You might also like
- Biochemistry 1Document4 pagesBiochemistry 1Beatriz SilvaNo ratings yet
- New Indian Era: Biology Class 12 Maharashtra Board New Syllabus 2020 Chapter - 1 Part - 1Document53 pagesNew Indian Era: Biology Class 12 Maharashtra Board New Syllabus 2020 Chapter - 1 Part - 1Samar Mane100% (2)
- Cell CycleDocument11 pagesCell CycleewbjerywegnfsujyekwrhbnfwfNo ratings yet
- 10 Cell Cycle N Cell Division-NotesDocument4 pages10 Cell Cycle N Cell Division-NotesAnanth Dharanidharan100% (1)
- Chapter 10 BIO 1510Document25 pagesChapter 10 BIO 1510Chachi CNo ratings yet
- Cell DivisionDocument4 pagesCell DivisionMhariane MabborangNo ratings yet
- 10 Cell Cycle N Cell Division-NotesDocument4 pages10 Cell Cycle N Cell Division-NotesWrestling Communication100% (1)
- Control Points in The Cell CycleDocument3 pagesControl Points in The Cell CyclelibbyNo ratings yet
- LS 6-Cell-Cycle MitosisDocument20 pagesLS 6-Cell-Cycle MitosisseanNo ratings yet
- Genetic 1Document64 pagesGenetic 1Le Phuong LyNo ratings yet
- Guided By: DR - Jyoti Bhojwani Faculty, Genetics Davv, Indore Presented By: Avnish MishraDocument58 pagesGuided By: DR - Jyoti Bhojwani Faculty, Genetics Davv, Indore Presented By: Avnish MishraPRASHANT SOLANKINo ratings yet
- Gen Bio ReviewerDocument3 pagesGen Bio ReviewerJustin JaranillaNo ratings yet
- To Appreciate The Animations and Explanations, PLS. Download As A Power Point. ThanksDocument34 pagesTo Appreciate The Animations and Explanations, PLS. Download As A Power Point. ThanksPawpaw Chan0% (1)
- Bio LectureDocument4 pagesBio LectureAbegail TaarNo ratings yet
- What Is The Cell Cycle?Document21 pagesWhat Is The Cell Cycle?Aqsa YaminNo ratings yet
- Cell Cycle and Cell DivisionDocument7 pagesCell Cycle and Cell DivisionAicel AdoptanteNo ratings yet
- GENB4Document5 pagesGENB4Azeleah Nosil VilladiegoNo ratings yet
- Unit 1 - Cell DivDocument73 pagesUnit 1 - Cell DivMuhammad SwalihNo ratings yet
- The Cell CycleDocument33 pagesThe Cell CycleSean JonesNo ratings yet
- Lesson 6. Cell CycleDocument64 pagesLesson 6. Cell CycleSharon Nicole MasamocNo ratings yet
- W9 Mitosis Chapter11Document26 pagesW9 Mitosis Chapter11Ibrahim AlibrahimNo ratings yet
- How Cells DivideDocument53 pagesHow Cells Dividesimelanepamela0No ratings yet
- Mitosis SLDocument5 pagesMitosis SLYuvraj GuptaNo ratings yet
- Chapter 6 Cell DivisionDocument51 pagesChapter 6 Cell DivisionAA ProductionNo ratings yet
- Mitosis: Division Is Also MultiplicationDocument37 pagesMitosis: Division Is Also MultiplicationAldrich ArellanoNo ratings yet
- Cell Cycle and Cell Division: (With Revision Tracking)Document5 pagesCell Cycle and Cell Division: (With Revision Tracking)SAMPATH SPNo ratings yet
- Block 1 Lecture 2 Cell CycleDocument34 pagesBlock 1 Lecture 2 Cell CycleNahome YebassewNo ratings yet
- Cell Cycle and MitosisDocument16 pagesCell Cycle and MitosisElah TapangNo ratings yet
- Cellular ReproductionDocument24 pagesCellular Reproductionapi-292966101No ratings yet
- E-Tutor Session Theme10 31mayDocument14 pagesE-Tutor Session Theme10 31mayOmogau KekanaNo ratings yet
- Lab MitosisDocument4 pagesLab Mitosisnowlik56100% (7)
- Concept 3 Notes - Cell Cycle and CancerDocument26 pagesConcept 3 Notes - Cell Cycle and Cancerlolosargent05No ratings yet
- Topic 2 CellsDocument1 pageTopic 2 CellsKandy Anne Castillo AbuanNo ratings yet
- Mitosis: Samuel C. BrilloDocument20 pagesMitosis: Samuel C. BrilloSam BrilloNo ratings yet
- Gen-Bio-Reviewer-Padua-First GradingDocument4 pagesGen-Bio-Reviewer-Padua-First Gradingmailforprinting101No ratings yet
- Genetic Dna Replication Mitososis MeiosisiDocument54 pagesGenetic Dna Replication Mitososis Meiosisiptm2409tempNo ratings yet
- Lesson 7 MitosisDocument36 pagesLesson 7 MitosisMobileLegends SbNo ratings yet
- Cell CycleDocument46 pagesCell Cyclehobiforlife 22No ratings yet
- Cell CycleMitosis - ElisaDocument40 pagesCell CycleMitosis - ElisabenjaNo ratings yet
- General Biology Reviewer 2nd QRTRDocument6 pagesGeneral Biology Reviewer 2nd QRTRAlfer JaoNo ratings yet
- Lesson 6Document4 pagesLesson 6Thea MillanesNo ratings yet
- POSTGRADUATE TEACHING DEPARTMENT OF ZOOLOGY PPT NewDocument18 pagesPOSTGRADUATE TEACHING DEPARTMENT OF ZOOLOGY PPT NewChetan MasramNo ratings yet
- Cell Cycle & MitosisDocument42 pagesCell Cycle & Mitosismariam yahiaNo ratings yet
- Mitosis and Meiosis: Cell DivisionDocument36 pagesMitosis and Meiosis: Cell DivisionHeart VidalNo ratings yet
- 6 Cell Division Cell Cycle 1633188130443Document25 pages6 Cell Division Cell Cycle 1633188130443Utkarsh AnandNo ratings yet
- Mitosis and Cell CycleDocument43 pagesMitosis and Cell CycleKavindu MunasingheNo ratings yet
- Cell Cycle and Cell DivisionDocument5 pagesCell Cycle and Cell DivisionParth GoyalNo ratings yet
- Cell Cycle - RSJ - 2020 - FINALDocument53 pagesCell Cycle - RSJ - 2020 - FINALArmy JmNo ratings yet
- L1 - Cell Cycle and MitosisDocument39 pagesL1 - Cell Cycle and MitosisTahmina MaryamNo ratings yet
- L 11 MeiosisDocument39 pagesL 11 MeiosissNo ratings yet
- Mitosis/Meiosis: How Cells Reproduce: Exercise IDocument10 pagesMitosis/Meiosis: How Cells Reproduce: Exercise ITAHA GABRNo ratings yet
- 10-Cell Cycle & DNA ReplicationDocument34 pages10-Cell Cycle & DNA Replicationwhether913No ratings yet
- 3 Cell Cycle and Mitosis Student VersionDocument35 pages3 Cell Cycle and Mitosis Student VersionIshu SainiNo ratings yet
- Lab Cell Division - Quinton WinekDocument10 pagesLab Cell Division - Quinton Winekapi-345219048No ratings yet
- Genetics - Chapter 2 - Mitosis and Meiosis-Cellular ReproductionDocument37 pagesGenetics - Chapter 2 - Mitosis and Meiosis-Cellular ReproductionDuy AnhNo ratings yet
- Cell Cycle: Histology Lab Mitosis and Meiosis 02/10/2021Document8 pagesCell Cycle: Histology Lab Mitosis and Meiosis 02/10/2021Danielle Anne Zamora-Matillosa LambanNo ratings yet
- The Cell Cycle and Cell Division: I G N MayunDocument39 pagesThe Cell Cycle and Cell Division: I G N Mayunigus696No ratings yet
- Cell Cycle and Cell Division - Vibares, Marquez,& Binavente - 12 MendeleevDocument23 pagesCell Cycle and Cell Division - Vibares, Marquez,& Binavente - 12 MendeleevMary Joy Purisima BinaventeNo ratings yet
- Chapter 9Document14 pagesChapter 9alaskamofoNo ratings yet
- The Importance of Cell Division: Grow and ReproduceDocument50 pagesThe Importance of Cell Division: Grow and ReproduceRidhima AroraNo ratings yet
- Human Chromosomes: An Illustrated Introduction to Human CytogeneticsFrom EverandHuman Chromosomes: An Illustrated Introduction to Human CytogeneticsRating: 5 out of 5 stars5/5 (1)
- Gene Editing, Epigenetic, Cloning and TherapyFrom EverandGene Editing, Epigenetic, Cloning and TherapyRating: 4 out of 5 stars4/5 (1)
- Bio 200 Spring 2015 SyllabusDocument3 pagesBio 200 Spring 2015 SyllabusEllen YinNo ratings yet
- Importance of Computer Education EssayDocument7 pagesImportance of Computer Education Essayeikmujnbf100% (2)
- Bougainvillea Glabra: Clonal Propagation of 'Magnifica' Through Shoot Apex CultureDocument6 pagesBougainvillea Glabra: Clonal Propagation of 'Magnifica' Through Shoot Apex CultureHuy NguyễnNo ratings yet
- Onion Root Mitosis LabDocument6 pagesOnion Root Mitosis Labapi-2469736100% (1)
- Science q4 ModuleDocument40 pagesScience q4 ModuleKesh LisabaNo ratings yet
- Adobe Scan Feb 25, 2024Document3 pagesAdobe Scan Feb 25, 2024mohitsorengts09No ratings yet
- Sexual Reproduction in Flowering PlantDocument92 pagesSexual Reproduction in Flowering PlantyerhERSNo ratings yet
- Genetics Growth and Development QuizDocument1 pageGenetics Growth and Development Quizapi-368213959No ratings yet
- Inheritance: IGCSE Biology (O610) WorkbookDocument16 pagesInheritance: IGCSE Biology (O610) WorkbookAnandNo ratings yet
- N20 H2 P2 Answers-1Document10 pagesN20 H2 P2 Answers-1Samuel TeohNo ratings yet
- Summative Test 2 Grade 8 Quarter 4Document4 pagesSummative Test 2 Grade 8 Quarter 4Anthony IlustreNo ratings yet
- Mol Bio SyllabusDocument2 pagesMol Bio SyllabusGandhiraj VNo ratings yet
- Botany - RespirationDocument17 pagesBotany - RespirationAira TanganNo ratings yet
- 1 3 2 1 3Document15 pages1 3 2 1 3Himanshu GuptaNo ratings yet
- Oxidative PhosphorylationDocument8 pagesOxidative PhosphorylationAISYAH NABILAH BINTI RAMLAN / UPMNo ratings yet
- DLP MitosisDocument6 pagesDLP MitosisReymark De Ocampo100% (1)
- Embryology Synopsis-Panineeya DemoDocument13 pagesEmbryology Synopsis-Panineeya DemoDr P N N ReddyNo ratings yet
- Human Reproduction PortfolioDocument19 pagesHuman Reproduction PortfolioIgnacio FelicityNo ratings yet
- Chapter 2 Sexual Reproduction in Flowering PlantsDocument10 pagesChapter 2 Sexual Reproduction in Flowering PlantsSuvidNo ratings yet
- Proses Perkembangan Embrio Pada Tahapan CleavageDocument43 pagesProses Perkembangan Embrio Pada Tahapan CleavageAnton TubagusNo ratings yet
- WBI02 01 MSC 20190307 PDFDocument25 pagesWBI02 01 MSC 20190307 PDFRadhiyaahNo ratings yet
- Kami Export - Cell Cycle Notes and VocabDocument5 pagesKami Export - Cell Cycle Notes and VocabVALERIA ACUNA VILLEGASNo ratings yet
- Disorders and Diseases That Result From The Malfunction of The Cell During The Cell Cycle NotesDocument3 pagesDisorders and Diseases That Result From The Malfunction of The Cell During The Cell Cycle NotesMischi Jeda ElumbaNo ratings yet
- G8 SCIENCE Learning Module Grade 8 1st QuarterDocument35 pagesG8 SCIENCE Learning Module Grade 8 1st QuarterMelanie TrinidadNo ratings yet
- Test Bank For Essentials of Genetics 9th Edition William S Klug Michael R Cummings Charlotte A Spencer Michael A PalladinoDocument13 pagesTest Bank For Essentials of Genetics 9th Edition William S Klug Michael R Cummings Charlotte A Spencer Michael A Palladinominhkhoit8cua3No ratings yet
- Biology The Essentials 1st Edition Hoefnagels Test BankDocument40 pagesBiology The Essentials 1st Edition Hoefnagels Test Bankblanchetoj0fg100% (26)
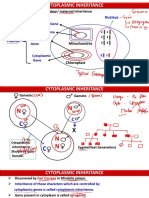
- It Is Also Called Extranuclear/ Maternal Inheritance: NucleusDocument9 pagesIt Is Also Called Extranuclear/ Maternal Inheritance: NucleusAyush bhadaniNo ratings yet
- Differences Between Mitosis and MeiosisDocument4 pagesDifferences Between Mitosis and MeiosisMeri SunderNo ratings yet