, 5-8 20
NH
Xipamid Al,
OH Xipamid beta,
O Ximpamid HEXAL i in.
H3C
CH3 klopamid Składnik Breserinu N, 4-8 20
NH
Viskaldix
N H
O
H3C
CH3 indapamid Indapamid-CT, 14-15 2,5
INDA-PUREN,
NH Natrilix
N
H
O
MUTSCHLER-2009.indd 732 2010-01-07 22:14:29
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 733
zaniach głównych. Ich szczególne znaczenie wynika efektywności) wszystkie diuretyki pętlowe są silnie
z długiego czasu działania, a przez to możliwości moczopędne.
przyjmowania tylko raz dziennie w leczeniu długo-
terminowym hipertonii i przewlekłej niewydolności Furosemid, prototyp diuretyków pętlowych, ma
serca. strukturę sulfonamidu z podstawnikiem przyciąga-
jącym elektrony w pozycji -o-grupy do grupy sulfo-
Dawkowanie. Typowe dawkowanie dobowe podano namidowej. W miejscu drugiej grupy sulfamidowej
w tab. B 7.4-1. Ponieważ zwiększenie dawek diurety- znajduje się grupa karboksylowa. Związki analogicz-
ku nie pociąga za sobą intensyfikacji ich pożądanego ne to bumetanid, piretanid i torasemid.
działania, a jedynie nasila działania niepożądane (diu-
retyki o umiarkowanej efektywności), nie jest wska- Bumetanid i piretamid są pochodnymi kwasu m-amino-
zane zwiększanie dawek przy braku skuteczności te- benzoesowego, a nie, jak w przypadku furosemidu, o-po-
chodnymi kwasu o-aminobenzoesowego. Torasemid jako
rapeutycznej. pochodna sulfonylomocznika wprawdzie odbiega znacznie
od furosemidu pod względem struktury chemicznej, posia-
da jednak prawie identyczny profil działania.
7.4.2. Diuretyki pętlowe
Działania. W porównaniu z tiazydami, diuretyki pęt-
Do grupy diuretyków pętlowych należą substancje lowe charakteryzuje krótsze, lecz nadzwyczaj silne
podobne do furosemidu (tab. B 7.4-2), inne związki, działanie (okresy półtrwania, por. tab. B 7.4-1 i B
np. kwas etakrynowy, wycofano. 7.4-2). Przy podawaniu pozajelitowym następujący
bezpośrednio po ich wstrzyknięciu wzrost wydzie-
Ze względu na rozwijanie działania w grubym wstę- lania sodu, chloru i wody jest bardziej znaczny niż
pującym ramieniu pętli Henlego (diuretyki o dużej w przypadku pozostałych dotychczas wymienionych
Tabela B 7.4-2. Diuretyki pętlowe
Wzór strukturalny Nazwa Nazwa handlowa Okres Średnia
międzynarodowa półtrwania dawka
(godz.) dobowa (mg)
furosemid Furo-CT, 0,5-1,5 20-40 (-80)
Cl NH Furorese,
O Furosemid-ratiopharm,
Lasix i in.
H2NO2S COOH
(CH2) 3CH3
bumetamid Burinex 1-1,5 0,5-2
HN
O
gosodarka wodno-elektr.
H2NO2S COOH Nerki i drogi moczowe,
piretanid Arelix 1-1,5 3-6 (-12)
N
O
H2NO2S COOH
CH3 torasemid Torasemid HEXAL, 3-6 2-5 (-10)
Torasemid-ratiopharm, B7
Torem,
Unat i in.
NH
N NH NH
S CH(CH3) 2
O O O
MUTSCHLER-2009.indd 733 2010-01-07 22:14:29
734 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
diuretyków. Poprzez zwiększenie dawki uzyskuje się (również u pacjentów dializowanych) mniej obcią-
zwiększenie efektu w szerokim zakresie dawek, te- żające dostarczanie wody i soli, poprawiając w ten
oretycznie przy odpowiednim dawkowaniu możliwe sposób jakość życia pacjentów. Mogą być ponadto
jest wydzielanie 30% przesączanych jonów sodo- stosowane w wymuszonej diurezie.
wych.
Ponieważ działanie utrzymuje się bardzo krótko, Dawkowanie. Zazwyczaj stosowane dawki doustne
obserwuje się, o ile diuretyk pętlowy nie będzie poda- (np. w nadciśnieniu, w niewydolności serca) są poda-
ny we właściwym czasie, stosunkowo szybki spadek ne w tab. B 7.4-2.
wydzielania poniżej wartości kontrolnych (zjawisko
odbicia).
Zarówno diuretyki pętlowe, jak i tiazydy zwięk- 7.4.3. Diuretyki oszczędzające potas
szają wydalanie jonów sodowych i chlorkowych
– w znacznie mniejszym stopniu działanie to dotyczy
Wspólną cechą dotychczas omówionych diuretyków
każdorazowo jonów sodowych – oraz jonów magne-
jest zwiększone wydalanie sodu, chloru i potasu, choć
zowych i potasowych. Intensywniej przebiega rów-
nasilenie i proporcje mogą być różne. Istnieję jednak
nież – inaczej niż w przypadku zastosowania tiazy-
związki, które pomimo zwiększenia (niezbyt wyraź-
dów – usuwanie jonów wapnia. Właściwość ta może
nego) wydalania NaCl ograniczają wydalanie jonów
być wykorzystywana przy hiperkalcemii.
potasu. Ze względu na swoje działanie są nazywane
W takiej sytuacji współczynnik przesączania kłę-
diuretykami oszczędzającymi potas. Rozróżnia się:
buszkowego nie zostanie wówczas obniżony.
■ antagonistów aldosteronu,
Mechanizm działania. Diuretyki pętlowe (furose-
■ cykliczne pochodne amidyny.
mid) ujawniają swoje działanie poprzez blokowa-
nie (szybko i odwracalnie) nośników Na+/K+/2Cl–
Efekt saluretyczny (około 2–4% przesączanego NaCl)
w świetle grubościennego odcinka ramienia wstę-
wymienionych związków działających w końcowym
pującego i hamowanie w ten sposób resorpcji jonów
odcinku kanalika dystalnego i w kanaliku zbior-
sodowych, potasowych i chlorkowych (ryc. B 7.4-3).
czym jest słabo zaznaczony. Odbywająca się bowiem
Ponadto wpływając na komórki plamki gęstej (macu-
w tych odcinkach resorpcja zwrotna jonów sodowych
la densa), osłabiają kanalikowo-kłębuszkowe sprzę-
i chlorkowych jest znacznie ograniczona, w związku
żenie zwrotne. Z tego powodu ta grupa diuretyków,
z czym regulacja wydzielania elektrolitów jest bardzo
w przeciwieństwie do tiazydów, nie obniża współ-
słaba. Diuretyki oszczędzające potas, za wyjątkiem
czynnika przesączania kłębuszkowego.
antagonistów aldosteronu (zob. niżej), nie są stoso-
wane samodzielnie, lecz w połączeniu z tiazydami
Farmakokinetyka. Aby diuretyki pętlowe (furo-
i diuretykami pętlowymi (tab. B 7.4-3).
semid) mogły zadziałać od strony światła kanalika,
muszą się przemieścić z krwiobiegu do płynu kanali-
kowego. Transport odbywa się, podobnie jak w przy-
padku tiazydów, z pominięciem filtracji kłębuszko- 7.4.3.1. Antagoniści aldosteronu
wej, przeważnie na drodze czynnego wydzielania
z kanalików proksymalnych przebiegającego przy W grupie steroidów 17-spirolaktonu znaleziono wie-
udziale przenośników kwasów. W pewnym stopniu le aktywnych antagonistów aldosteronu, spośród któ-
wyjaśnia to, dlaczego w niewydolności nerek, przy rych zastosowanie w leczeniu znalazł spironolakton
upośledzonych procesach wydzielniczych, niezbędne i jego metabolit kanrenon (ewentualnie kanrenoat po-
jest zwiększenie dawek leku, a rozpoczęcie jego dzia- tasu – zob. niżej), a także analog eplerenon.
łania następuje bardzo powoli. Ich działanie rozwija się stosunkowo wolno (nie-
kiedy dopiero dzień po zażyciu), ale utrzymuje się
Wskazania. Diuretyki pętlowe są szczególnie pożą- długo. Przy długotrwałym podawaniu należy kontro-
dane w przypadku, gdy konieczne jest szybkie i in- lować gospodarkę elektrolitową u pacjenta.
tensywne działanie, np. w obrzęku płuc. Podawane
są również pacjentom z niewydolnością serca, u któ- Mechanizm działania. W końcowym odcinku kana-
rych tiazydy przestały przynosić oczekiwane efekty lika dystalnego i w kanaliku zbiorczym antagoniści
terapeutyczne, oraz pacjentom z niewydolnością ne- aldosteronu blokują jego przyłączanie do receptora
rek. Nie zwiększają one jednak wydalania substancji, cytoplazmatycznego (ryc. B 7.4-3). Ponieważ unie-
które powinny znaleźć się w moczu, lecz umożliwiają możliwia to wniknięcie aldosteronu do jądra komórki,
MUTSCHLER-2009.indd 734 2010-01-07 22:14:30
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 735
Tabela B 7.4-3. Diuretyki – preparaty łączone (wybór) Farmakokinetyka. Spironolakton i eplerenon wchła-
niane są szybko i dobrze, a jako steroidy podlegają
Składniki Nazwa handlowa biotransformacji. Podstawowym metabolitem spiro-
(nazwa zarejestrowana) nolaktonu jest 7a-tiometylospironolakton, kolejnym
bardziej skutecznym jest natomiast wspomniany
amilorid, Diaphal
furosemid już kanrenon, powstający ze spironolaktonu na drodze
hydrolizy grupy tiooctowej i oddzieleniu bezwodnika
amilorid, Amiloretik,
hydrochlorotiazyd Amilorid comp.-ratiopharm, kwasu siarkowego. Pierścień laktonowy kanreno-
Diursan, nu może się otworzyć bez utraty aktywności i z po-
Moduretik i in. wstałym hydroksykwasem wytworzyć rozpuszczalną
triamteren, dehydro-sanol tri, sól potasową, kanrenoat potasu. W przeciwieństwie
bemetizyd diucomb
do trudno rozpuszczalnego spironolaktonu związek
triamteren, Dytide H, ten może być stosowany dożylnie. Ciekawe jest nie-
hydrochlorotiazyd Triampur compositum, wątpliwie to, że właśnie z kanrenonu, a nie spirono-
Triamteren comp.-CT,
Triamteren comp.-ratiopharm i in. laktonu, w którym brak jest podwójnego wiązania,
powstają rakotwórcze nadtlenki. Zastosowanie kan-
triamteren, Neotri renonu ewentulanie kanrenonu potasu jest w związku
ksypamid
z tym bardzo ograniczone.
Eplerenon metabolizowany jest przede wszystkim
przez CYP3A4; aktywnych metabolitów nie znale-
ziono.
ograniczona jest przez to synteza tzw. białek induko- Okres półtrwania w osoczu spironolaktonu wyno-
wanych aldosteronem (AIPs), tj. kanałów potasowych si około 1,4 godz., pochodnych tiometylu około 14
i Na+/K+-ATPazy. Skutkiem tego jest zmniejszenie godz., kanrenonu około 17 godz. i eplerenonu około
resorpcji sodu i jednoczesne obniżenie wydzielania 3–5 godz. Wydalanie antagonistów aldosteronu oraz
potasu. Opisany mechanizm działania wyjaśnia rów- ich metabolitów odbywa się drogą nerkową.
nież, dlaczego antagoniści aldosteronu zaczynają
działać dopiero po okresie utajenia, tj. po kilku godzi- Wskazania. Wskazaniem do podania spironolak-
nach i wyłącznie w obecności aldosteronu, a nie uak- tonu lub kanrenonu potasu jest obrzęk w przebiegu
tywniają się u zwierząt pozbawionych nadnerczy. hiperaldosleronemii (np. u pacjentów z marskością
wątroby i wodobrzuszem).
HO O O
CH2OH
O H3C
O
CH3 H
gosodarka wodno-elektr.
CH3 H
H H H H Nerki i drogi moczowe,
O O SCOCH3
aldosteron (postać półacetalowa) spironolakton
O
O – +
H3C O K
H3C
O OH B7
CH3 H CH3 H
H H H H
O O
kanrenon kanrenonat potasu
MUTSCHLER-2009.indd 735 2010-01-07 22:14:30
736 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
Eplerenon stosowany jest w lewokomorowej nie- niane z przewodu żołądkowo-jelitowego. Działa-
wydolności serca oraz wspomagająco w leczeniu beta nie diuretyczne rozpoczyna się po godzinie i osiąga
blokerem po niedawno przebytym zawale w przypad- po 3–4 godzinach wartość maksymalną.
ku objawów niewydolności serca. Triamteren ulega bardzo szybko biotransforma-
cji do działającego dosyć ciekawie metabolitu II
Dawkowanie. Spironolakton początkowo powinien fazy, hydroksytriamterenu, który następnie łączy się
być przyjmowany w dawkach 100–200 (–400) mg/ z (pół)estrem kwasu siarkowego. Amilorid jest nato-
dzień, w leczeniu przewlekłym 50–100 (–200) mg/ miast metabolizowany w bardzo niewielkim stopniu.
dzień Kanrenon potasu podawany jest w dawkach Okres półtrwania triamterenu w osoczu wynosi 4–6
200–400 mg/dzień dożylnie powoli lub w formie godz., amiloridu 18–20 godz. Obydwa związki wy-
szybkich wstrzyknięć. Leczenie eplerenonem należy dalane są przez nerki i wątrobę.
rozpocząć w ciągu 3–14 dni po przebytym zawale
mięśnia sercowego od dawki 25 mg/dzień i pod stała Dawkowanie. Dawka dobowa triamterenu wynosi
kontrolą poziomu potasu zwiększać ją do dawki do- 25–50 (–100/mg) a amiloridu 2,5–5 (–10) mg.
celowej i podtrzymującej wynoszącej 50 mg/dzień.
Działania niepożądane. Oprócz hiperkalemii mogą
Działania niepożądane. Oprócz już wspomnianej pojawić się zaburzenia żołądkowo-jelitowe i zawroty
hiperkaliemii mogą wystąpić zaburzenia ośrodkowe, głowy. U pacjentów szczególnie predysponowanych
zaburzenia ze strony układu żołądkowo-jelitowe- (np. z ciężką marskością wątroby) podanie triamtere-
go, a także wysypka. Działanie antyandrogeniczne, nu, ze względu na jego słabe działanie antagonistycz-
o wiele słabsze w przypadku eplerenonu niż w przy- ne w stosunku do kwasu foliowego, może prowadzić
padku spirolaktonu, u mężczyzn może prowadzić do rozwoju niedokrwistości megaloblastycznej.
do ginekomastii i zaburzeń potencji, u kobiet do za-
burzeń miesiączkowania, nadmiernego owłosienia, Interakcje. Podobnie jak w przypadku antagonistów
uczucia napięcia piersi, a także obniżenia głosu za- aldosteronu istnieje niebezpieczeństwo hiperkalie-
równo u mężczyzn, jak i u kobiet. mii, które wynika z jednoczesnego podawania inhi-
bitorów ACE.
Interakcje. Analgetyki niesteroidowe oraz inhibito-
ry ACE podnoszą ryzyko wystąpienia hiperkaliemii;
równoczesne podawanie digoksyny oraz antagoni-
stów aldosteronu skutkuje wzrostem poziomu digok-
syny we krwi. Stosowanie eplerenonu z substancjami
czynnymi, hamującymi CYP3A4 (np. klarytromycy-
ną, ketokonazolem, nelfinawirem lub ritonawirem)
zwiększa jego AUC; do zmniejszenia AUC prowa- H2N N N NH2
dzi natomiast podawanie jednoczesnie z eplereno-
N
nem substancji czynnych indukujących CYP3A4 N
(np. karbamazepiny, wyciągu z dziurawca lub feno- NH2
barbitalu).
triamteren
7.4.3.2. Pochodne cykloamidyny
Do diuretyków o strukturze cykloamidyn stosowanych
w stałym połączeniu z tiazydami lub diuretykami pętlo- H2N N NH2
wymi należą triamteren i amilorid. W przeciwieństwie H2N NH
do spironolaktonu pochodne cykloamidyny nie działa- N Cl
ją antagonistycznie do aldosteronu, ale blokują kanały NH O
sodowe w końcowym odcinku kanalika dystalnego
amilorid
i w kanaliku zbiorczym (ryc. B 7.4-3).
Farmakokinetyka. Po podaniu doustnym triamteren
i amilorid są szybko i w przeważającej części wchła-
MUTSCHLER-2009.indd 736 2010-01-07 22:14:30
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 737
7.4.4. Inhibitory anhydrazy części rdzeniowej nerki utrudnia utrzymanie zazwyczaj
wysokich gradientów stężeń, przez co prowadzi do zwięk-
węglanowej szonej diurezy. Ponieważ działanie pochodnych ksantyny
przy długotrwałym stosowaniu ulega osłabieniu i lecze-
Inhibitory anhydrazy węglanowej prawie nie są już sto- nie przestaje przynosić oczekiwane efekty, substancje te
nie są stosowane jako diuretyki. Ogólnie znane działanie
sowane jako leki moczopędne, podawane są natomiast
diuretyczne kawy i herbaty należy łączyć z zawartymi
w leczeniu jaskry oraz choroby wysokościowej. w nich metyloksantynami.
W roku 1940 zaobserwowano, że sulfanilamid i inne sul-
fonamidy hamują działanie enzymu zwanego anhydrazą
węglanową. Istotnym elementem struktury inhibitorów 7.4.6. Diuretyki osmotyczne
anhydrazy węglanowej jest niepodstawiona grupa sulfo-
namidowa (-SO2NH2) przyłączona do aromatycznego lub Diuretykami osmotycznymi, do których należą alkohole
heteroaromatycznego układu pierścieniowego. cukrowe mannitol i rzadziej stosowany sorbitol, określa się
Zablokowanie anhydrazy węglanowej zmniejsza kanali- substancje przesączane wprawdzie – po podaniu dożylnym
kową resorpcję zwrotną jonów sodowych, ponieważ mniej
jonów wodorowych oddawanych jest do kanalika. Skut-
kiem tego jest zwiększenie wydzielania nerkowego jonów
sodowych, potasowych i węglowodorowych, a tym samym
wody. Utrata zasad prowadzi do kwasicy krwi, co bardzo CH2OH CH2OH
szybko osłabia działanie inhibitorów anhydrazy węglano- HO H H OH
wej.
HO H HO H
W celach terapeutycznych stosowany jest systemowo
inhibitor anhydrazy węglanowej acetazolamid pierwszy H OH H OH
sulfonamid o działaniu moczopędnym, oraz miejscowo do- H OH H OH
rzolamid i brinzolamid w leczeniu jaskry. Za pomocą ace- CH2OH CH2OH
tazolamidu można leczyć również chorobę wysokościową,
w której na skutek hiperwentylacji dochodzi do powstania
zasadowicy. mannitol sorbitol
Acetazolamid jest dobrze wchłaniany z jelita cienkie-
go i w niezmienionej formie prawie całkowicie wydalany
jest drogą nerkową. Jego działanie rozpoczyna się po około
6 godz. i utrzymuje się przez około 4–7 godz.
W leczeniu obrzęków zazwyczaj podaje się średnio 250 – w kłębuszkach, ale niewchłaniane zwrotnie w kanalikach.
mg/dzień Poprzez jednoczesne podawanie wodorowęglanu W zależności od swojego ciśnienia osmotycznego zatrzy-
potasu można odtworzyć prawidłową rezerwę alkaliczną. mują wodę w świetle kanalika, przez co zwiększają diu-
rezę. W bardzo niewielkim stopniu wzmagają wydzielanie
elektrolitów.
Diuretyki osmotyczne mogą być stosowane w celu pod-
7.4.5. Pochodne ksantyny trzymania przepływu moczu w zagrażającej niewydolności
nerek, obniżenia podwyższonego ciśnienia wewnątrzgałko-
Pochodne ksantyny: kofeina, teofilina i teobromina, są diu- wego, wywołania diurezy oraz w leczeniu obrzęku mózgu.
retykami o zakresie działania od słabego do średnio mocne- Mannol powinien być podawany w dawce 0,3 g/kg
masy ciała/godz., sorbitol 0,6 g/kg masy ciała/godz. – cza-
gosodarka wodno-elektr.
go. Poprzez blokowanie receptora adezynowego substancje
Nerki i drogi moczowe,
te zwiększają przepływ nerkowy, zwłaszcza w części rdze- sowo i w miarę potrzeby – 0,3 g/kg masy ciała/godz. kilka
niowej nerki. Ponieważ opór naczynia doprowadzającego razy dziennie.
(vasa afferentia) zmniejsza się bardziej niż opór naczynia W przypadku skąpomoczu/bezmoczu należy poprzez
odprowadzającego (vasa efferentia), zwiększa się współ- wstrzyknięcie próbne sprawdzić, czy zaczęła się diureza.
czynnik przesączania kłębuszkowego. Pochodne ksantyny Brak diurezy jest wskazaniem do przerwania wstrzyknięć
są właściwie jedynymi diuretykami, które zwiększają GFR (niebezpieczeństwo przemieszczenia się płynów z prze-
i których działanie polega w pewnym stopniu na zwiększo- strzeni pozakomórkowej do przestrzeni wewnątrzkomór-
nym wytwarzaniu moczu pierwotnego. Lepsze ukrwienie kowej).
B7
MUTSCHLER-2009.indd 737 2010-01-07 22:14:30
738 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
7.5. Antydiuretyki
Antydiuretyki są substancjami czynnymi stosowanymi W razie konieczności, tj. kiedy podaż dużej ilości
w celu zmniejszenia (patologicznie) wysokich wydala- płynów staje się niemożliwa lub niewystarczająca,
nych ilości moczu. Głównym wskazaniem do ich po- ośrodkową moczówkę prostą można leczyć substy-
dania jest moczówka prosta (diabetes insipidus). tucyjnie za pomocą desmopresyny (dawkowanie
5–20 μg/2 × dzień donosowo, 0,2–1,2 mg/dzień do-
U podłoża moczówki prostej ośrodkowej (diabetes insipi- ustnie, w trzech pojedynczych dawkach lub 1–4 mg/
dus centralis), neurohormonalnej, jest produkcja zbyt małej dzień domięśniowo ew. dożylnie). Zaletą prepara-
ilości adiuretyny lub jej wydzielanie. Przyczyną mogą być
guzy mózgu lub przysadki, hipofizektomia lub choroby za- tów w formie kropli jest ich wchłanianie przez błonę
palne (np. zapalenie opon mózgowych), rzadziej wrodzone, śluzową nosa, co umożliwia pominięcie pasażu żo-
dziedziczne zaburzenia wytwarzania wazopresyny. U około łądkowo-jelitowego, w czasie którego białka są hy-
50% pacjentów przyczyna jest nieznana (diabetes insipidus drolizowane jeszcze przed dotarciem do krwiobiegu
idiopathicus). (por. znacznie wyższe dawki przy stosowaniu doust-
Na skutek zmniejszonej resorpcji zwrotnej wody w ka-
nalikach nerki wydalane są znaczne ilości (z reguły 4–12 nym).
l/dzień, a w przypadkach skrajnych do 40 1/dzień) moc- W przypadku nerkowej moczówki prostej stosuje
no rozcieńczonego moczu (gęstość < l,008 g/ml). Utrata się, ze względu na nieskuteczność analogów wazo-
wody i wynikające z tego zwiększenie osmolarności oso- presyny, diuretyki tiazydowe. Zasady działania tego
cza zwiększa uczucie pragnienia: utracona woda musi być (paradoksalnego) mechanizmu nie są jeszcze jedno-
uzupełniona przez zwiększone picie, w przeciwnym razie
bardzo szybko może bowiem dojść do hipertonicznego od- znacznie wyjaśnione. Z jednej strony, mówi się bo-
wodnienia organizmu. wiem o zwiększonym przenikaniu płynów w kanali-
kach proksymalnych na skutek zmniejszenia objęto-
Ośrodkową moczówkę prostą należy odróżnić od nerko- ści pozakomórkowej, z drugiej strony, o zwiększonej
wej moczówki prostej (diabetes insipidus renalis). W tej przepuszczalności wody w kanaliku dystalnym oraz
postaci nie jest upośledzone wytwarzanie wazopresyny
czy jej wydzielanie, lecz możliwości reagowania końco- kanaliku zbiorczym w wyniku działania podobne-
wych odcinków kanalików i kanalika zbiorczego na uwol- go do działania wazopresyny. (Poprzez blokowanie
nioną wazopresynę. Przyczyną jest albo wrodzone uszko- fosfodiesterazy tiazydy zwiększają, podobnie jak
dzenie kanalikowego receptora wazopresyny (receptora wazopresyna, stężenie cAMP). Ponadto na skutek
V2) na chromosomie X, (przeważnie) autosomalnej mutacji zmniejszenia się stężenia jonów sodowych w osoczu
recesywnej akwaporyny 2, albo nabytym uszkodzeniem ka-
nalików (np. na skutek przewlekłych chorób nerek, zatruć, następuje zmniejszenie uczucia pragnienia.
niedokrwistości sierpowatej).
MUTSCHLER-2009.indd 738 2010-01-07 22:14:31
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 739
7.6. Odprowadzające drogi moczowe
Z miedniczki nerkowej mocz przepływa moczowo- niówki gładkiej pęcherza stają się grubsze, tworząc zwie-
dem do pęcherza moczowego, skąd przez cewkę mo- racz wewnętrzny (sphincter internus). Dodatkowe za-
mknięcie cewki moczowej stanowi zwieracz zewnętrzny
czową wydalany jest na zewnątrz.
(sphincter externus), który tworzą mięśnie poprzecznie
prążkowane wychodzące z przepony miednicy.
Moczowody. Moczowody są cienkimi przewodami
o długości ok. 30 cm, biegnącymi pozaotrzewnowo
Pojemność pęcherza u osoby dorosłej wynosi 0,6–1 1.
(retroperitoneale) do miednicy małej i od tyłu ucho-
Zanim jednak osiągnie taką objętość, po zebraniu ok.
dzącymi do pęcherza moczowego. Ujście każdego
150–300 ml moczu pojawia się uczucie wypełnienia
z dwóch moczowodów zamyka przypominający za-
pęcherza. W początkowym okresie wypełnienia ciś-
stawkę fałd błony śluzowej. Te swoiste zawory ślu-
nienie wewnątrz pęcherza wzrasta, a przy dalszym
zówki oraz ukośne przejście moczowodu przez ścia-
zwiększaniu objętości gromadzonego moczu pozosta-
nę mięśniową pęcherza utrudniają wsteczny odpływ
je stałe. Zanim jednak ciśnienie wzrośnie do stałego
moczu podczas skurczu pęcherza.
poziomu, rozpoczyna się czynne opróżnianie pęche-
rza (zob. niżej). Mięśniówka pęcherza dostosowuje
Pęcherz moczowy. Pęcherz moczowy, jako na-
się tym samym do każdorazowego stanu wypełnienia
rząd jamisty posiadający w środku pustą przestrzeń
w taki sposób, aby możliwe było utrzymanie możli-
do gromadzenia moczu (ryc. B 7.6-1), jest położony
wie stałego napięcia pęcherza.
z przodu miednicy małej, bezpośrednio za kością ło-
nową.
Cewka moczowa. Męska cewka moczowa ma
Warstwa wewnętrzna pęcherza moczowego wyścielona 20 cm długości i w części początkowej otoczona
jest nabłonkiem przejściowym, który w opróżnionym pę- jest gruczołem krokowym (stercz, prostata, por. ni-
cherzu tworzy fałdy. Ściana pęcherza składa się z trzech żej). W obszarze tym do cewki moczowej uchodzą
nieznacznie odgraniczonych od siebie warstw mięśniówki również przewody wytryskowe (ductuli ejaculatorii).
gładkiej, które łącznie określane są jako mięsień wypieracz Od tego miejsca staje się ona wspólnym przewodem
moczu (detrusor vesicea).
W obrębie dna pęcherza znajduje się mały trójkątny dla moczu i wydzieliny gruczołu krokowego.
obszar zwany trójkątem moczowym (trigonum vesicea), Żeńska cewka moczowa nie przekracza nigdy 5 cm
w którego górnych kątach uchodzą obydwa moczowo- długości. Przebiega za spojeniem łonowym i kończy
dy, a z dolnego ostrego kąta odchodzi cewka moczowa w przedniej części przedsionka pochwy.
(urethra). W początkowym odcinku cewki włókna mięś-
Gruczoł krokowy. Nieparzysty narząd wielkości
kasztana leżący między dnem pęcherza moczowego
a mięśniówką przepony miednicy. Składa się z 30–50
moczowód
(ureter) rozgałęzionych kanalikowo-pęcherzykowatych gru-
gosodarka wodno-elektr.
czołów, które w okolicy wzgórka nasiennego (colli-
culus seminalis) uchodzą do cewki 15–30 przewoda- Nerki i drogi moczowe,
mi wyprowadzającymi i wytwarzają płynną, reagują-
cą słabo zasadowo wydzielinę o charakterystycznym
zapachu (sperma).
Mikcja. Opróżnianie pęcherza moczowego w począt-
ujście kowej fazie jest procesem dowolnym, w kolejnych
moczowodu zaś przebiega mimowolnie, a jego przebieg polega
trójkąt moczowy
(trigonum vesicea) na skurczu mięśnia wypieracza moczu, rozszerzeniu
prostata
cewki moczowej w obrębie zwieraczy wewnętrz- B7
nych, rozluźnieniu zwieraczy zewnętrznych oraz
wzgórek nasienny cewka skurczu mięśni brzucha i miednicy przy unierucho-
(colliculus seminalis) moczowa mionej przeponie. Jeżeli wypełnienie przekroczy
(urethtrea)
150 ml, wówczas na skutek działania bodźców prze-
noszonych drogami nerwowymi wstępującymi po-
Ryc. B 7.6-1. Przekrój podłużny pęcherza moczowego mężczy-
jawia się uczucie parcia na mocz. Parasympatyczne
zny (wg Leonhardta).
MUTSCHLER-2009.indd 739 2010-01-07 22:14:31
740 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
włókna zstępujące nerwu trzewnego miednicznego pęcherza moczowego, cewki moczowej i prostaty.
(n. splanchnicus pelvinus) wyzwalają skurcz mięśnia Poprzez blokowanie adrenoreceptorów α1 doprowa-
wypieracza moczu a reagujący na bodźce dowolne dza się do zmniejszenia siły oporu cewki moczowej,
nerw sromowy (n. pudendus) powoduje rozluźnienie a tym samym zapewnia ułatwiony odpływu moczu.
zwieracza zewnętrznego (sphincter externus). Chociaż w leczeniu PBS skuteczne mogą być w zasa-
Odruch oddawania moczu może być powstrzymy- dzie blokery α1, to najczęściej zaleca się substancje,
wany dzięki bodźcom pochodzącym ze śródmózgo- które wykazują powinowactwo do podtypu α1A adre-
wia, a zwłaszcza z kory mózgowej. noreceptora i w związku z tym w mniejszym stopniu
obniżają ciśnienie krwi. Do substancji tych należą
alfuzosyna.
7.6.1. Objawowy łagodny rozrostu Zaletą inhibitorów α1, dającą im przewagę nad
gruczołu krokowego (PBS) innymi lekami stosowanymi w łagodnym rozroście
stercza, jest działanie szybkie i przynoszące pra-
wie natychmiastowe złagodzenie objawów, które
7.6.1.1. Podstawy patofizjologiczne
utrzymuje się nawet w leczeniu długoterminowym.
U znacznej części mężczyzn w starszym wieku (powyżej Stosowanie leków z tej grupy nie prowadzi jednak
55–60 r.ż.) dochodzi do łagodnego powiększenia prostaty do zmniejszenia przerośniętego narządu.
(prostaty), spowodowanego rozrostem nabłonka oraz tkan- Jak dowodzą dotychczasowe obserwacje, inhibi-
ki włóknisto-mięśniowej wewnątrz gruczołu; powierzchnia tory α1 są wskazane przede wszystkim w przypadku
zewnętrzna jest natomiast wypierana przez tworzące się
liczne guzki i zgniatana do cienkiej warstwy tkanek. Cho-
pacjentów młodszych z przeważającymi objawami
roba, w przeszłości często nazywana łagodnym rozrostem podrażnienia. W dalszej kolejności można je poda-
gruczołu krokowego (BPH), obecnie najczęściej objawo- wać pacjentom z odroczoną lub niemożliwą operacją
wym łagodnym rozrostem gruczołu krokowego (PBS), prostaty, a także tym, u których do czasu przeprowa-
dotyka wprawdzie 75–100% ogółu mężczyzn powyżej 75 dzenia operacji konieczne jest złagodzenie dolegli-
roku życia, ale tylko w 30–40% przypadków pojawiają się
wyraźne dolegliwości o różnym czasie trwania i stopniu na-
wości bólowych.
silenia. Jej etiologia i patogeneza nie są jeszcze dokładnie
poznane. Za dosyć istotne czynniki patogenetyczne uznano Inhibitory 5α-reduktazy. Finasteryd oraz jego ana-
dotychczas dihydrotestosteron i estradiol. Dihydrotestoste- log dutasteryd blokują przekształcenie testosteronu
ron (DHT) pobudza bowiem wzrost komórek nabłonka pro- w 5α-dihydrotestosteron. Finasteryd wchodzi jednak
staty, a estrogeny nasilają to działanie. (U mężczyzn w po-
deszłym wieku stężenie estradiolu we krwi jest wyraźnie
w interakcję wyłącznie z 5α-reduktazą, dutasteryd
wyższe niż u młodych mężczyzn.) hamuje natomiast zarówno typ 1, jak i mniej istotny
W zależności od objawów rozróżnia się dolegliwości w przypadku BPH typ 2 5α-reduktazy. Po podaniu
ze strony prostaty wynikające z podrażnienia lub z niedroż- blokera 5α-reduktazy stężenie dihydrotestosteronu
ności dróg moczowych. W rozroście prostaty wyróżnia się w prostacie przy stężeniu testosteronu w surowicy
trzy etapy: na etapie 1. intensywność strumienia moczu
jest wyraźnie obniżona i rozpoczęcie mikcji opóźnione. Pa-
zbliżonym do stałego obniża się o około 70–80%,
cjent uskarża się na częste parcie na pęcherz w ciągu dnia wartość swoistego antygenu sterczą (PSA) obniża się
i w nocy. Na etapie 2., pomimo wciąż zwiększającej się jednocześnie o około 50%. (Ocena zmiany tego nie-
częstotliwości mikcji, pojawia się niemożność całkowitego zwykle ważnego parametru laboratoryjnego musi być
opróżnienia pęcherza, co prowadzi to do zalegania moczu uwzględniana w badaniu diagnostycznym, szczegól-
(około 100–150 ml). Na etapie 3. dochodzi do bezwiedne-
go oddawania moczu kroplami przy wypełnionym pęche-
nie w rozpoznawaniu raka prostaty.)
rzu albo całkowite zatrzymanie moczu przy utrzymującym Pożądany efekt leczenia inhibitorami 5α-reduktazy,
się parciu na pęcherz, ponadto pojawiają się objawy nerki zmniejszenie objętości prostaty o 20–30% oraz spo-
zastoinowej (postępująca niewydolność nerek). wolnienie ew. utrudnienie dalszego rozrostu gruczołu,
Stężenie antygenu prostaty (PSA) jest w objawowym ła- pojawia się dopiero po kilku miesiącach ich stosowa-
godnym rozroście gruczołu krokowego nieznaczne zwięk-
szone, w przeciwieństwie do stężenia w przypadku raka
nia i nie u wszystkich pacjentów. Działanie kliniczne
prostaty. (wpływ w przypadku dolegliwości ze strony prostaty
wynikających z niedrożności lub podrażnienia dróg
moczowych, poprawa strumienia moczu, zmniejsze-
nie ilości zalegającego moczu) jest jednak ogólnie
7.6.1.2. Środki stosowane mniejsze niż początkowo oczekiwano.
w leczeniu prostaty Dostępność biologiczna obu substancji wynosi 60–
65%. Dwa hydroksylowane metabolity finastery-
Inhibitory receptora α1-adrenergicznego. Bloka- du oraz wolne kwasy węglowe powstałe w wyniku
da receptorów α1 rozluźnia mięśnie gładkie szyjki hydrolizy jego grup amidowych zachowują jeszcze
MUTSCHLER-2009.indd 740 2010-01-07 22:14:31
Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa 741
20% właściwości substancji macierzystej. Dutaste- Leczenie farmakologiczne jest próbą obniżenia we-
ryd jest natomiast biotransformowany przede wszyst- wnątrz/gruczołowego napięcia mięśni i/lub zmniej-
kim przez CYP3A4 do znacznie słabiej działających szenia objętości prostaty. Osiągnięcie tego pozwala
hydroksymetabolitów. Okres półtrwania finasterydu nie tylko złagodzić dolegliwości, ale również uniknąć
wynosi 6–8 godzin, dinasterydu do 5 tygodni. Wy- powikłań (infekcje, uszkodzenia wynikające z zale-
dalanie finasterydu odbywa się w 40% przez nerki gania moczu).
(z moczem) i w 60% przez jelita (z kałem), dutasteryd
jest w ilości mniejszej niż 1% w formie niezmienio- Użycie inhibitorów receptora α1-adrenergicznego
nej usuwany przez nerki oraz w około 5% w formie umożliwia, jak już wspomniano, bardzo szybkie zła-
niezmienionej i w 40% w formie metabolitów przez godzenie objawów, a przez to zwiększenie objętości
jelita (z kałem). oddawanego moczu o około 20–35%. W niektórych
Podawanie inhibitorów 5α-reduktazy wskazane badanych przypadkach stwierdzono nawet niewielką
jest przede wszystkim w przypadku pacjentów w po- progresję choroby. Podobną skuteczność wykazują
deszłym wieku z wyraźnie powiększoną prostatą. adekwatne dawki pozostałych inhibitorów α (w pre-
Finasteryd należy podawać w ilości 5 mg/dzień, duta- paratach o przedłużonym uwalnianiu alfuzosyna 10
steryd 0,5 mg/dzień. mg/dzień i doksazosyna 4–8 mg/dzień oraz tamsulo-
Działaniami niepożądanymi powodowanymi przez syna 0,4 mg/dzień, terazosyna 5–10 mg/dzień), przy
finasteryd są zaburzenia potencji (zmniejszone libi- czym stosowane pierwotnie w leczeniu nawadniają-
do, zmniejszenie objętości ejakulatu, impotencja), cym doksazosyna i terazosyna, jak już wspomniano,
oraz stosunkowo rzadko ginekomastia oraz reakcje są nieco gorzej znoszone. Ze względu na niebezpie-
nadwrażliwości. czeństwo wystąpienia działań niepożądanych ze stro-
Przeciwwskazaniem do podania finasterydu są zabu- ny układu sercowo-naczyniowego, przed rozpoczę-
rzenia czynności wątroby. ciem leczenia należy wziąć pod uwagę ewentualne
przyjmowanie przez pacjenta leków obniżających
Inhibitory CYP3A4 np. itrakonazol, ketokonazol, indinawir ciśnienie krwi (inhibitorów ACE, β-blokerów, anta-
oraz ritonawir zwiększają stężenie dutasterydu w osoczu. gonistów kanałów wapniowych).
Finasteryd stosowany jest w leczeniu nie tylko łagodnego
przerostu prostaty, ale również łysienia androgenicznego. Leczenie inhibitorami 5α-reduktazy jest efektywne
tylko wtedy, gdy przed jego rozpoczęciem objętość
prostaty przekracza 40 ml. Wyraźna poprawa nastę-
Fitofarmaceutyki. W Niemczech, w przeciwień-
puje przeważnie w ciągu 3–6 miesięcy i utrzymuje
stwie do wielu krajów, fitofarmaceutyki stosowane
się. Badania długoterminowe wykazują spowolnienie
są w leczeniu łagodnego rozrostu prostaty. Do tej
postępu dolegliwości ze zmniejszeniem ryzyka bez-
grupy preparatów należą ekstrakty z pokrzywy, olej
moczu lub konieczności przeprowadzenia zabiegu
z palmy sabal, nasiona dyni, pyłek żyta, także mie-
operacyjnego z 13 do 7%. Podawanie substancji z tej
szanki fitosteroli z β-sitosterolem jako składnikiem
grupy pozwala zapobiec hematurii skojarzonej z PBS.
podstawowym. Istnieją wprawdzie nowsze badania
Zwiększenie maksymalnej ilości oddawanego moczu
gosodarka wodno-elektr.
odpowiadające aktualnym standardom i potwierdza-
jest jednak w porównaniu z placebo niewielkie.
jące działanie określonych wyciągów, jednak ogólna Nerki i drogi moczowe,
ilość posiadanych informacji nie jest jeszcze wystar-
Dotychczas najlepsze wyniki leczenia PBS uzyskano
czająca. Najistotniejszy jest brak kontrolowanych ba-
poprzez łączenie inhibitorów receptora α1-adrener-
dań przedłużonych.
gicznego (doksazosyny) z inhibitorem 5α-reduktazy
(finasteryd).
7.6.1.3. Główne kierunki – leczenie W przypadku zalegania moczu, nawracających infek-
konformacyjne łagodnego cji dróg moczowych lub makrohematurii, rozszerzenia
objawowego przerostu prostaty górnego odcinka dróg moczowych i zaburzeń funkcji B7
nerek spowodowanych PBS wskazane jest przepro-
W przypadku niewielkich dolegliwości leczenie wadzenie zabiegu operacyjnego, np. przezcewko-
nie jest konieczne, ponieważ u wielu pacjentów prze- wego usunięcia prostaty. Leczenie farmakologiczne
biegają one naprzemiennie z okresami bez zaburzeń jest w takich przypadkach niewskazane.
mikcji. Pacjenci z tej grupy powinni być co 6 miesię-
cy poddawani badaniom kontrolnym.
MUTSCHLER-2009.indd 741 2010-01-07 22:14:31
742 Nerki i drogi moczowe, gospodarka wodno-elektrolitowa i kwasowo-zasadowa
7.6.2. Nietrzymanie moczu Nietrzymanie moczu spowodowane wzmożonym
spowodowane obciążeniem parciem farmakologicznie leczy się przeważnie
za pomocą substancji działających antagonistycznie
i naglącym parciem na receptory M-cholinergiczne (spazmolityki neuro-
Nietrzymanie moczu (bezwiedne oddawanie moczu) tropowe) lub spazmolityków neurotropowych i mu-
jest objawem różnych zaburzeń czynności zwieraczy skulotropowych. Obniżają one parasympatykomi-
pęcherza moczowego i/lub mięśnia wypieracza moczu. metyczne napięcie mięśnia wypieracza i zwiększają
Szczególnie często występuje u kobiet, zwłaszcza po okre- (pośrednio) napięcie zwieracza spowodowane bloko-
sie menopauzy. waniem receptorów α-andrenergicznych.
W przypadku nietrzymania moczu spowodowanego obcią- Z grupy substancji działających antagonistycznie
żeniem osłabione są mięśnie przepony miednicy – najczęś- na receptory M-cholinergiczne zalecane są oprócz
ciej w wyniku ciąży. Obciążenie (np. przy kichaniu, kaszlu, oksybutyniny, która przez długi czas uważana była
skakaniu, wchodzeniu po schodach, noszeniu zbyt dużych za substancję standardową, także chlorek trospium,
ciężarów) powoduje bezwiedne wydalenie moczu. tolterodyna, a od niedawna także daryfenacyna i so-
Nietrzymanie moczu spowodowane wzmożonym par-
ciem jest efektem nagłego, niedającego się powstrzymać lifenacyna. Nie wiadomo jednak, czy dwie ostatnie
skurczu mięśnia wypieracza i spowodowanego tym odda- substancje będące antagonistami receptorów M3,
nia moczu. Z dolegliwością tą często jest związane mo- mają więcej zalet niż pozostałe spazmolityki.
czenie nocne (nykturia) lub częste oddawaniem moczu Dawkowanie oksybutyniny powinno wynosić
(polikisuria), a jej podłożem są zaburzenia neurologiczne 2,5–5 mg/3 × dzień, chlorku trospium 10–15 mg/3 ×
(np. choroba Parkinsona lub stwardnienie rozsiane).
dzień, tolterodyny (1-)2 mg/2 × dzień lub (2-)4 mg/
dzień w preparacie o przełdużonym uwalnianiu dosy-
W leczeniu nietrzymania moczu spowodowanego ob- fenacyny 7,5–15 mg/dzień i solifenacinsolifenacyny
ciążeniem główną rolę – oprócz zabiegu operacyjne- (5-)10 mg/dzień.
go w ciężkich przypadkach – ogrywa gimnastyka lub
elektrostymulacja mięśni przepony miednicy. Wspo- Spośród substancji neurotropowych i muskulotropo-
magająco można zastosować również leczenie farma- wych wskazana jest przede wszystkim propiweryna
kologiczne za pomocą agonistów α-adrenoreceptorów, w dawce 30–45 mg/dzień, podzielonej na 2–3 poje-
inhibitorów acetylocholinoesterazy, a także diulekse- dyncze dawki.
tyny, podawanych również w stanach depresyjnych.
MUTSCHLER-2009.indd 742 2010-01-07 22:14:31
Oko 743
8. Oko
W oku dzięki układowi optycznemu (aparatowi diop- magnetyczne o zakresie długości 400–750 nm) zosta-
trycznemu) na siatkówce zostają odwzorowane obiek- ją przy udziale procesów fotochemicznych zamienio-
ty ze środowiska zewnętrznego. Do części oka słu- ne w pobudzenia, które przez nerw wzrokowy bądź
żących do projekcji obrazu zaliczamy rogówkę oraz drogą wzrokową, są przesyłane do głównego ośrodka
soczewkę, które spełniają również funkcję ośrodków wzrokowego w płacie potylicznym kory mózgowej.
załamujących światło. Częścią oka odbierającą obraz
jest siatkówka. W niej bodźce fizyczne (fale elektro-
8.1. Anatomia oka
Gałka oczna (bulbus oculi), patrząc na rogówkę, ma Zewnętrzna błona gałki ocznej. Zewnętrzna błona gałki
kształt zbliżony do kuli (zob. ryc. B 8.1-1). Jej ściana ocznej składa się ze zwartej, nieprzezroczystej twardówki
składa się z trzech głównych warstw: oraz z przezroczystej rogówki.
Biała twardówka jest grubą, zbudowaną głównie z włó-
■ zewnętrznej błony gałki ocznej (tunica externa), kien kolagenowych, odporną na rozciąganie torebką łącz-
notkankową, która wspomagana przez ciśnienie śródgałko-
■ środkowej błony gałki ocznej (tunica media), we odpowiada za kształt gałki ocznej. W przednim odcinku
jest pokryta spojówką.
■ wewnętrznej błony gałki ocznej (tunica interna). Niepokrytą spojówką przednią cześć gałki ocznej stano-
wi przezroczysta rogówka. Jest ona, podobnie jak szkieł-
Wnętrze gałki ocznej składa się z leżących za sobą ko-
ko zegarkowe, osadzona w twardówce i ma jak ta ostatnia
lejno trzech przepuszczających światło ośrodków, któ- zwartą strukturę. Zrąb rogówki, który pokryty jest z obu
re stanowią: ciecz wodnista, soczewka i ciało szkliste. stron nabłonkiem, składa się z włókien kolagenowych, uło-
spojówka ciało szkliste
kanał Schlemma
tęczówka oś optyczna
oka
rogówka soczewka
K dołek środkowy
tarcza nerwu plamka żółta
K K
wzrokowego
blaszka sitowa
kości sitowej
przednia komora oka pasma więzadełkowe
tylna komora oka obwódki rzęskowej nerw wzrokowy
mięsień rzęskowy
siatkówka
Oko
naczyniówka
twardówka
Ryc. B 8.1-1. Schemat przekroju prawego oka (według Thewsa, Mutschlera, Vaupela).
B8
MUTSCHLER-2009.indd 743 2010-01-07 22:14:31
744 Oko
żonych na podobieństwo krzyżujących się blaszek. Regu- ■ część tylną, wrażliwą na światło – część wzrokową siat-
larny układ włókien kolagenowych, osadzonych w bogatej kówki (pars optica).
w glikoproteiny międzykomórkowej spoinie, oraz brak na-
czyń krwionośnych decydują o przezroczystości tej tkanki. Część ślepa siatkówki pokrywa ciało rzęskowe oraz tylną
Na granicy między twardówką a rogówką przebiega pier- powierzchnię tęczówki, z kolei część wzrokowa siatkówki
ścieniowato rozszerzające się naczynie krwionośne, kanał wyścieła nie tylko tylną połowę gałki ocznej, lecz sięga aż
Schlemma, który jest połączony z układem żylnym gałki do jej przedniej połowy. Składa się ona z dwóch warstw,
ocznej i umożliwia odpływ cieczy wodnistej. warstwy barwnikowej (nabłonka barwnikowego) oraz war-
Rogówka jest odżywiana substancjami przedostającymi stwy nerwowej.
się na drodze dyfuzji z cieczy wodnistej, cieczy łzowej oraz Główną funkcją warstwy barwnikowej jest odżywianie
z przyrogówkowo przebiegających naczyń tętniczych. komórek czuciowych.
Warstwa nerwowa, czyli właściwa, wrażliwa na światło
Środkowa błona gałki ocznej. Środkowa błona gałki ocz- część siatkówki, jest zbudowana z trzech warstw komórek:
nej składa się z trzech części: ■ warstwy nabłonka nerwowego (warstwy receptorów
■ błony naczyniowej światła),
■ ciała rzęskowego, ■ warstwy zwojowej siatkówki (warstwy dwubiegunowych
komórek nerwowych),
■ tęczówki.
■ warstwy zwojowej nerwu wzrokowego (warstwy dużych
Terminem naczyniówka określa się tę część środkowej bło- komórek zwojowych, których aksony tworzą nerw wzro-
ny gałki ocznej, która przylega do twardówki. Składająca kowy).
się z wielu warstw błona naczyniowa zawiera obok blasz-
kowato uporządkowanej tkanki łącznej z licznymi szcze- Pomiędzy tymi komórkami znajdują się komórki glejowe
linami również rozgałęzienia naczyń naczyniówki, które siatkówki – komórki podporowe Müllera.
przede wszystkim służą odżywianiu przyległych warstw.
W przednim odcinku oka środkowa błona gałki ocznej Wrażliwe na światło komórki czuciowe warstwy nabłon-
tworzy ciało rzęskowe, które w przeciwieństwie do gład- ka nerwowego, pręciki i czopki (zob. poniżej), znajdują się
kiej naczyniówki charakteryzuje się wyniosłościami, za- w położonej najbardziej zewnętrznie i graniczącej z nabłon-
głębieniami i wyrostkami. Ciało rzęskowe zawiera pasma kiem barwnikowym części siatkówki, tzn. są one oddalone
włókien mięśni gładkich mięśnia rzęskowego, który odpo- od strony, z której dociera światło i przykryte przez komór-
wiada za regulację promienia krzywizny soczewki. ki nerwowe i ich włókna (odwrócenie siatkówki). Światło
U podstawy ciała rzęskowego przyłączona jest od przo- musi zatem przeniknąć najpierw przez wszystkie warstwy
du tęczówka, która oddziela przednią komorę oka – prze- siatkówki, zanim dotrze do receptorów.
strzeń między tylną powierzchnią rogówki a przednią po-
wierzchnią tęczówki i soczewki wypełnioną cieczą wodni-
stą (zob. poniżej) – od mniejszej i pierścieniowato leżącej
wokół soczewki tylnej komory oka. naczynia
W środku tęczówki znajduje się kolista źrenica, której krwionośne
średnica może być zmieniana przez mięśnie gładkie tę- naczyniówka
czówki.
Tęczówka składa się głównie z luźnej, delikatnej tkanki nabłonek
łącznej (zrębu tęczówki), której tylna powierzchnia pokry- barwnikowy
ta jest przez nabłonek barwnikowy oraz pigmentową część pręciki
siatkówki (zob. poniżej). Ponadto w tęczówce znajdują się czopki
dwa mięśnie gładkie, mięsień zwieracz źrenicy i mięsień zewnętrzna
rozwieracz źrenicy, służące do odruchowej zmiany szero- warstwa ziarnista
kości źrenicy przy zmianie ilości docierającego światła.
Barwa tęczówki, czyli również kolor oczu, zależy od za-
wartości melaniny w jej zrębie. Jeżeli tęczówka ma barwę
niebieską, to nie znajdują się w niej niemal żadne komórki
barwnikowe, natomiast przy barwie tęczówki od zielonej
do brązowej obecnych jest w niej wiele takich komórek. wewnętrzna
Określeniem kąt przesączania opisuje się miejsce styku warstwa ziarnista
przedniej powierzchni brzegu rzęskowego tęczówki z twar-
dówką, bezpośrednio przed jej przejściem w rogówkę.
Tutaj znajdują się szczeliny (przestrzenie kąta tęczówko-
wo-rogówkowego), przez które ciecz wodnista z przedniej komórki zwojowe
komory oka może odpływać do kanału Schlemma.
warstwa włókien
Wewnętrzna błona gałki ocznej. Wewnętrzna błona gałki nerwowych
ocznej, siatkówka, dzieli się na dwie części:
■ część przednią, niewrażliwą na światło – część ślepą siat- Ryc. B 8.1-2. Warstwy siatkówki. Pionowy przekrój siatkówki
kówki (pars caeca), i części naczyniówki (według Thewsa, Mutschlera, Vaupela).
MUTSCHLER-2009.indd 744 2010-01-07 22:14:32
Oko 745
Nabłonek czuciowy zawiera około 6 milionów czop-
ków i w przybliżeniu 120 milionów pręcików. W plamce
nabłonek
żółtej, zwłaszcza w okolicy dołka środkowego, siatkówka barwnikowy
jest znacznie cieńsza i występują w niej wyłącznie czop- człon
ki, które przykryte są tylko cienką warstwą tkanki, tak zewnętrzny
że wpadające światło bezpośrednio dociera do komórek człon
czuciowych. W okolicy dołka środkowego na dwa pręciki wewnętrzny
przypada jeden czopek. Obwodowo od tego miejsca liczba jądro komórki jądro komórki
czopków zmniejsza się i dlatego w zewnętrznym obszarze wzrokowej wzrokowej
pręcikonośnej czopkonośnej
siatkówki przeważają liczbowo pręciki. W miejscu, w któ-
rym nerw wzrokowy wychodzi z gałki ocznej, czyli w za-
głębieniu tarczy nerwu wzrokowego, nie ma komórek czu- komórka nerwowa
ciowych („plamka ślepa”). pozioma
komórka
dwubiegunowa
W warstwie zwojowej siatkówki znajdują się dwubiegu- jądro komórki
nowe komórki sprzęgające, które posiadają synaptyczny z włóknami komórka
kontakt z komórkami czuciowymi. Poza tym aksony tych promieniowymi amakrynowa
komórek biegną do dużych neuronów warstwy zwojowej
nerwu wzrokowego (zob. poniżej). Ciała dwubiegunowych komórka zwojowa
komórek nerwowych znajdują się w tzw. wewnętrznej war- wielobiegunowa
stwie ziarnistej (zob. ryc. B 8.1-2).
W dołku środkowym każda komórka czuciowa jest po- włókna oboczne tętniczka
łączona z komórką dwubiegunową, która z kolei ma kon- stożek włókien
takt ze ściśle jej przyporządkowaną komórką zwojową promieniowych
znajdującą się w warstwie zwojowej nerwu wzrokowego.
Natomiast w pozostałej części siatkówki zawsze kilka ko-
mórek czuciowych jest połączonych z jedną komórką dwu- Ryc. B 8.1-3. Schemat receptorów i neuronów siatkówki (we-
biegunową, a znowu kilka tych komórek łączy się z jedną dług Buchera).
komórką w warstwie zwojowej nerwu wzrokowego (zob.
ryc. 8.1-3).
W najbardziej zewnętrznej warstwie komórek, czyli Człon zewnętrzny stanowi odbiorczą (receptorową) część
w komórkach warstwy zwojowej nerwu wzrokowego, komórek czuciowych. Jego długość wynosi około 30 μm,
znajduje się wiele dużych komórek nerwowych, którymi grubość 2 μm, a zbudowany jest z podwójnych błon biał-
są komórki zwojowe nerwu wzrokowego (n. opticus). Spo- kowo-lipidowych, które mają formę czopu (pręciki) lub
śród wszystkich komórek są one położone najdalej w kie- zagłębień błony (czopki). W tych strukturach zmagazyno-
runku ciała szklistego. Aksony tych komórek przebiegają wane są barwniki wzrokowe.
jako bezrdzenne włókna nerwowe do miejsca zbiorcze-
go, znajdującego się w odległości mniej więcej 3–4 mm
od tylnego bieguna gałki ocznej, tarczy nerwu wzrokowe- Dno oka. Dno oka, które za pomocą oftalmoskopu
go. Po przejściu przez mającą w tym miejscu sitowate ot- możemy zobaczyć bezpośrednio przez załamujące
wory twardówkę (pole sitowe twardówki) włókna nerwo- światło ośrodki gałki ocznej, ma barwę czerwonopo-
we w dalszym swym przebiegu są otaczane przez osłonkę
marańczową. W jego przynosowej połowie znajduje
rdzenną.
się tarcza nerwu wzrokowego (zob. poniżej). W niej
z kolei rozgałęzia się tętnica środkowa siatkówki
Struktura fotoreceptorów. Znajdujące się w naj- na wiele odnóg, natomiast żyły łączą się, tworząc
bardziej zewnętrznej warstwie siatkówki i zwró- żyłę środkową siatkówki. Liczne odgałęzienia na-
cone w kierunku twardówki fotoreceptory, czyli czyniowe biegną do plamki żółtej, będącej central-
pręciki i czopki, wykazują w swej budowie daleko nym obszarem najostrzejszego widzenia. W środku
idące podobieństwa. Składają się one z zewnętrz- plamki żółtej znajduje się dołek środkowy, niewiel-
nego członu, który jest wsunięty pomiędzy długie kie zagłębienie, w którym oś optyczna oka styka się
wypustki komórek nabłonka barwnikowego, oraz z dnem oka.
z członu wewnętrznego (zob. ryc. B 8.1-3). Oba
człony są połączone za pomocą cienkiego łącz- Soczewka i ciało szkliste. Dwuwypukła, niezawiera-
nika. jącą naczyń ani nerwów soczewka (zob. ryc. 8.1-1),
której przednia powierzchnia jest słabiej zakrzywiona
Człon wewnętrzny pręcików stanowi „centrum przemiany niż tylna, jest naciągnięta przez wieniec więzadeł wie-
Oko
materii” komórki czuciowej i zawiera wiele mitochondriów szadłowych zwanych pasmem więzadłowym obwód-
i rybosomów. Zwęża się on w kierunku ciała szklistego, sta-
jąc się aksonem. Jądro komórkowe fotoreceptorów znajduje ki rzęskowej, przyczepionym do ciała rzęskowego.
się albo w okolicy przejścia członu wewnętrznego w akson, Soczewka jest otoczona elastyczną torebką, w której
albo w samym aksonie. kończą się pasma więzadłowe obwódki rzęskowej.
B8
MUTSCHLER-2009.indd 745 2010-01-07 22:14:32
746 Oko
Galaretowate ciało szkliste (corpus vitreum), w du- ■ powieki,
żej mierze pozbawione komórek, wypełnia większą
■ spojówka,
część wnętrza gałki ocznej. Składa się ono w 98–99%
z wody, która jest związana z proteoglikanami. Prawie ■ narząd łzowy.
w każdym oku można znaleźć niewielkiego rozmiaru
zmętnienia w ciele szklistym (mouches volantes). Gałkę oczną osadzoną w tkance łącznej i tłuszczowej
oczodołu zakrywają powieki. Górna i dolna powieka, któ-
re stanowią granice szczeliny powiekowej, usztywniane
Droga wzrokowa. Zebrane w pęczek aksony dużych są przez miseczkowatą płytkę łącznotkankową (tarczkę
komórek zwojowych siatkówki tworzą nerw wzroko- powiekową, tarsus). W płytce tej znajdują się gruczoły
wy. U podstawy międzymózgowia oba nerwy wzro- łojowe (glandulae tarsales, gruczoły Meiboma) natłusz-
kowe tworzą skrzyżowanie wzrokowe (chiasma opti- czające brzeg powiek. Z zewnątrz powieki są pokryte
cum). W tym miejscu włókna przyśrodkowej (przy- wielowarstwowym, zrogowaciałym nabłonkiem płaskim
skóry. W przednim brzegu powiek osadzone są w kilku
nosowej) połowy siatkówki przechodzą na stronę szeregach rzęsy (cilia). W mieszkach włosowych rzęs
przeciwległą i wraz z nieskrzyżowanymi włóknami znajdują się ujścia gruczołów holokrynowych i apokryno-
zewnętrznej (skroniowej) połowy siatkówki tworzą wych. Tkanka podskórna powiek nie zawiera tłuszczów
pasmo wzrokowe (tractus opticus). i ma luźną strukturę, dzięki czemu łatwo rozpoznać tutaj
Około dwóch trzecich włókien pasma wzroko- ewentualne obrzęki.
Brwi (supercilia) wyznaczają górny brzeg oczodołów
wego kończy się w ciele kolankowatym bocznym oraz uniemożliwiają spływanie potu z czoła do szczeliny
(corpus geniculatum laterale), które jest pierwszym powiekowej.
ośrodkowym przekaźnikiem impulsu w drodze wzro-
kowej. Tutaj następuje przekazanie pobudzeń na dal- Wewnętrzną powierzchnię powiek wyścieła spojówka,
sze neurony, których aksony, tworząc promienistość której niezrogowaciały, wielowarstwowy nabłonek płaski
zachodzi w górnym i względnie dolnym fałdzie na przed-
wzrokową (radiatio optica), biegną do pierwotnej nią powierzchnię gałki ocznej i tam sięga aż do brzegu
kory wzrokowej (pola prążkowego kory wzrokowej) rogówki.
w korze płata potylicznego. Stąd odchodzą połącze-
nia nerwowe do wyższych ośrodków kory wzroko- Do narządu łzowego zaliczamy:
wej. Pozostałe włókna pasma wzrokowego biegną ■ gruczoł łzowy,
do wzgórków górnych blaszki czworaczej (colliculi ■ drogi odprowadzające łzy.
superiores) oraz do okolicy znajdującej się przed po-
krywą śródmózgowia. Te połączenia między droga- Gruczoł łzowy jest złożonym gruczołem cewkowym, któ-
mi nerwowymi pełnią funkcję dróg aferentnych od- rego przewody wyprowadzające mają swoje ujścia w ze-
ruchu źrenicznego i odruchu akomodacyjnego (zob. wnętrznym kącie oka w górnym fałdzie spojówki. Ich wy-
dzielina, czyli ciecz łzowa, to bezbarwny płyn ubogobiał-
poniżej). kowy o słonym smaku, którego zadaniem jest oczyszczanie,
nawilżanie i odżywianie rogówki. Ciecz łzowa rozprowa-
Zewnętrzne mięśnie gałki ocznej. Gałka oczna, któ- dzana jest ruchami powiek po całej powierzchni rogówki
ra osadzona jest w tkance łącznej i tłuszczowej oczodo- i dociera do tzw. jeziorka łzowego przy środkowym kącie
łu posiada ruchomość we wszystkich kierunkach. Ruchy oka.
te możliwe są dzięki aktywności sześciu zewnętrznych, Odpływ cieczy łzowej jest możliwy dzięki obydwóm
poprzecznie prążkowanych mięśni gałki ocznej, które kanalikom łzowym (canaliculi lacrimales) położonym rów-
w większości biorą swój początek w położonym głęboko nież przy środkowym kącie oka, które łączą się ze sobą
w oczodole pierścieniu ścięgnistym, tworząc następnie i uchodzą do woreczka łzowego (saccus lacrimalis). Stam-
stożek mięśniowy. tąd łzy spływają poprzez przewód nosowo-łzowy (ductus
nasolacrimalis) do znajdującego się pod dolną małżowiną
nosową otworu w jamie nosowej. Duża część cieczy łzo-
Aparaty pomocnicze i ochronne gałki ocznej. wej, która z powodu zawartości enzymów stanowi skutecz-
Do aparatów pomocniczych i ochronnych gałki ocz- ną ochronę przed zakażeniami, w wyniku parowania prze-
nej należą: chodzi do powietrza.
MUTSCHLER-2009.indd 746 2010-01-07 22:14:33
Oko 747
8.2. Układ odwzorowujący, reakcje źreniczne
i ciśnienie śródgałkowe
Dioptryka. Układ odwzorowujący oka (układ diop- do zmiany mocy łamiącej soczewki jest nieostre odwzoro-
tryczny) dokonuje projekcji na siatkówkę rzeczywi- wanie na siatkówce.
stego, odwróconego i pomniejszonego obrazu wi- Różnicę między mocą łamiącą oka przy nastawieniu
na punkt bliży a nastawieniem na punkt dali określa się
dzianych przedmiotów (zob. ryc. 8.1-1). Efekt ten jako amplitudę akomodacji. (Punkt dali, tzn. najbardziej
jest wynikiem załamania promieni światła na krzy- oddalony punkt odwzorowywany jako ostry obraz na siat-
wiznach powierzchni, które oddzielają od siebie kówce, w przypadku ludzi z prawidłowym wzrokiem
ośrodki o różnej gęstości (powietrze, rogówka, ciecz znajduje się w nieskończoności). U młodzieży amplitu-
wodnista, soczewka, ciało szkliste). da akomodacji wynosi około 10 dioptrii. Wraz z wiekiem
sprężystość soczewki zmniejsza się, w wyniku czego
Moc łamiącą układu optycznego podaje się w diop- dochodzi do postępującego zmniejszenia zdolności ako-
triach (dpt), tzn. jako odwrotną wartość ogniskowej modacyjnej soczewki bądź obniżenia amplitudy akomo-
mierzonej w metrach: dacji (presbyopia, starczowzroczność). U osób w wieku
powyżej 60 lat akomodacja jest możliwa już tylko w nie-
1 znacznym zakresie. Dlatego też osoby w starszym wieku,
zdolność łamiąca (dpt) = u których refrakcja jest prawidłowa, wymagają do ostrego
odległość ogniskowa (m)
widzenia mniejszej odległości od obserwowanych przed-
Całkowita moc łamiąca aparatu dioptrycznego ludz- miotów, a do czytania okularów z soczewkami skupiają-
cymi (okularów do czytania).
kiego oka w warunkach braku zmian akomodacji
(zob. poniżej) wynosi 1/0,017 = 59 dpt. Nieprawidłowa akomodacja. Przez pojęcie nieprawidło-
wej akomodacji rozumie się patologiczne zmiany układu
Operacyjne usunięcie soczewki (afakia = bezsoczewko- załamującego w oku, które prowadzą do odwzorowania
wość) z powodu zmętnienia soczewki (katarakta, zaćma) nieostrego obrazu na siatkówce. Zasadniczo rozróżnia się
powoduje zmniejszenie mocy łamiącej o 16 dioptrii, któ- trzy formy nieprawidłowej akomodacji:
re koryguje się przez wszczepienie soczewki z tworzywa
sztucznego. ■ astygmatyzm,
■ krótkowzroczność,
Akomodacja. Układ optyczny oka jest w stanie od- ■ nadwzroczność.
wzorować na siatkówce ostry obraz dowolnie odda-
W przypadku astygmatyzmu chodzi o wadę wzroku, w któ-
lonych przedmiotów. Jest to możliwe dzięki zmianom rej jedna z załamujących powierzchni, przede wszystkim
mocy łamiącej poprzez zmianę kształtu soczewki, przednia powierzchnia rogówki, posiada nierównomierną
przy czym zmianie ulega przede wszystkim promień krzywiznę. Jeżeli istnieją większe różnice krzywizny ro-
krzywizny przedniej powierzchni soczewki. gówki pomiędzy poszczególnymi jej częściami, to odwzo-
rowaniem kolistego przedmiotu na siatkówce będzie elip-
Przy akomodacji do dali (akomodacja spoczynkowa) tyczny, zniekształcony obraz.
mięsień rzęskowy nie kurczy się, tzn. pasma więzadłowe U podłoża krótkowzroczności (myopia) leży zazwy-
obwódki rzęskowej są napięte wskutek sprężystego nacią- czaj wydłużenie gałki ocznej lub rzadziej zwiększona moc
gu twardówki. To napięcie pasm więzadłowych obwódki łamiąca aparatu dioptrycznego. Skutkiem takich stanów
rzęskowej zostaje przeniesione na torebkę soczewki, przez jest w przypadku oka z akomodacją nastawioną do dali
co spłaszczeniu ulega elastyczna soczewka. Przy odległości skupianie się promieni przed siatkówką, w wyniku cze-
od obserwowanego przedmiotu przekraczającej 5 m mamy go powstaje na niej nieostry obraz. Znajdujące się blisko
do czynienia praktycznie z brakiem akomodacji. przedmioty mogą mimo wszystko przy odpowiedniej
Przy akomodacji do bliży, tzn. przy patrzeniu na przed- akomodacji zostać odwzorowane w postaci ostrego obra-
mioty znajdujące się w odległości mniejszej niż 5 m, na- zu na siatkówce. Ludzie z krótkowzrocznością wymagają
stępuje skurcz pierścieniowatego mięśnia rzęskowego. do patrzenia na oddalone przedmioty okularów z soczew-
W wyniku tego dochodzi do zmniejszenia naciągu pasm kami rozpraszającymi (wklęsłą krzywizną), które redukują
więzadłowych obwódki rzęskowej, przez co soczewka moc łamiącą aparatu dioptrycznego oka i przez to przesu-
wskutek własnej sprężystości przyjmuje kształt bardziej wają płaszczyznę odwzorowania obrazu na siatkówkę.
kulisty i tym samym zwiększa swoją moc łamiącą. Mak- Nadwzroczność jest spowodowana zbyt krótką gałką
symalna akomodacja do bliży pozwala osobom młodym oczną lub rzadziej za małą mocą łamiącą układu dioptrycz-
na odwzorowanie ostrego obrazu przedmiotów, które znaj- nego oka. Ponieważ w takim przypadku przy akomodacji
Oko
dują się w odległości około 10 cm od oka (tzw. odległość nastawionej do dali płaszczyzna powstającego obrazu znaj-
punktu bliży). duje się za siatkówką, odwzorowany na siatkówce obraz
Stan napięcia mięśnia rzęskowego i równocześnie moc jest nieostry. Aby mimo to móc ostro widzieć odległe
przedmioty, u osoby z nadwzrocznością musi ciągle za-
łamiąca soczewki są kontrolowane przez włókna przy-
chodzić akomodacja. Do zogniskowania blisko położonych
B8
współczulne nerwu okoruchowego. Właściwym bodźcem
MUTSCHLER-2009.indd 747 2010-01-07 22:14:33
748 Oko
przedmiotów nie wystarcza tutaj zdolność łamiąca aparatu racza źrenicy (m. sphincter pupillae) oraz mięśnia
dioptrycznego, tak więc punkt bliży jest bardziej oddalony rozwieracza źrenicy (m. dilatator pupillae). Poprzez
od oka niż u człowieka z prawidłowym widzeniem. Do ko-
skurcz mięśnia zwieracza źrenicy, źrenica staje się
rekcji tej wady wzroku potrzebne są okulary z soczewkami
skupiającymi (wypukłą krzywizną). węższa (mioza). Mięsień ten jest unerwiony przez
przywspółczulne włókna nerwowe. Skurcz mięśnia
Reakcje źrenic. Prawidłowo obie źrenice są okrągłe rozwieracza źrenicy powoduje natomiast rozszerze-
i tak samo szerokie. Szerokość źrenicy zależy od na- nie źrenicy (mydriasis). Unerwienie mięśnia rozwie-
tężenia wpadającego przez nią światła i – podobnie racza źrenicy odbywa się przez współczulne włókna
jak moc łamiąca soczewki – jest regulowana odru- nerwowe. Ogólne pobudzenie układu współczulne-
chowo. Szerokość źrenicy u młodzieży mieści się go, np. przy reakcji na zagrożenie, prowadzi często
w zakresie od 1,5 mm do 8,0 mm, natomiast odpo- do znacznego rozszerzenia źrenicy.
wiednie do tego względne natężenie docierającego
światła zmienia się trzydziestokrotnie. W podeszłym Ciśnienie śródgałkowe. Oprócz twardówki, która
wieku maksymalna możliwa szerokość źrenicy ulega charakteryzuje się mocną budową, również ciśnie-
znacznemu zmniejszeniu. nie śródgałkowe jest odpowiedzialne za zewnętrzny
Źrenice są tym szersze, im mniejsza jest luminacja kształt gałki ocznej. Zależy on przede wszystkim
(światłość powierzchniowa) otoczenia. Każde na- od ilości stale wytwarzanej i równocześnie odpływa-
silenie się strumienia światła docierającego do oka jącej cieczy wodnistej, która jest wydzielana przez
wywołuje zwężenie źrenic, w wyniku czego równo- nabłonek ciała rzęskowego za pomocą transportu
cześnie zwiększa się głębia ostrości odwzorowania aktywnego i biernego. Z tylnej komory gałki ocz-
(por. zmniejszenie szerokości przysłony w aparacie nej ciecz wodnista przepływa przez otwór źreniczny
fotograficznym). do przedniej komory. Stąd dostaje się ona poprzez kąt
Przy oświetleniu oka mamy do czynienia z reakcją tęczówkowo-rogówkowy do kanału Schlemma (zob.
– zwężeniem zarówno źrenicy oświetlanego oka (bez- ryc. 8.5-1) i następnie odpływa do małych żył. (Od-
pośrednia reakcja na światło), jak i z pośrednią reakcją pływ przez kanał Schlemma jest zapewniony szcze-
– zwężeniem źrenicy drugiego, nieoświetlanego oka gólnie wtedy, gdy źrenica jest zwężona i tym samym
(konsensualna reakcja na światło). Ponadto do zwęże- nie zawęża kąta tęczówkowo-rogówkowego.) W cią-
nia źrenic dochodzi przy akomodacji do bliży z powo- gu jednej doby objętość cieczy wodnistej jest wielo-
du dodatkowego unerwienia przez nerw okoruchowy, krotnie wymieniana na nową.
ponieważ nerw ten jest jednocześnie odpowiedzialny Ciśnienie śródgałkowe, którego wartość w zdro-
za zwężenie źrenic, akomodację do bliży oraz zbież- wym oku wynosi 10–21 mm Hg, pozostaje stałe, pod
ność gałek ocznych (reakcja zbieżności). warunkiem że ilość cieczy wodnistej odprowadzanej
Reakcje źrenic są realizowane, jak wspomniano przez kanał Schlemma w danym przedziale czasu
już we wstępie, dzięki działaniu dwóch mięśni gład- jest równa ilości cieczy wodnistej wytwarzanej de
kich znajdujących się w tęczówce – mięśnia zwie- novo (ok. 2 μl/min).
8.3. Czynność fotoreceptorów
Widzenie skotopowe i fotopowe. Obie formy re- Barwniki wzrokowe i potencjał receptorowy.
ceptorów siatkówki są przyporządkowane różnym W zewnętrznym członie pręcika znajduje się ok.
funkcjom zmysłu wzroku. Pręciki pośredniczą w wi- 1000 krążków błonowych, ułożeniem przypominają-
dzeniu w półmroku (widzeniu skotopowym), które cych rulony monet. W czopkach odpowiednie struk-
wprawdzie umożliwia odbieranie różnego natężenia tury zostają utworzone przez pofałdowanie błony ze-
światła, ale nie pozwala na rozróżnianie barw. wnętrznego członu czopka. W warstwach lipidowych
Czopki natomiast służą do widzenia przy świetle krążków błonowych (bądź w pofałdowaniach błony)
dziennym (widzenie fotopowe). Widzenie fotopowe są zmagazynowane fotopigmenty (barwniki wzroko-
charakteryzuje się możliwością rozróżniania barw, we), które pośredniczą w przekształcaniu bodźców
dużą ostrością wzroku w obrębie dołka środkowego świetlnych na zmiany potencjałów (potencjały re-
oraz dużą czułością kontrastową. ceptorowe). Barwniki wzrokowe są związkami, które
MUTSCHLER-2009.indd 748 2010-01-07 22:14:33
Oko 749
podczas absorpcji światła zmieniają swoją strukturę cji oka jest szczególnie duża, jednak w celu dosto-
chemiczną. sowania się do niewielkiej luminacji (jaskrawości)
Światłoczułym barwnikiem pręcików jest rodop- światła wymaga stosunkowo długiego czasu.
syna. Składa się ona z glikoproteiny – opsyny oraz Jeżeli w nocy wyjdzie się z oświetlonego pomieszczenia
z barwnika – 11-cis-retinalu (aldehydu witaminy A. na zewnątrz, to można jedynie stopniowo rozpoznać kontu-
Absorpcja fotonów (maksymalna przy długości ry przedmiotów znajdujących się w otoczeniu (adaptacja
fali równej 500 nm) wywołuje izomeryzację retina- do ciemności). W wyniku adaptacji aparatu czopkowego
lu, podczas której forma 11-cis ulega przekształceniu wrażliwość na światło (światłoczułość) wzrasta w ciągu
około 8 minut o współczynnik wynoszący 500, a z kolei
w formę all-trans. W wyniku tego rodopsyna w ciągu dużo wolniej postępująca adaptacja pręcików (zakończenie
kilku milisekund zostaje przekształcona poprzez kil- dopiero po upływie 40–50 minut) prowadzi do dalszego
ka form pośrednich w metarodopsynę II, która zapo- wzrostu światłoczułości o współczynnik równy 2000.
czątkowuje zmianę potencjału błonowego komórek. W przypadku utraty zdolności pręcików do adaptacji
mówi się o hemeralopii (tzw. ślepota zmierzchowa, kurza
ślepota). Tego rodzaju zaburzenie, powodujące znaczne
Czopki możemy podzielić na trzy typy, których trudności w orientacji po zmierzchu, może być skutkiem
barwniki wzrokowe selektywnie pochłaniają światło wrodzonej lub nabytej choroby. Jako przyczynę nabytej
w zakresie widma czerwonego, zielonego albo niebie- ślepoty zmierzchowej bierze się pod uwagę przede wszyst-
skiego. Również te fotolabilne barwniki składają się kim niedobór witaminy A, rzadziej zwyrodnienie nabłonka
z białka, tzw. opsyny czopkowej, oraz 11-cis-retinalu. barwnikowego lub atrofię nerwu wzrokowego.
W przeciwieństwie do adaptacji do ciemności, procesy
W porównaniu z rodopsyną dochodzi tutaj do szyb- adaptacyjne podczas przejścia z ciemnego do jasnego oto-
szego rozpadu przy oświetleniu, a z kolei w ciemnoś- czenia (adaptacja do jasności) przebiegają stosunkowo
ci do szybszej resyntezy barwników czopkowych. szybko. W przypadku nagłego, intensywnego oświetlenia
oka zaadaptowanego do widzenia w ciemności początkowo
Adaptacja do jasności i ciemności. Podobnie jak następuje przejściowe oślepienie, podczas którego zdol-
ność wzrokowego postrzegania kształtów jest zniesiona
inne narządy zmysłów, także wzrok posiada zdolność lub w dużym stopniu ograniczona. Następnie wrażliwość
adaptacji, tzn. dostosowywania się do różnej inten- aparatu odbierającego bodźce dostosowuje się w ciągu 15–
sywności napływających bodźców. Zdolność adapta- 60 sekund do nowych warunków oświetlenia.
8.4. Pole widzenia i widzenie przestrzenne
Pole widzenia. Terminem pole widzenia określa się i innych informacji pomocnych w orientacji. Przy patrzeniu
cały obszar otoczenia, który można objąć za pomo- obuocznym wrażenie głębi przestrzeni powstaje w wyni-
cą nieruchomego oka, tzn. przy skupieniu wzroku ku odbioru otoczenia przez każde z oczu pod trochę innym
kątem, co powoduje, że powstające na siatkówce obrazy
na punkcie centralnym. Zakres pola widzenia dla są nieznacznie przesunięte w stosunku do siebie. Ostatecz-
jednego oka jest ograniczony przez rozmiar siat- nie podczas ośrodkowego przetwarzania sygnałów ulegają
kówki, właściwości aparatu dioptrycznego oraz one połączeniu, tworząc wrażenie przestrzeni (zespolenie
przez zewnętrzne, anatomiczne bariery (nos i brwi). obuoczne).
Przy widzeniu barwnym widziany obszar otoczenia Bezpośrednio widziany obraz otoczenia powstaje tyl-
ko wówczas, gdy punkty jego odwzorowania znajdują się
jest mniejszy, ponieważ liczba czopków zmniejsza na określonych, odpowiadających sobie wzajemnie miej-
się w kierunku obwodu siatkówki. Przy obuocznym scach obu siatkówek (korespondujące ze sobą miejsca
patrzeniu na ten sam punkt pola widzenia obu oczu na siatkówkach obu oczu). Jeżeli jednak punkty odwzoro-
zachodzą na siebie z wyjątkiem obszarów skronio- wanego obrazu danego przedmiotu powstają w miejscach
wych. siatkówek znacznie od siebie odbiegających, to powstaje
obraz podwójny.
Widzenie przestrzenne. Widzenie obuoczne pozwala
Oko
na uzyskanie przestrzennego wrażenia głębi oraz ocenę Zez. Wadę wzroku, która jest spowodowana silniej-
odległości. Wprawdzie osoba widząca tylko na jedno oko
również jest w stanie prawidłowo ocenić głębię przestrzeni, szym odchyleniem obu osi gałek ocznych od siebie
jednak musi przy tym korzystać ze swojego doświadcze- i która prowadzi przez to do powstania obrazów po-
nia dotyczącego nakładania się konturów, różnic wielkości dwójnych, określa się jako zezowanie lub zez.
B8
MUTSCHLER-2009.indd 749 2010-01-07 22:14:33
750 Oko
8.5. Leki stosowane w terapii chorób oczu
W okulistyce leki stosuje się głównie miejscowo w po- dłowej, niosąc niemałe ryzyko układowych działań
staci kropli lub maści do oczu, jak również płynów niepożądanych. Na przykład po podaniu kropli ocz-
wstrzykiwanych podspojówkowo, przy- lub zagałko- nych zawierających lek blokujący receptory β (zob.
wo oraz iniekcji doszklistkowych. Znacznie rzadziej poniżej) opisywano u wielu pacjentów wystąpienie
celowe jest podawanie ogólne, np. przy stosowaniu działań niepożądanych typowych dla tej grupy sub-
środków immunosupresyjnych. Podanie miejscowe stancji, ze zgonem włącznie.
leków pozwala często na uzyskanie wyższego stęże-
nia substancji czynnej w miejscu działania, czyli gał-
ce ocznej, niż przy podaniu układowym. 8.5.1. Jaskra
Krople do oczu są sterylnymi, wodnymi lub olejowymi
roztworami albo też zawiesinami, które zakrapia się do wor-
8.5.1.1. Patofizjologia
ka spojówkowego. Aby uniknąć podrażnień, powinny one
być izotoniczne z płynem łzowym i w miarę możliwości Pojęcie jaskry obejmuje różne, szczególnie zna-
mieć wartość pH zbliżoną do fizjologicznej (izohydria). czące i częste formy choroby oczu, która prowadzi
Za pomocą substancji zwiększających lepkość (np. mety-
locelulozy, alkoholu poliwinylowego) można przedłużyć do uszkodzenia tarczy nerwu wzrokowego z powsta-
czas utrzymywania się wodnych roztworów kropli do oczu niem ubytków pola widzenia. Głównym czynnikiem
w miejscu podania i tym samym przedłużyć czas ich dzia- ryzyka w powstawaniu uszkodzeń związanych z ja-
łania. Pojedyncza dawka powinna być zawarta w objętości skrą jest zbyt wysokie osobnicze ciśnienie śródgałko-
jednej kropli (ok. 50 μl), ponieważ objętość cieczy łzowej we. Gdy jaskra wystąpi bez poprzedzającej ją cho-
w oku wynosi tylko około 10 μl (maksymalnie 30 μl). Dla-
tego też podanie objętości większej od jednej kropli pro- roby oczu, mamy wtedy do czynienia z jaskrą pier-
wadzi do jej wypływania na skórę lub odpływ do nosa, wotną. Natomiast jaskra wtórna jest następstwem
co w drugim przypadku wiąże się ze zwiększonym nie- istniejącej lub uprzednio występującej choroby oczu,
bezpieczeństwem wystąpienia ogólnoustrojowych działań np. wewnątrzgałkowego stanu zapalnego, lub choro-
niepożądanych. by ogólnej.
Maści do oczu stanowią alternatywną postać leków
w stosunku do kropli ocznych. Nie powinny one wywo-
ływać podrażnień, natomiast powinny ulegać szybkiemu W jaskrze z otwartym kątem przesączania (jaskrze pro-
rozprowadzeniu w postaci cienkiego filmu na powierzchni stej), najczęstszej postaci jaskry (ponad 90% przypadków),
gałki ocznej, jak również utrzymywać się na niej i możli- ciecz wodnista poprzez szeroki kąt tęczówkowo-rogów-
wie w małym stopniu upośledzać zdolność widzenia. Maści kowy dociera bez trudności do beleczkowania (zob. ryc.
do oczu są najczęściej aplikowane na noc. B 8.5.-1), jednak z powodu zmian strukturalnych utkania
beleczkowego z trudnością przez nie przenika. Inną przy-
czyną utrudnionego odpływu cieczy wodnistej może być
wzmożony opór w kanale Schlemma, jak również wzrost
Szczególne właściwości farmakokinetyczne leków ciśnienia w żyłach odprowadzających ciecz wodnistą. Roz-
stosowanych w okulistyce. Jeżeli chcemy, by poda- różnia się przy tym jaskrę z wysokim oraz z normalnym
wane miejscowo leki stosowane w leczeniu chorób (niskim) ciśnieniem śródgałkowym. Przy jaskrze z wyso-
kim ciśnieniem śródgałkowym ciśnienie to jest wyższe niż
oczu nie wnikały (lub tylko w ograniczonym stopniu) 21 mm Hg, a z kolei w jaskrze normociśnieniowej zawiera
do wnętrza gałki ocznej, to muszą one być hydrofilne, się w przedziale 10–21 mm Hg. Prawdopodobnie u podłoża
ewentualnie mogą posiadać jedynie nieznaczne właś- jaskry z normalnym ciśnieniem śródgałkowym leży szcze-
ciwości lipofilne. Do takich wskazań wybiera się za- gólna wrażliwość włókien nerwu wzrokowego, np. na za-
tem z określonej grupy leków tylko związki rozpusz- burzenia w mikrokrążeniu, tak że typowe uszkodzenia tar-
czy nerwu wzrokowego i ubytki w polu widzenia powstają
czalne w wodzie. Z drugiej strony leki, które mają już przy wartościach ciśnienia śródgałkowego niższych niż
działać wewnątrz gałki ocznej, powinny posiadać 20 mm Hg.
właściwości amfifilne, aby móc przenikać zarówno
przez kompartmenty hydrofilne, jak i lipofilne (m.in. W ostrej, zdecydowanie rzadszej (mniej niż 5% wszyst-
rogówka, przednia komora oka). kich przypadków) jaskrze z wąskim kątem przesącza-
Ponieważ przy aplikacji kropli ocznych stosun- nia wzrost ciśnienia śródgałkowego jest spowodowany,
kowo duża część podanej dawki leku odpływa przez jak wynika z samej nazwy, zwężeniem kąta tęczówkowo-
rogówkowego i zablokowaniem w wyniku tego odpływu
kanał nosowo-łzowy do nosa i – z ominięciem wystę- cieczy wodnistej. Mamy wtedy do czynienia z szybko roz-
pującego przy podaniu doustnym efektu pierwszego wijającym się ciężkim obrazem choroby z wartościami ciś-
przejścia – może wchłonąć się z jamy nosowo-gar- nienia śródgałkowego rzędu 60–80 mm Hg, silnymi bólami
MUTSCHLER-2009.indd 750 2010-01-07 22:14:33
Oko 751
na podstawie badań profilaktycznych oraz od odpo-
A rogówka wiednio wcześnie podjętego leczenia.
beleczkowanie
8.5.1.2. Leki stosowane w jaskrze
We wczesnym postępowaniu w jaskrze sięga się za-
żyła
zwyczaj do farmakoterapii. Jej celem jest zmniejsze-
kanał
Schlemma nie produkcji cieczy wodnistej, poprawa jej odpły-
wu przez beleczkowanie lub kanał Schlemma i/lub
zwiększenie naczyniówkowo-twardówkowego trans-
portu cieczy wodnistej. Celom tym służą:
ciało rzęskowe tęczówka
■ leki blokujące receptory β-adrenergiczne (β-bloke-
ry, β-adrenolityki),
B rogówka
■ sympatykomimetyki,
beleczkowanie
■ parasympatykomimetyki,
■ inhibitory anhydrazy węglanowej,
żyła ■ pochodne prostaglandyn,
kanał
Schlemma ■ diuretyki osmotyczne – jako leki stosowane w sta-
nach nagłych.
Dopiero gdy leczenie farmakologiczne okaże się nie-
ciało rzęskowe tęczówka wystarczające do opanowania choroby, wymagane
jest postępowanie chirurgiczne.
Ryc. B 8.5-1. Szeroki (A) i wąski (B) kąt tęczówkowo-rogówkowy
(przesączania) (według Leydheckera i Krieglsteina). Sieć bele-
czek (beleczkowanie) jest tworem gąbczastym, wyściełającym 8.5.1.2.1. Leki blokujące receptory
kąt tęczówkowo-rogówkowy. Przez pory w beleczkowaniu ciecz β-adrenergiczne
wodnista przepływa do kanału Schlemma i stamtąd dalej do
naczyń żylnych. Leki blokujące receptory β-adrenergiczne są prze-
de wszystkim stosowane w leczeniu jaskry z otwar-
tym kątem przesączania. Ich główne zalety polegają
głowy, nudnościami i często także wymiotami (ostry atak na tym, że przy skutecznym działaniu obniżającym
jaskry). Z powodu obrzęku nabłonka rogówki oraz niedo- ciśnienie śródgałkowe nie wpływają na szerokość
statecznego ukrwienia siatkówki wskutek podwyższonego źrenic ani na akomodację, a także z powodu względ-
ciśnienia śródgałkowego dochodzi do znacznego pogorsze-
nie długiego czasu działania pozwalającego na poda-
nia zdolności widzenia.
wanie tylko dwa razy na dobę. Mechanizm działania
tej grupy leków polega na zmniejszaniu wytwarzania
Duże znaczenie jaskry jako choroby wynika z tego, cieczy wodnistej.
że razem ze zwyrodnieniem plamki żółtej i retinopa- W tab. B 8.5-1 podano przykłady preparatów blo-
tią cukrzycową należy do trzech najczęstszych przy- kujących receptory β stosowanych w okulistyce. Naj-
czyn ślepoty w krajach wysoko uprzemysłowionych. lepiej udokumentowane są doświadczenia związane
Jaskra dotyka 1–2% populacji w wieku powyżej 40 ze stosowania tymololu. Nie wykazano wyższości
lat i wraz z wiekiem częstość tej choroby się zwięk- innych, stosowanych w terapii jaskry β-blokerów nad
sza. Na początku choroby zazwyczaj nie występują timololem.
żadne subiektywne objawy, więc jeśli pacjent nie zo- Mimo stosowania miejscowego (w postaci 0,1–0,5-
stanie w porę przebadany przez lekarza, zauważa on -proc. roztworu) mogą wystąpić, jak już wspomnia-
Oko
u siebie chorobę dopiero po pojawieniu się znacz- no, typowe dla β-blokerów ogólne działania niepożą-
nych ubytków w polu widzenia. Z tego powodu ro- dane (zaburzenia przewodnictwa przedsionkowo-ko-
kowanie istotnie zależy od wczesnego rozpoznania morowego, bradykardia, dolegliwości astmatyczne).
B8
MUTSCHLER-2009.indd 751 2010-01-07 22:14:33
752 Oko
Tabela B 8.5-1. Leki blokujące receptory β-adrenergiczne sto- Tabela B 8.5-2. Sympatykomimetyki stosowane w leczeniu ja-
sowane w leczeniu jaskry (przykłady) skry
Nazwa Preparat handlowy Nazwa Preparat handlowy
międzynarodowa międzynarodowa
betaksolol Betoptima, Optibetol, Betoptic, Betabion apraklonidyna Iopidine
karteolol Arteoptic, Cartebak, Carteol LP brimonidyna Alphagan, Luxfen, Clarix
lewobunolol Vistagan klonidyna Clonid-Ophtal, Dispaclonidin,
Isoglaucon
metipranolol Betamann
dipiwefryna d Epifrin, Glaucothil
tymolol Arutimol, Timo-COMOD, TimoEDO, Tim-
-Ophtal, Apo-Timol, Cusimolol, Oftan
Timolol, Oftensin, Timohexal, Timoptic
(EDO = Ein-Dosis-Ophtiole = pojemniki jednodawkowe = miniomsy) występującemu początkowi działania w postępowa-
niu w ostrym napadzie jaskry.
Roztwór 0,5-proc. apraklonidyny stosowany jest
Z tego też powodu krople oczne zawierające β-bloke- trzy razy na dobę, winian brimonidyny w postaci
ry są przeciwwskazane przy współistniejącej astmie 0,2-proc. roztworu dwa razy na dobę, a 0,0625–
oskrzelowej, spastycznym zapaleniu oskrzeli, zabu- 0,125-proc. roztwór klonidyny dwa do trzech razy
rzeniach przewodnictwa przedsionkowo-komorowe- na dobę.
go oraz w ciężkim alergicznym nieżycie nosa. Jako działania niepożądane wystąpić mogą przede
wszystkim zaburzenia dotyczące gałki ocznej (m.in.
pieczenie oczu, uczucie ciała obcego w oku) i dalej
8.5.1.2.2. Sympatykomimetyki już jako efekty ogólnoustrojowe sporadycznie mogą
pojawić się uczucie zmęczenia, zawroty głowy, spa-
Z grupy sympatykomimetyków jako leki przeciw- dek ciśnienia krwi i inne.
jaskrowe są stosowane: będąca α2- agonistą klonidy- Przeciwwskazaniem do stosowania tej grupy le-
na i jej analogi – apraklonidyna oraz brimonidyna, ków są ciężkie dolegliwości sercowo-krążeniowe,
jak również pochodna adrenaliny dipiwefryna (zob. jak również równoczesna farmakoterapia z użyciem
tab. B 8.5-2). inhibitorów MAO, innych sympatykomimetyków
lub trójpierścieniowych leków przeciwdepresyjnych.
Agoniści receptorów α2, podobnie jak β-blokery, Leki te nie powinny też być stosowane u małych dzie-
obniżają ciśnienie śródgałkowe w wyniku zmniej- ci z powodu opisanych działań niepożądanych.
szenia wytwarzania cieczy wodnistej. Podczas gdy
klonidyna i brimonidyna – w monoterapii lub w połą- Dipiwefryna, która jako diester łatwiej penetruje
czeniu z β-blokerem – stosowane są w długotermino- przez rogówkę oka niż adrenalina, ulega szybkiej hy-
wym leczeniu pacjentów chorych na jaskrę, apraklo- drolizie do właściwej substancji czynnej, czyli właś-
nidyna nadaje się przede wszystkim do zapobiegania nie adrenaliny. Za mechanizm jej działania przyjmu-
wzrostowi ciśnienia śródgałkowego po zabiegach je się poprawę odpływu cieczy wodnistej w wyniku
chirurgicznych w obrębie gałki ocznej (np. lasero- stymulacji receptorów β2- adrenergicznych znajdują-
wym nacięciu tęczówki), jak również dzięki szybko cych się w beleczkowaniu. Wskazaniem do stosowa-
OH
(H3C) 3C O NH
Cl Br CH3
N NH N N NH O
O
HN HN
H2N Cl N O C(CH3) 3
apraklonidyna brimonidyna dipiwefryna
MUTSCHLER-2009.indd 752 2010-01-07 22:14:34
Oko 753
nia dipiwefryny jest jaskra z otwartym kątem prze- Tabela B 8.5-3. Parasympatykomimetyki stosowane w leczeniu
sączania. jaskry
Dawkowanie stanowi jedna kropla 0,1-proc. roz-
tworu aplikowana co 12 godzin. Nazwa Preparat handlowy
międzynarodowa
Jako działania niepożądane opisywano pieczenie
oczu, odczynowe zaczerwienienie spojówek, nie- karbachol Carbamann, Isopto-Carbachol, Miostat
wyraźne widzenie, rozszerzenie źrenicy, zatkanie
pilokarpina Pilocarpin ankerpharm, Pilomann, Pilopos,
przewodów nosowych i uszkodzenie błony śluzowej Spersacarpin, Pilocarpinum 3% HEC,
nosa. Pilocarpinum WZF 2%
Dipiwefryna jest przeciwwskazana u pacjentów
z jaskrą z wąskim kątem przesączania, ponieważ sama
może prowadzić do zwężenia kąta przesączania.
postępowania w ostrym napadzie jaskry z wąskim ką-
tem przesączania, znacznie się zmniejszyło.
8.5.1.2.3. Parasympatykomimetyki (miotyki) Przy spowodowanej zmianami soczewki jaskrze
wtórnej, ostrym zapaleniu tęczówki oraz uszkodze-
Parasymaptykomimetyki przy podaniu miejscowym niach rogówki stosowanie parasympatykomimety-
do gałki ocznej wywołują długotrwały skurcz mięś- ków jest przeciwwskazane.
nia zwieracza źrenicy i mięśnia rzęskowego. W wyni-
ku tego z jednej strony zwężeniu ulega źrenica (dzia-
łanie miotyczne), a z drugiej strony w następstwie 8.5.1.2.4. Inhibitory anhydrazy węglanowej
rozszerzenia przewodów odprowadzających ciecz
wodnistą (rozszerzenie otworów w sieci beleczkowa- Anhydraza węglanowa ma istotny udział w wydziela-
nia) dochodzi do kilkugodzinnego spadku ciśnienia niu cieczy wodnistej. Poprzez zablokowanie funkcji
śródgałkowego. To odpowiada za korzystne działa- tego enzymu można doprowadzić do efektywnego
nie parasympatykomimetyków u pacjentów z jaskrą, zmniejszenia wytwarzania cieczy wodnistej.
zwłaszcza gdy mamy do czynienia z jaskrą z wąskim Początkowo jedynym stosowanym inhibitorem an-
kątem przesączania. hydrazy węglanowej był acetazolamid przeznaczony
W tab. B 8.5-3 zestawiono parasympatykomime- do stosowania ogólnego, dopiero później wprowa-
tyki stosowane w farmakoterapii jaskry. Zdecydo- dzono do terapii nadające się do miejscowego stoso-
wanie najczęściej stosowaną substancją jest pilo- wania dorzolamid i brinzolamid.
karpina. Acetazolamid jest praktycznie stosowany jeszcze
Karbachol jest stosowany 3 razy dziennie w po- tylko w ostrym napadzie jaskry w dawce wynoszącej
staci 1–3-proc. roztworu, a pilokarpina 4 razy dzien- od 500 do 1000 mg podawanej dożylnie (razem z kro-
nie jako roztwór 0,5–2(3)-proc. plami ocznymi zawierającymi pilokarpinę, β-bloker
Niekorzystnym efektem jest to, że wskutek skurczu i/lub apraklonidynę, a także z wlewem dożylnym
mięśnia rzęskowego oko akomoduje do bliży, przez 20-proc. mannitolu; zob. poniżej).
co przede wszystkim u młodszych pacjentów wy- Dorzolamid i brinzolamid nadają się do stosowa-
stępują przejściowe zaburzenia widzenia w postaci nia w monoterapii albo w połączeniu z inhibitorami
krótkowzroczności. Ponadto z powodu długotrwałe- receptorów β-adrenergicznych w przewlekłym lecze-
go zwężenia źrenicy pogorszeniu ulega zdolność wi- niu wszystkich postaci jaskry. Podobnie jak β-bloke-
dzenia o zmierzchu i nocą, co szczególnie u starszych ry, nie powodują one żadnych zmian źrenic lub zabu-
pacjentów wraz z początkami zmętnienia soczewki rzeń akomodacji.
może być bardzo uciążliwe. Dorzolamid stosowany jest w postaci 2-proc. roz-
Jako kolejne działania niepożądane mogą wy- tworu aplikowanego 3 razy na dobę, a brinzolamid
stąpić: wzmożone wydzielanie cieczy łzowej, za- jako 1-proc. roztwór 2 razy na dobę.
czerwienienie oczu oraz kurcz mięśnia rzęskowego Jako działania niepożądane przy stosowaniu brin-
z bólem oczu i głowy. Poza tym przy długotrwałym zolamidu i dorzolamidu mogą pojawić się: przej-
stosowaniu istnieje ryzyko rozdarcia lub odklejenia ściowo zamglone widzenie, nieprawidłowe odczucia
się siatkówki. Z powodu tych działań niepożądanych, dotyczące gałek ocznych, zaburzenia smaku, bóle
Oko
jak również dzięki wprowadzeniu nowych leków głowy, parestezje i reakcje alergiczne.
przeciwjaskrowych (zob. poniżej) stosowanie para- Inhibitory anhydrazy węglanowej nie powinny
sympatykomimetyków w terapii jaskry, z wyjątkiem być stosowane przy nadwrażliwości na sulfonamidy
B8
MUTSCHLER-2009.indd 753 2010-01-07 22:14:34
754 Oko
O O O O
S S CH3 S S
N OCH3
H2NO2S H2NO2S
HN HN
C2H5 C2H5
dorzolamid brinzolamid
w wywiadzie, jak również w ciężkich zaburzeniach Bimatoprost, choć także jest spokrewniony chemicz-
czynności nerek oraz w kwasicy hiperchloremicznej. nie z prostaglandyną F2α nie jest estrem, lecz amidem
i działa jak ten ostatni. Nie wiąże się także z recepto-
rami dla prostaglandyn, lecz z receptorami prostami-
8.5.1.2.5. Analogi prostaglandyn dowymi. (Prostamidy są dopiero niedawno odkrytymi
związkami endogennymi, które biorą udział w regu-
Analogi prostaglandyny F2α latanoprost i trawo- lacji ciśnienia śródgałkowego).
prost, będące równocześnie prolekami w posta-
ci estrów, obniżają ciśnienie śródgałkowe poprzez
usprawnienie naczyniówkowo-twardówkowego od- HO
pływu cieczy wodnistej. (Drogą tą odpływa w przypad- O
CH(CH3) 2
ku młodych osób do 30% cieczy wodnistej, natomiast
O
wraz z wiekiem ilość ta ulega znacznemu obniżeniu,
w wyniku czego wzrasta ryzyko wystąpienia jaskry). HO OH
Pochodne prostaglandyn są stosowane u pacjentów
z jaskrą z otwartym kątem przesączania. Ze względu
na korzystne dawkowanie – 1 raz na dobę, jak rów-
latanoprost
nież z powodu silnego działania obniżającego ciśnie-
nie śródgałkowe leki te są często stosowane w terapii
pierwszego rzutu. HO
Oba leki aplikowane są jednokrotnie na wieczór O
CH(CH3) 2
(latanoprost w postaci 0,005-proc. roztworu, a trawo- O
prost w postaci 0,004-proc. roztworu). O
HO OH
Charakterystycznym działaniem niepożądanym jest
(powolny) utrzymujący się wzrost ilości brązowego
pigmentu w tęczówce wskutek zwiększenia zawar- CF3
tości melaniny w melanocytach tęczówki, będący
szczególnie widoczny u pacjentów z mieszanym za- trawoprost
barwieniem (np. niebiesko-, szaro- lub zielonobrązo-
wym) tęczówki. Do innych działań niepożądanych
HO
możemy zaliczyć: uczucie ciała obcego w oku, prze-
NH
krwienie spojówki, stopniowe zmiany rzęs, obrzę- C2H5
ki okołooczodołowe oraz obrzęk rogówki, a także O
(rzadko) zapalenie tęczówki i wysypkę. HO OH
Latanoprostu nie mogą stosować pacjenci noszą-
cy soczewki kontaktowe. Również przy aplikacji
trawoprostu soczewki kontaktowe muszą być zdjęte
i mogą być ponownie założone dopiero po 15 minu- bimatoprost
tach od zakroplenia.
MUTSCHLER-2009.indd 754 2010-01-07 22:14:35
Oko 755
Bimatoprost poprawia odpływ cieczy wodnistej z 500 miligramami acetazolamidu podawanego do-
odbywający się zarówno przez beleczkowanie, jak żylnie oraz 20-proc. roztwór mannitolu we wlewie
i przez przestrzenie kąta tęczówkowo-rogówkowe- z szybkością 1,5 ml/kg m.c./godz. Następnie bierze
go i w wyniku tego obniża ciśnienie śródgałkowe się pod uwagę zastosowanie β-blokera i/lub apra-
w trochę większym stopniu niż latanoprost czy tra- klonidyny. Gdy napad zostanie opanowany, można
woprost. wdrożyć postępowanie operacyjne polegające na chi-
Jest on również aplikowany podobnie jak oba po- rurgicznej irydektomii albo laserowej irydotomii.
zostałe analogi prostaglandynowe, czyli raz na dobę
wieczorem (w postaci 0,03-proc. roztworu).
Działania niepożądane bimatoprostu odpowiadają 8.5.2. Mydriatyki
w dużej mierze tym, które są charakterystyczne dla (środki rozszerzające źrenicę)
pochodnych prostaglandyny F2α.
Mydriatyki stosowane są w celu diagnostycznego
rozszerzenia źrenicy oraz unieruchomienia tęczówki
8.5.1.2.6. Diuretyki osmotyczne
i ciała rzęskowego w przypadku wewnątrzgałkowych
Za pomocą osmotycznego środka diuretycznego np. 20-proc. stanów zapalnych. Mydriatycznie działają (zob. tab.
roztworu mannitolu można uzyskać szybki wzrost osmo- B 8.5-4) parasympatykolityki poprzez wpływ na mię-
larności osocza o 20–30 miliosmoli i przez to odprowa- sień zwieracz źrenicy oraz α-sympatykomimetyki,
dzenie wody z wnętrza oka do naczyń krwionośnych oka. których punktem uchwytu jest z kolei mięsień roz-
W wyniku tego dochodzi do szybkiego spadku ciśnienia wieracz źrenicy.
śródgałkowego. Ponadto zmniejszenie objętości ciała szkli-
stego i uwarunkowane tym przemieszczenie soczewki oraz
tęczówki do tyłu prowadzi do otwarcia kąta teczówkowo- Tropikamid jest stosowany szczególnie wtedy, gdy
-rogówkowego. To wyjaśnia korzystne efekty działania jak w przypadku diagnostycznego rozszerzania źrenic
diuretyków osmotycznych (stosowanych równocześnie wymagany jest stosunkowy krótki czas tego działa-
z ww. substancjami) w terapii ostrego napadu jaskry z za- nia. Natomiast jeżeli potrzebne jest uzyskanie szcze-
mkniętym kątem przesączania.
gólnie dużego rozszerzenia źrenicy, to tropikamid
podawany jest razem z fenylefryną. Cyklopentolat
jest stosowany przede wszystkim w celu uzyskania
8.5.1.3. Wytyczne w terapii jaskry krótkotrwałego porażenia akomodacji (cykloplegii)
przy badaniu refrakcji (doborze szkieł okularowych)
Celem farmakologicznej terapii jaskry z otwartym u dzieci. Atropina i skopolamina wykazują szcze-
kątem przesączania jest zmniejszenie lub przynaj- gólnie długi czas działania, który jest pożądany przy
mniej opóźnienie progresji choroby przez efektywne stanach zapalnych przebiegających we wnętrzu oka
i trwałe obniżenie ciśnienia śródgałkowego do war- (zapalenie tęczówki, zapalenie tęczówki i ciała rzęs-
tości docelowej właściwej dla danego pacjenta, za- kowego).
leżnej od tego, czy mamy do czynienia z jaskrą prze-
biegającą z wysokim czy też z niskim ciśnieniem Tabela B 8.5-4. Środki rozszerzające źrenicę (mydriatyki)
śródgałkowym.
Standardowo leczenie zaczynamy od monoterapii. Nazwa Preparat handlowy
Jeśli tylko nie ma przeciwwskazań (takich jak astma międzynarodowa
oskrzelowa), lekiem pierwszego rzutu, jak już wspo- I. Parasympatykolityki
mniano, jest β-bloker. Alternatywnie zastosowany
atropina Atropin EDO, Atropin-POS,
może być analog prostaglandyny lub (miejscowo) Atropinum sulfuricum WZF 1%,
inhibitor anhydrazy węglanowej. Jeśli tym sposobem
nie uzyskamy wystarczającego obniżenia ciśnienia cyklopentolat Cyclopentolat Alcon, Zyklolat EDO
śródgałkowego, to stosujemy farmakoterapię łączo-
ną β-blokera (tymolol) z inhibitorem anhydrazy wę- skopolamina Boro-Scopol N
glanowej, np. dorzolamidem, β-blokera z analogiem
tropikamid Mydriaticum Stulln, Mydrum, Tropicabion,
prostaglandyny, np. latanoprostem, czy też β-blokera Tropicamidum WZF
Oko
z sympatykomimetykiem, np. brimonidyną.
W jaskrze z wąskim kątem przesączania, która wy- II. α
α-Sympatykomimetyki
-Sympatykomimetyki
maga nagłego postępowania, stosuje się najczęściej fenylefryna Neosynephrin-POS, Visadron
pilokarpinę (miejscowo 1–2-proc. roztwór) razem B8
MUTSCHLER-2009.indd 755 2010-01-07 22:14:35
756 Oko
Jako działania niepożądane najczęściej opisywane 8.5.4. Przeciwzakaźne
są: zaburzenia akomodacji, podrażnienie spojówek leki okulistyczne
oraz suchość błony śluzowej nosa.
Przeciwwskazaniami do stosowania mydriatyków
W stanach zapalnych oka wywołanych zakażeniem
są jaskra z wąskim kątem przesączania oraz suche,
bakteryjnym, wirusowym czy grzybiczym stosowane
zanikowe zapalenie błony śluzowej nosa (rhinitis sic-
są te same leki, co przy innych chorobach zakaźnych
ca). Również w jaskrze z otwartym kątem przesącza-
(zob. rozdz. B 11.2).
nia parasympatykolityki podwyższają ciśnienie śród-
gałkowe poprzez zwiotczenie mięśnia rzęskowego.
Ze względu na szeroki zakres działania przeciwbak-
teryjnego aplikowane miejscowo gentamycyna i to-
bramycyna należą do antybiotyków standardowo sto-
8.5.3. Leki okulistyczne o działaniu sowanych w okulistyce (zob. tab. B 8.5-5). Również
miejscowo znieczulającym chloramfenikol, jako antybiotyk o szerokim zakresie
działania, jest jeszcze stosunkowo często ordynowa-
Krople do oczu zawierające środki miejscowo znie- ny miejscowo, w przeciwieństwie do prawie niesto-
czulające (anestetyki lokalne) pozwalają na uzyskanie sowanej już terapii ogólnej z użyciem tego antybio-
zniesienia czucia bólu przez spojówkę oraz rogów- tyku.
kę. Dzięki anestezji z użyciem kropli ocznych można
przeprowadzać operacje oczu w obrębie ich przednie-
go odcinka, np. usunięcie niezbyt głęboko tkwiącego
ciała obcego, jak również operację usunięcia zaćmy.
Tabela B 8.5-5. Leki działające przeciwbakteryjnie stosowane
Często stosowaną substancją czynną jest oksybupro-
w okulistyce (przykłady)
kaina. Przy operacji usunięcia zaćmy preferencyjnie
stosowana jest silniej działająca tetrakaina. Kokai-
Nazwa Preparat handlowy
na, która była pierwszym wprowadzonym do użycia międzynarodowa
anestetykiem lokalnym, jest stosowana nadal, gdy
zachodzi potrzeba uzyskania szczególnie silnej an- bibrokatol Noviform, Posiformin
estezji powierzchniowej. W celu przeprowadzenia chloramfenikol Posifenicol, Detreomycyna,
Cusi-Chloramfenikol
większych zabiegów wymagane są typowe anestetyki
powierzchniowe stosowane podczas znieczulenia na- cyprofloksacyna Ciloxan, Proxacin
siękowego lub przewodowego.
Ponieważ wielokrotne użycie kropli do oczu za- gentamycyna Gentamicin-POS, Gentamytrex, Gent-Ophtal,
Refobacin, Gentamicin WZF
wierających środki znieczulające miejscowo powo-
duje rozluźnienie nabłonka rogówki (z niebezpie- kanamycyna Kanamycin-POS, Kanamytrex,
Kana-Stulln, Kan-Ophtal
czeństwem perforacji rogówki i utraty oka), nie wol-
no ich przepisywać do samodzielnego stosowania lewofloksacyna Oftaquix
przez pacjenta.
lomefloksacyna Okacin
norfloksacyna Chibroxin
ofloksacyna Floxal
tobramycyna Tobramaxin, Tobrex, Tobrexan
O C2H5
O N
H3C(CH2) 3 O C2H5
W ostatnich latach na znaczeniu zyskały inhibitory
H2N gyrazy. Tetracykliny są stosowane m.in. w leczeniu
zakażeń chlamydiami, przede wszystkim w jagli-
okybuprokaina cy u dorosłych. W podobnych zakażeniach u dzieci
podaje się natomiast erytromycynę. Środek antysep-
tyczny – bibrokatol (4,5,6,7-tetrabromo-2-hydroksy-
-l,3,2-benzodioksabizmol) używany jest w leczeniu
jęczmienia (hordeolum), a także w stanach zapalnych
powiek i spojówki o podłożu infekcyjnym. Jodopo-
MUTSCHLER-2009.indd 756 2010-01-07 22:14:35
Oko 757
widon służy do rutynowego odkażania worka spo- Wskazaniem do stosowania leków z tej grupy
jówkowego podczas zabiegów operacyjnych. są np. alergiczne zapalenie spojówek, zapalenie na-
czyniówki i siatkówki, zapalenie tęczówki, zapalenie
Spośród leków przeciwwirusowych (zob. tab. B 8.5- tęczówki i ciała rzęskowego, zapalenie błony na-
6), do leczenia zakażeń oczu wywołanych wirusem czyniowej oka lub też pooperacyjne stany zapalne.
opryszczki nadaje się acyklowir oraz triflurydyna. Triamcinolon (w dawce 8–25 mg) jest stosowany do-
Triflurydyna jest wprawdzie skuteczna w zakażeniach szklistkowo (w iniekcji do ciała szklistego) w obrzęku
powierzchniowych oka, najczęściej jednak w takich plamki żółtej o podłożu cukrzycowym lub innym, jak
wypadkach stosuje się acyklowir, ponieważ może również razem z werteporfiną (zob. poniżej) w wysię-
on być używany zarówno w powierzchniowych, jak kowym (mokrym, neowaskularnym) zwyrodnieniu
i w głębokich infekcjach. plamki żółtej. Preparaty handlowe glukokortykoste-
roidów zestawiono w tab. B 8.5-7.
Tabela B 8.5-6. Leki przeciwwirusowe stosowane w okulistyce
(przyklady) Tabela B 8.5-7. Leki okulistyczne zawierające glukokortykoste-
roidy (przykłady)
Nazwa Preparat handlowy
międzynarodowa Nazwa Preparat handlowy
międzynarodowa
acyklowir Acic-Ophtal, Virupos, Zoliparin,
Zovirax, Cusiviral, Virolex deksametazon Dexamethason-Augensalbe JENAPHARM,
Isopto-Dex, Dexamethason WZF
fomiwirsen Vitravene
fosforan disodowy Dexagel, Dexa-sine, Spersadex,
triflurydyna Triflumann deksametazonu Totocortin, Dexafree
fluorometolon Efflumidex, Fluoro-Ophtal,
Fluoropos, Flucon, Flarex
Fomiwirsen, pierwszy wprowadzony do obrotu śro- octan hydrokortyzonu Ficortril, Hydrocortison-POS N
dek antysensowny, nadaje się do miejscowego stoso-
wania w wywołanym wirusem cytomegalii zapaleniu octan prednizolonu Inflanefran, Predni-Ophthal, Predni-POS,
Prednisolonum WZF
siatkówki u pacjentów z AIDS.
piwalan prednizolonu Ultracortenol
Jako leki przeciwgrzybicze stosowane miejscowo
do oka powszechnie stosuje się nystatynę, amfotery-
cynę B oraz natamycynę.
Stosowanie glukokortykosteroidów jest przeciw-
wskazane przy istniejących urazach i owrzodzeniach
8.5.5. Steroidowe rogówki, w jaskrze, w wysychającym zapaleniu spo-
(glukokortykosteroidowe) jówek, przy dużej krótkowzroczności oraz w stanach
lub niesteroidowe zapalnych wywołanych przez drobnoustroje.
leki przeciwzapalne
stosowane w okulistyce Niesteroidowe leki przeciwzapalne, podobnie jak glu-
kokortykosteroidy, stosowane są w stanach zapalnych
gałki ocznej niespowodowanych infekcją, szczegól-
Glukokortykosteroidy są wprawdzie skuteczne w sta-
nie w podostrych fazach choroby.
nach zapalnych gałki ocznej niewywołanych przez
drobnoustroje, jednak stosowanie ich obarczone
jest pewnym ryzykiem, ponieważ podwyższają one
ciśnienie śródgałkowe i przy dłuższym stosowaniu 8.5.6. (Inne) leki okulistyczne
prowadzą do powstania jaskry i zaćmy. Poza tym o działaniu przeciwalergicznym
Oko
z powodu ich działania immunosupresyjnego istnie-
je również niebezpieczeństwo powstania owrzodzeń W profilaktyce oraz leczeniu ostrych i przewlekłych
rogówki w wyniku wtórnych zakażeń bakteryjnych, zapaleń spojówki o podłożu alergicznym sprawdza-
wirusowych lub grzybiczych. ją się leki blokujące receptory histaminowe H1, jak
B8
MUTSCHLER-2009.indd 757 2010-01-07 22:14:36
758 Oko
również kromoglikan disodowy, nedokromil oraz lo- 8.5.9. Leki stosowane w terapii
doksamid. zwyrodnienia plamki żółtej
W tab. B 8.5-8 podano przykłady preparatów handlo-
wych.
Zwyrodnienie plamki żółtej związane z wiekiem
(AMD), w którym w wyniku procesów degeneracyj-
Jako działania niepożądane mogą się pojawić m.in.:
nych uszkodzeniu ulega centralna część siatkówki,
przejściowe pieczenie oczu, suchość oka, uczucie cia-
jest najczęstszą przyczyną znacznego ubytku wzro-
ła obcego w oku, obrzęk powiek lub światłowstręt.
ku (aż do ślepoty włącznie) u pacjentów powyżej 65
roku życia.
Tabela B 8.5-8. Leki przeciwalergiczne stosowane w chorobach Możemy przy tym rozróżnić dwie formy AMD,
oczu (przykłady) tzw. suchą oraz mokrą (wysiękową, neowaskularną).
W formie suchej, z którą mamy do czynienia
Nazwa Preparat handlowy w 80% zachorowań, w wyniku procesów starzenia
międzynarodowa
dochodzi do atrofii nabłonka barwnikowego oraz
azelastyna Allergodil, Vividrin akut Azelastin siatkówki.
kromoglikan disodowy Allergo-Comod, CromoHEXAL, Opticrom, Z kolei w formie mokrej dochodzi do wysięku
Vividrin, Allergocrom, Cusicrom, Lecrolyn, z naczyń włosowatych naczyniówki (stąd forma „mo-
Polcrom
kra) jak również do poddołkowej neowaskularyzacji
emedastyna Emadine naczyniówki (tworzenie nowych naczyń pod siatków-
epinastyna Relestat ką centralną) z równoczesnymi krwawieniami z tych
nieprawidłowych, kruchych naczyń.
lewokabastyna Livocab, Histimet
lodoksamid Alomide Farmakoterapia suchej formy AMD jest jak dotych-
czas niezadowalająca. Zaleca się stosowanie diety
nedokromil Irtan, Tilavist
bogatej w witaminy oraz w cynk w celu profilaktyki
olopatadyna OPATANOL
przejścia suchej formy w wysiękową formę zwyrod-
nienia plamki żółtej i zmniejszenia progresji choroby,
chociaż korzystne efekty są dyskusyjne. Podobnie
wygląda sytuacja z dogałkowym podawaniem gluko-
kortykosteroidów.
8.5.7. Leki okulistyczne działające
wazokonstrykcyjnie W leczeniu mokrej formy AMD stosuje się tzw. tera-
pię fotodynamiczną (PDT) z użyciem werteporfiny
(naczyniokurcząco)
– aktywowanej promieniami światła pochodnej ben-
Innymi związkami stosowanymi przy alergicznym zapa- zoporfiryny.
leniu spojówek i stanach podrażnień gałki ocznej, często
w połączeniu z innymi środkami, są α-sympatykomimetyki. Werteporfina jest podawana we wlewie dożylnym (6
Z powodu niebezpieczeństwa powstania przyzwyczajenia
bądź przekrwienia czynnego przy wielokrotnym poda- mg/m2 powierzchni ciała), we krwi wiąże się z lipo-
waniu tych leków przeznaczone są one jedynie do terapii proteinami o małej gęstości (LDL) i odkłada się w po-
krótkotrwałej. Najczęściej stosowaną substancją czynną staci kompleksów z LDL przede wszystkim w szybko
jest tetryzolina. proliferujących komórkach, a więc także śródgał-
Działania niepożądane i przeciwwskazania odpowia- kowo w śródbłonku nowo tworzących się naczyń.
dają w dużej mierze tym, które są charakterystyczne dla
mydriatyków. Po 15 minutach od rozpoczęcia infuzji barwnik zma-
gazynowany w oku zostaje aktywowany za pomocą
czerwonego światła laserowego (50 J/cm2). Energia
uwalniana przy jego powrocie do stanu wyjściowe-
8.5.8 Dekspantenol go prowadzi do powstania tlenu singletowego, który
przez lokalne uszkodzenia komórek i następowe two-
Dekspantenol, jeden z najczęściej ordynowanych rzenie zakrzepów powoduje zamknięcie nieprawidło-
preparatów okulistycznych, znajduje zastosowanie wych naczyń.
w terapii wspomagającej zmian skóry i spojówek W ciągu dwóch dni po tym zabiegu oczy i skóra
oka, np. ubytków rogówki lub stanów zapalnych ro- muszą być chronione przed promieniowaniem świetl-
gówki. nym.
MUTSCHLER-2009.indd 758 2010-01-07 22:14:36
Oko 759
CH3 CH3
H3COOC CH2 H3COOC CH2
H3COOC N HN H3COOC N HN
H3C H3C
NH N NH N
H3C CH3 H3C CH3
O O O O
OCH3 OH OH OCH3
werteporfina
(2 stereoizomery w stosunku 1:1)
Werteporfina jest metabolizowana przez esterazy Octan anekortawu jest znajdującą się jeszcze w fazie
wątrobowe i osoczowe do kwasu dikarboksylowego badań klinicznych angiostatycznie działającą substan-
pochodnego benzoporfiryny, a wydalana jest przede cją aktywną o budowie chemicznej podobnej do kor-
wszystkim z kałem. tyzolu. Związek ten jednak nie wykazuje właściwo-
Jako działania niepożądane opisywano zaburzenia ści typowych dla glukokortykosteroidów, szczególnie
widzenia (m.in. niewyraźne, zamazane widzenie), działania podwyższającego ciśnienie śródgałkowe.
nudności, świąd, bóle pleców w czasie infuzji leku Prawdopodobnie wyprze on dotychczas stosowane
oraz reakcje alergiczne.
Przeciwwskazaniami do zastosowania werteporfi-
ny są porfiria oraz ciężkie zaburzenia funkcji wątroby.
Nową, uzupełniającą metodą w leczeniu mokrej
formy zwyrodnienia plamki żółtej stało się zasto-
sowanie inhibitorów angiogenezy, tzn. substan-
cji, które w wyniku interakcji z czynnikiem wzrostu
śródbłonka naczyniowego (VEGF) uniemożliwiają
lub przynajmniej zmniejszają powstawanie nowych O
naczyń w naczyniówce oka. Do tak działających O
O CH3
związków należą: pegaptanib – pegylowany oligo- CH3
OH
nuklueotyd, bewacizumab – rekombinowane, huma- CH3 H
nizowane przeciwciało skierowane przeciw VEGF
oraz ranibizumab – fragment przeciwciała przeciw H
VEGF (rhuFab V2). Leki te są podawane doszklist- O
kowo w odstępie od 4 do 6 tygodni (dawkowanie:
0,3 mg pegaptanibu co 6 tygodni, 1–1,25 mg bewa- octan anekortawu
cizumabu co 4 tygodnie lub 0,5 mg ranibizumabu
co 4 tygodnie).
Najczęściej opisywanymi działaniami niepożą-
danymi były m.in. podrażnienia oczu, podwyższone
Oko
ciśnienie śródgałkowe, krwotoki spojówkowe lub
obrzęk rogówki.
Inhibitory angiogenezy są przeciwwskazane w in-
fekcjach oczu.
B8
MUTSCHLER-2009.indd 759 2010-01-07 22:14:37
760 Oko
w zwyrodnieniu plamki żółtej glukortykosteroidy jak Tabela B 8.5-9. Sztuczne łzy, środki nawilżające (przykłady)
np. triamcinolon (zob. powyżej).
Nazwa Preparat handlowy
międzynarodowa
karbomer Liposic, Siccapos, Vidisic,
Visc-0phthal, Oftagel
8.5.10. Środki tworzące warstwę
karmeloza Cellufresh, Cellumed, Celluvisc
ochronną na powierzchni
rogówki i spojówki kwas hialuronowy HYLO-COMOD, Hylovision,
Vislube, Vismed
Leki okulistyczne zawierające środki tworzące film hypromeloza Artelac, Sicca-Stulln, Sic-Ophthal N,
Sic-Ophthal sine, Isopto Tears, Tears
ochronny na powierzchni oka są stosowane jako naturale II, Tears naturale Free
sztuczne łzy w zespole suchego oka (keratocon-
junctivitis sicca, kseroftalmia) oraz w pielęgnacji ro- alkohol poliwinylowy Liquifilm N, Vistil, Lacrimal
gówki i spojówki podczas gojenia się powierzchnio-
wych uszkodzeń, a także jako środek do nawilżania poliwidon Arufil, Lacophthal, Oculotect fluid,
soczewek kontaktowych. Odpowiednie preparaty za- Vidisept EDO
wierają różne substancje nieczynne farmakologicznie sine = bez dodatku środków konserwujących
(zob. tab. B 8.5-9).
MUTSCHLER-2009.indd 760 2010-01-07 22:14:37
Skóra 761
9. Skóra
Skóra jest największym narządem. Tworzy ona ze- ■ bierze udział w regulacji temperatury ciała przez
Skóra
wnętrzną powierzchnię organizmu i tym samym sta- zwężanie lub rozszerzanie naczyń krwionośnych,
nowi barierę między otoczeniem oraz środowiskiem jak również przez parowanie potu,
wewnętrznym. Skóra:
■ jako narząd zmysłów przekazuje za pomocą licz- B9
■ chroni tkanki przed uszkodzeniami chemicznymi nych receptorów wrażenie ucisku, temperatury
oraz fizycznymi, w tym głównie mechanicznymi, oraz bodźce bólowe.
a także przed wtargnięciem mikroorganizmów,
■ zapobiega nadmiernemu wysuszeniu, umożliwiając
jednak fizjologiczne parowanie wody (przeznaskór-
kowa utrata wody),
9.1. Budowa skóry
Skóra składa się z: Warstwy naskórka. Keratynocyty powstające w war-
stwie podstawnej (zob. poniżej) wędrują w stronę po-
■ naskórka (epidermis) wraz z jego przydatkami (gru-
wierzchni naskórka, ulegając stopniowemu różni-
czoły, włosy, paznokcie),
cowaniu, spłaszczeniu oraz rogowaceniu. Odzwier-
■ skóry właściwej (dermis). ciedleniem tego procesu są poszczególne warstwy
naskórka (zob. ryc. B 9-1). Od zewnątrz wyróżnia się
Naskórek wraz ze skórą właściwą określa się łącz- kolejno:
nie mianem skóry (cutis) (zob. ryc. B.9-1). Pod skó-
■ warstwę rogową (stratum corneum),
rą znajduje się tkanka podskórna (tela subcutanea).
która bez ostrej granicy przechodzi w skórę właściwą ■ warstwę ziarnistą (stratum granulosum),
i wiąże ją z podłożem.
■ warstwę rozrodczą (stratum germinativum), która
dzieli się na warstwę kolczystą (stratum spinosum)
oraz warstwę podstawną (stratum basale).
9.1.1. Naskórek (epidermis)
Warstwa rogowa składa się ze spłaszczonych, całko-
Naskórek składa się z wielowarstwowego, rogowa- wicie zrogowaciałych, bezjądrzastych komórek (kor-
ciejącego nabłonka płaskiego o grubości od 40 μm neocytów), które nieustannie oddzielają się od po-
do 1,6 mm. Najcieńszy naskórek występuje na powie- wierzchni skóry w postaci drobnych łusek.
kach, natomiast najgrubszy w miejscach szczególnie Warstwa rogowa stanowi barierę oraz system
narażonych na działanie sił mechanicznych (dłonie, spichrzowy skóry. Zapobiega ona nadmiernemu pa-
podeszwy stóp). Naskórek jest zakotwiczony w skó- rowaniu wody oraz wnikaniu substancji obcych, a po-
rze właściwej za pośrednictwem kulistych brodawek nadto wiąże liczne substancje, które następnie ulegają
skórnych oraz gruczołów i mieszków włosowych. powolnemu uwalnianiu.
Skóra właściwa jest również źródłem substancji od- Wartość pH skóry, wynosząca w jej powierzchnio-
żywczych dla naskórka. wych warstwach około 5, ma szczególne znaczenie dla
Naskórek zbudowany jest głównie z keratynocy- mikroflory bakteryjnej i grzybiczej, składającej się głów-
tów, wśród których ulokowane są melanocyty wytwa- nie z gronkowców, bakterii z rodzaju Propionibacterium
rzające melaninę, komórki Langerhansa prezentujące oraz drożdżaków z rodzaju Pityrosporum, których zna-
antygen i inne. czenie zależy od okolicy skóry.
MUTSCHLER-2009.indd 761 2010-01-07 22:14:37
762 Skóra
włos ujście gruczołu potowego
włos
warstwa
naskórek rogowa
(epidermis) warstwa
rozrodcza
włókna nerwowe
z komórkami Merkela
gruczoł
skóra łojowy gruczoł łojowy
właściwa naczynia chłonne
(corium, mięsień
dermis) mieszka
włosowego
ciałka Ruffiniego
tkanka tłuszczowa
tkanka włókna nerwowe
podskórna ciałek Ruffiniego
(subcutis)
ciałka Paciniego
brodawka włosa gruczoł potowy żyła tętnica
Ryc. B 9.1-1. Pionowy przekrój przez skórę (według Thewsa, Mutschlera, Vaupela).
Warstwa ziarnista obejmuje zaledwie 2–5 warstw Warstwa siateczkowata składa się z wytrzymałych,
płaskich komórek z małymi jądrami. W tej warstwie splecionych pęczków włókien kolagenowych oraz
do przestrzeni międzykomórkowej wydzielane są ke- włókien sprężystych (elastyna i mikrofibryle) zapew-
ratynosomy. Zawierają one ceramidy, ważne składni- niających elastyczność skóry, a także z fibroblastów.
ki lipidów naskórkowych odpowiedzialnych za funk-
cje warstwy rogowej jako bariery skórnej.
W 4–8 warstwach warstwy kolczystej znajdują się Tkanka podskórna. Skóra właściwa przechodzi bez
obkurczone, wielokątne komórki połączone za po- ostrej granicy w tkankę podskórną – luźną, pasmowa-
średnictwem desmosomów. Dodatkowe wzmocnienie to ułożoną tkankę łączną, wśród której w postaci pła-
połączeń międzykomórkowych stanowią tonofibryle. cików rozmieszczona jest różnie rozwinięta tkanka
Regeneracja naskórka zachodzi w warstwie pod- tłuszczowa (panniculus adiposus). Podskórna tkanka
stawnej, warstwie cylindrycznych komórek z owal- tłuszczowa służy głównie do ochrony przed zimnem,
nymi jądrami. Kompartment naskórkowy cechuje się a ponadto stanowi zapas energii.
znaczną wydolnością metaboliczną, np. w związku
z obecnością esteraz.
Unaczynienie. W podskórnych pasmach tkanki łącz-
Keratynocyty i fibroblasty skóry właściwej wy-
nej przebiegają tętnice i żyły (oraz naczynia chłonne)
twarzają także inne enzymy metaboliczne, np. sulfo-
tworzące w dolnej warstwie skóry właściwej splot
transferazy czy izoformy cytochromu P-450.
głęboki. Z tego splotu wychodzą naczynia (tętniczki
i żyłki), które w górnej części warstwy siateczkowatej
tworzą splot naczyniowy powierzchowny, zaopatru-
9.1.2. Skóra właściwa (dermis)
jący drogą naczyń włosowatych ciała brodawkowate.
oraz tkanka podskórna Stąd na drodze dyfuzji odżywiany jest niezawierający
naczyń naskórek.
Skóra właściwa (dermis). Skóra zwierzęca, będąca
także surowcem w przemyśle garbarskim, składa się
z dwóch warstw: brodawkowatej (stratum papillare) Unerwienie. Układ nerwowy skóry jest bardzo zło-
oraz siateczkowatej (stratum reticulare). W warstwie żony, a funkcja niektórych struktur nerwowych po-
brodawkowatej występują liczne drobne fibryle, jak zostaje nieznana. W skórze zlokalizowany jest zmysł
również komórki (histiocyty, komórki tuczne) oraz dotyku i położenia, a także odczuwanie ucisku, wi-
naczynia włosowate. bracji, temperatury, bólu i świądu.
MUTSCHLER-2009.indd 762 2010-01-07 22:14:37
Skóra 763
9.1.3. Przydatki skóry Łysienie najczęściej wiąże się z całkowitą atrofią
brodawek włosowych. Typową przyczyną łysienia
jest genetycznie uwarunkowane wzmożenie prze-
Do przydatków skóry zalicza się:
miany testosteronu do dihydrotestosteronu, a także
■ paznokcie, wzmożone wytwarzanie receptorów androgenowych
w miejscu łysienia.
■ włosy,
■ gruczoły łojowe, Gruczoły łojowe. Większość gruczołów łojowych
Skóra
powstaje z nabłonka narządu włosowego i uchodzi
■ gruczoły apokrynowe (zapachowe),
do mieszka włosowego (zob. ryc. B 9.1-1). Dlatego
■ gruczoły potowe. określa się je mianem gruczołów łojowych mieszko-
wych. Niezwiązane z mieszkami włosowymi, wol- B9
Paznokcie. Najważniejszą częścią narządu paznok- ne gruczoły łojowe, występują w przedsionku nosa,
ciowego są lekko wypukłe płytki paznokciowe pokry- na czerwieni wargowej oraz na narządach płciowych.
wające strony grzbietowe dystalnych paliczków rąk Gruczoły łojowe składają się z dużych, zawierających
i stóp, stanowiące ochronę oraz oparcie dla opuszek liczne kropelki tłuszczu (lipidy łoju skórnego), wielo-
palców. Płytka paznokciowa leży na łożysku paznok- kątnych komórek, które ulegają rozpadowi, w całości
cia, odrost płytki odbywa się w macierzy paznokcia przekształcając się w wydzielinę gruczołów łojowych.
znajdującej się pod tylnym brzegiem płytki paznok- Wydzielina ta przedostaje się do mieszka włosowego,
ciowej (obłączek). a stamtąd na powierzchnię skóry, pokrywając ją wraz
z włosami ochronną warstwą tłuszczową. Czynność
Włosy. Włosy są giętkimi i wytrzymałymi na rozcią- gruczołów łojowych, która jest stymulowana przez
ganie nitkowatymi strukturami z keratyny o grubości hormony androgenne, ma istotne znaczenie w der-
5–200 μm. Część włosa wystającą ze skóry okre- matologii: pacjenci z nadprodukcją tych gruczołów
śla się mianem łodygi włosa, część ukryta w skórze (łojotok) chorują po części na zupełnie inne choroby
to korzeń włosa. Osadzony jest on w mieszku wło- skóry i wymagają innego leczenia niż pacjenci ze zbyt
sowym, będącym wgłobieniem nabłonka powierzchni skąpą produkcją łoju (sebostaza).
skóry (zob. ryc. B 9-1). Dolna część włosa rozszerza
się w cebulkę włosową, osadzoną na brodawce wło- Gruczoły apokrynowe, zapachowe. Gruczoły te
sowej odpowiedzialnej za odżywienie włosa. Z ce- również uchodzą do mieszka włosowego. U ludzi wy-
bulki włosowej, składającej się z niezróżnicowanych stępują one tylko w dołach pachowych, przewodach
komórek macierzystych oraz melanocytów, wyrasta słuchowych zewnętrznych, brodawkach sutkowych,
dziennie około 0,4 mm włosa. Rozróżnia się mieszki powiekach oraz narządach płciowych. Wytwarzają
włosów końcowych, które sięgają aż do tkanki pod- tłustą, zasadową wydzielinę.
skórnej i tworzą grube włosy, oraz mieszki włosów
meszkowych, które kończą się w skórze właściwej Gruczoły potowe. Gruczoły te rozmieszczone są na
i z których wychodzą włosy krótkie i cienkie (me- całej powierzchni ciała, a ich łączną liczbę szacuje się
szek). Siwienie włosów jest spowodowane niedosta- na 2 miliony. Występują one niezależnie od miesz-
tecznym wytwarzaniem barwnika lub pojawieniem ków włosowych (zob. ryc. B 9.1-1). Wydzielina tych
się przestrzeni powietrznych w strukturze włosa. gruczołów – pot zawiera sole, kwasy oraz mocznik.
MUTSCHLER-2009.indd 763 2010-01-07 22:14:38
764 Skóra
9.2. Skórne objawy chorobowe
W celu rozpoznania choroby skóry oraz wynikających tej książki. Dlatego (zob. rozdz. B 9.4 i kolejne) zo-
z niego działań leczniczych nieodzowne jest określe- staną tutaj krótko omówione jedynie przykłady kil-
nie klinicznie wykrywalnych zmian skórnych, czyli ku typowych chorób wraz z odpowiednimi lekami
wykwitów. Rozróżnia się przy tym wykwity pierwotne i schematami leczenia. Ze względu na to, że niektóre
oraz wtórne (zob. tab. B 9.2-1). Wykwity pierwotne substancje lecznicze lub przedstawiciele niektórych
powstają na skórze zdrowej, natomiast wykwity wtór- klas leków są środkami pierwszego wyboru w lecze-
ne rozwijają się na skórze zmienionej chorobowo. niu wielu dermatoz, zostaną one omówione w jednym
Przedstawienie nawet tylko najważniejszych cho- miejscu z uwzględnieniem wskazań.
rób skórnych (dermatoz) wykraczałoby poza ramy
Tabela B 9.2-1. Objawy chorób skóry
Wykwity pierwotne Wykwity wtórne
plama (macula): łuska (squama):
ograniczona zmiana zabarwienia, np. spowodowana oddzielające się fragmenty warstwy rogowej naskórka
rozszerzeniem naczyń, wylewem krwi do tkanek,
hiper- lub depigmentacją
bąbel pokrzywkowy (urtica): strup (crusta):
wywołany ograniczonym obrzękiem, zmiana wystająca powstaje przez zaschnięcie wydzieliny, ropy, krwi lub leków,
ponad poziom otaczającej skóry o barwie zwykle przykrywa zmiany skórne, np. nadżerki lub owrzodzenia
od jasnoczerwonej do białawej
grudka (zależnie od wielkości papula lub nodulus): nadżerka (erosio):
zmiana o wielkości od główki szpilki do ziarna soczewicy powierzchniowe uszkodzenie skóry w obrębie naskórka, goi się
spowodowana miejscowym zgrubieniem naskórka bez tworzenia blizn
lub namnożeniem komórek w skórze właściwej
guz (tuber, nodus): owrzodzenie (ulcus):
jak grudka, ale zdecydowanie większy głęboko sięgający ubytek skóry, następuje uszkodzenie naskórka,
skóry właściwej oraz przydatków skórnych,
goi się zawsze z tworzeniem blizny
pęcherzyk (vesicula): rozpadlina (rhagas):
zmiana o wielkości od główki szpilki do ziarna grochu, jedno- ubytek skóry w postaci szczeliny, najczęściej powstaje w miejscach
lub wielokomorowa przestrzeń wypełniona płynem, szczególnie narażonych na urazy, na szorstkiej skórze
umiejscowiona w naskórku lub pod naskórkiem (np. wnętrze dłoni, kąciki ust, przestrzenie międzypalcowe stóp)
pęcherz (bulla): zanik (atrophia):
jak pęcherzyk, ale większa i najczęściej jednokomorowa wszystkie warstwy skóry stają się cieńsze, zmniejszona liczba gruczołów
łojowych i potowych, brak włosów, skóra jest pomarszczona
i łatwo ją odciągnąć od podścieliska
krosta (pustula): blizna (cicatrix):
pęcherzyk wypełniony ropą, zwykle na skórze zmienionej efekt niecałkowitej naprawy ubytku tkankowego przez wypełnienie
zapalnie lub w mieszku włosowym tkanką łączną, która kurczy się w miarę starzenia, czasem może
jednak ulegać nadmiernemu rozrastaniu (blizna przerostowa, keloid)
MUTSCHLER-2009.indd 764 2010-01-07 22:14:39
Skóra 765
9.3. Podstawy leczenia chorób skóry
Oprócz właściwej substancji leczniczej, istotną rolę długotrwałego stosowania istnieje ryzyko przesu-
w leczeniu chorób skóry odgrywają substancje po- szenia.
mocnicze, forma leku oraz sposób aplikacji. Na przy-
kład maść musi być dopasowana do typu skóry (su- Oleje w postaci czystej (olej z oliwek, olej z orzesz-
cha czy łojotokowa). Postać leku (puder, zawiesina, ków ziemnych i inne) albo jako olejowe zawiesi-
Skóra
pasta itd.) musi być wybrana odpowiednio do sytu- ny tlenku cynku, talku lub tlenku tytanu stosowane
acji (zmiany sączące, swędzące, ostre, przewlekłe). są do oczyszczania strupów oraz w leczeniu rozle-
Uwzględnić należy również zróżnicowane wchłania- głych zmian skórnych.
nie leku w zależności od zastosowanego opatrunku B9
(np. okłady lub opatrunki okluzyjne). W przypadku stosowania maści, kremów (= maści
Również uwalnianie oraz penetracja do skóry o większej zawartości wody) i past (= maści o dużej
substancji czynnej w dużej mierze zależy od formy zawartości pudru) istotny jest dobór podłoża (waze-
galenowej preparatu. Uwalnianie jest najlepsze, je- lina, olej parafinowy, oleje lub tłuszcze zwierzęce,
żeli substancja czynna lepiej rozpuszcza się lub ma nieorganiczne i organiczne substancje żelujące) oraz
wyższe powinowactwo do struktur skóry niż do pod- wybór między emulsjami typu woda/olej i olej/woda.
łoża leku. Dlatego różne postacie tej samej substan- Emulsje woda/olej są przede wszystkim natłuszcza-
cji czynnej nie mogą być automatycznie traktowane jące i kryjące, jednak oddają również pewną ilość
jako biologicznie równoważne, co więcej, niekiedy wody. Maści tego typu są odpowiednie dla osób o su-
różnice w sile działania są bardzo znaczne. W przy- chej skórze, podczas gdy pacjenci z łojotokiem lepiej
padku leków miejscowych silnie działających utrud- tolerują emulsje olej/woda. Kremy tego typu są ła-
nia to zastąpienie jednego leku gotowego innym lub two zmywalne, szybko wchłaniają się w skórę i mają
zamianę na wykonany w aptece środek recepturowy. działanie chłodzące.
Poza tym w przypadku leków dermatologicznych
(jak również generyków) do rejestracji jako środka Maści tłuste składają się najczęściej z węglowo-
leczniczego konieczne jest kliniczne potwierdzenie dorów oraz triglicerydów. Hamując utratę wilgoci
skuteczności. Jedynie dla glukokortykosteroidów ze skóry, zwiększają nawilżenie warstwy rogowej na-
będących preparatami naśladowczymi jako parametr skórka, co poprawia penetrację substancji czynnych
zastępczy akceptuje się badanie aktywności w teście zawartych w maści.
płowienia.
Do zewnętrznych postaci leków zalicza się ponadto
Okłady wilgotne w związku z intensywnym pa- żele, które dzieli się na lipożele (zawierające oleje lub
rowaniem działają chłodząco, ograniczają nasi- substancje oleiste), żele mikroemulsyjne oraz hydro-
lenie zapalenia oraz świądu. Nadają się ponadto żele. Te ostatnie stosuje się w produkcji preparatów,
do zmiękczania strupów oraz pobudzają odtwarza- w których substancja czynna umieszczona jest w li-
nie naskórka. posomach.
Również pudry wykazują niewielkie działanie chło- Nawet proste podłoża bez substancji czynnej mogą wpły-
dzące, przeciwświądowe oraz wysuszające i zapobie- wać na przebieg chorób skóry. Na przykład syntetyczne
opatrunki (tzw. opatrunki hydrokoloidowe, wielowarstwo-
gają tarciu w okolicach wyprzeniowych. Ponieważ we układy z tworzyw nieprzepuszczalnych dla pary wod-
w przypadku silnie sączących chorób skóry pudry nej lub o dużych możliwościach chłonnych) wspomagają
mogą tworzyć nieprzyjemne strupy, nie powinny być leczenie ran oparzeniowych i owrzodzeń podudzi poprzez
stosowane na obszary skóry objęte ostrym stanem za- regulację miejscowej temperatury i wilgotności (analogicz-
palnym. nie do opatrunków okluzyjnych).
Materiał opatrunkowy wchłania wydzieliny z rany, mar-
twe tkanki oraz zanieczyszczenia, które są usuwane przy
Zawiesiny mają również działanie chłodzące, a po- zmianie opatrunku. Takie systemy, działające wyłącznie fi-
nadto ściągające oraz przeciwzapalne. W przypadku zycznie, mogą zostać zarejestrowane jako produkty lecznicze.
MUTSCHLER-2009.indd 765 2010-01-07 22:14:39
766 Skóra
9.4. Łuszczyca i jej leczenie
9.4.1. Podstawy patofizjologiczne tującym antygen. Proces ten jest następnie wspo-
magany przez interakcję białek powierzchniowych
komórek dendrytycznych, LFA-3 (antygen związa-
Łuszczyca (psoriaris vulgaris) jest dermatozą ru-
ny z czynnością limfocytów) oraz ICAM-1 (czą-
mieniowo-złuszczającą o prewalencji wynoszącej
steczka adhezji międzykomórkowej) z domenami
2%, charakteryzującą się znacznym przyspieszeniem
wiążącymi na limfocytach T (CD2, CD28 i LFA-1)
proliferacji i zaburzeniem różnicowania keratynocy-
(zob. ryc. B 9.4-1). Aktywowane limfocyty T wę-
tów. W skórze stwierdza się ponadto naciek zapalny.
drują do obszarów skóry, gdzie komórki śródbłonka
Jest to choroba dziedziczna wieloczynnikowa, na któ-
naczyń krwionośnych oddziałują poprzez znajdują-
rą istotny wpływ mają także czynniki środowiskowe.
ce się na ich powierzchni ICAM-1 z LFA-1 obec-
Wykwitami pierwotnymi są najczęściej czerwone,
ne na limfocytach T. Dzięki temu wzrasta prędkość
płaskowyniosłe ogniska zapalne pokryte białawymi
przepływu limfocytów T i staje się możliwe prze-
łuskami, które powiększają się i zlewają. Jednocześnie
chodzenie przez ściany naczyń do tkanki. W skó-
mogą powstawać nowe ogniska. Przy próbie cał-
rze limfocyty T wydzielają interferon γ (INF-γ),
kowitego usunięcia łusek dochodzi do punktowego
czynnik nekrozy nowotworów-α (TNF-α) i inne
krwawienia. Gojenie ognisk zapalnych zachodzi bez
cytokiny. TNF α wspomaga powstawanie kolejnych
pozostawienia blizn, jednak mogą powstawać od-
cytokin zapalnych i podział komórek podstawnych.
barwienia lub przebarwienia. Łuszczyca pojawia się
Dochodzi do zapalenia i nadmiernej proliferacji
szczególnie często na łokciach, kolanach, owłosionej
naskórka. INF-γ hamuje apoptozę keratynocytów,
skórze głowy i paznokciach. W 5–10% przypadków
co zaburza proces rogowacenia naskórka. Komórki
przebiega ona razem ze zniekształcającym zapale-
nie tracą swoich jąder – taki stan nazywany jest pa-
niem stawów palców oraz innych stawów (psoriaris
rakeratozą.
arthropathica).
Do określania stopnia ciężkości choroby służy tzw.
Psoriaris Area and Severity Index (PASI). Parametr
ten łączy stopień ciężkości objawów oraz wielkość
zmienionej powierzchni skóry.
9.4.2. Leki przeciwłuszczycowe
W patogenezie łuszczycy uczestniczą makrofagi
W leczeniu łuszczycy stosuje się:
i limfocyty T. Ze względu na to, że w uszkodzonych
miejscach skóry naciekanie komórek zapalnych po- ■ ditranol,
przedza przyspieszenie proliferacji keratynocytów,
■ psoraleny (terapia PUVA),
zakłada się, że najpierw limfocyty T ulegają akty-
wacji przez komórki dendrytyczne dzięki interakcji ■ retinoidy – acytretynę i tazaroten,
receptora limfocytów T z białkiem MHC prezen-
■ analogi witaminy D3,
■ ester kwasu fumarowego,
■ leki immunosupresyjne (metotreksat, cyklosporynę
komórka i tzw. biofarmaceutyki).
prezentująca
antygen (APC) limfocyt T
TNF-α Dotychczas w wielu przypadkach nie jest możli-
LFA-3 CD2, CD28 we trwałe wyleczenie łuszczycy. Zwykle po fazach
INF-γ bezobjawowych dochodzi do ponownego wystąpie-
MHC TCR
nia zmian skórnych. Niejednokrotnie zmiany skórne
ICAM-1 LFA-1 tylko częściowo zanikają pod wpływem leczenia.
IL-2
Obecnie za przejaw dobrej reakcji na leczenie uważa
Ryc. B 9.4-1. Aktywacja limfocytów T stanowi wczesny etap pa- się zmniejszenie PASI (zob. powyżej) o co najmniej
togenezy łuszczycy. Celem dla leków są przede wszystkim ko- 75%. Stopień reakcji na leczenie różnymi środkami
stymulujące białko powierzchniowe LFA-1 oraz cytokina TNF-α, przeciw łuszczycy podano na ryc. B 9.4-2.
która przyspiesza podziały komórkowe i działa zapalnie. Więcej Mimo swojej skuteczności glukokortykosteroidy
informacji w tekście. nie są obecnie uważane za środek pierwszego wyboru
MUTSCHLER-2009.indd 766 2010-01-07 22:14:39
Skóra 767
RePUVA
infliksimab
PUVA + kalcypotriol
cyklosporyna
Skóra
PUVA
metotreksat
B9
fototerapia UVB
acytretyna
etanercept
efalizumab
0 10 20 30 40 50 60 70 80 90 100
PASI
Ryc. B 9.4-2. Stopień reakcji na leczenie łuszczycy, zmierzony na podstawie zmniejszenia się obszaru objętego łuszczycą oraz indek-
su PASI. Terapia RePUVA = retynoid + PUVA (zmodyf. wedługg Koo i Khera).
w leczeniu łuszczycy, ponieważ sprzyjają występo-
waniu nawrotów (tzw. rebound-phenomen).
OH O OH OH O OH
9.4.2.1. Ditranol O2
.
Ditranol stosowany jest w leczeniu łuszczycy zwy- ditranol H
kłej oraz łysienia plackowatego. Substancja ta działa +
na skórę silnie drażniąco, w ogniskach łuszczyco-
O2*
.O – .OH
2
wych obniża wskaźnik mitotyczny oraz zmniejsza
nacieki zapalne. Działanie leku polega na tworzeniu
aktywnych rodników tlenowych (zob. ryc. B 9.4-3).
O2
Działanie ditranolu jest silniejsze w przypadku sto- OH O OH
sowania w postaci preparatów zawierających mocz-
nik lub kwas salicylowy. Przyjmuje się, że przyczyną
jest lepsza penetracja ditranolu do skóry. Ponadto OH O OH
kwas salicylowy ma go chronić przed utlenieniem
i inaktywacją.
Leczenie rozpoczyna się bardzo niskimi stężenia-
mi ditranolu (0,05%), w związku z rozwojem toleran- OH O OH O
cji konieczne jest w przebiegu leczenia zwiększanie
dimer ditranolu (nieaktywny) dantron (nieaktywny)
stężenia do l (–3)%.
Ditranol wchłaniany jest lepiej przez ogniska łusz-
czycowe niż przez zdrową skórę. W tak zwanej tera- Ryc. B 9.4-3. Powstawanie rodnika ditranolowego i reaktyw-
pii minutowej preparat zmywa się ze skóry po 10–30 nych cząstek tlenu z ditranolu pod wpływem O2; inaktywacja
min. W ten sposób ogranicza się działanie drażnią- poprzez dimeryzację i utlenianie do dantronu.
MUTSCHLER-2009.indd 767 2010-01-07 22:14:39
768 Skóra
ce oraz zabarwienie skóry i ubrania przez produkty
oksydacji ditranolu (,,opalenizna cygnolinowa”) bez
utraty skuteczności leczniczej.
Z powodu nasilonego działania drażniącego zasto-
O O O
sowanie ditranolu jest przeciwwskazane na twarzy,
w fałdach skórnych oraz pod opatrunkami okluzyj- OCH3
nymi.
metoksalen
9.4.2.2. Psoraleny
Psoraleny są fotoreaktywnymi pochodnymi furoku- sposób działają antymitotycznie. Hamują np. prolife-
maryny, które występują w licznych roślinach (np. rację komórek naskórka i limfocytów. W tak zwanej
baldaszkowatych). Pod wpływem światła reagują one terapii PUVA oprócz psoralenów stosuje się dodat-
z cząsteczkami zasady pirymidynowej w DNA, two- kowo naświetlanie długofalowym promieniowaniem
rząc monoaddukty oraz wiązania poprzeczne i w ten ultrafioletowym (UV-A).
Tabela B 9.4-1. Witamina D3 i jej analogi w leczeniu łuszczycy
Wzór strukturalny Nazwa Preparat handlowy Stężenie
międzynarodowa
H3C CH3 kalcytriol Silkis 3 μg/g kremu
CH3 lub maści
H3C OH
CH2
HO OH
OH kalcypotriol Calcipotriolpsorcutan 50 μg/g kremu
H3C Sandoz, Daivonex, lub emulsji
CH3 Psorcutan
CH2
HO OH
OH takalcytol Curatoderm 4 μg/g kremu
lub emulsji
H3C
CH3 CH(CH3) 2
CH2
HO OH
MUTSCHLER-2009.indd 768 2010-01-07 22:14:40
Skóra 769
Wskazaniem do terapii PUVA są różne choroby, Od kalcytriolu, aktywnej formy witaminy D3, kal-
głównie łuszczyca oraz chłoniak skórny (ziarniniak cypotriol i takalcytol różnią się tylko jednym zmie-
grzybiasty). nionym łańcuchem bocznym. Obie pochodne mają
porównywalny do kalcytriolu wpływ na proliferację
Najczęściej stosowaną substancją jest metoksalen i różnicowanie komórek, jednak ich wpływ na go-
(amoidyna, 8-metoksypsoralen; w tabletkach lub roz- spodarkę wapniową jest przy podaniu miejscowym
tworze). Przy stosowaniu doustnym dostępność biolo- znacznie mniejszy, ponieważ po wchłonięciu szybko
giczna podlega znacznej zmienności wewnątrz- oraz ulegają biotransformacji.
Skóra
międzyosobniczej, co wynika ze słabej rozpuszczal- Wskazaniem dla kalcypotriolu i takalcytolu
ności w wodzie, a zatem ograniczonego uwalniania, jest miejscowe leczenie łuszczycy. Stężenie substancji
a ponadto z efektu pierwszego przejścia w wątrobie. czynnych w maściach wynosi odpowiednio 50 μg/g
W przypadku stosowania miejscowego metoksalen i 4 μg/g. B9
penetruje do naskórka i jest niewykrywalny we krwi. Spośród działań niepożądanych mogą występo-
W przypadku rozległych zmian skórnych metok- wać miejscowe podrażnienia, szczególnie w przy-
salen można stosować wewnętrznie (0,6 mg/kg m.c. padku stosowania na twarzy. Ryzyko hiperkalcemii
2 godz. przed napromienieniem) lub w postaci krót- spowodowanej wchłanianiem obu preparatów ogra-
kiej kąpieli (0,4 mg/1 bezpośrednio przed napromie- nicza dawka.
nieniem). Przy stosowaniu zewnętrznym mniejsze
jest ryzyko systemowych działań niepożądanych (zob.
poniżej), a efekt leczniczy jest niezależny od bardzo 9.4.2.4. Estry kwasu fumarowego
zmiennej dostępności biologicznej. Dlatego obecnie
preferuje się leczenie miejscowe. Do leczenia łuszczycy stosuje się doustnie podawany
Przy ogólnym podaniu metoksalenu stosunko- kwas fumarowy, metabolit cyklu kwasu cytrynowego.
wo częstym objawem niepożądanym są nudności. W celu zmniejszenia drażniącego działania na prze-
W związku z ryzykiem uszkodzenia oczu (zaćmy), wód pokarmowy aplikuje się go w formie zestryfiko-
przez 6–8 godz. po zabiegu pacjenci powinni nosić wanej w postaci tabletek odpornych na działanie soku
okulary przeciwsłoneczne, które blokują również żołądkowego. Powoduje on poprawę symptomatyki
długofalowe promieniowanie UV. ciężkiej łuszczycy, prowadząc do obniżenia PASI
Na skutek nasilonej wrażliwości na światło, nawet o 50–80% w ciągu 3–4 miesięcy. Możliwe jest prze-
stosunkowo niewielkie przedawkowanie promienio- rwanie terapii bez wystąpienia nawrotu choroby.
wania UV prowadzi do ciężkich, w pojedynczych Mechanizm działania przeciwzapalnego lub im-
przypadkach wręcz śmiertelnych, oparzeń. munosupresyjnego nie został jeszcze całkowicie po-
Szczególnie w długoterminowej terapii PUVA nie znany. Dyskutuje się, czy hamowanie translokacji
można wykluczyć efektu mutagennego, a nawet ra- NFκB w jądrze komórkowym zmniejsza produkcję
kotwórczego, dlatego wskazania do leczenia należy cytokin zapalnych i cząsteczek adhezyjnych, zaburza
ustalać bardzo ostrożnie. Konieczna jest ponadto sta- różnicowanie komórek dendrytycznych i indukuje
ranna obserwacja chorego. apoptozę przy stosowaniu wysokich dawek leku.
Estry kwasu fumarowego są prawie całkowicie
Stosowanie w leczeniu fototoksycznych farmaceuty- wchłaniane z jelita. Dimetylofumaran jest szybko
ków, np. fenotiazyn, oleju z dziurawca, tetracyklin, przekształcany drogą enzymatyczną do monoestru.
sulfonamidów, zwiększa wrażliwość na światło sło- Monometylofumaran jest następnie dalej redukowa-
neczne. ny do kwasu fumarowego i CO2 (zob. ryc. B 9.4-4).
Przeciwwskazaniami są przebyte choroby, takie jak Kwas fumarowy w formie estrów di- i monomety-
skóra pergaminowa (xeroderma pigmentosum) i nowo- lowych ma zastosowanie w doustnej terapii ciężkiej
twory skóry oraz dermatozy indukowane światłem. łuszczycy oraz w zewnętrznej terapii niedostępnej
lub trudno dostępnej łuszczycy.
W leczeniu doustnym stosuje się dawkę dzienną
9.4.2.3. Analogi witaminy D3 wynoszącą 30 mg dimetylofumaranu oraz 67 mg
wodorofumaranu etylu i każdorazowo po siedmiu
Pochodnymi witaminy D3 stosowanymi w leczeniu dniach podnoszoną powoli aż do osiągnięcia wystar-
łuszczycy są: czającej skuteczności lub granicy tolerancji. Dawka
maksymalna wynosi 1,2 g estru kwasu fumarowego
■ kalcypotriol,
dziennie. Jeśli estry kwasu fumarowego mają być
■ takalcytol. stosowane w profilaktyce nawrotów choroby, daw-
MUTSCHLER-2009.indd 769 2010-01-07 22:14:41
770 Skóra
O O
H3CO esterazy H3CO
OCH3 OH
O O
– CH3OH
dimetylofumaran monometylofumaran
– CH3OH esterazy
O O
H5C2O esterazy HO
OH OH
O – C2H5OH O
monoetylofumaran kwas fumarowy
Ryc. B 9.4-4. Biotransformacja estrów kwasu fumarowego.
ka podtrzymująca zostaje osiągnięta przez powolne sze niż w leczeniu onkologicznym czy po transplan-
zmniejszanie dawki. tacjach. Mimo to również w leczeniu małymi dawka-
Częstymi działaniami niepożądanymi są zaburze- mi metotreksatu może dojść do uszkodzenia wątroby,
nia żołądkowo-jelitowe (szczególnie biegunki), ru- szczególnie u pacjentów z cukrzycą, zaburzeniami
mień i dolegliwości ośrodkowego układu nerwowe- gospodarki tłuszczowej oraz nadużywających alko-
go, a także leukopenia czy zaburzenia funkcji nerek. holu.
Przed oraz w trakcie leczenia wskazana jest kontrola Poza tymi niskocząsteczkowymi środkami lecz-
obrazu krwi i funkcji nerek. niczymi obecnie w leczeniu łuszczycy stawowej
Leczenie doustne jest przeciwwskazane przy zabu- oraz ciężkiej postaci łuszczycy płytkowej stosuje się
rzeniach obrazu krwi oraz w czasie ciąży i laktacji, również wysokocząsteczkowe immunosupresanty
natomiast miejscowe leczenie pochodnymi kwasu fu- pochodzenia biologicznego (tzw. biofarmaceutyki).
marowego jest dopuszczalne. Poza przeznaczonymi pierwotnie do leczenia chorób
reumatoidalnych inhibitorów TNF-α – infliksimabu
Obecnie pracuje się nad przygotowaniem monopreparatu i etanerceptu – należy do nich także efalizumab, któ-
zawierającego dimetylofumaran. ry skierowany jest specyficznie przeciw łuszczycy.
Europejski urząd dopuszczający preparaty lecznicze
odmówił natomiast rejestracji innej substancji (do-
puszczonej w USA) – alefaceptu. Skuteczność efali-
9.4.2.5. Leki immunosupresyjne zumabu i alefaceptu jest jednak mniejsza niż etaner-
ceptu.
W najcięższych, opornych na leczenie postaciach
łuszczycy (np. w łuszczycy stawowej) skuteczne oka- Efalizumab jest ludzkim przeciwciałem monoklonal-
zały się metotreksat oraz cyklosporyna. Ich działanie nym skierowanym przeciw cząsteczce powierzchnio-
polega na hamowaniu komórek T, których cytokiny wej CD11a, która jest ważnym składnikiem antygenu
stymulują w przebiegu łuszczycy nadmierną prolife- związanego z czynnością limfocytów LFA-1 odpo-
rację keratynocytów. wiadającym za adhezję limfocytów do innych komó-
W związku ze stosowaniem mniejszych dawek me- rek. Poprzez wiązanie z ICAM-1 bierze ona udział
totreksatu (10–15 mg raz w tygodniu) oraz cyklospo- w interakcji limfocytów z makrofagami, komórka-
ryny (maksymalna dawka dzienna wynosi 5 mg/kg mi śródbłonka i keratynocytami (zob. ryc. B 9.4-1).
m.c.) ryzyko uszkodzenia wątroby i nerek jest mniej- W ten sposób efalizumab znacząco zaburza komuni-
MUTSCHLER-2009.indd 770 2010-01-07 22:14:41
Skóra 771
Retinoidy stosuje się głównie w leczeniu nadmier-
efalizumab nego oraz zaburzonego rogowacenia (np. łuszczycy,
limfocyt rybiej łuski, trądziku). Tretynoinę stosuje się ponadto
anty-CD11a
Fc (lg) w leczeniu białaczek. Zastosowanie terapeutyczne
(zob. tab. B 9.4-2) znajdują retinoidy:
LFA-1
■ generacji – kwas witaminy A (tretynoina) oraz jego
hamowanie izomer 13-cis izotretynoina (kwas 13-cis-retino-
Skóra
migracji
1
M-
wy),
IC A
■ generacji – acytretyna,
■ generacji – adapalen i tazaroten. B9
komórka śródbłonka
W związku ze stosunkowo dobrą tolerancją izotre-
Ryc. B 9.4-5. Mechanizmy działania efalizumabu. Szczegóły
tynoina i acytretyna mogą być stosowane układowo,
w tekście.
natomiast pozostałe retinoidy stosuje się wyłącznie
w leczeniu miejscowym.
kację komórkową. Limfocyty biorące udział w pro-
cesie łuszczycowym nie mogą zostać zaktywowane Działanie. Podobnie jak witamina A, retinoidy nor-
w węzłach chłonnych i skórze. Zaburzona jest także malizują wzrost i różnicowanie komórek skóry i błon
migracja z krwi do skóry (zob. ryc. B 9.4-5). śluzowych. Dochodzi do obniżenia współczynnika
Efalizumab jest wskazany do leczenia ciężkich po- podziałów stwierdzanego np. w komórkach naskórka
staci łuszczycy. Po podaniu pierwszej dawki wyno- w obrębie ognisk łuszczycowych. Wpływając na pro-
szącej 0,7 mg/kg wstrzykuje się podskórnie 1 mg/kg ces keratynizacji, leki te powodują ponadto rozluźnie-
raz w tygodniu. U 25% pacjentów po 12 tygodniach nie warstwy rogowej, dzięki czemu powierzchowne
następuje obniżenie PASI o 75%. Stan ten może się keratynocyty łatwiej się złuszczają. Obniżeniu ulega
poprawiać przy kontynuacji leczenia. występujące w trądziku wzmożone wiązanie keraty-
Do działań niepożądanych należą objawy grypopo- nocytów w ujściach gruczołów łojowych.
dobne pojawiające się po pierwszej iniekcji. Ich cięż- Retinoidy działają ponadto immunomodulująco
kość ulega znacznemu obniżeniu już przy drugiej oraz przeciwzapalnie. Izotretynoina zmniejsza także
aplikacji leku. Po odstawieniu efalizumabu stopień wydzielanie łoju poprzez wydłużenie fazy spoczyn-
ciężkości choroby może się jednak nasilić ponad stan kowej sebocytów.
wyjściowy (rebound-phenomen, zob. powyżej).
Mechanizm działania. Retinoidy – podobnie jak
Alefacept jest białkiem fuzyjnym składającym się z ludz- steroidy, hormony tarczycy i witamina D – wiążą się
kiego IgG i domeny wiążącej LFA-3. Wiązanie domeny z receptorami wewnątrzkomórkowymi. Obecnie znane
LFA-3 do białka CD2 na powierzchni limfocytów T hamuje
aktywację i proliferację tych komórek pamięci. Następnie są dwa receptory kwasu retinowego, RAR i RXR, każ-
za pośrednictwem części Fc IgG alefacept prowadzi do apo- dy z nich ma po 3 podtypy (α, β, γ). W naskórku prze-
ptozy limfocytów T poprzez aktywację komórek NK. Te waża RAR-γ. Podczas gdy tretynoina i izotretynoina
efekty warunkują długo utrzymujące się działanie cyklu le- mają takie samo powinowactwo do wszystkich recepto-
czenia przy ciężkiej postaci łuszczycy. Reakcja jest jednak rów, retinoidy III generacji wiążą się głównie z RAR-γ
słabsza niż w odpowiedzi na efalizumab.
oraz RAR-β. Receptory RXR wiążą natomiast inną
izoformę kwasu retinowego – kwas 9-cis-retinowy.
Po związaniu ligandu z receptorem tworzą się homo-
9.4.2.6. Retinoidy (RAR-RAR) lub heterodimery (RAR-RXR), które
jako czynniki transkrypcyjne wpływają na ekspresję
Do grupy retinoidów zalicza się naturalne (np. tre- genów. Na tej drodze retinoidy obniżają wzmożoną
tynoina i izotretynoina) oraz syntetyczne pochodne proliferację, blokując białko aktywatora 1 (AP-1) oraz
witaminy A (retinol). Związki nieposiadające budowy hamują produkcję cytokin (zob. ryc. B 9.4-6). W two-
aromatycznej – np. retinoidy naturalne – określa się rzeniu wiązań retinoidów z receptorami RAR i RXR
mianem retinoidów I generacji, związki monoaro- kluczową rolę odgrywa grupa karboksylowa. Związek
matyczne – retinoidów II generacji, a związki polia- estrowy tazaroten jest prekursorem leku, z którego
romatyczne zalicza się do retinoidów III generacji. w skórze uwalniany jest kwas tazarotenowy.
MUTSCHLER-2009.indd 771 2010-01-07 22:14:41
772 Skóra
Farmakokinetyka. Spośród obecnie stosowanych re- ny skórne znikają, a skóra wygląda jak zregenerowa-
tinoidów żaden nie podlega w organizmie wielotygo- na. Należy jednak unikać dodatkowego drażnienia
dniowej kumulacji. Jednakże acytretyna jest częścio- skóry, np. intensywnego nasłonecznienia lub innych
wo metabolizowana do estru etylowego acytretyny, leków zewnętrznych o działaniu drażniącym.
czyli etretynatu, który w związku z wysoką lipofil- Pod względem tolerancji miejscowej adapalen wy-
nością wykazuje znaczne powinowactwo do tkanki raźnie przewyższa tretynoinę.
tłuszczowej i jest z niej eliminowany wyjątkowo po-
woli. Retinoidy stosowane w leczeniu doustnym. O ile
w przypadku witaminy A możliwe jest tylko stoso-
Początkowo sam etretynat stosowano w leczeniu łuszczy- wanie miejscowe, o tyle izotretynoinę i acytretynę
cy. Później jednak zastąpiono ten związek estrowy wolnym stosuje się doustnie. Wskazaniem do stosowania izo-
kwasem, który ma znacznie krótszy okres półtrwania (50
godz. w porównaniu ze 120 dniami). Ponieważ acytretyna tretynoiny są szczególnie ciężkie postacie trądziku
jest częściowo metabolizowana do etretynatu, nie spełniły (np. acne conglobatd), a acytretyny – ciężkie postacie
się nadzieje na znaczną poprawę bezpieczeństwa leczenia. łuszczycy oraz inne poważne zaburzenia rogowace-
nia (np. rybia łuska).
Leki miejscowe. Tretynoina, izotretynoina i adapa- Dawkowanie należy dostosować do efektów kli-
len stosuje się miejscowo w leczeniu trądziku po- nicznych oraz tolerancji leku (średnie dawki pod-
spolitego w stężeniu 0,025–0,1%. W leczeniu lekkiej trzymujące izotretynoiny to 0,5 mg/kg m.c./dzień,
do średnio ciężkiej łuszczycy stosuje się tazaroten a acytretyny 30–75 mg/dzień. Dawka całkowita
w żelu 0,05- i 0,1-proc. 720 mg/kg m.c. izotretynoiny powoduje długotrwałą
W związku z zapalną przemianą zaskórników, remisję choroby. Porównywalnego efektu nie da się
w pierwszych tygodniach leczenia trądziku retinoida- osiągnąć żadną inną metodą leczenia trądziku.
mi często tworzy się więcej krostek i grudek, może
ponadto dochodzić do podrażnień skóry. Mimo to le- Działania niepożądane i przeciwwskazania przy
czenie należy konsekwentnie prowadzić dalej, ewen- leczeniu systemowym. W związku z poważnymi
tualnie zmniejszając dawki. Po pewnym czasie zmia- działaniami niepożądanymi izotretynoinę i acytrety-
retinoid
RAR AP-1
retinoid
retinoid
RAR
RAR
AP-1
czynnik odpowiedzi na retinoid brak wiązania AP-1 do DNA
ekspresja genów ekspresja genów
zależnych od retinoidów ↑ zależnych od AP-1 ↓
synteza białek ↑ synteza białek ↓
różnicowanie ↑ proliferacja, zapalenie ↓
Ryc. B 9.4-6. Mechanizmy działania retinoidów. Szczegóły w tekście.
MUTSCHLER-2009.indd 772 2010-01-07 22:14:41
Skóra 773
nę można stosować wyłącznie po starannym ustale- chorych ryzyko jest mniejsze w leczeniu trądziku niż
niu wskazań. Oprócz niemal zawsze występujących łuszczycy czy rybiej łuski.
zapalenia czerwieni wargowej (cheilitis), świądu, Podobnie jak witamina A, stosowane doust-
złuszczania skóry na dłoniach i podeszwach stóp, nie retinoidy również mają działanie teratogenne.
a także podrażnienia i suchości skóry i błon śluzo- Dlatego stosowanie ich u kobiet w wieku rozrodczym
wych, w 30% przypadków obserwuje się zależne jest (względnie) przeciwwskazane. Jeżeli leczenie
od dawki, odwracalne wypadanie włosów. Niekiedy jest niezbędne, konieczne jest ścisłe stosowanie an-
dochodzi do podwyższenia stężenie transaminaz i li- tykoncepcji w trakcie leczenia oraz dłużej, aż do eli-
Skóra
pidów w surowicy. W związku z młodszym wiekiem minacji substancji aktywnej z organizmu. Dlatego
Tabela B 9.4-2. Retinoidy
B9
Wzór Nazwa Preparaty handlowe Okres Średnia
strukturalny międzynarodowa półtrwania dawka
(godz.) pojedyncza
Retinoidy I generacji
CH3 CH3 CH3 O tretynoina Airol, 0,5 mg/g kremu,
Cordes VAS roztworu
OH
CH3
CH3
CH3 CH3 CH3 izotretynoina ISOTREX 19 0,05-0,1-proc.
krem, żel
CH3 Aknefug Iso, 0,5-1 mg/kg
CH3 O OH Aknenormin, dziennie doustnie
Isotret-HEXAL,
ISOTRETIONIN-ISIS
Retinoidy II generacji
CH3 CH3 CH3 O acytretyna Neotigason 50 30-75 mg
H3C dziennie doustnie
OH
H3CO CH3
Retinoidy III generacji
O adaptalen Differin 1 mg/g kremu,
żelu
OH
H3CO
O tazaroten Zorac 0,05-0,1-proc. żel
N OC2H5
H3C CH3 C
C
CH2 beksaroten TARGETIN 1-3 300 mg/m2
H3C CH3 pow. ciała
dziennie doustnie
OH
CH3
H3C CH3
O
MUTSCHLER-2009.indd 773 2010-01-07 22:14:42
774 Skóra
nie wolno dopuszczać do zajścia w ciążę w przypad- 9.4.2.7. Uzupełnienie: Leki o działaniu
ku podawania izotretynoiny do końca pełnego cyklu keratolitycznym i żrącym
menstruacyjnego po zakończeniu leczenia, a w przy-
padku acytretyny nawet przez 2 lata od zakończenia
Substancją najczęściej używaną do złuszczania
kuracji.
skóry i zmiękczenia mas rogowych jest kwas sa-
licylowy. W użyciu znajduje się maść salicylowa
Interakcje. W przypadku łącznego podawania izo-
na wazelinie, spirytus salicylowy oraz kolodium
tretynoiny i tetracyklin wzrasta ryzyko wystąpienia
salicylowe (stężenie 2–10%). W leczeniu brodawek
wzmożonego ciśnienia śródczaszkowego (pseudotu-
ważne miejsce przypada plastrom z kwasem salicy-
mor cerebri). Dlatego jednoczesne podawanie obu
lowym. Działanie keratolityczne wykazują również
leków jest niedopuszczalne.
rezorcyna i mocznik w wyższych stężeniach (odpo-
Stosowanie preparatu złożonego zawierającego
wiednio 5–20 i 20–40%), a także precypitat siarko-
tretynoinę i erytromycynę przewyższa pod względem
wy. Do przyżegania przerastającego ziarninowania
skuteczności i tolerancji monoterapię tretynoiną.
ran lub rozpadlin stosuje się azotan srebra (lapis)
Łączona terapia retynoidami i PUVA (RePUVA)
i różnorodne kwasy (kwas trichlorooctowy, kwas
jest dopuszczalna w leczeniu ciężkiej postaci łusz-
mlekowy).
czycy.
9.5. Trądzik i leki stosowane w leczeniu trądziku
9.5.1. Podstawy patofizjologiczne lity zaczyna się w okresie dojrzewania i występuje
w postaci lekkiej u 40%, a w postaci ciężkiej u 30%
Kolejną dermatozę hiperproliferacyjną i jednocześ- młodzieży. Najczęściej zanika samoistnie w trzeciej
nie zależną od androgenów stanowią różne formy dekadzie życia.
trądziku. Przez pojęcie trądziku rozumie się choroby W przypadku trądziku egzogennego należy przede
gruczołów łojowych. Kiedy nadmierne rogowacenie wszystkim wymienić trądzik chlorowy, który jest po-
ujść mieszków włosowych utrudnia odprowadzenie wodowany przez chlorowane węglowodory aroma-
łoju przy jednoczesnej nasilonej produkcji łoju (ło- tyczne, trądzik kosmetyczny oraz trądzik polekowy.
jotok), jako pierwotny wykwit trądzikowy powstają Ostatnia forma trądziku może wystąpić np. po lecze-
zaskórniki. Następnie pojawiają się wykwity zapalne niu związkami bromu (trądzik bromowy), izoniazy-
– grudki, krosty oraz cysty. dem, fenytoiną albo glukokortykosteroidami (trądzik
Skłonność do trądziku jest dziedziczna, jednak steroidowy).
w powstawaniu objawów istotną rolę odgrywa-
ją również czynniki dodatkowe (zob. ryc. B 9.5-1).
Proliferację nabłonka gruczołów łojowych stymu- 9.5.2. Leki przeciwtrądzikowe
lują androgeny, natomiast hamują estrogeny. Mimo
że trądzik nie jest pierwotnie ropną chorobą skóry,
W leczeniu trądziku stosuje się:
jednak w jego patogenezie biorą udział bakterie,
przede wszystkim Propionibacterium acnes, ponie- ■ nadtlenek benzoilu,
waż bakteryjne produkty przemiany materii sprzyjają
■ kwas azelainowy,
przemianie zapalnej zaskórników. Trądzik może być
poza tym spowodowany działaniem czynników che- ■ leki przeciwinfekcyjne,
micznych lub fizycznych.
■ retinoidy – tretynoinę, izotretynoinę i adaptalen,
Odpowiednio wyróżnia się trądzik:
■ antyandrogeny.
■ endogenny,
■ egzogenny. Nadtlenek benzoilu. Nadtlenek benzoilu w stężeniu
3–10-procentowym występuje w popularnych prepa-
Trądzik endogenny dzieli się natomiast na trądzik ratach handlowych. Uwalniając wolne atomy tlenu
pospolity (acne vulgaris) oraz różne formy specjalne działa on odkażająco, szczególnie na beztlenowce,
(np. acne conglobata, acne inversa). Trądzik pospo- do których zalicza się również Propionibacterium ac-
MUTSCHLER-2009.indd 774 2010-01-07 22:14:42
Skóra 775
retinoidy,
BPO, hiperkeratoza retencyjna
kwas azelainowy
+
antyandrogeny produkcja androgenów ↑ zaskórnik
Skóra
+
środki
dezynfekujące, propionibakterie, ziarniaki grudka, krosta B9
przeciwinfekcyjne,
tretynoina +
uraz mechaniczny blizny
Ryc. B 9.5-1. Patogeneza i leczenie trądziku.
nes. Nadtlenek benzoilu powoduje ponadto odczyn za-
palny w skórze z wtórnym złuszczaniem. Podobnie jak
witamina A, jednak słabiej, działa rozpuszczająco. HOOC COOH
kwas azelainowy
O
O
O
O
Leki przeciwzapalne. W leczeniu miejscowym
nadtlenek benzoilu
stosuje się głównie erytromycynę w połączeniu
z witaminą A lub jonami cynkowymi oraz klinda-
mycynę. Narastająca oporność i oporność krzyżowa
Wchłonięty nadtlenek benzoilu ulega biotransfor-
Propionibacterium acnes zmusza jednak w dalszej
macji do kwasu benzoesowego i po koniugacji wyda-
perspektywie do zrewidowania stosunku korzyści do
lany jest w formie kwasu hippurowego.
ryzyka przy stosowaniu erytromycyny w leczeniu trą-
Spośród działań niepożądanych obserwowano
dziku. Enteralnie, oprócz erytromycyny, stosuje się
występowanie świądu i pieczenia skóry. Nie można
tetracykliny. Również w tej grupie leków skuteczność
z całkowitą pewnością wykluczyć działania sprzyja-
w leczeniu trądziku wynika z redukcji liczby bakterii
jącego nowotworom.
w gruczołach łojowych.
Nie można dopuszczać do kontaktu nadtlenku
benzoilu z oczami oraz błonami śluzowymi.
Kwas azelainowy. W przypadku dwuwęglanowego 9.5.3. Podstawowa pielęgnacja skóry
kwasu azelainowego, który również redukuje licz- skłonnej do trądziku
bę bakterii w gruczołach łojowych, za skuteczność
w trądziku odpowiada prawdopodobnie hamowanie Podobnie jak w przypadku atopowego zapalenia skó-
hiperkeratozy w ujściach gruczołów łojowych. ry, odpowiednia pielęgnacja skóry skłonnej do trądzi-
Wchłonięta substancja czynna zostaje częściowo ku ma ogromne znaczenie. Preparaty do mycia skóry
wydalona przez nerki, a ponadto ulega rozkładowi powinny posiadać fizjologiczne pH 5,5. Preparaty
w procesie β-oksydacji do kwasu octowego i malo- o odczynie zasadowym ułatwiają namnażanie się
nowego. Propionibacterium.
Objawem niepożądanym może być przemijające Efekty zapalnych uszkodzeń skóry można natomiast
podrażnienie miejscowe. zwalczać, stosując 4-proc. żel z nikotynoamidem.
MUTSCHLER-2009.indd 775 2010-01-07 22:14:42
776 Skóra
9.6. Atopowe lub alergiczne choroby skóry
9.6.1. Podstawy patofizjologiczne gotrwałego wyprysku dochodzi do lichenizacji, tzn.
do pogrubienia poletek skórnych.
Do atopowych lub alergicznych chorób zapalnych
Wyprysk atopowy. Atopowe zapalenie skóry
skóry należą różne formy wyprysku (egzemy), po-
jest w przeciwieństwie do wyprysku kontaktowego
krzywka i obrzęk naczynioruchowy. Mimo różnic
konstytucjonalną, tzn. uwarunkowaną genetycznie,
w patogenezie tych chorób, ze względu na domina-
chorobą skóry. Choroba rozpoczyna się zwykle u nie-
cję reakcji zapalnej, leczenie za pomocą leków dzia-
mowląt endogennym wypryskiem skóry twarzy, tzw.
łających antyflogistycznie lub immunosupresyjnie
skazą mleczną, i w miarę dojrzewania przekształca się
jest podobne. Duży udział reakcji zapalnej charakte-
w wyprysk okolic zgięciowych ze zlichenizowanymi
ryzuje także opisaną wcześniej łuszczycę.
ogniskami w obrębie dołów łokciowych i kolano-
wych. W wieku dorosłym choroba przechodzi w sta-
dium świerzbiączkowe charakteryzujące się obecnoś-
cią swędzących guzków. W tym samym czasie albo
w innym okresie życia u chorego może pojawić się
9.6.1.1. Wyprysk
astma lub alergiczny nieżyt nosa. Zazwyczaj po 5
roku życia dochodzi do decydującej poprawy w prze-
Rozróżnia się następujące formy wyprysku:
biegu choroby. Ponadto poprawę choroby obserwuje
■ wyprysk kontaktowy alergiczny lub z podrażnienia się zazwyczaj w lecie, a pogorszenie – w zimie.
(kontaktowe zapalenie skóry),
Wyprysk łojotokowy. W tej formie wyprysku za-
■ wyprysk atopowy (atopowe zapalenie skóry),
zwyczaj stwierdza się ostro ograniczone okrągłe
■ wyprysk łojotokowy. lub owalne ogniska rumieniowe ze złuszczaniem,
mogą ponadto występować grudki. Zmiany naj-
Histologicznie wyprysk cechuje się obecnością reak- częściej pojawiają się na owłosionej skórze głowy
cji zapalnej w warstwie brodawkowatej skóry właś- (seborrhoea capitis), fałdach nosowo-wargowych,
ciwej oraz w naskórku. Procesy zapalne w naskórku w okolicy mostkowej oraz na grzbiecie w okolicy
polegają na obrzęku komórkowym oraz międzyko- kręgosłupa. Przyczyną choroby jest nieprawidłowa
mórkowym (spongioza) oraz napływie komórek za- reakcja na drożdżaka Malassezia furfur (synonim
palnych (głównie limfocytów). Pityrosporum ovale). Drożdżak ten występuje rów-
nież na skórze osób zdrowych, a powstawanie zmian
Wyprysk kontaktowy. W wyprysku kontaktowym chorobowych tłumaczy się częściowym defektem
należy rozróżniać formę toksyczną oraz alergiczną. immunologicznym.
Wyprysk toksyczny powstaje na skutek albo krótko-
trwałego nasilonego uszkodzenia skóry, albo na sku-
tek długotrwałego działania bodźców powodujących
z osobna tylko niewielkie uszkodzenia. Przykładowe 9.6.1.2. Pokrzywka
substancje powodujące uszkodzenie podprogowe i obrzęk naczynioruchowy
to rozcieńczone zasady, gips, cement lub rozpusz-
czalniki organiczne (wyprysk ze skumulowanego Mianem pokrzywki (urticaria) określa się wysiew
działania toksycznego). U podłoża alergicznego wy- swędzących zazwyczaj bąbli. Pojedyncze wykwity
prysku kontaktowego leży alergia typu późnego, nie- szybko powstają i znikają. Objawy powstają na sku-
kiedy poprzedzana uszkodzeniem warstwy rogowej tek degranulacji komórek tucznych w skórze, która
naskórka. Na skutek tego uszkodzenia alergeny mogą może być zależna od IgE, może również nastąpić
wnikać do znajdujących się w głębszych warstwach na skutek aktywacji dopełniacza, a także pod wpły-
skóry komórek układu immunologicznego i powodo- wem cytokin, czynników nerwowych (substancja P)
wać uczulenie. lub substancji powodujących uwalnianie histaminy
W wyprysku kontaktowym występują jednocześnie na drodze bezpośredniej. Uwolnione mediatory po-
lub na przemian rumień, grudki i pęcherzyki, w dal- wodują powstanie bąbla.
szej kolejności może dochodzić również do tworze- Pokrzywka ostra ustępuje, zwykle samoistnie,
nia się strupów oraz złuszczania. W przypadku dłu- w ciągu 1–2, najwyżej 6 tygodni. Jest to jedna z naj-
MUTSCHLER-2009.indd 776 2010-01-07 22:14:43
Skóra 777
częstszych chorób skóry – około 25% wszystkich ■ przeciwświądowe,
ludzi co najmniej raz w życiu chorowało na tę cho-
■ zwężające naczynia.
robę. Pokrzywkę ostrą mogą wywoływać pokarmy
i używki, użądlenia owadów oraz leki – szczególnie
Związki między strukturą i działaniem. Wolna gru-
antybiotyki β-laktamowe, sulfonamidy, preparaty
pa –OH w pozycji C-11 ma zasadnicze znaczenie
krwiopochodne oraz ekstrakty alergenowe. W wielu
dla aktywności glukokortykosteroidów. Zwiększenie
przypadkach czynnik prowokujący pozostaje nieroz-
siły działania stosunkowo słabego hydrokortyzo-
poznany. Lek, który przyjęty w przebiegu infekcji
Skóra
nu, oprócz sposobów zwiększenia powinowactwa
spowodował wystąpienie pokrzywki, często nie bę-
do receptora kortyzolowego wykorzystanego również
dzie powodował objawów niepożądanych po zakoń-
w przypadku systemowych kortykosteroidów, można
czeniu choroby.
O pokrzywce przewlekłej mówi się w przypadku
osiągnąć za pomocą estryfikacji grupy –OH w pozy- B9
cji C-17. Większa lipofilność estrów w pozycjach C-l
trwania choroby ponad 6 tygodni. Zwykle występuje
7 i C-21 wzmaga wnikanie glukokortykosteroidów
ona u osób dorosłych, dokuczliwy świąd może utrzy-
do skóry i tym samym zwiększa ich działanie.
mywać się całymi miesiącami i latami. Im dłużej trwa
choroba, tym mniejsze są szansę na wyleczenie.
Podział. W zależności od siły działania miejscowe
glukokortykosteroidy dzieli się na klasy (zob. tab.
Występujący najczęściej u młodych kobiet obrzęk
B 9.6-1). Nasilenie efektów pożądanych i niepo-
naczynioruchowy (obrzęk Quinckego) polega
żądanych jest policzalne. Na podstawie algorytmu
na ostrym, ustępującym w ciągu 72 godzin obrzęku
można określić stosunek korzyści do ryzyka (indeks
tkanki podskórnej, zwykle w przebiegu alergicznej
terapeutyczny). Glukokortykosteroidy o szczególnie
reakcji natychmiastowej. Często obrzęk naczynio-
korzystnym stosunku działań pożądanych i niepożą-
ruchowy współ występuje z pokrzywką, może rów-
danych (zob. poniżej), tzn. indeks terapeutyczny > 2
nież występować w przebiegu reakcji anafilaktycz-
(glukokortykosteroidy kategorii 2), to prednikarbat,
nych. Zmiany chorobowe typowo umiejscawiają się
aceponat metyloprednizolonu, aceponat i maślan hy-
na powiekach, wargach oraz narządach płciowych.
drokortyzonu oraz pirośluzan mometazonu. Natomiast
W przypadku obrzęku głośni lub krtani może dojść
hydrokortyzon, walerianian betametazonu, acetonid
do uduszenia. Częstymi czynnikami prowokującymi
triamcynolonu i propionian klobetazolu to glukokor-
są pokarmy, dodatki spożywcze oraz leki (np. inhi-
tykosteroidy kategorii 1 (indeks terapeutyczny ok. 1),
bitory ACE, blokery receptorów β-adrenergicznych,
które wykazują mniej więcej wyrównany stosunek
kwas acetylosalicylowy, leki uwalniające histaminę).
działań pożądanych i niepożądanych.
Szczególnie istotne dla lepszej tolerancji są różni-
ce w biotransformacji tych leków w keratynocytach
9.6.2. Środki o działaniu i fibroblastach, najważniejszych komórkach skóry.
przeciwzapalnym Zróżnicowany jest wpływ na syntezę interleukiny-1
(IL-1) w tych komórkach. W keratynocytach glukor-
9.6.2.1. Glukokortykosteroidy tykosteroidy klasy 2, określane dawniej jako „łagod-
ne steroidy”, hamują wyraźnie produkcję IL-1, która
Glukokortykosteroidy stosowane są w wielu choro- działa prozapalnie. Hamowanie jest natomiast znacz-
bach skóry. Ich właściwości farmakologiczne zostały nie mniejsze w fibroblastach, w których IL-1 stymu-
omówione w rozdz. B 2.7. W tym miejscu zostaną luje proliferację (zob. ryc. B 9.6-1). Tłumaczy się tym
omówione tylko szczególne aspekty miejscowego mniejsze ryzyko zaniku skóry (zob. poniżej).
stosowania glukokortykosteroidów.
Glukokortykosteroidy stanowią jedną z najważ-
Farmakokinetyka. Jak już wspomniano, wchłania-
niejszych grup substancji stosowanych w miejsco-
nie miejscowych glukokortykosteroidów zależy
wym (i ogólnym) leczeniu chorób skóry. W ich zasto-
w znacznym stopniu od rejonu skóry (zob. ryc. B
sowaniu podstawowe znaczenie ma efekt:
9.6-2) oraz postaci galenowej preparatu. W przy-
■ przeciwzapalny, padku maści najczęściej wchłania się więcej sub-
stancji czynnej niż z kremu czy żelu. Ogólnie
■ antyproliferacyjny (antymitotyczny).
wchłonięciu ulega 1–10% substancji czynnej, która
na wiele dni gromadzi się w warstwie rogowej na-
Ponadto wykorzystuje się działanie:
skórka. Wiązania estrowe już w skórze ulegają częś-
■ immunosupresyjne, ciowemu rozkładowi hydrolitycznemu. Wchłonięta
MUTSCHLER-2009.indd 777 2010-01-07 22:14:43
778 Skóra
Tabela B 9.6-1. Glukokortykosteroidy miejscowe, podział według Niednera
Wzór strukturalny Nazwa Preparat handlowy Dawkowanie
międzynarodowa (%)
I. Słabe działanie
O hydrokortyzon Ebenol, 0,25-0,5
CH2OH Fenistil Hydrocort,
CH3 Hydrogalen,
HO OH
CH3 H
Systral Hydrocort
H H
O
II. Średnie działanie
O CH2Cl butanal klobetazonu Emovate 0,05
CH3 O CH3
O
CH3 H O
CH3
F H
O
O 21-piwalan klokortolonu Kaban, 0,1
O
Kabanimat
CH3
CH3 O
HO CH3
CH3
CH3 H CH3
Cl H
O
F
O 21-piwalan flumetazonu Cerson, 0,02
O CH3 LOCACORTEN
CH3 O
HO OH CH3
CH3
CH3 H CH3
F H
O
F
O 21-octan fluprednidenu Decoderm 0,1
O
CH3 O CH3
HO OH
CH3 H CH2
F H
O
O aceponat hydrokortyzonu Retef AP 0,1
O
CH3 O CH3
HO O
CH3 H CH3
O
H H
O
MUTSCHLER-2009.indd 778 2010-01-07 22:14:43
Skóra 779
Tabela B 9.6-1. Glukokortykosteroidy miejscowe, podział według Niednera (kontynuacja)
Wzór strukturalny Nazwa Preparat handlowy Dawkowanie
międzynarodowa (%)
II. Średnie działanie
O buteprat hydrokortyzonu Pandel 0,1
O
Skóra
CH3
CH3 O
HO O CH3
CH3 H
O
H H B9
O
17-butaran hydrokortyzonu Alfason, 0,1
O CH2OH Laticort
CH3
HO O CH3
CH3 H
O
H H
O
O CH2OH acetonid triamcinolonu Delphicort, 0,1
CH3
Korticoid-ratiopharm,
CH3 O Volon,
HO CH3 Volonimat
CH3 O
H
H
F H
O
III. Silne działanie
O amcynonid Amciderm 0,1
CH3
O O
CH3 O
HO
CH3 O
H
H
F H
O
17, 21-dipropionian Diprosis, 0,1
O betametazonu Diprosone
O CH3
CH3 O
HO O
CH3
CH3 H O
CH3
F H
O
O 17-walerianian BEMON, 0,1
CH2OH betametazonu Betnesol-V,
CH3 O Celestan-V,
HO CH3
Cordes-Beta
CH3 H O
CH3
F H
O
MUTSCHLER-2009.indd 779 2010-01-07 22:14:43
780 Skóra
Tabela B 9.6-1. Glukokortykosteroidy miejscowe, podział według Niednera (kontynuacja)
Wzór strukturalny Nazwa Preparat Dawkowanie
międzynarodowa handlowy (%)
III. Silne działanie
O CH2OH dezoksymetazon Topisolon 0,25
CH3
HO
CH3 H CH3
F H
O
O walerianian Nerisona 0,1
O CH3 21-diflukortolonu
CH3 O
HO
CH3 H CH3
F H
O
F
O CH2OH acetonid fluocynolonu Flucinar, 0,25
CH3 O CH3 Jellin
HO CH3
CH3 O
H
H
F H
O
F
O fluocynonid Topsym 0,05
CH3
O O
CH3 O CH3
HO CH3
CH3 O
H
H
F H
O
F
O S CH2F 17-propionian flutikazonu Flutivate 0,05
CH3 O C2H5
HO
CH3 H O
CH3
F H
O
F
O aceponat Ecural 0,1
O metyloprednizolonu
CH3 O CH3
HO O
CH3 H CH3
O
H H
O
CH3
MUTSCHLER-2009.indd 780 2010-01-07 22:14:44
Skóra 781
Tabela B 9.6-1. Glukokortykosteroidy miejscowe, podział według Niednera (kontynuacja)
Wzór strukturalny Nazwa Preparat handlowy Dawkowanie
międzynarodowa (%)
III. Silne działanie
O CH2Cl mometasonfuroat Ecural 0,1
Skóra
CH3 O
HO O
CH3 H O
CH3
Cl H
O B9
O prednikarbat Dermatop, Prednitop 0,25
O CH3
CH3 O
HO O O CH3
CH3 H
O
H H
O
IV. Bardzo silne działanie
O 17-propionian klobetazolu Clobegalen, 0,05
CH2Cl
CH3
Dermoxin,
O Karison
HO CH3
CH3 H O
CH3
F H
O
substancja wydzielana jest przede wszystkim przez Tabela B 9.6-2. Skuteczność glukokortykosteroidów w miejsco-
nerki. wym leczeniu dermatoz
Szybkie Często powolne
Wskazania i dawkowanie. Miejscowe leczenie glu- wyleczenie wyleczenie
kokortykosteroidami jest wskazane w wielu chorobach
I. Wyprysk
skóry, przede wszystkim w niezakażonym wyprysku,
łuszczycy, chorobach autoimmunologicznych (toczeń alergiczny
wyprysk kontaktowy
rumieniowaty, choroby pęcherzowe), zapalnych foto-
dermatozach i chorobach świerzbiączkowych (zob. wyprysk atopowy u dzieci wyprysk atopowy
tab. B 9.6-2). W celu uniknięcia działań niepożąda- u dorosłych
nych należy zawsze wybierać kortykosteroidy o moż- wyprysk łojotokowy
liwe najsłabszym działaniu. Dlatego w atopowym za-
II. Łuszczyca
paleniu skóry należy stosować glukokortykosteroidy
o średniej sile działania. Należy przy tym możliwie łuszczyca zwykła łuszczyca dłoni,
(zwłaszcza zmiany na twarzy podeszew stóp,
szybko przechodzić z leczenia ciągłego (smarowanie i w okolicach wyprzeniowych) łokci i kolan
jeden lub kilka razy dziennie) na leczenie przerywane III. Inne choroby skóry
lub naprzemienne (stosowanie na przemian preparatu
glukokortykosteroidowego i odpowiedniego prepara- oparzenie słoneczne łysienie plackowate
tu zawierającego tylko podłoże, tzw. maści lub kremu
bazowego, np. co drugi dzień lub tydzień). świąd okolic płciowych przerośnięte blizny
świerzbiączka pospolita toczeń rumieniowaty
Działania niepożądane. Zależnie od czasu trwania przewlekły
leczenia, siły działania glukokortykosteroidu oraz
MUTSCHLER-2009.indd 781 2010-01-07 22:14:44
782 Skóra
%
– stosowanych pierwotnie w medycynie transplan-
50 tacyjnej oraz innych substancji o podobnym mecha-
nizmie działania (np. pimekrolimus). Wymienione
hamowanie produkcji IL-1α w keratynocytach
PC immunoterapeutyki zostały szczegółowo opisane
40 w rozdz. B 13.4, dlatego tutaj zajmiemy się jedynie
BMV
ich zastosowaniem w dermatologii.
Masa cząsteczkowa tych cyklicznych peptydów
30 lub makrocyklicznych laktonów (> 800) znacz-
PD nie utrudnia resorpcję skórną, ponieważ powyżej
BM DM masy 500 Da resorpcja przez nieuszkodzoną skórę
20 jest znacznie utrudniona.
Wskazania do zastosowania inhibitorów NF-AT
zależą od siły działania oraz drogi aplikacji. Miejsco-
10 % we leczenie takrolimusem i pimekrolimusem, któ-
0 10 20 30 40
re nie wiąże się z silnymi układowymi działaniami
hamowanie produkcji IL-1α w fibroblastach
niepożądanymi można stosować w lekkich do umiar-
Ryc. B 9.6-1. Stosunek korzyści do ryzyka przy miejscowym
kowanie ciężkich (tylko takrolimus) formach ato-
stosowaniu glukokortykosteroidów: oznaczenie in vitro na pod- powego zapalenia skóry. Takrolimus jest stosowany
stawie hamowania produkcji IL-1 w keratynocytach (korzyści) w postaci 0,03- lub 0,1-procentowej maści, nato-
i fibroblastach (ryzyko). Najlepsze glukokortykosteroidy znaj- miast pimekrolimus jako 1-procentowy krem, o ile
dują się w górnym lewym kwadrancie (silna inhibicja w keraty- nie jest możliwe inne leczenie. Dotyczy to przede
nocytach = silnie przeciwzapalne, słaba inhibicja fibroblastów
= słabo antyproliferacyjne, mało atrofogenne). BM – betame-
tazon, BMV – 17-walerianian betametazonu, DM – dezoksyme-
tazon, PC – prednikarbat, PD – prednizolon (na podst. Gysler owłosiona skóra głowy 3,5%
i Schäfer-Korting).
czoło 6%
powieka 42%
sposobu stosowania miejscowe działania niepożą-
plecy 1,7%
dane mogą obejmować zaniki skórne (np. w posta-
ci rozstępów), teleangiektazje, okołoustne zapalenie
skóry oraz zaburzenia barwnikowe. Niekiedy wystę-
pują plamica, zapalenie mieszków włosowych (po
opatrunkach okluzyjnych) oraz uczulenie kontakto-
we. Steroidy łagodne nie powodują nasilenia zaniku przedramię 1,1%
skóry.
Istnieje ponadto ryzyko układowych działań niepo-
żądanych w formie supresji osi przysadkowo-nadner-
powierzchnia dłoni 0,8%
czowej lub zespołu Cushinga, zwłaszcza podczas sto-
sowania silnych glukokortykosteroidów, smarowania
dużych powierzchni ciała oraz przy uszkodzeniach moszna 42%
warstwy rogowej naskórka. U dzieci może czasami
dochodzić do zahamowania wzrostu.
Za najlepiej tolerowane, jednak mało efektywne,
uważa się dostępne bez recepty preparaty hydrokor-
tyzonu o stężeniu 0,25%.
9.6.2.2. Inhibitory czynnika jądrowego
pobudzonych limfocytów T (NF-AT) podeszwa stopy 0,14%
W zapalnych chorobach skóry pochodzenia immuno-
patologicznego można stosować leczenie za pomocą Ryc. B 9.6-2. Penetracja hydrokortyzonu w skórze ludzkiej
immunosupresantów – cyklosporyny i takrolimusu (według Feldmanna i Maibacha).
MUTSCHLER-2009.indd 782 2010-01-07 22:14:45
Skóra 783
wszystkim uszkodzeń przy stosowaniu glukokorty- o działaniu chłodzącym (kremy, mleczka), w celu
kosteroidów w obszarach zwiększonego ryzyka atro- ograniczenia lub likwidacji świądu stosuje się inne
fii skóry i teleangiektazji (np. na twarzy). Za pomocą środki, takie jak:
immunosupresantów można leczyć dzieci w wieku
■ mocznik powodujący zwiększoną zdolność wiąza-
powyżej dwóch lat.
nia wody w warstwie rogowej naskórka,
Układowe (doustne) podawanie cyklosporyny jest ■ środki powierzchniowo znieczulające, np. polido-
wskazane w najcięższych, opornych na leczenie kanol, obniżające wrażliwość pobudzonych ner-
Skóra
formach łuszczycy i atopowego zapalenia skóry. wów skórnych.
Szczególnie dobrze działa ona na pacjentów z łusz-
czycą płytkową. Przeciwświądowo działa również podawana miejsco-
Optymalne dawkowanie wynosi 2,5 mg/kg w dwóch wo izoprenalina, jej tolerancja jest jednak gorsza niż B9
osobnych dawkach. w przypadku wcześniej wymienionych leków.
Alternatywnie można rozważyć układowe podanie
leków przeciwhistaminowych blokujących receptor
9.6.2.3. Inne substancje o działaniu H1 (np. hydroksyzyny).
przeciwzapalnym
Oprócz glukortykosteroidów w leczeniu nieinfek-
cyjnych stanów zapalnych skóry stosuje się również 9.6.4. Kryteria leczenia zapalnych
inne substancje. Należą do nich przede wszystkim ta- chorób skóry
kie fitofarmaceutyki, jak rumianek i przetwory ocza-
ru, jak również syntetyczny garbnik – tamol, poli- Atopowe zapalenie skóry. Leczenie atopowego za-
kondensat fenolowo-formaldehydowo-mocznikowy. palenia skóry wymaga stosowania określonych środ-
Skuteczność tych środków nie jest jednak wystarcza- ków zapobiegawczych przez poszczególnych pacjen-
jąca w przypadku silnego zapalenia. tów. Należą do nich:
Ze względu na wysokie ryzyko działań niepożądanych (po- ■ przebywanie w otoczeniu ubogim w alergeny,
tencjalnie kancerogennych) nie poleca się stosowania pre- ■ podstawowe leczenie objawowe z prawidłowym
paratów z dziegciu, jak również słabo działającego, ale czę-
sto prowadzącego do reakcji alergicznych niesteroidowego dobraniem nośnika (emulsje woda/olej lub olej/
leku przeciwzapalnego – bufeksamaku. woda), szczególnie z kwasami nienasyconymi,
Specjalnie do leczenia dziedzicznego obrzęku naczynio- np. z kwasem linolowym,
ruchowego zaprojektowano antagonistę receptora bradyki-
niny typu 2 – ikatybant. ■ emulsje zawierające mocznik,
■ zastosowanie specyficznych substancji czynnych,
takich jak polidokanol, sztuczne garbniki (w okoli-
cach wyprzeniowych, na dłoniach i stopach), kwas
NH salicylowy (w rejonach silnie rogowaciejących),
OH
środki przeciwgrzybicze (szczególnie w obecności
H3C(CH2) 3 O drożdżaków), kwas fuzydynowy lub erytromycyna
O
przy zliszajowaceniu.
bufeksamak
Przy manifestacji objawów skórnych wskazane
jest stosowanie miejscowych glukokortykosteroidów
klasy I i II, przy egzemie na powierzchni dłoni rów-
nież glukokortykosteroidów klasy III albo miejsco-
wych inhibitorów NF-AT (zob. powyżej).
Tylko objawy występujące na dużej powierzchni
9.6.3. Leki przeciwświądowe wymagają stosowania układowego leczenia gluko-
kortykosteroidami (leczenie dużymi dawkami).
Świąd jest objawem wielu chorób skóry. Dla chorego Atopowe egzemy mają skłonność do nawrotów,
jest on często bardziej dokuczliwy od bólu. W przy- często już w ciągu 4 tygodni po wyleczeniu. Możliwa
padku gdy nie wystarczają leki na bazie podłoży i znacznie bardziej efektywna niż opisana tera-
MUTSCHLER-2009.indd 783 2010-01-07 22:14:46
784 Skóra
pia podstawowa jest profilaktyka z zastosowaniem Pokrzywka. Poza unikaniem czynników wyzwa-
0,005-proc. kremu z propionianem flutikazonu dwa lających (np. leków, zimna, niektórych produktów
razy w tygodniu. spożywczych) do środków terapeutycznych przy po-
Przy silnych objawach świądu można podawać krzywce należy stabilizacja komórek tucznych, ha-
leki przeciwhistaminowe H1, w ciągu dnia takie, mowanie działania mediatorów na narządy docelowe
które nie mają właściwości uspokajających. Można przez leki przeciwhistaminowe H1 (najczęściej w du-
równie brać pod uwagę fototerapię, przy czym należy żych dawkach), jak również – jako ostateczny środek
zachować szczególną ostrożność w przypadku dzieci – układowe zastosowanie glukokortykosteroidów.
poniżej 12 lat.
9.7. Choroby infekcyjne skóry i ich leczenie
9.7.1. Podstawy patofizjologiczne ki otoczonych wąską obwódką zapalną. Pokrywy
pęcherzyków szybko pękają, a ich dno pokrywa się
grubym strupem o kolorze miodowożółtym. Ogniska
są ostro odgraniczone i goją się, nie pozostawiając
9.7.1.1. Piodermie
blizny. Zmiany są zazwyczaj umiejscowione na twa-
rzy. Zakaźność choroby jest wysoka.
Piodermie są to choroby skóry spowodowane przez Drugą częstą formą jest liszajec gronkowcowy,
gronkowce i paciorkowce, przede wszystkim przez w którym pęcherze są znacznie większe, a ich pokry-
Staphylococcus aureus i Streptococcus pyogenes. wy bardziej wytrzymałe. Po ich usunięciu ukazują się
U niektórych ludzi są one stale obecne na błonach zaczerwienione nadżerki, które wyglądają jakby były
śluzowych nosa i gardła. Do choroby infekcyjnej do- polakierowane.
chodzi w przypadku obniżenia odporności (np. nie-
dożywienie, zespół niedoboru przeciwciał lub gluko-
kortykoterapia) albo niewielkich zranień. W zależ-
9.7.1.2. Grzybice skóry
ności od umiejscowienia i rozległości rozróżnia się
piodermie przymieszkowe oraz powierzchniowe.
Wśród grzybiczych zakażeń skóry należy wymienić:
Piodermie przymieszkowe. Zazwyczaj wywołują ■ dermatofitozy (tinea),
je gronkowce i występują w obrębie mieszków wło-
■ drożdżyce (kandydiozy),
sowych oraz gruczołów potowych.
■ łupież pstry (pityriasis versicolor).
Czyrak jest głęboko penetrującym zapaleniem miesz-
ka włosowego i tkanek otaczających przebiegającym Dermatofitozy. Ten termin obejmuje wszystkie istot-
z tworzeniem ropnia. Rozpoczyna się jako krostka ne zakażenia grzybami dermatofitowymi skóry, pa-
przymieszkowa albo od początku powstaje w głębszych znokci i włosów. Najczęstszymi czynnikami choro-
warstwach skóry i przyjmuje postać bolesnego guzka. botwórczymi są Trichophyton rubrum, Trichophyton
mentagrophytes i Microsporum canis. Typowe dla
W przebiegu ropnego zapalenia gruczołów poto- dermafitozy są pojedyncze lub mnogie ogniska ru-
wych (hidradenitis suppurativa axillaris) w dołach mieniowe ze złuszczającym się i zazwyczaj ostro od-
pachowych tworzą się skupiska guzków skórnych graniczonym brzegiem. W obrębie tych ognisk mogą
wielkości orzecha laskowego, których ropna zawar- się pojawiać krostki, które czasami wskazują na wtór-
tość przdostaje się na zewnątrz. Ten obraz chorobo- ne zakażenie bakteryjne.
wy uważa się obecnie za szczególną postać trądziku Szczególnie częsta jest grzybica stóp (tinea pedis).
(acne inversa). Wspólne dla poszczególnych form (międzypalcowej,
rogowaciejącej i potnicowej) jest okresowe pojawia-
Piodermie powierzchniowe. Do tych chorób, wy- nie się świądu oraz częstsze występowanie w lecie.
stępujących przeważnie w dzieciństwie, należy m.in. Zakażeniom sprzyja noszenie butów nieprzepuszcza-
liszajec paciorkowcowy. Choroba rozpoczyna się jących pary wodnej. Do zakażenia dochodzi w miej-
od pojawienia się pęcherzyków wielkości łebka szpil- scach, gdzie jest wilgoć, np. na basenach.
MUTSCHLER-2009.indd 784 2010-01-07 22:14:46
Skóra 785
Drożdżyca. Należące do grupy grzybów drożdżopo- Typowym umiejscowieniem półpaśca jest tułów, stąd
dobnych gatunki z rodzaju Candida albicans wywo- też wywodzi się jego nazwa. Poważną komplikacją
łują zakażenia o zróżnicowanym obrazie z rumienia- jest neuralgia popółpaścowa pojawiająca się często
mi, krostami oraz nadżerkami. Poza skórą zakażeniu po wyleczeniu pęcherzyków w efekcie niewystarcza-
mogą ulegać także błony śluzowe. jącej terapii antywirusowej.
Postać wyprzeniowa występuje typowo w okolicy
narządów płciowych i charakteryzuje się rumieniem Opryszczka zwykła. W zakażeniach wirusami z gru-
w kolorze czerwonego wina, o drobno ząbkowanym py Herpes simplex obserwuje się grupy pęcherzyków
Skóra
brzegu i obecnością niewielkich satelit. Szczególnie wielkości łebka szpilki na zaczerwienionym podłożu,
częsta jest drożdżyca jamy ustnej (tzw. pleśniawka) które w dalszym przebiegu choroby mogą przekształ-
u niemowląt oraz osób w podeszłym wieku. W prze- cać się w krostki oraz ulegać zlewaniu. Uczucie roz-
biegu tego zakażenia język, dziąsła oraz podniebie- pierania powiązane ze świądem i mrowieniem jest ty- B9
nie pokryte są drobnymi plamkami białawego nalotu powe w fazie powstawania wykwitów. W związku
na zaczerwienionym podłożu. Na drożdżycę choru- z niewielką immunogennością antygenów wirusa
ją szczególnie często osoby zakażone wirusem HIV ograniczone jest wytwarzanie przeciwciał odpor-
i chore na cukrzycę. nościowych, dlatego częste są nawroty choroby.
Zakażenie lokalizuje się szczególnie często w miej-
Łupież pstry. Zakażenie wywoływane jest przez scach przejścia skóry w błonę śluzową (opryszczka
grzyb Malassezia furfur (Pityrosporum ovale) i za- wargowa i nosowa powodowane najczęściej przez
zwyczaj obejmuje górną połowę tułowia. W przebie- wirusy Herpes simplex typu I oraz opryszczka narzą-
gu zakażenia pojawiają się żółtawe lub brązowawe dów płciowych wywoływana przez wirusy typu II).
plamy wielkości od ziarna soczewicy do monety,
w obrębie których stwierdza się otrębiaste złuszcza- Brodawki wirusowe. W przebiegu brodawek wiruso-
nie albo białawe plamy. wych dochodzi do przerostu naskórka spowodowane-
go działaniem ludzkiego wirusa brodawczaka (HPV,
wirusy Papova). Ogólnie znane są brodawki pospolite
(verrucoe vulgares), okrągłe twory wielkości od łeb-
9.7.1.3. Zakażenia wirusowe ka szpilki do ziarna grochu o szorstkiej, często rów-
nież poletkowanej powierzchni, które mogą występo-
Do najczęstszych zakażeń wirusowych skóry należą: wać pojedynczo lub w grupach na rękach, stopach lub
na głowie.
■ zakażenia Varicella-Zoster, ospa wietrzna (varicel- Brodawki płaskie młodzieńcze (verrucae planae
la) oraz półpasiec (zoster), juveniles) poza rękami i stopami występują przważ-
nie na skórze twarzy w postaci płaskich, zazwyczaj
■ opryszczka (Herpes simplex) wywoływana przez
słabo widocznych grudek.
wirusy z grupy Herpes simplex,
Kłykciny kończyste (condylomata acuminata)
■ brodawki powodowane przez ludzkie wirusy bro- występują zazwyczaj u dorosłych w rejonie odby-
dawczaka (Papilloma). tu i narządów płciowych. Na początku są to grudki
wielkości łebka szpilki, które stopniowo zlewają się
Ospa wietrzna i półpasiec. Pierwotne zakażenie wi- w większe blaszki i mogą tworzyć kalafiorowate two-
rusem Varicella-Zoster powoduje ospę wietrzną, bar- ry. U kobiet może dojść do przemiany tej choroby
dzo zakaźną chorobę dziecięcą, która charakteryzuje przenoszonej drogą płciową w raka szyjki macicy.
się jednoczesnym występowaniem grudkopęcherzy- Zdarza się to przede wszystkim w przypadku zakaże-
ków w różnym stadium rozwoju. nia wirusem typu HPV-16 i HPV-18. W przyszłości,
Półpasiec jest efektem wtórnego zakażenia, wy- dzięki stosowaniu od niedawna szczepień, częstość
stępującego zazwyczaj na skutek reaktywacji laten- zachorowań powinna zmaleć.
tnego wirusa przetrwałego w obwodowych włóknach
nerwowych. W obszarze unerwienia jednego lub
kilku nerwów rdzeniowych zaczynają się pojawiać
ułożone połowicznie lub w obrębie poszczególnych 9.7.1.4. Pasożytnicze choroby skóry
segmentów pęcherzyki na podłożu zaczerwienio-
nej i obrzękniętej skóry, którym mogą towarzyszyć Świerzb (scabies) wywoływany jest przez samice roz-
bóle neuropatyczne. Po pewnym czasie pęcherzy- tocza (Sarcoptes scabiei), które w warstwie rogowej
ki zasychają i goją się, często pozostawiając blizny. naskórka drążą ślepo zakończone korytarze. Choroba
MUTSCHLER-2009.indd 785 2010-01-07 22:14:46
786 Skóra
obejmuje najczęściej przestrzenie międzypalcowe, Działaniem niepożądanym mogą być podrażnienia
fałdy pachowe, brodawki sutkowe i męski członek. skóry.
W związku z silnym świądem i drapaniem dochodzi U kobiet ciężarnych i karmiących oraz u małych
do wtórnych zakażeń bakteryjnych. dzieci świerzb można leczyć za pomocą benzoesa-
Wszy głowowe, ubraniowe i łonowe są ssący- nu benzylu (emulsja przeciwświerzbowa). Roztwór
mi krew ektopasożytami człowieka powodującymi (25-proc. dla dorosłych i 10-proc. dla dzieci) nanosi
wszawicę (pediculosis). Przenoszenie pasożytów na- się na całą powierzchnię ciała przez trzy kolejne dni.
stępuje podczas kontaktów pomiędzy ludźmi.
Oprócz leczenia świerzbu, benzoesan benzylu stosuje się
do likwidacji roztoczy kurzu domowego w meblach po-
krytych tapicerką, dywanach itp. Za pomocą takich działań
9.7.2. Substancje czynne stosowane można uzyskać poprawę w przypadku alergii na roztocze.
w leczeniu zakażeń skóry
9.7.2.1. Środki odkażające
O
Substancje mające zastosowanie m.in. w leczeniu O
zakażeń bakteryjnych skóry omówiono w rozdz. B
11.1.
benzoesan benzylu
9.7.2.2. Leki przeciwgrzybicze
Substancje stosowane w leczeniu zakażeń grzybiczych Jako środki przeciw wszom mają zastosowanie
zostały przedstawione w rozdz. B 11.4. Szczególnymi permetryna oraz alletryna. Kolejny środek na bazie
postaciami galenowymi stosowanymi w grzybicach pyretryny zawiera ekstrakt złocienia (Pyrethrum).
skóry są np. preparat liposomowy z ekonazolem oraz Preparaty nanosi się na umytą pokrytą włosami skórę
połączenie bifonazolu z mocznikiem stosowane w le- głowy albo inne zaatakowane miejsca na ciele, a na-
czeniu grzybicy paznokci. Pod wpływem mocznika stępnie zmywa po upływie podanego czasu działania.
w opatrunku okluzyjnym dochodzi do rozpuszczenia Leczenie należy powtórzyć po 8–10 dniach, jednak
zmienionej chorobowo płytki paznokciowej w ciągu w przypadku permetryny najczęściej już jednokrotne
1–2 tygodni, co ułatwia penetrację leku do zakażo- zastosowanie zwalcza pasożyty.
nego łożyska paznokcia. W przypadku grzybicy stóp
możliwe jest jednokrotne zastosowanie bioadhezyj-
nego roztworu terbinafiny. 9.7.2.4. Środki stosowane
Leczenie łupieżu pstrego różni się od leczenia w leczeniu brodawek płciowych
innych grzybic. Tradycyjnie stosuje się jeszcze siar-
czek selenu w formie pasty. Obecnie jednak również
W leczeniu brodawek płciowych i okołoodbytowych
w tej chorobie preferowane są leki przeciwgrzybicze
(Condylomata acuminata) stosowano dotychczas
z grupy imidazoli (np. ketokonazol, często w postaci
25-proc. alkoholowy roztwór podofiliny, wyciągu
szamponów.
z Podophyllum peltatum lub główną substancję czyn-
ną podofilotoksynę.
Imikwimod charakteryzuje się nowym sposobem
9.7.2.3. Leki przeciwpasożytnicze działania. Ten miejscowy immunomodulator wzmaga
odporność przeciwwirusową gospodarza poprzez na-
W leczeniu świerzbu stosuje się: siloną syntezę cytokin. Mechanizm działania polega
na stymulacji receptorów Toll-podobnych, głównie
■ permetrynę,
receptora TLR-7 (zob. ryc. B 9.7-1). Receptory zlo-
■ alletrynę. kalizowane na powierzchni komórek prezentujących
antygen (komórek dendrytycznych, makrofagów,
W większości przypadków wystarczające jest jedno- monocytów) są stymulowane przez odpowiednie
krotne leczenie zaatakowanych obszarów skóry. struktury mikroorganizmów (np. lipopolisacharydy)
MUTSCHLER-2009.indd 786 2010-01-07 22:14:46
Skóra 787
I imikwimod
I
TLR-7
T
Y
R kinazy IκB
Skóra
P P
IκB-α IκB-α
p50 p65 proteoliza B9
jądro
komórkowe p50 p65 NF-κB
p50 p65
IFN-α ↑
mRNA TNF-α ↑
IL-2 ↑
efekt antywirusowy
Ryc. B 9.7-1. Mechanizm działania imikwimodu. Stymulacja receptora Toll-podobnego 7 (TLR-7) za pośrednictwem NFκB prowadzi
do migracji i dojrzewania limfocytów prezentujących antygen. Uwolnione cytokiny aktywują następnie limfocyty T, m.in. limfocyty T
pomocnicze typu 1 (zmodyf. według Saudera). IκB-α – inhibitor aktywacji NFκB.
i należą do wrodzonej odpowiedzi immunologicznej, biet niż u mężczyzn (72% w stosunku do 36% wy-
ale wspomagają także odpowiedź nabytą. Aktywacja leczeń w ciągu 16 tygodni), co wskazuje na różnice
TLR-7 prowadzi do uwalniania INF-α, TNF-α i in- w penetracji leku.
nych cytokin inflamatoryjnych aktywowanych przez
NF-κB. W ten sposób aktywacji ulegają limfocyty T Badania kliniczne wykazały, że imikwimod jest również
pomocnicze typu 1 oraz limfocyty T cytotoksyczne, skuteczny w leczeniu raka jasnokomórkowego skóry (ke-
ratozy aktyniczne) – 45% całkowitych oraz 14% częścio-
niedojrzałe komórki dendrytyczne wędrują do wę- wych wyleczeń uszkodzeń spowodowanych keratozą akty-
złów limfatycznych i rozwijają się w dojrzałe komór- niczną.
ki Langerhansa prezentujące antygen.
Lek stosuje się 3 razy w tygodniu. Skuteczność Lek jest dobrze tolerowany, a jako działanie niepożą-
w przypadku brodawek płciowych jest większa u ko- dane może wystąpić miejscowe podrażnienie.
MUTSCHLER-2009.indd 787 2010-01-07 22:14:46
788 Skóra
9.8. Choroby nowotworowe skóry
i fotostarzenie, leczenie i profilaktyka
9.8.1. Podstawy patofizjologiczne Czerniak złośliwy. Istnieje kilka form czerniaka,
np. czerniak guzkowy, czerniak szerzący się po-
9.8.1.1. Nowotwory skóry wierzchownie, czerniak o typie plamy soczewico-
watej złośliwej. Zmiany nowotworowe, zazwyczaj
w kolorze od ciemnego brązu do czerni, rozwija-
Najważniejsze złośliwe nowotwory skóry powstają
ją się najczęściej w obrębie zdrowej skóry, jednak
w wyniku transformacji komórek nabłonka lub mela-
w 30% przypadków powstają na bazie istniejącego
nocytów. Należą do nich:
od dawna znamienia (znamię z komórek barwniko-
■ nabłoniak podstawnokomórkowy, wych). Ze względu na znaczną i wcześnie występu-
jącą skłonność do przerzutów drogą chłonną i/lub
■ rak kolczystokomórkowy,
naczyń krwionośnych (głównie do płuc i mózgu)
■ czerniak złośliwy. czerniaka zalicza się do nowotworów o szczególnie
dużej złośliwości. Może się do tego przyczyniać fakt,
Wszystkie wymienione nowotwory są najczęściej in- że prawidłowe melanocyty są komórkami migrujący-
dukowane przez promieniowanie ultrafioletowe (in- mi, nietworzącymi struktur tkankowych ani połączeń
tensywne działanie słońca na skórę z częstymi opa- międzykomórkowych.
rzeniami słonecznymi). Rosnący dobrobyt w krajach Obecnie zapadalność na tę chorobę w Europie
uprzemysłowionych i wynikające stąd zmiany w spo- wynosi 6–14 nowych przypadków na 100 000 miesz-
sobie spędzania wolnego czasu powodują wzrost kańców rocznie. W przeciwieństwie do wcześniej
liczby tych nowotworów. omówionych nowotworów skóry, które rozwijają się
głównie u osób starszych, czerniak występuje naj-
Nabłoniak podstawnokomórkowy. Ten stosun- częściej u ludzi w średnim wieku.
kowo często występujący nowotwór skóry po-
wstaje z warstwy komórek podstawnych naskórka
oraz mieszków włosowych. Jest to powoli rosnący,
płaski lub zagłębiony w środkowej części guzek 9.8.1.2. Uszkodzenia świetlne,
o zwartej konsystencji. Widać w nim teleangiektazje fotostarzenie
(przeświecające przez naskórek drobne naczynka
wypełnione krwią). Nabłoniaki podstawnokomór- Ostre uszkodzenia skóry (dermatits solaris, oparzenie
kowe rosną naciekając i niszcząc otaczające tkanki, słoneczne) są efektem absorpcji światła UV, szcze-
nie dając jednak najczęściej przerzutów (nowotwo- gólnie UV-B o długości fali w zakresie 280–320 nm.
ry miejscowo złośliwe). Występują przede wszyst- Prowadzi to do fotochemicznych reakcji wtórnych,
kim u osób ze skórą wrażliwą na słońce. Poza tym przy czym wolne rodniki mogą wchodzić w reakcje
rozwój nabłoniaków mogą również prowokować ze składnikami DNA. Na ryc. B 9.8-1 przedstawio-
związki arsenu. no pojawianie się rumienia w zależności od długości
fali.
Rak kolczystokomórkowy skóry. Oprócz światła W niektórych chorobach skóry spowodowanych
słonecznego może go również indukować promienio- działaniem światła (zob. tab. B 9.8-1) poza UV-B
wanie rentgenowskie lub długotrwała terapia PUVA. szkodliwy wpływ wywierają także dłuższe fale UV-A
Nowotwór ten, jeśli nie jest leczony, prowadzi oraz światło widzialne. Niektóre substancje endo-
do śmierci. Rozpoczyna się zwykle jako keratoza ak- genne (np. porfiryny) i egzogenne (np. tetracykliny,
tyniczna (inaczej: rogowacenie słoneczne, stan przed- fenotiazyna, olejek z dziurawca) mogą powodować
rakowy wywołany promieniowaniem). Pierwszym fotouczulenie. Uważa się, że przyczyną jest nasilone
objawem tej coraz częściej występującej choroby powstawanie reaktywnych form tlenu (ROS) spowo-
jest niepozorne, lekko wyniosłe, podobne do bro- dowane działaniem UV-B. ROS aktywują metalopro-
dawki ognisko rogowacenia. Prewalencja w Europie teazy macierzy, które degradują kolagen.
w roku 2000 wynosiła dla ponadsiedemdziesięciolet- Od właściwego (wewnętrznego) starzenia się skó-
nich mężczyzn 34%, a dla kobiet 18%. ry należy odróżnić starzenie się skóry przyspieszone
MUTSCHLER-2009.indd 788 2010-01-07 22:14:47
Skóra 789
unikanie nadmiernego przebywania na dworze w go-
1
dzinach południowych, okrywające ubranie, unikanie
korzystania z solarium, w razie konieczności ko-
rzystanie ze środków chroniących przed działaniem
światła na skórę.
względny efekt rumienia
Skóra
0,5 9.8.2. Leczenie nowotworów skóry
Do leczenia raka skóry stosuje się:
B9
■ fluorouracyl,
■ imikwimod,
280 300 320 350 ■ diklofenak,
długość fali (nm) ■ metyloester kwasu aminolewulinowego.
Ryc. B 9.8-1. Powstawanie rumienia w zależności od długości fali.
W keratozach aktynicznych skuteczny jest inhibitor
syntezy tymidylanu – 5-fluorouracyl, który stosuje
się miejscowo dwa razy dziennie jako krem 5-proc.
przez działanie promieni UV (zewnętrzne). Efektami
Bardzo częstymi działaniami niepożądanymi
tego procesu, nazywanego fotostarzeniem (fotoage-
są miejscowe dolegliwości, takie jak bóle, pieczenie,
ing), poza niektórymi formami raka skóry, jest także
zaczerwienienie.
pojawienie się zmarszczek w młodym wieku oraz sła-
ba, wiotka skóra z plamami pigmentacyjnymi i zgru-
białą powierzchnią. Leczenie ran jest spowolnione.
Skóra postarzała pod wpływem światła jest, w przeci- 9.8.3. Substancje chroniące
wieństwie do naturalnie zestarzałej skóry, zgęstniała
przed działaniem światła
we wszystkich warstwach¸ melanocyty są rozmiesz-
czone nierównomiernie, ilość włókien łączących na-
Do zapobiegania ostrym i przewlekłym uszkodze-
skórek ze skórą właściwą jest tak samo zmniejszona
niom skóry w sytuacji wyraźnego w ostatnich latach
jak ilość kolagenu.
zanikania warstwy ozonowej chroniącej ziemię przed
Najlepszą profilaktyką fotostarzenia jest dbanie
promieniowaniem ultrafioletowym zaleca się ograni-
o ochronę przed promieniami UV, najlepiej poprzez
czenie ekspozycji na słońce (ochronę przed światłem
słonecznym).
Tabela B 9.8-1. Zakresy długości fal promieniowania wywołują- Naturalna ochrona przed promieniowaniem pole-
cego poszczególne fotodermatozy (według Bandmanna) ga na zwiększeniu grubości warstwy rogowej naskór-
ka (hiperkeratoza) oraz przede wszystkim na wytwa-
Dermatoza UV-B UV-A Światło IR rzaniu melaniny w skórze. Sztuczna ochrona przed
widzial-
ne promieniowaniem, poza noszeniem odpowiedniego
ubrania, jest możliwa również dzięki stosowaniu sub-
pokrzywka świetlna + + + +
stancji:
wielopostaciowe + +
osutki świetlne ■ pochłaniających światło (chemicznych) lub
fototoksyczne (+) + ■ odbijających światło (fizycznych).
zapalenie skóry
fotoalergiczne + (+) Substancje pochłaniające światło, nazywane rów-
zapalenie skóry nież filtrami przeciwsłonecznymi, dzieli się na różne
porfirie + + rodzaje, w zależności od maksimum absorpcji bądź
pasma absorpcji:
skóra +
pergaminowa ■ filtry UV-B (kwas paraaminobenzoesowy, kwas cy-
namonowy oraz pochodne benzoimidazolu),
MUTSCHLER-2009.indd 789 2010-01-07 22:14:47
790 Skóra
■ filtry UV-A (substancje o strukturze dibenzoilome- – można wystawiać na działanie promieniowania
tanu), słonecznego skórę pokrytą filtrem, zanim pojawi się
rumień. Środki o współczynniku ochrony większym
■ filtry szerokopasmowe (pochodne benzofenonu).
od 15 nazywane są blokerami promieniowania.
W tab. B 9.8-2 zestawiono środki chroniące przed Wadą czystych filtrów UV-B jest to, że ze względu
światłem oraz informacje na temat ich zakresu ab- na zmniejszenie ryzyka oparzenia słonecznego wy-
sorpcji. dłuża się ekspozycja słoneczna. W ten sposób wzra-
Działanie odbijające światło wykazuje szczegól- sta ryzyko uszkodzenia skóry przez typowo mniej
nie dwutlenek tytanu, a także tlenek cynku. szkodliwe promieniowanie UV-A. Dlatego nowo-
czesne środki chronią również przed promieniowa-
Filtry UV-B od dawna są stosowane w preparatach niem UV-A.
chroniących przed światłem. Ich zadaniem jest ab- Przykładem nowoczesnego filtru jest środek
sorpcja promieniowania wywołującego rumień w ta- ochronny zawierający 2-etyloheksyloester kwasu
kim zakresie, że powstaje tylko rumień podprogowy, p-metoksycynamonowego oraz 2-hydroksy-4-metok-
konieczny do powstania pożądanej opalenizny. sybenzofenon. Liposomy stosowane jako nośniki sub-
Działanie ochronne określa się za pomocą współ- stancji ochronnych zapewniają ich obecność w war-
czynnika ochrony. Współczynnik ten określa, ile stwie rogowej przez cały dzień, nawet w przypadku
razy dłużej – w porównaniu ze skórą niechronioną kontaktu z wodą (pocenie, kąpiele itp.).
Tabela B 9.8-2. Substancje stosowane w fotoprotekcji (według Gloora)
Wzór Nazwa chemiczna Zakres absorpcji
chemiczny
O kwas p-aminobenzoesowy i jego estry UV-B
OH
H2N
O ester izoamylowy kwasu p-di- UV-B
CH(CH3) 2 rnetyloaminobenzoesowego (padymat)
O
H3C
N
CH3
O ester 2-etyloheksylowy UV-B (+ UV-A)
kwasu p-metoksycynamonowego
(CH2) 3CH3
O
C2H5
H3CO
HO3S N kwas 2-fenylobenzimidazolo-5-sulfonowy UV-B (+ UV-A)
NH
O O 1-(4-izopropylofenylo)-3-(tert-butylofenylo)- UV-A
-1,3-propanodion (awobenzon)
(H3C) 3C OCH3
OH O 2-hydroksy-4-metoksybenzofenon UV-A i UV-B
(oksybenzon)
H3CO
MUTSCHLER-2009.indd 790 2010-01-07 22:14:48
Skóra 791
9.9. Zaburzenia pigmentacji
9.9.1. Podstawy patofizjologiczne nych zawierających dihydroksyaceton, który łączy
się kowalencyjnie z grupami aminowymi keratyny
lub wolnymi aminokwasami.
Zaburzenia pigmentacji mogą niekorzystnie wpływać
Pacjentom poleca się jednak przede wszystkim
na wygląd, jednak skórę można leczyć za pomocą
staranną ochronę przed promieniowaniem słonecz-
Skóra
substancji wspomagających pigmentację lub depig-
nym, ponieważ dzięki temu depigmentacja jest mniej
mentujących. Do zaburzeń pigmentacji należą:
widoczna.
■ bielactwo idiopatyczne z występowaniem plami-
stych obszarów odbarwionej skóry, B9
■ ostuda, O
HO OH
■ hiperpigmentacja wywołana działaniem ACTH
w chorobie Addisona,
dihydroksyaceton
■ hiperpigmentacja w wyniku stosowania perfum
na skórę eksponowaną na słońce (tzw. berlock-der-
matitis),
■ piegi (wrodzona hiperpigmentacja).
9.9.3. Substancje powodujące
depigmentację
9.9.2. Środki wspomagające
pigmentację W leczeniu przebarwień skóry nabytych lub wro-
dzonych (piegi) stosuje się hydrochinon, szczególnie
Stymulacji wytwarzania barwnika w skórze można w połączeniu z tretynoiną. Hydrochinon odwracalnie
próbować w bielactwie idiopatycznym. W leczeniu hamuje syntezę melaniny w melanosomach. Ponieważ
stosuje się metoksalen, który omówiono w rozdz. B substancja nie działa na już wytworzoną melaninę,
9.4.2.2. Możliwe jest również uzyskanie sztucznego depigmentacja pojawia się dopiero po jej rozkładzie,
zabarwienia skóry za pomocą środków kosmetycz- czyli po 2–4 miesiącach od rozpoczęcia leczenia.
MUTSCHLER-2009.indd 791 2010-01-07 22:14:48
792 Skóra
9.10. Ostre i przewlekłe rany oraz ich leczenie
9.10.1. Podstawy patofizjologiczne żej oraz po przebyciu zakrzepicy żylnej w kończy-
nach dolnych mogą się czasem pojawiać źle gojące
9.10.1.1. Oparzenia się rany. W owrzodzeniu podudzi pojawiają się bla-
de obszary uszkodzonej tkanki na zaczerwienionym
Uszkodzenie tkanki spowodowane działaniem wyso- podłożu.
kiej temperatury nazywane jest oparzeniem. Rozleg-
łość i siła uszkodzenia termicznego zależy od wyso- Owrzodzenie stóp (ulcus plantae). Problematyczne
kości temperatury i czasu jej działania (objawy od- rany podeszwy stopy są objawem zespołu stopy cuk-
mrożeń są bardzo zbliżone do oparzeń). rzycowej.
Stopnie oparzeń. Efekty oparzeń dzieli się na 4 stop-
nie: 9.10.2. Farmakoterapia ostrych
1. stopień – oparzenia z odwracalnym zaczerwie- i przewlekłych ran
nieniem i niewielkim obrzękiem skóry, związany
z uczuciem napięcia i bólem, Podstawę leczenia ran stanowi obecnie stosowa-
nie okładów, tzw. opatrunków hydrokoloidowych.
2. stopień z powstawaniem pęcherzy na skutek dzia-
Ze względu na ich przeważające działanie fizyczne
łania mediatorów zapalnych, które zostają uwol-
wprowadza się je na rynek jako produkty medyczne.
nione z uszkodzonych komórek i powodują wzrost
Odzwierciedlają dotychczasowy paradygmat „lecze-
przepuszczalności naczyń i przenikanie osocza
nia ran na mokro”. Poza preparatami, które nie za-
do tkanek (ekstrawazacja osocza),
wierają substancji czynnych, są też takie z substancją
3. stopień z trwałym uszkodzeniem naskórka i przy- czynną. Bardzo rozpowszechnione jest stosowanie
datków skóry (denaturacja białka oraz nekroza się- działającego przeciwbakteryjnie srebra. Jeśli chce się
gają do skóry właściwej, tkanki podskórnej albo zmniejszyć ból w trakcie zmiany opatrunku, można
jeszcze głębiej), zastosować okład zawierający ibuprofen.
Obecnie rzadziej stosuje się natomiast preparaty
4. stopień, w którym tkanki ulegają nie tylko koa-
enzymatyczne. Do leczenia owrzodzeń, w których
gulacji, ale także zwęgleniu na skutek działania
martwe tkanki razem z ropą i krwią sprzyjają zaka-
znacznej energii termicznej.
rzeniom, stosuje się leczenie miejscowe streptokina-
zą. Uważa się, że proteazy przyspieszają gojenie ran
Rozległość oparzeń. Oprócz stopnia oparzenia decy-
poprzez rozpuszczenie martwych tkanek.
dujące znaczenie dla rokowania ma jego rozległość.
Czynniki wzrostowe, np. czynnik wzrostowy ke-
W przypadku rozległych oparzeń ich skutki nie ogra-
ratynocytów, wspomagają granulację. Palifermina,
niczają się tylko do miejsca oparzenia. Wskutek sil-
analog otrzymywany techniką genową, jest wskaza-
nego bólu, zwiększonego wydzielania amin biogen-
na do leczenia zapalenia błony śluzowej jamy ustnej
nych, utraty osocza oraz zaburzeń mikrokrążenia
przy radio- lub chemioterapii.
rozwija się choroba oparzeniowa, której głównym
objawem jest wstrząs oparzeniowy.
Czynnik wzrostowy trombocytów (platelet-derived
Przy odpowiedniej intensywnej opiece medycz-
growth factor, PDGF) ma zastosowanie w owrzodze-
nej możliwe jest przeżycie oparzeń obejmujących
niu stóp, jednak skuteczność tej terapii jest ograni-
do 70% powierzchni ciała.
czona.
Epitalizację ran wspomaga dekspantenol.
9.10.1.2. Owrzodzenie podudzi i stóp
Owrzodzenie podudzi (ulcus cruris). W związku
z niewydolnością zastawek żyły odpiszczelowej du-
MUTSCHLER-2009.indd 792 2010-01-07 22:14:48
Skóra 793
9.11. Łysienie androgenne i hirsutyzm
Łysienie spowodowane działaniem androgenów ornityny w putrescynę. Ta stymulująca podziały ko-
jest najczęstszą przyczyną utraty włosów zarówno mórkowe poliamina uczestniczy również w regulacji
u mężczyzn, jak i u kobiet. Dotyka ono do 50% męż- wzrostu włosów. Zmniejszenie produkcji putrescyny
czyzn i kobiet powyżej 40 roku życia. odpowiednio redukuje wzrost włosów niezależnie
Skóra
Stosowanie finasterydu u mężczyzn, jako inhibi- od jego przyczyny. Efekt leczenia jest widoczny po 8
tora 5α-reduktazy typu II występującej w komórkach tygodniach, konieczne jest długoterminowe leczenie.
wypustek skórnych produkujących włosy, może spo- Krem nanosi się na skórę twarzy dwa razy dziennie.
walniać lub całkowicie hamować wypadanie wło- Jako działania niepożądane mogą się pojawiać B9
sów. Dawkowanie wynosi 1 mg dziennie. Substancja zapalenia mieszków włosowych i skóry oraz trądzik.
nie wykazuje jednak efektu przekraczającego okres Ze względu na toksyczny wpływ na reprodukcję
terapii. w badaniach na zwierzętach, eflornityna jest przeciw-
Nadmierny zarost na twarzy kobiet może być ob- wskazana w ciąży.
jawem towarzyszącym nasilonej produkcji androge-
nów w ramach policystycznych jajników i poprawia
O
się w wyniku leczenia. Do miejscowego leczenia hir-
sutyzmu, także formy wrodzonej, nadaje się eflorni- H2N OH
tyna. H2N CHF2
Eflornityna. Substancja przeznaczona pierwotnie
eflornityna
do leczenia śpiączki jest nieodwracalnym inhibito-
rem dekarboksylazy ornityny katalizującej przemianę
MUTSCHLER-2009.indd 793 2010-01-07 22:14:48
MUTSCHLER-2009.indd 794 2010-01-07 22:14:49
Witaminy 795
10. Mikroskładniki odżywcze:
witaminy i pierwiastki śladowe
Mikroskładniki odżywcze:
Poza białkami, węglowodanami i tłuszczami, tzw. stępują w niewielkich ilościach, ale są niezbędne dla
witaminy i pierwiastki
makroskładnikami odżywczymi, wykorzystywanymi ważnych do życia funkcji przemiany materii. Należą
do budowy struktur organizmu i uzyskania energii, do nich witaminy i pierwiastki śladowe, określane ter-
pożywienie zawiera także inne składniki, które wy- minem mikroskładniki odżywcze.
10.1. Witaminy
Witaminy są: B 10
dostarczone organizmowi. Z jednej strony te sub-
■ niezbędnymi do życia, stancje są dostarczane wraz z pożywieniem, a z dru-
giej strony niektóre witaminy są w pewnym zakresie
■ organicznymi substancjami działającymi fizjolo-
syntetyzowane przez bakterie jelitowe i wydzielane
gicznie w niewielkich dawkach (rzędu mg lub μg),
do jelit częściowo w postaci wchłanialnej. W przeci-
które
wieństwie do niezbędnych aminokwasów lub kwa-
w ludzkim organizmie albo nie mogą być produko- sów tłuszczowych witaminy nie mają znaczenia ani
wane, albo są wytwarzane jedynie w określonych jako elementy budulcowe, ani jako nośniki energii,
warunkach i w niedostatecznych ilościach (np. przy natomiast biorą udział w procesach katalitycznych
niedoborze światła UV). (jako koenzymy) lub w procesach regulacji (jako
prohormony).
Z powyższych powodów same witaminy lub pre-
Pełnowartościowe pożywienie, które – pominąw-
kursory witamin w postaci prowitamin muszą zostać
szy obszary głodu – jest obecnie dostępne, zawiera
stadium
1 2 3 4 5 6
całkowita ilość w organizmie
stężenie w osoczu krwi i moczu
aktywność enzymów zależnych od witamin
objawy kliniczne
utajony objawy objawy
niedobór niespecyficzne charakterystyczne
stan graniczny
Ryc. B 10.1-1. Stadia niedoboru witaminy (zmodyf. według Brubachera).
MUTSCHLER-2009.indd 795 2010-01-07 22:14:49
796 Witaminy
dostateczne ilości witamin. Osoby, które się prawid- ka niedobór witamin występuje zazwyczaj bardzo
łowo odżywiają, nie muszą przyjmować żadnych rzadko, ponieważ potrzebne ilości witamin są bar-
preparatów witaminowych. Wbrew wypowiedziom dzo małe. Niewłaściwe odżywianie się lub niewłaś-
w licznych reklamach nie istnieją przekonujące dane ciwe przygotowywanie pożywienia, a także palenie
świadczące o tym, że dodatkowe przyjmowanie wi- tytoniu czy nadużywanie alkoholu mogą się jednak
tamin przez osoby stosujące zrównoważoną, bogatą przyczynić do pojawienia się niedoboru. Najczęściej
w witaminy dietę może zapobiegać chorobom lub występuje niedobór witaminy A, D, Bl, B6 i kwasu
nawet je leczyć. Zawartość witamin w środkach foliowego.
spożywczych jest jednak bardzo zmienna i zależy Przyjmowanie niedostatecznych ilości witamin
od warunków produkcji, przechowywania i przygo- prowadzi do ich niedoboru (zob. ryc. B 10.1-1), któ-
towywania. Podczas gotowania niektóre witaminy ry zależnie od nasilenia podzielono na sześć stadiów.
(np. kwas foliowy i witamina C) w dużej mierze Poszczególne stadia nie są od siebie wyraźnie odgra-
ulegają zniszczeniu. U zdrowego dorosłego człowie- niczone.
Tabela B 10.1-1. Witaminy – nazwy, choroby z niedoboru oraz zalecane dawki dzienne według danych Niemieckiego Towarzystwa
Żywienia; w nawiasach podano optymalną dzienną dawkę
Litera Nazwa Choroba Średnie dzienne Potwierdzona
z niedoboru zapotrzebowanie (mg) nieszkodliwa
dawka dzienna (mg)
dorośli dzieci
I. Witaminy rozpuszczalne w tłuszczach
A retinol kseroftalmia, 1 (-3) 1 7,51)
hiperkeratoza
D kalcyferol krzywica 0,005 0,0125 0,025-0,05
E tokoferol nieznana 15 6-10
K zaburzenia krzepnięcia krwi 0,06 0,01-0,06
II. Witaminy rozpuszczalne w wodzie
B1 aneuryna, tiamina beri-beri, zapalenie 1,2 0,3-1,2 110
wielonerwowe
B2 ryboflawina zapalenie rogówki, 1,5 0,4-1,4 650
zapalenie skóry i in.
B6 pirydoksyna drgawki padaczkopodobne, 1,6 0,3-1,6 2002)
zapalenie skóry
amid kwasu pelagra 15 6-15 700-1400
nikotynowego,
nikotynamid, niacyna
kwas foliowy niedokrwistość megaloblastyczna, 0,4 0,03-0,3 8,16
w ciąży większa częstość
wad rozwojowych
kwas pantotenowy zespół piekących stóp 10 6 1000
biotyna zapalenie skóry nieznana 140
B12 kobalamina niedokrwistość złośliwa 0,003 0,0005-0,003 1
(czynnik
zewnątrzpochodny)
C kwas askorbinowy szkorbut 100 (-150) 35-45 5000
(witamina
przeciwszkorbutowa)
1) nie dotyczy kobiet ciężarnych 2) w szczególnych przypadkach do 1 g pod stałą kontrolą
MUTSCHLER-2009.indd 796 2010-01-07 22:14:49
Witaminy 797
Tabela B 10.1-2. Wskaźniki służące do oceny statusu witaminowego u dorosłych (zmodyf. według Biesalskiego)
Witamina Parametr Akceptowalna ilość Obniżona ilość lub stężenie (umiarkowane
lub stężenie ryzyko wystąpienia niedoboru)
A retinol (O, μg/ml) > 20 10-20
D 25-OH-D3 (O, ng/ml) > 10 3-10
Mikroskładniki odżywcze:
E α-tokoferol (O, mg/ml) >2 1-2
witaminy i pierwiastki
B1 tiamina (M, μg/24 godz.) > 100 40-100
B2 ryboflawina (M, μg/24 godz.) > 120 40-120
B6 fosforan pirydoksalu (O, nM) > 60 < 60
B12 witamina B12 (O, pg/ml) > 150 100-150
kwas foliowy kwas foliowy (O, ng/ml) >6 3-6
B 10
biotyna biotyna (M, μg/24 godz.) > 25 10-25
niacyna N-metylonikotynamid (M, μg/6 godz.) > 0,6 0,2-0,6
kwas pantotenowy kwas pantotenowy (O, μg/ml) > 0,6 < 0,6
C kwas askorbinowy (O, μg/ml) > 30 20-30
O – osocze, M – mocz
Przyjmowanie witamin w ilościach nieznacznie mniejszych pokarmowym tylko wtedy, kiedy tłuszcze wchłania-
od koniecznej (stadium l. i 2.) objawia się zmniejszeniem ją się prawidłowo, tzn. wydzielana jest dostateczna
ich stężenia w organizmie. W stadium 3. dochodzi do za-
burzenia czynności enzymów, których aktywność zależy
ilość żółci. Poza tym przedawkowanie witamin roz-
od danej witaminy (suboptymalne zaopatrzenie w wita- puszczalnych w tłuszczach, przede wszystkim wita-
minę). Stadium 4. odznacza się niecharakterystycznymi miny A i D, może spowodować poważne zaburzenia,
objawami niedoboru, takimi jak ograniczenie sprawności natomiast przedawkowanie witamin rozpuszczalnych
fizycznej i umysłowej. Jeszcze większy niedobór (hipo- w wodzie (przy prawidłowej czynności nerek) nie po-
witaminoza; stadium 5.) wywołuje zazwyczaj charaktery-
styczne objawy chorobowe, które ustępują po dostarczeniu
ciąga za sobą żadnych szkodliwych następstw.
potrzebnej witaminy. W końcowym stadium ciężkiego W tab. B 10.1-1 zestawiono najważniejsze witami-
niedoboru (awitaminoza; stadium 6.) mogą powstać nieod- ny. Optymalna ilość witamin, jaką należałoby dostar-
wracalne uszkodzenia (np. ślepota przy utrzymującym się czać organizmowi, jest najprawdopodobniej trochę
niedoborze witaminy A). większa niż podane wartości dziennego zapotrzebo-
Zdolność gromadzenia danej witaminy determinuje to,
jak szybko pojawią się objawy jej niedoboru. Na przykład
wania, które przedstawiają dolną granicę pozwalają-
czas upływający do wyczerpania zapasów witaminy B1 wy- cą uniknąć pojawienia się typowych objawów niedo-
nosi 4–10 dni, natomiast dla witaminy B12 – 3–5 lat. boru. Indywidualne zapotrzebowanie może także być
większe.
Zależnie od rozpuszczalności witaminy dzieli się
na dwie grupy:
Standaryzacja. Preparaty zawierające witaminy
■ rozpuszczalne w tłuszczach, można było początkowo standaryzować jedynie
przez wykonanie badań u zwierząt. Zwierzętom
■ rozpuszczalne w wodzie.
podawano karmę zawierającą w dostatecznej
ilości wszystkie witaminy poza testowaną. Na-
Taki podział, sprawiający początkowo wrażenie arbi-
stępnie ustalano:
tralnego, jest jednak uzasadniony, ponieważ informuje
o tym, w którym środku spożywczym przypuszczal- ■ jaką ilość testowanej witaminy należy dodać do tej
nie występuje dana witamina. Witaminy rozpuszczal- karmy, aby zapobiec wystąpieniu objawów niedo-
ne w tłuszczach ulegają wchłanianiu w przewodzie boru (dawka podtrzymująca),
MUTSCHLER-2009.indd 797 2010-01-07 22:14:49
798 Witaminy
■ jaka dawka jest potrzebna po pojawieniu się obja- Tabela B 10.1-3. Zalecane dodatkowe podawanie witamin
wów niedoboru, aby spowodować ich ustąpienie u kobiet ciężarnych (od 4. miesiąca) oraz w okresie laktacji (we-
(dawka lecznicza). dług danych Niemieckiego Towarzystwa Żywienia)
Po ustaleniu chemicznej budowy witamin dawkuje Witamina Zwiększenie dawki (%)
się je wagowo. Wykorzystywane jeszcze częściowo ciężarne karmiące piersią
jednostki międzynarodowe ustalono odpowiednio A 38 125
według ilości wagowych witamin.
D 100 100
Określenie stanu organizmu dotyczącego wita- E 17 42
min. W celu ustalenia zaopatrzenia organizmu w wi- tiamina 25 42
taminy można oznaczać różne witaminy lub ich me- ryboflawina 20 53
tabolity w materiałach biologicznych (np. w osoczu, niacyna 13 33
moczu, erytrocytach, leukocytach). W tab. B 10.1-2 pirydoksyna 63 38
podano wartości umożliwiające ocenę stanu organi- kwas foliowy 100 53
zmu co do zawartości witamin u dorosłych. kobalamina 33 33
kwas askorbinowy 33 67
Wskazania do dodatkowego podawania witamin.
Podanie witamin jest konieczne w przypadku wystę-
powania ujemnego bilansu witaminowego, spowodo-
wanego:
■ dostarczaniem niedostatecznych ilości witamin 10.1.1. Witaminy rozpuszczalne
(przy jednostronnym lub niedostatecznym odży- w tłuszczach
wianiu),
10.1.1.1. Witamina A
■ zwiększonym zapotrzebowaniem na witaminy (np. (akseroftol, retinol) i jej analogi
w wieku niemowlęcym, w okresie ciąży i laktacji
(zob. tab. B 10.1-3), także przy wyczynowym upra-
Witamina A (all-trans-witamina A) jest alkoholem diter-
wianiu sportu, gorączce i nadużywaniu alkoholu),
penowym składającym się z czterech jednostek izopre-
■ zmniejszonym wchłanianiem witamin (np. przy bra- nowych, wrażliwym na światło i tlen. Jest ona wytwa-
ku czynnika wewnątrzpochodnego; przy leczeniu rzana w ścianie jelit przez oksydacyjne rozszczepienie
antybiotykami o szerokim zakresie działania, które prowitamin — karotenów – z udziałem cząsteczkowe-
uszkadzają florę jelitową), go tlenu i wytworzeniem substancji pośredniej aldehy-
du lub przez hydrolizę estrów retinylowych. Witamina
■ wzmożoną biotransformacją witamin (np. u osób
A jest gromadzona głównie w wątrobie w postaci
palących tytoń),
estrów kwasów tłuszczowych. W razie potrzeby ulega
■ zaburzeniami aktywacji witaminy D (np. przy nie- ona biohydrolitycznemu uwalnianiu z zapasów i prze-
wydolności nerek). dostaje się do osocza krwi, gdzie jest transportowana
w formie związanej z α1-globuliną (białkiem surowicy
Dawka profilaktyczna trzykrotnie przekracza zapotrze- wiążącym retinol – RBP ) i transtyretyną (TBPA; zob.
bowanie dzienne, a dawka podawana przy objawowym ryc. B 10.1-2) lub albuminą. Produktami wydalania
niedoborze – dziesięciokrotnie (zob. tab. B 10.1-1). są m.in. glukuronid retinolu, a także wolny lub sprzę-
żony kwas (kwas witaminy A, kwas retinowy).
Pojedyncze witaminy, przede wszystkim witaminy C Najważniejszą prowitaminą jest β-karoten, z któ-
i E oraz prowitamina A, są także stosowane w znacz- rego mogą powstać dwie cząsteczki witaminy A.
nie większych dawkach (w tzw. megadawkach). Taki β-Karoten nie jest jednak wykorzystywany ilościowo
sposób postępowania opiera się na poglądzie, że przez (6 μg β-karotenu jest ilością równoważną z l μg reti-
podawanie dużych dawek witamin działających prze- nolu). Jeżeli są przyjmowane ilości β-karotenu prze-
ciwutleniająco można zapobiec następstwom wzmo- kraczające aktualne zapotrzebowanie na witaminę A,
żonego powstawania rodników. Warunkiem do stoso- to nie dochodzi do jego rozszczepiania, lecz jest groma-
wania takich dużych dawek witamin jako akceptorów dzony w tkankach. Z tego powodu β-karoten nie może
rodnikowych jest duży zakres terapeutycznych dawek wywołać hiperwitaminozy witaminy A. Obok β-karo-
danych witamin. Warunek ten spełniają wymienione tenu w naturze występuje wiele innych karotenów, spo-
substancje, ale nie sama witamina A. śród których jednak tylko niektóre tworzą witaminę A.
MUTSCHLER-2009.indd 798 2010-01-07 22:14:50
Witaminy 799
β-karoten ester retinylu retinol światło jelita
retinal retinol
jelito
Mikroskładniki odżywcze:
ester retinylu
witaminy i pierwiastki
chylomikrony,
ester retinylu chłonka,
osocze
ester retinylu
produkty wątroba
rokładu retinol ester retinylu B 10
(gromadzenie)
+ RBP,
+ TTR
retinol-RBP osocze
(-RBP, -prealbumina)
TTR-kompleks
+ CRBP
komórka docelowa
CRBP-retinol kwas retinowy
+ CRABP
DNA
jądro komórkowe
Ryc. B 10.1-2. Wchłanianie, dystrybucja i metabolizm witaminy A. CRABP – białko komórkowe wiążące kwas retinowy, CRBP – białko
komórkowe wiążące retinol, RBP – białko wiążące retinol, TTR – transtyretyna.
Karoteny znajdują się we wszystkich zielonych roślinach protein, CRBP). Po utlenieniu do kwasu all-trans-
oraz ich częściach, a także w większości części żółtych. i 9-cis-retinowego następuje wiązanie kolejnej czą-
Szczególnie dużą ich ilość zawierają jarmuż, szpinak i mar- steczki – komórkowego białka wiążącego kwas re-
chew.
Witamina A nie jest syntetyzowana w roślinach. Większe tinowy (CRABP), które reguluje wewnątrzkomór-
jej ilości zawierają tran rybi, wątroba, masło, mleko kową dostępność obu kwasów. Kwasy oddziałują
i jajka. z wewnątrzkomórkowymi receptorami – kwas all-
trans-retinowy przede wszystkim z receptorem kwa-
Obok all-trans witaminy A istnieje jeszcze wiele izo- su retinowego (receptor RA, RAR), natomiast kwas
merów cis-trans, które w organizmie ulegają prze- 9-cis-retinowy z receptorem RX (RXR), przy czym
kształceniu w formę all-trans. istnieją trzy podtypy każdego receptora. Kompleksy
kwas retinowy-receptor po dimeryzacji – RXR jako
Mechanizm działania. Jak przedstawiono na ryc. B heterodimer z innymi receptorami, np. z receptora-
10.1-2, po uwolnieniu z kompleksu z RBP i transty- mi witaminy D lub hormonów tarczycy, wpływają
reiną retinol wiąże się do specjalnego komórkowego na ekspresję genów oraz syntezę białek. W ten spo-
białka wiążącego retinol (cellular retinol binding sób jest regulowana synteza ponad 40 różnych białek.
MUTSCHLER-2009.indd 799 2010-01-07 22:14:50
800 Witaminy
H3C
CH3 CH3 CH3
H3C
CH3
CH3 CH3 CH3
CH3
β-karoten
CH3 CH3 CH3
CH2OH
CH3
CH3
witamina A
Należą do nich np. receptor czynnika wzrostowego W procesie widzenia następuje najpierw izomeryzacja wi-
naskórka, keratyny, białka osłonowe zrogowacia- taminy A z wytworzeniem 11-cis-retinolu, a potem odwo-
łych komórek, a także dekarboksylaza ornitynowa, dornienie z wytworzeniem odpowiedniego aldehydu, czyli
11-cis-retinalu. On natomiast łączy się z opsyną, cześcią
która za pośrednictwem tworzenia poliamin reguluje białkową czerwieni wzrokowej i wskutek tego dochodzi
wzrost komórek. W zależności od danego białka i sta- do wytworzenia czerwieni wzrokowej – rodopsyny.
nu wyjściowego komórki synteza białka może ulec Przy naświetlaniu, dzięki wielokrotnym zmianom kon-
zwiększeniu lub zahamowaniu. Natomiast w proce- formacji opsyny, dochodzi do krótkotrwałego powstawa-
sie widzenia czynnej formy witaminy A nie stanowi nia niestabilnej metarodopsyny II, która razem z transdu-
cyną wiąże się z białkiem G. Prowadzi to do stymulacji
kwas, lecz aldehyd – retinal (zob. ryc. B 10.1-3). fosfodiesterazy cGMP i spadku stężenia cGMP. W efekcie
dochodzi do zamykania kanałów Na+/Ca2+ w zewnętrz-
nej błonie komórkowej fotosensorów, wzrostu potencja-
łu błonowego o –40mV oraz zmniejszonego uwalniania
11-cis-retinol all-trans-retinol transmiterów na synapsach z komórkami efektorowy-
(witamina A) mi. Jednocześnie 11-cis-retinal zostaje przekształcony
w 11-trans-retinal, który odłącza się od opsyny (zob. ryc.
B 10.1-3).
Regeneracja czerwieni wzrokowej następuje w ciemnoś-
11-cis-retinal ci przez izomeryzację all-trans-retinalu do 11-cis-retinalu.
Poza tym retinal ulega redukcji do retinolu, który może ulec
wymianie na retinol znajdujący się w krwiobiegu.
opsyna all-trans-retinal Znaczenie fizjologiczne. Witamina A jest niezbędna
do:
■ wzrostu całego organizmu,
rodopsyna opsyna ■ wzrostu i różnicowania się komórek nabłonko-
wych,
światło
■ rozmnażania się (spermiogenezy),
batorodopsyna metarodopsyna II ■ rozwoju embrionalnego,
■ funkcji widzenia.
lumirodopsyna metarodopsyna I Witamina A chroni komórki nabłonkowe błon śluzo-
wych przed zrogowaceniem (funkcja ochrony nabłon-
Ryc. B 10.1-3. Funkcja witaminy A w procesie widzenia. ka). W wyniku uszczelnienia nabłonków i stymulacji
MUTSCHLER-2009.indd 800 2010-01-07 22:14:50
Witaminy 801
wytwarzania śluzu witamina A zapobiega wniknię- zatrucia występowały m.in. utrata apetytu, wysusze-
ciu drobnoustrojów do organizmu (funkcja ochrony nie skóry, wypadanie włosów, pęknięcia naskórka
przed zakażeniami). Natomiast w skórze witamina (ragady) w kącikach ust, bóle kości i stawów, opóź-
A wzmaga rogowacenie. nienie wzrostu, obrzęk okostnej i anoreksja.
Pod względem działania na komórki nabłonkowe Toksyczne dawki są tak duże, że przy typowym
retinol można zastąpić jego metabolitami, retinalem dawkowaniu witaminy A nie należy obawiać się prze-
i kwasem retinowym, a także wieloma syntetycznymi dawkowania.
Mikroskładniki odżywcze:
retinoidami. Oprócz niedoboru witaminy A wady rozwojowe
witaminy i pierwiastki
Retinoidy wpływają również na rozwój embrio- mogą także być uwarunkowane dostarczaniem nad-
nalny i płodność. miernych ilości witaminy A (> 3 mg/dzień). Aby
uniknąć uszkodzenia organizmu płodu, we wczes-
Niedobór witaminy A. Niedobór witaminy A może nym okresie ciąży nie należy przekraczać dawki
być spowodowany niedostateczną ilością witami- 3 mg/dzień Leczenie dawkami > 8 mg/dzień prowa-
ny A lub karotenów w pożywieniu, zaburzeniami dzone u dojrzałych płciowo kobiet wymaga stosowa-
wchłaniania tłuszczów (np. przy chorobach trzustki, nia pewnej metody antykoncepcji.
niedrożności dróg żółciowych, celiakii). Tego rodza-
ju niedobór objawia się początkowo upośledzeniem Kwas witaminy A. Substancję tę omówiono już
adaptacji wzroku przy zmianach oświetlenia i ślepotą w rozdz. B 9.4.2.6. B 10
zmierzchową. Jeżeli niedobór witaminy A nie zosta-
nie wyrównany, pojawiają się kolejne objawy będące β-Karoten. Podobnie jak witamina C i E, prowita-
efektem zwiększonej ilości podziałów komórkowych mina A również inaktywuje tlen w stanie cząstecz-
i niedostatecznego różnicowania się komórek: kowym i reaktywne formy tlenu. Ze względu na te
właściwości β-karoten przeciwdziała wywoływaniu
■ wysuszenie i zrogowacenie spojówki (kseroftal-
uczulenia na światło przez hematoporfirynę oraz po-
mia), a także rozmięknienie rogówki (keratomala-
prawia tolerancję na światło. Z tego powodu jest on
cja), które prowadzą do ślepoty,
stosowany przy protoporfirii, dermatozie wywołanej
■ wysuszenie i zrogowacenie błon śluzowych, któ- światłem, a ponadto przy nieprawidłowej pigmentacji
rym towarzyszą zmniejszenie zdolności węcho- (np. bielactwie nabytym).
wych, brak soku żołądkowego, biegunka i in., Dawkowanie wynosi 25–50 (–200) mg/dzień.
Działaniem niepożądanym jest czerwonawe zabar-
■ suchość, tworzenie łusek i zmarszczek skóry (hi-
wienie skóry, powstające w następstwie odkładania
perkeratoza).
się β-karotenu.
W następstwie niedostatecznego różnicowania się
komórek natomiast dochodzi do powstawania więk-
szej liczby nowotworów.
10.1.1.2. Witamina D (kalcyferol)
i jej pochodne
Zapotrzebowanie dzienne, dawkowanie. Zapo-
trzebowanie na witaminę A u osoby dorosłej wynosi Cholekalcyferol (witamina D3), fizjologiczna wi-
około l mg, a u dzieci w wieku do 10 lat 0,6–0,8 mg/ tamina D, może być syntetyzowany w organizmie.
dzień (l mg = 3000 IU). W wyniku oddziaływania promieni UV cholekalcyfe-
Do substytucyjnej terapii ślepoty zmierzchowej rol powstaje w keratynocytach skóry z 7-dehydrocho-
wystarcza podawanie 10–15 mg/dzień przez 2–3 ty- lesterolu, który jest wytwarzany w wątrobie z chole-
godnie. sterolu. Ilość produkowana w organizmie w okresie
pierwszych dwóch lat życia nie jest jednak zazwyczaj
Objawy przedawkowania. Rozróżnia się ostre i prze- wystarczająca: przyczynia się do tego z jednej strony
wlekłe formy przedawkowania witaminy A. Ostre duże zapotrzebowanie podczas fazy wzrostu, a z dru-
przedawkowanie obserwowano u ludzi (np. u polar- giej strony duża absorpcja promieni UV w naszej
ników), którzy odżywiali się jednostronnie wątrobą szerokości geograficznej uwarunkowana zanieczysz-
niedźwiedzia polarnego i fok. Obserwowane objawy czeniem powietrza. Poza tym ludzie najczęściej prze-
to nudności, wymioty, bóle głowy i uczucie ucisku bywają niedostateczną ilość czasu na świeżym powie-
na mózg. trzu. Z tego powodu witaminę D3 należy konsekwen-
Przy przewlekłym przedawkowaniu, przede tnie stosować u wszystkich niemowląt, aby zapobiec
wszystkim u dzieci (dziennie > 10 mg), jako objawy chorobie z niedoboru witaminy D, tj. krzywicy.
MUTSCHLER-2009.indd 801 2010-01-07 22:14:51
802 Witaminy
Znaczenie fizjologiczne. Witamina D3 lub produkty
cholesterol jej hydroksylacji są razem z parathormonem potrzeb-
ne do utrzymania fizjologicznego stężenia jonów
wątroba wapnia we krwi (zob. ryc. B 2.4-1). Wymienione sub-
stancje podwyższają stężenie wapnia we krwi przez
7-dehydrocholesterol
zwiększenie:
skóra
■ wchłaniania wapnia z jelit,
cholekalcyferol
(witamina D3) ■ zwrotnego wchłaniania jonów wapnia w kanali-
kach nerkowych,
wątroba
■ aktywności osteoklastów w tkance kostnej.
25-hydroksy-cholekalcyferol
(kalcyfediol)
Witamina D3, mimo mobilizacji jonów wapnia z tkan-
mniejsze ki kostnej w wyniku stymulacji aktywności osteokla-
nerka zapotrzebowanie
na kalcytriol stów, powoduje wzmożone tworzenie się tkanki kost-
nej. Można to wyjaśnić następująco: mineralizacja
1,25-dihydroksy- 24,25- substancji międzykomórkowej kości wytwarzanej
cholekalcyferol -dihydroksy- przez osteoblasty i przez to tworzenie sprawnych
(kalcytriol) cholekalcyferol czynnościowo kości zależy od dostatecznego stęże-
nia wapnia we krwi. Takie stężenie zostaje osiągnię-
Ryc. B 10.1-4. Synteza i metabolizm witaminy D3. te za pomocą opisanych mechanizmów. W wyniku
działania witaminy D3 powstaje więc więcej tkanki
kostnej, niż ulega rozkładowi.
Naturalnym źródłem witaminy D3 jest olej z wątroby ryb W nowszych badaniach wykazano, że witamina
oraz tkanka tłuszczowa zwierząt. Żółtko jajka, mleko
i masło zawierają niewielką ilość witaminy D3. Obecnie
D3 reguluje nie tylko stężenie wapnia, lecz odgrywa
jest ona produkowana z cholesterolu metodami częściowo także ważną rolę w prawidłowej proliferacji i róż-
syntetycznymi.
Poza witaminą D3 znanych jest jeszcze wiele innych wita-
min D, otrzymywanych przez napromienianie światłem UV
Δ5,7-dwunienasyconych steroli. Spośród nich przez pewien C
okres większe znaczenie miał ergokalcyferol (witamina
D2), ponieważ początkowo uważano go za właściwą wita-
minę, a ponadto łatwo otrzymuje się go z ergosterolu.
HSP
Witamina D przyjęta wraz z pożywieniem wchłania C + CR HSP
C
się przez pasywną dyfuzję z całego jelita cienkiego.
CR
Właściwą czynną postacią witaminy D3 jest 1,25- C
-dihydroksycholekalcyferol (kalcytriol, hormon D3). CR
Grupa hydroksylowa przy C-25 jest wprowadzana
w wątrobie, natomiast przy C-1 w nerkach i keraty-
nocytach. Parathormon przez stymulację hydroksyla- jądro komórkowe
transkrypcja
zy w nerkach wzmaga wytwarzanie kalcytriolu, który
działa mniej więcej 20–25 razy silniej od witaminy
D3. W stanach małego zapotrzebowania na kalcytriol
translacja białko
zamiast niego jest wytwarzany nieczynny 24,25-
-dihydroksycholekalcyferol (zob. ryc. B 10.1-4).
Ze względu na to, że kalcytriol jako właściwa sub- (np. kalbindina D, fosfataza
stancja czynna jest wytwarzany w specjalnym narzą- zasadowa, ATP-aza stymulowana
przez wapń)
dzie, uwalniany do krwi wykazuje swoje działanie
w miejscu odległym od miejsca wytwarzania, powin- Ryc. B 10.1-5. Mechanizm działania witaminy D3 lub kalcytriolu
no się go nazywać hormonem (hormon D3), a nie po- (hormon witaminowy). C – kalcytriol, CR – kalcytriol-receptor,
chodną witaminy. HSP – białko szoku cieplnego.
MUTSCHLER-2009.indd 802 2010-01-07 22:14:51
Witaminy 803
nicowaniu się komórek skóry. Ponadto witamina D3 oraz aktywnych limfocytach B i T wyjaśnia wpływ
działa immunomodulująco — hamuje aktywność lim- wywierany przez grupę witamin D na skórę i układ
focytów T i wzmaga aktywność makrofagów. immunologiczny.
Mechanizm działania. Witamina D3 i witamina A wy- Niedobór witaminy D. Niedobór witaminy D wywo-
kazują podobne mechanizmy działania. Po związaniu łuje krzywicę u niemowląt i małych dzieci. Wystę-
się ze swoistym cytozolowym receptorem i transferze pujący przy tym niedobór wapnia i nieprawidłowa
Mikroskładniki odżywcze:
kompleksu hormon-receptor do jądra komórkowego miękkość układu kostnego prowadzą do deformacji
witaminy i pierwiastki
cholekalcyferol, a w jeszcze większym stopniu 1,25- szkieletu.
-dihydroksycholekalcyferol, stymulują wytwarzanie
białek biorących udział w transporcie i przemianach Kręgosłup wygina się pod wpływem ucisku głowy i obrę-
wapnia (zob. ryc. B 10.1-5). W ten sposób dochodzi czy barkowej (krzywica, kifoskolioza), dolny otwór klatki
m.in. do indukcji syntezy białka wiążącego wapń, piersiowej ulega odgięciu (klatka dzwonowata), kości pod-
udzi wyginają się (kolana szpotawe lub koślawe), następuje
kalbindiny D, a także fosfatazy zasadowej i ATP-azy zgrubienie granicy części kostnej i chrzęstnej żeber (tzw.
stymulowanej przez jony wapnia. Duże stężenie kal- różaniec krzywiczy). Ząbkowanie i zarośnięcie ciemiączka
cytriolu hamuje ponadto uwalnianie parathormonu są opóźnione, kości czaszki dają się wciskać (craniolabes).
i w wyniku tego, poprzez ujemne sprzężenie zwrotne, Jeżeli stężenie wapnia we krwi obniża się jeszcze bardziej,
syntezę kalcytriolu w nerkach. dochodzi do kurczów tężyczkowych (spazmofilia = skłon- B 10
ność do tężyczki).
Oprócz komórek nabłonkowych nerek i jelit oraz
osteocytów wiele innych komórek posiada receptory
witaminy D3. Ich występowanie w keratynocytach, U dorosłych ciężki niedobór witaminy D wywołuje
fibroblastach, a także w monocytach, makrofagach rozmięknienie kości.
H3C H3C CH3 H3C
CH3 CH(CH3) 2 CH3 CH3 CH(CH3) 2
H3C OH
H H H
CH2 CH2 CH2
HO HO HO OH
cholekalcyferol 25-hydroksy-cholekalcyferol 1-hydroksy-cholekalcyferol
(witamina D3) (kalcyferol) (alfakalcydol)
CH3 CH3
H3C CH3 H3C H3C
CH3 CH3 CH(CH3) 2 CH3 CH(CH3) 2
H3C OH
H H H
CH2 CH2 H3C
HO OH HO OH
1,25-dihydroksy-cholekalcyferol ergokalcyferol dihydrotachysterol
(kalcytriol) (witaminaD2)
MUTSCHLER-2009.indd 803 2010-01-07 22:14:51
804 Witaminy
Dawkowanie. W profilaktyce krzywicy u niemowląt w jelitach, a w tkance kostnej może dojść do demine-
i małych dzieci podaje się przez 1–1,5 roku 500 IU = ralizacji (nerkowej osteopatii). Można ją leczyć, po-
0,0125 mg witaminy D3 dziennie (l IU = 0,025 μg). dając hydroksylowane pochodne witaminy D, przede
wszystkim kalcytriol. Potrzebną dawkę należy usta-
Ten sposób leczenia jest obecnie stosowany zamiast typo- lać odpowiednio do stężenia wapnia we krwi (średnia
wej w przeszłości profilaktyki uderzeniowej z trzykrotnym początkowa dawka wynosi 0,25 μg/dzień).
podaniem l0 mg w odstępie 3 miesięcy. Profilaktykę ude-
rzeniową powinno się jednak nadal stosować, gdy nie ma Innymi substancjami, które są odpowiednie
pewności co do codziennego dawkowania z powodu braku do leczenia nerkowej osteopatii i krzywicy opornej
odpowiedniej współpracy ze strony rodziców dziecka. na działanie witaminy D, są:
■ 25-hydroksycholekalcyferol (kalcyfediol),
W leczeniu krzywicy podaje się 10 tys. IU dzien-
nie lub jeden raz 15 mg jako dawkę uderzeniową. ■ 1-hydroksycholekalcyferol (alfakalcydiol).
Jednocześnie należy dostarczyć jony wapnia i fo- Pozostałe pochodne witaminy D. W terapii stosuje
sforanów, ponieważ ich wchłanianie było zaburzone się także dihydrotachysterol (DHT), który jest po-
i w takim stanie nie ma zapasów wystarczających
do zaspokojenia potrzeb szybko rozpoczynającego
się procesu kostnienia.
Przy jednocześnie prowadzonej profilaktyce próch-
nicy często podaje się witaminę D3 w połączeniu R3
z fluorkiem. R2 O CH3
CH3
CH3 CH3 CH3
Objawy przedawkowania. Przedawkowanie wita- HO
miny D3 może wywołać ciężkie (aż do najcięższych R1 R1 R2 R3
postaci) zatrucie, które jest spowodowane zbyt wy-
α-tokoferol CH3 CH3 CH3
sokim stężeniem wapnia we krwi (efekt wapnicy).
Takie zatrucie odpowiada obrazowi chorobowemu β-tokoferol CH3 H CH3
zatrucia parathormonem. Uruchomiony wapń od- γ-tokoferol H CH3 CH3
kłada się częściowo w nerkach i naczyniach, a także
w zwiększonych ilościach jest wydalany z moczem.
Do klinicznych objawów należą wymioty, biegunka,
bóle głowy i stawów. Niewydolność nerek może do-
prowadzić do zgonu. Wymienione objawy zatrucia
chodną ergosterolu podawaną w leczeniu niedoczyn-
ustępują w przypadku odstawienia we właściwym
ności przytarczyc, oraz kalcypotriol w miejscowym
czasie witaminy D3. Zwapnienie naczyń również
leczeniu łuszczycy.
ustępuje.
Hydroksylowane pochodne witaminy D. Przy prze-
wlekłej niewydolności nerek oraz genetycznie uwa- 10.1.1.3. Witaminy E
runkowanej tzw. krzywicy opornej na działanie wi- (tokoferole, tokotrienole)
taminy D występuje niedostateczne przekształcanie
cholekalcyferolu w jego właściwą formę czynną – kal- Termin zbiorczy witaminy E oznacza wszystkie na-
cytriol (zob. powyżej) lub brak takiego przekształca- turalne i syntetyczne tokoferole (mono-, di- i trimety-
nia. Wskutek tego zmniejsza się wchłanianie wapnia lotokole) oraz tokotrienole, które jakościowo działają
CH3 CH3
H3C O C16H33 . H3C O
+
C16H33
- e, - H
CH3 . CH3
+ e, + H
HO O
CH3 CH3
α-tokoferol chinon α-tokoferolu
Ryc. B 10.1-6. Utlenianie α-tokoferolu.
MUTSCHLER-2009.indd 804 2010-01-07 22:14:52
Witaminy 805
podobnie jak α-tokoferol (zob. poniżej). Są to po-
chodne 6-chromanolu, posiadające nasycony (toko- Niedobór witaminy E. Tak jak w przypadku innych
ferole) lub trójnienasycony (tokotrienole) boczny witamin rozpuszczalnych w tłuszczach, przyczyną
łańcuch fitolowy i różniące się od siebie tylko liczbą niedoboru witaminy E może być niedostateczne wy-
i położeniem grupy metylowej przy pierścieniu chro- dzielanie żółci do jelita cienkiego. Ponadto występu-
manolowym oraz właściwościami stereochemiczny- je on u pacjentów z A-β-lipoproteinemią lub prze-
mi obu grup metylowych w pozycji 4′ i 10′ w łańcu- wlekłymi zapalnymi chorobami jelit. Następstwem
Mikroskładniki odżywcze:
chu bocznym. niedoboru witaminy E jest skrócenie czasu życia
witaminy i pierwiastki
Najważniejszą występującą w naturze substancją erytrocytów, kreatynuria, wzmożone wytwarzanie
wykazującą aktywność witaminy E jest RRR-α-toko- lipofuscyny, osłabienie mięśni oraz zaburzenia ukła-
ferol (2R, 4′R, 8′R-α-tokoferol). Jeżeli przyjmie się, du nerwowego (ośrodkowe i obwodowe neuropatie).
że jego aktywność wynosi 100%, to względna aktyw- Zaburzenia układu nerwowego pojawiają się u doro-
ność różnych substancji z grupy witamin E jest nastę- słych dopiero po upływie ok. 10 lat, natomiast u dzie-
pująca: RRR-β-tokoferolu 50%, RRR-γ-tokoferolu ci już po 18–24 miesiącach stale utrzymującego się
10%, all-rac-α-tokoferolu 74%, octanu RRR-α-toko- złego wchłaniania tłuszczów i witaminy E. Ponadto
ferylu 91%, α-tokotrienolu 30% oraz R-β-tokotrieno- istnieją dane świadczące o częstszym występowaniu
lu 5%. miażdżycy tętnic lub choroby wieńcowej i nowotwo-
Jako biologiczny wzorzec do oznaczania witaminy rów przy odżywianiu się pożywieniem zawierającym B 10
E nie jest wykorzystywany RRR-α-tokoferol, lecz ra- małe ilości witaminy E.
cemiczna substancja, all-rac-α-tokoferol: l mg odpo-
wiada l międzynarodowej jednostce (IU). Zapotrzebowanie dzienne. Dokładne dzienne za-
potrzebowanie na witaminę E nie jest znane. Można
Według obecnego stanu wiedzy tokoferole i tokotrienole je jedynie oszacować. Przypuszczalnie jest ono
są syntetyzowane tylko przez rośliny. Do najbardziej wy- większe niż 15 mg, wartość zazwyczaj podawa-
dajnych źródeł witaminy E należą kiełki zbóż, orzechy
i oleje roślinne. Również warzywa liściaste zawierają dużą na przez Niemieckie Towarzystwo ds. Żywienia.
ilość witaminy E. Zapotrzebowanie na witaminę E wzrasta przy dostar-
czaniu organizmowi większej ilości nienasyconych
kwasów tłuszczowych oraz w stanach zwiększonego
Podobnie jak w przypadku innych witamin rozpusz-
obciążenia fizycznego lub umysłowego.
czalnych w tłuszczach, wchłanianie witaminy E
z przewodu pokarmowego zależy od rodzaju i ilości Podawanie witaminy E w dawce do 100 mg/dzień jest uzna-
tłuszczów w pożywieniu oraz od obecności kwasów wane (jeszcze) za fizjologiczne. Podawanie 100–400 mg/
żółciowych. Estry witaminy E przed wchłanianiem dzień jest tolerowane bez pojawiania się działań niepożą-
muszą najpierw ulec hydrolizie. Witamina E jest wy- danych. Jeszcze większe dawki dzienne nie wywołują żad-
dalana przede wszystkim w postaci glukuronidu, po- nych szkodliwych skutków, nie są jednak ani potrzebne, ani
użyteczne.
chodnej hydrochinonowej z żółcią, a także przez ner-
ki jako kwas tokoferonowy i jego lakton.
Znaczenie terapeutyczne. Skuteczność w przypad-
ku wielu wskazań do stosowania witaminy E, poda-
Znaczenie fizjologiczne. Witamina E bierze udział wanych przede wszystkim w reklamach przeznaczo-
w procesach utleniająco-redukcyjnych przemian po- nych dla laików (np. choroby wątroby, zaburzenia
średnich (zob. ryc. B 10.1-6). Jako lipofilny akcep- ukrwienia, miopatie), nie jest udokumentowana.
tor rodników zapobiega ona tworzeniu nadtlenków Pewne sukcesy odnotowano w leczeniu reumatoidal-
nienasyconych wyższych kwasów tłuszczowych nego zapalenia stawów (mniejsze zapotrzebowanie
w lipidach błonowych, w które zostaje wbudowa- na podawanie niesteroidowych leków przeciwzapal-
na z powodu posiadania długich łańcuchów bocz- nych). U pacjentów z A-β-lipoproteinemią stosowa-
nych. Tokoferol odkłada się także w lipoproteinach nie witaminy E powoduje poprawę pod względem
o małej gęstości (LDL). Chroni on również składniki objawów ze strony układu nerwowego lub zapobiega
tych lipoprotein przed oksydacyjnym zniszczeniem. ich wystąpieniu.
Dodatkowo – podobnie jak witamina C – tokoferol
hamuje tworzenie nitrozoamin. Poza tym rozważa się
wpływ witaminy E na płynność błon komórkowych,
aktywność różnych enzymów (m.in. hamowanie fo- 10.1.1.4. Witamina K
sfolipazy A2) oraz syntezę kwasów nukleinowych
i białek. Witaminę K omówiono już w rozdz. B 4.1.4.1.1.
MUTSCHLER-2009.indd 805 2010-01-07 22:14:52
806 Witaminy
18 12 9 18 15 12 9
COOH H3C COOH
H3C
kwas linolowy
– 2H kwas α-linolenowy
– 2H
18 12 9 6 18 15 12 9 6
H3C
H3C COOH COOH
kwas γ-linolenowy
+ C2 + C2
20 14 11 8 20 17 14 11 8
H3C
H3C COOH COOH
kwas eikozatrienowy
– 2H – 2H
20 14 11 8 5
COOH H20 17 14 11 8 5
COOH
H3C 3C
kwas arachidonowy kwas eikozapentaenowy
Ryc. B 10.1-7. Synteza kwasu arachidonowego i eikozapentaenowego.
10.1.1.5. Uzupełnienie: Kwas linolowy występuje w szczególnie dużym stężeniu
niezbędne kwasy tłuszczowe w roślinnych olejach z zarodków zbóż. Jest on składnikiem
acyloglukozyloceramidów, które są potrzebne przy tworze-
niu blaszek lipidowych w zrogowaciałej warstwie skóry,
W organizmie ssaków jest możliwa dehydrogena- zmniejszających wyparowywanie wody.
Również kwas α-linolenowy jest przyjmowany głównie
cja nasyconego kwasu tłuszczowego, kwasu stea-
z olejami roślinnymi. Jego najważniejszymi metabolitami
rynowego, z wytworzeniem jednonienasyconego są wymieniony kwas eikozapentaenowy, a także kwas do-
kwasu tłuszczowego, kwasu oleinowego (wiązanie kozaheksaenowy. Głównym bezpośrednim źródłem tych
podwójne w pozycji 9 od końca metylowego, kwas dwóch kwasów tłuszczowych są oleje rybne.
ω-9-tłuszczowy). Brakuje natomiast enzymów dla
przekształcenia kwasu oleinowego w dwunienasy-
cony kwas tłuszczowy, kwas linolowy (pierwsze wią-
zanie podwójne w pozycji 6 od końca metylowego, CH3
–
N
+ OH X
kwas ω-6-tłuszczowy) lub w trójnienasycony kwas N
tłuszczowy, kwas α-linolenowy (pierwsze wiązanie
H3C N NH2 S
podwójne w pozycji 3 od końca metylowego, kwas
ω-3-tłuszczowy). Te kwasy tłuszczowe mające izolo-
wane wiązania podwójne w konfiguracji cis są tzw. witamina B1
substancjami niezbędnymi dla ssaków.
W organizmie ssaków do kwasu linolowego
z udziałem mikrosomalnej desaturazy może zostać O
–
O
–
CH3
wprowadzone kolejne wiązanie podwójne w kierun- –
+ O P O P O
ku do końca karboksylowego z wytworzeniem kwasu N N
O O
γ-linolenowego (kwasu gamolenowego). W wyniku H3C N NH2 S
wydłużenia łańcucha o dwa atomy węgla i wprowa-
dzenia kolejnego wiązania podwójnego zostaje z nie-
go utworzony fizjologicznie ważny kwas C20, kwas difosforan tiaminy
arachidonowy, natomiast analogicznie z kwasu α-li-
nolenowego – kwas eikozapentaenowy.
MUTSCHLER-2009.indd 806 2010-01-07 22:14:52
Witaminy 807
Wielonienasycone kwasy tłuszczowe są składnika- niu wyprysku atopowego stosuje się olej z nasion wiesiołka
mi fosfolipidów, które, ze względu na swoje właści- (Oenothera biennis), zawierający dużą ilość tego nienasy-
conego kwasu tłuszczowego. Dłużej trwające podawanie
wości amfofilne, odgrywają istotną rolę w budowie
prowadzi do zmniejszenia świądu. Średnia dawka kwasu
podwójnej warstwy lipidowych błon biologicznych. γ-linolenowego wynosi 2 razy dziennie 200 mg.
Ich płynność jest tym większa, im większa jest liczba
podwójnych wiązań w kwasach tłuszczowych zawar- Kwasy ω-3-tłuszczowe. W przypadku spożywania poży-
tych w lipidach błonowych. Szczególnie duża płyn- wienia bogatego w kwasy ω-3-tłuszczowe, w organizmie,
Mikroskładniki odżywcze:
ność jest potrzebna przy szybkich procesach trans- poza typową prostacykliną (PGI2) i tromboksanem (TXA2)
powstającymi z kwasu arachidonowego, są wytwarzane
witaminy i pierwiastki
portowych.
PGI3 oraz TXA3 z kwasu eikozapentaenowego. Podczas
gdy agregacja trombocytów jest hamowana w taki sam spo-
sób przez te obie prostacykliny, TXA3, w przeciwieństwie
do TXA2, nieznacznie ją wzmaga. Z tego powodu kwasy
NH2 CH3 ω-3-tłuszczowe zmniejszają w pewnym stopniu ryzyko
S wystąpienia miażdżycy i mogą być stosowane w profilak-
N N tyce chorób sercowo-naczyniowych. Ich wartość lecznicza
O stanowi jednak kwestię sporną.
H3C N H O O
O P OH
OH B 10
10.1.2. Witaminy rozpuszczalne
benfotiamina w wodzie
10.1.2.1. Witamina B1 (aneuryna, tiamina)
Zapotrzebowanie na niezbędne kwasy tłuszczowe
jest oceniane różnie. Ogólnie podaje się, że wynosi Witamina B1 zawiera dwa heterocykliczne pierście-
ono 10–30 godz./dzień. nie – pirymidynowy i tiazolowy – które są ze sobą
połączone poprzez grupę metylenową. Grupa mety-
U szczurów niedobór wielonienasyconych kwasów tłusz- lenowa jest wrażliwa na działanie środków utlenia-
czowych prowadzi do wypadania włosów, zaburzeń gospo- jących i redukujących. W zasadowych roztworach
darki wodnej, bezpłodności i w końcu do śmierci. U ludzi
nie zaobserwowano jeszcze tego rodzaju objawów niedo- wodnych, pod wpływem oddziaływania nukleofil-
boru, ponieważ z pożywieniem zawsze jest przyjmowana nego, łatwo dochodzi do rozszczepienia pierścienia
pewna ilość wielonienasyconych kwasów tłuszczowych. tiazolowego.
U pacjentów z wypryskiem atopowym wskutek defektu
Δ6-desaturazy jest zmniejszona zawartość kwasu γ-linole- Tiamina znajduje się w owocni i kiełkach traw, a ponadto
nowego we krwi. Dlatego obecnie w podstawowym lecze- w drożdżach, warzywach i ziemniakach. Również u zwie-
+ +
N N
HO – CO2 H2N S
HO
S S
R R S
O
O OH H
produkt połączenia
ketokwasu i TDP aktywny aldehyd liponamid
CoA H2N SH
S
koenzym A S
O R O
O R
aktywowany kwas tłuszczowy ester dihydroliponamidu
Ryc. B 10.1-8. Oksydacyjna dekarboksylacja α-ketokwasów.
MUTSCHLER-2009.indd 807 2010-01-07 22:14:53
808 Witaminy
rząt występuje we wszystkich narządach. Największe pleksów 2-oksokwasy-dehydrogenaza. Są to kom-
jej stężenie stwierdza się w wątrobie, nerkach, sercu pleksy składające się z wielu enzymów, z udziałem
i mózgu.
których 2-oksokwasy (kwasy 2-oksokarboksylowe)
są przekształcane z dekarboksylacją w związki acy-
Po doustnym podaniu małych dawek (< l mg) tia- lokoenzymu A. Z powodu pełnienia tej funkcji difo-
mina za pomocą aktywnego transportu prawie sforan tiaminy ma duże znaczenie dla przemian wę-
w całości ulega wchłonięciu z przewodu pokarmo- glowodanów.
wego. Natomiast po podaniu dawki 5 mg wchłania
Podczas oksydacyjnej dekarboksylacji kwasów α-oksokar-
się już tylko 33% tego czwartorzędowego związku boksylowych kwas oksokarboksylowy ulega połączeniu
amonowego. Biologiczny okres półtrwania wyno- z TDP, odszczepiany jest CO2, a następnie reszta aldehy-
si 9,5–18,5 dnia. Tiamina jest wydalana w postaci dowa zostaje przeniesiona na liponamid, działający przy
niezmienionej bądź w postaci różnych metabolitów tym jako środek utleniający. W następnym etapie w reakcji
(m.in. estrów tiaminy z kwasem siarkowym, kwasu przeestryfikowania reszta acylowa zostaje dalej przekazana
na koenzym A (zob. ryc. B 10.1-8). W ten sposób aktyw-
tiaminowego) przez nerki. ny kwas tłuszczowy może zostać łatwo włączony w dalsze
Aby zapewnić dobrą biodostępność większych przemiany, np. cykl kwasu cytrynowego.
dawek (do 300 mg/dzień), wyprodukowano benfo-
tiaminę, czyli rozpuszczalną w tłuszczach pochodną Poza tym witamina B1 bierze udział w reakcji katali-
witaminy B1. Kontrolowane badania wykazały, że za- zowanej przez transketolazę, w której przenosi alde-
stosowanie substancji czynnej zawierającej otwarty, hyd glikolowy na cukier C5, np. rybozę lub erytrozę.
lipofilny łańcuch tiazolowy, który podczas powsta-
wania witaminy B1 w organizmie ulega zamknięciu, Znaczenie fizjologiczne. Witamina B1 w istotny spo-
doprowadziło do poprawy szybkości przewodnictwa sób bierze udział w przewodzeniu bodźców w obwo-
nerwowego u pacjentów z neuropatią cukrzycową. dowym układzie nerwowym, a także w przemianach
różnych neuroprzekaźników. (Wytwarzanie acetylo-
Mechanizm działania. W organizmie witamina B1 choliny, GABA, glutaminianu i asparaginianu jest po-
ulega fosforylacji z wytworzeniem czynnej postaci, wiązane z przemianami glukozy). Nie wyjaśniono
difosforanu tiaminy (TDP; dawne określenie: pirofo- jednak jeszcze w sposób jednoznaczny, czy i w jakim
sforan tiaminy, TPP). TDP stanowi koenzym dla kom- zakresie istnieje związek tych procesów z zaburze-
OH OH OH OH
HOH2C HOH2C H
OH N +
+ 2H H3C OH N N O
H3C N O
+
– 2H
NH NH
H3C N H3C N
O H O
ryboflawina dihydroryboflawina
NH2
N N
O O OH OH
N N O O P O P O
OH OH OH N
H3C N O
HO OH
NH
H3C N
O
dinukleotyd flawinoadeninowy (FAD)
MUTSCHLER-2009.indd 808 2010-01-07 22:14:53
Witaminy 809
niami układu nerwowego obserwowanymi przy nie- Poza beri-beri, awitaminozą bardzo rzadko wy-
doborze tiaminy. stępującą w krajach zachodnich, stosowanie tiaminy
jest wskazane tylko przy zwiększonym zapotrzebo-
Niedobór witaminy B1. Niedobór samej witaminy waniu na witaminę B1, np. w czasie ciąży, u alkoho-
B1 objawia się u ludzi: lików i przy jednostronnym odżywianiu się węglowo-
danami. Ogólnie w takich przypadkach wystarcza do-
■ zmniejszoną sprawnością umysłową i fizyczną,
ustne podawanie 10 (–40) mg witaminy B1 dziennie,
Mikroskładniki odżywcze:
■ brakiem apetytu, zmniejszeniem masy ciała i bra- ale krótkotrwale można podawać do 300 mg/dzień.
witaminy i pierwiastki
kiem wydzielania soku żołądkowego, co stanowi Przy zaburzeniach wchłaniania konieczne jest poda-
oznakę zaburzeń czynności żołądka i jelit, wanie pozajelitowe lub stosowanie rozpuszczalnej
w tłuszczach pochodnej witaminy B1 (zob. poniżej).
■ zanikiem mięśni, zwłaszcza w kończynach dol-
Z powodu niebezpieczeństwa reakcji rzekomo ana-
nych,
filaktycznych tiaminę powinno się podawać pozaje-
■ zmianami w EKG. litowo jedynie w przypadku dokładnie określonego
wskazania, monitorując przy tym pacjenta.
Beri-beri – obraz chorobowy obserwowany od sta-
rożytności u ludności krajów wschodnioazjatyckich
żywiącej się głównie ryżem. Częstość występowa- 10.1.2.2. Witamina B2 (ryboflawina) B 10
nia tej choroby wzrosła zatrważająco, kiedy zaczęto
Ryboflawina, pochodna izoalloksazyny [7,10-dime-
maszynowo zdejmować osłonki z ziaren ryżu. Beri-
tylo-10-(1′-D-rybitolo)-izoalloksazyna], jest bardzo
-beri jest złożoną awitaminozą uwarunkowaną, poza
wrażliwa na działanie światła, a w roztworze zasado-
niedoborem witaminy B1, także niedoborem innych
wym jest nietrwała również po podgrzewaniu.
witamin. (Polerowanie ryżu powoduje usuwanie wi-
tamin i pierwiastków śladowych). Pełnoobjawowa Ryboflawina występuje we wszystkich komórkach zwie-
choroba charakteryzuje się występowaniem zapale- rzęcych i roślinnych; najwięcej ryboflawiny znajduje się
nia wielonerwowego z parestezjami i porażeniami, w drożdżach, kiełkach zbóż, owocach strączkowych oraz
zmian psychicznych czasem obrzęków. wątrobie, nerkach, mleku i serze.
Niedobór witaminy B1 znacznego stopnia stwier-
dza się także przy polineuropatii i encefalopatii Małe dawki ryboflawiny są wchłaniane z przewodu
Wernickego uwarunkowanych nadużywaniem alko- pokarmowego za pomocą aktywnego transportu, na-
holu, a także (rzadziej) przy innych chorobach bę- tomiast podawana w większych stężeniach wchłania
dących następstwem nieprawidłowego odżywiania się drogą pasywnej dyfuzji. Przyjęcie pożywienia
się z niedowładem mięśni gałki ocznej, oczopląsem, i kwasy żółciowe zwiększają wchłanianie ryboflawi-
ataksją i zaburzeniami psychicznymi. ny. Jest ona wydalana przez nerki w postaci niezmie-
nionej, a także w postaci związków hydroksylowa-
Zapotrzebowanie dzienne, wskazania, dawkowa- nych i innych metabolitów.
nie. Dzienne zapotrzebowanie na witaminę B1 zależy
od składu przyjmowanego pożywienia: węglowoda- Mechanizm działania i znaczenie fizjologiczne.
ny i alkohol zwiększają zapotrzebowanie, natomiast Po wchłonięciu się z jelita cienkiego witamina B2
tłuszcze zmniejszają. Przy przyjmowaniu pożywienia ulega fosforylacji w błonie śluzowej jelit z wytwo-
o wyrównanym składzie uważa się, że wystarczającą rzeniem ryboflawiny-kwasu 5-fosforowego (FMN
ilość stanowi 1–2 mg/dzień. = mononukleotydu flawinowego). Mononukleotyd
OH NH2 H O
HOH2C OH HOH2C OH HOH2C OH
N CH3 N CH3 N CH3
pirydoksol pirydoksamina pirydoksal
MUTSCHLER-2009.indd 809 2010-01-07 22:14:53
810 Witaminy
flawinowy oraz dinukleotyd flawinowo-adeninowy rydoksol, ze względu na to, że jest odporny na ciepło,
(FAD), powstający z FMN i adenozynomonofosfora- zasady i kwasy.
nu, są koenzymami enzymów flawinowych (flawopro-
tein). Nukleotydy flawinowe są niezbędne do przeno- Witamina B6 znajduje się we wszystkich żyjących komór-
szenia wodoru w łańcuchu oddechowym, odwodor- kach,
You might also like
- Aldehydy I Ketony - ReakcjeDocument3 pagesAldehydy I Ketony - Reakcjeprskt100% (2)
- Kwasy KarboksyloweDocument42 pagesKwasy KarboksyloweAndrew LondonNo ratings yet
- Bas I, IIDocument10 pagesBas I, IIdarNo ratings yet
- Literatura - Technik FarmaceutycznyDocument3 pagesLiteratura - Technik Farmaceutycznylellek122No ratings yet
- Chemia Leków - Krótkie Wykłady PDFDocument322 pagesChemia Leków - Krótkie Wykłady PDFkulenowaNo ratings yet
- Ndigczas017194 1937 005 006Document36 pagesNdigczas017194 1937 005 006Nilka Ok100% (1)
- Chemia Organiczna ReakcjeDocument37 pagesChemia Organiczna Reakcjeapi-3849705100% (5)
- aminy notatkiDocument20 pagesaminy notatkipotteromaniaNo ratings yet
- Aromatyczne Związki HeterocykliczneDocument61 pagesAromatyczne Związki HeterocykliczneKoralinaNo ratings yet
- Hydroksylowe Pochodne Węglowodorów - Alkohole I FenoleDocument6 pagesHydroksylowe Pochodne Węglowodorów - Alkohole I FenoleMarcelina GrycNo ratings yet
- Aminy PDFDocument8 pagesAminy PDFKlaudia MajorNo ratings yet
- AcylacjaDocument11 pagesAcylacjaradek.witek54321No ratings yet
- MATURA 2015 Wybrane Wzory I Stałe FizykochemiczneDocument1 pageMATURA 2015 Wybrane Wzory I Stałe Fizykochemicznecivada6531No ratings yet
- Rozdzial 20Document14 pagesRozdzial 20MarcinAdamskiNo ratings yet
- Httpscke - Gov.plimages EGZAMIN MATURALNY OD 2023informatorywybrane Wzory Stale Fizykochemiczne EM2023 PDFDocument24 pagesHttpscke - Gov.plimages EGZAMIN MATURALNY OD 2023informatorywybrane Wzory Stale Fizykochemiczne EM2023 PDFolgapisarska17No ratings yet
- 38 Addycja Nukleofilowa Do Alfa-beta Nienasyconych Aldehydów, Ketonów i EstrówDocument9 pages38 Addycja Nukleofilowa Do Alfa-beta Nienasyconych Aldehydów, Ketonów i EstrówlaaamorozecNo ratings yet
- 11 - Benzen, Związki AromatyczneDocument22 pages11 - Benzen, Związki AromatyczneW WojcikNo ratings yet
- ZolcienDocument1 pageZolcienRobert KamińskiNo ratings yet
- Aldehydy I Ketony PDFDocument26 pagesAldehydy I Ketony PDFKlaudia MajorNo ratings yet
- Tablice Fizyczne Chemiczne Biologiczne 2023Document24 pagesTablice Fizyczne Chemiczne Biologiczne 2023Farkov GamesNo ratings yet
- Nukleozydy Nukleotydy I Kwasy NukleinoweDocument52 pagesNukleozydy Nukleotydy I Kwasy NukleinoweJan CzarnoleskiNo ratings yet
- Alkohole I FenoleDocument8 pagesAlkohole I FenoleEwciaNo ratings yet
- Substytucja NukleofilowaDocument6 pagesSubstytucja NukleofilowaBeataGajewskaNo ratings yet
- Podsumowanie WeglowodoryDocument14 pagesPodsumowanie Weglowodorygrazynasroka774No ratings yet
- chemiaDocument1 pagechemiadomzalskaannaaNo ratings yet
- Aminy I AmidyDocument18 pagesAminy I AmidyZosia WalczakNo ratings yet
- EM Komunikat o Dostosowaniach 2022Document34 pagesEM Komunikat o Dostosowaniach 2022patrycja.ol777No ratings yet
- Wyklad 3Document102 pagesWyklad 3Andrew LondonNo ratings yet
- Odżywianie Się Roślin - Fotosynteza 4-3Document1 pageOdżywianie Się Roślin - Fotosynteza 4-3juliablacha4No ratings yet
- Chemia Organiczna - Ćwiczenia - x8Document34 pagesChemia Organiczna - Ćwiczenia - x8Tetiana BalandaNo ratings yet
- Materiały - Ćw. Ch. Org. Semestr 2 - Część VII-2 - Reakcje Charakterystyczne (Właściwości) Aldehydów I KetonówDocument5 pagesMateriały - Ćw. Ch. Org. Semestr 2 - Część VII-2 - Reakcje Charakterystyczne (Właściwości) Aldehydów I KetonówTetiana BalandaNo ratings yet
- Zadania treningowe - aldehydy i ketonyDocument2 pagesZadania treningowe - aldehydy i ketonyGabrysia JesiołkiewiczNo ratings yet
- ChemiaDocument1 pageChemiaNorbert TwarógNo ratings yet
- Wyklad 5Document18 pagesWyklad 5karolNo ratings yet
- Chemia Organiczna - Ćwiczenia - x7Document19 pagesChemia Organiczna - Ćwiczenia - x7Tetiana BalandaNo ratings yet
- AldehydyDocument3 pagesAldehydyNikola JasionekNo ratings yet
- Węglowodory Aromatyczne PDFDocument8 pagesWęglowodory Aromatyczne PDFKlaudia MajorNo ratings yet
- Httpwww Witu Mil Plwwwbiuletynzeszyty20070103p129Document8 pagesHttpwww Witu Mil Plwwwbiuletynzeszyty20070103p129Adrianna BielińskaNo ratings yet
- CW - 1 - Biochemia Właściwości Aminokwasów I BiałekDocument9 pagesCW - 1 - Biochemia Właściwości Aminokwasów I BiałekNatalia PiłatNo ratings yet
- Czy Fenol Jest Mocnym KwasemDocument27 pagesCzy Fenol Jest Mocnym Kwasemrweronika2006No ratings yet
- Estry (Gotowa)Document3 pagesEstry (Gotowa)Aleksandra WojciechowskaNo ratings yet
- Estry (Gotowa)Document3 pagesEstry (Gotowa)Aleksandra WojciechowskaNo ratings yet
- Pospolite Utleniacze I Reduktory - Docx-1Document1 pagePospolite Utleniacze I Reduktory - Docx-1paulinabrz08No ratings yet
- Fenol1 IDocument10 pagesFenol1 ICristian CiubotaruNo ratings yet
- Czy Sole Kwasow Karboksylowych HydrolizujaDocument20 pagesCzy Sole Kwasow Karboksylowych HydrolizujaJan KłosekNo ratings yet
- GlikolizaDocument1 pageGlikolizaAnna SzyllerNo ratings yet
- Podstawy Genetyki - Slajdy Z WykładówDocument22 pagesPodstawy Genetyki - Slajdy Z WykładówKarolina KardynałNo ratings yet
- 53 Ob Etap SzkolnyDocument28 pages53 Ob Etap SzkolnyZosia JarosNo ratings yet
- ODP Zadania treningowe - aldehydy i ketonyDocument3 pagesODP Zadania treningowe - aldehydy i ketonyGabrysia JesiołkiewiczNo ratings yet
- Alkohole (G)Document2 pagesAlkohole (G)Aleksandra WojciechowskaNo ratings yet
- Alkohole (G)Document2 pagesAlkohole (G)Aleksandra WojciechowskaNo ratings yet
- Aminokwasy - PrezentacjaDocument22 pagesAminokwasy - PrezentacjaAneta SochaczewskaNo ratings yet
- Węglowodory Aromatyczne (Cz.2 - Otrzymywanie I Właściwości)Document4 pagesWęglowodory Aromatyczne (Cz.2 - Otrzymywanie I Właściwości)Marcelina GrycNo ratings yet
- Otrzymywanie Soli Roznymi MetodamiDocument26 pagesOtrzymywanie Soli Roznymi Metodami76hcx7hrtgNo ratings yet
- Diagram Alir AsamDocument1 pageDiagram Alir AsamAgustin R. RerungNo ratings yet
- Chemia Organiczna - Ćwiczenia - x5Document49 pagesChemia Organiczna - Ćwiczenia - x5Tetiana BalandaNo ratings yet
- Chemia Organiczna - Ćwiczenia - x9Document50 pagesChemia Organiczna - Ćwiczenia - x9Tetiana BalandaNo ratings yet
- AlkanyDocument18 pagesAlkanyprsktNo ratings yet
- AminokwasyDocument23 pagesAminokwasydaevaauwuNo ratings yet
- Węglowodory - Podsumowanie AktualneDocument1 pageWęglowodory - Podsumowanie AktualneAlehjandor MatadorNo ratings yet
- Kalendarz 2024Document1 pageKalendarz 2024Inférus AdvenæNo ratings yet
- Test Próbny (Odpowiedzi)Document6 pagesTest Próbny (Odpowiedzi)Michał GlondysNo ratings yet
- Calki MetodyDocument9 pagesCalki MetodyJakub JęśkowiakNo ratings yet
- Przemysł I Energetyka - Test Klasa IIIDocument3 pagesPrzemysł I Energetyka - Test Klasa IIIMaciek DąbrowskiNo ratings yet